

Abstract
Methods for the detection of RNA modifications are of fundamental importance for advancing epitranscriptomics. N 6‐methyladenosine (m6A) is the most abundant RNA modification in mammalian mRNA and is involved in the regulation of gene expression. Current detection techniques are laborious and rely on antibody‐based enrichment of m6A‐containing RNA prior to sequencing, since m6A modifications are generally “erased” during reverse transcription (RT). To overcome the drawbacks associated with indirect detection, we aimed to generate novel DNA polymerase variants for direct m6A sequencing. Therefore, we developed a screen to evolve an RT‐active KlenTaq DNA polymerase variant that sets a mark for N 6‐methylation. We identified a mutant that exhibits increased misincorporation opposite m6A compared to unmodified A. Application of the generated DNA polymerase in next‐generation sequencing allowed the identification of m6A sites directly from the sequencing data of untreated RNA samples.
Keywords: DNA polymerases, enzyme engineering, epitranscriptomics, N6-methyladenosine, RNA modification
Cellular RNAs are posttranscriptionally modified through the enzymatic introduction of more than 150 modifications.1 The research field of epitranscriptomics aims to investigate the role of these modifications, which possess functional importance but do not alter the RNA sequence itself.2 Therefore, reliable and straightforward methods to detect modifications in a transcriptome‐wide manner are required. However, while nucleic acid analysis in general has profited tremendously from the rise of next‐generation sequencing (NGS) technologies3,the enormous potential of these techniques has so far only rarely been adapted for the direct analysis of modified nucleotides. This is because modifications in an RNA template strand that do not alter the sequence are “erased” during reverse transcription (RT). Modifications located at the Watson–Crick face of the nucleobase constitute an exception to this rule since they affect RT, resulting in the appearance of “RT signatures” at modification sites.4 These signatures arise from increased incorporation of mismatched nucleotides and/or accumulated rates of RT abortion at modification sites. On this basis, direct prediction of N 1‐methyladenosine (m1A) sites from NGS sequencing data has been realized.5 This approach is, however, restricted to modifications that interfere with correct Watson–Crick base pairing. To overcome this limitation, we aimed to evolve a novel RT system that introduces signatures opposite a normally RT‐silent modification.
N 6‐methyladenosine (m6A) was chosen as target modification because it is a reversible6 and highly abundant7 modification in mammalian mRNA. m6A modification of cellular RNA has been demonstrated to affect mRNA splicing,8 nuclear export,6b, 9 translation,10 and degradation.11 Proposed functions include the generation of “translational pulses”,12 the control of the circadian clock,9 the initiation of the DNA damage response,13 and the clearance of maternal RNA14 and pluripotency factors.15 Furthermore, m6A modification can also be found in other cellular RNAs, including rRNA, tRNA, and lncRNA.1a, 2c Current methods to map m6A typically employ immunoprecipitation of m6A‐specific antibodies and covalent crosslinking of the antibody to the RNA molecule prior to analysis by NGS.7a,7b, 16 These methods are complex and laborious and the results may suffer from artifacts deriving from poor antibody specificity and cross‐reactivity.17 Therefore, novel m6A detection systems might benefit from reverse transcriptases (RTases) that sense m6A during cDNA synthesis and create a signal that can be passed on during PCR. The fact that certain RT‐active DNA polymerases are capable of discriminating m6A from unmodified A has been shown by a previous study.18 We were able to advance this feature by engineering an RT‐active DNA polymerase that features significantly increased error rates opposite m6A but not unmodified A. The enzyme was evolved from a thermostable KlenTaq DNA polymerase variant with RT activity (KlenTaq L459M S515R I638F M747K, henceforth referred to as RT‐KTQ).19
As a first step, the incorporation of complementary and non‐complementary nucleotides opposite m6A and unmodified A by RT‐KTQ was investigated. Single‐nucleotide incorporation was performed with each of the four dNTPs, employing a primer hybridized to two different RNA oligonucleotides of the same sequence carrying either A or m6A at the site of first incorporation (Figure S1 in the Supporting Information). As expected, misincorporation of dAMP, dCMP, and dGMP was considerably less efficient than incorporation of the complementary dTMP for both templates. Moreover, dTMP and dAMP incorporation differed only slightly between the A and m6A templates, whereas dCMP and dGMP incorporation was significantly hampered opposite m6A. Inspired by previous studies that utilized capillary electrophoresis (CE) to monitor the activity of DNA polymerases and other enzymes,20 we conceived an assay to screen for DNA polymerase variants with increased misincorporation opposite m6A. The screen involved extension of 5′‐fluorophore‐labeled primer strands through single‐nucleotide incorporation, followed by CE. Multiplexed analysis of several primer extension reactions could be achieved by employing primers of different lengths labeled with different fluorophores (Figure 1 a). More specifically, six primers were designed that possess the same 3′‐terminal 20 nucleotides complementary to the RNA template but differ in their length due to varying 5′‐overhangs (Table S1 in the Supporting Information). Additionally, primers were employed as 5′‐FAM‐ and 5′‐HEX‐labeled variants. The devised assay was applied to screen a library composed of RT‐KTQ single mutants created by site‐directed mutagenesis. Mutation sites were selected based on their vicinity to the nascent base pair in a crystal structure of an RT‐KTQ closed complex19a (Figure 1 b). For each site, all 19 mutants were generated and expressed in E.coli in 96‐well plates. Single‐nucleotide incorporation was then performed with the RT‐KTQ expression lysates after heat‐denaturation of the E.coli host proteins.21
Figure 1.

Screening for DNA polymerase variants with increased misincorporation rates opposite m6A. a) DNA polymerase expression lysates were applied to catalyze the incorporation of dTMP or dAMP opposite A and m6A. Utilization of primers with different length and fluorophores (FAM=6‐carboxyfluorescein; HEX=hexachlorofluorescein) enabled the joint analysis of 12 reaction mixtures in one capillary. b) Amino acids in proximity to the nascent base pair were chosen for saturation mutagenesis. Adapted from PDB ID: 4BWM19a using PyMOL (Schrödinger, LLC; New York, NY). c) Anticipated outcome for promising RT‐KTQ variants: high m6A discrimination for dTMP incorporation and high efficiency for dAMP misincorporation.
It was reported that low‐fidelity DNA polymerases differ from high‐fidelity DNA polymerases mainly in the efficiency of correct nucleotide incorporation, whereas the incorporation of incorrect nucleotides is comparable.22 Thus, we reasoned that enhanced error rates opposite m6A would probably derive from decreased dTMP incorporation rather than from increased misincorporation. For this reason, the developed screening assay was employed to monitor dTMP incorporation opposite m6A and unmodified A. Here, we looked for variants with considerably decreased incorporation of dTMP opposite m6A but not A (Figure S2). Furthermore, to ensure that only incorporation of the correct nucleotide was reduced and not overall activity, dAMP incorporation was monitored in an additional screening (Figure S3). Evaluation of the screening data was performed by qualitative assessment of extension peaks in the electropherogram (Figure 1 c). Significantly increased “dTMP discrimination” was achieved by many RT‐KTQ variants with mutations of G672, G668, Y671, or M673 and by some sporadic variants with mutations at other positions (Figure S2). Many variants with mutations of I614, A661, T664, G668, and Y671 featured comparatively high dAMP misincorporation (Figure S3). Mutations with the most prominent effect on m6A discrimination were combined with mutations exerting the greatest effect on dAMP misincorporation to create a second‐generation library containing all possible double mutants with one “discriminator” mutation (L616T, Y671A, G672H, G672A, G672K, M673T, R746K) and one “misincorporator” mutation (I614A, A661K, T664K, G668Y, Y671T, F749P). This library was screened in the same manner (Figure S4) and the most promising mutants from both libraries were affinity purified followed by evaluation of their error rates at m6A sites.
For this purpose, the selected RT‐KTQ variants were applied for the RT step in a previously published NGS library preparation method that includes RT‐stop products within the PCR amplified library.5 As a template, we employed the m6A‐containing RNA oligonucleotide used in the initial screening. After sequencing and data processing, sequences were mapped to the reference sequence and error‐rate signatures were extracted and visualized by employing CoverageAnalyzer.23 While most of the RT‐KTQ variants exhibited regular error rates at the m6A site, rates were considerably elevated for variants carrying mutations at amino acid Y671 (Table S2, Figure S5). Two single mutants (Y671A and Y671T) and one double mutant (G668Y Y671A) featured particularly prominent signatures. The highest overall error rate of about 15 % was measured for RT‐KTQ G668Y Y671A (Figure 2). Here, 0.1 % G‐reads (due to dCMP incorporation during cDNA synthesis), 10 % T‐reads (dAMP incorporation), and 4.7 % C‐reads (dGMP incorporation) were present at the modification site. Moreover, when looking at the overall sequencing profile for this enzyme, the m6A site was the only site with an error rate of more than 10 %, and error rates did not exceed 5 % for any of the unmodified adenosines in the template (Figure 2). Interestingly, the engineered DNA polymerases tend to stall after the misincorporation of non‐complementary nucleotides opposite m6A, resulting in cDNA termination directly adjacent to the modification site. Thus, the measured elevated error rates will only be observable when RT‐stop products are included within the library.
Figure 2.

RT‐KTQ G668Y Y671A features elevated error rates opposite m6A. a,b) Sequencing profiles of an m6A‐containing RNA oligonucleotide reverse transcribed by unmodified RT‐KTQ (a) and RT‐KTQ G668Y Y671A (b). Sites with error rates of more than 10 % are highlighted with yellow arrows, with colored bars indicating the nature of the reads. Mismatch rates are depicted as black crosses, arrest rates as red lines. The m6A site is indicated with a red underline. Figure created with CoverageAnalyzer.23 C) Mismatch signature of RT‐KTQ G668Y Y671A opposite m6A and all unmodified As present in the RNA oligonucleotide.
RT‐KTQ G668Y Y671A was further employed for the analysis of a known m6A site in E.coli tRNA Val.1a We employed isolated E.coli tRNA extracts as a template for library preparation. Once again, a significantly elevated error rate was observed at the m6A site (14.3 %; Figure 3). The only other sites with error rates of more than 10 % were located opposite another modified nucleotide (5‐methyluridine, T) or at the 5′‐end of the RNA molecule, where rates are inaccurate due to low coverage. The lower coverage derives from reduced activity of the enzyme (Table S3) and synthesis arrest on account of tRNA secondary structure and modifications, and it cannot be resolved decisively by altered reaction conditions.5 Another modification that affected RT by this enzyme was uridine‐5‐oxyacetic acid (V), which triggered high rates of RT arrest. In contrast, almost all unmodified A sites exhibited error rates below 5 %. Only A35 constituted an exception, with an error rate of 8.5 %. We assume that the increased error rate at this position might arise from the fact that this nucleotide is located directly adjacent to the RT‐affecting uridine‐5‐oxyacetic acid. The m6A signature was not observed when unmodified RT‐KTQ was applied (Figure S6). When comparing the two analyzed m6A sites, misincorporation and arrest patterns vary due to different sequence contexts, as has been observed previously for m1A signatures.5
Figure 3.
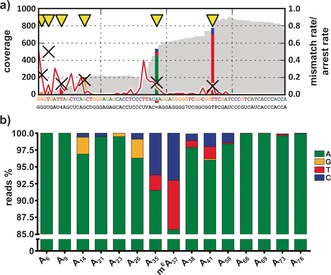
Analysis of a known m6A site in E.coli tRNA Val b applying RT‐KTQ G668Y Y671A. a) Sequencing profile of E.coli tRNA Val reverse transcribed by RT‐KTQ G668Y Y671A. Sites with error rates of more than 10 % are highlighted with yellow arrows, with colored bars indicating the nature of the reads. Mismatch rates are depicted as black crosses, arrest rates as red lines. The colored sequence at the top represents the expected cDNA sequence. The black sequence at the bottom is the actual sequence of tRNA Val containing all its modified nucleotides (′4′=4‐thiouridine; ′D′=dihydrouridine; ′V′=uridine‐5‐oxyacetic acid; ′=′=m6A; ′7′=7‐methylguanosine; ′T′=5‐methyluridine; ′P′=pseudouridine).1a Figure created with CoverageAnalyzer.23 b) Mismatch signature of RT‐KTQ G668Y Y671A opposite m6A and all unmodified As present in E.coli tRNA Val.
We aimed to investigate why the engineered RT‐KTQ G668Y Y671A double mutant exhibited an elevated error rate opposite m6A (and an increased amount of T‐reads in particular), whereas other mutations identified by the initial screening did not show this effect. Therefore, we determined the incorporation rates of dAMP and dTMP opposite A and m6A at a given dNTP concentration of 100 μm (Table S3, Figure S7, S8). For unmodified RT‐KTQ, the ratio of dTMP to dAMP incorporation rate was similar for A and m6A. For RT‐KTQ Y671A, dAMP misincorporation rates were comparable to the unmodified RT‐KTQ. However, dTMP incorporation was significantly reduced opposite m6A, whereas it decreased only slightly opposite A. A similar effect was observed for the G668Y mutation. It was necessary to combine both mutations in an RT‐KTQ double mutant to attain an increased amount of T‐reads at the m6A site. Correspondingly, RT‐KTQ G668Y Y671A featured an even further reduced incorporation rate of dTMP opposite m6A. For this enzyme, dTMP incorporation opposite m6A was only 1.6 times faster than dAMP misincorporation. In contrast, mutation of residues I614 and G672 did not result in elevated error rates opposite m6A. Whereas the G672H mutation delivered the most prominent discrimination of m6A during dTMP incorporation, it also hampered the misincorporation of dAMP tremendously. RT‐KTQ I614A featured significantly increased rates of dAMP misincorporation but lost m6A discrimination.
In this study we provide a novel engineering strategy to create reverse transcriptase variants exhibiting RT signatures as a response to encountering a specific RNA modification. The strategy to evolve an “m6A‐sensing” RT‐active DNA polymerase involved the generation of DNA polymerase libraries in combination with a primer‐extension‐based screening assay. Notably, the assay should also be suitable for other modifications and the throughput of the assay could be increased for future projects by employing more primers of different length, a greater variety of 5′‐fluorophores, and/or several orthogonal primer/template sequences. Qualitative examination of the screening data for variants with increased m6A discrimination during dTMP incorporation but with unaffected dAMP misincorporation delivered promising mutants. Interestingly, the identified Y671 residue is located directly at the C‐terminal end of the O‐helix and is known to undergo substantial conformational changes upon dNTP binding, thereby playing an important role in the selectivity of KlenTaq DNA polymerase and the homologous KF DNA polymerase.24
We have been able to show that the engineered RT‐KTQ G668Y Y671A delivers prominent RT signatures at m6A sites in different sequence contexts, without exerting elevated error rates opposite unmodified nucleotides and the majority of the other modified nucleotides present in the E.coli tRNA Val. Only uridine‐5‐oxyacetic acid and 5‐methyluridine resulted in the emergence of high arrest rates and increased dGMP misincorporation, respectively. However, these RT signatures are highly characteristic, which might enable their distinction from (and detection simultaneously to) m6A. RT‐KTQ G668Y Y671A could contribute to the development of new sequencing approaches to map m6A sites in cellular RNA in the future. Here, a technology that is orthogonal to the present antibody‐enrichment methodologies (MeRIP)7a,7b, 16 is desperately needed to validate candidate sites17 and to simplify detection procedures. The development of such assays necessitates algorithms to identify m6A‐specific RT signatures and to distinguish them from signals deriving from other sources. As already implemented for the detection of m1A, machine‐learning‐based algorithms can be trained to predict modification sites when fed with sufficient data for modification‐specific RT signatures.5 For this purpose, sequencing data from modified RNA oligonucleotides and/or validated m6A sites in rRNA, mRNA, or lncRNA could be utilized to generate training data sets.18, 25
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge support by the Deutsche Forschungsgemeinschaft (SPP 1784), the European Research Council (ERC Advanced Grant 339834), the Carl Zeiss Stiftung (stipend to J.A.), the ANR HTRNAMod (ANR‐13‐ISV8‐0001/HE 3397/8‐1; grant to Y.M.), and the Konstanz Research School Chemical Biology. We further thank the Next‐Generation Sequencing Core Facility, FR3209 BMCT CNRS‐UL, Lorraine University, Vandoeuvre‐les‐Nancy (France) for sequencing services.
J. Aschenbrenner, S. Werner, V. Marchand, M. Adam, Y. Motorin, M. Helm, A. Marx, Angew. Chem. Int. Ed. 2018, 57, 417.
References
- 1.
- 1a. Machnicka M. A., Milanowska K., Oglou O. O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., Helm M., Bujnicki J. M., Grosjean H., Nucleic Acids Res. 2013, 41, D262; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Helm M., Alfonzo J. D., Chem. Biol. 2014, 21, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Saletore Y., Meyer K., Korlach J., Vilfan I. D., Jaffrey S., Mason C. E., Genome Biol. 2012, 13, 175; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Frye M., Jaffrey S. R., Pan T., Rechavi G., Suzuki T., Nat. Rev. Genet. 2016, 17, 365; [DOI] [PubMed] [Google Scholar]
- 2c. Nachtergaele S., He C., RNA Biol. 2017, 14, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Goodwin S., McPherson J. D., McCombie W. R., Nat. Rev. Genet. 2016, 17, 333; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Reuter J. A., Spacek D. V., Snyder M. P., Mol. Cell 2015, 58, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Findeiss S., Langenberger D., Stadler P. F., Hoffmann S., Biol. Chem. 2011, 392, 305; [DOI] [PubMed] [Google Scholar]
- 4b. Ryvkin P., Leung Y. Y., Silverman I. M., Childress M., Valladares O., Dragomir I., Gregory B. D., Wang L. S., RNA 2013, 19, 1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Hauenschild R., Tserovski L., Schmid K., Thüring K., Winz M. L., Sharma S., Entian K. D., Wacheul L., Lafontaine D. L., Anderson J., Alfonzo J., Hildebrandt A., Jäschke A., Motorin Y., Helm M., Nucleic Acids Res. 2015, 43, 9950; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Tserovski L., Marchand V., Hauenschild R., Blanloeil-Oillo F., Helm M., Motorin Y., Methods 2016, 107, 110. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y. G., He C., Nat. Chem. Biol. 2011, 7, 885; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Zheng G., Dahl J. A., Niu Y., Fedorcsak P., Huang C. M., Li C. J., Vågbø C. B., Shi Y., Wang W. L., Song S. H., Lu Z., Bosmans R. P. G., Dai Q., Hao Y. J., Yang X., Zhao W. M., Tong W. M., Wang X. J., Bogdan F., Furu K., Fu Y., Jia G., Zhao X., Liu J., Krokan H. E., Klungland A., Yang Y. G., He C., Mol. Cell 2013, 49, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., Rechavi G., Nature 2012, 485, 201; [DOI] [PubMed] [Google Scholar]
- 7b. Meyer K., Saletore Y., Zumbo P., Elemento O., Mason C., Jaffrey S., Cell 2012, 149, 1635; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Desrosiers R., Friderici K., Rottman F., Proc. Natl. Acad. Sci. USA 1974, 71, 3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiao W., Adhikari S., Dahal U., Chen Y. S., Hao Y. J., Sun B. F., Sun H. Y., Li A., Ping X. L., Lai W. Y., Wang X., Ma H. L., Huang C. M., Yang Y., Huang N., Jiang G. B., Wang H. L., Zhou Q., Wang X. J., Zhao Y. L., Yang Y. G., Mol. Cell 2016, 61, 507. [DOI] [PubMed] [Google Scholar]
- 9. Fustin J. M., Doi M., Yamaguchi Y., Hida H., Nishimura S., Yoshida M., Isagawa T., Morioka M. S., Kakeya H., Manabe I., Okamura H., Cell 2013, 155, 793. [DOI] [PubMed] [Google Scholar]
- 10. Wang X., Zhao B. S., Roundtree I. A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C., Cell 2015, 161, 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X., Lu Z., Gomez A., Hon G. C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., He C., Nature 2014, 505, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao B. S., Roundtree I. A., He C., Nat. Rev. Mol. Cell. Biol. 2017, 18, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiang Y., Laurent B., Hsu C. H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S., Ling D., Hsu P. H., Zou L., Jambhekar A., He C., Shi Y., Nature 2017, 543, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao B. S., Wang X., Beadell A. V., Lu Z., Shi H., Kuuspalu A., Ho R. K., He C., Nature 2017, 542, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A. A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y. S., Ben-Haim M. S., Eyal E., Yunger S., Pinto Y., Jaitin D. A., Viukov S., Rais Y., Krupalnik V., Chomsky E., Zerbib M., Maza I., Rechavi Y., Massarwa R., Hanna S., Amit I., Levanon E. Y., Amariglio N., Stern-Ginossar N., Novershtern N., Rechavi G., Hanna J. H., Science 2015, 347, 1002. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Linder B., Grozhik A. V., Olarerin-George A. O., Meydan C., Mason C. E., Jaffrey S. R., Nat. Methods 2015, 12, 767; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Chen K., Lu Z., Wang X., Fu Y., Luo G. Z., Liu N., Han D., Dominissini D., Dai Q., Pan T., He C., Angew. Chem. Int. Ed. 2015, 54, 1587; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1607. [Google Scholar]
- 17. Helm M., Motorin Y., Nat. Rev. Genet. 2017, 18, 275. [DOI] [PubMed] [Google Scholar]
- 18. Harcourt E. M., Ehrenschwender T., Batista P. J., Chang H. Y., Kool E. T., J. Am. Chem. Soc. 2013, 135, 19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Blatter N., Bergen K., Nolte O., Welte W., Diederichs K., Mayer J., Wieland M., Marx A., Angew. Chem. Int. Ed. 2013, 52, 11935; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12154; [Google Scholar]
- 19b. Aschenbrenner J., Marx A., Nucleic Acids Res. 2016, 44, 3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Greenough L., Schermerhorn K. M., Mazzola L., Bybee J., Rivizzigno D., Cantin E., Slatko B. E., Gardner A. F., Nucleic Acids Res. 2016, 44, e15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Lohman G. J., Bauer R. J., Nichols N. M., Mazzola L., Bybee J., Rivizzigno D., Cantin E., T. C. Evans, Jr. , Nucleic Acids Res. 2016, 44, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Gloeckner C., Kranaster R., Marx A., Curr. Protoc. Chem. Biol. 2010, 2, 89; [DOI] [PubMed] [Google Scholar]
- 21b. Huber C., von Watzdorf J., Marx A., Nucleic Acids Res. 2016, 44, 9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beard W. A., Shock D. D., Vande Berg B. J., Wilson S. H., J. Biol. Chem. 2002, 277, 47393. [DOI] [PubMed] [Google Scholar]
- 23. Hauenschild R., Werner S., Tserovski L., Hildebrandt A., Motorin Y., Helm M., Biomolecules 2016, 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Li Y., Korolev S., Waksman G., EMBO J. 1998, 17, 7514; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Obeid S., Blatter N., Kranaster R., Schnur A., Diederichs K., Welte W., Marx A., EMBO J. 2010, 29, 1738; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Carroll S. S., Cowart M., Benkovic S. J., Biochemistry 1991, 30, 804; [DOI] [PubMed] [Google Scholar]
- 24d. Astatke M., Ng K., Grindley N. D. F., Joyce C. M., Proc. Natl. Acad. Sci. USA 1998, 95, 3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Liu N., Parisien M., Dai Q., Zheng G., He C., Pan T., RNA 2013, 19, 1848; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Narayan P., Rottman F. M., Science 1988, 242, 1159; [DOI] [PubMed] [Google Scholar]
- 25c. Piekna-Przybylska D., Decatur W. A., Fournier M. J., Nucleic Acids Res. 2008, 36, D178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary