

Supplemental digital content is available in the text.
Key Words: alcohol, experimental acute pancreatitis, mitochondrial permeability transition pore (MPTP), necrosis, TRO40303
Abstract
Objectives
Mitochondrial permeability transition pore inhibition is a promising approach to treat acute pancreatitis (AP). We sought to determine (i) the effects of the mitochondrial permeability transition pore inhibitor 3,5-seco-4-nor-cholestan-5-one oxime-3-ol (TRO40303) on murine and human pancreatic acinar cell (PAC) injury induced by fatty acid ethyl esters (FAEEs) or taurolithocholic acid-3-sulfate and (ii) TRO40303 pharmacokinetics and efficacy in experimental alcoholic AP (FAEE-AP).
Methods
Changes in mitochondrial membrane potential (Δψm), cytosolic Ca2+ ([Ca2+]c), and cell fate were examined in freshly isolated murine or human PACs by confocal microscopy. TRO40303 pharmacokinetics were assessed in cerulein-induced AP and therapeutic efficacy in FAEE-AP induced with palmitoleic acid and ethanol. Severity of AP was assessed by standard biomarkers and blinded histopathology.
Results
TRO40303 prevented loss of Δψm and necrosis induced by 100 μM palmitoleic acid ethyl ester or 500 μM taurolithocholic acid-3-sulfate in murine and human PACs. Pharmacokinetic analysis found TRO40303 accumulated in the pancreas. A single dose of 3 mg/kg TRO40303 significantly reduced serum amylase (P = 0.043), pancreatic trypsin (P = 0.018), and histopathology scores (P = 0.0058) in FAEE-AP.
Conclusions
TRO40303 protects mitochondria and prevents necrotic cell death pathway activation in murine and human PACs, ameliorates the severity of FAEE-AP, and is a candidate drug for human AP.
Acute pancreatitis (AP) occurs in up to 100 per 100,000 persons per year, mostly caused by cholelithiasis or excessive alcohol intake.1 Twenty percent of cases are severe, these being frequently complicated by pancreatic necrosis, multiple organ failure, prolonged hospitalization, and death.2 Despite several hundred randomized clinical trials to date, there is no internationally validated specific drug therapy for this common disease, one of the commonest gastrointestinal emergencies requiring hospitalization in the United States costing approximately $2.6 billion per annum.3
Pancreatic necrosis is characterized by severe pathophysiological changes that include mitochondrial swelling, plasmalemmal disruption, and leakage of activated digestive enzymes and other cellular contents, which trigger acute exudative inflammation of the surrounding tissue.1,4 Necrosis is a key predictor of disease outcome in patients with AP.2 Because clinical pancreatic necrosis develops typically over several days rather than hours, there is a therapeutic window that could be exploited to prevent necrosis if an appropriate mechanism can be targeted. Mitochondria have been increasingly implicated in pancreatic acinar cell (PAC) injury, necrosis, and AP.5–7 Pancreatitis toxins cause acinar cytosolic calcium overload through excessive release from the endoplasmic reticulum calcium store sustained by continued calcium entry.8 Cytosolic calcium overload causes mitochondrial matrix calcium overload, inducing the mitochondrial permeability transition pore (MPTP), a nonspecific inner mitochondrial membrane channel that results in loss of mitochondrial membrane potential (Δψm) essential for ATP production.9 Inhibition of the MPTP has been shown to sustain ATP production in PACs under stress and is potentially a suitable strategy for the treatment of AP.10
An important group of pancreatitis toxins are fatty acid ethyl esters (FAEEs), nonoxidative metabolites of ethanol formed in the pancreas more than any other organ per unit weight of tissue, reaching high concentrations following excess ethanol consumption.11 We have shown FAEEs to cause pancreatic acinar cytosolic calcium overload through induction of second messenger receptor calcium-channel release from the endoplasmic reticulum,12 leading to mitochondrial impairment and diminished ATP production,13 alongside premature intracellular digestive enzyme activation.14 We have since developed an in vivo AP model derived from these findings, induced by simultaneous administration of ethanol and fatty acid (FAEE-AP).15 Inhibition of FAEE synthesis or FAEE hydrolysis reduced the severity of AP, confirming FAEEs as the principal source of toxicity.15 Preliminary data using the cyclosporin A analog Debio-025 have indicated that inhibition of the MPTP is protective in FAEE-AP, consistent with findings in other models.10
TRO40303 (3,5-seco-4-nor-cholestan-5-one oxime-3-ol) is a cholesterol-oxime cytoprotective compound developed by Trophos (since acquired by Roche) that binds to the outer mitochondrial translocator protein (TSPO), delaying MPTP opening and cell death in rat cardiomyocytes, and has been shown to significantly reduce the extent of myocardial infraction in a rat model.16 TRO40303 protected HepG2 cells and primary mouse embryonic hepatocytes from palmitate intoxication and significantly reduced mortality in murine FAS-induced hepatotoxicity.17 Results from a randomized safety and tolerance phase 1 trial showed that TRO40303 can be safely administered intravenously in humans at doses expected to be pharmacologically active.18 We have previously demonstrated that 10 μM TRO40303 protects PACs from bile acid–induced mitochondrial injury and necrosis in vitro, whereas 3 mg/kg TRO40303 reduced the severity of cerulein (CER-AP)–induced and bile acid (taurolithocholic acid-3-sulfate [TLCS]–AP)–induced experimental AP in vivo.10 Here we have evaluated the effects of a range of concentrations of TRO40303 on murine and human PACs exposed to FAEEs or TLCS. Because cyclophilin D (Cyp-D) is an integral modulator of MPTP opening, we also evaluated whether Cyp-D is a potential target of TRO40303. To determine in vivo effects, we assessed pharmacokinetics of TRO40303 over 24 hours following administration of a range of doses in both naive mice and in murine experimental AP, to select a dose that we then tested in FAEE-AP.
MATERIALS AND METHODS
Human Pancreatic Sampling
Normal human pancreatic tissue samples were obtained from consenting patients with no history of jaundice or chronic pancreatitis who were undergoing elective left pancreatectomy, resections for duodenal tumors or nonobstructed right-sided cancer resections at the Royal Liverpool University Hospital (Liverpool Adult Local Research Ethics Committee approval 03/12/242/A). Isolation of human PACs was performed as previously described.19 The time from sampling to the start of cell isolation procedure was 10 minutes or less.
Animals
Wild-type 8- to 12-week-old CD1 mice, weighing approximately 30 g each, were used for PAC isolation and cell experiments. C57Bl/6 mice heavier than 25 g were used for in vivo experiments.
Confocal Microscopy
Acinar cells were loaded with appropriate dye(s) and imaged using Zeiss LSM 510 and 710 confocal microscopes (Jena, Germany), equipped with multiple laser lines. Cells were visualized with a C-Apochromat 63X water immersion objective. Between 150 and 180 μL of PACs suspended in Na HEPES were placed in a microplate, and healthy-looking duplets or triplets of acinar cells with intact loading of fluorescent dyes were selected for recording of Δψm and [Ca2+]c.10,12,13,15 To assess mitochondrial function, live acinar cells were loaded with 50 nM tetramethylrhodamine methyl ester (TMRM; excitation 543 nm, emission >550 nm), a dye used to measure Δψm. Similarly to measure changes in [Ca2+]c, cells were loaded with Fluo-4 (excitation 488 nm, emission 505 nm) by incubation in solution containing 3 μM Fluo-4 AM. Typically cells were pretreated for 30 minutes with TRO40303 or vehicle prior to the start of each experiment. After the assessment of baseline fluorescence levels (F0), cells were stimulated with 100 μM palmitoleic acid ethyl ester (POAEE) or 500 μM TLCS and at the end of each experiment perfused with 10 μM carbonyl cyanide m-chlorophenylhydrazone as a positive control. Cells were imaged every 5 to 10 seconds for measurement of Δψm and [Ca2+]c, in the absence or presence of TRO40303 at a concentration of 1 μM (T1), 3 μM (T3), or 10 μM (T10). In some experiments, cells were coloaded with TMRM and Fluo 4 to measure (Δψm) and [Ca2+]c respectively, using the multitrack configuration of the LSM510 system. Both TMRM and Fluo-4 fluorescent signals were normalized to baseline fluorescence (F/F0).
For assessment of necrotic cell death pathway activation, 1 μM propidium iodide (PI; excitation 543 nm, emission 630–693 nm) was used to evaluate plasma membrane rupture.10,12,13 Isolated PACs from each animal or human sample were divided into 3 groups and imaged: (i) PAC only (negative control): acinar cells incubated with Na HEPES for 30 minutes; (ii) PAC + toxin (500 μM TLCS or 100 μM POAEE); acinar cells incubated with toxin for 30 minutes; (iii) PAC + toxin + T1/T3/T10: acinar cells incubated with 1, 3, or 10 μM TRO40303 and toxin simultaneously. Sixteen randomly selected fields of view were taken of each mouse isolate, and the total number of cells displaying PI uptake was counted per field. This gave a percentage ratio for each field, averaged across fields, converted to a mean ± SEM for a minimum of 3 mice and 2 or 3 human samples per experimental group. Each experiment was performed in a blinded fashion, with the observer choosing the fields and the observer undertaking image analysis blind to the treatment groups. Necrotic cell death pathway activation induced by 50 μM POAEE was also evaluated using time-lapse recording of PI (2 μg/mL; excitation 543 nm, emission >640 nm) signals at 37°C for 13 hours using a plate reader. In each experiment, 6 wells (on a 96-well plate) were allocated to each treatment group from which an average was calculated. T1, T3, and T10 were added 30 minutes prior to the addition of 50 μM POAEE.
Surface Plasmon Resonance
The Surface Plasmon Resonance technology–based Biacore X100 (GE Healthcare, Amersham, United Kingdom) was used to evaluate the interaction of TRO40303 with Cyp-D.20 All experiments were carried out using buffer containing Tris-HCl pH 7.6, NaCl 150 mM, 0.05% surfactant p20, and 5% of dimethyl sulfoxide as a running buffer, at a constant flow rate of 30 μL/min at room temperature. The protein, Cyp-D, was immobilized directly and covalently on a CM5 chip (BIAcore, Amersham, United Kingdom) by using standard primary amine coupling. All data analyses were carried out using BIA evaluation software version 3.0 (GE Healthcare, Amersham, United Kingdom).
Tissue and Plasma TRO40303 Exposure
Pharmacokinetics were assessed following intraperitoneal (IP) administration of 3 or 10 mg/kg TRO40303 in a liposomal preparation to C57Bl/6 mice. A maximum of 0.25 mL of blood was collected into lithium heparin tubes at 15 and 30 minutes and 1, 2, 4, 8, and 24 hours after drug administration (3 mice per time point). Blood samples were cooled on ice, and plasma samples prepared within 60 minutes of sampling by centrifugation at 1500g at 4°C for 10 minutes then stored at −20°C until analysis. For measurement of pancreatic levels, TRO40303 was administered by IP injection to mice at 3 mg/kg in liposomes, then pancreata were isolated at 15 minutes, 2 hours, and 24 hours after injection (3 mice per time point) and washed with saline. Samples were frozen and stored at −20°C until analysis.
Because the experiments in naive mice found TRO40303 levels in the pancreas to be similar to those achieved in the heart using 1 mg/kg TRO40303 administered by an intravenous route,16 confirmation that adequate levels of TRO40303 could be achieved in experimental AP was required. TRO40303 levels were therefore measured in blood (plasma) and pancreata sampled 15 minutes after administration of the liposomal preparation of 1, 3, or 10 mg/kg of TRO40303 in cerulein-induced AP, the most widely used model of AP.1 Plasma obtained from blood by immediate centrifugation (2500 revolutions/min at 4°C for 15 minutes) and pancreatic samples were stored at −20°C for bioanalysis. Thawed samples were extracted with acetonitrile, centrifuged, purified on SPEC C2 cartridge (Varian, Palo Alto, Calif), and analyzed by high-performance liquid chromatography–tandem mass spectrometry together with calibration standards. Analysis was carried out on an Alliance 2695 (Waters, Guyancourt, France) system interfaced with an API Quattro Micro (Waters) mass spectrometry detector. Calculations were undertaken using Waters Quan Lynx software version 1.40.
Experimental Acute Pancreatitis
Alcoholic AP was induced by IP injection of ethanol and palmitoleic acid (FAEE-AP), a fatty acid that combines with ethanol in the pancreas to form FAEEs, the principal agents of PAC injury from ethanol excess.15 Mice received 2 IP injections of 1.32 g/kg ethanol and 1.5 mg/kg palmitoleic acid (dissolved in the ethanol), each 1 hour apart, to induce AP. For treatment, 3 mg/kg TRO40303 (selected on the basis of the pharmacokinetic analyses and prior experience with TLCS-AP and CER-AP) was administered intraperitoneally 1 hour after the last injection.
The experimental groups were (i) controls with no toxin or treatment; (ii) positive controls, that is, FAEE-AP alone; and (iii) treatment group, that is, FAEE-AP with 3 mg/kg TRO40303 (6 per group). The dose was optimized based on its efficacy in CER-AP and TLCS-AP.10 TRO40303 stock solution in liposomes (20 mg/mL) was diluted into saline and injected intraperitoneally using a constant volume of 2.5 mL/kg. Animals were killed humanely 24 hours after induction of FAEE-AP for assessment of biochemical and histological parameters of severity.
All in vivo experiments complied with the Animals (Scientific Procedures) Act 1986 (Project Licence PPL70/8109) as administered by the Home Office UK.
Biochemical Parameters of FAEE-AP Severity
The biochemical parameters of severity were serum amylase, pancreatic trypsin, pancreatic myeloperoxidase (MPO) activity, and lung MPO activity. Amylase levels were tested using a kinetic method with a Roche automated clinical chemistry analyzer (Burgess Hill, United Kingdom). Trypsin activity was measured by a fluorogenic assay, using Boc-Gln-Ala-Arg-AMC substrate converted by trypsin to a fluorescent product (excitation 380 nm, emission 440 nm).21 For MPO activity, 20 μL of extract was added to 200 μL phosphate buffer (100 mM, pH 5.4, with 0.5% HETAB) and 20 μL 3,3′,5,5′-tetramethylbenzidine (20 mM) in dimethyl sulfoxide. This mixture was incubated at 37°C for 3 minutes, followed by addition of 50 μL of H2O2 (0.01%) for further incubation over 3 minutes. The difference of absorbance between 0 and 3 minutes at 650 nm was calculated using a standard curve compiled using human MPO.22
Histopathologic Parameters of AP Severity
Pancreatic tissue was fixed in formaldehyde, and standard hematoxylin-eosin sections were prepared. Scoring of edema (0–3), leukocyte infiltration (0–3), and necrosis (0–3) was undertaken by 2 independent, blinded investigators in ×10 high-power fields per slide per mouse10,15; ×200 magnification was used throughout. Scores were summated, then means ± SEM histology scores were calculated for each experimental group.
Statistical Analysis
Data are presented as means ± SEM. Comparisons were performed using the unpaired Student’s t-test or χ2 test as appropriate, with P < 0.05 taken as the minimum level of statistical significance.
RESULTS
TRO40303 Reduces Loss of Δψm and Necrotic Cell Death Pathway Activation From POAEE and TLCS in a Concentration-Dependent Manner
POAEE (100 μmol/L with 850 mmol/L ethanol to maintain solubility) induced a sharp fall in Δψm in murine PACs and rise in PI uptake; pretreatment with 10 μM TRO40303 significantly reduced the extent of depolarization (Fig. 1) and PI uptake (Fig. 2A). Comparison of the effects of 1, 3, and 10 μM TRO40303 on PI uptake induced by 50 μmol/L POAEE over 13 hours demonstrated that the protection afforded by TRO40303 is concentration dependent, with increasing effect at increasing concentrations (Fig. 2B).
FIGURE 1.
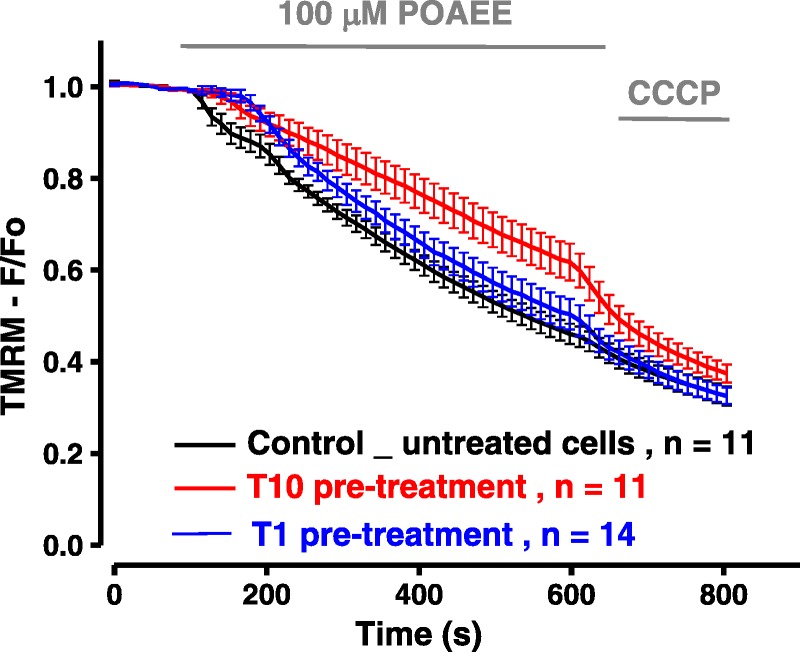
TRO40303 reduces loss of Δψm induced by FAEE. Palmitoleic acid ethyl ester (100 μM) induced a sharp fall in Δψm; this fall was reduced by pretreatment with 10 μM TRO40303 (T10), although there was no protection from pretreatment with 1 μM TRO40303 (T1).
FIGURE 2.
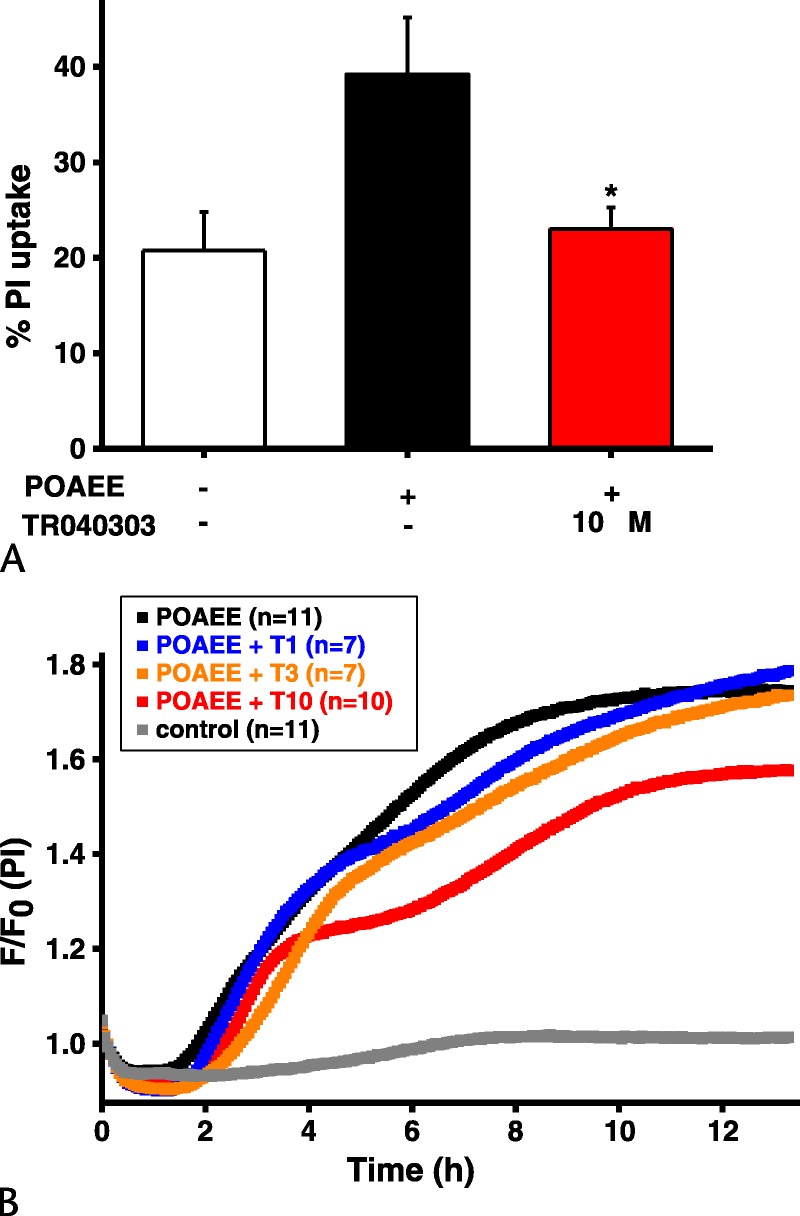
Effect of TRO40303 on necrotic cell death pathway activation induced by FAEE in PACs. A, Palmitoleic acid ethyl ester (100 μM) exposure increased PI uptake, whereas pretreatment with TRO40303 (10 μM) reduced necrotic cell death pathway activation in response to POAEE (POAEE vs POAEE plus TRO40303, *P < 0.05). B, Simultaneous comparison of PI uptake in response to POAEE (50 μM) alone or with 1, 3, or 10 μM TRO40303 pretreatment. Aggregate curves and paired analysis showed that all concentrations protected PACs against POAEE-induced cell death, in a concentration-dependent manner; n indicates number of independent experiments.
Because we previously found 10 μM TRO40303 inhibited falls in Δψm and prevented TLCS-induced necrotic cell death pathway activation in isolated murine and human PACs,10 but had not determined the concentration-dependence of these effects, we tested lower doses of TRO40303 on TLCS-induced loss of Δψm, [Ca2+]c, and PI update in murine and human PACs (Supplemental Figures 1 and 2, http://links.lww.com/MPA/A618). Pretreatment with TRO40303 inhibited depolarization, although 3 μM showed a trend to be more effective than 1 μM TRO40303. Consistent with this mitochondrial protection, we found TRO40303 reduced PI uptake in both murine and human PACs (Supplemental Figure 2, http://links.lww.com/MPA/A618) more effectively using 3 μM than 1 μM TRO40303. Protection from necrotic cell death pathway activation achieved statistical significance only at the higher concentration in both murine and human PACs but not to the same extent as observed with 10 μM TRO40303.10 In keeping with these effects, clearance of [Ca2+]c was more effective with higher concentrations of TRO40303 (Supplemental Figure 1, http://links.lww.com/MPA/A618).
TRO40303 Does Not Bind to Cyclophilin D
Using surface plasmon resonance, we evaluated the binding affinity of TRO40303 with Cyp-D, a regulator of the MPTP.9 TRO40303 did not bind to Cyp-D (Supplemental Figure 3, http://links.lww.com/MPA/A618). Moreover, no interaction between TRO40303 and Cyp-D was detected by nuclear magnetic resonance spectroscopy (data not shown). These findings indicate that TRO40303 inhibits the MPTP via a Cyp-D–independent mechanism.
TRO40303 Levels in Plasma and Pancreas
TRO40303 is distributed throughout the body with significant affinity for steroidogenic tissues and substantial levels in organs with high numbers of mitochondria.16 The liposomal preparation of TRO40303 was prepared for IP injection, an easier parenteral route than the previously studied intravenous route16 for administration in murine AP models. We considered determination of pancreatic levels to be critical as protection of the pancreas from opening of the MPTP is the primary purpose of this strategy.10 In naive mice receiving 3 mg/kg TRO40303 in the liposomal preparation, plasma levels reached approximately 5 μg/mL 2 hours after injection (Fig. 3A), with concentrations in the pancreas approximately 2.5 μg/g at the same time (Fig. 3B). This corresponded with the concentration achieved in the heart 5 minutes after intravenous injection of 1 mg/kg TRO40303 formulated into β-cyclodextrin in a rat model of coronary occlusion.16 Although 10 mg/kg TRO40303 in the liposomal preparation gave significantly higher plasma levels, levels were undetectable in plasma 24 hours following either dose. Nevertheless, at 24 hours after 3 mg/kg TRO40303 in the liposomal preparation, pancreatic levels were approximately 1 μg/g, indicative of a prolonged half-life (>12 hours) in the pancreas following a single dose and of potential efficacy in that organ (at approximately 2–5 μM) over 24 hours at that dose. Furthermore, plasma and pancreatic levels of TRO40303 at 15 minutes after injection were significantly higher in CER-AP (Figs. 3A, B; 1–10 mg/kg TRO40303). Therefore, 3 mg/kg TRO40303 was used to test the efficacy of this compound in FAEE-AP.
FIGURE 3.
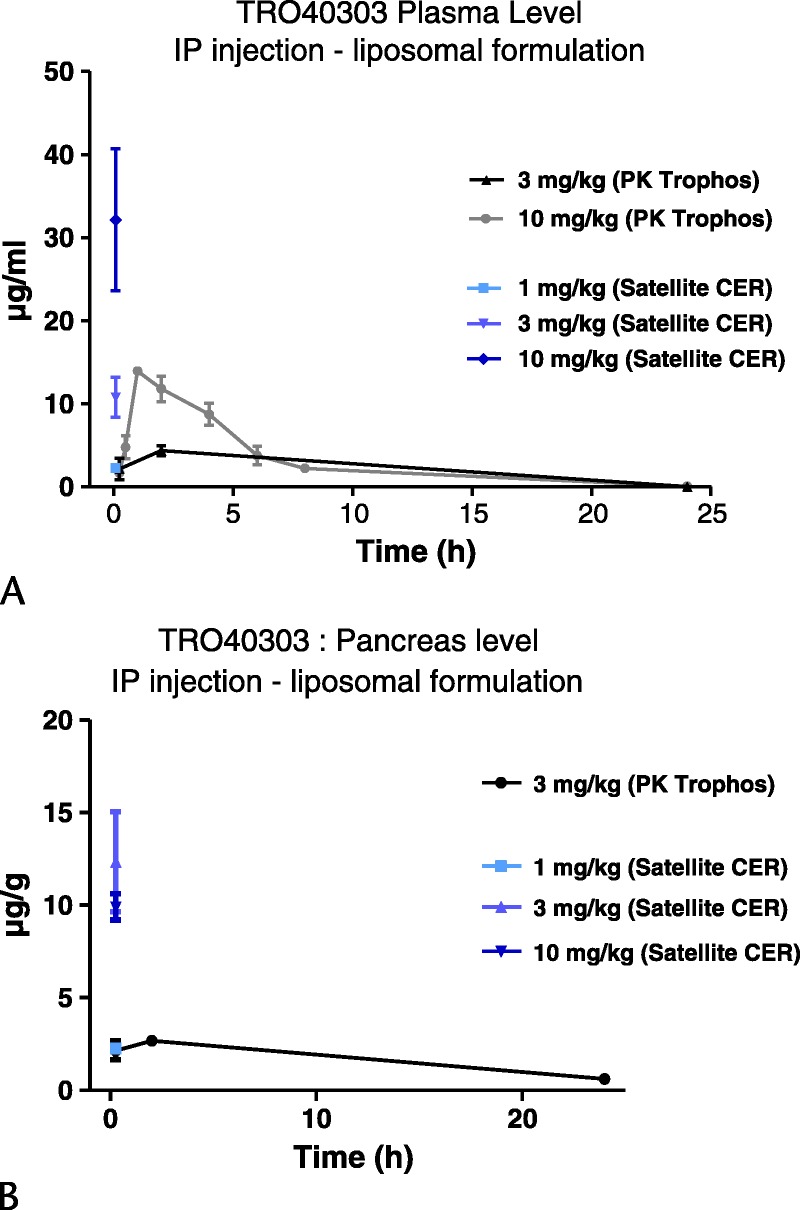
TRO40303 levels in plasma and pancreas following administration of a range of doses (1–10 mg/kg). A, Plasma level of TRO40303 expressed in μg/mL during 24 hours after IP administration of 3 or 10 mg/kg TRO40303 in liposomes contrasted with plasma level of TRO40303 15 minutes after IP administration in CER-AP. B, Pancreatic concentration of TRO40303 expressed in μg/g tissue during 24 hours after IP administration of 3 mg/kg TRO40303 in liposomes contrasted with plasma level of TRO40303 15 minutes after administration in CER-AP.
TRO40303 Reduces Biochemical Responses and Pancreatic Histopathology in FAEE-AP
Fatty acid ethyl ester–induced AP parallels acute alcoholic pancreatitis through in vivo formation of toxic ethanol metabolites that we have previously demonstrated to induce overload of [Ca2+]c and mitochondrial impairment in isolated PACs.12,13 Fatty acid ethyl ester–induced AP was found to cause significant elevations in serum amylase, pancreatic trypsin, and MPO, as well as lung MPO, indicating neutrophil infiltration (Fig. 4). Corresponding significant elevations in pancreatic edema, inflammatory cell infiltration, and parenchymal necrosis were confirmed through assessment of histological sections, reflected in significant elevation of the overall histology score (Fig. 5). As TRO40303 protects mitochondria and isolated PACs from POAEE-induced injury, we assessed the impact of the liposomal preparation of TRO40303 (3 mg/kg) on FAEE-AP. TRO40303 administered as a treatment 1 hour after induction of FAEE-AP significantly reduced FAEE-AP–induced increases in serum amylase, pancreatic trypsin, and MPO, as well as lung MPO (Fig. 4). There were concomitant significant reductions in edema, inflammation, necrosis, and overall histopathology scores as a result of TRO40303 in FAEE-AP (Fig. 5).
FIGURE 4.
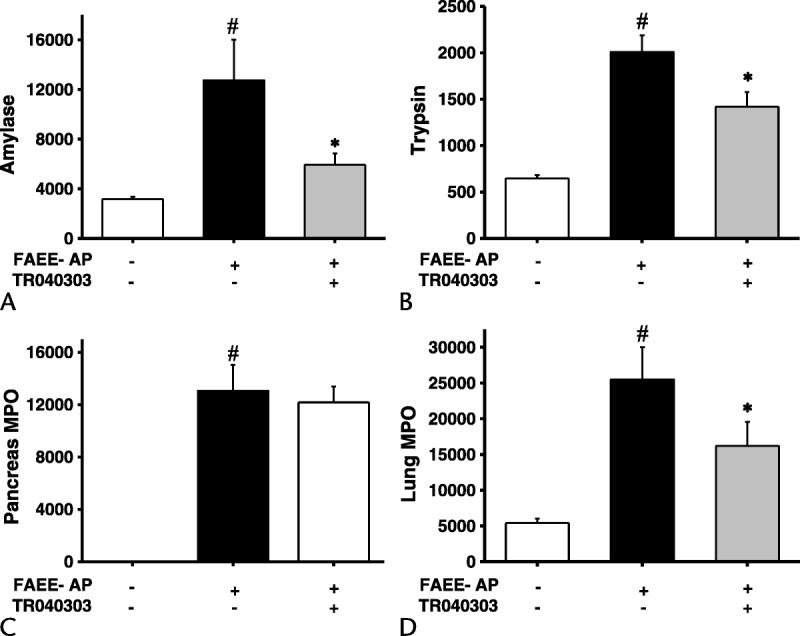
TRO40303 markedly reduced biochemical responses of FAEE-AP. Fatty acid ethyl ester–induced AP caused significant elevations in (A) serum amylase, (B) pancreatic trypsin activity, (C) pancreatic, and (D) lung MPO activity. Administration of the liposomal preparation of TRO40303 (3 mg/kg IP) reduced all biochemical parameters of severity (mean ± SEM >6 animals/group; #P < 0.05, control vs FAEE-AP; *P < 0.05 FAEE-AP vs FAEE-AP plus TRO40303).
FIGURE 5.
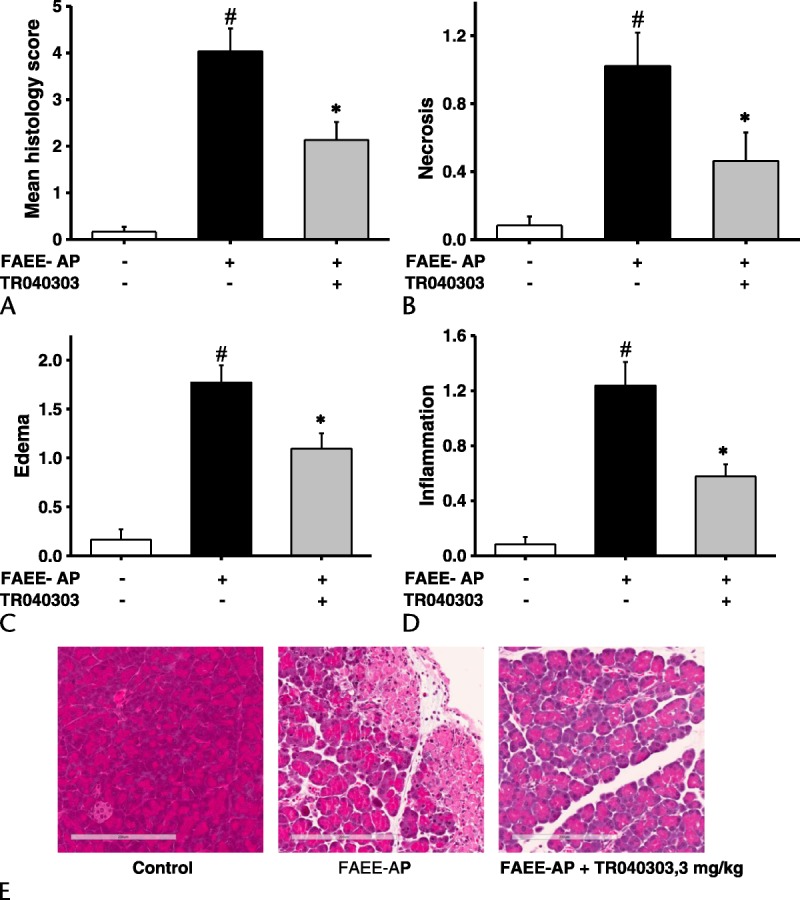
TRO40303 markedly reduced pancreatic histopathology in FAEE-AP. Fatty acid ethyl ester–induced AP led to significant elevations in (A) overall histology score, (B) necrosis, (C) edema, and (D) inflammatory cell infiltrate. Administration of the liposomal preparation of TRO40303 (3 mg/kg IP) markedly reduced all histopathologic markers of severity (mean ± SEM >6 animals/group; #P < 0.05, control vs FAEE-AP; *P < 0.05 FAEE-AP vs FAEE-AP plus TRO40303). E, Representative images showing normal pancreatic histology, typical histopathology from FAEE-AP, and typical histopathology after treatment with TRO40303.
DISCUSSION
This study has demonstrated the protective effects of TRO40303 on PAC injury developing in response to FAEEs in vitro and the efficacy of TRO40303 in a clinically relevant experimental model of alcoholic AP in vivo. The FAEE-AP model is clinically relevant as high levels of FAEEs accumulate in the pancreas following acute alcohol intoxication, in contrast to other organs commonly damaged by alcohol,11 and substantial evidence now implicates FAEEs in PAC,12,13,15 as well as pancreatic ductal cell injury.23,24 Epidemiological studies have also shown a marked increase in the level of alcohol consumption in recent decades, which parallels a sustained rise in the incidence of AP.25 Fatty acid ethyl esters as well as TLCS induce toxic sustained calcium overload in PACs, leading to mitochondrial dysfunction and necrosis,12,13 and the combination of ethanol with palmitoleic acid induces significant pancreatic damage with extensive acinar cell edema, neutrophil infiltration, and necrosis in vivo.15 Although not targeting calcium overload per se, TRO40303 inhibits critical downstream effects on the mitochondria controlling cell fate.16–18 The protective effects of TRO40303 on POAEE-induced PAC mitochondrial dysfunction and necrosis, as well as the attenuation of FAEE-AP severity when given after disease induction, demonstrate the potential applicability of TRO40303 as a treatment for human alcohol-induced AP. This study now adds a third in vivo AP model in which TRO40303 has been shown to be effective when administered after disease induction. The novel human data support translational potential for TRO4303; we have previously shown TRO40303 protects murine PACs from TLCS-induced injury by similar mechanisms, here confirmed in human PACs. The cumulative data on the effects of TRO40303 contrast with previous preclinical trials of drugs for AP, the majority of which have been conducted in a single model only (most often CER-AP)1 with treatment given prophylactically before disease induction. In the majority of patients developing AP, however, prophylactic administration of a drug is not possible.
The mitochondrion is the powerhouse of the cell. There is evidence indicating that sepsis-induced multiple organ failure depends on the development of mitochondrial dysfunction and consequent cellular energetic failure.26 Multiorgan failure is also the leading cause of death in patients with SAP.2 TRO40303 is an MPTP modulator that has been shown to be protective in multiple cellular and animal models characterized by stress-induced mitochondrial permeabilization,16–18,27,28 although whether TSPO binding is the primary mechanism of action remains to be confirmed.29 Pharmacokinetic profiles of TRO40303 have shown accumulation at substantial levels within the lungs, liver, heart, and kidneys,16 all organs contributing to organ failure in AP. Levels were higher in experimental AP than in naive animals, possibly due to reduction in the speed of metabolism and/or excretion of the drug, a consideration in the design of any future human trials. Nevertheless, the accumulation in vital organs suggests that TRO40303 might also usefully prevent or lessen systemic organ dysfunction in AP through direct effects on these organs, suggesting promise in this approach.
This study strengthens an established rationale for targeting the MPTP to treat a range of acute and chronic conditions in which necrotic cell death is a critical and/or prominent feature.9,29 Cyclosporin A is the only licensed drug used experimentally to inhibit formation of the MPTP, the mechanism of action being inhibition of Cyp-D, the most widely recognized regulator of the MPTP. Clinical trials testing the efficacy of cyclosporin A in this regard have been undertaken, with mixed results: a pilot study found reduction in the size and severity of myocardial infarction associated with cyclosporin A treatment prior to percutaneous coronary intervention,30 not replicated in a larger study.31 The immunosuppressive actions of cyclosporin A essential to its primary therapeutic use in controlling autoimmunity and allograft rejection, however, preclude widespread use in conditions where the MPTP is a target. For example, AP predisposes to local and systemic infection, which contributes significantly to mortality1,2; the powerful immunosuppressive actions of cyclosporin A are likely to impact negatively on infective outcomes in AP. There are continuing major efforts to develop new, specific inhibitors of Cyp-D, although none is ready for first-in-man study.32 TRO40303, which does not have the immunosuppressive actions of cyclosporin A but which has been shown experimentally to have significant impact on a number of forms of organ injury, has also been tested in a phase 2 trial of cardiac preservation following acute myocardial infarction, but was not found to have significant protective effects.33 While the mechanism of action of TRO40303 remains to be confirmed,29 our study shows that TRO40303 did not bind to Cyp-D. Nevertheless, because TRO40303 may have both pancreatic as well as systemic protective effects in AP, and the therapeutic window is potentially longer than in that in myocardial infarction, there is perhaps a higher chance of efficacy in AP than in myocardial infarction. While phenotypic screens have identified new MPTP inhibitors, and a number of powerful compounds have been identified, all present challenges: cinnamic anilides were found to be no more effective than cyclosporin A in experimental myocardial infarction, isoxazoles are unstable in vivo, and benzamides impair ATP generation.29 The small number of agents in development for the treatment of AP and the extensive work already undertaken to develop TRO40303 as a drug support the case to test TRO40303 in human AP.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Paula Ghaneh, Chris Halloran, John Neoptolemos, Michael Raraty, and Rajesh Satchidanand, who assisted in the provision of human pancreatic tissue samples.
Footnotes
This work was supported by CORE, United Kingdom (M.A.J.); the Royal College of Surgeons of England (M.A.J.); a Liverpool China Scholarship Council Award (L.W.); the UK National Institute for Health Research Biomedical Research Unit Funding scheme (M.A., D.L., W.H., A.T., D.N.C., R.S.); the UK Medical Research Council (A.T., D.N.C., R.S.); Trophos (S.S., T.B., M.M., and R.P.); and R.S is an NIHR Senior Investigator.
S.S., T.B., M.M., and R.P. contributed to this work as employees of Trophos. R.S. has advised and received funding from Calcimedica, Cypralis, Debiopharm, GlaxoSmithKline, and Novartis. The other authors declare no conflict of interest.
Supplemental digital contents are available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.pancreasjournal.com).
REFERENCES
- 1.Pandol SJ, Saluja AK, Imrie CW, et al. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;132:1127–1151. [DOI] [PubMed] [Google Scholar]
- 2.Petrov MS, Shanbhag S, Chakraborty M, et al. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology. 2010;139:813–820. [DOI] [PubMed] [Google Scholar]
- 3.Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149:1731–1741.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartwig W, Jimenez RE, Werner J, et al. Interstitial trypsinogen release and its relevance to the transformation of mild into necrotizing pancreatitis in rats. Gastroenterology. 1999;117:717–725. [DOI] [PubMed] [Google Scholar]
- 5.Sung KF, Odinokova IV, Mareninova OA, et al. Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp Cell Res. 2009;315:1975–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booth DM, Murphy JA, Mukherjee R, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140:2116–2125. [DOI] [PubMed] [Google Scholar]
- 7.Shalbueva N, Mareninova OA, Gerloff A, et al. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology. 2013;144:437–446.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen L, Mukherjee R, Huang W, et al. Calcium signaling, mitochondria and acute pancreatitis: avenues for therapy. July 30, 2016. Pancreapedia: Exocrine Pancreas Knowledge Base. Available at: https://www.pancreapedia.org/reviews/calcium-signaling-mitochondria-and-acute-pancreatitis-avenues-for-therapy. Accessed February 18, 2017.
- 9.Bernardi P, Rasola A, Forte M, et al. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. 2015;95:1111–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukherjee R, Mareninova OA, Odinokova IV, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. 2016;65:1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laposata EA, Lange LG. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science. 1986;231:497–499. [DOI] [PubMed] [Google Scholar]
- 12.Criddle DN, Raraty MG, Neoptolemos JP, et al. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci U S A. 2004;101:10738–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Criddle DN, Murphy J, Fistetto G, et al. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130:781–793. [DOI] [PubMed] [Google Scholar]
- 14.Gerasimenko JV, Lur G, Sherwood MW, et al. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci U S A. 2009;106:10758–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang W, Booth DM, Cane MC, et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut. 2014;63:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaller S, Paradis S, Ngoh GA, et al. TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J Pharmacol Exp Ther. 2010;333:696–706. [DOI] [PubMed] [Google Scholar]
- 17.Schaller S, Michaud M, Latyszenok V, et al. TRO40303, a mitochondrial-targeted cytoprotective compound, provides protection in hepatitis models. Pharmacol Res Perspect. 2015;3:e00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Lamer S, Paradis S, Rahmouni H, et al. Translation of TRO40303 from myocardial infarction models to demonstration of safety and tolerance in a randomized phase I trial. J Transl Med. 2014;12:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy JA, Criddle DN, Sherwood M, et al. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology. 2008;135:632–641. [DOI] [PubMed] [Google Scholar]
- 20.Guo HX, Wang F, Yu KQ, et al. Novel cyclophilin D inhibitors derived from quinoxaline exhibit highly inhibitory activity against rat mitochondrial swelling and Ca2+ uptake/release. Acta Pharmacol Sin. 2005;26:1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nathan JD, Romac J, Peng RY, et al. Transgenic expression of pancreatic secretory trypsin inhibitor-I ameliorates secretagogue-induced pancreatitis in mice. Gastroenterology. 2005;128:717–727. [DOI] [PubMed] [Google Scholar]
- 22.Dawra R, Ku YS, Sharif R, et al. An improved method for extracting myeloperoxidase and determining its activity in the pancreas and lungs during pancreatitis. Pancreas. 2008;37:62–68. [DOI] [PubMed] [Google Scholar]
- 23.Judák L, Hegyi P, Rakonczay Z, Jr, et al. Ethanol and its non-oxidative metabolites profoundly inhibit CFTR function in pancreatic epithelial cells which is prevented by ATP supplementation. Pflugers Arch. 2014;466:549–562. [DOI] [PubMed] [Google Scholar]
- 24.Maléth J, Balázs A, Pallagi P, et al. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology. 2015;148:427–439.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duran-Bedolla J, Montes de Oca-Sandoval MA, Saldaña-Navor V, et al. Sepsis, mitochondrial failure and multiple organ dysfunction. Clin Invest Med. 2014;37:E58–E69. [DOI] [PubMed] [Google Scholar]
- 27.de Tassigny Ad, Assaly R, Schaller S, et al. Mitochondrial translocator protein (TSPO) ligands prevent doxorubicin-induced mechanical dysfunction and cell death in isolated cardiomyocytes. Mitochondrion. 2013;13:688–697. [DOI] [PubMed] [Google Scholar]
- 28.Richter F, Gao F, Medvedeva V, et al. Chronic administration of cholesterol oximes in mice increases transcription of cytoprotective genes and improves transcriptome alterations induced by alpha-synuclein overexpression in nigrostriatal dopaminergic neurons. Neurobiol Dis. 2014;69:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Šileikytė J, Forte M. Shutting down the pore: the search for small molecule inhibitors of the mitochondrial permeability transition. Biochim Biophys Acta. 2016;1857:1197–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. [DOI] [PubMed] [Google Scholar]
- 31.Cung TT, Morel O, Cayla G, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–1031. [DOI] [PubMed] [Google Scholar]
- 32.Cypralis: novel medicines for degenerative diseases. Available at: http://www.cypralis.com/news/cypralis-has-launched-focus-developing-novel-medicines-degenerative-diseases. Accessed January 17, 2017.
- 33.Atar D, Arheden H, Berdeaux A, et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur Heart J. 2015;36:112–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.