


Abstract
Characterizing the binding behaviors of RNA-binding proteins (RBPs) is important for understanding their functional roles in gene expression regulation. However, current high-throughput experimental methods for identifying RBP targets, such as CLIP-seq and RNAcompete, usually suffer from the false negative issue. Here, we develop a deep boosting based machine learning approach, called DeBooster, to accurately model the binding sequence preferences and identify the corresponding binding targets of RBPs from CLIP-seq data. Comprehensive validation tests have shown that DeBooster can outperform other state-of-the-art approaches in RBP target prediction. In addition, we have demonstrated that DeBooster may provide new insights into understanding the regulatory functions of RBPs, including the binding effects of the RNA helicase MOV10 on mRNA degradation, the potentially different ADAR1 binding behaviors related to its editing activity, as well as the antagonizing effect of RBP binding on miRNA repression. Moreover, DeBooster may provide an effective index to investigate the effect of pathogenic mutations in RBP binding sites, especially those related to splicing events. We expect that DeBooster will be widely applied to analyze large-scale CLIP-seq experimental data and can provide a practically useful tool for novel biological discoveries in understanding the regulatory mechanisms of RBPs. The source code of DeBooster can be downloaded from http://github.com/dongfanghong/deepboost.
INTRODUCTION
RNA binding proteins (RBPs) play important roles in multiple aspects of gene expression regulation, such as alternative splicing, RNA modification, mRNA export and localization (1). Not only does the dysregulation of RBPs induce abnormality, but also the mutations in their binding targets have the potential to cause diseases (2). So, capturing the intrinsic binding preferences of RBPs and identifying their binding targets in a precise and large-scale manner are essential to understand the regulatory roles of RBPs and reveal their connections to the pathogenesis of complex diseases.
Before the development of high-throughput techniques for characterizing RNA-protein interactions, only a few RBPs were well studied based on the small-scale experiments, such as in vitro EMSA (3) and in vivo fluorescence methods (4). Recently, several high-throughput sequencing-based approaches, e.g. CLIP-seq (5–7), SELEX (8,9) and RNAcompete (10,11), have been proposed to measure RBP binding sites and binding affinities in a transcriptome-wide manner. However, despite the huge amount of data generated by these techniques, they still suffer from the false negative issue mainly due to experimental noise and bias (12). To overcome these drawbacks, various computational models (13–20) have been developed to learn RBP binding preferences and detect putative RBP targets based on abundant experimental data.
As many RBPs have been validated to recognize structured regions (21), there is a tendency in recent studies to incorporate the structural features of target RNAs into prediction models, such as MEMERIS (15), GraphProt (17) and our recent deep learning based model (19), where the integration of RNA structural information has been shown to largely boost the prediction performance. Nevertheless, the current transcriptome-wide experimental techniques for measuring RNA structures are far from maturity. On the other hand, predicting RNA structures using computational models usually requires a substantial amount of additional effort and time, and a predicted RNA structure is generally less accurate compared to that derived from experimental approaches. In addition, systematic integration of both sequence and structural information generally requires a more complex prediction model. So far, it remains largely unknown whether we can derive a sequence based prediction model that only takes RNA sequence as input, while still achieving prediction performance comparable to that of the state-of-the-art prediction methods that require both sequence and structural profiles. To fill this gap between modeling accuracy and computational complexity, we develop a deep boosting based model, called DeBooster, that requires only sequence information and can capture RBP binding preferences and predict binding sites from high-throughput CLIP-seq data with high accuracy and efficiency.
Through testing on 24 CLIP-seq datasets, we have shown that even without using RNA structural information, DeBooster can outperform the state-of-the-art methods that take both sequence and structural information as input, including both GraphProt (17) and our previous deep learning based model (19). In addition, we have performed comprehensive tests to validate the superiority of DeBooster: (i) DeBooster can accurately capture RBP binding preferences and generate RBP binding motifs that are consistent with previous studies in the literature; (ii) The predictions of DeBooster can be successfully validated through cross-platform datasets.
In addition to the above extensive validation tests, we have further demonstrated several new possible applications of DeBooster in studying the regulatory roles of RBPs. With an integrative analysis based on other types of data and our prediction results, we not only derive literature-consistent results concerning RBP regulation, but also hope to gain novel insights into the biological rationale of the regulatory roles of RBPs. In particular, we have confirmed that the binding targets of the RNA helicase MOV10 predicted by DeBooster are highly associated with the fold changes of mRNA half-lives, providing another evidence on the regulatory functions of RNA helicases on mRNA half-lives. In addition, it has been confirmed that a fraction of ADAR2 binding events are ‘non-productive’, i.e. these bindings may not trigger any RNA editing (22). We have also observed two potentially different types of ADAR1 binding sites which also show the similar ‘productive’ and ‘non-productive’ patterns, respectively. Moreover, we applied DeBooster to study the antagonizing effect of RBP binding on miRNA repression. In particular, it has been known that in the 3′ UTR of the oncogene ERBB2, the RBP ELAVL1 (also called HUR) antagonizes the repression effect of the miRNA miR-331-3p by binding to a U-rich element (URE) near the miRNA target region called miR-331b (23). With a mutant URE, we have observed that the new ELAVL1 binding sites predicted by DeBooster shift to a position more distant from the miR-331b region, which is largely consistent with the previous experimental studies. At last, we have used DeBooster to predict the effects of the single nucleotide variant (SNV) mutations on the RBP binding sites related to splicing events, which may provide useful hints for identifying pathogenic mutations and investigating their connections to the pathogenesis of complex diseases. Based on these test results, we expect that DeBooster will have great application potentials and be widely used by the community to analyze more CLIP-seq experimental data and discover more biologically relevant findings on the functional roles of RBPs in post-transcriptional gene regulation.
MATERIALS AND METHODS
The DeBooster framework
We have developed a deep boosting based approach, called DeBooster, to predict the sequence specificities of RNA-binding proteins (RBPs) from high-throughput CLIP-seq data (Figure 1). As RNA primary sequence can be viewed as a string over the alphabet {A, U, C, G}, we mainly use the basic bag-of-words model (24) as in the natural language processing field to encode the features of a given RNA sequence (Figure 1A). In particular, for each word of fixed length k, we count how many times it appears in the RNA sequence and store its frequency information in a vector of length 4k. We extract the word frequency information for both an RBP target region and its upstream and downstream flanking regions of 150 nucleotides each. We consider words (i.e. the substrings) of lengths 1, 2, 3, which results in 2 × (4 + 42 + 43) = 168 features in total.
Figure 1.

Schematic overview of DeBooster, a deep boosting approach for identifying the sequence specificities of RNA-binding proteins (RBPs). (A) Schematic illustration of the strategy for encoding the sequence features of RBP binding targets. The nucleotides in the target region of an input sequence are represented by capitalized letters while the extended regions on both sides are represented by lowercase letters. Each number within a box stands for the value of the corresponding feature. The numbers on the right side represent the total number of features in individual categories. (B) Schematic illustration of the prediction pipeline. More details can be found in the main text.
Note that the bag-of-words model mainly focuses on the occurrences of words and reflects little about the order of the letters in a sequence. In other words, if we swap the first half and the second half of an RNA sequence, the features provided by the bag-of-words model would roughly remain the same. To better incorporate the order of letters into the model, we further use the following scheme to extract the ‘second-order’ word count information. For a fixed stride m and a given RNA sequence a1a2⋅⋅⋅at, we count the words a1am + 1, a2am + 2, ..., at − mat and use a vector to record the corresponding count information. As before, we also consider both an RBP target region and the flanking regions of 150 nucleotides both upstream and downstream. We consider the stride lengths 4, 5 and 6, which generates 2 × 3 × 42 = 96 more features in total. Moreover, we consider five additional features, such as the length of the target region, whether the target length is a multiple of 3, whether the target region contains the stop codons UAG, UAA and UGA. Thus, overall we extract 168 + 96 + 5 = 269 features for a given RNA sequence.
We then apply a deep boosting based method, to learn a classification model from the above encoded features (Figure 1B). The deep boosting method (25), similar to other boosting methods like Adaboost (26), learns an ensemble of base classifiers. Here, a base classifier is a ‘weak’ classifier that classifies the samples in a moderate accuracy, but the combination of multiple base classifiers into a stronger one can achieve higher accuracy. In particular, the classifier employed in our deep boosting method is in the following form:
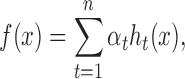 |
(1) |
where f(x) is the final classifier to output, each ht(x) is a base classifier (in our case a decision tree) and αt is the weight of the corresponding base classifier.
For the ith training example (xi, yi), the label yi is ±1 and we want the output of f(xi), namely 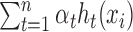 , to be as close to yi as possible, which is equivalent to requiring
, to be as close to yi as possible, which is equivalent to requiring 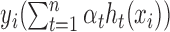 as close to 1 as possible. Therefore, we have the following objective function during the training process:
as close to 1 as possible. Therefore, we have the following objective function during the training process:
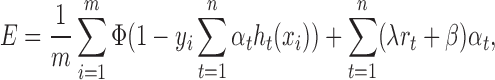 |
(2) |
where (xi, yi) stands for the ith training example, m stands for the total number of training examples, Φ stands for the loss function (e.g. the exponential (26) or logistic function (27)), rt is a regularization term for the tth decision tree classifier, and λ and β are two hyper-parameters to be determined. As the deeper decision trees can always fit the training examples better, but may lose the generalization for the unseen samples, the second term in Equation (2) serves as a regularization term to overcome this overfitting problem. Specifically, if ht(x) is a decision tree of depth d, then rt is the Rademacher complexity (28) of the set of all decision trees of depth d.
The above objective function can be optimized using the same techniques as in other boosting methods (26,29). After the training process, the learned model can be used to predict the binding specificities of RBPs and also generate the corresponding binding motifs.
DeBooster is implemented by a combination of C++ and Python. All our computational experiments were performed on a 64-bit version of CentOS server.
Training datasets
We used 24 CLIP-seq datasets to train and validate our prediction model. These datasets were preprocessed in (17) to construct both positive and negative samples. In particular, the CLIP-seq binding site was extended with 150 nt both upstream and downstream (i.e. resulting in sequences that were 300 nt longer than the binding sites) to generate positive samples. For negative samples, the unbound sites were selected by shuffling the coordinates of CLIP-seq binding sites among the genes with at least one CLIP-seq binding site. Then, these selected unbound sites were also extended with 150 nt both upstream and downstream to generate the negative samples. The list of all RBP names in these datasets can also be found in Figure 2A. Among these datasets, AGO1-4 contained the binding sites for four RBPs of the argonaute family, and IGF2BP1-3 contained the binding targets of three insulin-like growth factor 2 mRNA-binding proteins. ELAVL1 HITS-CLIP, ELAVL1 PAR-CLIP(A), ELAVL1 PAR-CLIP(B) and ELAVL1 PAR-CLIP(C) included the binding sites of the RBP ELAVL1 measured from different experimental platforms.
Figure 2.

Performance evaluation of DeBooster on 24 CLIP-seq datasets. (A) The comparisons of the area under receiver operator characteristic curve (AUROC) scores between different prediction approaches via a 10-fold cross-validation procedure. The best prediction result for each dataset is highlighted in bold. (B and C) The receiver operator characteristic (ROC) and precision-recall (PR) curves achieved by DeBooster for all 24 CLIP-seq datasets in the cross-validation results, respectively. (D) Examples of the sequence motifs of the RBP binding targets predicted by DeBooster.
Determination of hyperparameters
We use an independent validation dataset of RBP C22ORF28 to determine the optimal setting of the hyperparameters of DeBooster, including the type of the loss function (denoted by Φ), the number of the base decision tree classifiers (denoted by n), the maximum depth of these decision trees (denoted by k), and parameters λ, β controlling the relative importance of the complexity penalty. This process yields the following optimal setting of the hyperparameters: the exponential function as the loss function Φ, n = 200, k = 5, λ = 0.3 and β = 0.
Motif generation
We use the following procedure to generate representative motifs of the RBP binding sites predicted by DeBooster. First, we use the set of the weighted decision trees resulting from the deep boosting algorithm to evaluate the relative importance of each encoded feature. In particular, for each decision tree with weight ω in the model, we identify the feature ψ and the corresponding threshold τ used to split the root node for this feature. Suppose that at the root node a fraction p1 of all examples in the training set are positive, and at the right child of the root node (in which the value of feature ψ is larger than τ), a proportion p2 of all examples in the training set are positive. We then use (p1 − p2)ω to represent the importance of feature ψ. By doing so, we score each feature based on its contribution to RBP binding. A higher absolute value of a positive score means higher contribution to RBP binding, while a higher absolute value of a negative score means less contribution to RBP binding. We use a vector s to store the importance scores of all encoded features. Next, we go through all 8-mers and extract the feature vector vi for each of them. We then rank these 8-mers according to the inner product of vi and s, and we select the top 500 8-mers with the highest ranking scores. As the top 8-mers may come from shifts around the best one, we align all 8-mers with respect to the top one such that the largest number of base matchings is achieved. After that, we generate the binding motif based on this alignment step and visualize it using the WebLogo site (30).
Predicting the MOV10 targets along the 3′ UTRs
To predict the MOV10 binding sites along the 3′ UTRs of the genes whose fold-changes of mRNA half-lives were measured after MOV10 knock-down (31), we trained DeBooster using the same MOV10 dataset as in the training data for the validation test, and then performed the analysis on the same set of 7000 genes as in (31). The UTRs of these genes were obtained based on UCSC genome annotation (32). For a gene with two or more annotated 3′ UTRs, we chose the longest one within 3000 nucleotides. We then scanned the whole UTR using a sliding window whose length was equal to the average length of MOV10 binding sites in the training data (34 nt). The step size was about one-fourth of the average length of MOV10 binding sites.
Predicting ELAVL1 binding scores along the 3′ UTR of gene ERBB2
Both wild-type and mutant 3′ UTR sequences of gene ERBB2 were obtained from (23) (Supplementary Notes). The lengths of these sequences are all 119 nt. For each sequence, we took a window of length 41 nt (the average length of the ELAVL1 target regions over training samples) and slided this window along the 3′ UTR of mRNA ERBB2 with a stride length of 1 nt. For each sliding window, we assigned the resulting prediction score to the central nucleotide of this window. Overall, we obtained the prediction scores along positions 21-99 for each sequence (Figure 7), and the first and last 20 nucleotides were not included in our analysis.
Figure 7.

The predicted influence of ELAVL1 binding on the repression effect of miRNA miR-331-3p. (A) An illustrative model of the co-binding of RBP ELAVL1 and miRNA miR-331-3p on the 3′ UTR of gene ERBB2. miR-331b represents the binding region of miRNA miR-331-3p. The width of the arrow represents the relative strength of miR-331-3p binding. (B) The change of the predicted binding scores corresponded to the shift of ELAVL1 binding sites from the wild-type to the URE mutant on the 3′ UTR of gene ERBB2. The bottom shows the locations of URE and miR-331b regions, mutation positions in the URE region, mutation positions in the miR-331b region, mutation positions in both URE and miR-331b regions, and the experimentally detected shift of ELAVL1 binding resulting from the URE mutant, respectively. All mutation sites are represented by the inverted triangles. Abbreviation: WT, wild-type; MT, mutant; URE, U-rich element.
Studying the effects of mutations in RBP binding targets
The mutation data related to splicing events were derived from COSMIC (33). Sequences with mutation sites in the middle and lengths equal to those of the corresponding RBP binding targets were prepared as input samples to DeBooster. For both pathogenic or neutral mutations near 5′ or 3′ splice sites, we selected those single-nucleotide variant (SNV) mutations within 10 nt from splice sites. The lengths of RBP binding targets are usually larger than 20 nt, so generally splice sites were covered by samples centered at mutation positions. In total, we collected 7000 neutral mutations in both regions near 5′ and 3′ splice sites, and 4000 and 20 000 mutations in regions near 5′ and 3′ splice sites, respectively. In Figure 8, the change of the prediction score resulting from a mutation was calculated as ‘(prediction score for the mutant sequence)−(prediction score for the wild-type sequence)’.
Figure 8.

The comparisons between the overall changes of the predicted binding scores of individual RBPs after pathogenic or neutral mutations in regions near 5′ and 3′ splice sites. *P < 0.001, Student’s t test.
In Figure 9 and Supplementary Figure S3, the prediction scores for regions around the mutation sites along both wild-type and mutant sequences were shown. For each selected mutation, we showed the prediction scores for 41 positions, including the mutation site and the flanking regions of 20 nucleotides both upstream and downstream. For each site, its prediction score was calculated using the window centered at this position and of length equal to the average length of the corresponding RBP targets in the training data.
Figure 9.

Examples of the predicted effects on the potentially disease-causing mutations near splice sites or on exonic splicing enhancers (ESEs). (A) The exonic mutations of the SFRS1 binding sites near a 5′ splice site for gene CDH1. (B, C) The mutations of SFRS1 binding sites disrupting or creating exonic splicing enhancer (ESE) motifs for genes TCFIL2 and THRAP3, respectively. The ESE motifs were obtained from (54). (D, E) The exonic mutations of the TIA1 binding sites near the splice sites for genes TRRAP and KTN1, respectively. (F) A mutation near a 5′ splice site of gene ATM that changed the predicted binding scores of both QKI and FUS. Abbreviation: WT, wild-type; MT, mutant.
RESULTS
DeBooster captures the sequence preferences of RBP binding
We used 24 sets of CLIP-seq based data about RBP binding sites to train and validate our prediction model. Details about the datasets can be found in the Materials and Methods section. We first ran a 10-fold cross-validation procedure for each of 24 CLIP-seq datasets to evaluate the overall prediction performance of DeBooster. The hyperparameters in the deep boosting framework were determined using an independent dataset (Methods). We also compared the performance of DeBooster with the state-of-the-art approaches for predicting RBP target sites, including GraphProt (17) and the deep belief net (DBN) method (19). The comparison results (Figure 2A–C) showed that DeBooster can significantly outperform both GraphProt and the DBN method, with the increase of the area under receiver operator characteristic curve (AUROC) by up to 10.1%. Note that GraphProt and the DBN method integrate both RNA sequence and structural information (i.e. RNA secondary structural information (17) or both RNA secondary and tertiary structural profiles (19)) into the prediction framework, while DeBooster requires only RNA sequence information. We also performed additional tests to demonstrate that the performance improvement in DeBooster was attributed to both our new feature encoding scheme (see Figure 1A and Materials and Methods) and the better predictive power of the underlying deep boosting model (Supplementary Notes). We further showed that adding extra structural features did not improve the performance of DeBooster (Supplementary Notes).
Through a transcriptome-wide analysis on RBP binding targets, we also found that the difference in the predicted binding scores of DeBooster over different characterized genomic regions mostly reflected the known functions of individual RBPs (Supplementary Notes). In addition, we examined the sequence motifs of the RBP binding sites generated from training data (Methods). Our results indicated that the sequence motifs resulting from DeBooster agreed well with those reported in the literature (Figure 2D). For example, the binding sequence motif of AGO2 computed by DeBooster was enriched with A, U and C but depleted of G, which was consistent with the previous study (34). PTB, as indicated by its name (polypyrimidine tract-binding protein), mainly binds to the U/C-rich regions (35), which was also reflected in the sequence motif derived from DeBooster. EWSR1, FUS and TAF15 belong to the FET family. Although several works showed that they bind to the GU-rich motif (36,37), recent studies found that the FET protein family prefers binding to the AU-rich stem loops, and the AU-rich sequences achieve higher binding affinities than those enriched with G and U (38). Such an AU-rich pattern was also observed in the sequence motif generated by DeBooster. It has been found that the binding targets of QKI usually contain a core sequence NACUAAY (where Y stands for a pyrimidine) and a half-site UAAY (39). The binding motif of QKI identified by DeBooster also agreed well with such a pattern. DeBooster yielded a U-rich sequence motif for the binding sites of HNRNPC, which can also be supported by a known fact that HNRNPC generally binds to the poly-U tracts (40). According to the DeBooster prediction results, SFRS1 prefers binding to a GA-rich motif, which aligned well with the previous result (41). As shown in the previous study (7), PUM2 binds to a consensus motif UGUANAUA, which shared high similarity with the corresponding binding motif predicted by DeBooster. The majority of the TDP43 binding sites predicted by DeBooster contained the (UG)n motif and was relatively less enriched with A and C. Such an observation agreed well with the previous known result (42). Motifs for all the 24 training datasets and the comparisons between the motifs generated by DeBooster and GraphProt (17) are also provided in Supplementary Notes. Taken together, most of the sequence motifs of RBP binding sites captured by DeBooster were consistent with the previous known results in the literature.
The predictions of DeBooster can be validated through cross-platform datasets
It is well-known that different CLIP-seq experiments can yeild a large fraction of non-overlapping results and individual experiments may miss a vast number of true RBP binding sites (43,44). Here, we showed that the prediction results of DeBooster can be validated through cross-platform CLIP-seq datasets (Figure 3). In particular, we tested DeBooster on different cross-platform ELAVL1 datasets, which displayed a large degree of discrepancy between the original RBP binding targets measured from CLIP-seq experiments (Figure 3A). Such a large variation indicated that in general a single CLIP-seq experiment cannot cover all RBP binding sites and individual datasets may have high false negative rates in current experimental measurement. The tests on the cross-platform ELAVL1 datasets showed that the predictions of DeBooster from one dataset can be well validated by another one collected from a different platform, achieving both high AUROC scores and similar sequence motifs (Figure 3B). We also evaluated the cross-dataset AUROC scores based on the GraphProt (17) predictions, and showed that GraphProt performed less well than DeBooster on this task (Supplementary Notes). In addition, most of the sequence features encoded in DeBooster displayed highly correlated weights except the outliers G and UNNNNU (Figure 3C and D), which was probably due to experimental bias introduced from the original CLIP-seq data. These results implied that the predictions of DeBooster can be well validated through cross-platform CLIP-seq datasets.
Figure 3.

Performance validation of DeBooster through cross-platform CLIP-seq datasets. (A) The proportional Venn diagrams of four ELAVL1 CLIP-seq datasets collected from different experimental platforms. If binding region A from a dataset has at least one nucleotide overlap with binding region B from another dataset, we regarded A and B as a common element of these two datasets. The datasets ELAVL1, ELAVL1(A) and ELAVL1(C) were from the HEK293 cells, while the dataset ELAVL1(B) was from the HeLa cells. (B) The AUROC scores and binding sequence motifs computed by DeBooster using different combinations of training and test datasets. The diagonal scores shown in bold correspond to the cross-validation results in which both training and test datasets were collected from the same experimental platform. (C, D) The plots of the relative weights of individual sequence features computed by DeBooster for the ELAVL1 datasets collected from different experimental platforms, including ELAVL1(B) vs. ELAVL1 (C) and ELAVL1(A) versus ELAVL1 (D). (E, F) The plots of the DeBooster prediction scores for all 8-mers across different RBPs within the same family, including TAF15 versus EWSR1 (E) and FUS versus EWRS1 (F). TAF15, FUS and EWSR1 all belong to the FET family and generally share similar binding preferences.
We also investigated the agreement of the DeBooster prediction results between different RBPs from the same family. In particular, we examined the consistency between the DeBooster prediction scores of 8-mers for TAF15, FUS and EWSR1, all belonging to the FET family. Consistent with the previous results that these three RBPs have a large overlap in binding sites (38), our tests showed that the 8-mers from different RBPs exhibited highly correlated prediction scores (Figure 3E and F). Such observations further supported the above argument that the prediction results of DeBooster can be verified from cross-platform CLIP-seq datasets, even for different RBPs from the same family. These results suggested that DeBooster was not prone to overfitting, and may provide a practically useful tool to analyze high-throughput CLIP-seq data.
The binding scores predicted by DeBooster match the experimentally measured binding affinity data
To investigate whether the prediction results of DeBooster can truly reflect the RBP binding preferences, we further checked the agreement between the binding scores predicted by DeBooster and the experimentally determined binding affinity data. In particular, we checked the agreement between the prediction scores of DeBooster, which was trained using the in vivo CLIP-seq data, and the experimentally determined 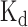 values for two RBPs, including SFRS1 and TDP43 (Figure 4A and B). Our comparison showed that for the 8-mers as the potential RNA targets of SFRS1, the prediction scores of DeBooster closely matched the in vivo measured
values for two RBPs, including SFRS1 and TDP43 (Figure 4A and B). Our comparison showed that for the 8-mers as the potential RNA targets of SFRS1, the prediction scores of DeBooster closely matched the in vivo measured 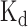 values (45) (Figure 4A). In addition, for the RNA nucleotides as the potential binding targets of TDP43, the prediction scores of DeBooster aligned well with the
values (45) (Figure 4A). In addition, for the RNA nucleotides as the potential binding targets of TDP43, the prediction scores of DeBooster aligned well with the 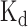 values experimentally measured from the electrophoretic mobility shift assay (EMSA) (46) (Figure 4B).
values experimentally measured from the electrophoretic mobility shift assay (EMSA) (46) (Figure 4B).
Figure 4.

The comparisons between the prediction scores derived by DeBooster and the experimentally determined binding affinity data. (A, B) The plots of the prediction scores derived by DeBooster (which was trained based on CLIP-seq data) versus the experimentally determined 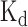 values of different 8-mers or RNA oligonucleotides for SFRS1 and TDP43, respectively. The
values of different 8-mers or RNA oligonucleotides for SFRS1 and TDP43, respectively. The 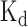 values of SFRS1 were measured in vivo (45), while the
values of SFRS1 were measured in vivo (45), while the 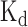 scores of TDP43 were acquired from the electrophoretic mobility shift assay (EMSA) (46). The same terminology as in (46) for the names of RNA oligonucleotides was used for the binding targets of TDP43 (Supplementary Notes).
scores of TDP43 were acquired from the electrophoretic mobility shift assay (EMSA) (46). The same terminology as in (46) for the names of RNA oligonucleotides was used for the binding targets of TDP43 (Supplementary Notes).
The predicted targets of RNA helicases may provide useful hints for understanding the regulation of mRNA degradation
RNA helicases, such as MOV10, regulate the life cycle of mRNAs and thus gene expression by remodeling RNA secondary structures and RNA-protein interactions (47). Here, we showed that the RNA targets of MOV10 predicted by DeBooster can be connected to the regulation of mRNA half-lives and thus may provide useful hints for understanding the functional roles of MOV10 in controlling gene expression. Our analysis was performed on a set of 7000 mRNAs, in which the fold changes of their half-lives had been measured after MOV10 knockdown (31). These mRNAs were basically divided into four groups according to the fold changes of their half-lives, i.e. top 25%, 25–50%, 50–75% and bottom 25%, which corresponded to Group 1, Group 2, Group 3 and Group 4, respectively. Only the bottom group (i.e. Group 4) contained those genes whose expression levels were unchanged or up-regulated after MOV10 knockdown.
Compared to the results derived directly from the original CLIP-seq data (Figure 5A), the fraction of UTRs with MOV10 binding resulting from DeBooster prediction displayed a more evident decreasing trend (Figure 5B). In addition, the sum of all positive prediction scores per UTR, which basically considered both binding strength and the number of hits for the MOV10 binding targets on individual genes, also exhibited the same decreasing order for four groups of genes that were divided and ranked according to the fold changes of mRNA half-lives (Figure 5C). Moreover, when we grouped all transcripts according to the DeBooster prediction scores, the resulting fold changes of mRNA half-lives also presented a similar decreasing trend (Figure 5D). Similar analysis was also conducted using GraphProt (17) for comparison (Supplementary Notes). Although both DeBooster and GraphProt performed comparably well on this task, there is still an advantage to use DeBooster, as it can run much faster than GraphProt, which requires the prediction of secondary structure for each input RNA sequence. Furthermore, the DeBooster prediction scores for seven genes also showed good agreement with the fold changes of mRNA half-lives experimentally measured by qRT-PCR (Figure 5E). Taken together, the above results demonstrated that the binding targets of the RNA helicase MOV10 predicted by DeBooster were associated with the changes of mRNA half-lives. Thus, the prediction results from DeBooster may provide useful clues for further understanding the regulatory mechanisms of RNA helicases on the life cycle of mRNAs.
Figure 5.

Understanding the predicted binding effects of MOV10 on mRNA degradation. (A, B) Fractions of 3′ UTRs with MOV10 binding for four groups classified according to the original CLIP-seq data (A) and the DeBooster prediction results (B), respectively. Genes were evenly separated into four groups according to the fold changes of their mRNA half-lives. Groups 1, 2, 3 and 4 corresponded to top 25%, 25–50%, 50–75% and bottom 25%, respectively. In the DeBooster prediction results, we only considered those robust binding targets with prediction scores > 0.2 (the default threshold was zero and the range of prediction scores was in [–1,1]). (C) The sum of positive prediction scores per UTR for four groups of genes, which were classified and ranked according to the fold changes of their mRNA half-lives in a descending order. *P value < 0.001, Wilcoxon rank sum test. (D) The cumulative distribution on the fold changes of mRNA half-lives for four groups of genes, classified and ranked according to the DeBooster prediction scores in a descending order. That is, Groups 1, 2, 3 and 4 corresponded to genes with top 25%, 25–50%, 50–75% and bottom 25% predicted scores, respectively. The P values were computed using the Wilcoxon rank sum test. (E) The plot of the DeBooster prediction scores versus the fold changes of mRNA half-lives measured by qRT-PCR for seven genes.
DeBooster may distinguish two potentially different types of ADAR1 binding patterns
ADARs are a family of homologous enzymes catalyzing adenosine-to-inosine (A-to-I) editing in the RNA, and have similar double-stranded RNA binding domains (dsRBDs) and a common deaminase domain (48). Despite their major role as RNA-editing enzymes, a fraction of ADAR2 binding events have been confirmed to be ‘non-productive’, that is, these bindings might not trigger any RNA editing (22). On the contrary, those ADAR2 binding events that indeed produce RNA editing were considered ‘productive’. Recent studies showed that many ADAR1 binding sites are distant from the editing sites (49). It was found that ADAR1 actually has diverse functions rather than simply catalyzing RNA editing, and some of these functions are independent of its editing activity (49). To investigate whether ADAR1 also has potentially different binding patterns, such as ‘productive’ and ‘non-productive’ binding modes like ADAR2, we compared the prediction results from three DeBooster models, which were trained using all, productive and non-productive ADAR1 binding sites, respectively.
We first introduced the concept of the binding-editing distance, which was defined as the genomic distance between a known or predicted ADAR1 binding position and its closest editing site. The known RNA editing sites were obtained from the RADAR database (50). Our first model, also called the all-binding model, was trained using all ADAR1 binding sites measured from CLIP-seq experiments (49) as the positive samples. The negative samples were defined as those unbound regions that were adjacent to the positive samples in transcripts and had the lengths equal to those of the corresponding positive samples. In our second model, also called the productive binding model, the CLIP-seq sites (i.e. the ADAR1 binding sites measured from CLIP-seq experiments) with small binding-editing distances (0−100 nt) were used as the positive samples, while the CLIP-seq sites with large binding-editing distances (>1000 nt) together with the adjacent unbound regions were used as the negative samples. In our third model, also called the non-productive binding model, the CLIP-seq sites with large binding-editing distances (>1000 nt) were used as the positive samples, while the CLIP-seq sites with small binding-editing distances (0−100 nt) together with the adjacent unbounded regions were used as the negative samples. We then used the three trained models to search novel ADAR1 binding sites in the human transcriptome. The distances from the three groups of the new predicted binding sites to editing sites were calculated and shown in Figure 6A. The median of the binding-editing distances resulting from the all-binding model was 814 nt (Figure 6A), which was roughly on the same scale as from the original CLIP-seq data (606 nt). The median of the binding-editing distances from the productive binding model was zero (i.e. the ADAR1 binding region contained at least one editing site), which was significantly different from that of the non-productive binding model (4665 nt, Figure 6A). The above results showed that DeBooster may be able to learn the difference between different groups of ADAR1 binding sites, and such difference may be possibly related to the editing activity of the enzyme.
Figure 6.

The comparison results on three different DeBooster models, which were trained using all ADAR1 binding sites identified by CLIP-seq experiments, productive ADAR1 binding sites (i.e. triggering A-to-I editing), and non-productive ADAR1 binding sites (i.e. without triggering A-to-I editing), respectively. (A) The boxplot of the binding-editing distances, which were defined as the genomic distances between the new predicted ADAR1 binding sites and the closet editing sites, for three different DeBooster models. *P value < 0.001, Wilcoxon rank sum test. (B) The sequence motifs of the ADAR1 binding sites identified by three different DeBooster models. More details can be found in the main text.
We also examined the sequence motifs of the ADAR1 binding sites identified by three different DeBooster models (Figure 6B). Although all three sequence motifs showed high GC content, the motif generated by the productive-binding model had relatively higher frequencies of As and Us than those from the other two models. This observation indicated that those ADAR1 binding sites with relatively lower GC content might be more prone to being edited. This result was also in agreement with the known evidence that the published motif of the ADAR1 binding sites (49) contained relatively higher GC content than that of the genomic regions near the editing sites (51). We also trained GraphProt (17) on the same three different datasets, and compared the analysis result to that of DeBooster. More details about the comparison can be found in Supplementary Notes.
Although the results shown in Figure 6 may be caused by the definition of productive and non-productive binding sites in our problem setting, we argued that this is very unlikely the case because of the following reasons. First, although we labeled productive and non-productive binding sites mainly based on the binding-editing distances, such a distance feature was not fed into our model as input data. Once the training data had been selected, DeBooster only used the sequence features of these training data. On the other hand, we showed that our trained model can well capture the intrinsic difference between these two binding types, including the binding-editing distances and the binding motifs (Figure 6). Second, the ADAR binding sites measured from CLIP-seq experiments are generally relatively long (average 190 nt in our training data) compare to those of other RBPs. The difference between the percentages of As in productive and non-productive binding sites were almost negligible (23.4% versus 22.9%). In addition, among all As inside the ADAR1 binding sites, only a small fraction of them were edited (about 2.88% in our training data). Thus, it unlikely that the motif difference shown in Figure 6B was introduced by the bias from our original definition of productive and non-productive binding sites. Therefore, most likely our model can capture and distinguish the intrinsic sequence features of productive and non-productive binding sites in our setting.
To summarize, we can seperate the ADAR1 binding sites into two groups. One contained the binding sites close to the editing sites, while the other covered the binding sites that are thousands of nucleotides away from the editing sites. The different patterns between these two types of binding sites can be learned by DeBooster and reproduced in the DeBooster predictions, which indicated that there might be different regulatory mechanisms underlying these two different groups of ADAR1 binding sites. However, it will still need more comprehensive investigation to study whether these two different groups of binding sites actually truly reflect the different binding behaviors of the RNA editing enzyme, and whether this difference embodies the diverse regulatory roles of ADAR1.
The shift of the predicted RBP binding scores from mutations may predict the antagonizing effect of RBP binding on miRNA repression
RBPs and miRNAs are two classes of essential regulators controlling mRNA degradation and expression, and they often interplay with each other to display co-regulatory effects (52). For example, in the 3′ UTR of an oncogene ERBB2, the RBP ELAVL1 (also called HUR) antagonizes the repression effect of the miRNA miR-331-3p by binding to a U-rich element (URE) near the miRNA target region called miR-331b (23). With a mutant URE, the repression effect of ELAVL1 binding on miR-331-3p is weakened, since the new ELAVL1 binding sites shift to a position that is more distant from the miR-331b region (Figure 7A), and also reduces the binding affinity of ELAVL1 (the magnitude of the experimentally measured 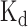 values change from 10−8 M to 10−7 M) (23). Here, we showed that DeBooster can successfully identify this mutational effect that was consistent with the previous experimental observation.
values change from 10−8 M to 10−7 M) (23). Here, we showed that DeBooster can successfully identify this mutational effect that was consistent with the previous experimental observation.
We used the CLIP-seq dataset of ELAVL1 measured from the Hela cells (53) as training data (those overlapping records about the measured binding sites in the 3′ UTR of gene ERBB2 were removed) and performed a comparative study on the predicted binding scores of four cases, i.e. WT-URE/WT-331b, MT-URE/WT-331b, WT-URE/MT-331b and MT-URE/MT-331b, which represented the wild-type sequence, a URE mutant with the wild-type miR-331b region, the wild-type URE with a miR-331b mutant, and a sequence with mutations in both URE and miR-331b regions, respectively (Figure 7B). All the binding scores predicted by DeBooster showed obvious peaks near the URE, indicating the high-affinity binding of ELAVL1 in this region. More importantly, the prediction results of DeBooster displayed a clear position-shifted and affinity-decreased pattern of ELAVL1 binding on a URE mutant (Figure 7B). The curves of the predicted binding scores for WT-URE/WT-331b (i.e. wild-type) and WT-URE/MT-331b (i.e. only mutations in the miR-331b region) had similar shapes, which was consistent with the previous experimental result that the mutations in the miR-331b region rarely affect ELAVL1 binding (23). In addition, the peaks of these two curves were approximately located in positions 50-90 along the 3′ UTR of ERBB2, while the peaks of the other two curves with mutations in the URE region (i.e. MT-URE/WT-331b and MT-URE/MT-331b) were located around positions 45-60. Such a position shift of the ELAVL1 binding sites identified by DeBooster in fact agreed with the previous experimental RNA footprinting results (see Figure 7B in (23)). Moreover, the decrease of the binding scores predicted by DeBooster was also consistent with the loss of the experimentally-determined 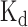 values with respect to the same mutations (23). Taken together, these results indicated that DeBooster can successfully identify the changes of the RBP binding scores caused by the mutations in binding targets which may be used to predict the antagonizing effect of RBP binding on miRNA repression.
values with respect to the same mutations (23). Taken together, these results indicated that DeBooster can successfully identify the changes of the RBP binding scores caused by the mutations in binding targets which may be used to predict the antagonizing effect of RBP binding on miRNA repression.
The prediction scores of DeBooster may provide a useful index to study pathogenic mutations affecting RNA splicing
Recent studies revealed that abnormal splicing play a vital role in development of many human diseases, such as cancer and neurological disorders (55–57). The mutations near splice sites or on splicing regulatory elements, such as exonic splicing enhancers (ESEs) and exonic splicing silencers (ESSs), may influence RNA splicing and cause human diseases by disrupting RBP binding (2). Here, we were particularly interested in whether DeBooster can provide a useful tool to study the mutational effects of sequence variants related to splicing events. We first examined the overall changes of the predicted binding scores of individual RBPs with respect to the sequence variants of their binding targets near 5′ and 3′ splice sites (Methods) and checked whether the DeBooster prediction results can reflect the difference between pathogenic mutations and neutral sequence variants. Our comparisons showed that the changes of the binding scores predicted by DeBooster for a majority of pathogenic sequence variants in regions near 5′ and 3′ splice sites were significantly different from those of neutral mutations (Figure 8). We also confirmed that if the mutations occurred outside the RBP binding sites, the predicted RBP binding scores were only affected to a much smaller extent (Supplementary Notes). In addition, almost all of these pathogenic mutations displayed relatively larger changes in the predicted binding scores than neutral variants. On the other hand, most of the neutral mutations near 5′ and 3′ splice sites displayed similar effects (with only 4 among 20 RBPs showing significant difference with P < 0.001 in the Student’s t test). Furthermore, the pathogenic mutations near splice sites generally showed a greater extent of difference in the predicted binding scores than those pathogenic mutations randomly chosen from the COSMIC records (33) (Supplementary Figure S2). For instance, among 20 RBPs, 15 and 18 proteins exhibited significantly different mutational effects on the pathogenic variants near 5′ and 3′ splice sites, respectively, compared to only seven RBPs in those pathogenic mutations randomly selected from COSMIC (Figure 8 and Supplementary Figure S1). Such an observation implied that the sequence disruptions of the RBP binding targets around splice sites may generally play a more important role in the pathogenesis of a disease. Overall, our studies indicated that the binding scores derived from DeBooster may provide an effective indicator for distinguishing pathogenic mutations from neutral variants in RBP binding targets near splice sites.
Next, we further analyzed the mutational effects predicted by DeBooster for a number of known pathogenic single-nucleotide variants (SNVs) obtained from COSMIC (33). Below we describe several examples (Figure 9). First, a synonymous substitution of the last base in Exon 7 (G to A) of gene CDH1 (which encodes the E-cadherin protein) led to an increase in the SFRS1 binding scores predicted by DeBooster near a 5′ splice site (Figure 9A), which may be related to the dysregulation of CDH1 that causes tumor metastasis (58). Such an observation may also be supported by a previous experimental validation study that this mutation can actually alter splicing by causing intron retention to various extents (59).
As a second example, a mutation from G to A in a TCF7L2 exon (60) disrupted the ESE motifs (which are 6 nt motifs located in exons and bound by SR proteins to promote exon splicing (61)) and suppressed SFRS1 binding (Figure 9B), while a mutation from U to A in a THRAP3 exon (60) enriched the ESE motifs and thus enhanced SFRS1 binding (Figure 9C). Such disruptions in those disease-relevant genes may influence the binding behaviors of the important splicing regulator SFRS1, and thus may be related to the tumorigenesis associated with aberrant splicing (45).
In our third example, the mutation from U to C near a 3′ splice site of gene TRRAP (60) weakened TIA1 binding (Figure 9D). TRRAP interacts with oncoproteins MYC and E2A (62), and its mis-regulation can be heavily related to various types of cancers (63). On the other hand, another mutation from C to U near a 3′ splice site of gene KTN1 (60) strengthened TIA1 binding (Figure 9E). KTN1 encodes kinectin 1, and has been shown to display different splicing patterns in cancers (64). Thus, these two sequence variants in the binding sites of TIA1 may be associated with cancer pathogenesis by changing the alternative splicing modes of its target genes.
Another interesting example is the intronic mutation near a 5′ splice site of gene ATM (60), which increased the binding scores of both FUS and QKI (Figure 9F). Such a mutation may influence the splicing result of this tumor suppressor (i.e. ATM) (65) by creating new potential binding sites for both splicing regulators (i.e. FUS and QKI).
In addition to the above cases, there were other examples to demonstrate that the prediction scores of DeBooster may reflect the pathogenic effects of sequence disruptions in RBP binding. For instance, a substitution from C to U near a 5′ splice site of gene NF1 (60) enhanced HNRNPC binding (Supplementary Figure S3A), which may be associated with the known related neurologic disorders (66). On the other hand, a mutation from U to C near a splice site of the proto-oncogene BRAF (60) decreased the HNRNPC binding score (Supplementary Figure S3B). In addition, a mutation from A to G (60) near a splice site of gene TET2 may help form a novel GU-repeat region for strong TDP43 binding (Supplementary Figure S3C), and thus influence the splicing process. Moreover, the SMAD4 splicing site may be disrupted by the mutation from G to U (60) that may increase the PTB binding score (Supplementary Figure S3D) and thus alter the corresponding splicing result. Both TET2 and the SMAD4 genes act as tumor suppressors (67,68), so the inhibition of their normal splicing may thus facilitate cancer formation.
Taken together, the above examples illustrated that the RBP binding scores predicted by DeBooster may offer a useful index to investigate the pathogenic effects of sequence disruptions related to RNA splicing.
CONCLUSION
We developed DeBooster, a deep boosting based framework to model the sequence binding specificities of RNA-binding proteins (RBPs) from high-throughput CLIP-seq data. Compared to the state-of-the-art methods which usually require both sequence and structure profiles, DeBooster uses only sequence information as input. Tests on 24 CLIP-seq datasets demonstrated that DeBooster can achieve better prediction performance than previous methods. In addition, the binding sites predicted by DeBooster can be validated through cross-platform datasets. Moreover, the prediction scores of DeBooster agreed with the experimentally-determined binding affinity scores, such as in vivo measured 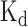 values.
values.
We further showed the great application potentials of DeBooster by applying it to study the regulatory roles of several important RBPs. In particular, we demonstrated that the predicted targets of the RNA helicase MOV10 can better explain its binding effects on the regulation of mRNA degradation than the original CLIP-seq data. In addition, the predicted RBP binding sites may help understand the potentially different binding patterns of the RNA-editing enzymes ADARs. We also showed that a shift of the predicted ELAVL1 binding scores from wild-type to mutant in a U-rich element (URE) region of gene ERBB2 can effectively predict the antagonizing effect of RBP binding on miRNA regulation. Moreover, DeBooster may be used as an effective index to identify pathogenic mutations from normal sequence variants and study the effects of potential disease-causing mutations in RBP binding sites related to splicing. Based on these test results and analyses, we expect that DeBooster will provide a promising tool to analyze more large-scale CLIP-seq data and gain more biological insights related to RBP regulation.
The training datasets used in our study were originally prepared in GraphProt (17), in which the negative data was randomly selected from the unbound regions of target genes. Through this manner, the constructed negative data can include comprehensive information of the background. However, to better control the local sequence bias in the prediction results, using the upstream/downstream regions near target sites as negative samples might be also worthy of consideration. Further work will be needed to improve the quality of training and test datasets to achieve better predictions.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Qiangfeng Zhang and Mr. Hailin Hu, Mr. Bin Zhou and Mr. Xuan He for their helpful discussions about this work. They thank the anonymous reviewers for their helpful comments and suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [61472205]; US National Science Foundation [DBI-1262107, IIS-1646333]; China’s Youth 1000-Talent Program; Beijing Advanced Innovation Center for Structural Biology. Funding for open access charge: China’s Youth 1000-Talent Program.
Conflict of interest statement. None declared.
REFERENCES
- 1. Glisovic T., Bachorik J.L., Yong J., Dreyfuss G.. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008; 582:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scotti M.M., Swanson M.S.. RNA mis-splicing in disease. Nat. Rev. Genet. 2016; 17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dahlberg A.E., Dingman C.W., Peacock A.C.. Electrophoretic characterization of bacterial polyribosomes in agarose-acrylamide composite gels. J. Mol. Biol. 1969; 41:139. [DOI] [PubMed] [Google Scholar]
- 4. Czworkowski J., Odom O.W., Hardesty B.. Fluorescence study of the topology of messenger RNA bound to the 30S ribosomal subunit of Escherichia coli. Biochemistry. 1991; 30:4821–4830. [DOI] [PubMed] [Google Scholar]
- 5. Licatalosi D.D., Mele A., Fak J.J., Ule J., Kayikci M., Chi S.W., Clark T.A., Schweitzer A.C., Blume J.E., Wang X. et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008; 456:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Konig J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J.. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010; 17:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jungkamp A., Munschauer M. et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818. [DOI] [PubMed] [Google Scholar]
- 9. Stoltenburg R., Reinemann C., Strehlitz B.. SELEX-A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007; 24:381. [DOI] [PubMed] [Google Scholar]
- 10. Ray D., Kazan H., Chan E.T., Castillo L.P., Chaudhry S., Talukder S., Blencowe B.J., Morris Q., Hughes T.R.. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009; 27:667. [DOI] [PubMed] [Google Scholar]
- 11. Ray D., Kazan H., Cook K., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Yang A., Na H. et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013; 499:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reyesherrera P.H., Ficarra E.. Computational methods for CLIP-seq data processing. Bioinformatics Biol. Insights. 2014; 2014:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bailey T.L., Boden M., Buske F.A., Frith M.C., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foat B.C., Morozov A.V., Bussemaker H.J.. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Nucleic Acids Res. 2006; 22:doi:10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- 15. Hiller M., Pudimat R., Busch A., Backofen R.. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res. 2006; 34:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kazan H., Ray D., Chan E.T., Hughes T.R., Morris Q.. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLOS Computat. Biol. 2010; 6:e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maticzka D., Lange S.J., Costa F., Backofen R.. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 2014; 15:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831. [DOI] [PubMed] [Google Scholar]
- 19. Zhang S., Zhou J., Hu H., Gong H., Chen L., Cheng C., Zeng J.. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res. 2015; 44:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu B., Yang Y.T., Huang Y., Zhu Y., Lu Z.J.. POSTAR: a platform for exploring post-transcriptional regulation coordinated by RNA-binding proteins. Nucleic Acids Res. 2016; 45:D104–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buratti E., Baralle F.E.. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell. Biol. 2004; 24:10505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klaue Y., Kallman A.M., Bonin M., Nellen W., Ohman M.. Biochemical analysis and scanning force microscopy reveal productive and nonproductive ADAR2 binding to RNA substrates. RNA. 2003; 9:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Epis M.R., Barker A., Giles K.M., Beveridge D.J., Leedman P.J.. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J. Biol. Chem. 2011; 286:41442–41454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saeys Y., Inza I., Larranaga P.. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007; 23:2507–2517. [DOI] [PubMed] [Google Scholar]
- 25. Cortes C., Mohri M., Syed U.. Deep boosting. Proceedings of the 31st International Conference on Machine Learning. 2014. [Google Scholar]
- 26. Freund Y., Schapire R.E.. A decision-theoretic generalization of on-line learning and an application to boosting. 1995.
- 27. Walker S.H., Duncan D.B.. Estimation of the probability of an event as a function of several independent variables. Biometrika. 1967; 54:167–179. [PubMed] [Google Scholar]
- 28. Bartlett P.L., Mendelson S.. Rademacher and gaussian comlexities: risk bounds and structural results. J. Mach. Learn. Res. 2001; 3:463–482. [Google Scholar]
- 29. Friedman J.H. Greedy function approximation: a gradient boosting machine. Ann. Stat. 2001; 29:1189–1232. [Google Scholar]
- 30. Crooks G.E., Hon G.C., Chandonia J., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregersen L.H., Schueler M., Munschauer M., Mastrobuoni G., Chen W., Kempa S., Dieterich C., Landthaler M.. MOV10 Is a 5′ to 3′ RNA helicase contributing to UPF1 mRNA target degradation by translocation along 3′ UTRs. Mol. Cell. 2014; 54:573–585. [DOI] [PubMed] [Google Scholar]
- 32. Raney B.J., Dreszer T.R., Barber G.P., Clawson H., Fujita P.A., Nguyen N., Paten B., Zweig A.S., Karolchik D., Kent W.J.. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2013; 30:1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Forbes S., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S. et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J., Kim T., Nutiu R., Ray D., Hughes T.R., Zhang Z.. Identifying mRNA sequence elements for target recognition by human Argonaute proteins. Genome Res. 2014; 24:775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xue Y., Zhou Y., Wu T., Zhu T., Ji X., Kwon Y., Zhang C., Yeo G.W., Black D.L., Sun H. et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol. Cell. 2009; 36:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lerga A., Hallier M., Delva L., Orvain C., Gallais I., Marie J., Moreaugachelin F.. Identification of an RNA binding specificity for the potential splicing factor TLS. J. Biol. Chem. 2001; 276:6807. [DOI] [PubMed] [Google Scholar]
- 37. Takahama K., Arai S., Kurokawa R., Oyoshi T.. Identification of Ewing’s sarcoma protein as a G-quadruplex DNA- and RNA-binding protein. FEBS J. 2011; 278:988–998. [DOI] [PubMed] [Google Scholar]
- 38. Larsson E.G., Runge S., Nusbaum J.D., Duggimpudi S., Farazi T.A., Hafner M., Borkhardt A., Sander C., Tuschl T.. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011; 18:1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galarneau A., Richard S.. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat. Struct. Mol. Biol. 2005; 12:691. [DOI] [PubMed] [Google Scholar]
- 40. Cienikova Z., Damberger F.F., Hall J., Allain F. H.T., Maris C.. Structural and mechanistic insights into poly(uridine) tract recognition by the hnRNP C RNA recognition motif. J. Am. Chem. Soc. 2014; 136:14536–14544. [DOI] [PubMed] [Google Scholar]
- 41. Sanford J.R., Wang X., Mort M., Vanduyn N., Cooper D.N., Mooney S.D., Edenberg H.J., Liu Y.. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009; 19:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Colombrita C., Onesto E., Megiorni F., Pizzuti A., Buratti E., Ratti A.. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012; 287:15635–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blencowe B.J., Ahmad S., Lee L.J.. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev. 2009; 23:1379–1386. [DOI] [PubMed] [Google Scholar]
- 44. Derrien T., Estelle J., Sola S.M., Knowles D.G., Raineri E., Guigo R., Ribeca P.. Fast computation and applications of genome mappability. PLOS ONE. 2012; 7:e30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anczukow O., Akerman M., Clery A., Wu J., Shen C., Shirole N.H., Raimer A., Sun S., Jensen M.A., Hua Y. et al. SRSF1-regulated alternative splicing in breast cancer. Mol. Cell. 2015; 60:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhardwaj A., Myers M.P., Buratti E., Baralle F.E.. Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. 2013; 41:5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bourgeois C.F., Mortreux F., Auboeuf D.. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat. Rev. Mol. Cell Biol. 2016; 17:426. [DOI] [PubMed] [Google Scholar]
- 48. Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010; 79:321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bahn J.H., Ahn J., Lin X., Zhang Q., Lee J., Civelek M., Xiao X.. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat. Commun. 2015; 6:6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramaswami G., Li J.B.. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014; 42:D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greene T., Bass B.L.. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011; 2:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ciafrè S.A., Galardi S.. microRNAs and RNA-binding proteins: a complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013; 10:934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lebedeva S., Jens M., Theil K., Schwanhäusser B., Selbach M., Landthaler M., Rajewsky N.. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell. 2011; 43:340–352. [DOI] [PubMed] [Google Scholar]
- 54. Yeh R., Sharp P.A., Burge C.B.. Predictive identification of exonic splicing enhancers in human genes. Science. 2002; 297:1007–1013. [DOI] [PubMed] [Google Scholar]
- 55. Oltean S., Bates D.O.. Hallmarks of alternative splicing in cancer. Oncogene. 2014; 33:5311–5318. [DOI] [PubMed] [Google Scholar]
- 56. Pajares M.J., Ezponda T., Catena R., Calvo A., Pio R., Montuenga L.M.. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol. 2007; 8:349–357. [DOI] [PubMed] [Google Scholar]
- 57. Dredge B.K., Polydorides A.D., Darnell R.B.. The splice of life: alternative splicing and neurological disease. Nat. Rev. Neurosci. 2001; 2:43–50. [DOI] [PubMed] [Google Scholar]
- 58. Onder T.T., Gupta P.B., Mani S.A., Yang J., Lander E.S., Weinberg R.A.. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008; 68:3645–3654. [DOI] [PubMed] [Google Scholar]
- 59. Oda T., Kanai Y., Oyama T., Yoshiura K., Shimoyama Y., Birchmeier W., Sugimura T., Hirohashi S.. E-cadherin gene mutations in human gastric carcinoma cell lines. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K. R.M., Ozenberger B., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013; 45:1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhu J., Krainer A.R.. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell. 2001; 8:1351–1361. [DOI] [PubMed] [Google Scholar]
- 62. Mcmahon S.B., Van Buskirk H.A., Dugan K.A., Copeland T.D., Cole M.D.. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998; 94:363–374. [DOI] [PubMed] [Google Scholar]
- 63. Murr R., Vaissiere T., Sawan C., Shukla V., Herceg Z.. Orchestration of chromatin-based processes: mind the TRRAP. Oncogene. 2007; 26:5358–5372. [DOI] [PubMed] [Google Scholar]
- 64. Wang H., Su Y., Han K., Pang X., Peng J., Liang B., Wang S.G., Chen W.. Multiple variants and a differential splicing pattern of kinectin in human hepatocellular carcinoma. Biochem. Cell Biol. 2004; 82:321–327. [DOI] [PubMed] [Google Scholar]
- 65. Morgan S.E., Kastan M.B.. p53 and ATM: cell cycle, cell death, and cancer. Adv. Cancer Res. 1997; 71:1–25. [DOI] [PubMed] [Google Scholar]
- 66. Viskochil D.H. Review article : genetics of neurofibromatosis 1 and the NF1 gene. J. Child Neurol. 2002; 17:562–570. [DOI] [PubMed] [Google Scholar]
- 67. Delhommeau F., Dupont S., James C., Masse A., Couedic J. P.l., Valle V.D., Alberdi A., Dessen P., Fontenay M., Casadevall N., Soulier J., Bernard Olivier, Vainchenker W.. TET2 is a novel tumor suppressor gene inactivated in myeloproliferative neoplasms: identification of a pre-JAK2 V617F event. Blood. 2008; 112:lba-3. [Google Scholar]
- 68. Schwartewaldhoff I., Volpert O.V., Bouck N., Sipos B., Hahn S.A., Kleinscory S., Luttges J., Kloppel G., Graeven U., Eilertmicus C. et al. Smad4/DPC4-mediated tumor suppression through suppression of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.