

Abstract
Programmed DNA elimination is a developmentally regulated process leading to the reproducible loss of specific genomic sequences. DNA elimination occurs in unicellular ciliates and a variety of metazoans, including invertebrates and vertebrates. In metazoa, DNA elimination typically occurs in somatic cells during early development, leaving the germline genome intact. Reference genomes for metazoa that undergo DNA elimination are not available. Here, we generated germline and somatic reference genome sequences of the DNA eliminating pig parasitic nematode Ascaris suum and the horse parasite Parascaris univalens. In addition, we carried out in-depth analyses of DNA elimination in the parasitic nematode of humans, Ascaris lumbricoides, and the parasitic nematode of dogs, Toxocara canis. Our analysis of nematode DNA elimination reveals that in all species, repetitive sequences (that differ among the genera) and germline-expressed genes (approximately 1000–2000 or 5%–10% of the genes) are eliminated. Thirty-five percent of these eliminated genes are conserved among these nematodes, defining a core set of eliminated genes that are preferentially expressed during spermatogenesis. Our analysis supports the view that DNA elimination in nematodes silences germline-expressed genes. Over half of the chromosome break sites are conserved between Ascaris and Parascaris, whereas only 10% are conserved in the more divergent T. canis. Analysis of the chromosomal breakage regions suggests a sequence-independent mechanism for DNA breakage followed by telomere healing, with the formation of more accessible chromatin in the break regions prior to DNA elimination. Our genome assemblies and annotations also provide comprehensive resources for analysis of DNA elimination, parasitology research, and comparative nematode genome and epigenome studies.
Genome maintenance and stability are paramount for organisms. Major genome changes and genome instability can lead to abnormalities, disease, and lethality. While a variety of cellular processes have evolved to ensure genome stability, some organisms have developed processes that lead to regulated genome changes during their life span. These include the genome rearrangements that are critical for generating diversity in immunoglobulins and T-cell receptors in lymphoid lineages of vertebrates (Bassing et al. 2002). More extreme examples include the phylogenetically diverse process of programmed DNA elimination (Wang and Davis 2014b). Programmed DNA elimination occurs in unicellular ciliates where it is involved in generating the macronucleus (the somatic genome) from the micronucleus (the germline genome) during sexual reproduction (Chalker and Yao 2011; Bracht et al. 2013). Many metazoans, including some nematodes, arthropods, cartilaginous fishes, hagfish and lampreys, and other vertebrates, also carry out programmed DNA elimination (Wang and Davis 2014b).
In metazoa, two major forms of programmed DNA elimination have been observed: (1) loss of an entire chromosome or portions of a chromosome contributing to sex determination or (2) chromosome breakage and loss of chromosome regions in somatic precursor cells during early development while the genome in germline cells remains intact.
A number of fundamental questions regarding programmed DNA elimination in multicellular organisms (Wang and Davis 2014b) remain outstanding, including (1) what function(s) does DNA elimination serve, (2) is the function the same in different organisms carrying out this process, (3) what determines where chromosomes break, (4) what determines which portions of chromosomes are kept and which are eliminated, (5) what sequences are eliminated, and (6) how conserved and regulated is this process. Answers to these questions are currently limited. The generation of reference genomes and comparative genome analysis of DNA elimination can provide key information to address these questions.
Programmed DNA elimination has been studied in parasitic nematodes since its discovery by the cell biologist Theodor Boveri in 1887 (Boveri 1887). Boveri observed that the single pair of large chromosomes of the horse parasite Parascaris univalens break in somatic precursor cells during the first few embryonic divisions. Many new chromosomes are formed concomitant with the loss of large portions of the somatic chromosomes during early embryo division. Chromosome breakage and loss does not occur in germline cells, thus generating two separate genomes in the organism: a germline and a somatic genome. A similar process was observed in other ascarid nematodes, including the nematodes of pigs, Ascaris suum, and dogs, Toxocara canis (Meyer 1895; Bonnevie 1902; Walton 1918). A variety of cytological and genomic data (Streit et al. 2016) suggest the following working model for nematode DNA elimination: (1) Chromosomes undergo replication; (2) chromosomes align at the metaphase plate during mitosis; (3) chromosome breaks occur; (4) chromosome fragments are differentially segregated to daughter nuclei based on centromeres/kinetochores localization (Kang et al. 2016); and (5) chromosome fragments that are not segregated into daughter nuclei are relegated to the cytoplasm, where they undergo degradation in subsequent cell cycles, and thereby are eliminated from the somatic genome. This process occurs independently in five precursor somatic cells during early development.
Assembly and annotation of draft genomes of the germline and somatic genomes of A. suum revealed that ∼13% of the A. suum germline genome is eliminated in somatic cells (Wang et al. 2012). Sites were identified where apparent chromosome breaks occur and the ends of retained chromosomes are healed by telomere addition. While the majority of eliminated DNA consists of a 121-bp repetitive sequence (Muller et al. 1982; Streeck et al. 1982; Niedermaier and Moritz 2000), about 700 genes primarily expressed in the germline and early embryo are eliminated in somatic cells (Wang et al. 2012). These data suggest that DNA elimination in A. suum may be a mechanism to silence germline-expressed genes. While the A. suum draft genome sequences provide valuable insights into DNA elimination, the fragmented nature of the genome assemblies results in a limited view of the genome changes that occur and does not offer a large-scale overview of the location of chromosome breaks or their relationship to each other, or the overall organization of germline chromosomes and newly formed somatic chromosomes.
We used optical mapping, Pacific Biosciences (PacBio) sequencing, fosmid end sequencing, and additional short-read sequencing to generate chromosome assemblies of the A. suum germline and somatic genomes to gain additional insight into programmed DNA elimination. To carry out a comparative analysis of DNA elimination in nematodes, we have also assembled and annotated the germline and somatic genomes of the horse parasitic nematode P. univalens. We also defined the chromosomal breaks and DNA loss in the related human nematode parasite Ascaris lumbricoides, which is estimated to infect upward of 1 billion people worldwide (de Silva et al. 2003; Bethony et al. 2006; Hotez et al. 2008; Pullan et al. 2014), as well as in the dog nematode T. canis. These data and analyses provide the most comprehensive comparative analysis of the genome changes that occur in metazoan programmed DNA elimination.
Our improved reference genome assemblies, gene annotations, and transcriptomes in A. suum, a new reference genome for P. univalens, and reanalysis of the T. canis genomes provide new insights into programmed DNA elimination and also comprehensive genome and transcriptome resources for these important parasitic nematodes.
Results
A. suum reference genomes
In-depth analysis of programmed DNA elimination requires comprehensive germline and somatic genome assemblies and their annotation. To improve the germline and somatic genome sequences of A. suum, we carried out additional sequencing and mapping, including 62× coverage of PacBio sequencing, 41× coverage of fosmid end sequencing, four independent Bionano optical mapping analyses (with an average N50 = 2.4 Mb), and 160× Illumina long (2 × 250 bp) reads (see Supplemental Material and Supplemental Table S1). By using these new data, we generated reference-quality (N50 = 4.6 Mb) A. suum germline and somatic genomes (Table 1; Fig. 1; Supplemental Table S2). They represent a 16-fold improvement in germline scaffold N50, from 290 kbp to 4.6 Mbp, and a 65-fold improvement in somatic scaffold N50, from 70 kbp to 4.6 Mbp. The new assemblies increase the Ascaris germline genome size by ∼32.5 Mbp (11%) and the somatic genome by ∼28.4 Mbp (10%) (Table 1). Over 99% of these genome assemblies were validated using long scaffolding data (Table 1). The strategy and approaches used to generate the final assemblies are described in the Supplemental Text (see Improvement of Genome Assemblies).
Table 1.
Ascaris and Parascaris genome assemblies
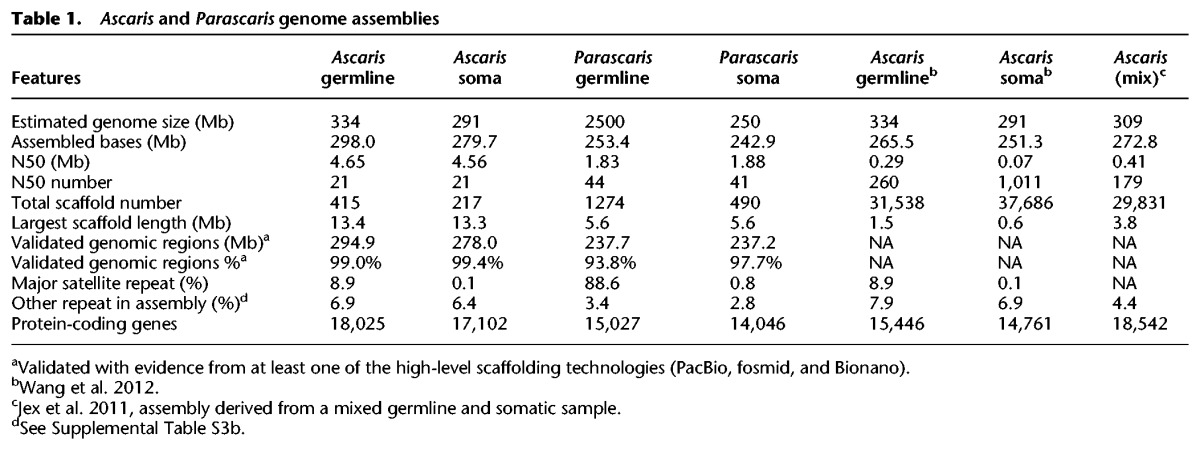
Figure 1.

Ascaris chromosome landscapes and genome annotation. (A) Landscape of three Ascaris chromosomes. Illustrated along the length of three assembled Ascaris somatic chromosomes with centromeric regions (defined by CENP-A ChIP-seq; blue), actively transcribed regions (defined by H3K36me3 ChIP-seq; red), heterochromatic regions (defined by H3K9me3 ChIP-seq; green dots), and putative transposon elements illustrated (TEs defined by sequence homology; orange dots). (B) Ascaris genome browser view. An expanded view of the gene models, histone marks, RNA-seq, small RNA data, and 5′ ends of mRNA from the shaded area of chromosome AsR004 in Figure 1A. ChIP-seq data are from 5 d (32–64 cell) embryos. Units for all tracks are normalized to 10× genome coverage (3 Gbp). (SL) spliced leader sequence.
We have incorporated new comprehensive gene models (see below), genome-wide histone modification ChIP-seq analyses, and RNA expression data (mRNAs and small RNAs) into an accessible genome browser for the A. suum genomes (Fig. 1B; Supplemental Text). Gene density is in general uniformly distributed along chromosomes, with no apparent gene-rich or gene-desert regions (Fig. 1A), and extensive heterochromatic regions in the assembly defined by H3K9me3 marks appear absent. These chromosome level assemblies—along with the improved gene models, histone modification ChIP-seq data, and the developmental expression profiling of mRNAs, lncRNAs, and small RNAs—provide a comprehensive view of gene organization, structure, and expression in A. suum.
Ascaris genes, alternative splicing, and RNA expression
The gene annotations of two previous A. suum draft genomes were mainly based on ab initio gene prediction methods (Jex et al. 2011; Wang et al. 2012). We generated and used additional in-depth RNA-seq and transcriptome data from 25 different tissues or developmental stages to refine gene models in A. suum (see Supplemental Methods). The new genome assembly and transcriptome data significantly improved the gene models. Many previously annotated independent genes (fragments) were merged into single genes (Fig. 2A,B; Supplemental Table S7). We defined or extended mRNA untranslated regions to generate more full-length transcripts (Fig. 2A). We used mRNA 5′ end and spliced leader enriched libraries to define the 5′ ends of mRNAs (see Fig. 1B; Supplemental Fig. S1; Supplemental Text). We found that as many as ∼85% of Ascaris transcripts are trans-spliced, consistent with previous estimates (Maroney et al. 1995). Some genes appear to be organized into polycistronic loci that are resolved into independent mRNAs by spliced leader trans-splicing. However, these loci are relatively infrequent in A. suum compared with Caenorhabditis elegans (Guiliano and Blaxter 2006; Allen et al. 2011). We also defined alternatively spliced transcripts from comprehensive transcriptome data (Fig. 2C; Supplemental Table S4). In addition, we identified more than 5500 new genes in the current genome, a 30% increase over previous assemblies (Fig. 2B; Supplemental Table S7). The extensive additional transcriptome data have also enabled us to define the expression of Ascaris genes through male and female gametogenesis, development, and tissue-specific gene expression (Fig. 2D; Supplemental Table S4). As previously described, extensive transcription occurs prior to pronuclear fusion and in early development (Wang et al. 2014).
Figure 2.

Improved Ascaris gene models, alternative splicing, and RNA expression profiles. (A) Improved Ascaris gene models. Illustrated is a genome browser locus view for selected RNA-seq tracks and a comparison of gene models defined in previous papers (Jex et al. 2011; Wang et al. 2012) and here defined with a gene prediction program (AUGUSTUS) (Stanke et al. 2006) and with new RNA-seq data (Current Gene Models) (see Supplemental Material). Comprehensive RNA-seq data from 25 different developmental stages were used to refine Ascaris gene models. (B) Refined and new gene models. Comparison of the gene models between previous studies and the current version reveals that (1) many previously defined independent genes correspond to single genes and have been merged in the current genome annotations, and (2) an additional approximately 5500 (30%) new genes were identified and annotated in our revision. (C) Alternative splicing in Ascaris and Parascaris. Alternatively spliced isoforms were identified using comprehensive RNA-seq data sets in Ascaris and Parascaris (see Supplemental Material). (D) Ascaris developmental RNA expression profiles. The heatmap illustrates the dynamic expression of 10,874 genes (with average RPKM ≥5 or max RPKM ≥20) across the 25 different developmental stages of Ascaris (see Supplemental Material), including regions of the male germline (1, mitotic region; 2, spermatogenesis; 3, post-meiotic region; 4, seminal vesicle; and 5, spermatids), regions of the female germline (1, mitotic region; 2, early pachytene; 3, late pachytene; 4, diplotene; and 5, oocyte), zygote maturation stages prior to pronuclear fusion isolated from the uterus (z1-4) (see Wang et al. 2014), early development stages (1c, 1-cell [24 h of development at 30°C]; 2c, 2-cell [46 h]; 4c, 4-cell [64 h]; 16c, 16-cell [96 h]; 5d, 5-day [32–64 cells]; and 7 d, 7-d [about 256 cells]), larvae (L1 and L2), and adult somatic tissues. (Mu) Muscle; (In) intestine; (Ca) carcass, which includes the cuticle, hypodermis, muscle, nervous system, and pharynx.
Histone ChIP-seq analysis and annotation
Mapping of the data from ChIP-seq of CENP-A and other histone marks (Kang et al. 2016) onto the new chromosomal level genome assemblies confirm that centromere/kinetochore regions (defined by CENP-A and CENP-C colocalization) are negatively correlated with transcription along Ascaris holocentric chromosomes. The Ascaris centromere/kinetochore regions are not associated with H3K9me3, repetitive sequences, or potential transposable elements (Fig. 1A). H3K9me3 localizes primarily to repetitive sequences, H3K4me3 localizes primarily to putative promotor regions, H3K36me3 marks actively transcribed genes, and H3K27me3 likely marks facultative heterochromatin.
Ascaris sex chromosomes
Ascaris has an XO sex-determination system, as in most other nematodes. Consequently, we predicted that genomic read coverage for the sex chromosomes in a female (XX) would be twofold higher compared with the coverage observed in a male (X). We used this difference in read coverage to identify at least 23 scaffolds (spanning 62.4 Mb or ∼23% of the sequences) that appear to belong to A. suum sex chromosomes (A. suum have eight haploid sex chromosomes in the somatic cells) (Supplemental Fig. S2). Analysis of RNA expression data from these genome scaffolds demonstrated that these scaffolds are silenced during spermatogenesis, supporting their identification as sex chromosomes (Supplemental Fig. S3A; Supplemental Table S6; Supplemental Text). RNA expression analysis indicates that genes derived from sex chromosomes are expressed at a higher level (more than 2×) than autosomal genes during Ascaris oocyte maturation (Supplemental Fig. S3B; Supplemental Table S6). The identification of Ascaris sex chromosomes and characterization of their expression during development provide a resource to study Ascaris sex chromosome gene expression and regulation, as well as data for comparative analysis of sex chromosomes in nematodes (Supplemental Fig. S3C,D; Supplemental Table S6; see Supplemental Text).
Parascaris genome assemblies
P. univalens is a related ascarid parasite of horses that also undergoes programmed DNA elimination during early development (Boveri 1887; Niedermaier and Moritz 2000; Bachmann-Waldmann et al. 2004). The P. univalens germline genome comprises a single pair of chromosomes with an estimated haploid genome size of ∼2.1 Gb (Moritz and Roth 1976). Our sequencing data suggest the germline genome is ∼2.5 Gb. DNA elimination results in loss of large terminal portions of the chromosomes and the generation of ∼35 (1n) smaller chromosomes (Muller and Tobler 2000; Niedermaier and Moritz 2000). We applied a strategy similar to the one used in Ascaris to sequence and de novo assemble the Parascaris germline and somatic genomes, with the goal of comparing DNA elimination and its evolution in these parasitic nematodes. Since the large Parascaris germline consists of ∼89% repetitive sequence (Supplemental Table S3), we were not able to obtain high-quality scaffolding data for the germline genome. The germline genome instead was built using a high-quality somatic genome as the reference (see Supplemental Material). The assemblies are at the chromosomal level with N50 = 1.8 Mb, and ∼95% of the assembled sequences were validated with PacBio reads or optical mapping data (Table 1; Supplemental Table S1). DNA elimination in P. univalens leads to the loss of ∼89% of the germline genome consisting of primarily short satellite repeats (∼2.2 GB) but significantly also the loss of ∼10 Mb of unique sequence (Fig. 3B).
Figure 3.

Ascaris and Parascaris programmed DNA elimination genome changes. (A) Breakpoints and eliminated sequences in Ascaris. Illustrated are Ascaris genomic regions (scaffolds) that are partially or completely eliminated (blue in germline scaffold ring). Scaffolds with DNA breakpoints are shown in red within the germline scaffolds ring (the largest outer circle). The positions for the 40 identified DNA break regions are shown as black lines connecting the largest and smallest circles. Genomic regions (scaffolds) eliminated were concatenated for illustration and are shown as a blue block (ring). The somatic scaffolds track indicates the retained somatic sequences. Telomeres are indicated as yellow boxes. Note that all breaks are healed by new telomere addition. The syntenic regions and conserved genes between Ascaris and Parascaris are illustrated in the light blue ring/track. Gene transcript levels (derived from RNA-seq data) for the germline, two-cell embryo, and several somatic tissues are illustrated in the inner circles. Note the high level of RNA expression in the testis corresponding to DNA eliminated regions. In addition, a few of the eliminated genes (six out of 921) appear to be expressed in the soma due to paralogous genes that are retained or to low level contamination of highly expressed germline genes (see Supplemental Text). (B) Breakpoints and eliminated sequences in Parascaris. Presentation is the same as in Figure 3A. Circle plots are not drawn to scale. (C) DNA elimination at the chromosome level in Ascaris and Parascaris (see text).
Parascaris transcriptome
We generated and used transcriptome data to annotate the Parascaris genome as described for the A. suum genomes (see Supplemental Material) and compared the developmental expression profiles between Ascaris and Parascaris during early development (Wang and Davis 2014a; Wang et al. 2014). Early development in the two nematodes appears identical except that cell cycle length is about 10 times shorter in Parascaris. Interestingly, we found that Parascaris exhibits a larger number of maternally contributed transcripts and appears to rely on fewer zygotically transcribed genes for early development compared to A. suum (Supplemental Fig. S4; Supplemental Table S5; see Supplemental Text). These data suggest that Parascaris embryos may be more dependent on maternally deposited RNA than A. suum to drive early development.
Ascaris and Parascaris comparative genomics
Ascaris and Parascaris exhibit very strong synteny in their genome structures (Fig. 3A,B), with 86% of the assembled Ascaris genome and 88% of the assembled Parascaris genome in syntenic blocks (Supplemental Figs. S5, S6; Supplemental Table S8). Parascaris somatic genome assemblies have ∼40 Mb less sequence than Ascaris (Table 1). The larger A. suum somatic genome size appears largely due to more gene insertions observed in the syntenic blocks (Supplemental Fig. S6). We also identified a core set of orthologous gene groups (2623) that are shared among the three related ascarid nematodes (genes in A. suum, P. univalens, and T. canis that are absent in the free-living nematode C. elegans) (Supplemental Fig. S7A; Supplemental Table S9). This data set provides a framework for future work to identify genes associated with this clade of nematodes and possible genes associated with parasitism.
T. canis genomic sequences and DNA elimination
T. canis is a parasite of dogs that undergoes programmed DNA elimination (Walton 1918; Muller and Tobler 2000). We analyzed publicly available raw genomic reads from an immature (Zhu et al. 2015) and a mature adult Toxocara male (50 Helminth Genome Initiative at the Wellcome Trust Sanger Institute; http://www.sanger.ac.uk/science/collaboration/50hgp). Analysis of publicly available Toxocara RNA expression data and differences in genomic reads between the mature adult and the immature male indicates that the immature male genome reads correspond primarily to the somatic genome, whereas the mature male genome reads correspond primarily to the germline genome (Supplemental Fig. S8). By using the two sets of raw genomic data, we created a revised Toxocara germline genome assembly for our use to analyze DNA elimination in Toxocara (see Supplemental Material). By using read depth and new telomere addition sites in the somatic reads (Wang et al. 2012), we identified 32 sites where chromosomal breaks and telomere healing occur. We also identified 29 Mb (8.8%) of repetitive sequence and ∼20 Mb (6%) of unique DNA (about 2000 genes, 10% of all genes) that are eliminated from the Toxocara germline to form the somatic genome (Supplemental Fig. S8; Supplemental Table S3; Supplemental Text).
A. lumbricoides genome and DNA elimination
A. lumbricoides is an important parasite of humans, infecting upward of 1 billion people (de Silva et al. 2003; Bethony et al. 2006; Hotez et al. 2008; Pullan et al. 2014). We sequenced and assembled the germline and somatic genomes (intestine) of two female A. lumbricoides worms expelled from humans following a single 400-mg dose of albendazole (Easton et al. 2016). The genomes of A. suum and A. lumbricoides are highly similar with nucleotide identify of >99%. Virtually all A. lumbricoides reads can be mapped to the A. suum germline assembly; the two genomes appear to differ only in SNPs. The sequences eliminated in A. lumbricoides and the DNA break regions involved in DNA elimination are the same as observed in A. suum (see below). Several studies indicate cross-infectivity between Ascaris worms from pigs and humans can occur, and there has been considerable discussion over whether A. suum and A. lumbricoides should be considered distinct species (Betson et al. 2013; Betson and Stothard 2016; da Silva Alves et al. 2016; Soe et al. 2016). The A. lumbricoides genome will enable additional in-depth studies to help determine if the two nematodes are independent species or strains of the same species.
Nematode DNA elimination: repetitive sequences lost
We identified both unique and repetitive sequences eliminated in these nematodes. Satellite sequences represent the major sequences lost but differ in composition among the genera (see Supplemental Table S3). In Ascaris, the dominant eliminated sequence is a 120-bp satellite sequence corresponding to 9% (∼ 30 Mb) of the germline, as previously described (Roth and Moritz 1981; Streeck et al. 1982). (This repeat was previously described as 121 bp; however, our analysis indicates that the major form is 120 bp, while the 121 bp is a variant that constitutes 10% of the satellite sequences.) In Parascaris, two short tandemly repeated satellites (a pentanucleotide, TTGCA, and a decanucleotide, TTTGTGCGTG) are the primary sequences eliminated as previously described (Niedermaier and Moritz 2000). We estimate these two repeats represent ∼50% and ∼38% of the germline genome, respectively, corresponding to a loss of ∼2.2 Gb (89% of the germline genome) during DNA elimination. A GA-rich repeat comprising ∼20 Mb (∼1%) of the Parascaris germline genome is also eliminated (Supplemental Table S3). In Toxocara, a 49-bp satellite sequence (9% of the genome) and a 32-bp interspersed repeat (0.4% of the genome) are the two main repetitive elements eliminated (Supplemental Table S3). In all three nematodes, almost all (>99%) of these satellite repeats are eliminated to form the somatic genomes. The eliminated repeats are primarily present as long tandem repeats (>100 kb) in the Ascaris and Parascaris genomes. The long repetitive segments make it very difficult to incorporate them accurately into the genome assemblies (see Discussion and Supplemental Text). In Ascaris, the satellite repeats are typically not located within 50 kb of the chromosomal breakage regions (see below).
Nematode DNA elimination: loss of approximately 1000–2000 germline-expressed genes
All the nematodes eliminate unique sequences. These sequences represent approximately 1000 genes in Ascaris (an increase of 300 genes over the previous genome assembly) and Parascaris (13 Mb for Ascaris and 10 Mb for Parascaris), corresponding to ∼5% of germline genes (Supplemental Tables S4, S5, S11). Fifty percent of the eliminated genes are conserved between Ascaris and Parascaris. In T. canis, we estimate that ∼20 Mb of DNA and approximately 2000 genes (10% of germline genes) are eliminated (Supplemental Fig. S8; Supplemental Table S11 and Supplement Text). In all the nematodes, the eliminated genes are primarily expressed in the germline and early embryo (Fig. 4A,B; Supplemental Table S11). These data reinforce the view that DNA elimination serves to silence a group of germline-expressed genes in nematodes (Wang et al. 2012).
Figure 4.

Conservation of eliminated nematode genes and their expression during spermatogenesis. (A) Conservation of eliminated genes among Ascaris, Parascaris, and Toxocara (see Supplemental Material). The color-coded values illustrate the direct gene comparison for each genus. Note that there is more than one value as not all gene comparisons among these nematodes correspond to a 1:1 match. (B) Expression of Ascaris genes that are eliminated in all three nematodes, eliminated in two nematodes, or the eliminated genes unique to Ascaris (for description of the stages, see Fig. 2D). (C) Heatmap showing the expression of conserved versus nonconserved eliminated Ascaris genes in different developmental stages (for description of the stages, see Fig. 2D).
Conservation of eliminated genes and function
Thirty-five percent of the eliminated genes are conserved among all the nematodes (Fig. 4A). These genes likely represent a core set of the key eliminated genes. Our Gene Ontology analysis revealed that these conserved genes are highly enriched in biological processes related to gamete generation and germ cell development, regulation of translation, protein modification, and the Wnt signaling pathway (see Supplemental Table S12). Many have protein kinase activity, are RNA 3′ UTR binding proteins, or bind ribonucleotides. The encoded proteins are predicted to be found in P granules, germ plasm, ribonucleoprotein complexes, and the synaptonemal complex (Supplemental Table S12). Interestingly, the gene expression pattern of genes eliminated in Ascaris that are conserved in all three nematodes is that they are mainly expressed during Ascaris male spermatogenesis. The nonconserved eliminated genes are more typically expressed in the ovary and early embryos (Fig. 4B,C; Supplemental Table S11). Consistent with this, we also found the conserved eliminated genes in Parascaris and Toxocara are preferentially expressed in male germline tissues (Supplemental Fig. S9; Supplemental Table S11). This suggests that a common function for nematode DNA elimination is to remove genes that are specifically involved in the development and maturation of the male germ cells.
DNA elimination: identification of chromosome break regions
We defined chromosomal breaks as locations in the germline genome that correspond to sites of telomere addition in the somatic genome with the concomitant loss of sequences to one side of the break (Wang et al. 2012). The improved Ascaris genome assembly enabled us to further characterize 40 DNA sites where chromosomal breakage occurs within 36 Ascaris scaffolds (Fig. 3A). These telomere addition sites become the ends of the new somatic chromosomes. These break sites are limited to specific locations (∼3–6 kb; see below) within chromosomes, revealing high fidelity of break site selection. These breaks occur at the same location in all five presomatic cells that undergo elimination during early development and are the same in males and females across different individuals. However, within these regions the exact position of telomere addition exhibits some heterogeneity (see below). Therefore, these regions will be called chromosome breakage regions (CBRs) as previously described (Muller et al. 1991; Jentsch et al. 2002; Bachmann-Waldmann et al. 2004). Our Ascaris somatic assembly contains nine fully assembled chromosomes with eight of them derived from DNA breaks in germline chromosomes followed by new telomere addition (Fig. 3A; Supplemental Table S10). We also identified 46 and 32 CBRs in the Parascaris and Toxocara germline chromosomes, respectively (Fig. 3B; Supplemental Fig. S8; Supplemental Table S10).
DNA elimination: CBRs are not conserved among all nematodes
When we compare the CBRs between Ascaris and Parascaris, we found that 28 (70%) of the Ascaris breaks can be matched to 26 (57%) Parascaris breaks (Supplemental Table S10). This suggests that many of the breakpoints are conserved between these two nematodes. However, comparison of the Toxocara CBRs with Ascaris and Parascaris indicated that only a few are conserved among all these nematodes. Notably, the sequences for most of the nonconserved CBRs in these nematodes can readily be mapped onto the other ascarid genomes. We analyzed the SNP density and insertion/deletion rate within CBR regions and compared them to other regions of the genome (see Supplemental Table S10). Notably, CBR regions have a higher density of SNPs and indels compared with the exons, the genes, and the whole genome. These data suggest that the CBR regions are not under high selective pressure and indicate that they are some of the more variable regions in the genomes. Overall, sequence analysis suggests there is less selective pressure to retain specific sequences within the Ascaris CBRs compared with other non-CBR regions of the Ascaris genome. We speculate that other characteristics of the regions, such as epigenetic features, are more important than the actual sequence in the CBR for DNA elimination process.
DNA elimination: Telomere addition sites are heterogeneous
We previously showed that the A. suum chromosomal breakage regions are conserved in all five independent somatic precursor cells in the embryos that undergo DNA elimination as well as among different individuals (Wang et al. 2012). From this limited sampling, we found that the telomere addition sites for each chromosome DNA elimination event are heterogeneous over a 500- to 2000-bp region. With the updated Ascaris and the new Parascaris genomes, we analyzed the CBRs more comprehensively to obtain additional insights into how the breaks and telomere healing are generated. In the adult intestine of a single worm, where all cells are derived from a single DNA elimination event, we typically observed two sites where a new telomere is added (see Fig. 5A,B; Supplemental Fig. S10). We interpret these distinct events as the telomere healing for each of the two homologous chromosomes. In tissues derived from several different presomatic cells that each undergo independent DNA elimination (such as the carcass of a single nematode derived from five different cells undergoing elimination at the four- to 16-cell stage of development), the number of telomere addition sites increases (up to 10), presumably reflecting the healing events associated with each chromosome pair that has undergone DNA elimination.
Figure 5.

Heterogeneity of telomere addition sites. (A) Telomere addition sites on somatic chromosomes following DNA elimination. A telomere addition site in A. suum (break_a16) and the corresponding telomere addition sites in A. lumbricoides or in P. univalens (break17) are illustrated. The shaded area corresponds to the defined CBRs (determined by the breadth of telomere addition sites at a break in a population of cells), and the red ticks are the frequency and position of the observed telomere addition sites. The center of the CBR is defined as where the highest density of observed telomere addition sites is found in the population. The regions to the left (negative) correspond to retained DNA, while regions to the right (positive) correspond to eliminated DNA. The read frequency was normalized to 50× genome coverage (with 100-bp read length). Note that in an individual there are a limited number of sites that undergo telomere healing. In contrast, in a population, the breadth of telomere addition sites observed is the sum of the independent events in each individual. (B) Compilation of the number of observed telomere addition sites for all 40 breakpoints in Ascaris and 46 breakpoints in Parascaris. (C) Chromosomal breakage region size defined for Ascaris and Parascaris. The region is defined by the extent of all telomere addition sites at a break area.
We next generated and analyzed sequences derived from 60 individual A. lumbricoides intestinal samples and millions of A. suum 7-d embryos (256-cell embryos) and Parascaris L1 larvae (both these embryos and larvae are derived from five independent DNA eliminations in precursor somatic cells). These data reveal the breadth of telomere healing sites randomly located within a 3- to 6-kb region for all Ascaris and Parascaris CBRs (Fig. 5; Supplemental Fig. S10). This rather narrow and consistent window size for telomere addition suggests a constrained mechanism for telomere healing and/or the chromosome breaks during DNA elimination. Additional analyses of the telomere healing events indicates (1) there is no sequence bias for where the telomere sequence is added within the CBRs; (2) telomere priming for healing typically uses only 1 or 2 nucleotides (nt), and this can occur from any position within the telomere sequence (TTAGGC); (3) there is a variant (TTAAGC, 22%) for the first telomere unit added, but all subsequently added sequences are (TTAGGC)n repeats; and (4) no additional sequences are added at the healing sites between the germline and the added telomeric sequences, suggesting healing likely occurs independently of any DNA repair activity (Supplemental Fig. S11).
Changes in DNA accessibility in the chromosomal breakage regions
Despite having identified a large set of CBRs, we were not able to identify any clear sequence or structural features (including using sequence motif prediction by MEME Suite, Z-DNA prediction by Z-Hunt, palindromes, repetitive sequences, etc.) associated with the CBRs and the sites of telomere healing. However, we cannot rule out the presence of degenerate motifs that are difficult to identify. We hypothesized that DNA/chromatin regions where the chromosome breaks occur might be more accessible at the onset of DNA elimination. To explore this possibility, we examined the accessibility of DNA at break sites to transposon insertion using transposase-accessible chromatin sequencing (ATAC-seq) (Buenrostro et al. 2013). This method measures the accessibility of DNA to transposon insertion, providing a relative measure of DNA accessibility in different regions of the genome. ATAC-seq peaks (sites of DNA accessibility) are enriched at the promoter regions of active genes, where H3K4me3 is enriched (Fig. 6A). As gene promoters typically have accessible DNA, these data suggest that ATAC-seq can identify accessible chromatin in A. suum (Fig. 6A, see red arrows). Notably, for all 40 Ascaris break sites, the CBR regions become more accessible just prior to DNA elimination (four-cell stage at 60 h) compared with earlier embryo stages (one-cell) or germline tissues (testis and ovary) (Fig. 6B). These open regions correspond to the 3- to 6-kb chromosomal regions where telomere healing occurs (see Fig. 5). These chromosomal breakage regions remain open for a few cell cycles after DNA elimination (to 5 d, 32–64 cells) but then become no longer accessible in 7-d (256 cells) embryos and in somatic tissues (Fig. 6B,C). We speculate that more accessible chromatin in the CBRs just before elimination might be due to reduced number or compactness of nucleosomes or other epigenetic changes. However, we observed no differences in the histone modifications analyzed, including H3K4me3, H3K36me3, H3K27me3, H4K20me1, CENP-A, and H3K9me2/3 compared with other regions of the genome. We speculate that other epigenetic factors, such as specialized histone modifications, small RNAs, or the 3D organization of the chromosomes, might lead to the more open chromatin observed. These unknown epigenetic factors might recruit the telomere addition machinery to the regions and thus further open the CBRs in the subsequent cell cycles after elimination. Finally, following the breaks and telomere addition, we observed that H3K9me3 increases in the region adjacent to the telomere addition sites. Overall, these data indicate that chromosomal breakage regions in Ascaris are more accessible just before and during DNA elimination.
Figure 6.

Increased chromatin accessibility is associated with Ascaris CBRs. (A) ATAC-seq identifies accessible chromatin at Ascaris promoters. Gene models, transcripts (red, plus strand; blue, minus strand) and H3K4me3 ChIP-seq illustrated in a genome region of 5-d embryos (32- to 64-cell stage). For ATAC-seq data, red arrows indicate peaks of more accessible chromatin near transcriptional start sites, while the blue arrow points to open chromatin within a gene. Note the ATAC-seq signal is enriched at promoters/transcription start sites. Also illustrated are RNA-seq data for 5-d embryos. (B) Ascaris CBRs exhibit increased chromatin accessibility just prior to DNA elimination. The developmental ATAC-seq profile for a DNA breakpoint region (from AgB03) is illustrated. Note that in addition to the transcription start site–associated ATAC-seq peaks, a broad area of open chromatin appears within the break region at the four-cell stage (60 h) immediately prior to DNA elimination. This region remains open through early development (5 d; gastrulation) but is closed in late embryos (7 d; morphogenesis) and somatic tissues (intestine and muscle). (C) Chromatin accessibility for 40 Ascaris breakpoint regions. Illustrated are the breakpoints with their 5-kb flanking regions. Upstream of the breakpoints (−5 kb to 0) are the retained DNA, while downstream regions (0 to 5 kb) are the eliminated regions. Note the open chromatin at the breakpoints in 60 h (immediately prior to DNA elimination) that increases and persists in 5-d embryos. In addition, note that the open regions correspond exactly to the CBRs defined by chromosomal breaks and telomere addition.
DNA elimination does not lead to a telomere position effect
Changes in the chromosomal location of a gene, including a move to near a telomere, alter gene expression (Huang et al. 1996; Ottaviani et al. 2008). The improved Ascaris genome assembly and additional transcriptome data enabled us to further examine whether genes whose chromosomal positions move to the end of the chromosome, adjacent to a telomere, undergo changes in gene expression following DNA elimination. We asked whether we could identify a pattern of gene silencing or activation for genes within 50 kb of the new chromosome ends. Overall, expression of genes within 50 kb of the new chromosome ends does not change significantly, though a few genes immediately adjacent to the new telomere exhibit a trend toward gene silencing (Supplemental Table S13). However, it remains to be determined if the removal of repetitive sequences from chromosomes through DNA elimination may impact gene expression and 3D chromosomal organization.
Discussion
DNA elimination in nematodes occurs primarily in the parasitic nematodes of the order Ascaridida, where the process has been described in 10 species (Muller and Tobler 2000; Streit et al. 2016) since its discovery in Parascaris in 1887 (Boveri 1887). Here, we sequenced, assembled, and annotated or reassembled the genomes of four of these parasitic nematodes, including A. suum (pigs), A. lumbricoides (humans), P. univalens (horses), and T. canis (dogs) with the goal of carrying out a comparison of DNA elimination among these nematodes.
Conservation of eliminated sequences and DNA breaks among ascarid nematodes
Eliminated repetitive sequences
Comparison of DNA elimination among the three ascarid genera (Ascaris, Parascaris, and Toxocara) reveals that most of the sequence eliminated are repetitive sequences. Notably, the eliminated repetitive sequences differ in all the three genera and include pentanucleotide (TTGCA, 941.9 Mb), decanucleotide (TTTGTGCGTG, 1253.1 Mb), or GA-rich (GAGAGTGAGA, 20.1 Mb) tandem repeats in Parascaris (89% of the germline genome), 49-bp tandem repeats (28.8 Mb) in Toxocara (8.8% of the germline genome), and a 120-bp tandem repeat (29.7 Mb) in Ascaris (8.9%) (see Supplemental Table S3). Over 99% of these repeats are eliminated from the germline genome to form the somatic genomes in each nematode.
Repetitive sequences can cause problems in DNA replication and can be deleterious to the genome. Thus, their maintenance in the germline remains enigmatic, particularly in the extreme case where 89% of the germline genome is repetitive and eliminated from the somatic genome of Parascaris. Their retention in the germline suggests a key property or function in the germline that must outweigh potential deleterious consequences for their maintenance and replication. The repeats may facilitate homologous recombination during meiosis, enable genome evolution, or serve as spacer or scaffolding. Notably, the elimination of repetitive sequences is a common feature of DNA elimination in all organisms examined to date (ciliates, nematodes, copepod crustaceans, hagfish, and lampreys) (Kubota et al. 1997; Kohno et al. 1998; Degtyarev et al. 2004; Drouin 2006; Zagoskin et al. 2008; Smith et al. 2009, 2012; Chalker and Yao 2011; Nowacki et al. 2011; Coyne et al. 2012; McKinnon and Drouin 2013; Grishanin 2014; Sun et al. 2014).
One goal of improving the germline genome of Ascaris was to provide a chromosomal context for the location of the repetitive elements that are eliminated and to gain insight into their potential contribution to the mechanisms of DNA elimination. While the PacBio and fosmid sequencing and optical mapping greatly improved the genome assembly, the context and organization of most of the 120-bp repetitive elements remain a challenge. Several lines of evidence suggest that the majority of these elements are in tandem repeats of >100 kb. First, FISH labeling of the 120-bp repeats in Ascaris previously showed they exist in large blocks on many chromosomes, with many present in megabase blocks (Niedermaier and Moritz 2000). Second, sequencing reads with the 120-bp repeats, including PacBio reads as long as 40 kb, consist primarily only of 120-bp repeat sequences with very few reads corresponding to the junction of 120-bp repeat and unique sequences. Third, there remains an estimated 150 unscaffolded large gaps in our germline genome assembly. If these gaps are predominantly due to the 30 Mb of the 120-bp repeat, we estimate that each block of the 120-bp repeat is ∼200 kb. The length for these large satellite repeats prohibits their incorporation into the genome assembly. Since the satellite repeats are mostly not within 50 kb of the CBR regions, it seems unlikely that they help directly to identify where the breaks occur. These repetitive sequences are preferentially marked with H3K9me3, suggesting they are heterochromatic.
Eliminated genes
The most striking aspect of DNA elimination is the loss of unique sequences from the germline to form the somatic genome. This leads to elimination of 1000 (∼5% of the genes in Ascaris and Parascaris) to 2000 genes (∼10% of the genes in Toxocara). The eliminated genes in all three nematodes are primarily expressed in the germline and early embryo, supporting the view that DNA elimination in nematodes is a mechanism for silencing germline-expressed genes (Wang et al. 2012). It should be noted that not all germline-expressed genes are eliminated in these nematodes. Over 50% of the eliminated genes are conserved between Ascaris and Parascaris, ∼35% among all three nematodes. These data indicate that we have identified a conserved, core set of genes that are targeted for elimination. Expression analysis suggests that this core set of conserved genes is preferentially expressed during male spermatogenesis. This suggests that one function for DNA elimination in nematodes is to regulate genes that are specifically involved in the development and maturation of the male germ cells. Growing evidence indicates that elimination of germline-expressed genes may also be a common feature of all organisms with programmed DNA elimination (Smith et al. 2012; Chen et al. 2014; Bryant et al. 2016; Lin et al. 2016).
Chromosomal break regions (CBRs)
Our analysis of the genome changes resulting from DNA elimination indicates that chromosomes break at specific genomic locations. Specific chromosome regions are then retained, while others are eliminated. We have previously shown in A. suum that differential CENP-A localization on chromosomal regions defines which portions of chromosomes are retained or eliminated (Kang et al. 2016). A. suum chromosomes are holocentric in the germline with CENP-A and kinetochores distributed along the length of the chromosomes. During early development, CENP-A becomes reduced or absent in regions of the chromosomes that will be lost during DNA elimination. Unique sequences and genes lost are associated with chromosome regions that have lost CENP-A and centromeres/kinetochores. Thus, both the generation of chromosome breaks and the relocalization of centromeres/kinetochores play key roles in DNA elimination. We note that most of the eliminated genes are clustered together in the genome. These clustered genes are interspersed within large blocks of the 120-bp eliminated repetitive sequences.
We have defined the genomic regions where the ends of new chromosomes are formed and new telomere sequences are added in somatic cells. However, our current data do not necessarily identify where double-stranded break(s) occurs in the chromosome during DNA elimination. Following a double-stranded break, the DNA might be resected before telomere healing occurs or internal telomerase priming sites may be used for telomere addition. Thus, sites of telomere healing may not be coincident with double-strand break locations. In addition, in contrast with what has been observed in ciliate programmed DNA rearrangements (Betermier and Duharcourt 2014; Yao et al. 2014; Yerlici and Landweber 2014), we did not observe any chromosomal fusions or rearrangements associated with DNA elimination in any of the nematodes. We conclude that complex removal of internally eliminated sequences or the sequence rearrangements that have been observed in ciliates does not appear to occur in these nematodes.
A key feature of the nematode chromosomal breakpoint regions first characterized by Muller and colleagues is their heterogeneous nature, leading to the term chromosomal break regions, or CBRs (Muller et al. 1991; Jentsch et al. 2002; Bachmann-Waldmann et al. 2004). We have extended these observations and extensively characterized the precise positions where telomere healing occurs within CBRs for all the breaks identified, providing chromosomal context for CBRs in three different nematodes. The chromosomal breakage regions are 3–6 kb in length and occur in precise locations within the megabase chromosomes in all five precursor somatic cells that undergo elimination. Telomere addition sites occur throughout the entire CBR region and are randomly distributed within the region. Analysis of the sequences at and around the telomere addition sites indicates there is no sequence bias for where telomere healing occurs; SNP and indel analysis suggests that the CBR sequences are some of the more variable regions in the genome. Our data further suggest that telomere healing typically occurs by priming using 1 to 2 nt from within the telomere sequence. The lack of any new or altered sequences at the sites of healing suggests that DNA repair does not occur prior to telomere healing.
In addition, no clear sequence motifs, sequence bias, or structural features appear to be associated with the CBRs. This suggests that the mechanism of chromosomal breakage is sequence independent. Just prior to DNA elimination, our ATAC-seq data indicate that these chromosomal regions become more accessible. These regions of more accessible chromatin correspond exactly to the CBRs defined by where telomere healing is observed. How this more accessible chromatin leads to or facilitates chromosomal breakage remains to be determined. The open chromosomal regions identified by ATAC-seq are only present at the CBRs where telomere healing occurs and are not found throughout the eliminated regions, suggesting that DNA breaks may only occur within the CBRs. While we assume that single break sites occur during elimination, we cannot exclude the possibility that the entire region of a chromosome that is destined for elimination shatters or is fragmented into many pieces. Notably, chromosomal breakage regions occur both between and within genes. Many of the breaks occur between genes with opposing transcription units. It remains to be determined whether DNA replication, transcription complex interference, and/or replication and transcription collisions or other specific cellular processes contribute to the breaks in cells undergoing DNA elimination.
Earlier work by Muller et al. demonstrated that several CBRs were conserved between Ascaris and Parascaris (Bachmann-Waldmann et al. 2004). Our analyses extend these observations and demonstrate that >50% of the chromosomal breakpoint regions are conserved between Ascaris and Parascaris. However, conservation of the location of break sites drops dramatically with evolutionary divergence, as very few of the breakpoints are conserved among all three nematodes. This supports the idea that the mechanism of chromosome breakage is likely sequence independent and not strictly constrained by chromosomal contexts. In contrast, DNA break regions in some ciliates occur at conserved sequences. A 15-bp conserved motif is used in Tetrahymena (Yao et al. 1990), whereas a different 10-bp sequence in Euplotes defines where chromosomal breaks will occur (Baird and Klobutcher 1989). However, in other ciliates (Paramecium and isotrichs) no sequences or structures that guide chromosome breakage are apparent (Betermier and Duharcourt 2014; Yerlici and Landweber 2014). Finally, a palindrome was observed at one break site in the sea lamprey (Smith et al. 2012). Thus, there appears to be significant variation in the sequence requirements for chromosome breakage in different organisms, suggesting potential diverse mechanisms for the generation of chromosomal breaks during DNA elimination.
Increased number of chromosomes in the somatic genomes and number of CBRs
A clear contrast in the genomes of A. suum and P. univalens is the chromosomal organization of the germline genomes. P. univalens has one haploid chromosome of ∼2.5 Gb. In contrast, the A. suum germline genome consists of 19 haploid autosomes and five sex chromosomes. Strikingly, the somatic genomes of the two nematodes following DNA elimination are similar: 36 for Ascaris and 35 for Parascaris. T. canis has 18 haploid germline chromosomes (12 autosomes and six sex chromosomes), which increases to 36 following DNA elimination (Walton 1918). As previously noted, we observed no fusion or recombination of chromosomes resulting from nematode DNA elimination. DNA breaks that occur at the end of a chromosome with the loss of the terminal sequences do not increase the number of the somatic chromosomes (see Fig. 3C). In the Parascaris genome, 70 [2 + (34 × 2)] CBRs in its single chromosome are required to produce the 34 new somatic cell chromosomes. In our current Parascaris germline sequence assembly, we identified 46 (66%) CBRs. It is likely the other 24 CBRs are either in poorly assembled and repetitive-rich regions or within repeats that cannot be identified. In Ascaris we identified 40 CBRs. Our data suggest that CBRs are located within multiple Ascaris germline chromosomes, with many of these breaks leading to the loss of the ends of chromosomes without increasing chromosome number (Fig. 3C). The number of CBRs (32) identified in Toxocara is likely underestimated due to the limited number of somatic reads and the quality of the draft genome. An open question is whether one function of DNA elimination in these nematodes is to generate a consistent set of somatic chromosomes (35–36) from different numbers of germline chromosomes. Interestingly, various Parascaris species that produce similar numbers of somatic chromosomes have been described with one, two, or three haploid germline chromosomes, further illustrating the plasticity of the germline chromosomes and a constant number of somatic chromosomes.
DNA elimination in other nonascarid nematodes
Previous studies suggested that DNA elimination likely does not occur in the nematode C. elegans or in Panagrellus redivivus (Emmons et al. 1979; de Chastonay et al. 1990). In a separate study, we sequenced a wild-type C. elegans N2 strain and a glp-4 mutant that lacks a germline (J Seydoux, D Rasoloson, RE Davis, S Gao, J Wang, unpubl.). Comparison of the two genome sequences did not identify differences between the glp-4 mutant, which consists almost entirely of somatic cells, and the wild-type N2 strain. This further supports the conclusion that large-scale DNA elimination does not occur in C. elegans. However, we cannot exclude the possibility that some genome changes might occur in a few specific cells in C. elegans.
Comprehensive genome resources and annotations for A. suum and P. univalens
The improved A. suum genomes have an N50 of 4.6 Mb, with several chromosomes fully assembled. The improved genome annotations include an additional 5500 genes; comprehensive developmental profiles of mRNAs, lncRNAs, and small RNAs; alternative splicing isoforms; characterization of the 5′ ends of the mRNAs; and genome-wide histone modifications (H3K4me3, H3K36me3, H4K20me1, H3K9me2, H3K9me3, H3K27me3, and CENP-A). We believe this is the broadest and most comprehensive set of annotations for a parasitic nematode. The P. univalens genomes are also reference quality with annotations from comprehensive transcriptome data. Finally, we have generated a new germline genome assembly for T. canis for comparative genome analyses. These data sets offer important resources for the parasitology community.
Conclusions
Programmed DNA elimination in several presomatic cells during nematode early development involves specific chromosome breaks, telomere healing of these breaks, and the retention or elimination of specific chromosomal regions. We assembled and compared chromosomal level genomes (germline and somatic) of a group of parasitic nematodes that undergo programmed DNA elimination. The sequences eliminated include repeats (9%–90% of the germline genome) that are different in each nematode genus, as well as 1000–2000 genes (5%–10% of the germline genome). The eliminated genes of these three nematode genera include a core set of eliminated genes that are primarily expressed during and contribute to spermatogenesis. The chromosomal break regions are not well conserved, the breaks are not sequence dependent, and the chromatin around the break regions becomes more accessible just prior to DNA elimination. Overall, these findings suggest that nematode DNA elimination is a conserved mechanism for silencing germline-expressed genes in the soma. In addition, DNA elimination is a highly precise and regulated process in presomatic cells of the early embryo that specifically acts on defined target regions of chromosomes in a developmentally regulated manner. Our work provides a comprehensive analysis of programmed DNA elimination in nematodes, as well as genome and transcriptome resources for these important parasitic nematodes.
Methods
Parasite material
Collection of A. suum tissues, zygotes, and zygote embryonation were as previously described (Wang et al. 2011, 2014). Parascaris material was collected from young foals; methods for Parascaris tissues, zygotes, and zygote embryonation were carried out as for Ascaris except embryonation was at 37°C. A. lumbricoides adult females were obtained from stool after a single 400-mg dose of albendazole (Easton et al. 2016). Additional information on parasite samples can be found in the Supplemental Materials and Methods.
Genome sequencing, assembly, and annotation
Illumina paired-end and mate-pair libraries were constructed and sequenced using standard protocols. PacBio data were obtained using P6-C4 chemistry at the University of Washington PacBio Sequencing Services facility (https://pacbio.gs.washington.edu/). The fosmid library was made using a pNGS FOS vector (Lucigen) and end sequenced with Illumina. Optical mapping was carried out on a Bionano Genomics Irys. The genomes were assembled through iterations of processes including de novo assembly, step-wise scaffolding, validation with optical maps, and then followed by reassembly of unvalidated regions. In brief, initial contigs were assembled with Illumina reads, and these contigs were scaffolded with mate-paired, PacBio, and/or fosmid end reads. The scaffolds were mapped to Bionano optical maps, and those mapped regions were kept as confirmed scaffolds. Genomic reads not matching the confirmed scaffolds were subject to reassembly using the same process to yield additional confirmed scaffolds. The cycle was repeated until no further scaffolds could be confirmed. A final scaffolding step was performed to generate the final assemblies. The genomes were annotated mainly using RNA-seq data. The annotations were further improved with ab initio gene prediction tools. Detailed procedures for the libraries construction, sequencing, assembly, and annotation are described in the Supplemental Material.
Transcriptome sequencing, histone ChIP-seq, and ATAC-sequencing
Transcriptomes for Ascaris germline tissues and Parascaris samples were done as previously described (Wang et al. 2014). Histone ChIP-seq was carried out as previously described (Kang et al. 2016). ATAC-seq was carried out as previously described (Buenrostro et al. 2013). Additional information can be found in the Supplemental Material.
Data analysis
Bioinformatic analyses, including 5′ mRNA analysis, RNA-seq and expression analysis, ChIP-seq analysis, sex chromosome identification and expression analysis, genome synteny analysis, orthologous genes groups and analysis, CBR identification, motif analysis, comparative genomic analysis, and ATAC-seq analysis are described in the Supplemental Material.
Data access
The Whole Genome Shotgun projects for the A. suum germline, A. suum somatic, P. univalens germline, and P. univalens somatic from this study have been submitted to GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers AEUI00000000, ANBK00000000, NJFU00000000, and NINM00000000, respectively. The versions described in this paper are versions AEUI03000000, ANBK02000000, NJFU01000000, and NINM01000000, respectively. The raw genomic sequencing reads from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) and can be found under the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) accession numbers PRJNA62057 for Ascaris and PRJNA386823 for Parascaris. The RNA-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE99523 for Ascaris and GSE99524 for Parascaris. Data are also made available in the UCSC Genome Browser track data hubs (https://genome.ucsc.edu/cgi-bin/hgHubConnect) using the “My Hubs” tab with the following link: http://amc-sandbox.ucdenver.edu/User14/genomes.txt. Data will also be made available in the WormBase ParaSite (http://parasite.wormbase.org/index.html).
Supplementary Material
Acknowledgments
We thank Richard Komuniecki, Jeff Myers, and Routh Packing Co. for their support and hospitality in collecting Ascaris material and Mark Johnson and Jay Hesselberth for comments on the manuscript. S.G. was supported in part by the China Scholarship Council (201604910424). This work was supported in part by National Institutes of Health grants to R.E.D. (AI049558 and AI114054) and to J.W. (AI125869).
Author contributions: J.W. and R.E.D. conceived and designed the project. J.W. carried out DNA isolation, nuclei isolation, and Illumina genome library construction; Y.S., B.Z., and S.H. prepared fosmid libraries and carried out their sequencing. Y.M. and P-Y.K. carried out optimal mapping. J.W. carried out RNA isolation and prepared the RNA-seq libraries. M.Z. constructed the 5′ mRNA libraries. Y.K. carried out ChIP-seq experiments. J.W. carried out ATAC-seq. J.W., S.G., and L.K.W. carried out bioinformatic analyses. M.K.N. provided P. univalens material. A.E. and T.B.N. provided A. lumbricoides sequence data. J.W. and R.E.D. wrote the paper and all authors read and edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.225730.117.
References
- Allen MA, Hillier LW, Waterston RH, Blumenthal T. 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann-Waldmann C, Jentsch S, Tobler H, Muller F. 2004. Chromatin diminution leads to rapid evolutionary changes in the organization of the germ line genomes of the parasitic nematodes A. suum and P. univalens. Mol Biochem Parasitol 134: 53–64. [DOI] [PubMed] [Google Scholar]
- Baird SE, Klobutcher LA. 1989. Characterization of chromosome fragmentation in two protozoans and identification of a candidate fragmentation sequence in Euplotes crassus. Genes Dev 3: 585–597. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Swat W, Alt FW. 2002. The mechanism and regulation of chromosomal V(D)J recombination. Cell 109(Suppl: S45–S55. [DOI] [PubMed] [Google Scholar]
- Betermier M, Duharcourt S. 2014. Programmed rearrangement in ciliates: paramecium. Microbiol Spectr 2 10.1128/microbiolspec.MDNA3-0035-2014. [DOI] [PubMed] [Google Scholar]
- Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ. 2006. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet 367: 1521–1532. [DOI] [PubMed] [Google Scholar]
- Betson M, Stothard JR. 2016. Ascaris lumbricoides or Ascaris suum: What's in a name? J Infect Dis 213: 1355–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betson M, Nejsum P, Stothard JR. 2013. From the twig tips to the deeper branches: new insights into evolutionary history and phylogeography of Ascaris. In Ascaris: the neglected parasite (ed. Holland CL), pp. 265–285. Academic Press, London. [Google Scholar]
- Bonnevie K. 1902. Über Chromatindiminution bei Nematoden. Jena Z Naturwiss 36: 275–288. [Google Scholar]
- Boveri T. 1887. Über Differenzierung der Zellkerne wahrend der Furchung des Eies von Ascaris megalocephala. Anat Anz 2: 688–693. [Google Scholar]
- Bracht JR, Fang W, Goldman AD, Dolzhenko E, Stein EM, Landweber LF. 2013. Genomes on the edge: programmed genome instability in ciliates. Cell 152: 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant SA, Herdy JR, Amemiya CT, Smith JJ. 2016. Characterization of somatically-eliminated genes during development of the sea lamprey (Petromyzon marinus). Mol Biol Evol 33: 2337–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Yao MC. 2011. DNA elimination in ciliates: transposon domestication and genome surveillance. Annu Rev Genet 45: 227–246. [DOI] [PubMed] [Google Scholar]
- Chen X, Bracht JR, Goldman AD, Dolzhenko E, Clay DM, Swart EC, Perlman DH, Doak TG, Stuart A, Amemiya CT, et al. 2014. The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell 158: 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne RS, Lhuillier-Akakpo M, Duharcourt S. 2012. RNA-guided DNA rearrangements in ciliates: Is the best genome defence a good offence? Biol Cell 104: 309–325. [DOI] [PubMed] [Google Scholar]
- da Silva Alves EB, Conceicao MJ, Leles D. 2016. Ascaris lumbricoides, Ascaris suum, or “Ascaris lumbrisuum”? J Infect Dis 213: 1355. [DOI] [PubMed] [Google Scholar]
- de Chastonay Y, Muller F, Tobler H. 1990. Two highly reiterated nucleotide sequences in the low C-value genome of Panagrellus redivivus. Gene 93: 199–204. [DOI] [PubMed] [Google Scholar]
- de Silva NR, Brooker S, Hotez PJ, Montresor A, Engels D, Savioli L. 2003. Soil-transmitted helminth infections: updating the global picture. Trends Parasitol 19: 547–551. [DOI] [PubMed] [Google Scholar]
- Degtyarev S, Boykova T, Grishanin A, Belyakin S, Rubtsov N, Karamysheva T, Makarevich G, Akifyev A, Zhimulev I. 2004. The molecular structure of the DNA fragments eliminated during chromatin diminution in Cyclops kolensis. Genome Res 14: 2287–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin G. 2006. Chromatin diminution in the copepod Mesocyclops edax: diminution of tandemly repeated DNA families from somatic cells. Genome 49: 657–665. [DOI] [PubMed] [Google Scholar]
- Easton AV, Oliveira RG, O'Connell EM, Kepha S, Mwandawiro CS, Njenga SM, Kihara JH, Mwatele C, Odiere MR, Brooker SJ, et al. 2016. Multi-parallel qPCR provides increased sensitivity and diagnostic breadth for gastrointestinal parasites of humans: field-based inferences on the impact of mass deworming. Parasit Vectors 9: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons SW, Klass MR, Hirsh D. 1979. Analysis of the constancy of DNA sequences during development and evolution of the nematode Caenorhabditis elegans. Proc Natl Acad Sci 76: 1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishanin A. 2014. Chromatin diminution in Copepoda (Crustacea): pattern, biological role and evolutionary aspects. Comp Cytogenet 8: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiliano DB, Blaxter ML. 2006. Operon conservation and the evolution of trans-splicing in the phylum Nematoda. PLoS Genet 2: e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotez PJ, Brindley PJ, Bethony JM, King CH, Pearce EJ, Jacobson J. 2008. Helminth infections: the great neglected tropical diseases. J Clin Invest 118: 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Stoffel R, Tobler H, Mueller F. 1996. A newly formed telomere in Ascaris suum does not exert a telomere position effect on a nearby gene. Mol Cell Biol 16: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch S, Tobler H, Muller F. 2002. New telomere formation during the process of chromatin diminution in Ascaris suum. Int J Dev Biol 46: 143–148. [PubMed] [Google Scholar]
- Jex AR, Liu S, Li B, Young ND, Hall RS, Li Y, Yang L, Zeng N, Xu X, Xiong Z, et al. 2011. Ascaris suum draft genome. Nature 479: 529–533. [DOI] [PubMed] [Google Scholar]
- Kang Y, Wang J, Neff A, Kratzer S, Kimura H, Davis RE. 2016. Differential chromosomal localization of centromeric histone CENP-A contributes to nematode programmed DNA elimination. Cell Rep 16: 2308–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno S-H, Kubota S, Nakai Y. 1998. Chromatin diminution and chromosome elimination in hag fishes. In The biology of hagfishes (ed. Jorgensen JM, et al. ). Chapman & Hall, London. [Google Scholar]
- Kubota S, Ishibashi T, Kohno S. 1997. A germline restricted, highly repetitive DNA sequence in Paramyxine atami: an interspecifically conserved, but somatically eliminated, element. Mol Gen Genet 256: 252–256. [DOI] [PubMed] [Google Scholar]
- Lin CG, Lin IT, Yao MC. 2016. Programmed minichromosome elimination as a mechanism for somatic genome reduction in Tetrahymena thermophila. PLoS Genet 12: e1006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney PA, Denker JA, Darzynkiewicz E, Laneve R, Nilsen TW. 1995. Most mRNAs in the nematode Ascaris lumbricoides are trans-spliced: a role for spliced leader addition in translational efficiency. RNA 1: 714–723. [PMC free article] [PubMed] [Google Scholar]
- McKinnon C, Drouin G. 2013. Chromatin diminution in the copepod Mesocyclops edax: elimination of both highly repetitive and nonhighly repetitive DNA. Genome 56: 1–8. [DOI] [PubMed] [Google Scholar]
- Meyer OTB. 1895. Cellulare Untersuchungen an Nematoden-Eiern. Jena Z Naturwiss 29: 391–410. [Google Scholar]
- Moritz KB, Roth GE. 1976. Complexity of germline and somatic DNA in Ascaris. Nature 259: 55–57. [DOI] [PubMed] [Google Scholar]
- Muller F, Tobler H. 2000. Chromatin diminution in the parasitic nematodes Ascaris suum and Parascaris univalens. Int J Parasitol 30: 391–399. [DOI] [PubMed] [Google Scholar]
- Muller F, Walker P, Aeby P, Neuhaus H, Felder H, Back E, Tobler H. 1982. Nucleotide sequence of satellite DNA contained in the eliminated genome of Ascaris lumbricoides. Nucleic Acids Res 10: 7493–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, Wicky C, Spicher A, Tobler H. 1991. New telomere formation after developmentally regulated chromosomal breakage during the process of chromatin diminution in Ascaris lumbricoides. Cell 67: 815–822. [DOI] [PubMed] [Google Scholar]
- Niedermaier J, Moritz KB. 2000. Organization and dynamics of satellite and telomere DNAs in Ascaris: implications for formation and programmed breakdown of compound chromosomes. Chromosoma 109: 439–452. [DOI] [PubMed] [Google Scholar]
- Nowacki M, Shetty K, Landweber LF. 2011. RNA-mediated epigenetic programming of genome rearrangements. Annu Rev Genomics Hum Genet 12: 367–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Gilson E, Magdinier F. 2008. Telomeric position effect: from the yeast paradigm to human pathologies? Biochimie 90: 93–107. [DOI] [PubMed] [Google Scholar]
- Pullan RL, Smith JL, Jasrasaria R, Brooker SJ. 2014. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit Vectors 7: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth GE, Moritz KB. 1981. Restriction enzyme analysis of the germ line limited DNA of Ascaris suum. Chromosoma 83: 169–190. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Antonacci F, Eichler EE, Amemiya CT. 2009. Programmed loss of millions of base pairs from a vertebrate genome. Proc Natl Acad Sci 106: 11212–11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Baker C, Eichler EE, Amemiya CT. 2012. Genetic consequences of programmed genome rearrangement. Curr Biol 22: 1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soe MJ, Kapel CM, Nejsum P. 2016. Ascaris from humans and pigs appear to be reproductively isolated species. PLoS Negl Trop Dis 10: e0004855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. 2006. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34: W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeck RE, Moritz KB, Beer K. 1982. Chromatin diminution in Ascaris suum: nucleotide sequence of the eliminated satellite DNA. Nucleic Acids Res 10: 3495–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit A, Wang J, Kang Y, Davis RE. 2016. Gene silencing and sex determination by programmed DNA elimination in parasitic nematodes. Curr Opin Microbiol 32: 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wyngaard G, Walton DB, Wichman HA, Mueller RL. 2014. Billions of basepairs of recently expanded, repetitive sequences are eliminated from the somatic genome during copepod development. BMC Genomics 15: 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton AC. 1918. The oogenesis and early embryology of Ascaris canis werner. J Morphol 30: 527–603. [Google Scholar]
- Wang J, Davis RE. 2014a. Contribution of transcription to animal early development. Transcription 5: e967602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Davis RE. 2014b. Programmed DNA elimination in multicellular organisms. Curr Opin Genet Dev 27C: 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Czech B, Crunk A, Mitreva M, Hannon G, Davis RE. 2011. Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res 21: 1462–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mitreva M, Berriman M, Thorne A, Magrini V, Koutsovoulos G, Kumar S, Blaxter ML, Davis RE. 2012. Silencing of germline-expressed genes by DNA elimination in somatic cells. Dev Cell 23: 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Garrey J, Davis RE. 2014. Transcription in pronuclei and one- to four-cell embryos drives early development in a nematode. Curr Biol 24: 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MC, Yao CH, Monks B. 1990. The controlling sequence for site-specific chromosome breakage in Tetrahymena. Cell 63: 763–772. [DOI] [PubMed] [Google Scholar]
- Yao MC, Chao JL, Cheng CY. 2014. Programmed genome rearrangements in Tetrahymena. Microbiol Spectr 2 10.1128/microbiolspec.MDNA3-0012-2014. [DOI] [PubMed] [Google Scholar]
- Yerlici VT, Landweber LF. 2014. Programmed genome rearrangements in the ciliate Oxytricha. Microbiol Spectr 2 10.1128/microbiolspec.MDNA3-0025-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagoskin M, Grishanin A, Korolev A, Palenko M, Mukha DV. 2008. Characterization of Cyclops kolensis inter-simple sequence repeats in germline and postdiminution somatic cells. Doklady Biochem Biophys 423: 337–341. [DOI] [PubMed] [Google Scholar]
- Zhu XQ, Korhonen PK, Cai H, Young ND, Nejsum P, von Samson-Himmelstjerna G, Boag PR, Tan P, Li Q, Min J, et al. 2015. Genetic blueprint of the zoonotic pathogen Toxocara canis. Nat Commun 6: 6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.