

SUMMARY
Retrograde signaling systems are fundamental modes of communication that synapses utilize to dynamically and adaptively modulate activity. However, the inductive mechanisms that gate retrograde communication in the postsynaptic compartment remain enigmatic. We have investigated retrograde signaling at the Drosophila neuromuscular junction, where three seemingly disparate perturbations to the postsynaptic cell trigger a similar enhancement in presynaptic neurotransmitter release. We show that the same presynaptic genetic machinery and enhancements in active zone structure are utilized by each inductive pathway. However, all three induction mechanisms differ in temporal, translational, and CamKII activity requirements to initiate retrograde signaling in the postsynaptic cell. Intriguingly, pharmacological blockade of postsynaptic glutamate receptors, and not calcium influx through these receptors, is necessary and sufficient to induce rapid retrograde homeostatic signaling through CamKII. Thus, three distinct induction mechanisms converge on the same retrograde signaling system to drive the homeostatic strengthening of presynaptic neurotransmitter release.
ETOC BLURB
Retrograde signaling systems stabilize neurotransmission at synapses. Goel et al. find that disparate inductive processes in the postsynaptic compartment ultimately converge on a unitary retrograde signaling system to homeostatically modulate presynaptic neurotransmitter release. This might ensure coherent trans-synaptic communication while enabling adaptations to diverse postsynaptic challenges to excitability.
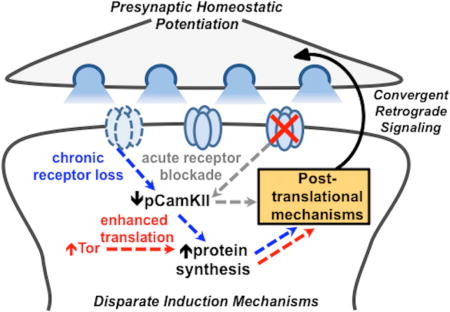
INTRODUCTION
Retrograde feedback systems operate over distinct spatial and temporal scales to stabilize synaptic strength. These synaptic signaling systems are triggered following perturbations to activity in the postsynaptic cell, where retrograde feedback adjusts presynaptic efficacy, tuning neurotransmitter release to potentiate or inhibit transmission in diverse organisms (Regehr et al., 2009). The induction and expression of retrograde signaling can occur over varying time scales, and can exhibit distinct dependencies on gene transcription, protein synthesis, and post-translational modifications (Jakawich et al., 2010). How specific induction mechanisms are integrated and coordinated to ultimately enable precise and adaptive modulations to presynaptic function remains enigmatic.
The Drosophila neuromuscular junction (NMJ) is an established system to study retrograde synaptic signaling. At this model glutamatergic synapse, genetic or pharmacological perturbations to postsynaptic receptor functionality triggers retrograde signaling that instructs the neuron to precisely increase presynaptic neurotransmitter release, maintaining stable levels of synaptic strength (Frank, 2013). This process is termed presynaptic homeostatic potentiation (PHP), and can be induced through two distinct disruptions to postsynaptic glutamate receptor functionality. First, acute pharmacological blockade of receptors reduces miniature EPSP (mEPSP) amplitude, initiating the rapid expression of PHP (increase in quantal content) within 10 mins (Frank et al., 2006). Second, genetic loss of the postsynaptic glutamate receptor subunit GluRIIA leads to a similar reduction in mEPSP amplitudes over chronic time scales (days), and a similar expression of PHP (Petersen et al., 1997). Although these perturbations each disrupt receptors and lead to adaptive increases in presynaptic neurotransmitter release, PhTx- and GluRIIA-mediated PHP signaling exhibit important differences. First, some genes have been identified that are only necessary for GluRIIA-dependent PHP expression, while PHP is robustly expressed following acute PhTx application in larvae with mutations in these genes (Frank et al., 2009; Kauwe et al., 2016; Penney et al., 2016; Spring et al., 2016; Tsurudome et al., 2010). In addition, PhTx-induced PHP expression is translation-independent (Frank et al., 2006), while GluRIIA-induced PHP is blocked by inhibitions to postsynaptic translation through loss of the translational regulator Target of Rapamycin (Tor) (Kauwe et al., 2016; Penney et al., 2012). Although several genes and mechanisms necessary for the expression of PHP in the presynaptic neuron have been identified (Dickman and Davis, 2009; Frank, 2013; Kiragasi et al., 2017), far less is known about the mechanistic differences in postsynaptic transduction between PhTx- and GluRIIA-induced PHP signaling.
Recently, a novel manipulation to the postsynaptic muscle that does not affect glutamate receptors was demonstrated to induce retrograde PHP signaling at the Drosophila NMJ. This was accomplished by postsynaptic overexpression of the non-specific translational regulator Tor (Tor-OE) (Penney et al., 2012), which leads to a chronic, global increase in muscle protein synthesis (Chen and Dickman, 2017). While Tor-OE does not functionally impact glutamate receptors, somehow the increased muscle protein synthesis is converted into an instructive retrograde signal that appears to induce an enhancement in presynaptic glutamate release of comparable magnitude to that observed in PhTx- and GluRIIA-mediated PHP (Penney et al., 2012). Although PhTx application, loss of GluRIIA, and Tor-OE each induce a similar enhancement in presynaptic release, to what extent they utilize separate or shared postsynaptic induction pathways, retrograde signaling systems, and modulations to presynaptic function is not known.
Here, we have characterized PHP signaling and expression when induced through PhTx application, loss of GluRIIA, and Tor-OE. This analysis has revealed that a common retrograde signaling system drives similar homeostatic adaptations in the presynaptic terminal, but that separate inductive pathways differentially respond to glutamate receptor perturbation, Ca2+/calmodulin-dependent protein kinase II (CamKII) activity, and protein synthesis.
RESULTS
Presynaptic potentiation driven by postsynaptic Tor-OE cannot be further potentiated by acute receptor blockade
To determine whether separate or shared retrograde signaling systems operate at the Drosophila NMJ, we induced and expressed PHP using three distinct perturbations to the postsynaptic cell. The control condition was wild type (WT; w1118), the isozygous genetic background in which other genotypes were bred in this study. First, we acutely induced PHP by application of philanthotoxin-433 (PhTx), an irreversible non-competitive antagonist of postsynaptic glutamate receptors (Figure 1A; (Frank et al., 2006)). This led to the expected reduction in postsynaptic mEPSP amplitude, but EPSP amplitude was maintained at wild-type levels because of a homeostatic increase in presynaptic release (quantal content; Figure 1A–1E). Second, to induce chronic PHP expression, we utilized a genetic mutation in the GluRIIA subunit (GluRIIA), which exhibits a reduction in mEPSP amplitude over days similar to acute PhTx application, and also a similar increase in quantal content that maintains the stable EPSP amplitude (Figure 1A–1E). Third, we overexpressed the translational regulator Tor in the postsynaptic muscle (Tor-OE). As expected, Tor-OE does not change mEPSP amplitude but leads to a retrograde potentiation in presynaptic release, enhancing EPSP amplitude (Figure 1A–1E; (Penney et al., 2012)). Thus, three perturbations to the postsynaptic muscle - PhTx application (acute pharmacological receptor blockade), GluRIIA mutation (chronic absence of a receptor subunit), and Tor-OE (chronic elevation in postsynaptic protein synthesis) - are sufficient to individually drive similar enhancements in presynaptic release.
Figure 1. Acute receptor perturbation fails to trigger additional neurotransmitter release following PHP induction by Tor-OE.
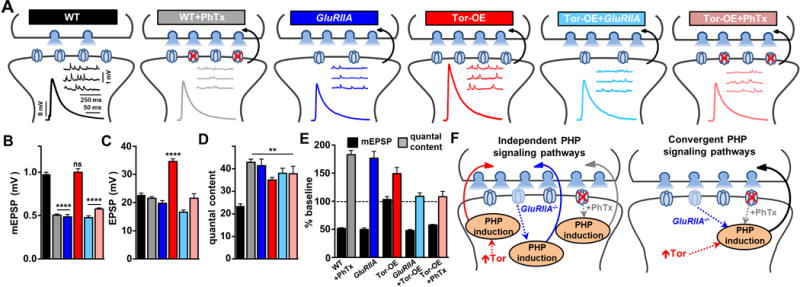
(A) Schematic depicting pharmacological and genetic manipulations to the Drosophila NMJ that induce a retrograde increase in presynaptic release. WT synapses (w1118) serve as the baseline control condition. Presynaptic homeostatic potentiation (PHP) is observed when mEPSP amplitudes are reduced due to acute blockade of postsynaptic glutamate receptors following PhTx application (WT+PhTx) or from chronic loss of receptors due to mutations in GluRIIA (GluRIIA: w;GluRIIASP16). Postsynaptic overexpression of Tor (Tor-OE: w;UAS-Tor/MHC-Gal4) results in an increase in presynaptic neurotransmitter release independently of receptor perturbation. No additional increase in release is observed in Tor-OE when combined with GluRIIA mutations (GluRIIA+Tor-OE: w; GluRIIASP16; UAS-Tor/MHC-Gal4) or PhTx application (Tor-OE+PhTx). Representative traces of electrophysiological recordings (mEPSP and EPSP) are shown in the indicated manipulations. Quantification of average mEPSP amplitude (B), EPSP amplitude (C), and quantal content (presynaptic release) (D) is shown for each genotype. (E) mEPSP amplitude and quantal content for the indicated genotypes are normalized as a percentage of baseline values. Note that WT is the baseline for all conditions except Tor-OE+GluRIIA and Tor-OE+PhTx, which are normalized to Tor-OE to demonstrate that no additional increase in quantal content is observed. (F) Schematic illustrating distinct PHP induction mechanisms ultimately converge on the same retrograde signaling system to increase presynaptic neurotransmitter release.
We next determined whether an additional enhancement in presynaptic release could be expressed when two of these manipulations are combined at a single synapse. An additional increase in release would suggest that separate signaling pathways independently modulate presynaptic function (schematized in Figure 1F). A previous report demonstrated that overexpression of Tor in GluRIIA mutants fails to further potentiate release (Penney et al., 2012), a result that we also confirmed (Tor-OE+GluRIIA; Figure 1A–1E). This is consistent with GluRIIA and Tor-OE each requiring Tor-dependent modulations in protein synthesis. In contrast, PhTx was reported to induce PHP expression independently of translation (Frank et al., 2006), suggesting that the acute induction of PHP may operate through a distinct signaling system. However, application of PhTx to GluRIIA-mutant synapses fails to further reduce mEPSP amplitude (Frank et al., 2006), likely because GluRIIA-containing receptors are specifically targeted by PhTx (Kiragasi et al., 2017). Although we could not test this hypothesis by applying PhTx to GluRIIA-mutant synapses, we did apply PhTx to Tor-OE NMJs. PhTx application resulted in reduced mEPSP amplitude at Tor-OE NMJs (Tor-OE+PhTx; Figure 1A and 1B), but no change in presynaptic release was observed (Figure 1E). This demonstrates that synapses potentiated by Tor-OE are incapable of further potentiating presynaptic neurotransmitter release, even when glutamate receptors are acutely perturbed, which normally induces a translation-independent form of PHP signaling.
Together, these results suggest two possibilities: First, Tor-OE induces a potentiated synapse that is operating at a maximally elevated state through a novel mechanism, and therefore occludes further potentiation by conventional PHP signaling through loss of GluRIIA or PhTx application (Figure 1F). Alternatively, Tor-OE, GluRIIA, and PhTx each ultimately converge to drive the same retrograde signaling system and trigger modulations that enable PHP expression in the presynaptic neuron (Figure 1F). We went on to distinguish between these models.
PhTx, GluRIIA, and Tor-OE homeostatically enhance presynaptic active zone structure
We considered the possibility that Tor-OE transforms the presynaptic neuron into a novel potentiated state, distinct from the shared PHP expression mechanisms that have been defined for PhTx application and GluRIIA mutants (Muller and Davis, 2012; Weyhersmuller et al., 2011). It was previously shown that the presynaptic active zone, labeled by the scaffold Bruchpilot (BRP), is enhanced following PHP induction by either PhTx or loss of GluRIIA (Weyhersmuller et al., 2011), interpreted to be a biomarker for potentiated synaptic strength in the presynaptic terminal. Indeed, other studies have shown that increased BRP levels strengthen individual release sites by changing release probability through adaptive increases in calcium influx and the readily releasable vesicle pool (Tsurudome et al., 2010). Consistently, both calcium influx and the readily releasable vesicle pool are increased after PHP induction by PhTx or GluRIIA (Kiragasi et al., 2017; Muller and Davis, 2012). However, there is no precedent for a nonspecific increase in postsynaptic protein synthesis to induce active zone remodeling, so we tested whether Tor-OE led to any changes in synaptic growth or BRP levels indicative of or distinct from conventional PHP induction. We found no significant differences in synaptic growth or morphology following immunostaining of the NMJ in PhTx, GluRIIA, and Tor-OE conditions (Figure 2A and 2B), consistent with previous studies (Frank et al., 2006; Penney et al., 2012). Further, we found no significant changes in BRP puncta number or density, consistent with unperturbed synaptic growth (Figure 2A, 2C, and 2D), and indicating that the potentiation in presynaptic release observed in PhTx, GluRIIA, and Tor-OE does not result from the recruitment of additional BRP-positive active zones.
Figure 2. PHP induction by PhTx, GluRIIA, and Tor-OE leads to similar enhancements in presynaptic active zone structure.
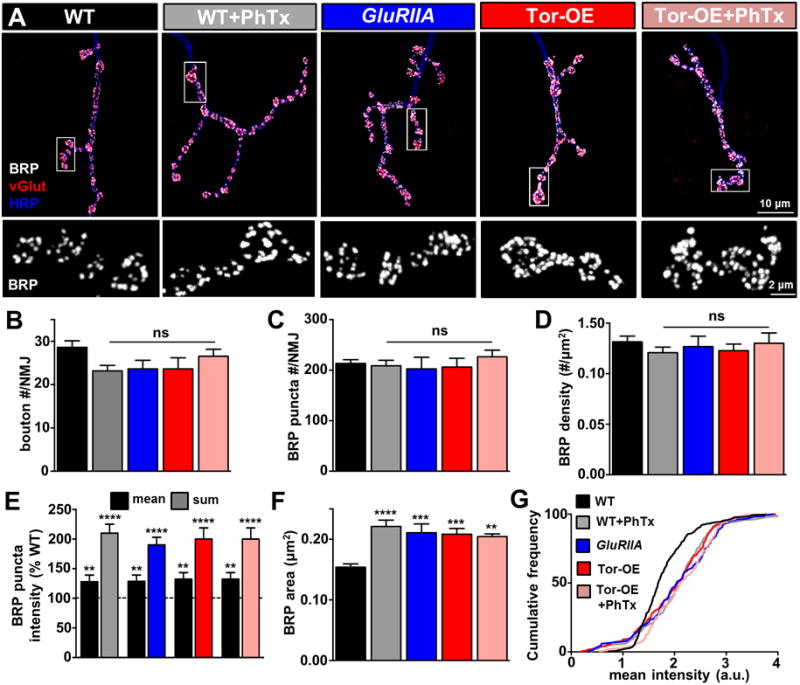
(A) Representative images of NMJs immunostained with antibodies that recognize the presynaptic active zone scaffold Bruchpilot (BRP; white), the synaptic vesicle marker vGlut (red), and the neuronal membrane (HRP; blue). Lower panels: representative images of BRP immunostaining at individual boutons, demonstrating an increase in intensity and size of individual puncta. No significant changes in total bouton number (B), BRP puncta per NMJ (C), or BRP density (D) are observed. Quantification of mean and sum fluorescence intensity of individual BRP puncta (E) and size of individual BRP puncta (F) reveals increased BRP levels at individual active zones following PHP induction by PhTx, GluRIIA, and Tor-OE. (G) Cumulative frequency distribution of BRP puncta mean intensity shows a significant rightward shift, indicative of an overall increase in mean intensity in all conditions compared to wild-type NMJs. No additional increase in BRP area or intensity is observed in Tor-OE+PhTx.
We next confirmed the previously reported increase in BRP intensity at NMJs potentiated by PhTx application and in GluRIIA mutants. We observed an increase in the average mean BRP fluorescence intensity at individual active zones, as well as an apparent increase in the size of individual BRP puncta (Figure 2A and 2E–2G). Together, this remodeling of active zones induced by retrograde PHP signaling led to a large enhancement in the sum intensity of individual BRP puncta (Figure 2E and Table S1). Importantly, we found an increase in both the intensity and size of individual BRP puncta at synapses potentiated by Tor-OE that was indistinguishable from PhTx- and GluRIIA-mediated PHP conditions (Figure 2A and 2E–2G). Finally, we observed no additional increase in BRP intensity following PhTx application to Tor-OE (Figure 2A and 2E–2G), consistent with Tor-OE terminals not being further potentiated by glutamate receptor perturbation. Thus, Tor-OE, PhTx, and GluRIIA induces identical increases in both presynaptic release and active zone remodeling, consistent with a common retrograde homeostatic signaling system being induced by each of these perturbations to the postsynaptic muscle.
Postsynaptic overexpression of Tor requires presynaptic dysbindin to potentiate release
Although the results thus far demonstrate that Tor-OE induces retrograde enhancements in presynaptic release and active zone intensity that cannot be further potentiated when combined with homeostatic challenges to postsynaptic glutamate receptors, it is possible that Tor-OE happens to potentiate presynaptic terminals in similar ways to conventional PHP but through a completely independent mechanism. If this were the case, then core genes needed for the specific expression of PHP, but not for baseline neurotransmission, should not be required for the enhancement in presynaptic release induced by Tor-OE.
The schizophrenia susceptibility gene dysbindin (dysb) is necessary in motor neurons for the expression of PHP following both PhTx application and loss of GluRIIA (Dickman and Davis, 2009). Further, dysb has no known roles in active zone structure or remodeling, which we confirmed (Table S1). PHP expression was blocked in dysb mutants following PhTx application (dysb+PhTx) and when combined with loss of GluRIIA (dysb+GluRIIA; Figure 3A–3E), as expected. Importantly, loss of dysb in larvae overexpressing Tor in the muscle (dysb+Tor-OE) exhibited no change in mEPSP amplitude, but EPSP amplitudes were now reduced to wild type levels due to a failure to potentiate presynaptic release that was normally induced by Tor-OE (Figure 3A–3E). This demonstrates that Tor-OE in the muscle drives a retrograde potentiation in presynaptic function, enhances active zone remodeling, and utilizes the same genetic mechanisms necessary for conventional PhTx and GluRIIA-mediated PHP expression. Thus, PhTx, GluRIIA, and Tor-OE trigger the same retrograde signaling system that converges upstream of dysb to ultimately drive the homeostatic enhancement in presynaptic release.
Figure 3. dysbindin is required for Tor-OE-mediated PHP expression.
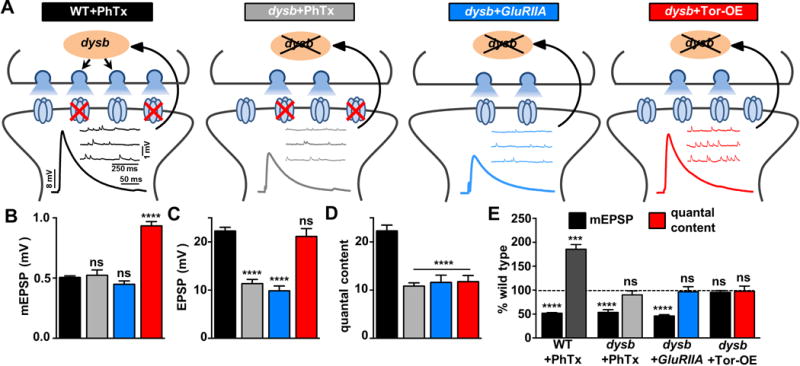
(A) Schematic illustrating the requirement of dysbindin (dysb: w; dysb1), necessary in motor neurons, to enhance presynaptic neurotransmitter release following PHP induction. PHP fails to be expressed following application of PhTx to dysb mutants (dysb+PhTx), loss of GluRIIA in dysb mutants (dysb+GluRIIA: w; GluRIIASP16; dysb1), and overexpression of Tor in dysb mutants (dysb+Tor-OE: w; G14-Gal4/UAS-Tor; dysb1). Representative traces of mEPSP and EPSP recordings in the indicated conditions are shown. Quantification of mEPSP amplitude (B), EPSP amplitude (C), and quantal content (D) in the indicated conditions. Although mEPSP amplitude is reduced in dysb+PhTx and dysb+GluRIIA, there is no homeostatic increase in quantal content, leading to reduced EPSP amplitude. Tor-OE, which normally leads to elevated EPSP amplitude, fails to increase EPSP amplitude when combined with dysb mutations (dysb+Tor-OE). (E) Quantification of mEPSP amplitude and quantal content, normalized to wild type values, demonstrating a failure to express PHP in PhTx, GluRIIA, and Tor-OE when combined with loss of dysb.
PHP induction has distinct dependencies on protein synthesis, CamKII activity, and extracellular calcium
Given the apparent common signaling system underlying PHP expression shared between PhTx, GluRIIA, and Tor-OE conditions, we sought to distinguish between the postsynaptic induction mechanisms. One obvious distinction is the involvement of postsynaptic translation. While PhTx application was reported to induce PHP expression independently of new protein synthesis (Frank et al., 2006), Tor overexpression involves the activation of protein synthesis through cap-dependent mechanisms, and GluRIIA mutants were reported to require new protein synthesis through the Tor pathway (Penney et al., 2012). We first confirmed this previously published work, finding that application of the protein synthesis inhibitor cycloheximide (CHX) has no impact on PHP expression following PhTx application (Figure 4A and 4B), while loss of one copy of Tor in a GluRIIA mutant background (GluRIIA+Tor+/−) did indeed disrupt the expression of PHP (Figure 4C). However, PHP was robustly expressed following application of PhTx to heterozygous Tor mutants (Figure 4C), further underscoring the differential dependency on Tor signaling between PhTx- and GluRIIA-dependent PHP signaling. We next asked whether acutely disrupting protein synthesis by CHX application was sufficient to block PHP expression in GluRIIA mutants and Tor-OE. We applied CHX to both preparations for 20 mins and then recorded. PHP was still robustly expressed in both GluRIIA mutants and Tor-OE despite the block in new protein synthesis (Figure 4A and 4B), demonstrating that ongoing protein synthesis is not required to maintain PHP expression in GluRIIA and Tor-OE. Together, this suggests that although new protein synthesis appears to be involved in GluRIIA and Tor-OE-dependent PHP signaling, post-translational mechanisms are sufficient to initiate and maintain PHP expression.
Figure 4. Protein synthesis, CamKII activity, and glutamate receptor blockade distinguish PHP induction mechanisms.
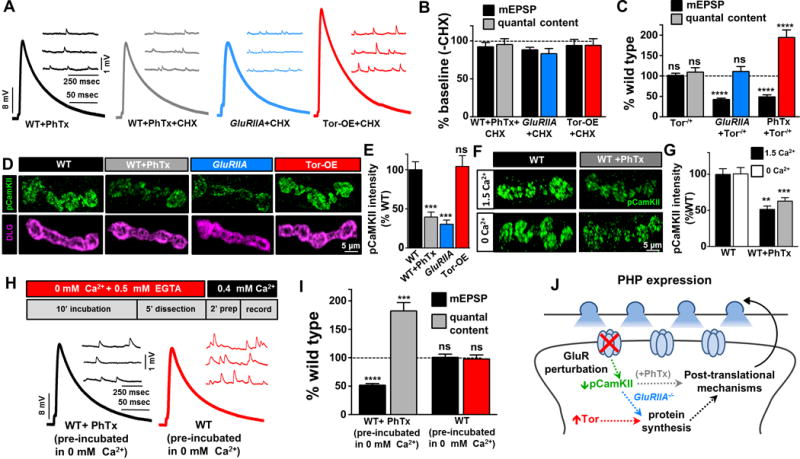
(A) Representative electrophysiological traces of NMJ recordings after pre-incubation in cyclohexamide (CHX) for 20 mins in PhTx (WT+PhTx+CHX), GluRIIA mutant (GluRIIA+CHX), or Tor-OE (Tor-OE+CHX) conditions. (B) Quantification of mEPSP amplitude and quantal content values, normalized to baseline values (−CHX), reveals that acute inhibition of protein synthesis has no effect on PHP expression. (C) Loss of one copy of Tor (Tor−/+: w;torΔP/+) does not impact synaptic physiology, but disrupts PHP expression when combined with GluRIIA mutations (GluRIIA+Tor−/+: w;GluRIIASP16, TorΔP/GluRIIASP16). PHP is robustly expressed in Tor−/+ following PhTx application (Tor−/++PhTx), demonstrating a differential requirement for protein synthesis between PhTx, GluRIIA, and Tor-OE-mediated PHP induction. (D) Representative images of NMJs immunostained with antibodies that recognize the active (phosphorylated) form of CamKII (pCamKII; green) and the postsynaptic scaffold Discs Large (DLG; magenta). (E) Quantification of total pCamKII intensity per muscle 6/7 NMJ reveals a reduction at synapses treated with PhTx or loss of GluRIIA. However, no change in pCamKII levels are observed in Tor-OE. (F–G) Pharmacological perturbation of postsynaptic glutamate receptors is sufficient to acutely reduce pCamKII levels independently of extracellular Ca2+. PhTx was incubated for 10 mins in the presence or absence of 1.5 mM extracellular calcium, and pCamKII levels were quantified. (H–I) Acute incubation in 0 mM extracellular Ca2+ saline does not induce PHP expression. NMJs were incubated in 0 mM calcium + 0.5 mM EGTA saline for 10 mins, and exposed to calcium only in the final 2 mins before recording. While PhTx application in this condition induced robust PHP expression, no change was observed in NMJs incubated without PhTx. (J) Model illustrating the differences in postsynaptic induction mechanisms driving retrograde homeostatic signaling.
We next considered possible post-translational mechanisms that may contribute to PHP induction in the postsynaptic muscle. Genetic evidence has suggested that a reduction in postsynaptic CamKII activity is involved in GluRIIA-dependent PHP signaling (Haghighi et al., 2003). In particular, postsynaptic overexpression of a constitutively active form of CamKII blocks PHP expression in GluRIIA mutants. More recently, a local reduction in phosphorylated (active) CamKII (pCamKII) in the postsynaptic compartment was observed in GluRIIA mutants (Newman et al., 2017). However, pCamKII levels have never been examined in the context of PhTx or Tor-OE forms of PHP signaling. We first confirmed a reduction in pCamKII immunostaining in the postsynaptic compartment of GluRIIA mutants compared to wild type (Figure 4D and 4E). Next, we found a similar decrease in pCamKII following 10 min application of PhTx to wild-type NMJs, but no significant change in pCamKII levels in Tor-OE NMJs (Figure 4D and 4E). This demonstrates that a reduction in pCamKII correlates with the induction of PHP signaling following glutamate receptor loss or pharmacological perturbation, but that a non-specific elevation in postsynaptic protein synthesis through Tor-OE can activate PHP signaling independently of a change in pCamKII levels.
CamKII is responsive to local calcium signaling (Hell, 2014), so we next turned our attention to the role of calcium in the postsynaptic muscle during PHP induction. The obvious hypothesis is that a reduction in postsynaptic calcium influx through GluRIIA-containing receptors, triggered by PhTx application or loss of GluRIIA, leads to reduced CamKII activity and initiates PHP signaling. Given that an acute 10 min application of PhTx is sufficient to both reduce pCamKII levels and initiate PHP signaling, we next probed the role of extracellular calcium in this process. First, we incubated wild-type NMJs in the presence or absence of PhTx, as well as in 0 mM or 1.5 mM extracellular calcium concentrations, followed by immunostaining of pCamKII as a readout of activity. Surprisingly, we found that PhTx application was itself sufficient to induce the reduction in pCamKII, independently of extracellular calcium (Figure 4F and 4G). This demonstrates that a pharmacological perturbation to postsynaptic glutamate receptors per se, and not an acute reduction in postsynaptic calcium influx from extracellular sources, is necessary and sufficient to diminish pCamKII levels over rapid time scales.
We next investigated calcium influx into the postsynaptic compartment during miniature activity at the Drosophila NMJ. mEPSP activity alone over 10 mins is sufficient to induce PHP signaling and expression (Frank et al., 2006), and GluRIIA-containing receptors are calcium permeable. However, we found that neither mEPSP amplitude nor frequency is significantly changed when calcium is absent or calcium concentrations are elevated in the extracellular saline (Figure S1). Next, we imaged postsynaptic calcium influx using a genetically encoded calcium indictor (SynapGCaMP6f) targeted to postsynaptic glutamate receptors at the Drosophila NMJ (Newman et al., 2017). This reporter is capable of detecting calcium transients during individual mEPSP events (Newman et al., 2017), but the response during mEPSP events across varying ranges of extracellular calcium has not been determined. We therefore imaged the SynapGCaMP6f response during miniature activity in four extracellular calcium concentrations: 0 mM, 1.5 mM, 5 mM, and 10 mM (Figure S2). This analysis revealed no detectable calcium signal in 0 mM extracellular calcium, while the signal was enhanced across increasing extracellular calcium concentrations (Figure S2). Thus, while calcium influx through glutamate receptors does not measurably contribute to voltage changes in the postsynaptic cell, these receptors are highly calcium permeable and conduct large transients during miniature events at physiologic calcium concentrations.
Although an acute reduction in calcium influx is not sufficient to diminish pCamKII levels, we asked whether this condition is sufficient to induce PHP expression. We therefore incubated dissected larvae in saline lacking calcium and supplemented with 0.5 mM EGTA for 10 mins (with or without PhTx), then quickly perfused saline containing 0.4 mM calcium and recorded in less than 2 mins (Figure 4H). We found that PHP was robustly expressed at NMJs incubated in PhTx, while synaptic physiology in larvae pre-incubated in 0 mM calcium alone was indistinguishable from controls (Figure 4H). There was, by necessity, calcium present in the final 2 mins before recording, however, PHP induction requires a minimum of 8 mins to be fully expressed (Frank et al., 2006), indicating that pharmacological blockade of glutamate receptors, and not a reduction in calcium influx from extracellular sources, is sufficient to acutely initiate PHP signaling in the postsynaptic cell. We illustrate a schematic to summarize the requirements of protein synthesis and pCamKII activity in the postsynaptic cell during PHP induction (Figure 4I).
DISCUSSION
Here we demonstrate that distinct postsynaptic transduction pathways that individually induce enhanced presynaptic efficacy but differ in their timescales, genetic requirements, necessity for new protein synthesis, and dependence on pCamKII levels, ultimately converge on a common retrograde signaling system to enhance neurotransmitter release at the Drosophila NMJ. Together, this underscores the diverse sensors that integrate and transduce changes in cellular excitability and metabolism to gate the initiation of homeostatic retrograde signaling, highlighting the robustness with which this homeostat stabilizes neurotransmission.
There appear to be a core set of genes necessary for both acute and chronic PHP expression, including ones involved in the homeostatic modulation of synaptic vesicle trafficking, presynaptic excitability, calcium channel activity, and active zone remodeling (Dickman and Davis, 2009; Frank et al., 2006; Muller et al., 2015; Younger et al., 2013). However, other genes appear to be dispensable for this core program, and may rather be involved in secondary functions, such as maintaining PHP expression over chronic time scales or supporting other aspects of homeostatic adaptation (Frank et al., 2009; Penney et al., 2012; Penney et al., 2016; Spring et al., 2016; Tsurudome et al., 2010). Interestingly, the existence of multiple retrograde signaling pathways may be one reason for the failure of forward genetic approaches to identify any individual genes required in the muscle for the core process of PHP induction (Dickman and Davis, 2009; Younger et al., 2013), suggesting some level of redundancy. This convergence of diverse induction mechanisms in the postsynaptic cell enables multiple pathways to detect and respond to homeostatic challenges by feeding into a unitary retrograde signaling system that potentiates presynaptic neurotransmitter release to stabilize synaptic strength.
CamKII activity plays a crucial role in gating diverse forms of synaptic plasticity (Hell, 2014). At the Drosophila NMJ, transgenic manipulations that impact postsynaptic CamKII activity have been reported to modulate the expression of PHP in GluRIIA mutants (Haghighi et al., 2003; Newman et al., 2017). Our results indicate that pCamKII levels are reduced to similar levels in GluRIIA mutants (Figure 4; (Newman et al., 2017)) or following acute pharmacological receptor blockade, consistent with CamKII activity being capable of modulation in seconds at postsynaptic compartments (Hell, 2014). An attractive model would be that a reduction in calcium influx, either over 10 min or during chronic time scales, triggers diminished pCamKII levels and activates PHP signaling, as proposed by several labs (Frank et al., 2006; Haghighi et al., 2003; Newman et al., 2017). However, we found that pharmacological blockade of receptors is necessary and sufficient to reduce pCamKII levels at postsynaptic densities, independently of extracellular calcium, and that incubation in calcium free saline alone is not sufficient to acutely induce PHP expression (Figure 4F–I). Although there are several indications that reduced calcium influx in the postsynaptic muscle over chronic time scales likely contributes to PHP signaling, perhaps necessitating translation-dependent pathways (Haghighi et al., 2003; Newman et al., 2017; Penney et al., 2012; Penney et al., 2016), a calcium-independent system drives the acute expression of PHP following PhTx application, implying a distinct mechanism.
We consider two possibilities to explain how PhTx application to glutamate receptors is transduced into PHP retrograde signaling without requiring calcium signaling through extracellular sources. First, PhTx binding to receptors may induce a conformational perturbation, distinct from ion influx through the receptor, to initiate PHP signaling. Such a mechanism could operate through a metabotropic mechanism, which would be unanticipated but not unprecedented. For example, at mammalian central synapses, NMDA receptor-dependent LTD induction does not require calcium influx through NMDA, but rather pharmacological perturbation to the receptor is sufficient (Aow et al., 2015). Here, a metabotropic pathway has been proposed. Further, mammalian kainate receptors, to which the Drosophila glutamate receptors are homologous, are also capable of signaling through metabotropic mechanisms (Lerma and Marques, 2013). Thus, pharmacological perturbation to GluRIIA-containing receptors could, in principle, initiate PHP signaling through an undefined metabotropic mechanism. However, at present, there is no evidence for such a mechanism in Drosophila.
Alternatively, pharmacological disruption of glutamate receptors may lead to local signaling at the NMJ through interactions with scaffolds such as DLG/PSD-95 and CASK. These scaffolds are known to be in complexes with CamKII and capable of modulating CamKII activity and phosphorylation at the subsynaptic reticulum (SSR) (Lu et al., 2003). Intriguingly, defects in the elaboration of the SSR have recently been reported to disrupt retrograde homeostatic plasticity (Koles et al., 2015). CamKII signaling during PHP appears to be restricted to postsynaptic densities of type 1b boutons (Newman et al., 2017), suggesting that compartmentalized signaling at the SSR orchestrates local PHP signal transduction. However, Tor-OE is capable of initiating PHP signaling independently of pCamKII reduction, where it promotes translation throughout the cell. This implies that protein synthesis modulates retrograde signaling downstream of or in parallel to CamKII signal transduction, yet ultimately feeds back into local post-translational signaling pathways. Future experiments probing the interactions between glutamate receptors, postsynaptic scaffolds, translation, and CamKII activity will clarify the signaling at this compartmentalized synapse.
The finding that PHP can be acutely induced by pharmacological perturbation of glutamate receptors, and not through reductions in calcium influx over rapid time scales, may help to explain perplexing observations about the phenomenology of PhTx-mediated PHP. For example, it was noted that PHP can be induced and expressed by a 10 min incubation of PhTx with only mEPSP events occurring (Frank et al., 2006). Although a reduction in calcium during these mEPSP events was discussed as a possible induction mechanism, estimates are that, at most, six mEPSP events occur per active zone during this induction time (Frank et al., 2006), a very low level and frequency of activity to reliably and robustly produce PHP expression. Indeed, a recent study demonstrated that mEPSP events account for a very small fraction (<1%) of the total postsynaptic calcium signal at individual NMJs (Newman et al., 2017), making a reduction in calcium even more implausible to explain acute PHP induction. Hence, pharmacological perturbation of postsynaptic glutamate receptors, rather than a reduction in calcium through these receptors, is an attractive mechanism to explain the characteristics of the acute induction of PHP by PhTx and raises interesting questions for future studies about how pharmacological receptor perturbation is transduced into PHP induction.
Why might a single retrograde signaling system exist to homeostatically stabilize synaptic strength at the Drosophila NMJ? In central neurons, diverse forms of synaptic plasticity, including Hebbian and homeostatic, dynamically operate over multiple timescales to bi-directionally adjust synaptic strength (Regehr et al., 2009; Tao and Poo, 2001). Further, translation-dependent and independent processes also contribute to retrograde homeostatic signaling in the hippocampus following AMPA receptor blockade (Henry et al., 2012). In contrast, the NMJ is built for stable excitation and is acutely sensitive to reductions in receptor function (Frank et al., 2006; Petersen et al., 1997). However, when neurotransmitter sensitivity in muscle in enhanced by increased receptor expression, no retrograde signaling system exists to homeostatically downregulate presynaptic efficacy (Petersen et al., 1997). Thus, the muscle is endowed with multiple signaling systems to respond to perturbations but appears limited to signal retrograde increases in neurotransmitter release. Hence, a single retrograde signaling system might provide an efficient means to ensure non-additive potentiation in synaptic strength and prevent hyper-excitation when conflicting signals and multiple inductive mechanisms are simultaneously activated.
MATERIALS AND METHODS
Fly Stocks
Drosophila stocks were raised at 25°C on standard molasses fo od. The w1118 strain is used as the wild type control unless otherwise noted, as this is the genetic background of the transgenic lines and other genotypes used in this study. A complete list of stocks can be found in the Supplemental Experimental Procedures.
Immunocytochemistry
Third-instar larvae were dissected in ice cold 0 Ca2+ HL-3 and immunostained as described (Kikuma et al., 2017). Details of specific antibodies can be found in Supplementary Experimental Procedures.
Imaging and analysis
Samples were imaged using a Nikon A1R Resonant Scanning Confocal microscope equipped with NIS Elements software and a 100× APO 1.4NA oil immersion objective using separate channels with four laser lines (405 nm, 488 nm, 561 nm, and 637 nm) as described (Perry et al., 2017). Additional details can be found in Supplemental Experimental Procedures.
Electrophysiology
All dissections and recordings were performed in modified HL3 saline as described in (Kiragasi et al., 2017) containing (in mM): 70 NaCl, 5 KCl, 10 MgCl2, 10 NaHCO3, 115 Sucrose, 5 Trehelose, 5 HEPES, and 0.4 CaCl2 (unless otherwise specified), pH 7.2. Miniature excitatory postsynaptic potentials (mEPSPs) were recorded in the absence of any stimulation, and cut motor axons were stimulated to elicit excitatory postsynaptic potentials (EPSPs). Semi-intact larvae were incubated with or without philanthotoxin-433 (Santa Cruz Biotechnology; 80 μM) and resuspended in HL-3 for 10 mins, as described (Frank et al., 2006). Pre-incubation in cyclohexamide (50μg/ml; Sigma) was performed for 20 mins, as described (Frank et al., 2006).
Statistical Analysis
All data are presented as mean +/−SEM with varying levels of significance assessed as p<0.05 (*), p<0.01 (**), p<0.001 (***), p<0.0001 (****), ns=not significant. Detailed statistical information (mean values, SEM, n, p) for all data is shown in Table S1.
Supplementary Material
Highlights.
A single retrograde signaling system exists at the Drosophila neuromuscular junction
Disparate inductive pathways operate in the postsynaptic compartment
Distinct temporal and translational processes can induce retrograde signaling
Postsynaptic CamKII rapidly responds to pharmacological receptor blockade
Acknowledgments
We thank Pejmun Haghighi (Buck Institute, CA, USA) and Graeme Davis (UCSF, CA, USA) for sharing Drosophila stocks. We also thank Bernardo Sabatini (Harvard, MA, USA) for helpful comments and discussions. PG was supported in part by a USC Provost Graduate Research Fellowship. This work was supported by a grant from the National Institutes of Health (NS091546) and research fellowships from the Alfred P. Sloan, Ellison Medical, Whitehall, Mallinckrodt, and Klingenstein-Simons Foundations to DKD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
PG and XL obtained all experimental data. PG, XL, and DKD analyzed and interpreted all data. The manuscript was written by PG and DKD.
References
- Aow J, Dore K, Malinow R. Conformational signaling required for synaptic plasticity by the NMDA receptor complex. PNAS. 2015;112:14711–14716. doi: 10.1073/pnas.1520029112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Dickman D. Ribosome profiling reveals post-translational signaling mechanisms drive the retrograde enhancement of presynaptic efficacy. bioRxiv 2017 [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA. Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology. 2013;78:63–74. doi: 10.1016/j.neuropharm.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS. Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron. 2003;39:255–267. doi: 10.1016/s0896-6273(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Hell JW. CaMKII: claiming center stage in postsynaptic function and organization. Neuron. 2014;81:249–265. doi: 10.1016/j.neuron.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry FE, McCartney AJ, Neely R, Perez AS, Carruthers CJ, Stuenkel EL, Inoki K, Sutton MA. Retrograde changes in presynaptic function driven by dendritic mTORC1. J Neurosci. 2012;32:17128–17142. doi: 10.1523/JNEUROSCI.2149-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 2010;68:1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauwe G, Tsurudome K, Penney J, Mori M, Gray L, Calderon MR, Elazouzzi F, Chicoine N, Sonenberg N, Haghighi AP. Acute Fasting Regulates Retrograde Synaptic Enhancement through a 4E-BP-Dependent Mechanism. Neuron. 2016;92:1204–1212. doi: 10.1016/j.neuron.2016.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuma K, Li X, Kim D, Sutter D, Dickman DK. Extended Synaptotagmin Localizes to Presynaptic ER and Promotes Neurotransmission and Synaptic Growth in Drosophila. Genetics. 2017 doi: 10.1534/genetics.117.300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiragasi B, Wondolowski J, Li Y, Dickman DK. A Presynaptic Glutamate Receptor Subunit Confers Robustness to Neurotransmission and Homeostatic Potentiation. Cell Rep. 2017;19:2694–2706. doi: 10.1016/j.celrep.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koles K, Messelaar EM, Feiger Z, Yu CJ, Frank CA, Rodal AA. The EHD protein Past1 controls postsynaptic membrane elaboration and synaptic function. Mol Biol Cell. 2015;26:3275–3288. doi: 10.1091/mbc.E15-02-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Marques JM. Kainate receptors in health and disease. Neuron. 2013;80:292–311. doi: 10.1016/j.neuron.2013.09.045. [DOI] [PubMed] [Google Scholar]
- Lu CS, Hodge JJL, Mehren J, Sun XX, Griffith LC. Regulation of the Ca2+/CaM-Responsive Pool of CaMKII by Scaffold-Dependent Autophosphorylation. Neuron. 2003;40:1185–1197. doi: 10.1016/s0896-6273(03)00786-4. [DOI] [PubMed] [Google Scholar]
- Muller M, Davis GW. Transsynaptic control of presynaptic Ca(2)(+) influx achieves homeostatic potentiation of neurotransmitter release. Curr Biol. 2012;22:1102–1108. doi: 10.1016/j.cub.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Genc O, Davis GW. RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron. 2015;85:1056–1069. doi: 10.1016/j.neuron.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman ZL, Hoagland A, Aghi K, Worden K, Levy SL, Son JH, Lee LP, Isacoff EY. Input-Specific Plasticity and Homeostasis at the Drosophila Larval Neuromuscular Junction. Neuron. 2017;93:1388–1404. doi: 10.1016/j.neuron.2017.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Elazzouzi F, Livingstone M, Gonzalez M, Sonenberg N, Haghighi AP. TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron. 2012;74:166–178. doi: 10.1016/j.neuron.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Kauwe G, Gray L, Yanagiya A, M RC, Sonenberg N, Haghighi AP. LRRK2 regulates retrograde synaptic compensation at the Drosophila neuromuscular junction. Nat Commun. 2016;7:12188. doi: 10.1038/ncomms12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S, Han Y, Das A, Dickman DK. Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Human Mol Genet. 2017 doi: 10.1093/hmg/ddx304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 2009;63:154–170. doi: 10.1016/j.neuron.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring AM, Brusich DJ, Frank CA. C-terminal Src Kinase Gates Homeostatic Synaptic Plasticity and Regulates Fasciclin II Expression at the Drosophila Neuromuscular Junction. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1005886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao HW, Poo M. Retrograde Signaling at central synapses. PNAS. 2001;98:11009–11015. doi: 10.1073/pnas.191351698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurudome K, Tsang K, Liao EH, Ball R, Penney J, Yang JS, Elazzouzi F, He T, Chishti A, Lnenicka G, et al. The Drosophila miR-310 cluster negatively regulates synaptic strength at the neuromuscular junction. Neuron. 2010;68:879–893. doi: 10.1016/j.neuron.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmuller A, Hallermann S, Wagner N, Eilers J. Rapid active zone remodeling during synaptic plasticity. J Neurosci. 2011;31:6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger MA, Muller M, Tong A, Pym EC, Davis GW. A presynaptic ENaC channel drives homeostatic plasticity. Neurn. 2013;79:1183–1196. doi: 10.1016/j.neuron.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.