

Abstract
Pseudomonas aeruginosa is an opportunistic human pathogen which is prevalent in hospitals and continues to develop resistance to multiple classes of antibiotics. Historically, β-lactam antibiotics have been the first line of therapeutic defense. However, the emergence of multidrug-resistant (MDR) strains of P. aeruginosa, such as AmpC β-lactamase overproducing mutants, limits the effectiveness of current antibiotics. Among AmpC hyper producing clinical isolates, inactivation of AmpG, which is essential for the expression of AmpC, increases bacterial sensitivity to β-lactam antibiotics. We hypothesize that inhibition of AmpG activity will enhance the efficacy of β-lactams against P. aeruginosa. Here, using a highly drug resistant AmpC inducible laboratory strain PAO1, we describe an ultra-high throughput whole cell turbidity assay designed to identify small molecule inhibitors of the AmpG. We screened 645K compounds to identify compounds with the ability to inhibit bacterial growth in the presence of Cefoxitin; an AmpC inducer, and identified 2,663 inhibitors which were also tested in the absence of Cefoxitin to determine AmpG specificity. The Z′ and S:B were robust at 0.87 ± 0.05 and 2.2 ± 0.2, respectively. Through a series of secondary and tertiary studies, including a novel luciferase based counterscreen, we ultimately identified 8 potential AmpG specific inhibitors.
Keywords: Pseudomonas, AmpG, Gram negative bacteria
Introduction
The rise in microbial resistance and emergence of pan-resistant bacterial strains demands urgent attention and great efforts to produce new antibiotics that can help overcome this critical problem. Recognizing the severity of this issue and hoping to encourage accelerated R&D efforts, in February 2017 the World Health Organization (WHO) released a list of antibiotic-resistant priority pathogens that agency officials believe are the greatest threat to human health.1 Highest priority are three types of gram-negative bacteria that are resistant to multiple antibiotics, including carbapenems: carbapenem-resistant Acinetobacter baumanni, carbapenem-resistant Pseudomonas aeruginosa, and carbapenem-resistant, extended-spectrum beta lactamase (ESBL)–producing Enterobacteriaceae. At present the number of new antibiotics in the discovery pipeline is roughly a tenth of what it was in the 1980s.2 The CDC reports >2 million infections per year in the US are resulting from multidrug resistant (MDR) bacteria and while the economic impact on our health care system is difficult to accurately estimate, the per person cost increase for treatment of MDR is tremendous.3 Often the available antibiotics are failing and in many cases, including hospital-acquired infections, there are no effective treatments.
Gram-negative bacteria are a major cause of nosocomial infections in immunocompromised patients, whereas in particular Pseudomonas aeruginosa causes serious infections.4, 5 For example, nearly 80% of patients with Cystic Fibrosis become infected with P. aeruginosa by early adulthood, and a majority of them succumb to the infection caused by this microorganism.6 It is also commonly found in patients with diabetes and military personnel who suffer from chronic wounds7, 8. Fortunately, we now have a better understanding of the underlying principles of bacterial resistance mechanisms and have the methods and tools to overcome some of these problems.9
β-lactams are important anti-pseudomonas antibiotics and P. aeruginosa readily becomes resistant to all of the widely-used antipseudomonal β-lactams, leading to therapeutic failure. The most common mechanism of resistance involves overexpression of a chromosomal gene (AmpC) which encodes a class C β-lactamase. Unfortunately, currently available β-lactamase inhibitors are ineffective for this class of enzymes.
We had previously identified and characterized an AmpG gene in this bacterium, which encodes a membrane protein that is essential for the expression of AmpC.10 The AmpG is a trans-membrane permease for the peptidoglycan building blocks used in cell wall recycling, thus it is highly conserved among Gram negative bacteria. Interestingly, P. aeruginosa with an AmpG mutation is much more sensitive to β-lactams than the AmpC mutant, indicating that the AmpG is required for multiple mechanisms of β-lactam resistance.10 Consistent with this, sensitivity to β-lactams can be restored among pan-β-lactam resistant clinical isolates of P. aeruginosa by knocking out their AmpG genes, demonstrating that AmpG is an ideal target for the control of resistance against β-lactam antibiotics.11
In this study, we have focused on P. aeruginosa and specifically on the PAO1 strain because it exhibits multi-drug resistance through overexpression of AmpC β-lactamase.12 The complete genome of PAO1 has been elucidated which also makes it a good candidate organism for drug discovery.13 Treatment of PAO1 with Cefoxitin rapidly induces AmpC in vitro, and resistance is discernable within a few hours10. This same phenomenon is also observed clinically and establishes a clear need for better use and more effective antibiotics.14
Our previous work demonstrated that inactivation of AmpG gene leads to the loss of AmpC β-lactamase expression and therefore loss of drug resistance10. A later study with multidrug resistant clinical isolates further proved the importance of AmpG in the β-lactam antibiotic resistance.11 Recently, a structure function analysis of AmpG characterized this protein in the context of mutational analysis of the transmembrane domain and found conserved amino acids, which when disrupted, affected AmpG function and β-lactamase production, suggesting it is an appropriate target for small molecule intervention.15 This laid the groundwork and provided the tools necessary to attack AmpG as a novel target for drug discovery.
With these methods available, we aimed to develop an HTS-amenable 1536-well format anti-microbial assay for the identification of novel small molecule inhibitors of the AmpG in PAO1. Here, we describe the phenotypic whole cell turbidity assay development and ultra-high throughput screen (uHTS) campaign to identify small molecule inhibitors that may serve as potential leads for further development of drugs to counter bacterial β-lactam resistance.
Materials and Methods
Minimum Inhibitory Concentration (MIC) and Synergy Determination
Laboratory strain PAO1 and its AmpG deletion mutant (PAO1ΔG) have previously been described.10 Bacterial pharmacology was confirmed and antibiotic MICs were determined by the standard broth dilution method according to M7-A7 Clinical and Laboratory Standards Institute (CLSI), with Mueller Hinton Cation Adjusted Broth (CAMHB). 106 CFU/mL was inoculated into CAMHB containing an equal volume of two fold serial dilutions of antibiotics. Cultures were incubated for 17 h at 37°C. To aid in visualization of the MIC in 1536 well format we applied an imaging technique to monitor all sample wells simultaneously using the HIAPI.16 In this sense, the HIAPI was only used as a qualitative visual confirmation that, per CLSI, bacterial growth matched the expected MIC and also matched the MIC as determined using the absorbance readout described below. The MIC was defined as the lowest drug concentration that prevented visible bacterial growth (Figure 1).
Figure 1.

Confirmation of PAO1 pharmacology in 1536wpf. 0.5 E6 CFU/mL bacteria cultured for 17h at 37C in CAMHB exposed to 2 fold dilutions of Cefoxitin antibiotic (A). Digital images acquired on Brooke’s Plate Auditor (aka HIAPI) representing bacterial growth (brown wells) and death (clear wells). Representative Cefoxitin MIC of PAO1 vs PAO1ΔG. Each dilution encompasses two columns (B). Deletion of the AmpG gene renders PAO1 susceptible to Cefoxitin (MIC 62.5ug/mL; n=16 replicates/concentration). Absorbance measurements graphed as OD600 vs PAO1 and PAO1ΔG in the presence of 62.5ug/mL Cefoxitin – S:B 3.3, Z′ 0.88 (C).
For the synergy assay determination the same CLSI approved MIC assay format was applied but, in 96 well format which, incorporated 25ug/mL of test compound in combination with the antibiotics at as a dilution series.
Bacterial Culture and 1536-well Turbidity Assay
PAO1 was prepared according to the CLSI techniques.17 Bacteria was inoculated in CAMHB and incubated overnight at 37°C. On the day of screening, CAMHB media was added to 1,536-well black, clear bottom, untreated plates (part 789173-F, Greiner Bio-one, Monroe, NC) (2.5 μl/well). This was followed by pin-tool addition of test compounds and controls (positive control of 30μg/mL Ciprofloxacin and negative control of DMSO only) at 100nl of DMSO. Overnight bacterial cultures were quantified via spectrophotometer, diluted to 1e6 cfu/mL (OD600) and spiked with Cefoxitin (200μg/mL) and dispensed to assay plates (2.5μL/well). The plates were incubated for 17 h at 37°C. After incubation, absorbance at 600nm was measured using a PerkinElmer Envision plate imager (PerkinElmer Lifesciences, Waltham, MA). The optimized counterscreen was identical to the primary screen, except that the bacteria were cultured in the absence of Cefoxitin. The final HTS protocol is summarized in (Table 1).
Table 1.
AmpG Absorbance 1536wpf Primary HTS protocol
| Order | Step | Condition | Comments |
|---|---|---|---|
| 1 | Measure absorbance of PAO1 bacteria stock (overnight culture) | Optical Density | Dilute 1:10 in CAMHB and measure OD600 on spectrophotometer |
| 2 | Dilute PAO1 to 10^6 CFU/mL | CAMHB + 200ug/mL Cefoxitin | |
| 3 | Dispense CAMHB | 2.5μL/well | Greiner black w/clear bottom 1536 well plate |
| 4 | Pin compounds/controls | 100nL | Compounds pinned at 18.6uM final concentration in 2% DMSO |
| 5 | Dispense PAO1 | 2.5μl/well | |
| 6 | Incubation | 17 hours | 37C in humidified chamber |
| 7 | Read Absorbance | Absorbance | 600nm on Envision (Perkin Elmer) |
PAO1 PampC-lux 1536-well Luciferase Assay
Strain PAO1ampC-lux is a PAO1 derivative with ampC-lux reporter fusion integrated into the chromosome. The ampC-lux reporter fusion was integrated into the attT7 site on the PAO1 chromosome using a method described previously.18 Bacterial cultures were prepared and stored according to CLSI techniques.17 On the day of the assay, cultures were quantified and 2e6 cfu/mL spiked into CAMHB. First, 2.0μL CAMHB was dispensed into each of the 1,536-well white solid bottom plates (Greiner, #789175), followed by pin-tool addition of test compounds and controls. 2.0μL of bacterial culture (2e6 cfu/mL) was dispensed to assay plates and then incubated for 2 hours at 37°C. A Cefoxitin dose dependent AmpC induction experiment was done to better understand the optimal condition for AmpC induction and subsequent timing for ampC-lux detection. In this system Cefoxitin can induce AmpC-lux to maximum within one hour at a concentration of 50ug/mL. The apparent concentration for half maximal induction as measure one hour after addition is ~25ug/mL. Ultimately, we used 10ug/ml as it did not inhibit the growth of ampG mutant. With the addition of AmpG inhibitor, the PAO1 should theoretically behave like ampG mutant. Hence, 1μL of Cefoxitin (10μg/mL final) was dispensed and the plates were incubated for 4 hours at 37°C. Luminescence reads were taken on the PerkinElmer ViewLux plate reader and relative light units (RLU’s) were reported.
Chemical Library Screening
The Scripps Diversity Drug Library (SDDL), used for this HTS campaign, currently consists of 646,275 unique compounds, representing a diversity of drug-like small molecules relevant to traditional and non-traditional drug-discovery biology. This library has been previously described in detail.19 All samples in the SDDL were confirmed for purity and mass via LC-MS to provide adequate QA/QC after completion of an HTS campaign.
Screening data acquisition, normalization, representation and analysis
All data files were uploaded into the Scripps institutional HTS database as previously described.20 Activity for each well was normalized on a per-plate basis using the following equation:
| Eq (1) |
where “High Control” represents wells containing no bacteria, “Low Control” represents wells containing DMSO + PAO1 + 100μg/mL Cefoxitin, and “Test Well” contain the same, including tested compounds. The Z′ and S:B were calculated using the High Control and Low Control wells. In each case, a Z′ value greater than 0.5 was required for a plate to be consider valid.21
In addition, we included a “Reference Control” which monitors the effect of Ciprofloxacin in this assay at 30ug/mL, a concentration well above the MIC of 2μg/mL. It is implemented to add confidence in the pintool transfer and to show that the bacteria elicited the appropriate plate to plate and day to day response to a known drug. When included and analyzed as a “Test Well” it provides complete killing of PAO1 at level equivalent to the OD600 observed for the “High Control”.
Results
Assay principle and screening strategy
AmpG is necessary for the AmpC-mediated β-lactam resistance in PAO110 and cephalosporin-based antibiotics such as Cefoxitin (sold under the brand name Mefoxin®) are known to induce AmpC in PAO1. Using the CLSI-approved procedures, in order to confirm the pharmacology of PAO1, we cultured PAO1 and PAO1ΔG (in which the ampG gene was deleted) in the presence of varying concentrations of Cefoxitin in 96-well plate format (wpf) and measured the optical density at 600nm (OD600). In this assay, Cefoxitin achieved an MIC of >800μg/mL against PAO1, while PAO1ΔG scored an MIC of 100μg/mL (data not shown). These data matched the expected MIC’s for Cefoxitin in these microorganisms.17 The 96-wpf assay was miniaturized into 1536-wpf and Cefoxitin MIC determination was repeated to compare PAO1 versus PAO1ΔG (Figure 1). A detailed assay protocol can be found in Table 1. In this assay, the MIC of Cefoxitin on PAO1 and PAO1ΔG were >1000μg/mL and 62.5μg/mL (S:B=3.29, Z′=0.88, n=16 replicates), respectively. These MIC values are well within the CLSI approved 2-fold day-to-day variance.
In order to further verify the 1536-wpf assay pharmacology of PAO1, we experimentally determined the MIC’s of several other well-studied antibiotics.17, 22 We cultured PAO1 in the presence of various concentrations of ciprofloxacin, erythromycin, or aztreonam and compared our in-house observed MIC values to their expected MICs (Supplemental Table 1). Based on the above data, we chose to use 30μg/mL ciprofloxacin as a reference control for the HTS.
Because all SDDL compounds were dissolved in DMSO we assessed DMSO tolerance by pintool transfer using the Kalypsys GNF unit outfitted with 30nL pins asserting 1–3 transfer events and determined that the assay would tolerate up to 2X pinning (~2% or nominally 20μM screening concentration).
To determine the overall readiness of the 1536-wpf assay for HTS prior to robotic validation for fully automated uHTS and also to determine the hit rate and reproducibility of the hits, we tested 3,446 compounds against PAO1, at 10μM nominal concentration, in triplicate using standalone equipment. This included a 1,280-compound library of pharmacologically active compounds (LOPAC) and libraries from Prestwick and Tocris. Of the 3,446 compounds tested, we observed 77 actives (2.23% hit rate), which had an activity greater than the cutoff of 46.01%; a cutoff = 3 × stdev. + average activity of all samples tested. We applied this cut-off because the general activity of the tested sample population was relatively high, and as such it was not necessary to designate a cut-off close the sample “noise” of the assay in order to identify hits. Experimental reproducibility was monitored using ciprofloxacin as a reference at a high concentration of 30μg/mL and also by verifying its MIC.
Primary HTS
In fully automated fashion on our robotics platform, a small pilot validation run commenced surveying 36 randomly chosen diversity SDDL plates containing approximately 46K compounds. These were tested at 10μM nominal screening concentration and, again using a 3 × stdev. + average activity cut-off (11.3%), 103 compounds were found to be active (0.19% hit rate); quite divergent from what we observed using the LOPAC and drug-like collections. While not totally surprising, the number of hits was low which prompted us to test 17 of the 36 “matched” plates (almost 22K compounds) using a double pintool transfer method, achieving ~20μM screening concentration, to determine if the hit rate would improve. Using the same method for hit cut-off determination which equaled 13.48% in this case, we observed 56 active compounds (0.23% hit rate) and good reproducibility in activity of compounds found in the previous run with a correlation coefficient of 0.82 and a two tailed P-value of <0.0001 (Supplemental figure 1). While the most active compounds reproduced activity, we also found some that were weakly active that now had slightly increased in their activity. Taken together, this was considered a modest improvement and we advanced to the primary HTS utilizing a ~20μM screening concentration.
In the primary screen, 646,275 SDDL compounds were tested in singlicate at a final nominal test concentration of 18.6 μM using the optimized conditions described in Table 1. The results of the primary screen are shown in Figure 2 and the statistics can be found in Table 2. The average Z′ for the primary screen was excellent at 0.87±0.05 with a S:B of 2.2±0.2. Day-to-day variance and pharmacologic reproducibility was monitored using ciprofloxacin MIC determinations, which were stable at 2.42±0.79 μg/mL over 15 plates throughout the primary screen; again well within the CLSI parameters. In addition to the performance measures indicated in table 2 and mentioned above, the primary HTS entailed 519 test compound plates interleaved with approximately 73 control plates and was completed in 9 fully automated campaigns across 9 success business days. The maximum number of plates tested per day was 117 equal to ~150K compounds per day throughput but, averaged ~80 per day as we ramped up to the full scale HTS which leveled off at 100 as a standard procedure. As is typical within large scale HTS efforts, we did observe some plates that failed Z′. All failed plates were rescheduled until they passed all criteria which in this assay resulted in retesting 15% of the total test plates; a number that was higher than normal due to a failed dispenser valve in one run. In summary, 2,663 compounds were found to be active, that is, they exhibited activity greater than a cutoff of 11.10%; a value obtained using an interval based mathematical algorithm described previously.20 By comparison, the hit cut-off using the 3 × stdev. + average of all samples tested in the primary assay was 12.37% inhibition. This would have identified 1,861 hits as compared to 2,663 using the interval based cut-off. Here, we justify using the interval cut-off method, one which is more aggressive in identifying hits because it sets the value closer to the sample noise, in order to preserve more hits when the overall activity of the sample population as tested was relatively low. In addition, as seen in figure 2, there was also negative activity in the sample field demonstrating lower than −20% inhibition, possibly indicating compounds that elicit bacterial proliferation or absorbance artifacts, both which would have increased the former cut-off value to the point of limiting the number of hits obtained beyond what we felt reasonable.
Figure 2.

AmpG primary HTS scattergram of 646,275 compounds screened in singlicate at ~18.2 μM; Test wells (black dots), reference control (blue dots), and control wells as described in the text (red and green dots). Data are shown normalized to percent max response of the high control. The hit cutoff is shown as the red line at 11.10%. (B) Example MIC of the control antibiotic Ciprofloxacin as tested during HTS graphed as OD600 vs log ug/mL of Ciprofloxacin. The average MIC is equal to 2.42± 0.79ug/mL; N = 15 plates with 16 replicates/dilution.
Table 2.
Summary and results of AmpG ultra-HTS campaign
| Step | Screen Type | Readout | Target | Number of Compounds Tested | Selection Criteria | Number of Selected Compounds (Hit Rate) | Z′ | S/B |
|---|---|---|---|---|---|---|---|---|
| 1a | Primary screen | Absorbance | AmpG | 646,275 | 11.10%a | 2663 (0.41%) | 0.87±0.05 | 2.22±0.15 |
| 2a | Confirmation screen | Absorbance | AmpG | 2,660 | 11.10%b | 851 (31.99%) | 0.86±0.02 | 4.33±0.46 |
| 2b | Counterscreen screen | Absorbance | AmpG No Cefoxitin |
2,660 | 12.70%c | 864 (32.48%) | 0.88±0.03 | 4.53±0.32 |
| 3a | Dose-response screen | Absorbance | AmpG | 110 | IC50 < 10uM | 1 (0.09%) | 0.84±0.02 | 4.36±0.31 |
| 3b | Dose-response counterscreen | Absorbance | AmpG No Cefoxitin |
110 | IC50 < 10uM | 1 (0.09%) | 0.84±0.02 | 3.99±0.31 |
| 4a | PampC Lux dose response | Luciferase | AmpG | 110 | Chemistd | 8 (7.27%) | 0.59±0.17 | 112.75±9.23 |
| 4b | PampC Lux dose response | Absorbance | AmpG | 110 | Chemist | 0 (0.00%) | 0.79±0.04 | 2.88±0.23 |
The primary screen hit cutoff was calculated using the interval-based cutoff.
The primary hit cutoff was used.
The counterscreen hit cutoff was calculated by using the average + three standard deviations of the DMSO sample field plates, n=7.
Due to the novelty of this target, compounds with any observable activity in either the primary assay, the Lux assay, but not in the absorbance assay, were of interest for follow-up.
Confirmation and Counterscreen Assays
Active compounds were selected for confirmation screening, which used the same reagents and detection system as the primary screening assay, but tested each of the 2,660 available compounds at a single concentration (nominally 18.6 μM) in triplicate. The PAO1 plus Cefoxitin inhibitor confirmation assay performance was similar to the primary campaign and yielded an average Z′ of 0.86±0.02 and a S:B of 4.3±0.5. Using the primary HTS assay cut-off, 851 hits confirmed with activity equal to or above 11.10%, equating to a reasonable confirmation rate of 31.99%. This was nearly identical to the confirmation rate achieved when analyzing the average pilot HTS triplicate hit data to the overlapping compound data in the primary HTS, whereby we achieved a 36.36% confirmation rate.
A counterscreen assay, similar in format to the PAO1 anti-microbial assay, was then performed against the 2,660 available primary hits. This assay involved running the PAO1 bacteria without the addition of Cefoxitin and hence induction of AmpC. Thus any compounds that were active in this screen would have activity independent of AmpG. The PAO1 minus Cefoxitin counterscreen assay performance had an average Z′ of 0.88±0.03 and a S:B of 4.5±0.3. Using a standard method for the counterscreen hit cut-off (the average of the DMSO samples + 3SD), which equates to 12.7%, 864 hits were found. These 864 actives presumably hit off target or were identified as known antibiotics within the screening system and, in the scope of the current research, were not of further interest.
Following comparison of the actives, removal of PAINs23,24 as well as compounds that have shown activity in more than five other assays tested at Scripps Florida, a total of 110 active compounds were further investigated. These compounds were found to have a significant activity index; i.e., a separation in potency favoring more potency in the primary assay over the counterscreen (Supplemental table 2). Reproducibility amongst these 110 compound replicate data was quite good with the majority of the compounds (88%) demonstrating 2.5% to 49% coefficient of variation in their triplicate data. Additionally the reproducibility of the same set of actives vs. their primary HTS data, demonstrated that 96% confirmed activity within two fold of the primary response. These 110 compounds were selected to be tested in concentration response studies, of which all were available. A summary of the secondary assay data is shown in Table 2.
Concentration Response Assays
The concentration response curve (CRC) assays were run following the confirmation and counterscreening assay protocols but, with 110 compounds tested at 10 doses (3-fold dilutions) in triplicate. The PAO1 anti-microbial CRC assay performance had an average Z′ of 0.84 and a S:B of 4.4±0.3 and the counterscreen CRC assay had an average Z′ of 0.84 and a S:B of 4.0±0.3. For each compound tested, percent activation was plotted against compound concentration. A four parameter equation describing a sigmoidal concentration response curve was then fitted with adjustable baseline using Assay Explorer software (Symyx Technologies Inc.). The reported IC50 values were generated from fitted curves by solving for the X-intercept value at the 50% activity level of the Y-intercept value. One compound elicited an IC50 <10 μM in both assays. Notably, antimicrobial drugs typically act in an “all or nothing” like manner and as such drug responses in this sense generate very steep hillslopes. In our case, we are experimentally determining efficacy in a whole cell phenotypic fashion similar to typical MIC determination assays and we also observe multiple compounds that elicit steep hillslope responses which, while not a bad outcome, may limit the ability to identify weak AmpG specific inhibitors.
Ultimately we wanted to identify compounds that were efficacious in the primary assay and that specifically affect AmpG. Next, we aimed to develop a more sensitive assay as another means of triaging hits with AmpG specificity by integrating an ampC-lux fusion reporter gene into PAO1 and employing a luciferase based readout. We then tested all 110 active compounds with 10 dose titrations in triplicate. The PAO1ampC-lux CRC assay had an average Z′ of 0.59 and a S:B of 112.7±9.2. IC50 values were determined as described in the previous CRC assays. From this assay, there were 2 active compounds with IC50 <10μM and 6 more compounds with favorable physiochemical properties which we selected for further persecution. As an additional measure we tested PAO1ampC-lux using a whole cell turbidity readout which would confirm that the increased activity in the ampC-lux assay was due to an AmpG related mechanism and not the result of cytotoxicity. The PAO1ampC-lux turbidity counterscreen CRC assay had an average Z′ of 0.79 and S:B 2.9±0.2. IC50 values were determined as described in the previous CRC assays. None of the compounds resulted in an IC50 <10μM.
Taken together, in consideration of the novelty of AmpG as a target and the modest activity in mind, we initiated some preliminary effort to determine if compound numbers 2 and 6 in Table 3 elicited any enhancement in antibiotic efficacy when tested in combination against PAO1. First both compounds were titrated to determine their own MIC values vs PAO1. Both MICs appeared at 50ug/ml which equates to 324 μM for compound 2 and 156 μM for compound 6 so we adjusted the test concentration of each compound to be 25ug/mL to keep the synergy test concentration as high as reasonably possible without eliciting an MIC on its own. We then tested them separately but in combination with individual known drugs including Ampicillin, Cefoxitin, and Ticarcillin. We did observe a 2 fold enhancement in the activity of the test antibiotics in the presence of compounds 2 and 6 when tested on the same microtiter plate in the same assay. While this wouldn’t be construed as a synergistic result because we didn’t observe the requisite 4 fold enhancement in potency, it does indicate some advancement in potency.
Table 3.
Compounds which exhibit a measurable AmpG-specific activity index.
| entry | ID | structure | analysis |
|---|---|---|---|
| 1 | SR-01000076250-2 |
|
Inorganic dye: poor drug-likeness and poorly-suited for follow-up |
| 2 | SR-05000002238-3 |
|
Patulin: natural product mycotoxin produced by a variety of molds. In ring-opened form it is chemically reactive, forming Schiff bases with biological amines. |
| 3 | SR-01000320782-1 |
|
A synthetic compound: phenolic hydrazones are structure alerts and thus modifications to the structure in probe development would be desirable |
| 4 | SR-05000002074-1 |
|
Natural product Polymyxin B, primarily used for resistant gram-negative infections. It is derived from the bacterium Bacillus polymyxa. |
| 5 | SR-01000226538-1 |
|
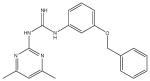
A synthetic compound in the aryl guanidine class. No stability or reactivity concerns. |
| 6 | SR-01000115944-1 |
|
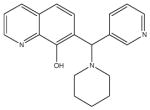
A synthetic compound in the 7-aminomethyl 8-hydroxyquinoline class. These are known as “Betti bases” and may be chemical unstable to retro-Michael addition. More chemically stable analogs would be preferred. |
| 7 | SR-01000115950-1 |
|
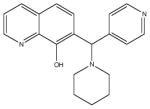
A structural isomer also in the 7-aminomethyl 8-hydroxyquinoline class. More chemically stable analogs would be preferred. |
| 8 | SR-01000520919-1 |
|
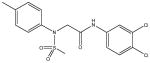
A synthetic compound in the N-aryl sulfonamide class. No stability or reactivity concerns. |
Discussion
Increased production of β-lactamases, biofilm formation, activation of efflux pumps, and intrinsically low outer membrane permeability are some of the features that contribute to antibiotics resistance in P. aeruginosa and all of these factors have historically made it a difficult target for drug discovery.25–27 Here, we focused on AmpG-mediated mechanisms of action for therapeutic development of inhibitors. AmpG is responsible for AmpC induction and overexpression in PAO1 as well as other gram-negative bacteria, which leads to acquired resistance to β-lactams through the overproduction of β-lactamases.10, 12, 28–30 We proposed that an AmpG-specific inhibitor would have a synergistic effect by reducing AmpC expression and thereby render PAO1 susceptibility (susceptible) to β-lactams10.
In the current experiments, we aimed to discover small molecule inhibitors of AmpG using an uHTS campaign. To our knowledge, this is the first report on the successful phenotypic 1536-well formatted uHTS on large collection (>640K compounds) against P. aeruginosa. By utilizing a whole cell phenotypic screening approach, we were able to identify compounds which inhibit PAO1 and delineate those acting through AmpG by Cefoxitin-mediated induction of AmpC. We successfully completed the 645K uHTS, found 2,663 active compounds and further validated the assay through the recognition of several relevant antibiotics in the initial list of hits. Out of the 2,663 active compounds, 851 were shown to be active in our confirmation assays, with 110 of those appearing to have some AmpG selectivity in our counterscreen assay, however, a small but significant activity index was noted when comparing the two assays. Here, the activity index is described as the relative separation in the IC50 values and activities for a particular compound as determined in the CRC assays in the AmpG antimicrobial primary screening assay versus the counterscreen assays. A graphical example of a desired compound’s activity index is illustrated in Figure 3A. Ciprofloxacin, a DNA gyrase inhibitor, as expected, exhibited an identical MIC (~2μg/ml) in the primary and counterscreen assays, thus equating to no activity index at all with regards to this study.
Figure 3.

Theoretical AmpG CRC assay graphical representation of desired compound activity index. The ideal compound would inhibit AmpG activity in the PAO1/PampC-Lux assay (pink line) to a greater extent than the PAO1 viability assay (blue line) and have no activity in the no Cefoxitin counterscreen (green line) ultimately bearing a selectivity index of at least >10 fold. FOX=Cefoxitin (B) Examples of the concentration response curves for the 8 molecules initially of interest following the HTS. The color legend for each of the assays is per part A. Points on each curve are derived from an N=3 with error bars shown. The SR# can be matched to the structure in Table 3 and also to activity in the supplement.
Follow-up CRC assays revealed a predominant overlap in the potencies of the confirmed active compounds against PAO1 and the counterscreen (PAO1 minus Cefoxitin) (Figure 3B). We hypothesized that this was the result of a lack in assay sensitivity inherent to using a phenotypic screening approach. This led us to develop and implement an AmpC-luciferase reporter assay to further triage these hits. Notably, this assay was also successfully miniaturized and yields passing Z′ values when cultured for only 4 hours. Ultimately, this method increased the opportunity to observe selectivity toward AmpG-specific activity while limiting the effect of the cytotoxicity profile of compounds thus resulting in 8 potential AmpG-specific PAO1 inhibitors (Figure 3B, 4 and Supplemental Data). Antibiotics known to induce AmpC were subjected to combination testing in the presence of two of these compounds (Table 3, #2 and #6). The test condition was optimized to allow for compounds to be at maximal concentration yet below their individual MICs. Concentrations at 50ug/mL were toxic on their own while lower than 25ug/mL were tested and were found ineffective. Two fold enhancement is not synergistic by default; a compound or antibiotic MIC is still valid if within two fold day to day. Still this was consistent for multiple experiments when tested on the same day and same plate (see supplemental figure 2). We hypothesize that PAO1, although quite capable of AmpC induction, may not have been the appropriate strain to test for synergy but, by implementing other, perhaps clinical isolates, with higher and lower levels of AmpC resistance, this may allow for a more sensitive version of the assay; a system we plan to test in the future.
These eight compounds together represent new chemical space that differ from traditional antibiotics. While some are ill-suited for probe optimization (see the inorganic dye in entry 1) several of these compounds may be suitably drug-like for optimization and will be subjected to further SAR studies. Compound-specific medicinal chemistry analysis is provided in Table 3. While it is true that the activity of these hits in the described assays is generally low and there is some diversity in the structural outcomes, we do show concentration dependent activity in the ampC-Lux assay, a system that has been definitively shown to require AmpG in order to achieve AmpC induction. Note, each of these analogs shown in figure 3B, including compound 6, has been tested on at least three separate occasions and again reproduce activity in each case. Activity of these eight hits has also been confirmed by testing of independent second samples in independent labs (Scripps and University of Florida) across all assays mentioned. An internal assessment identified that these compounds were found as a hit in less than 6 other assays out of an average of 75 total, indicating that the compounds are not promiscuously active. Compounds 2 and 4 have reported antibiotic activities, though no reports of anti-Amp G activity have appeared for these or for any of the other compounds shown. No reports on the structural optimization of these chemotypes for other targets have appeared, to our knowledge. The luminescence readout for the ampC-Lux fusion reporter further narrows the potential targets of the newly identified chemical components to the AmpG-AmpC signaling pathway, including AmpG, NagZ and AmpR. More sensitive downstream assays are being developed to further narrow down the exact targets. The possible influence of the chemical compounds directly on the luciferase enzyme has been eliminated by using PAO1 strain expressing Lux under a constitutive lac promoter (data not shown). Identity and purity of all samples was also verified via LC-MS.
This information serves as another starting point for further SAR optimization against a novel, unique and until recently an unexplored HTS target in microbiology. In the future we plan to further develop one or more these hits, verifying to what degree they interact with AmpG, with the intention of identifying a chemical probe acting by a novel mechanism, targeting Pseudomonas infections, and ultimately with promise for clinical translation.
Supplementary Material
Acknowledgments
We thank Pierre Baillargeon and Lina DeLuca (Lead Identification, Scripps Florida) for compound management.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grant R21 AI119043.
Footnotes
Declaration of Conflicting Interests
There are no conflicts of interest amongst any of the authors and the work pertained in this manuscript.
References
- 1.http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/. WHO publishes list of bacteria for which new antibiotics are urgently needed. 2017.
- 2.Ventola CL. The antibiotic resistance crisis: part 1: causes and threats. P T. 2015;40(4):277–83. [PMC free article] [PubMed] [Google Scholar]
- 3.Gandra S, Barter DM, Laxminarayan R. Economic burden of antibiotic resistance: how much do we really know? Clin Microbiol Infect. 2014;20(10):973–80. doi: 10.1111/1469-0691.12798. [DOI] [PubMed] [Google Scholar]
- 4.Pagani L, Colinon C, Migliavacca R, et al. Nosocomial outbreak caused by multidrug-resistant Pseudomonas aeruginosa producing IMP-13 metallo-beta-lactamase. J Clin Microbiol. 2005;43(8):3824–8. doi: 10.1128/JCM.43.8.3824-3828.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romling U, Wingender J, Muller H, et al. A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl Environ Microbiol. 1994;60(6):1734–8. doi: 10.1128/aem.60.6.1734-1738.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pier GB. Pulmonary disease associated with Pseudomonas aeruginosa in cystic fibrosis: current status of the host-bacterium interaction. J Infect Dis. 1985;151(4):575–80. doi: 10.1093/infdis/151.4.575. [DOI] [PubMed] [Google Scholar]
- 7.Arivett BA, Ream DC, Fiester SE, et al. Draft Genome Sequences of Pseudomonas aeruginosa Isolates from Wounded Military Personnel. Genome Announc. 2016;4(4) doi: 10.1128/genomeA.00829-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serra R, Grande R, Butrico L, et al. Chronic wound infections: the role of Pseudomonas aeruginosa and Staphylococcus aureus. Expert Rev Anti Infect Ther. 2015;13(5):605–13. doi: 10.1586/14787210.2015.1023291. [DOI] [PubMed] [Google Scholar]
- 9.Meeske AJ, Riley EP, Robins WP, et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature. 2016 doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Bao Q, Gagnon LA, et al. ampG gene of Pseudomonas aeruginosa and its role in beta-lactamase expression. Antimicrob Agents Chemother. 2010;54(11):4772–9. doi: 10.1128/AAC.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamorano L, Reeve TM, Juan C, et al. AmpG inactivation restores susceptibility of pan-beta-lactam-resistant Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother. 2011;55(5):1990–6. doi: 10.1128/AAC.01688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korfmann G, Sanders CC. ampG is essential for high-level expression of AmpC beta-lactamase in Enterobacter cloacae. Antimicrob Agents Chemother. 1989;33(11):1946–51. doi: 10.1128/aac.33.11.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406(6799):959–64. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 14.Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009;22(4):582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li P, Ying J, Yang G, et al. Structure-Function Analysis of the Transmembrane Protein AmpG from Pseudomonas aeruginosa. PLoS One. 2016;11(12):e0168060. doi: 10.1371/journal.pone.0168060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baillargeon P, Scampavia L, Einsteder R, et al. Monitoring of HTS compound library quality via a high-resolution image acquisition and processing instrument. J Lab Autom. 2011;16(3):197–203. doi: 10.1016/j.jala.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wikler MA. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard. 7. 7. Wayne, PA: Clinical and Laboratory Standards Institute; United States: 2006. p. c2006. [Google Scholar]
- 18.Choi KH, Schweizer HP. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc. 2006;1(1):153–61. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 19.Jambrina E, Cerne R, Smith E, et al. An Integrated Approach for Screening and Identification of Positive Allosteric Modulators of N-Methyl-D-Aspartate Receptors. J Biomol Screen. 2016;21(5):468–79. doi: 10.1177/1087057116628437. [DOI] [PubMed] [Google Scholar]
- 20.Smith E, Janovick JA, Bannister TD, et al. Identification of Potential Pharmacoperones Capable of Rescuing the Functionality of Misfolded Vasopressin 2 Receptor Involved in Nephrogenic Diabetes Insipidus. J Biomol Screen. 2016;21(8):824–31. doi: 10.1177/1087057116653925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 22.Fass RJ, Barnishan J. Minimal inhibitory concentrations of 34 antimicrobial agents for control strains Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853. Antimicrob Agents Chemother. 1979;16(5):622–4. doi: 10.1128/aac.16.5.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jasial S, Hu Y, Bajorath J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J Med Chem. 2017;60(9):3879–3886. doi: 10.1021/acs.jmedchem.7b00154. [DOI] [PubMed] [Google Scholar]
- 24.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–40. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 25.Nikaido H. Role of permeability barriers in resistance to beta-lactam antibiotics. Pharmacol Ther. 1985;27(2):197–231. doi: 10.1016/0163-7258(85)90069-5. [DOI] [PubMed] [Google Scholar]
- 26.Nikaido H. Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science. 1994;264(5157):382–8. doi: 10.1126/science.8153625. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura F, Nikaido H. Permeability of Pseudomonas aeruginosa outer membrane to hydrophilic solutes. J Bacteriol. 1982;152(2):636–42. doi: 10.1128/jb.152.2.636-642.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bush K. New beta-lactamases in gram-negative bacteria: diversity and impact on the selection of antimicrobial therapy. Clin Infect Dis. 2001;32(7):1085–9. doi: 10.1086/319610. [DOI] [PubMed] [Google Scholar]
- 29.Takeda S, Nakai T, Wakai Y, et al. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2007;51(3):826–30. doi: 10.1128/AAC.00860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angus BL, Carey AM, Caron DA, et al. Outer membrane permeability in Pseudomonas aeruginosa: comparison of a wild-type with an antibiotic-supersusceptible mutant. Antimicrob Agents Chemother. 1982;21(2):299–309. doi: 10.1128/aac.21.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.