

Abstract
The incidence of nephrolithiasis continues to rise. Previously, we showed that a monogenic cause could be detected in 11.4% of individuals with adult-onset nephrolithiasis or nephrocalcinosis and in 16.7-20.8% of individuals with onset before 18 years of age, using gene panel sequencing of 30 genes known to cause nephrolithiasis/nephrocalcinosis. To overcome the limitations of panel sequencing, we utilized whole exome sequencing in 51 families, who presented before age 25 years with at least one renal stone or with a renal ultrasound finding of nephrocalcinosis to identify the underlying molecular genetic cause of disease. In 15 of 51 families, we detected a monogenic causative mutation by whole exome sequencing. A mutation in seven recessive genes (AGXT, ATP6V1B1, CLDN16, CLDN19, GRHPR, SLC3A1, SLC12A1), in one dominant gene (SLC9A3R1), and in one gene (SLC34A1) with both recessive and dominant inheritance was detected. Seven of the 19 different mutations were not previously described as disease causing. In one family a causative mutation in one of 117 genes that may represent phenocopies of nephrolithiasis-causing genes was detected. In nine of 15 families the genetic diagnosis may have specific implications for stone management and prevention. Several factors that correlated with the higher detection rate in our cohort were younger age at onset of nephrolithiasis/nephrocalcinosis, presence of multiple affected members in a family, and presence of consanguinity. Thus, we established whole exome sequencing as an efficient approach towards a molecular genetic diagnosis in individuals with nephrolithiasis/nephrocalcinosis who manifest before age 25 years.
Keywords: Nephrolithiasis, nephrocalcinosis, monogenic cause, whole exome sequencing
Introduction
Nephrolithiasis (NL) is a highly prevalent condition affecting up to 10% of individuals worldwide.1 It is associated with high morbidity, high recurrence rate as well as high economic cost.2 Although NL is less common among children compared to adults, the incidence of NL and nephrocalcinosis (NC) in the pediatric age group has been rising over the past 10 years.3 The causes of NL are not well understood. Formerly monogenic causes of NL were thought to be restricted to rare tubulopathies and genetic syndromes. However, we recently revealed that a causative monogenic mutation can be detected in one of 30 known NL causing genes in 20.8% of patients with onset of NL before age of 18 yrs.4 We subsequently confirmed the high rate of a molecular diagnosis in 16.7% of early-onset NL in a three-center cohort and found that important therapeutic and preventative measures may result from mutation detection.5 These previous studies employed exon sequencing in gene panels, rather than whole exome sequencing (WES). Because WES offers the opportunity to detect dozens of additional genes at a rather low cost and has not yet been applied to patients with NL, we here employed WES in 51 families (65 individuals) with at least one episode of renal stone or evidence of NC on renal ultrasound before the age of 25 yrs, in order to identify monogenic causes in the 30 NL/NC known genes. We also evaluated WES data for 117 additional phenocopy genes (30 known renal tubulopathy genes, 87 renal ciliopathy genes) and 16 hypothesized candidate genes. We confirm the high rate of detection (29.4%) of causative mutations in one of the 30 known NL/NC genes. We study genotype-phenotype correlations, determine factors such as early onset and familial disease correlating with high mutation detection rate, and determine that a molecular genetic diagnosis allows for finely tailored treatment plans that may prevent recurrent disease or delay progression to ESRD.
Results
We performed WES in 65 individuals from 51 families with nephrolithiasis and/or a finding of nephrocalcinosis on renal ultrasound, who manifested before the age of 25 years. Of the 65 individuals, 32 had isolated NL, 22 had isolated NC, and 11 had both NL and NC. No affected individual had hypercalciuria in the absence of NL or NC (Suppl. Fig. 1). When evaluating WES data for 30 genes known to cause NL or NC when mutated (Suppl. Table 1), we identified a mutation in 15 of 51 families (29.4%) (Fig. 1). Recessive or dominant causative mutations were detected in 9 of the 30 genes (Table 1, 2). Pathogenic mutations were detected in 8 recessive genes in 17 individuals from 12 families: AGXT (4 individuals, 3 families), ATP6V1B1 (1 individual, 1 family), CLDN16 (1 individual, 1 family), CLDN19 (3 individuals, 1 family), GRHPR (2 individuals, 1 family), SLC3A1 (1 individual, 1 family), SLC12A1 (3 individuals, 2 families), and SLC34A1 (2 individuals, 2 families), (Table 1). Pathogenic mutations were detected in 2 dominant genes in 5 individuals from 3 families: SLC9A3R1 (1 individual, 1 family) and SLC34A1 (4 individuals, 2 families) (Table 2). SLC34A1 gene mutations may follow an autosomal recessive mode of inheritance causing infantile hypercalcemia6 or an autosomal dominant mode of inheritance causing nephrolithiasis or nephrocalcinosis.5,7. The family history, status of consanguinity and detailed phenotype of individuals is shown (Table 1, 2). The pedigrees of all 15 families show family history and consanguinity (Suppl. Fig. 2).
Figure 1. Flow diagram on detection by WES of causative monogenic mutations in 30 NL/NC genes in 51 families with NL/ NC.

51 families of 100 available families with NL/NC were tested by WES for detection of monogenic causation of stone disease. A causative mutation was detected in a known NL/NC gene in 15 of 51 families (29.4%). A causative mutation was detected in a phenocopy gene (CTNS) in one patient with nephrocalcinosis. 35 families remain unsolved.
Table 1. Monogenic causative mutations of 8 recessive genes detected in 17 individuals from 12 families with NL/NC.
Mutated genes and related diseases (OMIM nomenclature) are shown in sub headers to the table.
| Family Individual |
Nucleotide change |
Amino acid change |
State | PPh2 | Evolutionary conservation |
Ref. | Ethnicity (Sex) |
Age of Onset |
Family history |
Consangu nity |
Phenotype prior to WES |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| AGXT, Primary hyperoxaluria type I | |||||||||||
|
| |||||||||||
| B1468 21 | c.1079G>C | p.Arg360Pro | het | 1 | C. elegans | Novela | Egyptian (M) | 6 | Y (1 sibling affected) | N | NL/NC, CKD, hyperoxaluria NC, microscopic hematuria |
| c.121G>A | p.Gly41Arg | het | 0.99 | D. melanogaster | Y24 | ||||||
| B1468 24 | c.1079G>C | p.Arg360Pro | het | 1 | C. elegans | Novela | Egyptian (F) | 2.5 | |||
| c.121G>A | p.Gly41Arg | het | 0.99 | D. melanogaster | Y24 | ||||||
| B1230_21 | c.731T>C | p.Ile244Thr | hom | 0.4 | M. musculus | Y25 | Egyptian (M) | 1 | Y | Y | NL, CKD, hyperoxaluria |
| B1646_21 | c.481G>A | p.Gly161Ser | hom | 0.99 | C. elegans | Y26 | Middle Eastern (M) | 3 | Y (cousin) | Y | Recurrent bilateral NL, CKD, hyperoxaluria |
|
| |||||||||||
| ATP6V1B1, Renal Tubular acidosis with deafness | |||||||||||
|
| |||||||||||
| B1121 | c.242T>C | p.Leu81Pro | hom | 1 | C. elegans | Y27 | European (M) | 1.7 | N | N | NC, RTA, neurodevelopmental delay |
|
| |||||||||||
| CLDN16, Hypomagnesemia, familial with hypercalciuria and nephrocalcinosis | |||||||||||
|
| |||||||||||
| B1645_21 | c.695T>G | p.Phe232Cys | hom | 0.99 | D. rerio | Middle Eastern (F) | 4 | Y (1 sibling affected) | Y | Bilateral NL, hypomagnesemia | |
|
| |||||||||||
| CLDN19, Hypomagnesemia/renal/ophthalmological abnormalities | |||||||||||
|
| |||||||||||
| A4283_21 | c.535G>A | p.Gly179Ser | hom | 0.99 | D. rerio | Novela | South Asian (F) | 11 | Y (2 siblings affected) | Y | NC, CKD |
| A4283_22 | c.535G>A | p.Gly179Ser | hom | 0.99 | D. rerio | Novela | South Asian (M) | 5 | NC, CKD | ||
| A4283_23 | c.535G>A | p.Gly179Ser | hom | 0.99 | D. rerio | Novela | South Asian (M) | 4 | NC, CKD | ||
|
| |||||||||||
| GRHPR, Primary hyperoxaluria type II | |||||||||||
|
| |||||||||||
| B35 21 | c.103del | p.Asp35Thrfs*11 | het | NA | NA | Y28 | American (M) | 0.08 | Y (1 sibling affected) | N | NL/NC, hyperoxaluria, hypercalciuria |
| c.404+5G>A | Splice site | het | NA | NA | Y29 | American (M) | |||||
| B35_22 | c.103del | p.Asp35Thrfs*11 | het | NA | NA | Y28 | 3 | ||||
| c.404+5G>A | Splice site | het | NA | NA | Y29 | NL/NC, hyperoxaluria. | |||||
|
| |||||||||||
| SLC3A1, Cystinuria, type A | |||||||||||
|
| |||||||||||
| B1648_21 | c.592delG | p.Ala198fs | hom | NA | NA | Y30 | Middle Eastern (M) | 3 | N | N | NL, 100% cysteine stone on analysis |
|
| |||||||||||
| SLC12A1, Bartter syndrome type 2 | |||||||||||
|
| |||||||||||
| B917_21 | c.769G>A | p.Gly257Ser | het | 1 | C. elegans | Y31 | Caucasian (M) | 1 | N | N | NL/NC, polyuria, polydipsia, hypercalciuria |
| c. 1424G>A | p.Cys475Tyr | het | 0.99 | D. melanogaster | Novela | ||||||
| B1508_21 | c.1157delT | p.Ile386fs | hom | NA | NA | Novela | European (F) | 0.8 | Y (1 sibling affected) | Y | NC, hypercalciuria, failure to thrive, metabolic derangements NC, hypercalciuria, failure to thrive, metabolic derangements |
| B1508_22 | c.1157delT | p.Ile386fs | hom | NA | NA | Novela | European (M) | 1.2 | |||
|
| |||||||||||
| SLC34A1, Infantile hypercalcemia/hypophosphatemia/nephrolithiasis | |||||||||||
|
| |||||||||||
| B1437_21 | c. 644+1 G>A | Splice site | het | NA | NA | Y6 | Egyptian (M) | 3 | Y-(relatives) | N | NC, hypercalciuria |
| c.1204G>C | p.Gly402Arg | het | 1 | D. rerio | Novela | ||||||
| B1602_21 | c.1724C>T | p.Thr575Ile | hom | .61 | D. melanogaster | Novela | Caucasian (M) | 0.2 | Y | Y | NC, hypercalcemia |
AGXT, Alanine-glyoxalate aminotransferase; ATP6V1B1, ATPase, H+ transporting, lysosomal, 56/58-KD, V1 Subunit B, Isoform 1; BS1, Bartter syndrome type 1; CKD, chronic kidney disease; CLDN16, claudin16; CLDN19, claudin19; DRTAD, distal renal tubular acidosis with progressive deafness; F, female; GRHPR Glyoxalate Reductase/hydroxypyruvate reducatse; het, heterozygous; hom, homozygous; hx, history; M, male; N, No; NA, not applicable; NC, nephrocalcinosis; NL, nephrolithiasis; Novel, mutation detected for the first time in this study; PPh2, PolyPhen-2; RTA, renal tubular acidosis; SLC3A1, solute carrier family 3; SLC12A1, Solute carrier family 12; SLC34A1, Solute carrier family 34; Y, Yes.
Different mutations at this position are reported.
Table 2. Monogenic causative mutations detected in 2 dominant genes in 5 individuals from 3 families with NL/NC.
Mutated genes and related diseases (OMIM nomenclature) are shown in subheaders to table.
| Family Individual |
Nucleotide change |
Amino acid change |
State | PPh2 | Evolutionary conservation |
Ref. | Ethnicity (Sex) |
Age of onset (yr) |
Family history |
Consan guinity |
Phenotype prior to WES |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| SLC9A3R1, Nephrolithiasis/osteoporosis (NPHLOP2) | |||||||||||
|
| |||||||||||
| B969-21 | c.673G>A | p.Glu225Lys | het | 0.82 | D. rerio | Y32 | American (F) | 7 | Y | N | NL, hypophosphatemia |
|
| |||||||||||
| SLC34A1, Infantile hypercalcemia/hypophosphatemia/nephrolithiasis | |||||||||||
|
| |||||||||||
| B1301 21 | c.398C>T | p.Ala133Val | het | 0.99 | C. intestinalis | Y33 | European (F) | 6.5 | Y (1 sibling affected) | N | NC |
| B1301_22 | c.398C>T | p.Ala133Val | het | 0.99 | C. intestinalis | Y33 | European (M) | 10.5 | NC | ||
| B986_21 | c.536T>C | p.Leu179Pro | het | 1 | C. intestinalis | Novel | American (F) | 1.5 | Y (1 sibling affected) | N | NC |
| B986_22 | c.536T>C | p.Leu179Pro | het | 1 | C. intestinalis | Novel | American (F) | 1.5 | NC | ||
F, female; het, heterozygous; hx, history; M, male; N, No; NC, nephrocalcinosis; NL, nephrolithiasis;NPHLOP2, Nephrolithiasis/osteoporosis, hypophosphatemic, 2; PPh2, PolyPhen-2; SLC9A3R1, solute carrier family 9, member 3, regulator 1; SLC34A1, solute carrier family 34; Y, Yes.
Of the 22 individuals, in whom we detected causative mutations in this study, 9 presented with NL and 13 presented with NC on a renal ultrasound (Table 1, 2). The radiographic evidence of NC in 6 of 13 individuals affected with NC is shown (Fig. 2). The clinical characteristics of individuals with an identified monogenic cause for NL/NC are described (Suppl. Table 3). Seven of 19 detected mutations (36.8%) were novel pathogenic variants that have not been previously reported in databases of human disease causing mutations.
Figure 2. Renal ultrasound and CT scan images of patients with NC, in whom causative mutations were detected (Mutated genes are given in parenthesis).
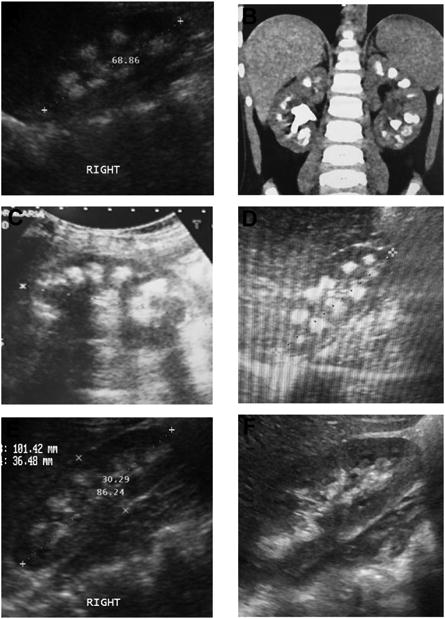
(A) Image from B1121 (ATP6V1B1). (B) Image from B1465_22 (CLDN16). (C) Image from A4283 (CLDN19). (D) Image from B1437_21 (SLC34A1). (E) Image from B1301_21 (SLC34A1). (F) Image from B986_21 (SLC34A1).
Detection rate of causative mutations was not different between sexes (14/32 in males and 8/33 in females, p = 0.27). Median age of onset was significantly lower (Z score: 2.6, p= 0.009) in patients with a monogenic cause (3 yrs) vs. those without detection of monogenic cause (7 yrs) (Suppl. Fig 3). We evaluated our cohort for differences regarding disease (NL/NC) at presentation, age of onset of disease, and causative mutation detection (Fig. 3). Individuals with NC presented before the age 15 yrs, whereas individuals with NL presented from 0 to 25 years (Fig. 3). Causative mutations were detected in 13 of 29 NC individuals and in 9 of 36 NL individuals (Fig. 3). We did not detect a causative mutation in any individual who presented after the age of 15 yrs (Fig. 3).
Figure 3. Distribution of number of affected individuals with NL/NC by age of onset and by the criterion if a causative mutation was detected.
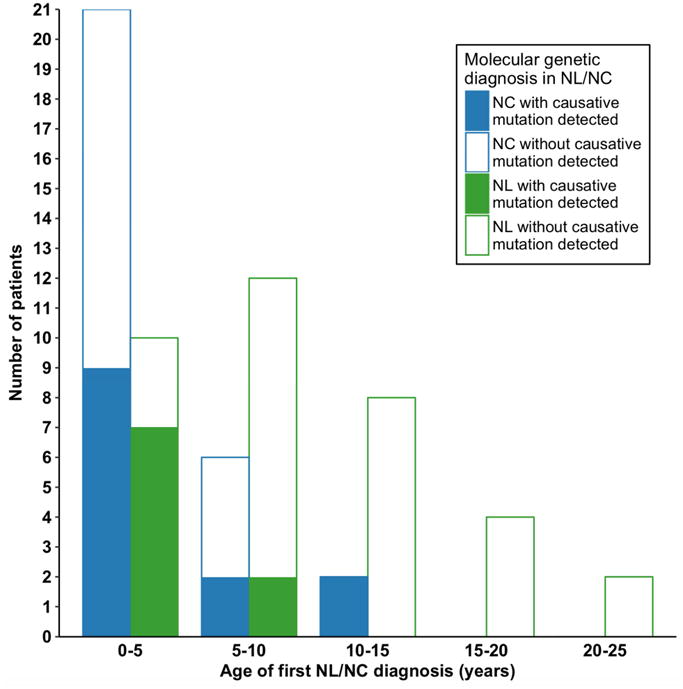
In a total of 65 affected individuals, 36 presented with NL, and 29 presented with NC. A causative mutation was detected in 9/36 (25%) individuals who presented with NL, and in 13/29 (44.8%) individuals presenting with NC. Affected individuals with NC presented before the age of 15 yrs.
In 14 of the 24 individuals (58%) who presented before age 3 yrs, a monogenic mutation was detected (Suppl. Table 4A), indicating a higher mutation detection rate in early onset disease. In 12 of the 29 families (41%) with multiple affected individuals included in our cohort we detected a causative genetic mutation (Suppl. Table 4B), indicating a higher likelihood of detecting a monogenic cause in familial cases. A monogenic cause was detected in 5 of 29 (17%) American, 3 of 11 (27%) European, 3 of 4 (75%) Middle Eastern, and 3 of 5 (60%) Egyptian families, likely reflecting higher monogenic mutation detection rate in regions with higher consanguinity. Monogenic causation was detected in 75% of consanguineous families and in 21% of families, which were either non-consanguineous (Suppl. Table 4C, 4D).
We evaluated the age of onset of NL/NC in relation to the 9 genes, in which we detected causative mutations in this cohort, and compared the data with our previously published data4,5 (Fig. 4). The median age of onset of disease for mutated recessive genes SLC34A1, SLC12A1, GRHPR, and AGXT is below the age of 5 yrs in the current and previously published studies4,5. The median age for recessive genes ATP6V1B1, CLDN19, SLC3A1, and CLDN16 as well as for dominant genes SLC34A1 and SLC9A3R1 is greater than 5 yrs in the current and previously published studies4,5.
Figure 4. Distribution of age of first diagnosis of NL/NC for each mutated gene detected by WES in 22 individuals from 15 families with NL/NC in this paper (filled circles), and from previously published 13 families 5 (hollow squares), and from 7 previously published families 4 (hollow triangles).
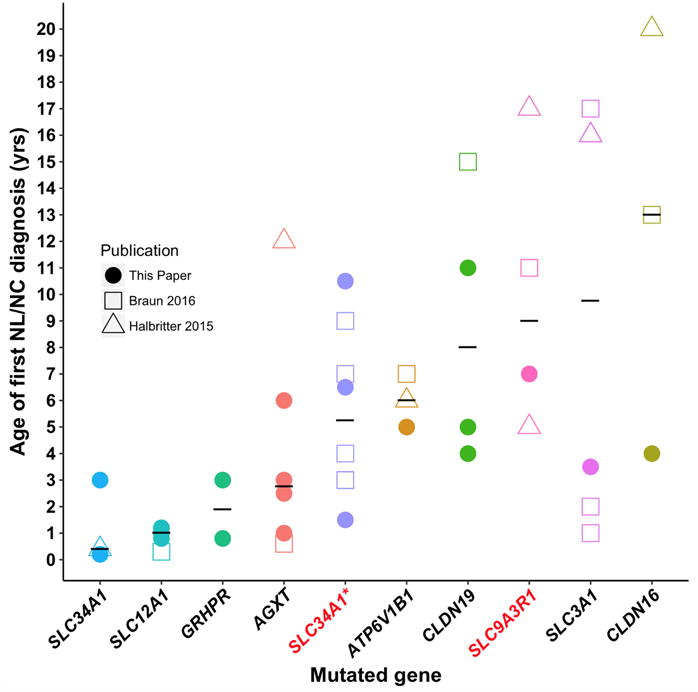
Medians are depicted as black horizontal lines. Gene symbols are given on the X-axis for recessive genes (black) and dominant genes (red). Note that median age of onset of disease for mutated recessive genes SLC34A1, SLC12A1, GRHPR, AGXT is below the age of 5 yrs. The median age for recessive genes ATP6V1B1, CLDN19, SLC3A1, CLDN16, and for dominant genes SLC34A1 and SLC9A3R1 is greater than 5 yrs. SLC12A1, Solute carrier family 12 (sodium/potassium/chloride transporter) member 1, SLC34A1, Solute carrier family 34 (Type II Sodium/Phosphate cotransporter) member 1; GRHPR, Glyoxalate Reducatse/Hydroxypyruvate Reducatse; AGXT, Alanine-Glyoxalate Aminotransferase; ATP6V1B1, ATPase, H+ transporting, lysosomal, 56/58-KD, V1 subunit B, isoform 1; SLC9A3R1, Solute Carrier Family 9, Member 3, Regulator 1; CLDN19, Claudin 19
WES data of 36 unsolved families were evaluated for mutations in 117 genes known to represent phenocopies of NL or NC. This included genes causing renal ciliopathies and tubulopathies (Suppl. Table 2). These genes, if mutated, may represent phenocopies of NC as they cause increased echogenicity on renal US.8 Among these phenocopy genes (Suppl. Table 2) we detected a previously reported homozygous frameshift mutation (c.828_829insA) in CTNS gene9 in one affected individual with nephrocalcinosis and chronic kidney disease of unclear etiology. This affected individual presented at the age of 2 yrs without a classic cystinosis phenotype at initial presentation. The monogenic causative mutation was detected 6 months after his initial presentation at which point he had developed few cystinosis features such as failure to thrive, polyuria and polyuria. We performed homozygosity mapping for this affected individual and the detected homozygous CTNS mutation was located in a segment of homozygosity10 (Suppl Fig. 4). The detection of a mutation in a phenocopy gene provides an unequivocal diagnosis and demonstrates the advantage of WES because more genes than the 30 known NL/NC genes can be evaluated.
In 7 of the 15 families (B1468, B1230, B1646, B1645, B35, B1508, B1648) in whom a causative mutation was detected, the clinician suspected the genetic diagnosis based on biochemical testing performed prior to undergoing WES. Interestingly in family B1468 with both siblings diagnosed with PH1, the index case presented with recurrent stones, CKD, and hyperoxaluria, which raised the suspicion of PH1, however the index patient's younger brother only manifested with microscopic hematuria and nephrocalcinosis where PH1 was not suspected prior to WES.
In remaining 8 of the 15 families in whom a causative mutation was detected, the genetic diagnosis was not suspected prior to WES because the subject did not manifest classic or complete phenotype at the time of genetic testing. In family B1121 in whom a mutation in ATP6V1B1 (distal RTA gene with deafness) was detected, the patient had bilateral nephrocalcinosis and renal tubular acidosis but did not manifest deafness prior to WES. In family A4283, the siblings presented with nephrocalcinosis and CKD without evidence of hypomagnesemia or ocular findings, and therefore mutation in CLDN19 was not suspected prior to WES. Post WES, the data for all three siblings is not available. In family B917, the clinical presentation included polyuria, polydipsia, and intermittent metabolic derangements raising the suspicion of a tubulopathy, not specifically of Bartter's disease. WES detected a mutation in SLC12A1, a bartter's type 1 gene. In both families B1437 and B1602, idiopathic hypercalcemia due to SLC34A1 gene mutation was not suspected by the clinician due to evidence of mild hypophosphatemia in the former, and no hypophosphatemia in the latter. In family B969, due to intermittent hypophosphatemia and no evidence of osteoporosis, the suspicion of nephrolithiasis/osteoporosis due to SLC9A3R1 gene mutation was not present prior to WES. Similarly, in families B1301, B989, who did not present with hypophosphatemia, the genetic diagnosis of SLC34A1 was not clinically suspected prior to WES. Therefore, WES allows definitive diagnosis even when all symptoms have not manifested and can suggest gene specific diagnostic and treatment approaches.
We also generated a list of 16 hypothetical candidate genes11 and evaluated WES data of 51 families for these genes. However, no mutation was detected in any of these candidate genes.
Discussion
Frequency of monogenic NL/NC
Here, we performed WES in 65 individuals from 51 families with NL or NC with onset before 25 years of age. We identified causative mutations in 15 of 51 families (29.4%) in one of 30 genes known to cause monogenic NL/NC. This percentage is higher than seen previously, when we identified a monogenic cause of NL/NC in 20.8% of childhood onset, and 11% of adult onset NL/NC in a separate mixed pediatric and adult cohort using a targeted sequencing panel.4 Our rate of identifying a causative mutation in a monogenic gene is also higher than in our previous panel based study, in which we identified a monogenic cause of NL/NC in 16.7% of children who presented with NL/NC before the age of 18.5 This higher mutation detection rate may be due to inclusion of more early onset and familial cases in this study that were recruited from tertiary stone clinics. So far, WES had not been applied to patients with NL/NC. Our data show that WES is an efficient tool for molecular diagnostics in NL/NC.
Phenocopies
We evaluated 36 families without a molecular genetic diagnosis in any of the 30 NL/NC genes for mutations in phenocopy genes. Renal ciliopathies and tubulopathies may represent phenocopies of NC, because they may generate a renal US finding of increased echogenicity.12 In one family with NC and chronic kidney disease, we detected a mutation in a phenocopy gene (CTNS) that causes nephropathic cystinosis. This molecular genetic diagnosis was not suspected clinically, since the affected individual had not developed any features of cystinosis at initial presentation. Therefore, in this individual, detection of the causative CTNS mutation by WES established an etiologic diagnosis before the correct clinical diagnosis was made, demonstrating that WES may help to distinguish between NL/NC and other kidney diseases that may phenocopy the presentation of NC on renal ultrasound. The unbiased detection of causative mutations by WES can be particularly advantageous in very early stages of disease progression, in which characteristic symptoms might not yet be present, as well as in very rare genetic syndromes. The ability to evaluate a large number of phenocopy genes represents an additional benefit of WES. Because WES evaluates all 20,000 human genes, mutation detection is not restricted to a limited subset of monogenic diseases that are suspected based on the clinical presentation, but allows taking a broader spectrum of monogenic causes into consideration.
Although a phenocopy/candidate gene panel can be used in cases that test negative for mutations in known NL/NC genes, the gene panels may not necessarily be updated to keep up with novel genes causing NL/NC or tubulopathies being discovered. Additionally, although specific gene panels may still be less expensive, the cost of WES has been declining, and the rate of decline of the WES service cost may make it comparable to gene panel costs. Today the average commercial cost of WES ranges from $1,000-$2,100 per sample in the United States, and the costs for WES in a research study is even less ∼$500. The average commercial gene panels cost ranges from $1,000-$1,900 for NL/ NC panels. Often, the turnaround time for results is comparable between these two techniques.
High mutation detection rate
The ability to identify causative mutations by WES is a function of not only the genetic technique employed but also of the population studied. Therefore, we performed post-hoc analysis of the frequency of causative mutations identified within subgroups of our cohort (Suppl. Table 4A-C). While the collective rate of mutation detection is 29.4% (15/51), several factors correlated with a higher percentage of detecting causative mutations. The factors with higher detection rate were: younger age of onset of NL/NC, presence of multiple affected in a family, ethnic background, and presence of consanguinity (Suppl. Table 4A-E). The respective rates of detecting the causative mutation were: 58% in age of onset < 3 yrs, 41% in positive family history with multiple family members with NL/NC, 75% in descent from the Middle East, 60% in descent from Egypt, and 75% in case with reported consanguinity. Nonetheless, the mutation detection rate for subjects outside of these groups was also notable: 20% for subjects older than 3 years old, 14% for those lacking a family history, 17% and 27% for North Americans and Europeans respectively, and 21% for those without a clear history of consanguinity. Because consanguinity is not always revealed when taking the family history, we performed homozygosity mapping from subject exomes on 45 of 51 families.10 Consistent with the postulated relationship to consanguinity, the mutation detection rate correlated with the amount of homozygosity. The mutation detection rate was 11% at < 10 Mb, 40% at 10-100 Mb, and 50% at > 100 Mb of total homozygosity. (Suppl. Table 4D-E). Therefore, the higher percentage of monogenic causative mutational detection in this cohort is likely due to patients being referred from tertiary centers, and higher number of early onset disease, and familial cases.
SLC34A1
We identified mutations in SLC34A1 in 6 individuals from 4 families in this study (Table 1A-B). The individuals harboring recessive mutations in this study exhibited nephrocalcinosis, hypercalcemia, hypercalciuria, and hypophosphatemia, consistent with renal phosphate wasting (Table 1A). On the other hand, affected individuals bearing dominant mutations in this study had nephrocalcinosis and hypercalciuria (Table 1B). Upon reviewing all published pediatric cases with SLC34A1 mutations (Table S5), the phenotypic spectrum observed mirrors our findings. All cases exhibit nephrolithiasis or nephrocalcinosis. However, recessive cases typically exhibit hypercalcemia, hypercalciuria, and hypophosphatemia whereas dominant cases exhibit hypercalciuria and rarely include hypercalcemia (Tables S5, S6). Moreover, there is also a dichotomy in SLC34A1 mutations between reported recessive and dominant cases based on age of onset, as recessive cases typically present in infancy (median 0.25 years) whereas dominant cases tend to present later in childhood (median 3.5 years) (Suppl. Fig. 5).
Clinical implications
A molecular genetic diagnosis may have implications for both the affected individual and apparently asymptomatic family members. As addressed in Table 2, information resulting from mutation analysis by WES will guide clinicians to monitor individuals for development of disease and to institute preventative treatment when possible. Although individual treatment continued in all families prior to WES, a molecular diagnosis resulted in precise treatment options in 3 families (B1437, B1602, B785) of the 15 families in which genetic mutation was detected (Table 2). Two families (B1437 and B1602) were receiving treatment for their hypercalcemia with diuretics and bisphosphonates prior to WES. However, a molecular diagnosis of SLC34A1 gene mutation causing idiopathic infantile hypercalcemia detected by WES in these two families prompted the physician to consider phosphorous supplement to treat infantile hypercalcemia6. In family B785, detection of CTNS mutation causing cystinosis, allowed for the possibility of treatment with cysteamine, which was not considered prior to WES, allowing molecular diagnosis to lead to disease specific treatment, and possibly delay in progression to ESRD13. In 3 other families (B1121, A4238, B969); the molecular diagnosis led to precise screening as shown in clinical Table 2, which was not performed prior to WES. Families B1121, A4283 with recessive mutations in ATP6V1B1 and CLDN19 may benefit from hearing and ophthalmology testing respectively, and from early treatment for growth retardation, metabolic correction and close monitoring which can delay progression to ESRD.14 Individual B969, who has a SLC9A3R1 mutation will benefit from close monitoring and treatment for hypophosphatemia. Recessive AGXT mutations were detected in B1468 family in which one sibling presented with severe hyperoxaluria type 1 and another younger sibling exhibited a milder phenotype of nephrocalcinosis. This genetic diagnosis in the younger sibling may allow early chronic kidney disease preventative care. 14
Future directions/limitations
35 of 51 families in our cohort (68%) remained without a molecular diagnosis after WES evaluation. In recessive monogenic diseases about 85% of all causative mutations are located within the coding sequence or the adjacent splice sites.15 The remaining 15% however, may represent genetic variants that are difficult to detect by WES such as complex deletion-insertion variants, copy-number variants, or variants residing within a promotor or other intronic region. This technical limitation might explain why some cases remain without a molecular diagnosis after WES. In addition, causative mutations may be missed because the stringent criteria that we currently use to assess for deleteriousness may not include mild, hypomorphic mutations. WES might also miss a subset of causative variants due to low coverage in the respective target region. Additionally, we speculate that some remained without a molecular diagnosis secondary to genetic stone disease caused by a novel gene which is yet to be functionally studied. The WES data for these unsolved patients allows discovery of novel causative genes. And in some cases without a molecular diagnosis, we speculate the stone disease to be secondary to other non-genetic factors such as environmental effects.
We here present WES as a rapid and reliable tool for molecular diagnostics in individuals with a history of at least one renal stone or NC on a renal ultrasound in 29% of affected individuals. The higher percentage of monogenic causative mutational detection in this cohort correlates with a higher number of early onset disease, and familial cases. This study represents a major advance in providing a specific etiologic diagnosis in NL/NC, delaying progression to CKD/ESRD, and enabling personalization of the treatment plan for each stone former with a detectable monogenic cause of disease.
Materials and Methods
Study participants
The study was approved by the institutional review board (IRB) of Boston Children's Hospital (BCH). We obtained informed consent, clinical data, pedigree information, and DNA samples from subjects who had at least one occurrence of nephrolithiasis or demonstration of nephrocalcinosis on renal ultrasound, manifesting before the age of 25 years. Subjects with a potential secondary cause of NL such as hyperparathyroidism or use of loop diuretics were excluded from the study. WES was performed in 65 individuals from 51 families. 8 families were consanguineous, and 29 families had more than one affected child. The cohort selected for WES was enriched for families with multiple affected members and individuals with recurrent disease. The families were selected for study as follows (Suppl. Fig. 6): We enrolled 339 families affected with NL or NC from November 2013 to May 2016 (31 months). Of the 339 families, 239 families were previously screened by multiplex PCR of a 30-gene panel4,5. Of the 100 patients in our cohort with nephrolithiasis and nephrocalcinosis, we initially selected all families with multiple affected family members (n= 49). Of these 49 families, we selected 29 families where DNA was available for more than one affected individual for WES. Of the remaining 51 families where one individual was affected, we selected families with recurrent and/or early onset disease (n= 7), and subsequently families where samples of both parents were available for trio analysis (parents and patient, n = 15) for improved interpretation and analysis of WES data.
Whole exome sequencing and mutation calling
In brief, genomic DNA was isolated from blood lymphocytes and subjected to exome capture using Agilent SureSelect™ human exome capture arrays (Life technologies™) followed by next generation sequencing on the Illumina HighSeq™ sequencing platform. Sequence reads were mapped to the human reference genome assembly (NCBI build 3/hg19) using CLC Genomics Workbench™ (version 6.5.2) software (CLC bio, Aarhus, Denmark). Following alignment to the human reference genome, variants were filtered as previously described 8 and as summarized in Suppl. Fig. 7. For homozygosity mapping, downstream processing of aligned BAM files was done using Picard and samtools16. SNV calling was performed using GATK17 and the generated VCF file was subsequently used in homozygosity mapper.18 In the first step variants with minor allele frequencies (MAF) >1% in the dbSNP (version 142) were excluded. In the second step, synonymous variants and intronic variants that were not located within splice site regions were excluded. In step 3, variants were evaluated for mutations in 30 known NL/NC genes (Suppl. Table 1). In step 4, remaining variants were ranked based on their probable impact on protein sequence and function considering evolutionary conservation among orthologs across phylogeny, as well as web-based prediction programs (PolyPhen-219, SIFT20 and MutationTaster21). In step 5, remaining variants were searched further evaluated by reviewing the existing literature and determining phenotypic match (Suppl. Fig. 7). Clinician scientists and geneticists, who had knowledge of the clinical phenotypes and pedigree structure, as well as experience with exome evaluation performed mutation calling. Remaining variants were confirmed in original patient DNA by Sanger sequencing as previously described.22 Whenever parental DNA was available, segregation analysis was performed.
In a second evaluation process, variants in all 117 genes (Suppl. Table 2) that are known monogenic, recessive causes of renal ciliopathies8 and tubulopathies23 that may phenocopy the clinical presentation of NC/NL were systematically evaluated. In the third evaluation process, variants in 16 genes that were deemed potential novel candidate genes to cause NL/NC11 if mutated (evidence with mouse models or NL risk allele) were systematically evaluated. These genes were ADCY6, CHD1, CLDN2, CLDN10, CLDN14, DMP1, ENPP1, FETUB, GALNT3, ITPKC, KL, MGP, ORAl1, PHEX, SLC13A2, SPP1.11
Supplementary Material
Table 3.
Clinical implications following detection of a monogenic cause of NL/NC.
| Gene (Family number) | Clinical diagnosis (Pre-screening) | Genetic diagnosis (Post-screening) | Practical implications |
|---|---|---|---|
| AGXT (B1468) (B1230) (B1646) | NL, hyperoxaluria | Primary hyperoxaluria I | GC, Consider pyridoxine trial, Potential enrollment in trials of new therapy for hyperoxaluria for future stone prevention |
| ATP6V1B1 (B1121) | NC, RTA | Distal renal tubular acidosis, deafness | GC, Hearing test, ESRD progression prevention |
| CLDN19 (A4283) | NC, CKD | Hypomagnesemia 5, renal with ocular disease | GC, Ophthalmology test |
| SLC34A1 (B1427) (B1602) | NC | Hypercalcemia, infantile, type 2 | GC, Phosphate supplementation |
| SLC9A3R1 (B969) | NL | Nephrolithiasis, osteoporosis, hypophosphatemia | GC, Screening for hypophosphatemia, Screening for abnormal bone density, Osteoporosis prevention |
| CTNS (B785) | NC | Cystinosis | GC, Cysteamine therapy, ESRD progression prevention |
AGXT, Alanine-glyoxalate aminotransferase; ATP6V1B1, ATPase, H+ transporting, lysosomal, 56/58-KD, V1 subunit B, isoform 1; CLDN19, Claudin 19; CTNS, Cystinosis; GC, genetic counseling; NC, nephrocalcinosis; NL, nephrolithiasis, RTA, renal tubular acidosis; SLC34A1, solute carrier family 34; SLC9A3R1, solute carrier family 9, member 3, regulator 1.
Acknowledgments
We thank the physicians and the participating families for their contribution. We thank Leslie Speanas, Brittany Fisher, and Kassaundra Amann for recruitment of study participants. F.H. is the William E. Harmon Professor of Pediatrics. This research was supported by grants from the National Institutes of Health DK1069274, DK1068306, and DK064614 to FH and 5U54HG006504 to RPL. H.Y.G. was supported by the Basic Science Research Program through National Research Fund of Korea, funded by Ministry of Education. Tilman Jobst Schwan is supported by the Deutsche Forschungsgemeinschaft (Jo 1324/1-1). WES performed at Yale Center for Mendelian Genomics.
Footnotes
Disclosure Statement: None of the authors has competing financial interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scales CD, Jr, Smith AC, Hanley JM, Saigal CS. Prevalence of kidney stones in the United States. Eur Urol. 2012 Jul;62(1):160–165. doi: 10.1016/j.eururo.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rule AD, Bergstralh EJ, Melton LJ, 3rd, Li X, Weaver AL, Lieske JC. Kidney stones and the risk for chronic kidney disease. Clin J Am Soc Nephrol. 2009 Apr;4(4):804–811. doi: 10.2215/CJN.05811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dwyer ME, Krambeck AE, Bergstralh EJ, Milliner DS, Lieske JC, Rule AD. Temporal trends in incidence of kidney stones among children: a 25-year population based study. J Urol. 2012 Jul;188(1):247–252. doi: 10.1016/j.juro.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halbritter J, Baum M, Hynes AM, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol. 2015 Mar;26(3):543–551. doi: 10.1681/ASN.2014040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braun DA, Lawson JA, Gee HY, et al. Prevalence of Monogenic Causes in Pediatric Patients with Nephrolithiasis or Nephrocalcinosis. Clin J Am Soc Nephrol. 2016 Apr;0711(4):664–672. doi: 10.2215/CJN.07540715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlingmann KP, Ruminska J, Kaufmann M, et al. Autosomal-Recessive Mutations in SLC34A1 Encoding Sodium-Phosphate Cotransporter 2A Cause Idiopathic Infantile Hypercalcemia. J Am Soc Nephrol. 2016 Feb;27(2):604–614. doi: 10.1681/ASN.2014101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prie D, Huart V, Bakouh N, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med. 2002 Sep 26;347(13):983–991. doi: 10.1056/NEJMoa020028. [DOI] [PubMed] [Google Scholar]
- 8.Gee HY, Otto EA, Hurd TW, et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014 Apr;85(4):880–887. doi: 10.1038/ki.2013.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Besouw MT, Van Dyck M, Francois I, Van Hoyweghen E, Levtchenko EN. Detailed studies of growth hormone secretion in cystinosis patients. Pediatr Nephrol. 2012 Nov;27(11):2123–2127. doi: 10.1007/s00467-012-2213-x. [DOI] [PubMed] [Google Scholar]
- 10.Hildebrandt F, Heeringa SF, Ruschendorf F, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009 Jan;5(1):e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gee HY, Jun I, Braun DA, et al. Mutations in SLC26A1 Cause Nephrolithiasis. Am J Hum Genet. 2016 Jun;0298(6):1228–1234. doi: 10.1016/j.ajhg.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braun DA, Schueler M, Halbritter J, et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016 Feb;89(2):468–475. doi: 10.1038/ki.2015.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gahl WA. Early oral cysteamine therapy for nephropathic cystinosis. Eur J Pediatr. 2003 Dec;162(Suppl 1):S38–41. doi: 10.1007/s00431-003-1349-x. [DOI] [PubMed] [Google Scholar]
- 14.Jungers P, Joly D, Barbey F, Choukroun G, Daudon M. ESRD caused by nephrolithiasis: prevalence, mechanisms, and prevention. Am J Kidney Dis. 2004 Nov;44(5):799–805. [PubMed] [Google Scholar]
- 15.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009 Nov 10;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;4311(10):11–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009 Jul 1;37(Web Server issue):W593–599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010 Apr;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 21.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014 Apr;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 22.Otto EA, Ramaswami G, Janssen S, et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2010 Nov 10; doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. 2016 Mar;12(3):133–146. doi: 10.1038/nrneph.2015.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danpure CJ. Primary hyperoxaluria type 1 and peroxisome-to-mitochondrion mistargeting of alanine:glyoxylate aminotransferase. Biochimie. 1993;75(3-4):309–315. doi: 10.1016/0300-9084(93)90091-6. [DOI] [PubMed] [Google Scholar]
- 25.von Schnakenburg C, Rumsby G. Primary hyperoxaluria type 1: a cluster of new mutations in exon 7 of the AGXT gene. J Med Genet. 1997 Jun;34(6):489–492. doi: 10.1136/jmg.34.6.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lage MD, Pittman AM, Roncador A, Cellini B, Tucker CL. Allele-specific characterization of alanine: glyoxylate aminotransferase variants associated with primary hyperoxaluria. PLoS One. 2014;9(4):e94338. doi: 10.1371/journal.pone.0094338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999 Jan;21(1):84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 28.Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet. 1999 Oct;8(11):2063–2069. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- 29.Hopp K, Cogal AG, Bergstralh EJ, et al. Phenotype-Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. J Am Soc Nephrol. 2015 Oct;26(10):2559–2570. doi: 10.1681/ASN.2014070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brauers E, Hozyasz K, Golabek B, et al. Identification of novel cystinuria mutations in pediatric patients. J Pediatr Urol. 2006 Dec;2(6):575–578. doi: 10.1016/j.jpurol.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Adachi M, Asakura Y, Sato Y, et al. Novel SLC12A1 (NKCC2) mutations in two families with Bartter syndrome type 1. Endocr J. 2007 Dec;54(6):1003–1007. doi: 10.1507/endocrj.k06-204. [DOI] [PubMed] [Google Scholar]
- 32.Karim Z, Gerard B, Bakouh N, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med. 2008 Sep 11;359(11):1128–1135. doi: 10.1056/NEJMoa0802836. [DOI] [PubMed] [Google Scholar]
- 33.Lapointe JY, Tessier J, Paquette Y, et al. NPT2a gene variation in calcium nephrolithiasis with renal phosphate leak. Kidney Int. 2006 Jun;69(12):2261–2267. doi: 10.1038/sj.ki.5000437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.