

Abstract
We report the first chemical syntheses of both (−)-majucin and (−)-jiadifenoxolane A via 10 net oxidations from the ubiquitous terpene (+)-cedrol. Additionally, this approach allows for access to other majucin-type sesquiterpenes, like (−)-jiadifenolide, (−)-jiadifenin, and (−)-(1R,10S)-2-oxo-3,4-dehydroxyneomajucin (ODNM) along the synthetic pathway. Site-selective aliphatic C(sp3)-H bond oxidation reactions serve as the cornerstone of this work which offers access to highly oxidized natural products from an abundant and renewable terpene feedstock.
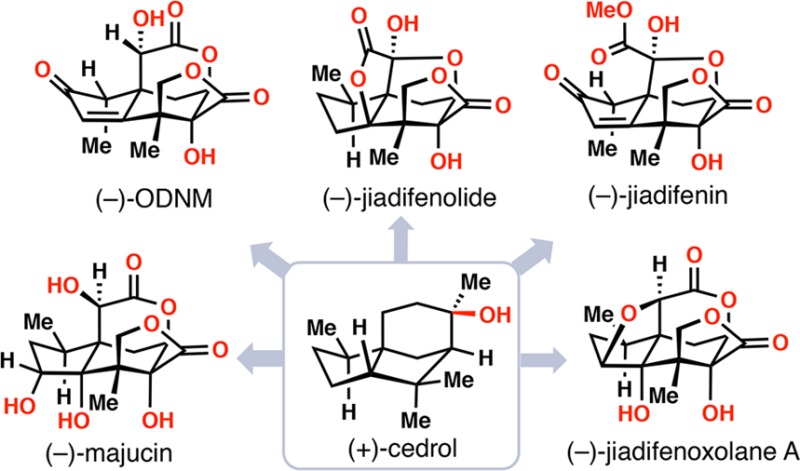
Exclusive to the Illicium species of plants, seco-prezizaane sesquiterpenes are noted for their highly oxidized polycyclic architectures. Within this family, over 20 members possess the majucin core, including the eponymous member (−)-majucin (1), first isolated in 1988 from pericarps of the Chinese flowering plant Illicium majus (Figure 1A).1,2 Majucinoids are among the most highly oxidized members of the seco-prezizaane family, and 1 in particular features a complex scaffold containing both a bridging δ-lactone and a fused γ-lactone, along with four stereodefined hydroxyl groups in close proximity.2 These attributes loom as challenges to chemical synthesis efforts, and indeed, majucin stands as one of the few flagship Illicium sesquiterpenes yet to succumb to chemical synthesis. Work toward the synthesis of other majucin-type natural products, however, has been prolific. Jiadifenin (4), the first member of this subtype to fall to synthetic efforts, has been prepared on multiple occasions.3 (−)-(1R,10S)-2-Oxo-3,4-dehydroxyneomajucin (ODNM, 3) serves as the direct precursor to 4 in the aforementioned syntheses and thus has also been synthesized.3 By far though, (−)-jiadifenolide (5) has received the most attention from the synthetic community with a multitude of impressive formal and total syntheses reported.4,5 No doubt such interest has arisen from the long-known GABA-modulatory properties of members of this Illicium class and especially the ability of 2–5 (and derivatives) to promote neurotrophic phenotypes in both cultured rat cortical neurons and, more recently, human induced pluripotent stem cells.3a,3d,5k,6−8 To the best of our knowledge, the neurotrophic activity of 1 has not been reported.
Figure 1.
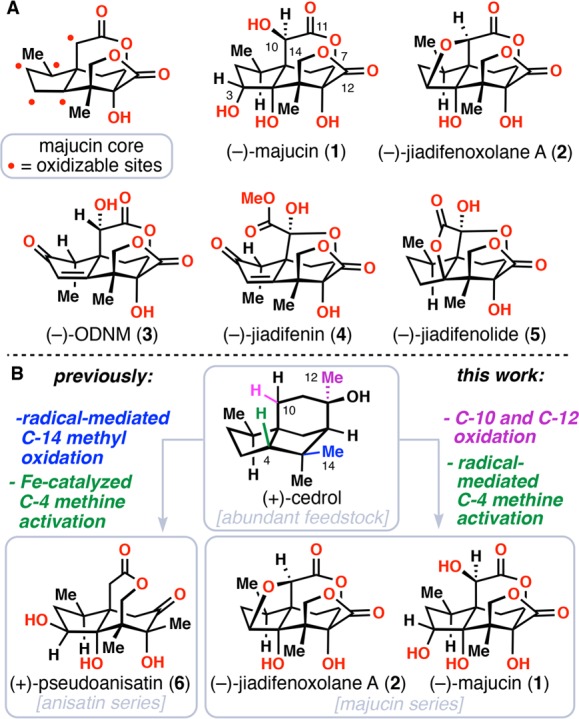
(A) Complex majucinoids from Illicium sp. (B) An oxidative strategy for the construction of Illicium sesquiterpenes from the feedstock chemical (+)-cedrol.
As part of our continued efforts directed toward a unifying synthesis of all Illicium sesquiterpenes using C(sp3)–H activation strategies,9 we recently disclosed an oxidative approach to the simpler family member (+)-pseudoanisatin (6) from the abundant sesquiterpene (+)-cedrol (Figure 1B).10 During our work, iron- and radical-mediated oxidations of the cedrol C-4 methine (shown in green) and C-14 methyl positions (shown in blue) respectively were employed as key steps. In extending this strategy to the more highly oxidized majucinoids, such as 1 and 2, it is also necessary to oxidize all the C–H bonds of the C-12 methyl group and the indicated C–H bond of the C-10 methylene (both shown in magenta, seco-prezizaane numbering). Moreover, the low yields encountered in previous studies using acid-directed Fe-catalysis prompted us to seek alternative solutions to the C-4 methine oxidation problem. Herein, we present our studies toward realizing these–and other–goals which have culminated in the first chemical syntheses of (−)-majucin (1) and (−)-jiadifenoxolane A (2) via 10 net oxidations from (+)-cedrol. Moreover, formal syntheses of (−)-ODNM (3), (−)-jiadifenin (4), and (−)-jiadifenolide (5) have also been accomplished along the way.
We began our synthetic studies in analogy to previous work on 6, but found that the strained THF ring formed in the initial Suárez oxidation (I2, PhI(OAc)2, hν) could be conveniently converted to acetoxy cedrene (7) simply by adding acetic anhydride and phosphoric acid directly to the reaction mixture (Scheme 1). This procedure delivered 7 in 67% yield and on 120 mmol scale.11 Diverging from past work, in which an olefin oxidative cleavage reaction was used to generate a keto acid,10 we found that 7 could be converted into ketone 8 directly via a hydroboration/double oxidation sequence (BH3·THF/CrO3·2pyr.). The use of Collin’s reagent avoided hydrolysis of the acetate protecting group as compared to many other oxidants examined. Ketone 8 was then easily reduced (NaBH4) from the convex face to give alcohol 9 in 72% yield over 2 steps as essentially a single diastereomer. Taking inspiration from the work of Waegell on simpler cedrene scaffolds,12 we explored the use of Suárez conditions to oxidize the C-4 methine position at this stage. Owing to the very close spatial proximity of the secondary hydroxyl to the ring junction C-4 methine, an exceptionally high yielding (93%) C–H functionalization ensued (I2, PhI(OAc)2, hν). In contrast to our previous work employing iron complexes to oxidize this position, the presence of preexisting C-14 oxidation had little impact on this transformation. Next, prolonged stirring with in situ generated RuO4 (RuCl3·xH2O, KBrO3) accomplished a remarkably clean triple oxidation reaction, cleaving the C-6/C-11 bond and delivering ketone lactone 11.12,13 While not optimized, we were also able to isolate quadruple oxidation product 12 wherein an additional C–H bond has been hydroxylated.14
Scheme 1. Synthesis of Complex Majucinoids.

Reagents and conditions: (a) PhI(OAc)2 (1.1 equiv), I2 (0.4 equiv), cyclohexane, hν (visible), 1.5 h then Ac2O (10.0 equiv), H3PO4 (2.0 equiv), 67%; (b) BH3·THF (1.3 equiv), THF, 1.5 h then CrO3·2pyr (25.0 equiv), DCM, 30 min; (c) NaBH4 (1.5 equiv), MeOH, 30 min, 72% over two steps; (d) PhI(OAc)2 (3.0 equiv), I2 (1.0 equiv), DCM, hν (visible), 0 °C, 1.5 h, 93%; (e) KBrO3 (2 × 5.0 equiv), RuCl3·xH2O (3 × 0.03 equiv), MeCN/CCl4/H2O (2:2:3), 75 °C, 3 d, 72% of 11, 7% of 12; (f) SeO2 (3.5 equiv), 4 Å MS (1.0 mass equiv), diglyme, 130 °C, 4 h then K2CO3 (3.0 equiv), Me2SO4 (1.5 equiv), 1 h; (g) l-selectride (1.2 equiv), THF, −78 °C, 30 min then KOH (10.0 equiv), MeOH, 0 °C, 30 min, 50% over two steps; (h) DMDO (1.5 equiv), 12 h; (i) PhCF3, 170 °C, 2 h; (j) Me4NBH(OAc)3 (7.0 equiv), MeCN/AcOH (3:1), – 40 °C, 16 h, 64% over three steps; (k) TsOH·H2O (2.2 equiv), n-BuOH, 150 °C, 26 h, 71%; (l) LHMDS (3.0 equiv), MoOPH (5.0 equiv), THF, −78 → 0 °C, 2.5 h, 65%; (m) [Ru2(PEt3)6(OTf)3](OTf) (0.1 equiv), NMM (0.2 equiv), TFE/dioxane (1:1), 120 °C, 18 h then i-PrOH (3.0 equiv), 120 °C, 5 h, 75%; (n) OsO4·TMEDA (1.0 equiv), DCM, −78 → 0 °C, 2 h then NaHSO3 (10.0 equiv), H2O, 16 h, 61%; (o) MsCl (5.0 equiv), pyr. (10.0 equiv), DCE, rt →80 °C, 15 h, 92%. DMDO = dimethyldioxirane, LHMDS = lithium bis(trimethylsilyl)amide, MoOPH = oxodiperoxymolybdenum(pyridine)-(hexamethylphosphoric triamide).
With the construction of the tricyclic propellane-like core accomplished in five steps, we were poised to address the crucial majucin-type γ-lactone which called for the exhaustive oxidation of the cedrol C-12 methyl group (vide supra). In a single, remarkable step, we found that a quadruple oxidation of all C–H bonds α to the ketone group in 13 was achieved under anhydrous Riley oxidation conditions (SeO2, 4 Å MS, Δ).15 In order to facilitate handling of this compound, it proved essential to methylate the intermediate acid (K2CO3, Me2SO4) prior to workup, thus delivering unsaturated keto ester 13. Upon treatment of 13 with L-selectride, a diastereomeric mixture of allylic alcohols was obtained via 1,2-reduction of the ketone moiety. When the reaction was quenched with basic methanol, both intermediates converged to enol lactone 14, presumably via an acetate cleavage/translactonization/alkene isomerization cascade process. Overall 14, whose rigid tetracyclic skeleton was confirmed by X-ray crystallographic analysis, was obtained in 50% yield from 11 without intermediate silica-gel purification.
In order to rearrange the 5,5-fused ring system found in tetracycle 14 into the majucinoid 5,6-fused core typified by 1–5, we envisioned employing an α-ketol rearrangement. However, unlike our previous work on 6, this system required a transannular bond migration event (Scheme 2). Experimentally, we found that enol lactone 14 could be oxidized to a single, somewhat unstable, α-hydroxyketone diastereomer (see 19) using DMDO. A solvent swap from acetone to trifluorotoluene followed by heating to 170 °C elicited clean bond reorganization to the majucin core. Precedented directed reduction (Me4NBH(OAc)3) of the α-ketol group then furnished known tetracyclic diol 15 in 76% yield over 3 steps, without the need for intermediate silica-gel purification, and as essentially a single diastereomer. The structure of 15, which completes a formal synthesis of 5,3d,4,16 was secured by X-ray crystallographic analysis.
Scheme 2. Stereochemical Considerations for the α-Ketol Rearrangement of Enol Lactone 14.
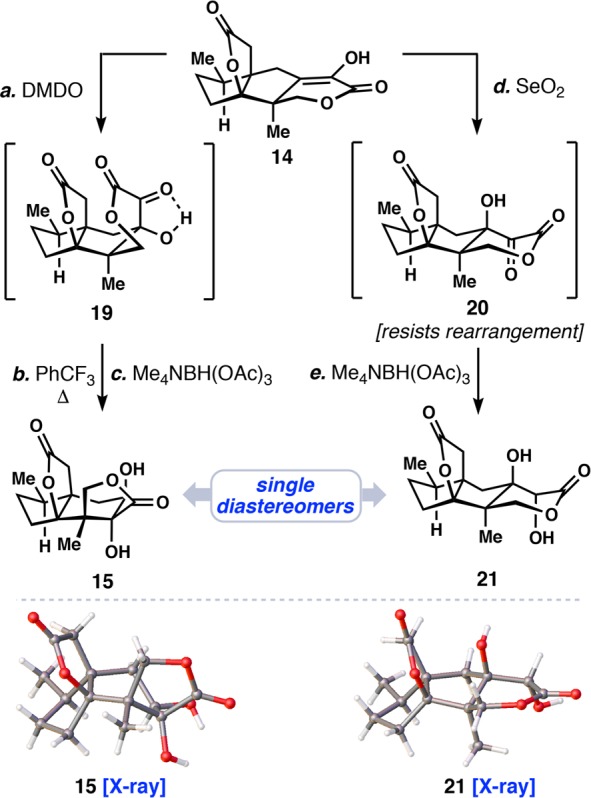
Reagents and conditions: (a–c) See Scheme 1; (d) SeO2 (2.0 equiv), pyr, 110 °C, 16 h; (e) Me4NBH(OAc)3 (7.0 equiv), MeCN/AcOH (3:1), −40 °C, 16 h, 51% over two steps.
Exploration into the diastereoselective oxidation of the enol lactone 14 revealed that while DMDO gave α-ketol 19 selectively, formation of the epimeric α-ketol 20 could be accomplished with SeO2 (Scheme 2).17 Reduction of 20 gave diol 21 and X-ray crystallographic analysis unambiguously assigned its stereochemistry. Although the rearrangement of α-ketol 19 was facile, 20 did not rearrange under a variety of conditions.18
To gain access to 1 and 2, we first converted the jiadifenolide-type γ-lactone ring system into the δ-lactone system via simple treatment with acid (TsOH/n-BuOH, Δ) which unveiled trisubstituted alkene-containing 16 in 71% yield (Scheme 1). Theodorakis and co-workers have demonstrated the synthesis of (−)-ODNM (3) and (−)-jiadifenin (4) in two and three steps, respectively, from 16.3d,16 Direct α-hydroxylation of δ-lactone 16 from the convex face had been reported using the Davis oxaziridine, although reagent byproduct removal proved problematic.3d Seeking an alternative method, we found that enolate oxidation with the molybdenum(VI) reagent MoOPH led to the isolation of clean hydroxy lactone 17.19 We viewed 17 as an excellent testing grounds for Hartwig’s recently reported epimerization methodology via Ru-catalyzed transfer hydrogenation.20 Much to out delight, the catalyst [Ru2(PEt3)6(OTf)3][OTf] in combination with isopropanol delivered diastereomer 18, a recently isolated natural product itself,21 cleanly in 75% yield. To complete the synthesis of majucin (1) a challenging dihydroxylation was required. While we observed no desired reactivity of 18 with OsO4, application of precomplexed osmium tetroxide and tetramethylethylenediamine (OsO4·TMEDA) ultimately provided (−)-majucin (1) in 61% yield.22 X-ray crystallographic analysis unambiguously confirmed the structure of 1. Finally, selective monomesylation (MsCl, pyr.) of (−)-majucin followed by heating elicited a high-yielding intramolecular etherification to give neurotrophic natural product (−)-jiadifenoxolane A (2) in 92% yield. While the enzymatic pathways to 1–5 are not known, this facile displacement could have relevance to the biosynthetic connection between 1 and 2.
In summary, we have demonstrated the first chemical synthesis of the complex majucin-type natural products (−)-majucin (1) and (−)-jiadifenoxolane A (2) in 14 and 15 steps, respectively, and the formal synthesis of (−)-ODNM (3), (−)-jiadifenin (4), and (−)-jiadifenolide (5) from the chiral-pool feedstock (+)-cedrol, a building block available for ∼$0.05 USD/gram.23 During this process, 13 oxidations were employed; however, 3 reduction steps were necessary for oxidation state and stereochemical adjustments,24 highlighting existing gaps in the oxidative synthetic repertoire. Nevertheless, combined with our previous C–H hydroxylation strategy for the anisatin series, this work definitively establishes (+)-cedrol as a versatile platform for the synthesis of nearly all subtypes of seco-prezizaane natural products. Moreover, the formation of C–H hydroxylated intermediate 12 hints to the possibility of accessing even further oxidized, unnatural analogs of these sesquiterpenoids using similar chemistry.
Acknowledgments
In memory of Gilbert Stork and Ronald Breslow who deduced the structure of cedrol. Financial support is provided by the NIH (GM116952). T.J.M. is a Cottrell Scholar and acknowledges unrestricted support from Novartis, Bristol-Myers Squibb, Amgen, and Eli Lilly. M.L.C. acknowledges UC-Berkeley and the NSF for a Berkeley graduate fellowship and NSF predoctoral fellowship (DGE-1106400) respectively. K.H. thanks Eli Lilly for a graduate fellowship. S.J.H. thanks the UC-Berkeley College of Chemistry for a summer undergraduate research award. Mr. Chris Hill (Hartwig lab) is acknowledged for providing generous samples of transfer hydrogenation catalysts as well as helpful discussions. We are grateful to Dr. Hasan Celik and Dr. Jeffrey Pelton for NMR assistance wherein NIH grant GM68933 is acknowledged. Mr. Nicholas Settineri is acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11493.
The authors declare no competing financial interest.
Supplementary Material
References
- Fukuyama Y.; Huang J.-M.. Chemistry and neurotrophic activity of seco-prezizaane- and anislactone-type sesquiterpenes from Illicium species. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier B.V.:2005; Vol. 32, p 395. [Google Scholar]
- Yang C.-S.; Kouno I.; Kawano N.; Sato S. Tetrahedron Lett. 1988, 29, 1165. 10.1016/S0040-4039(00)86678-2. [DOI] [Google Scholar]
- a Cho Y. S.; Carcache D. A.; Tian Y.; Li Y.-M.; Danishefsky S. J. J. Am. Chem. Soc. 2004, 126, 14358. 10.1021/ja045939p. [DOI] [PubMed] [Google Scholar]; b Carcache D. A.; Cho Y. S.; Hua Z.; Tian Y.; Li Y.-M.; Danishefsky S. J. J. Am. Chem. Soc. 2006, 128, 1016. 10.1021/ja056980a. [DOI] [PubMed] [Google Scholar]; c Yang Y.; Fu X.; Chen J.; Zhai H. Angew. Chem., Int. Ed. 2012, 51, 9825. 10.1002/anie.201203176. [DOI] [PubMed] [Google Scholar]; d Trzoss L.; Xu J.; Lacoske M. H.; Mobley W. C.; Theodorakis E. A. Chem. - Eur. J. 2013, 19, 6398. 10.1002/chem.201300198. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cheng X.; Micalizio G. C. J. Am. Chem. Soc. 2016, 138, 1150. 10.1021/jacs.5b12694. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Trzoss L.; Xu J.; Lacoske M. H.; Mobley W. C.; Theodorakis E. A. Org. Lett. 2011, 13, 4554. 10.1021/ol201742j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xu J.; Trzoss L.; Chang W. K.; Theodorakis E. A. Angew. Chem., Int. Ed. 2011, 50, 3672. 10.1002/anie.201100313. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Siler D. A.; Mighion J. D.; Sorensen E. J. Angew. Chem., Int. Ed. 2014, 53, 5332. 10.1002/anie.201402335. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Paterson I.; Xuan M.; Dalby S. M. Angew. Chem., Int. Ed. 2014, 53, 7286. 10.1002/anie.201404224. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lu H. H.; Martinez M. D.; Shenvi R. A. Nat. Chem. 2015, 7, 604. 10.1038/nchem.2283. [DOI] [PubMed] [Google Scholar]; e Shen Y.; Li L.; Pan Z.; Wang Y.; Li J.; Wang K.; Wang X.; Zhang Y.; Hu T.; Zhang Y. Org. Lett. 2015, 17, 5480. 10.1021/acs.orglett.5b02845. [DOI] [PubMed] [Google Scholar]; f Gomes J.; Daeppen C.; Liffert R.; Roesslein J.; Kaufmann E.; Heikinheimo A.; Neuburger M.; Gademann K. J. Org. Chem. 2016, 81, 11017. 10.1021/acs.joc.6b02039. [DOI] [PubMed] [Google Scholar]
- Additionally, many total and formal syntheses of other Illicium sesquiterpenes have been reported: For anisatin type:; a Niwa H.; Nisiwaki M.; Tsukada I.; Ishigaki T.; Ito S.; Wakamatsu K.; Mori T.; Ikagawa M.; Yamada K. J. Am. Chem. Soc. 1990, 112, 9001. 10.1021/ja00180a067. [DOI] [Google Scholar]; b Kende A. S.; Chen J. J. Am. Chem. Soc. 1985, 107, 7184. 10.1021/ja00310a076. [DOI] [Google Scholar]; c Loh T.-P.; Hu Q.-Y. Org. Lett. 2001, 3, 279. 10.1021/ol006918c. [DOI] [PubMed] [Google Scholar]; d Ogura A.; Yamada K.; Yokoshima S.; Fukuyama T. Org. Lett. 2012, 14, 1632. 10.1021/ol300390k. [DOI] [PubMed] [Google Scholar]; For other non-seco-prezizaane Illicium sesquiterpenes:; e Cook S. P.; Polara A.; Danishefsky S. J. J. Am. Chem. Soc. 2006, 128, 16440. 10.1021/ja0670254. [DOI] [PubMed] [Google Scholar]; f Birman V. B.; Danishefsky S. J. J. Am. Chem. Soc. 2002, 124, 2080. 10.1021/ja012495d. [DOI] [PubMed] [Google Scholar]; g Inoue M.; Sato T.; Hirama M. J. Am. Chem. Soc. 2003, 125, 10772. 10.1021/ja036587+. [DOI] [PubMed] [Google Scholar]; h Mehta G.; Singh S. R. Angew. Chem., Int. Ed. 2006, 45, 953. 10.1002/anie.200503618. [DOI] [PubMed] [Google Scholar]; i He W.; Huang G.; Sun X.; Frontier A. J. Am. Chem. Soc. 2007, 129, 498. 10.1021/ja068150i. [DOI] [PubMed] [Google Scholar]; j Chen J.; Gao P.; Yu F.; Yang Y.; Zhu S.; Zhai H. Angew. Chem., Int. Ed. 2012, 51, 5897. 10.1002/anie.201200378. [DOI] [PubMed] [Google Scholar]; k Ohtawa M.; Krambis M. J.; Cerne R.; Schkeryantz J. M.; Witkin J. M.; Shenvi R. A. J. Am. Chem. Soc. 2017, 139, 9637. 10.1021/jacs.7b04206. [DOI] [PubMed] [Google Scholar]
- Shenvi R. A. Nat. Prod. Rep. 2016, 33, 535. 10.1039/C5NP00160A. [DOI] [PubMed] [Google Scholar]
- For biological studies on majucinoids, see:; a Kouno I.; Baba N.; Hashimoto M.; Kawano N.; Takahashi M.; Kaneto H.; Yang C.-S.; Sato S. Chem. Pharm. Bull. 1989, 37, 2448. 10.1248/cpb.37.2448. [DOI] [PubMed] [Google Scholar]; b Kouno I.; Baba N.; Hashimoto M.; Kawano N.; Takahashi M.; Kaneto H.; Yang C.-S. Chem. Pharm. Bull. 1990, 38, 422. 10.1248/cpb.38.422. [DOI] [PubMed] [Google Scholar]; c Yokoyama R.; Huang J.-M.; Yang C.-S.; Fukuyama Y. J. Nat. Prod. 2002, 65, 527. 10.1021/np010571k. [DOI] [PubMed] [Google Scholar]; d Kubo M.; Okada C.; Huang J.-M.; Harada K.; Hioki H.; Fukuyama Y. Org. Lett. 2009, 11, 5190. 10.1021/ol9021029. [DOI] [PubMed] [Google Scholar]; e Shoji M.; Nishioka M.; Minato H.; Harada K.; Kubo M.; Fukuyama Y.; Kuzuhara T. Biochem. Biophys. Res. Commun. 2016, 470, 798–803. 10.1016/j.bbrc.2016.01.092. [DOI] [PubMed] [Google Scholar]
- For biological studies on Illicium sesquiterpenoid analogs, see:; a Inoue M.; Lee N.; Kasuya S.; Sato T.; Hirama M.; Moriyama M.; Fukuyama Y. J. Org. Chem. 2007, 72, 3065. 10.1021/jo0700474. [DOI] [PubMed] [Google Scholar]; b Richers J.; Pöthig A.; Herdtweck E.; Sippel C.; Hausch F.; Tiefenbacher K. Chem. - Eur. J. 2017, 23, 3178. 10.1002/chem.201605362. [DOI] [PubMed] [Google Scholar]
- For recent examples of C(sp3)–H oxidation in total synthesis, see:; a Yuan C.; Jin Y.; Wilde N. C.; Baran P. S. Angew. Chem., Int. Ed. 2016, 55, 8280. 10.1002/anie.201602235. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chu H.; Smith J. M.; Felding J.; Baran P. S. ACS Cent. Sci. 2017, 3, 47. 10.1021/acscentsci.6b00313. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kawamata T.; Nagatomo M.; Inoue M. J. Am. Chem. Soc. 2017, 139, 1814. 10.1021/jacs.6b13263. [DOI] [PubMed] [Google Scholar]; d Loskot S. A.; Romney D. K.; Arnold F. H.; Stoltz B. M. J. Am. Chem. Soc. 2017, 139, 10196. 10.1021/jacs.7b05196. [DOI] [PMC free article] [PubMed] [Google Scholar]; e McCallum M. E.; Rasik C. M.; Wood J. L.; Brown M. K. J. Am. Chem. Soc. 2016, 138, 2437. 10.1021/jacs.5b13586. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Quinn R. K.; Könst Z. A.; Michalak S. E.; Schmidt Y.; Szklarski A. R.; Flores A. R.; Nam S.; Horne D. A.; Vanderwal C. D.; Alexanian E. J. J. Am. Chem. Soc. 2016, 138, 696. 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Hughes J. M. E.; Gleason J. L. Angew. Chem., Int. Ed. 2017, 56, 10830. 10.1002/anie.201706273. [DOI] [PubMed] [Google Scholar]
- Hung K.; Condakes M. L.; Morikawa T.; Maimone T. J. J. Am. Chem. Soc. 2016, 138, 16616. 10.1021/jacs.6b11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggaley K. H.; Erdtman H.; Norin T. Tetrahedron 1968, 24, 3399. 10.1016/S0040-4020(01)92637-7. [DOI] [Google Scholar]
- a Brun P.; Waegell B. Tetrahedron 1976, 32, 1137. 10.1016/0040-4020(76)85037-5. [DOI] [Google Scholar]; b Tenaglia A.; Terranova E.; Waegell B. J. Org. Chem. 1992, 57, 5523. 10.1021/jo00046a040. [DOI] [Google Scholar]
- See the Supporting Information for a scheme detailing a proposed mechanism for this transformation.
- The tertiary hydroxyl group stereochemistry was assigned based on the known preference for RuO4 to oxidize C–H bonds with retention of stereochemistry. See:McNeill E.; Du Bois J. J. Am. Chem. Soc. 2010, 132, 10202. 10.1021/ja1046999. [DOI] [PubMed] [Google Scholar]
- Chuang K. V.; Xu C.; Reisman S. E. Science 2016, 353, 912. 10.1126/science.aag1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for the precise routes to the indicated natural products.
- This reactivity is in contrast to the dehydrogenation seen in the conversion from 11 to 13 and is unusual for SeO2. For a discussion of these reactivity patterns, see:Nicolaou K. C.; Petasis N. A.. Selenium in Natural Product Synthesis (Philadelphia) 1984. [Google Scholar]
- Both 19 and 20 appear to be able to access stereoelectronically favorable bond migration conformations. Intramolecular hydrogen bonding, however, is proposed to facilitate thermal α-ketol rearrangements, and this effect potentially contributes to the observed difference in reactivity. For a comprehensive review, see:Paquette L. A.; Hofferberth J. E. Organic Reactions 2003, 62, 477. 10.1002/0471264180.or062.03. [DOI] [Google Scholar]
- Prolonged stirring of this reaction mixture led to a distribution of 17, 18, and a presumed keto lactone. Unfortunately, low mass recovery precluded exploitation of this reactivity for the preparative synthesis of 18.
- Hill C. K.; Hartwig J. F.. Nat. Chem. 2017, 10.1038/nchem.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Liu F.; Zhang N.; Wang Y.; Yang L.; Bi Y.; Zhang Y.; Liu M. Nat. Prod. Res. 2016, 30, 322. 10.1080/14786419.2015.1058793. [DOI] [PubMed] [Google Scholar]
- a Donohoe T. J.; Moore P. R.; Waring M. J.; Newcombe N. J. Tetrahedron Lett. 1997, 38, 5027. 10.1016/S0040-4039(97)01061-7. [DOI] [Google Scholar]; b Donohoe T. J.; Mitchell L.; Waring M. J.; Helliwell M.; Bell A.; Newcombe N. J. Tetrahedron Lett. 2001, 42, 8951. 10.1016/S0040-4039(01)01999-2. [DOI] [Google Scholar]; c Donohoe T. J. Synlett 2002, 8, 1223. 10.1055/s-2002-32947. [DOI] [Google Scholar]
- Brill Z. G.; Condakes M. L.; Ting C. P.; Maimone T. J. Chem. Rev. 2017, 117, 11753. 10.1021/acs.chemrev.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burns N. Z.; Baran P. S.; Hoffmann R. W. Angew. Chem., Int. Ed. 2009, 48, 2854. 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]; b Gaich T.; Baran P. S. J. Org. Chem. 2010, 75, 4657. 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.