

Abstract
Host shift events play an important role in epizootics as adaptation to new hosts can profoundly affect the spread of the disease and the measures needed to control it. During the late 1990s, an epizootic in Turkey resulted in a sustained maintenance of rabies virus (RABV) within the fox population. We used Bayesian inferences to investigate whole genome sequences from fox and dog brain tissues from Turkey to demonstrate that the epizootic occurred in 1997 (±1 year). Furthermore, these data indicated that the epizootic was most likely due to a host shift from locally infected domestic dogs, rather than an incursion of a novel fox or dog RABV. No evidence was observed for genetic adaptation to foxes at consensus sequence level and dN/dS analysis suggested purifying selection. Therefore, the deep sequence data were analysed to investigate the sub-viral population during a host shift event. Viral heterogeneity was measured in all RABV samples; viruses from the early period after the host shift exhibited greater sequence variation in comparison to those from the later stage, and to those not involved in the host shift event, possibly indicating a role in establishing transmission within a new host. The transient increase in variation observed in the new host species may represent virus replication within a new environment, perhaps due to increased replication within the CNS, resulting in a larger population of viruses, or due to the lack of host constraints present in the new host reservoir.
Keywords: lyssavirus, viral heterogeneity, whole genome sequencing, next generation sequencing, cross-species transmission, rabies virus
1. Introduction
The lyssavirus genus is a group of negative-strand RNA viruses characterised by their ability to cause fatal encephalitis. The type species for lyssaviruses, Rabies lyssavirus is transmitted by a wide range of mammalian hosts within the Carnivora and Chiroptera orders and has a global distribution. Unlike all other lyssaviruses, rabies viruses (RABVs) have established independent transmission cycles in a broad range of carnivore and meso-carnivore host reservoirs, where particular RABV lineages circulate within host conspecifics. Phylogenetic analyses of RABV sequences have demonstrated particular RABV lineages associate with individual host species (Nel et al. 1993; Kuzmin et al. 2008a; Hanke et al. 2016). Host shifts (where the virus is maintained in a new host) infrequently occur; however, a study in the USA determined that cross-species transmissions (CSTs, which do not result in sustained onward transmission within the new host) involving raccoon variant RABV increased over a 20-year period, in both wildlife and domestic animals posing a significant risk to public health (Wallace et al. 2014). Identifying successful host shifts using molecular inferences is a useful tool to understand RABV evolution (Daoust et al. 1996; Leslie et al. 2006; Kuzmin et al. 2012; Borucki et al. 2013). Importantly for lyssaviruses, the molecular mechanisms influencing virus spread between different species and adaptation to a new potential reservoir host are poorly understood, but it is clear that host, viral, and ecological factors all play important roles (Streicker et al. 2010; Mollentze et al. 2014).
RNA virus populations exhibit a heterogeneous population of viruses within single individuals, often referred to as ‘quasispecies’ but perhaps more accurately termed ‘viral heterogeneity’ (Holmes and Moya 2002). Viral heterogeneity, represented by nucleotide sequence variation of the viral RNA, was originally proposed for RABV as a mechanism for viral evolution and adaptation to a new host (Benmansour et al. 1992). The heterogeneous population of an RABV street strain (European fox isolate) has been investigated previously (Kissi et al. 1999) using cloned PCR products covering 19% of the genome. This study identified sub-consensus sequences and concluded no host-specific consensus level changes were observed, although the amount of heterogeneity observed varied depending on the host used to passage the virus. Furthermore, the fixed RABV strain CVS was passaged experimentally in BHK cells, resulting in selection of a dominant variant that differs genetically and phenotypically from the consensus sequence present in mice or neuroblastoma cell passaged CVS (Morimoto et al. 1998). Since the late 1990s until the development of next generation sequencing (NGS) techniques, very little progress in the area of viral heterogeneity was made.
NGS techniques generate unprecedented cost-effective large-scale data, enabling both the generation of complete genomes (Marston et al. 2013; McGinnis et al. 2016; Montmayeur et al. 2017; Parker and Chen 2017) and the analysis of viral sub-consensus populations (Eriksson et al. 2008; Wright et al. 2011; Poh et al. 2013; Raghwani et al. 2016). The inter-host dynamics of multiple different viral infections have been examined. These range from persistent infections, representing narrow transmission bottlenecks, where few virus particles are transmitted (Fischer et al. 2010), to acute infections of equine influenza virus and norovirus, which have broad transmission bottlenecks (Murcia et al. 2010; Bull et al. 2012). Detailed analysis of foot and mouth disease virus (FMDV) traced minor variants within and between animal hosts using NGS data (Morelli et al. 2013). In the lyssavirus field, a number of studies have used NGS to obtain consensus whole genome sequences for phylogenetic analysis (Hoper et al. 2015; Hanke et al. 2016; Troupin et al. 2016). The application of NGS to study lyssavirus host shift events is becoming more commonplace; however, utilising the deep sequence data to investigate viral heterogeneity is an area still largely unexplored (Borucki et al. 2013; Nadin-Davis et al. 2017).
Despite concerted control efforts, rabies remains a significant public health problem in many regions of Turkey (Johnson et al. 2010). The first phylogenetic study to focus on carnivore rabies in Turkey, compared a small conserved region of the RABV gene encoding the nucleoprotein (327 base pairs) of eighteen samples from across the country and identified three clades of RABV within Turkey that clustered geographically (Johnson et al. 2003). More recently, a comprehensive study of RABV in the Middle East, confirmed the clades and highlighted the importance of trans boundary movements in the epidemiology of rabies in the region (Horton et al. 2015).
The original phylogenetic study demonstrated that fox rabies was emerging in western Turkey and was related to rabies circulating in dogs from the same part of the country (Johnson et al. 2003). It is widely accepted that a similar host shift caused the fox enzootic in Europe in the second half of the twentieth century (McElhinney et al. 2011). Determining whether the virus represented a host shift from a dog RABV into a sylvatic species, multiple CSTs without a host shift, or had been circulating undetected in foxes was not possible previously using partial gene data and lack of genetic barcoding to confirm the host species. Understanding the source of infection had direct implications on the control strategy implemented, as wildlife reservoirs required a completely different approach to that used to control RABV in dog populations (Fusaro et al. 2013). Epidemiological records showed that rabies spread eastwards and southwards within the fox population from a presumed epicenter near the western city of Izmir prompting the introduction of oral rabies vaccination campaigns targeting wildlife species, covering the provinces of Izmir and Manisa in 2008 (Un et al. 2012). As a direct result, epidemiological records suggest that fox rabies was eliminated within the vaccination zone by 2010; however, re-emergence of the disease in the original vaccination zone was observed by 2012 (Un et al. 2012).
In the present study, a cohort of RABV samples from this Turkish fox host-shift dataset from western and central Turkey, including all available fox and dog samples between 1999 and 2015 (n = 21) were analysed. The principal objectives were to utilise whole genome sequencing (WGS) to (1) confirm the most likely source of the fox enzootic, (2) refine the date of the host shift, (3) infer the spatio-temporal phylogenetic relationships based on concatenated coding sequence data, and (4) investigate adaptation in the new fox host resulting in successful onward transmission in the Turkish fox population. Together, these data will provide information useful for rabies control policies and informing our understanding of virus emergence and maintenance in new host reservoirs.
2. Results
RABV fluorescent antibody test positive brain samples (n = 30) were obtained from rabies suspect animals in Turkey between 1999 and 2015 and one sample from Russia which is included as an outgroup for some analyses (Fig. 1 and Table 1). During this period, Turkey reported 434 fox rabies cases to Rabies Bulletin Europe (RBE) (RBE 2017). Twenty viruses represented the host-shift dataset which had spread as far East as Ankara and surrounding areas by 2015 including three dog samples (RV3163D, Sub 5791 D, and RV3161D) collected from canine rabies-free towns around Ankara, suspected to be infected with the fox RABV lineage. For further comprehensive analyses, the twenty viruses were divided into two different temporal phases: the early phase, 1999–2007 (n = 7) including RV1124F the first confirmed fox RABV in the area representing the early phase Clade 2 viruses and; RV1126F representing the early phase Clade 1 viruses (Fig. 2). The late phase comprises isolates from 2008 to 2015 [fox (n = 10) and dog (n = 3)] (Table 1). RV2975D represented the dog strain circulating in Izmir at the time of the documented host shift and is referred to as ‘pre-host shift’ in the analyses. Additionally, ten isolates, from geographic locations outside the host shift region were included (non-host-shift dataset) to provide some context to the wider RABV epidemiology in Turkey. The host species of all thirty-one samples was determined by mapping reads from each clinical sample against the Cytochrome C Oxidase 1 (COI) gene (Table 1).
Figure 1.

Map of Turkey and surrounding countries with location of the viruses from the Turkey dataset. Pre-host-shift canine RABV (*); early phase host-shift RABVs (red); late phase host shift (green); and non-host-shift RABVs (blue). RV303RG not included as outside of map area. See Table 1 for sample details.
Table 1.
Turkish RABV isolates analysed in this study.
| ID | Area | Host | Year | viral reads | % viral reads | Genbank accession |
|---|---|---|---|---|---|---|
| RV2975D* | Izmir | Dog | 1999 | 48,801 | 0.532 | KY860583 |
| RV1124F | Izmir | Fox | 1999 | 17,010 | 0.24 | KY860584 |
| RV1126F | Izmir | Fox | 2001 | 27,740 | 0.26 | KY860585 |
| RV1127F | Manisa | Fox | 2001 | 125,950 | 5.395 | KY860586 |
| RV1128F | Manisa | Fox | 2001 | 15,435 | 0.18 | KY860587 |
| RV2976F | Denizli | Fox | 2004 | 30,993 | 1 | KY860588 |
| RV2977F | Izmir | Fox | 2006 | 20,791 | 0.31 | KY860589 |
| RV2984F | Izmir | Fox | 2007 | 45,162 | 4.771 | KY860590 |
| RV2979F | Kutahya | Fox | 2008 | 31,332 | 0.257 | KY860591 |
| RV2980F | Manisa | Fox | 2009 | 10,845 | 0.22 | KY860592 |
| RV2981F | Izmir | Fox | 2010 | 113,385 | 5.526 | KY860593 |
| RV2982F | Ankara | Fox | 2012 | 54,074 | 0.83 | KY860594 |
| RV2983F | Manisa | Fox | 2012 | 11,565 | 0.438 | KY860595 |
| RV3162F | Aksaray | Fox | 2014 | 77,904 | 3.5 | KY860596 |
| RV3163D | Aksaray | Dog | 2014 | 30,321 | 1.45 | KY860597 |
| Sub5790F | Ankara-Cubuk | Fox | 05/05/14 | 79,651 | 3.17 | KY860598 |
| Sub 5791D | Ankara-Cubuk | Dog | 16/05/14 | 318,799 | 17.65 | KY860599 |
| Sub 5792F | Kirkkale-Yahsiyan | Fox | 2014 | 43,663 | 2.01 | KY860600 |
| RV3160F | Konya-Eregli | Fox | 27/05/14 | 95,237 | 3.65 | KY860601 |
| RV3161D | Konya-Eregli | Dog | 26/05/14 | 21,225 | 1 | KY860602 |
| RV3166F | Kirsehir-Boztepe | Fox | 21/05/15 | 11,945 | 0.26 | KY860603 |
| RV1145F | Erzurum | Fox | 2000 | 13,552 | 0.35 | KY860604 |
| RV1144F | Erzurum | Fox | 2001 | 33,268 | 0.79 | KY860605 |
| RV1129F | Erzurum | Fox | 2001 | 8,408 | 0.19 | KY860606 |
| RV1136D | Bursa | Dog | 2001 | 27,835 | 1.2 | KY860607 |
| RV1137D | Bursa | Dog | 2001 | 43,891 | 1.36 | KY860608 |
| RV1142D | Istanbul | Dog | 2001 | 140,830 | 3.3 | KY860609 |
| RV1133D | Ardahan | Dog | 2001 | 450,449 | 8.7 | KY860610 |
| RV1134D | Ardahan | Dog | 2001 | 169,262 | 3.15 | KY860611 |
| RV201D | Yavuzeli | Dog | 1989 | 333,831 | 9.4 | KY860612 |
| RV303RD | Russia (East) | Raccoon Dog | 1980 | 107,066 | 3.52 | KY860613 |
Samples are listed in date/location order and divided as follows: RV2975* pre-host-shift canine RABV; host-shift dataset, twenty isolates (seventeen fox and three dog) subdivided to indicate early (green) and late (red) phases of the host shift; and non-host shift (blue) from other regions of Turkey with Russia as outgroup.
Figure 2.

Bayesian maximum clade credibility phylogeny of Turkey concatenated coding sequences. Branch coloured according to reservoir host species: dog (red) or fox (green). Posterior probabilities are indicated at each node. The node for the MRCA of the host-shift event is enlarged and the dates indicated (HPD 95%). The fox host shift viruses are divided into two distinct clades indicated, clade 1 genome length is 11,923* and clade 2 genome length is 11,924 or 11,925^ (Supplementary Table S2). SNPs between clades 1 and 2 are described in Supplementary Table S5. #The position where the non-synonymous changes are observed.
2.1 Whole genome sequencing from clinical brain samples provides adequate reads to obtain consensus sequences
All thirty-one brain samples were sequenced directly from extracted RNA that were subsequently depleted of gDNA and rRNA. The total number of RABV reads, average coverage, and percentage to total reads varied between samples (Table 1 and Supplementary Table S1). The lowest number of specific RABV viral reads obtained for a sample was 8,408 reads representing 0.19 per cent of total reads, which was sufficient to obtain a full genome consensus sequence (Table 1). The coverage of reads across the genome was not uniform, but was consistent between different samples, suggesting physical constraints might affect the evenness of the coverage, including conformation of the native RNA molecules (Supplementary Table S1 and Fig. S1).
2.2 The source of the fox host shift was locally circulating dog RABV resulting in a successful host shift event
The RABVs circulating in Turkey all cluster within previously described Middle East clades (Supplementary Text S1 and Fig. S2) (Horton et al. 2015). The RABVs circulating in Turkey cluster according to geographic location and the host shift RABV sequences cluster with RV2975D in two distinct clades from a common ancestor (Fig. 2, inset). Comparison of models implemented in BEAST gave very similar values for a modified Akaike’s information criterion (AIC), indicating mean substitution rates at 3.10 × 10−4 subs/site/year [95 per cent highest posterior density (HPD) 2.68–3.56 × 10−4 subs/site/year) (Table 2). The maximum clade credibility (MCC) trees from these analyses had similar topologies, and the times to the most recent common ancestors (TMRCAs) of key clades were also similar.
Table 2.
Assessment of model fit using a modified AIC for RABV full genome concatenated coding sequences.
| Molecular clock | Population prior | AICM | Mean substitution rate (95% HPD) |
|---|---|---|---|
| Strict | Bayesian skyline | 50,443 | 3.10 (2.68–3.56) x 10−4 |
| Strict | Constant | 50,444 | 3.11 (2.69–3.58) x 10−4 |
| Uncorrelated lognormal | Constant | 50,450 | 3.21 (2.69–3.79) x 10−4 |
| Uncorrelated lognormal | Bayesian skyline | 50,449 | 3.14 (2.68–3.67) x 10−4 |
An MCMC chain length of ten million iterations using thirty-one representative sequences in BEAST. AICM compared using Tracer (v1.6). Lower AICM values indicate better fit.
All models support the occurrence of a common ancestor virus of the fox clades that first emerged in the fox population (probability 98%) between 1996 and 1998 (Fig. 2). These data strongly support circulation of rabies in dogs, followed by a spill over into foxes at that time. The emergent fox clade is genetically distinct from contemporaneous fox viruses isolated from other regions in Turkey (Erzurum). The closest viruses to this ancestor were detected in Izmir/Manisa, corroborating epidemiological information, indicating that this location is likely to be the region where the host shift occurred. Furthermore, the MCC analysis confirms the spread of rabies from Izmir and surrounding areas westwards to the Ankara region.
There was overall drop in the effective population size starting in the 1990s (Fig. 3a), which coincides with the reduced number of lineages observed in Turkey and reflects the concerted rabies control efforts in dogs at the time. In contrast, the effective population size of the host shift sequences alone (1999–2015) increased, reflecting the expansion of the virus in a new host reservoir (Fig. 3b). The increase in effective population size stabilises by 2001 and continues to the present day.
Figure 3.
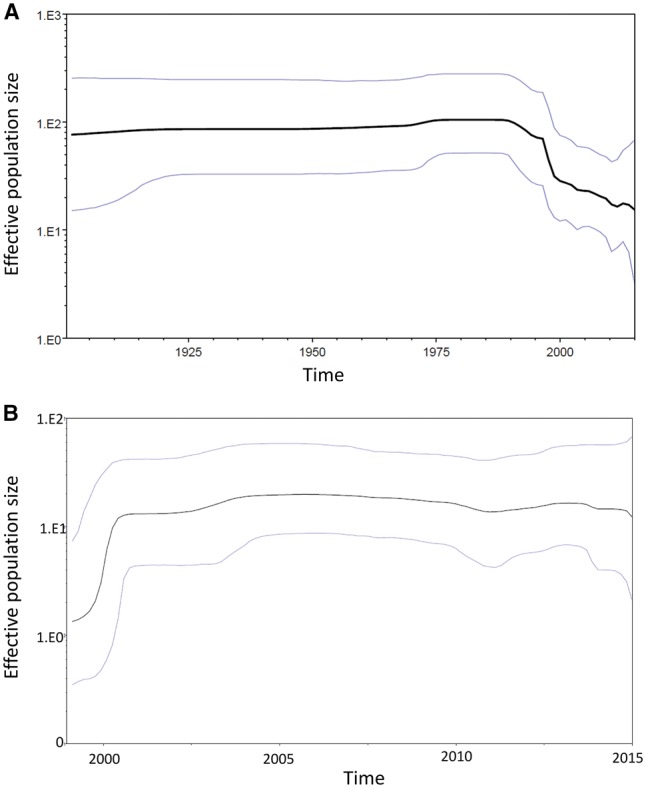
Bayesian skyline plots showing the effective population size over time of Turkey RABV population with HPD 95% limits for (a) whole Turkey dataset (1980–2015) and (b) fox host-shift dataset (1999–2015).
2.3 Analysis of non-coding regions does not reveal host specific viral adaptation
An alignment of all thirty-one complete genome sequences identified two intergenic regions with single nucleotide (nt) indels in homopolymer regions, most likely due to polymerase error. These indels result in differing genomic lengths between 11,923 and 11,925 depending on the presence or absence of one or both of these indels (Supplementary Table S2). The first indel (position 3192) is a homopolymeric region of either 5 or 6 adenosine residues (As) in the M-G intergenic region (Supplementary Table S2 and S3). This indel differentiates the two fox clades: clade 1 viruses (and RV2975D) are 11,923 nts, clade 2 viruses are 11,924–5 nts long (Fig. 2). The sequences from outside the host-shift dataset (n = 9) have 5 As.
The second indel (position 5348) is a homopolymeric region of 7 or 8 As in the poly-A signal of the glycoprotein gene in the G-L intergenic region (Supplementary Table S2 and S4). All but two sequences (sub5792D and RV3160D, both clade 2) have 7 As in the consensus. The additional ‘A’ at position 5348 in sub5792F and RV3160F result in a total genome length of 11,925 nts. The majority of clade 2 samples have sub-populations of both 7 and 8 As indicating that this region is not fixed, resulting in different length consensus sequences for highly related viruses (Supplementary Table S2).
2.4 Analysis of coding regions demonstrates no positive selection at consensus level
Concatenated genes (total length 10,815 nts) from the twenty host shift RABV samples were aligned with the pre-host shift RV2975D sequence to investigate single nucleotide polymorphisms (SNPs). Eight positions, two located in the nucleoprotein, one in the phosphoprotein, and five in the RdRp were identified (Table 3). Two of eight were non-synonymous, located in the RdRp (977Val/Ile and 1871Leu/Arg). However, these residues are unlikely to be species specific, as the majority of the dog sequences from outside the host-shift region had the fox SNP, rather than RV2975D SNP.
Table 3.
SNPs between pre-host shift canine RABV RV2975D Izmir 1999 and all host shift RABV sequences.
| Nuc position | AA position within gene | Gene | Codon RV2975D | Codon host shift | AA RV2975D | AA Host shift |
|---|---|---|---|---|---|---|
| 75 | 25 | N | CAG | CAA | Gln | Gln |
| 792 | 264 | N | AAT | AAC | Asn | Asn |
| 2182 | 277 | P | TTA | CTA | Leu | Leu |
| 5998 | 523 | L | CTG | TTG | Leu | Leu |
| 7360 | 977 | L | GTT | ATT | Val | Ile |
| 8604 | 1391 | L | TTG | TTA | Leu | Leu |
| 10043 | 1871 | L | CTA | CGA | Leu | Arg |
| 10050 | 1873 | L | TCT | TCC | Ser | Ser |
Nucleotide (Nuc) positions relate to concatenated sequences. Amino acid (AA) positions relate to specific gene. Nucleotides that differ are in bold and non-synonymous changes are underlined. N, nucleoprotein gene; P, phosphoprotein gene; L, large protein gene (RNA-dependent RNA polymerase - RdRp).
Due to the lack of host specific residues in the dataset, SNPs observed within the host-shift dataset were analysed. Across the concatenated genes, twenty-six SNPs were identified, which represent the clade 1 and clade 2 SNPs and subsequent substitutions accumulating over time (Fig. 2 and Supplementary Table S5). The majority of the substitutions are synonymous; of which one (382Leu RdRp) had three different nucleotides in the third position all encoding Leucine (CTG, CTT, and CTA). All four non-synonymous substitutions observed with the host-shift dataset were located where RV1124F clusters from the remaining clade 2 viruses. There is a 5-year time period between RV1142F and the next sampled clade 2 virus (RV2976F-2004) during which time the four non-synonymous substitutions occurred (Fig. 2, Supplementary Table S5). It is striking that all four non-synonymous changes occurred during the same time period; however, its importance is unclear. Taken together, the majority of substitutions observed within the consensus sequences over time are not due to positive selection after host switching, rather the result of genetic drift over time.
Selection pressure was evaluated by computing the non-synonymous to synonymous (dN/dS) ratio for the consensus sequence of each gene of the samples in this study. The dN/dS ratios of the five genes were generated from the dataset, using a reference sequence evolutionarily unrelated to the host shift viruses (RV303RD). Across the entire data set, the dN/dS ratios are substantially <1 with N, M, and L genes obtaining ratios below 0.02. P-gene had the highest observed ratio for all viruses (Supplementary Fig. S3A). The average dN/dS ratio across the five genes for all viruses analysed was between 0.02 and 0.025 regardless of the sample date (Supplementary Fig. S3B). Such low dN/dS ratios across the genes, across the dataset suggest that purifying selection is driving the consensus level variation observed, similar results were obtained when using RV1142D (Istanbul, 2001) as the reference sequence (data not shown). However, we note that dN/dS is a relatively conservative measure that may be unable to detect directional selection indicative of a host switch event.
Although the consensus sequences for all five genes, either separately or concatenated strongly suggested purifying selection as the main effect on the viruses, this did not take into account selection at specific residues, known to be important in the glycoprotein, which is the main target of the host antibody responses (Evans et al. 2012). Analysis of individual codons across the glycoprotein did not detect evidence for a change in selection pressure on the glycoprotein associated with the host shift event (data not shown).
2.5 Sub-viral population heterogeneity changes during the early phase of the fox epizootic
To further investigate the RABV host shift within the fox population, the deep sequence data were utilised, representing the sub-consensus viral population present in the brain samples. For each sample dataset, the viral reads were mapped against the corresponding consensus sequence. The mapped viral reads were analysed for alternative calls in relation to the reference sequence using an heterogeneity index (H Index). The H Index was calculated on the viral reads as the number of alternative calls per million base calls sequenced. The resulting calculation controls for the variation of the depth of coverage within and between virus populations, enabling a direct comparison between individual viruses.
The H Index for all sequences was calculated (Supplementary Table S6). No correlation was found between the H Index and the total number of viral reads obtained, for samples with total viral reads >25,000 (Table 1 and Supplementary Table S6). Samples with reads <25,000 had sufficient reads to provide complete genome consensus sequence data, but had a strong correlation between H Index and viral reads, possibly due to low frequency variants being missed, particularly using strict quality criteria. Therefore, heterogeneity and dN/dS analysis of the sub-viral population was undertaken on samples with >25,000 viral reads. Repeat H Index analysis on the same read dataset provided reproducible results [RV2975D: 713 (710); RV2983F: 427 (426)] as did analysis from an independent RNA extraction from same sample (RV1124F: 713 and 710). Shannon entropy (SE) has been previously used as a measure of diversity with FMDV intra-host data (Morelli et al. 2013). Independent analysis of the Turkey RABV read dataset to determine the entropy scores resulted in similar trends between both methods. Individual virus heterogeneity was measured using both methods with good correlation, albeit that SE scores had less range between the highest and lowest (SE range: 252–957; H Index range: 384–1841) (Supplementary Table S6 and Fig. S4). The only exception was RV2982F where the SE value was lower than expected compared to the H Index (Entropy 529; H Index 1048) (Supplementary Fig. S4). The average H Index scores were compared between the whole host-shift dataset, the early stage and late stages of the host-shift dataset, and the non-host-shift viruses (Fig. 4). The early and late virus phases were defined using the H Index scores and aligns with the start of the vaccination campaign in 2008. The highest heterogeneity was present in the sub-consensus population of the early stage viruses (average H Index 1523; SD 300.4), compared to the viruses within the late stage (861.5; 188.4, P = 0.0014). The average heterogeneity score of the host-shift dataset was still significantly higher than the non-host-shift viruses (P = 0.0112) (Fig. 4). To further investigate the differences in the heterogeneity present, the H Index of all viruses was plotted over time. These data indicated that the early phase viruses exhibited higher heterogeneity than those in the later phase (P = 0.0007) (Supplementary Fig. S5). Furthermore, the non-host-shift viruses and pre-host shift RV2975D heterogeneity scores were all significantly lower than the early phase viruses (P = 0.0002). The entropy data analysis from the same dataset was in concordance with the H Index results (data not shown). Due to the strong phylogenetic association between the early and late phase viruses, phylogenetic dependency was investigated using a phylogenetic generalised least squares regression using the Brownian model for correlation structure. The temporal phase and H Index scores were not considered significantly correlated (P = 0.2708). Together, these results indicate an expansion of the sub-viral population after the bottleneck of a host switch event, which continued until the virus was established in the fox population. The heterogeneity subsequently stabilised approximately 10 years after the initial host shift at a level observed in other RABVs sampled across Turkey.
Figure 4.

Graphical representation of sub consensus heterogeneity using H Index for all viruses with viral reads >25,000. All Turkey viruses grouped as follows: early phase (n = 4), late phase (n = 9; fox 7, dog 2), all host-shift dataset (n = 13), and non-host shift (n = 6, fox 1, dog 5). Sample standard deviation is indicated for each group, unpaired t-test P values provided.
2.6 Purifying selection in the sub-viral population during the early phase appeared more apparent than during the late phase host shift viruses
The dynamics of the heterogeneity observed was evaluated by computing the dN/dS ratio at each position for each gene of the samples in this study. Analysis of the dN/dS ratios observed within the NGS read data including all substitutions was undertaken (Supplementary Fig. S6). In comparison to non-host-shift viruses, which largely have a dN/dS = 1 (Supplementary Fig. S6B), lower dN/dS ratios (particularly in the N gene) were observed in the host-shift viruses in the early phase, which was not apparent in the later phase (Supplementary Fig. S6A). The matrix (M)-protein of RV2982F has a dN/dS ratio >1, indicating positive selection in the sub-consensus viral population, which is present in >0.5% of the reads (data not shown). Analysis using average dN/dS ratios across the entire concatenated coding region, revealed that the dN/dS ratio was lower in the early phase (three of the four viruses have dN/dS ratios lower than non-host-shift viruses) and increased over time to approximately 1, suggesting that the sub-viral population within the viruses in the early phase were under purifying selective pressure in the years after the host shift in comparison to the non-host shift and late phase RABVs (Supplementary Fig. S6C).
3. Discussion
RABV host-shift events have rarely been documented, and until recently, investigations have been mostly epidemiological in nature, with less emphasis on understanding the viral molecular adaptations required to establish an infection within a new host reservoir. This study has utilised WGS to investigate the host shift that occurred in Turkey at the end of the twentieth century, estimating the date of the host shift into foxes to within one year, and investigating viral adaptation at the sub-consensus population level. The preparation of the viral RNA for WGS, and the methods employed to obtain deep sequence data will inevitably affect the downstream analysis. Here, the samples were sequenced without amplification, resulting in an unbiased dataset without the introduction of PCR errors. A limitation to this approach is the reduction in depth of coverage across the genome due to the reduced number of viral reads in a clinical sample in comparison to cultured or PCR amplified sample.
Complete genome sequences from thirty-one RABV positive brain samples were obtained; twenty of which were RABV cases from the host-shift dataset covering the time period from its emergence in foxes to the present day. Clearly, although a geographically broad data set was available, only a proportion of an unknown total number of samples were available. Prior to 1999, fox rabies was rarely detected in Turkey; however, after 1999, the cases of fox rabies disproportionately increased and although control measures have reduced fox rabies, it continues to circulate (Johnson et al. 2010; RBE 2017). Analysis of the RABV dataset indicated that the virus effective population size in Turkey reduced from the 1990s (Fig. 3). This decrease corroborated the epidemiological data and coincided with the concerted rabies control efforts in dogs at the time. This overall reduction was also parsimonious with the suggestion from the Bayesian analysis (Fig. 2) that several clades detected in the early 2000s in Bursa, Istanbul and Ardahan were no longer detected.
Using BEAST, with host as a discrete trait, the most likely source of the fox host shift has been demonstrated to be from the locally circulating dog RABV variant with a TMRCA corroborating the epidemiological data (Fig. 2). The substitution rate 3.10 (2.68–3.56) × 10−4 subs/site/year was observed regardless of the model used (Table 2). This rate is higher than other rates calculated from similar (concatenated genome or complete genome) datasets, including a comprehensive canine RABV dataset 2.44 (2.10–2.80) × 10−4 subs/site/year (Troupin et al. 2016), and a fox RABV dataset from Greenland 2.5 (1.9–3.1) × 10−4 subs/site/year (Hanke et al. 2016). Higher substitution rates and larger variation in substitution rates were observed in viruses from mongoose and ferret badgers up to 7.82 (3.13–13.17) × 10−4 subs/site/year. The increased substitution rate observed in this dataset could be a consequence of the smaller dataset analysed; however, it may have also been influenced by the host shift. There are two highly related fox clades, rooted by RV2975D (Izmir 1999). The two clades circulated in the same area and time, including contemporary RABV samples (Fig. 2). Interestingly, viruses most closely related to the early host shift viruses continued to circulate during the vaccination campaign between 2008 and 2010 (Supplementary Text S2). These observations highlight the importance of maintaining rabies-free areas using continued vaccination campaigns. Data such as complete genome length, and SNP analysis provide the fine-resolution required to differentiate such highly related viruses confirming the continued presence of early phase variants during and after the vaccination campaigns.
The inclusion of dog RABV sequences from similar regions to the fox samples provided important data regarding the possible frequency of transmissions between fox and dog populations (Supplementary Text S3). All three RABV dog sequences from the same region as the fox outbreak were highly related to the fox sequences, but were distinct from dog RABVs in other regions of Turkey (Figs 1 and 2). As seen in other European countries, these dog cases are likely to be individual spill over cases in unvaccinated dogs from the ongoing fox enzootic, rather than a host shift to the dog population. In addition, despite consistent surveillance, the number of confirmed dog rabies cases in these regions is low (five in Eregi and ten in Aksaray in 2014). However, the re-emergence of this RABV lineage in dogs is possible if vaccination of dogs is not sustained and effective at local levels.
The high sequence identity between fox and dog (Sub5790F and Sub5791D) demonstrated transmission is not dependent on host-specific residues in the RABV genome. More importantly, comprehensive analysis of the fox host-shift sequences over 15 years has demonstrated a lack of evidence for positive selection at the consensus level for sustained onward transmission. A number of fixed substitutions were identified in the sequence dataset, although none were unique to specific hosts, and are therefore unlikely to be responsible for host species switching. A comprehensive analysis using WGS of RABVs from carnivores also concluded that positive selection was not clearly associated with host switching (Troupin et al. 2016). Furthermore, similar observations have been documented previously for host switching events between bats and recipient species (Kuzmin et al. 2012) and skunks and foxes (Borucki et al. 2013).
It was generally believed that non-fox RABV lineages need higher infecting doses than fox RABVs to productively infect foxes, indicating a species specificity of the respective reservoir host strain encrypted in the prevailing genetic setting (Blancou and Aubert 1997). In this study, however, the results imply that consensus level ‘fixed’ adaptations in the virus genome are not necessary for successful onward maintenance in new host species, i.e. foxes. Perhaps the generally high susceptibility of foxes to RABV infection as experimentally demonstrated (Blancou and Aubert 1997) is one reason for successful host switching events. This is further supported by an extensive analysis of RABV isolates from the Middle East identifying regular host switching and trans-boundary movements of RABV (Horton et al. 2015). This implies that any rabies control effort in this region should consider wildlife reservoirs as a potential source of host switching and vice versa, i.e. that if the reservoir in dogs is not eliminated by sufficient population immunity, there is the risk of establishment of a subsequent wildlife RABV reservoir as observed in Mexico (Garces-Ayala et al. 2017).
Viral heterogeneity has been long proposed as a mechanism for viruses to adapt to new environments. Therefore, deep sequence data from the WGS analysis were used to investigate the heterogeneity in the sub-consensus viral population, and to investigate whether sub-consensus level changes were driving adaptation of RABV in a new host. Utilising NGS data from clinical samples is advantageous for a number of reasons including lack of introduced error due to amplification, a methodology which is applicable to all viruses regardless of homology (because no primers are required), and utilising host data to confirm the host species. However, the depth of coverage across the genome is not uniform (Supplementary Fig. S1), and is lower than the coverage obtained by amplification (Borucki et al. 2013; Stapleford et al. 2016). The H Index was developed to analyse this clinical dataset. Although directly sequencing clinical material may not be appropriate for investigating low level variants, the data demonstrated that assessment of population heterogeneity is reliable with samples containing >25,000 viral reads. Utilising a H Index, RABV heterogeneity was compared across all the Turkey RABVs. SE scores indicated that H Index and entropy analyses were comparable (Supplementary Table S6). Analysis of the H Index dataset demonstrated that the combined H Index values from outside the study area were significantly lower than the host-shift RABVs, and higher H Index values were observed in the early phase viruses compared to the late phase viruses.
Due to the complexities surrounding surveillance of wildlife, including the geography and timescales involved, the sample numbers in this dataset are less than optimum. Although there are only four viruses representing the early phase host-shift viruses, statistically the difference between this group and the other groups is significant. Furthermore, the number of samples within the late phase and non-host-shift groups is larger, with statistically significant differences in the H Index values. Despite the smaller than optimum sample sizes, this valuable dataset, representing one of the few recorded, sampled RABV host shifts. With the exception of a comprehensive analysis investigating bat host shifts, which found evidence for positive selection in a number of genes (Streicker et al. 2010); analysis of host shifts involving non-volant mammals has failed to provide definitive evidence for positive selection at the consensus level related to the host shift event (Kuzmin et al. 2012; Borucki et al. 2013; Troupin et al. 2016). Therefore, investigation of the sub-consensus data, and the spectrum of viral heterogeneity contained within, is a logical progression. The mechanisms involved in modulating the diversity observed at the sub-consensus level are currently undefined; however, there are a number of plausible options. Firstly, the increase in heterogeneity observed in the early phase viruses could be a result of an increased incubation time within the new host, resulting in the generation of a diverse virus population. Although it is generally understood that replication rate is low during this time, the diversity within the virus population would be predicted to be greater given an extended incubation period. A previous study comparing European fox RABVs, isolated 10 years apart, showed the more recently isolated RABV had a decreased time in the onset of clinical disease when infected into naïve foxes suggesting adaptation of the virus over time in that population (Aubert et al. 1991). We suggest that increased incubation (either at the site of inoculation, within the CNS or both) early on in a host shift could result in increased viral heterogeneity, which may reduce as the virus transmits within the new host, adapting over time. Although lyssaviruses are acute viral pathogens, due to a high mortality rate they are unlike other acute viruses such as Influenza A, which have been described to have a ‘smash and grab’ strategy for transmission (Lythgoe et al. 2017). Therefore, reducing viral heterogeneity in established host reservoirs such as bats and dogs might be a valid strategy to optimise transmission within the host population. After a CST event, it is hypothesised that the majority of RABVs do not continue within the new host population (Mollentze et al. 2014), which in part is likely due to the host succumbing to the disease before onward transmission, in addition to other ecological and host factors. When onward transmission does occur, reducing the virulence, by reducing the viral heterogeneity could aid the virus to establish within the new host population. The mechanism utilised by RABV to achieve this is unclear, perhaps involving the fidelity of the RdRp.
The variation observed in the viral sub-population in a successful RABV host shift, rather than at a consensus level, is worthy of further investigation, particularly where NGS data already exist (Borucki et al. 2013). In addition to viral genetic diversity, there are many other factors which influence the success of a host shift, including ecological and host factors. With the increased availability of NGS platforms and decrease in cost, WGS will be pivotal in investigating and unravelling the genetic mechanisms in which RABVs evolve and emerge in new host reservoirs.
4. Material and methods
4.1 RABV samples
The Turkey dataset brain samples (n = 30) used in this study were obtained from infected dogs (Canis lupus familiaris) and red foxes (Vulpes vulpes) submitted to the Etlik Central Veterinary Control and Research Institute (Ankara, Turkey), RV303RD was held within APHA archives and originated from Russia. The majority of samples (n = 21) were collected within the host-shift areas, another nine samples were located in other geographic regions of Turkey (Fig. 1) and one (RV303RD) from Russia. Total RNA from infected brain was extracted using TRIzol (Invitrogen, UK), following the manufacturer’s instructions, resuspending the RNA and adjusting to 1 µg/µl in molecular grade water. All samples were confirmed RABV positive using either a hemi-nested RT-PCR (Heaton et al. 1997) or a real-time TaqMan differential RT-PCR (Wakeley et al. 2005). The Ct values of the samples tested by real-time TaqMan RT-PCR range from 19 to 26, consistent with similarly high viral load.
4.2 Preparation of total RNA for WGS
Total RNA was depleted of host genomic DNA (gDNA) and ribosomal RNA (rRNA) following methods described previously (Marston et al. 2013, 2015). Briefly, gDNA was depleted using the on-column DNase digestion protocol in RNeasy plus mini kit (Qiagen) following the manufacturer’s instructions, eluting in 30 µl molecular grade water. Subsequently, rRNA was depleted, using Terminator 5′-phosphate-dependent exonuclease (Epicentre Biotechnologies). Briefly, 30 µl of gDNA depleted RNA was mixed with 3 µl of Buffer A, 0.5 µl of RNAsin Ribonuclease inhibitor (20–40 U/µl) and incubated at 30 °C for 60 min. The depleted RNA was purified to remove the enzyme using the RNeasy plus mini kit as above, without the DNase digestion, eluting in 30 µl of molecular grade water. Double-stranded cDNA (ds-cDNA) was synthesised using random hexamers and a cDNA synthesis kit (Roche) following the manufacturer’s instructions. The resulting ds-cDNA was purified using AMPure XP magnetic beads (Beckman Coulter), quantified using Quantifluor (Promega) and approximately 1 ng of each sample was used in a ‘tagmentation’ reaction mix using a Nextera XT DNA sample preparation kit (Illumina) following the manufacturer’s instructions—without the bead normalisation step. DNA libraries were quantified using Quantifluor (Promega). Individual libraries were pooled (approximately 8–12 samples per MiSeq run) and normalised to equimolar concentrations. Libraries were sequenced as 2 × 150 bp paired-end reads on an Illumina MiSeq platform.
4.3 Mapping to reference sequence to obtain complete genome sequences
Short reads from the first sample to be sequenced (RV1124F) were mapped to the most genetically related RABV available (RV427—Estonian Raccoon dog RABV—KF154997). The resulting RV1124F full genome sequence was subsequently used to remap the RV1124F raw data, and to map all the Turkey dataset. Reads were mapped using the Burrow-Wheeler Aligner (BWA version 0.7.5a-r405) (Li and Durbin 2010) and were visualised in Tablet (Milne et al. 2013). A modified SAMtools/vcfutils (Li et al. 2009) script was used to generate an intermediate consensus sequence in which any indels and SNPs relative to the original reference sequence were appropriately called. The intermediate consensus sequence was used as the reference for four subsequent iterations of mapping and consensus calling. Sequencing resulted in 98–100 per cent coverage for all samples, with an average depth of coverage of 82,588 (range 1,523–450,449) (Supplementary Table S1). Due to the transposase-based method of the Nextera XT kit, and the complementary nature of the lyssavirus genomic ends, the genome ends were not obtained for all samples. However, as the lyssavirus genomic ends are highly conserved (Marston et al. 2007; Kuzmin et al. 2008b), and the Turkish RABV sequence dataset revealed a large degree of homology at the genomic termini, genomic termini with no mapped reads were added using a representative sequence, resulting in a dataset of complete genome sequences. Consensus sequences were submitted to Genbank (accession numbers KY860583–KY860613) and raw data deposited in European Nucleotide Archive (Accession number: PRJEB22173).
4.4 Multiple alignments
Complete genome sequences and concatenated sequences for each RABV were aligned using Megalign Pro (DNAStar) and saved as multiple sequence formats (msf). The msf files were inspected for accuracy, including correct open reading frames (ORFs) and presence of indels using GeneDoc to confirm indels and identify positions of variation.
4.5 COI analysis
Sequence reads for each sample were mapped to COI reference sequences obtained from Barcode of Life Data Systems [http://www.boldsystems.org/; C. lupus familiaris: GTENK021-11 and V. vulpes: GBMA1928-09 with BWA (Li and Durbin 2010)]. Consensus sequences were called and taxonomic assignment confirmed by using the Identification feature of the BOLD website (http://www.boldsystems.org/index.php/IDS_OpenIdEngine).
4.6 Bayesian reconstructions for host and temporal analysis
Analysis was undertaken to infer evolutionary relationships and the probable host and date of ancestral viruses. Each concatenated sequence was given a discrete trait corresponding to the species in which the virus sequences were detected (dog or fox). Phylogenetic inference was implemented using Bayesian Markov Chain Monte Carlo simulation, without partitioning, in the BEAST package v1.8.1 (Drummond et al. 2012). A TN93 nucleotide substitution model with rate variation among sites was chosen as the best nucleotide substitution model available using Bayesian Information Criterion in MEGA 6.0. Markov Chain Monte Carlo (MCMC) simulations were undertaken with strict and relaxed molecular clock models, and with constant or skyline population priors for 10,000,000 iterations, sampling every 1,000 states to give effective sample sizes of over 200. Molecular clock and population coalescent models were compared using a modified AIC in Tracer for computational efficiency, as described previously (Baele et al. 2012). Maximum clade credibility trees were then annotated using TreeAnnotator (v1.8.1) after 10% of trees were discarded and were then visualised using Fig Tree (v1.4.0) (Fig. 3). Branches are coloured by most probable host at the ancestral node, and node labels are posterior probabilities.
In addition to the concatenated coding sequence analysis, partial N-gene sequences (400 bp) from a wide range of RABV cases from the Middle East (Horton et al. 2015) were analysed alongside the same region from the Turkey sequence dataset to provide context to the samples (Supplementary Fig. S2 and Text S1). The same conditions were used as described for coding region sequences above.
Bayesian skyline plot analyses were conducted as above on two datasets: whole Turkey dataset (1980–2015) and whole host-shift dataset (1999–2015), using the Coalescent Bayesian Skyline as the tree prior (ESS > 200) (Drummond et al. 2005).
4.7 H Index analysis
For each NGS sample, short reads were filtered to discard duplicates in order to reduce the chances of having overrepresented reads due to the library preparation PCR. Additionally, in order to reduce sequencing errors, the sort reads were end trimmed when the quality score of the base call dropped below 20 (range 0–40) (Bolger et al. 2014). Reads shorter than 36 bases were also discarded. The resulting set of reads was mapped onto the homologous viral consensus genome using SMALT (SANGER Institute). Only reads with a top mapping quality of 60 (range 0–60) were considered from this point.
An H Index was calculated on the remaining viral reads as the number of alternative calls per million of base calls sequenced. This is the ratio of the sum of the number of alternative calls for every read divided by the sum of the number of calls of each read. This value is then multiplied by 1 × 106. In order to minimise false alternative calls due to sequencing errors, the base calls with a sequencing score lower than a threshold equal to 30 (0–40) are ignored during the calculation. Additionally, alternative base calls at a given genomic position are ignored if the proportion of reads agreeing with that particular call is lower than 5%.
The thresholds used during data processing were set to minimise incorporating sequencing errors without being too restrictive. The purpose of this H Index is not the calculation of the absolute heterogeneity within the sample, rather the H Index is comparable from sample to sample and alterations on the thresholds do not significantly affect the relativeness of the H Index between samples (particularly because all the samples have been sequenced using the same equipment). The two-tail Student’s t-tests were performed based the assumptions that H indices are normally distributed with equal variances.
4.8 Entropy and dN/dS analysis
The heterogeneity in each virus sample was characterised by computing the SE at each site and then averaging over every site in the genome as described previously (Morelli et al. 2013). The resulting value was multiplied by 100,000 to obtain equivalent values to H index. Similarly to H Index, in order to minimise false alternative calls due to sequencing errors, the base calls with a sequencing score lower than a threshold equal to 30 (0–40) are ignored during the calculation.
For each sample’s consensus sequence, the dN/dS ratio for each codon in the five RABV ORFs was determined by counting the number of synonymous and non-synonymous mutations with respect to the consensus sequence of sample RV303RD which is an evolutionary distinct lineage of RABV to the host-shift samples, using the program SNAP (https://www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html) (Korber 2000). The dN/dS ratio of the sub-consensus sequences was obtained as described previously (Morelli et al. 2013). Briefly, in order to estimate the dN/dS ratio from NGS data, for each codon the observed number of synonymous mutations are counted (only considering reads that cover the codon entirely), divided by the expected number of mutations for the codon, and subsequently divided by the read coverage to give a pS value for each codon; a similar process is used for non-synonymous mutations to give a pN value for each codon. Individual pN and pS values for each codon are then summed and averaged to give an overall pN and pS for the ORF. dN/dS ratio was determined from pN and pS as described previously (Nei and Gojobori 1986).
Supplementary Material
Acknowledgements
The authors wish to thank Emma Wise (Public Health England), Isobel Jarvis, Dr. Guanghui Wu, and Adam Ashton (APHA) for technical support, Ad Vos for his invaluable help and knowledge and Kirstyn Brunker (University of Glasgow) for constructive and informative discussions.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
This work was supported by an initiative of an OIE funded laboratory twinning project on rabies between the FLI and the Etlik Veterinary Control and Research Institute Ankara (File Ref: GKB/KH/2009/22) and was financially supported by the UK Department for Environment, Food and Rural Affairs (Defra), Scottish Government, and Welsh Government by grants (SE0427, SE0431); the European Union H2020-funded Research Infrastructure Grant ‘European Virus Archive Global (EVAg)’ [H2020—grant agreement n°653316] and by an intramural collaborative research grant from the Friedrich-Loeffler-Institut.
Conflict of interest: None declared.
References
- Aubert M. F. et al. (1991) ‘Transmissibility and Pathogenicity in the Red Fox of Two Rabies Viruses Isolated at a 10 Year Interval’, Annales De Recherches Veterinaires. Annals of Veterinary Research, 22: 77–93. [PubMed] [Google Scholar]
- Baele G. et al. (2012) ‘Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty’, Molecular Biology and Evolution, 29: 2157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmansour A. et al. (1992) ‘Rapid Sequence Evolution of Street Rabies Glycoprotein Is Related to the Highly Heterogeneous Nature of the Viral Population’, Virology, 187: 33–45. [DOI] [PubMed] [Google Scholar]
- Blancou J., Aubert M. F. (1997) ‘Transmission of Rabies Virus: Importance of the Species Barrier’, Bulletin de L'Académie Nationale de Médecine, 181: 301–11 discussion 311-302. [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics (Oxford, England), 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borucki M. K. et al. (2013) ‘Ultra-Deep Sequencing of Intra-Host Rabies Virus Populations during Cross-Species Transmission’, PLoS Neglected Tropical Diseases, 7: e2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull R. A. et al. (2012) ‘Contribution of Intra- and Interhost Dynamics to Norovirus Evolution’, Journal of Virology, 86: 3219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoust P. Y., Wandeler A. I., Casey G. A. (1996) ‘Cluster of Rabies Cases of Probable Bat Origin among Red Foxes in Prince Edward Island, Canada’, Journal of Wildlife Diseases, 32: 403–6. [DOI] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2005) ‘Bayesian Coalescent Inference of past Population Dynamics from Molecular Sequences’, Molecular Biology and Evolution, 22: 1185–92. [DOI] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2012) ‘Bayesian phylogenetics with BEAUti and the BEAST 1.7’, Molecular Biology And Evolution, 29: 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson N. et al. (2008) ‘Viral Population Estimation Using Pyrosequencing’, PLoS Computational Biology, 4: e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. S. et al. (2012) ‘Rabies Virus Vaccines: Is There a Need for a Pan-Lyssavirus Vaccine?’, Vaccine, 30: 7447–54. [DOI] [PubMed] [Google Scholar]
- Fischer W. et al. (2010) ‘Transmission of Single HIV-1 Genomes and Dynamics of Early Immune Escape Revealed by Ultra-Deep Sequencing’, PLoS One, 5: e12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro A. et al. (2013) ‘The Introduction of Fox Rabies into Italy (2008-2011) Was Due to Two Viral Genetic Groups with Distinct Phylogeographic Patterns’, Infect Genet E, 17: 202–9. [DOI] [PubMed] [Google Scholar]
- Garces-Ayala F. et al. (2017) ‘Molecular Characterization of Atypical Antigenic Variants of Canine Rabies Virus Reveals Its Reintroduction by Wildlife Vectors in Southeastern Mexico’, Archives of Virology, 162: 3629–37. [DOI] [PubMed] [Google Scholar]
- Hanke D. et al. (2016) ‘Spatio-Temporal Analysis of the Genetic Diversity of Arctic Rabies Viruses and Their Reservoir Hosts in Greenland’, PLoS Neglected Tropical Diseases, 10: e0004779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton P. R. et al. (1997) ‘Heminested PCR Assay for Detection of Six Genotypes of Rabies and Rabies-Related Viruses’, Journal of Clinical Microbiology, 35: 2762–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C., Moya A. (2002) ‘Is the Quasispecies Concept Relevant to RNA Viruses?’, Journal of Virology, 76: 460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoper D. et al. (2015) ‘High Definition Viral Vaccine Strain Identity and Stability Testing Using Full-Genome Population Data–the Next Generation of Vaccine Quality Control’, Vaccine, 33: 5829–37. [DOI] [PubMed] [Google Scholar]
- Horton D. L. et al. (2015) ‘Complex Epidemiology of a Zoonotic Disease in a Culturally Diverse Region: Phylogeography of Rabies Virus in the Middle East’, PLoS Neglected Tropical Diseases, 9: e0003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N. et al. (2003) ‘Rabies Emergence among Foxes in Turkey’, Journal of Wildlife Diseases, 39: 262–70. [DOI] [PubMed] [Google Scholar]
- Johnson N. et al. (2010) ‘Rabies Epidemiology and Control in Turkey: Past and Present’, Epidemiology and Infection, 138: 305–12. [DOI] [PubMed] [Google Scholar]
- Kissi B. et al. (15510 co-authors). (1999) ‘Dynamics of Rabies Virus Quasispecies during Serial Passages in Heterologous Hosts’, Journal of General Virology, 80: 2041–50. [DOI] [PubMed] [Google Scholar]
- Korber B. (2000) ‘HIV Signature and Sequence Variation Analysis’, in Rodrigo AG, Learn GH (eds) Computational Analysis of HIV Molecular Sequences. Netherlands: Kluwer Academic Publishers; pp. 55–72. [Google Scholar]
- Kuzmin I. V. et al. (2008a) ‘Arctic and Arctic-like Rabies Viruses: Distribution, Phylogeny and Evolutionary History’, Epidemiology and Infection, 136: 509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin I. V. et al. (2012) ‘Molecular Inferences Suggest Multiple Host Shifts of Rabies Viruses from Bats to Mesocarnivores in Arizona during 2001–2009’, PLoS Pathogens, 8: e1002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin I. V. et al. (2008b) ‘Complete Genomes of Aravan, Khujand, Irkut and West Caucasian Bat Viruses, with Special Attention to the Polymerase Gene and Non-Coding Regions’, Virus Research, 136: 81–90. [DOI] [PubMed] [Google Scholar]
- Leslie M. J. et al. (2006) ‘Bat-Associated Rabies Virus in Skunks’, Emerging Infectious Diseases, 12: 1274–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2010) ‘Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform’, Bioinformatics (Oxford, England), 26: 589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) ‘The Sequence Alignment/Map Format and SAMtools’, Bioinformatics (Oxford, England), 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lythgoe K. A. et al. (2017) ‘Short-Sighted Virus Evolution and a Germline Hypothesis for Chronic Viral Infections’, Trends in Microbiology, 25: 336–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston D. A. et al. (2013) ‘Next Generation Sequencing of Viral RNA Genomes’, BMC Genomics, 14: 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston D. A. et al. (2007) ‘Comparative Analysis of the Full Genome Sequence of European Bat Lyssavirus Type 1 and Type 2 with Other Lyssaviruses and Evidence for a Conserved Transcription Termination and Polyadenylation Motif in the G-L 3' Non-Translated Region’, Journal of General Virology, 88: 1302–14. [DOI] [PubMed] [Google Scholar]
- Marston D. A. et al. (2015) ‘Complete Genomic Sequence of Rabies Virus from an Ethiopian Wolf’, Genome Announcements, 3: e00157-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinney L. M. et al. (2011) ‘Molecular Diversity and Evolutionary History of Rabies Virus Strains Circulating in the Balkans’, The Journal of General Virology, 92: 2171–80. [DOI] [PubMed] [Google Scholar]
- McGinnis J. et al. (2016) ‘Next Generation Sequencing for Whole Genome Analysis and Surveillance of Influenza a Viruses’, Journal of Clinical Virology: The Official Publication of the Pan American Society for Clinical Virology, 79: 44–50. [DOI] [PubMed] [Google Scholar]
- Milne I. et al. (2013) ‘Using Tablet for Visual Exploration of Second-Generation Sequencing Data’, Briefings in Bioinformatics, 14: 193–202. [DOI] [PubMed] [Google Scholar]
- Mollentze N., Biek R., Streicker D. G. (2014) ‘The Role of Viral Evolution in Rabies Host Shifts and Emergence’, Current Opinion in Virology, 8: 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montmayeur A. M. et al. (2017) ‘High-Throughput Next-Generation Sequencing of Polioviruses’, Journal of Clinical Microbiology, 55: 606–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli M. J. et al. (2013) ‘Evolution of Foot-and-Mouth Disease Virus Intra-Sample Sequence Diversity during Serial Transmission in Bovine Hosts’, Veterinary Research, 44: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K. et al. (1998) ‘Rabies Virus Quasispecies: Implications for Pathogenesis’, Proceedings of the National Academy of Sciences of the United States of America, 95: 3152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcia P. R. et al. (2010) ‘Intra- and Interhost Evolutionary Dynamics of Equine Influenza Virus’, Journal of Virology, 84: 6943–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadin-Davis S. A. et al. (2017) ‘Application of High-Throughput Sequencing to Whole Rabies Viral Genome Characterisation and Its Use for Phylogenetic Re-Evaluation of a Raccoon Strain Incursion into the Province of Ontario’, Virus Research, 232: 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M., Gojobori T. (1986) ‘Simple Methods for Estimating the Numbers of Synonymous and Nonsynonymous Nucleotide Substitutions’, Molecular Biology and Evolution, 3: 418–26. [DOI] [PubMed] [Google Scholar]
- Nel L. H., Thomson G. R., Von Teichman B. F. (1993) ‘Molecular Epidemiology of Rabies Virus in South Africa’, The Onderstepoort Journal of Veterinary Research, 60: 301–6. [PubMed] [Google Scholar]
- Parker J., Chen J. (2017) ‘Application of Next Generation Sequencing for the Detection of Human Viral Pathogens in Clinical Specimens’, Journal of Clinical Virology: The Official Publication of the Pan American Society for Clinical Virology, 86: 20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh W. T. et al. (2013) ‘Viral Quasispecies Inference from 454 Pyrosequencing’, BMC Bioinformatics, 14: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghwani J. et al. (2016) ‘Exceptional Heterogeneity in Viral Evolutionary Dynamics Characterises Chronic Hepatitis C Virus Infection’, PLoS Pathogens, 12: e1005894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RBE reported rabies cases in Europe database [Internet] (2017) cited 14/01/2017. Available from http://www.who-rabies-bulletin.org/site-page/queries.
- Stapleford K. A. et al. (2016) ‘Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements’, PLoS Neglected Tropical Diseases, 10: e0004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicker D. G. et al. (2010) ‘Host Phylogeny Constrains Cross-Species Emergence and Establishment of Rabies Virus in Bats’, Science (New York, N.Y.), 329: 676–9. [DOI] [PubMed] [Google Scholar]
- Troupin C. et al. (2016) ‘Large-Scale Phylogenomic Analysis Reveals the Complex Evolutionary History of Rabies Virus in Multiple Carnivore Hosts’, PLOS Pathogens, 12: e1006041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Un H. et al. (2012) ‘Oral Vaccination of Foxes against Rabies in Turkey between 2008 and 2010’, Berliner Und Munchener Tierarztliche Wochenschrift, 125: 203–8. [PubMed] [Google Scholar]
- Wakeley P. R. et al. (2005) ‘Development of a Real-Time, TaqMan Reverse Transcription-PCR Assay for Detection and Differentiation of Lyssavirus Genotypes 1, 5, and 6’, Journal of Clinical Microbiology, 43: 2786–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace R. M. et al. (2014) ‘Right Place, Wrong Species: A 20-Year Review of Rabies Virus Cross Species Transmission among Terrestrial Mammals in the United States’, PLoS One, 9: e107539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C. F. et al. (2011) ‘Beyond the Consensus: Dissecting within-Host Viral Population Diversity of Foot-and-Mouth Disease Virus by Using Next-Generation Genome Sequencing’, Journal of Virology, 85: 2266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.