

Abstract
Parkinson's disease (PD) is associated with the formation of α-synuclein amyloid fibrils. Elucidating the role of these β-sheet-rich fibrils in disease progression is crucial; however, collecting detailed structural information on amyloids is inherently difficult because of their insoluble, non-crystalline, and polymorphic nature. Here, we show that Raman spectroscopy is a facile technique for characterizing structural features of α-synuclein fibrils. Combining Raman spectroscopy with aggregation kinetics and transmission electron microscopy, we examined the effects of pH and ionic strength as well as four PD–related mutations (A30P, E46K, G51D, and A53T) on α-synuclein fibrils. Raman spectral differences were observed in the amide-I, amide-III, and fingerprint regions, indicating that secondary structure and tertiary contacts are influenced by pH and to a lesser extent by NaCl. Faster aggregation times appear to facilitate unique fibril structure as determined by the highly reproducible amide-I band widths, linking aggregation propensity and fibril polymorphism. Importantly, Raman spectroscopy revealed molecular-level perturbations of fibril conformation by the PD–related mutations that are not apparent through transmission electron microscopy or limited proteolysis. The amide-III band was found to be particularly sensitive, with G51D exhibiting the most distinctive features, followed by A53T and E46K. Relating to a cellular environment, our data would suggest that fibril polymorphs can be formed in different cellular compartments and potentially result in distinct phenotypes. Our work sets a foundation toward future cellular Raman studies of amyloids.
Keywords: amyloid, electron microscopy (EM), fibril, mutant, Parkinson's disease
Introduction
Parkinson's disease (PD)2 is a chronic and progressive movement disorder, with symptoms of resting tremor, slowness of movement, rigidity of limbs, and impaired balance that continue to develop and worsen over time. These debilitating symptoms are caused by the death of dopamine-producing cells in the substantia nigra region of the brain (1, 2), but the mechanism responsible for cell death is not known. A diagnostic hallmark of PD in patient brain tissue is the presence of Lewy bodies, cellular inclusions composed of α-synuclein (α-syn) amyloid fibrils (3).
Although most instances of PD are sporadic, genetic factors are known to play important roles in PD etiology, and the cytosolic accumulation of insoluble α-syn fibrils is a common feature for both sporadic and familial PD (4). Currently, six missense mutations (A30P, E46K, H50Q, G51D, A53T, and A53E) in the SNCA gene that encodes α-syn, as well as gene duplication and triplication, are known to cause early-onset PD (5, 6). Individuals with any one of these point mutations generally experience rapid disease progression (5), and in vitro studies have shown missense mutations of α-syn affect the rate of α-syn aggregation (7–14), suggesting that amyloid formation kinetics play an important role in disease development.
Experimental observations suggest that conformational changes in α-syn can dictate pathology and are important in disease pathways (15–17). Studies using in vitro and in vivo models show that aggregation and accumulation of α-syn can have harmful consequences for many cellular processes (6, 18). Moreover, treatments with α-syn fibrils have also been shown to be toxic to cells (19). For these reasons, there is great interest in structurally characterizing amyloid fibrils and understanding the molecular mechanisms that lead to cell death and disease (20).
In solution, α-syn is intrinsically disordered (21) but spontaneously aggregates to form fibrils in vitro that resemble fibrils isolated from patients (22). In this fibrillar form, α-syn is assembled into well-ordered β-sheets, with β-strands running perpendicular to the fibril axis and hydrogen bonds between β-strands running parallel to the fibril axis (Fig. 1A) (3, 7, 23). This distinctive pattern is referred to as “cross-β” and has been observed in X-ray diffraction patterns of full-length α-syn fibrils (24). Site-directed spin labeling and electron paramagnetic resonance spectroscopy have shown that α-syn β-strands are parallel in-register, with side chains of the amino acids aligning on top of equivalent sites in neighboring α-syn monomers (25).
Figure 1.

α-Synuclein amyloid formation probed by Raman spectroscopy. A, representative schematic of Raman excitation of parallel in-register amyloid structure adopted by α-syn. Hydrogen bonds are indicated by red dashed lines. Scale bar, 100 nm. B, comparison of Raman spectra of WT-α-syn (500 μm) before (black) and after (cyan) 4 h incubation at 37 °C in pH 5 buffer (10 mm NaOAc, 100 mm NaCl). The spectra were collected at room temperature and normalized to Phe band at 1003 cm−1.
The insoluble, non-crystalline nature of α-syn fibrils excludes the use of many traditional methods for structural characterization. The best structural models currently available have utilized electron diffraction, solid-state NMR, and cryo-EM techniques (23, 26, 27), but because of the polymorphic nature of α-syn fibrils, other structures are possible and remain to be determined (27–29). Recently, polymorph-dependent synucleinopathies and toxicity have been observed in animal models (30), highlighting the role fibril polymorphism may play in PD development and clinically observed phenotypes.
For basic identification of amyloid, there are several characterization techniques used, such as transmission electron and atomic force microscopy, to evaluate macroscopic feature of non-branching fibrillar structures, positive staining with amyloid-specific small molecules like Congo Red and thioflavin T (ThT), CD or infrared spectroscopy to quantify β-sheet signatures, and limited proteolysis to identify the amyloid core region (31–35). Although broadly used, these techniques lack sensitivity to detect structural differences in fibril polymorphs. Thus far, only more sophisticated and time-intensive approaches have been successful in revealing molecularly distinct polymorphs such as solid-state NMR and cryo-EM (27, 29). Unfortunately, the preparation of fibril material for these experiments can be costly, and the necessary instruments are not widely accessible (23, 36).
In this work, we have employed Raman spectroscopy as a robust technique to obtain structural insights into α-syn amyloid fibrils. The flexibility of Raman instrumentation allows us to easily collect spectroscopic signals for both soluble α-syn monomers and insoluble aggregated amyloid fibrils by using the amide-I band and the full fingerprint region of the spectrum. Because we are measuring intrinsic molecular vibrations, this method directly reports on amyloid structure, without need of additional probes or pretreatment of the sample. The technique is fast, nondestructive, requires minimal sample amounts (<10 μl of ∼15 μm protein fibrils in solution), and can be done in water, an important solvent for modeling physiological conditions (37, 38).
Despite these advantages, Raman spectroscopy has been relatively under-utilized compared with other techniques for characterizing amyloids (39, 40). Prior Raman studies on α-syn have been particularly limited. Two investigations have studied soluble α-syn in organic solvents (41, 42) and a third reported the Raman spectrum of WT α-syn fibrils, with analysis of the amide-I band (43). Here, we use Raman spectroscopy to probe α-syn fibril polymorphism and investigate its relationship to aggregation propensity, an issue not yet addressed by Raman spectroscopy. Specifically, we report on structural details of WT α-syn amyloid fibrils prepared under several solution conditions: pH 5 versus 7.4 to compare characteristic pH of the acidic lysosome to the neutral cytosol and low versus high ionic strengths, which is known to cause α-syn fibril polymorphism. Importantly, we carried out a detailed comparison of four PD–related missense mutations (A30P, E46K, G51D, and A53T) by combining Raman spectroscopy and aggregation kinetics, along with complementary transmission electron microscopy (TEM) and limited proteolysis. We carefully consider the amide-I, amide-II, and amide-III regions to reveal secondary structure differences, as well as the CH deformation stretching region of the spectrum to gain insight into fibril structure.
Results
Observing amyloid formation by Raman spectroscopy
Upon aggregation of WT-α-syn, we observe a clear narrowing of the amide-I band, which arises from the C=O stretch of the peptide backbone and has an intensity maximum at 1667 cm−1 (Fig. 1B, cyan trace). The width and position of this band indicate β-sheet secondary structure consistent with the formation of amyloid as previously observed (37). The amide-III peak maximum, which is the result of a coupled C–N stretch and N–H bend, shifts from 1245 to 1228 cm−1, also corroborating the formation of β-sheet structure. Bands arising from side-chain vibrations (e.g. CO2− symmetric stretch near 1401 cm−1 and CH2CH3 deformations near 1450 cm−1) are very similar in both the soluble and aggregated forms. A new band for aggregated α-syn appears with a maximum at 1556 cm−1, whereas the band is absent for soluble α-syn (expanded view shown in Fig. S1). This band falls in the expected region for amide-II frequencies. The amide-II band arises from an out-of-phase combination of C–N stretching and N–H bending along the peptide backbone but is generally considered a weak vibration only observable under resonant excitation (39).
α-Synuclein amyloid formation kinetics are modulated by pH and ionic strength
ThT is a small molecule dye that becomes highly emissive upon binding to amyloid fibrils and is widely used to monitor amyloid formation (32, 44, 45). Typical in vitro aggregation kinetics exhibit a characteristic sigmoidal curve with an initial lag phase where little amyloid is formed, followed by a rapid growth phase during which many fibrils are formed, and ending with a saturation phase where fibril growth is at equilibrium. Monitoring the aggregation kinetics of α-syn by ThT emission shows that amyloid formation is dramatically different under neutral and acidic solution conditions (Fig. 2). Under acidic conditions, the large net negative charge of α-syn is neutralized (46), making it much more prone to aggregation (Fig. 2, red traces). From Table 1, α-syn rapidly forms amyloid in the absence of NaCl within our first measurement time point of 0.5 h and a t½ (the time required to reach half the maximum emission intensity) of 2.1 ± 0.6 h (n = 5). In the presence of 100 mm NaCl, α-syn aggregation is delayed slightly with a lag time (tlag) of 1.4 ± 0.2 h with a comparable growth phase and a t½ of 2.7 ± 0.4 h (n = 5). These differences in lag times can be discerned in Fig. 2B. Contrastingly, the kinetics at pH 7.4 are markedly slower. In the absence of NaCl, α-syn exhibits a longer and more variable tlag of 36 ± 13 h with a protracted t½ to 66 ± 8 h (n = 5). With the addition of 100 mm NaCl, α-syn aggregation is hastened (tlag of 21.0 ± 4.7 h and t½ of 47.0 ± 3.6 h, n = 5).
Figure 2.
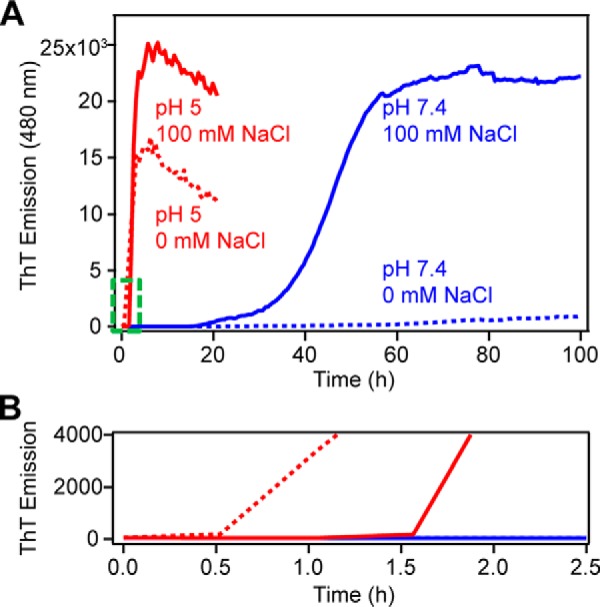
Aggregation kinetics of WT-α-syn monitored by ThT fluorescence. A, effect of pH (10 mm NaOAc, pH 5 (red) and 10 mm NaPi, pH 7.4 (blue)) and ionic strength (in the presence (solid line) and absence (dotted line) of 100 mm NaCl) on WT-α-syn (100 μm, 37 °C, average of n = 5). B, enlarged view of the area outlined in green in A.
Table 1.
Effect of pH and ionic strength on aggregation kinetics of WT-α-Syn measured by ThT fluorescence
[WT] = 100 μm and [ThT] = 10 μm at 37 °C.
| pH 5a |
pH 7.4b |
|||
|---|---|---|---|---|
| 0 mm NaCl | 100 mm NaCl | 0 mm NaCl | 100 mm NaCl | |
| tlag (h)c | ≤ 0.5 | 1.4 ± 0.2 | 36 ± 13 | 21 ± 5 |
| t½ (h)d | 2.1 ± 0.6 | 2.7 ± 0.4 | 66 ± 8 | 47 ± 4 |
a 10 mm NaOAc.
b 10 mm NaPi.
c Time before observing any ThT intensity increase.
d Time required to reach half of the maximum ThT intensity.
Effect of pH on α-synuclein fibril structure
To investigate whether there are structural differences that correlate with the observed kinetic differences, we utilized Raman spectroscopy to gain a molecular “fingerprint” of each fibril sample. Fig. 3A compares the Raman spectra of fibrils formed at pH 5 (red) and pH 7.4 (blue) under high salt concentrations (100 mm NaCl). Intensity differences can be observed for the amide-I (1667 cm−1), amide-III (1229 and 1275 cm−1), CH deformation stretches (1316 and 1342 cm−1), CO2− symmetric stretch (1402 cm−1), and CH2CH3 deformation stretches (1448 cm−1), whereas the amide-II band at 1556 cm−1 is very similar for both fibrils (band assignments from Refs. 41, 47, and 48)).
Figure 3.
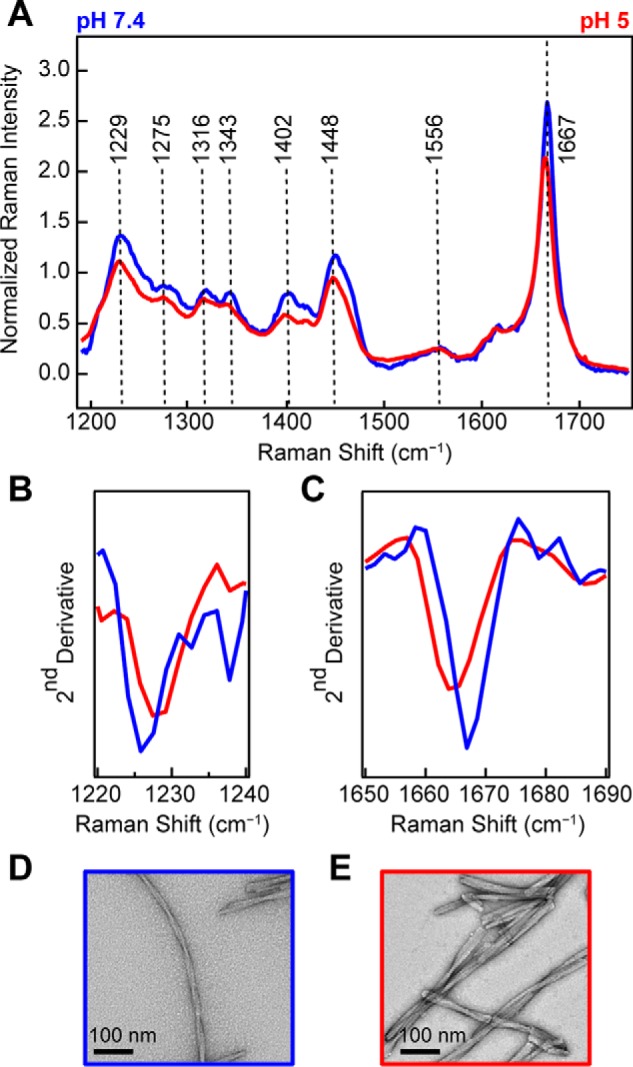
Comparison of WT-α-syn fibrils made at neutral versus acidic pH. A, Raman spectra of WT-α-syn fibrils (100 μm) formed at pH 7.4 (blue) and pH 5 (red) in the presence of 100 mm NaCl. The spectra were collected at room temperature and normalized to Phe band at 1003 cm−1. B, second derivative analysis of amide-III region. C, second derivative analysis of amide-I region. D and E, representative negatively stained TEM images of WT-α-syn fibrils formed at pH 7.4 (D) and pH 5 (E).
We chose second derivative analysis to objectively identify changes from positive to negative slope, enabling characteristic spectral component differences in the vibrational bands to be precisely located (Fig. 3, B and C). From this analysis, the negative maximum of the second derivative identifies the maximum intensity frequency for a spectral region. Here, we closely consider the amide-III (1220–1240 cm−1; Fig. 3B) and amide-I (1650–1690 cm−1; Fig. 3C) regions. We observe that for pH 5 fibrils the amide-III band is shifted to higher energies, whereas the amide-I is shifted to lower energies. Both pH 5 fibril peaks are well-resolved from the peak of fibrils formed at pH 7.4. We can also see that fibrils formed at pH 7.4 have a slightly higher intensity in the amide-I band (Fig. 3A). Collectively, these differences indicate distinct secondary structure (amide bands) and tertiary contacts by aliphatic side chains (carboxylate and deformation stretches) for the two fibril types. TEM images show that the fibrils are also morphologically distinct (Fig. 3, D and E). At neutral pH (Fig. 3D), fibrils appear to be formed by a pair of twisted protofibrils (protofibril is defined here as the narrowest observed fibrillar structure by TEM). At acidic pH (Fig. 3E), fibrils formed are shorter and have many lateral associations, which appear to be from bundling of several protofibrils.
Effect of solution ionic strength on α-synuclein fibril structure
When WT-α-syn is aggregated at pH 7.4 (10 mm NaPi) under conditions of high (100 mm NaCl) or low (0 mm NaCl) ionic strength, we again observe a narrow amide-I peak at 1667 cm−1 (Fig. 4A). Here, more subtle differences are observed in the Raman spectra. The second derivatives of amide-III region indicate some secondary structure differences (Fig. 4B), whereas the amide-I region shows little change (Fig. 4C). TEM images show at high ionic strength (100 mm NaCl), and the fibrils formed appear as a pair of twisted protofibrils (Fig. 4D), whereas at low ionic strength (0 mm NaCl), the fibrils appear as much shorter and laterally associated fragments (Fig. 4E), much like those fibrils formed at pH 5.
Figure 4.
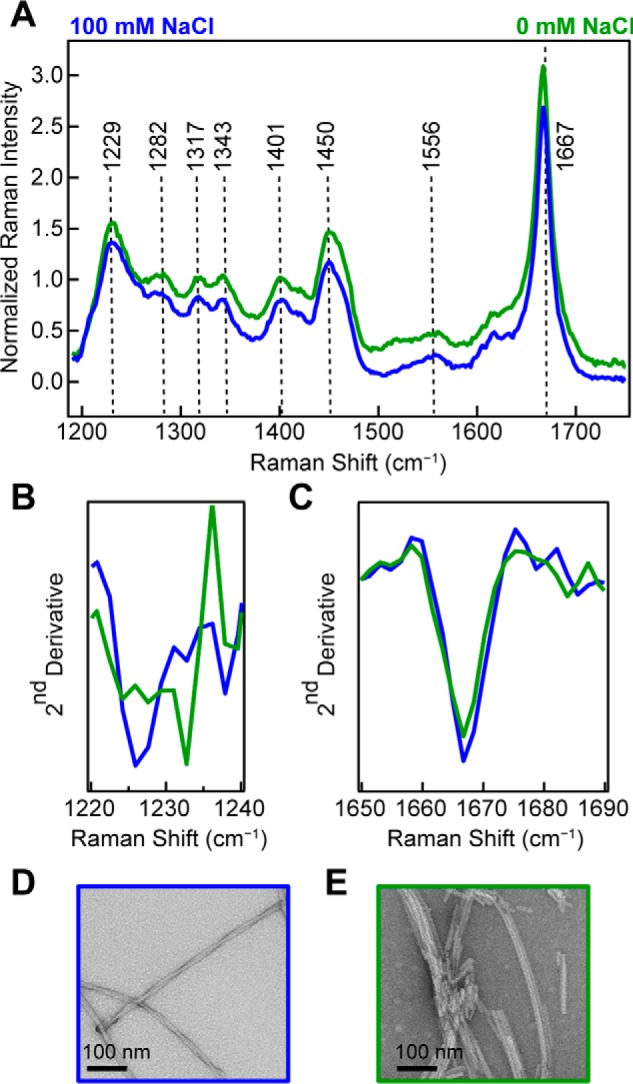
Effect of ionic strength on WT-α-syn fibrils made at neutral pH. A, Raman spectra of WT-α-syn fibrils (100 μm) formed at pH 7.4 in the presence (blue) and absence (green) of 100 mm NaCl. The spectra were collected at room temperature and normalized to Phe band at 1003 cm−1. B, second derivative analysis of amide-III region. C, second derivative analysis of amide-I region. D and E, representative negatively stained TEM images of WT-α-syn fibrils formed at 100 mm NaCl (D) and 0 mm NaCl (E).
Correlating amide-I band to aggregation propensity
Our Raman data show that the amide-I band of α-syn is sensitive to underlying molecular differences in the amyloid fibrils formed under different solution conditions, which were corroborated by TEM. Thus, we postulated that the reproducibility of the amide-I band could be indicative of unique molecular structures. Fig. 5 shows individual data sets for independently prepared fibril samples aggregated at pH 5 in the absence (n = 10) and presence (n = 11) of 100 mm NaCl and at pH 7.4 in the absence (n = 12) and presence (n = 13) of 100 mm NaCl. The mean spectrum of each data set is also shown (black curve). Fibrils were formed at a variety of protein concentrations (15–990 μm) and in several different vessels (Eppendorf tubes and 96-well microplates, with or without glass beads) to test the reproducibility of the amide-I band widths and the dependence on aggregation conditions. In this comparison, data have been normalized to the amide-I band to discern reproducibility of the band shape, because this should inform on the distribution of molecular conformations adopted by the protein.
Figure 5.
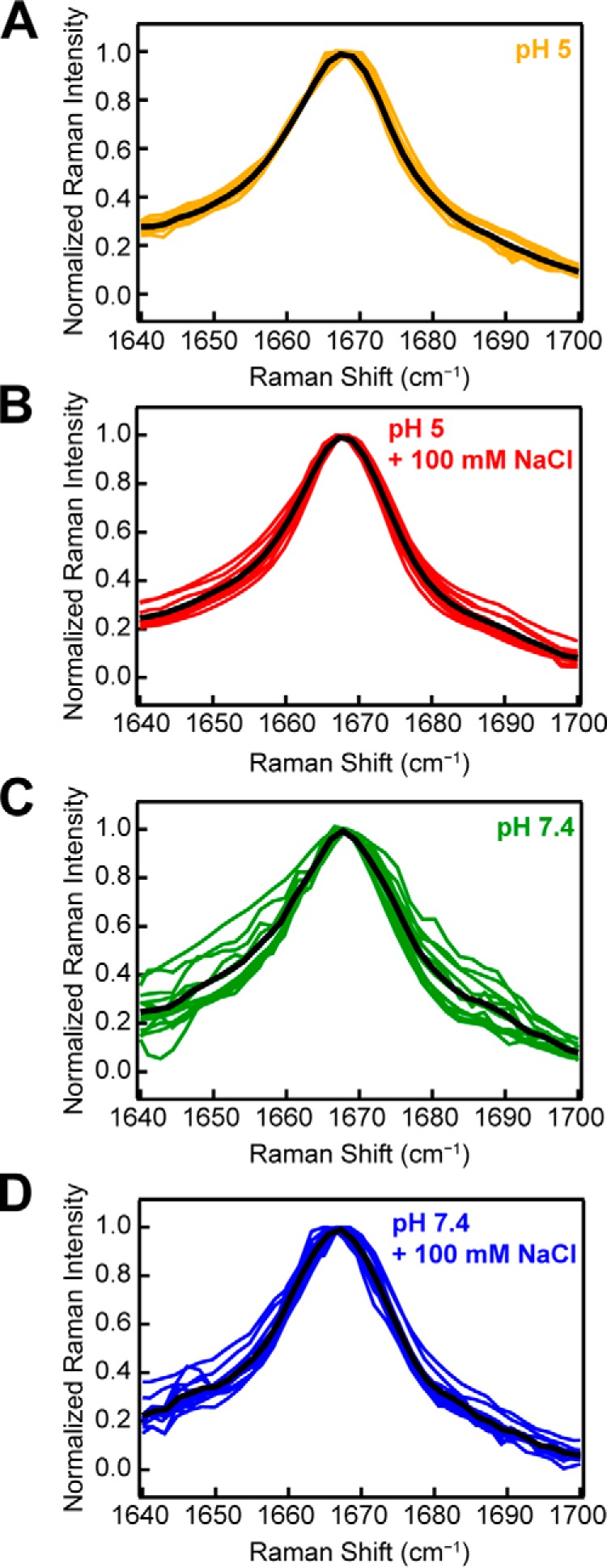
Consistency and reproducibility of amide-I peak for WT-α-syn. Samples were aggregated at several protein concentrations in a variety of aggregation vessels at pH 5 in the absence (A, n = 10) and presence (B, n = 11) of 100 mm NaCl and at pH 7.4 in the absence (C, n = 12) and presence (D, n = 13) of 100 mm NaCl. Individual spectra are plotted as colored curves, with mean curve of the distribution plotted in black. The data are normalized to the amide-I peak at 1667 cm−1.
Interestingly, it appears that under solution conditions that promote faster aggregation kinetics (Fig. 2A), the amide-I band is highly reproducible, regardless of the protein concentration or aggregation vessel. Fibrils formed at pH 5 and low ionic strength (0 mm NaCl) consistently aggregate the fastest (Fig. 2B) and yield an amide-I band centered at 1667 cm−1 with an average FWHM of 20 cm−1 (Fig. 5A). Fibrils formed at pH 7.4 and low ionic strength, however, aggregate the slowest and yield a broader amide-I band (Fig. 5C). The variability of the amide-I band may suggest that under slow aggregation conditions (pH 7.4, low ionic strength), α-syn can sample more conformations, leading to a distribution of polymorphs. Under conditions that encourage fast aggregation (pH 5, low ionic strength), however, the protein forms strong interactions that cannot be overcome and therefore adopts a more uniform secondary structure.
Revealing fibril conformational differences in PD–related α-syn mutants
Several missense mutations of α-syn that are linked to familial early-onset PD are known to affect the rate of α-syn aggregation and fibril formation in vitro (7–9, 49). Individuals with specific mutations experience unique symptoms and disease onset at different ages (4), suggesting a link between aggregation kinetics and disease etiology. A clear understanding of the relationship between mutant fibril structures and disease progression, however, has not yet been achieved. Therefore, we sought to characterize fibrils formed by four disease-related α-syn mutants (A30P, E46K, G51D, and A53T) by Raman spectroscopy to reveal mutant-dependent structural features.
Aggregation kinetics experiments (Fig. 6B) were performed, and mutations modulated aggregation times to varying degrees (9, 11–14). At pH 5, A53T, E46K, and G51D are significantly faster than WT-α-syn, whereas amyloid formation is delayed for A30P (Table 2). TEM images of WT and disease-related mutant α-syn fibrils were collected and revealed no major differences in fibril morphologies (Fig. S2). Further, cathepsin L (CtsL) limited-proteolysis experiments (34) revealed a common protease-resistant core, composed of residues 28–114 for all mutants and WT α-syn (Fig. 6A and Table S1). Raman spectra were then collected and revealed distinct structural features for each α-syn mutant, which were not apparent through TEM nor limited proteolysis.
Figure 6.
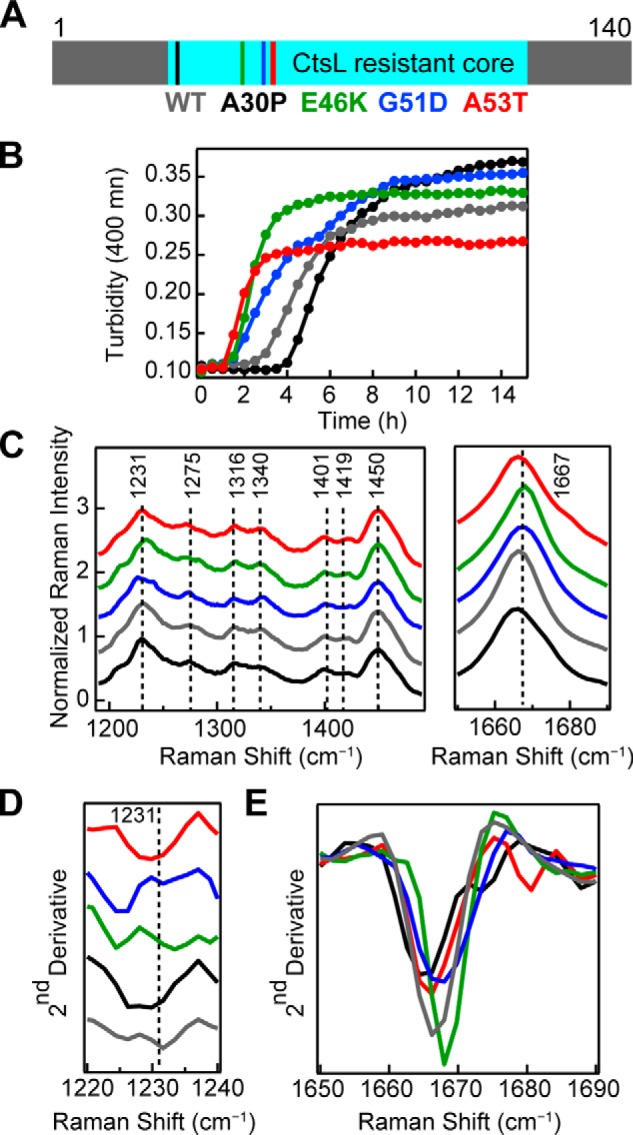
Aggregation kinetics and Raman spectra of PD–related mutants. A, primary sequence of WT-α-syn indicating the locations of PD–related mutations (A30P, E46K, G51D, and A53T) studied here and the common CtsL-resistant amyloid core (residues 28–114). B, comparison of aggregation kinetics (average of n = 10) of PD–related mutants to WT-α-syn (40 μm, 50 mm NaOAc, 20 mm NaCl, pH 5, 37 °C). Mutants are color-coded as in A. C, Raman spectra of fibrils formed from WT and disease-related mutants of α-syn (40 μm, 10 mm NaOAc, 100 mm NaCl, pH 5, 37 °C). The spectra were collected at room temperature and normalized to Phe band at 1003 cm−1. Mutants are color-coded as in A. Dashed lines are drawn as reference guides. The region between 1490 cm−1 and 1650 cm−1 is not shown, and the spectra are stacked for clarity. D, second derivative analysis of amide-III region. E, second derivative analysis of amide-I region. The spectra are overlaid for easy comparison of peak shifts.
Table 2.
Effect of PD mutations on aggregation kinetics measured by turbidity
[protein] = 40 μm in 50 mm NaOAc, 20 mm NaCl, pH 5, at 37 °C.
| WT | A30P | E46K | G51D | A53T | |
|---|---|---|---|---|---|
| tlag (h)a | 2.6 ± 0.5 | 3.6 ± 0.2 | 1.4 ± 0.2 | 1.3 ± 0.3 | 1.1 ± 0.2 |
| t½ (h)b | 3.7 ± 0.7 | 5.2 ± 0.6 | 1.9 ± 0.4 | 2.5 ± 0.5 | 1.5 ± 0.3 |
a Time before observing any turbidity increase.
b Time required to reach half of the maximum turbidity signal.
Several criteria were considered for distinguishing differences in WT and mutant Raman spectra. First, observable peak maximum and spectral width (Fig. 6C). Next, second derivative analysis was done to determine spectral components and peak shifts (Fig. 6, D and E). Finally, we examined relative peak intensity ratios to assess relative vibrational strengths (Table S2). From these comparisons, we observe several differences in the fingerprint region of the spectra (Fig. 6C and Fig. S3).
From second derivative analysis, it is clear the position and peak width of the amide-III peak (maximum at 1231 cm−1) are significantly different across the mutants (Fig. 6D). Most noticeably, G51D and E46K both have significantly shifted peaks to lower energies. Second derivative analysis also shows that mutant amide-III peaks near 1275 cm−1 have unique profiles when compared with WT-α-syn (Fig. S3). The peak near 1275 cm−1 is more prominent for A30P, G51D, and WT and is shifted to lower energies for A53T and E46K. The small shoulder near 1284 cm−1 is more defined for A30P, E46K and WT and broadened for A53T and G51D. For E46K, the ratio of these peaks is close to 1, splitting the band into two nearly equal intensity peaks. G51D has the strongest relative intensity at 1275 cm−1 in the series (Table S2). Differences in peak ratios for the CH deformations (1316 and 1340 cm−1) are also observed, with G51D having the weakest relative intensity at 1316 cm−1 (Table S2). Second derivative analysis of the amide-I band (Fig. 6E) reveals slight differences in peak maxima and widths across the mutants, as well as in the higher energy feature (∼1675 cm−1) for A30P and A53T. Taken together, these spectroscopic features indicate molecular differences in fibril structure of the individual mutants both in the overall β-sheet content of the fibrils and in the side chain packing within the fibril structure.
Discussion
In our work, we use spontaneous Raman spectroscopy, a relatively simple experiment with low excitation powers, to gain insight into structural features of α-syn fibrils. In the soluble form, we observe a broad amide-I band for α-syn, indicating an initial disordered conformation. Upon aggregation, the amide-I band dramatically narrows, indicating β-sheet structure and amyloid formation. Unique amide-I band widths and differences in the fingerprint region were observed for α-syn fibrils formed under different solution conditions, which we attribute to fibril polymorphism. One cause of fibril polymorphism is thought to result from different numbers of associated protofibrils (27), which involves side-chain packing, influencing side-chain environments, and could result in distinct molecular interactions. This is supported by the macroscopic differences in the fibrils visualized by TEM.
Unexpectedly, we also observe the amide-II band for α-syn amyloid (Fig. S1), which is typically only seen when the excitation light is in resonance with the amide bond. We hypothesize that the distinct alignment of parallel, in-register β-sheets in amyloid fibrils strengthens this frequency through vibrational coupling (orienting the C–N peptide bonds along the fibril growth axis; Fig. 1A) and allows its observation outside resonance excitation. Similar amide-II bands have been assigned for Raman optical activity spectra of prion proteins for unusually flat β-sheets (50).
Additionally, we found that as the propensity for aggregation increases (enhanced fibril formation kinetics observed by ThT either by lowering the pH or increasing ionic strength), the amide-I band of the Raman spectra is more reproducible, suggesting consistent formation of the same fibril structure and offering a new understanding of α-syn aggregation kinetics. Specifically, faster aggregation times appear to facilitate the formation of unique fibril structure, whereas the longer it takes for α-syn to aggregate, more polymorphs are populated, as indicated by broader amide-I bands.
Finally, differences observed in the Raman spectra of WT-α-syn and the four disease-related mutants further support the utility of spontaneous Raman spectroscopy as a powerful method for monitoring unique α-syn structural features. Although limited-proteolysis experiments showed indistinguishable amyloid cores across the four mutants and WT-α-syn, our Raman data revealed mutant-dependent spectra that indicate molecular differences in fibril conformations. Previous work has also suggested that mutations in α-syn can perturb fibril structure, although these studies were performed at neutral pH. Rienstra and co-workers (51) have shown that E46K and A53T mutations have measurable structural perturbations, whereas A30P does not, because of their respective positions in β-strands and in a loop region. Further, based on their recent NMR structural model of WT α-syn fibrils (23), the disruption of a salt bridge between Glu-46 and Lys-80 would likely change the E46K mutant fibril structure compared with the WT protein. Additionally, the atomic structure of an 11-residue segment of α-syn by Eisenberg and co-workers (26) suggests that the A53T mutation would influence side-chain packing and lead to a more stable β-sheet interface.
To our knowledge, our Raman study is the first to be reported for PD–related α-syn mutant fibrils. Further, unlike most Raman studies that focus on the amide-I band as a secondary structure reporter, we find that the amide-III region is particularly informative and sensitive to features of all four PD–related mutants. Interestingly, our data suggest that A30P, like the E46K and A53T fibrils, are altered from the WT fibrils. However, conformational differences for A30P were only seen in the amide-III band (Fig. 6C) and not in the CH deformation bands. This is in contrast for E46K, G51D, and A53T mutants, where differences were seen in both regions. In addition to alterations to secondary structural content, we interpret our data to indicate that mutations are causing side chain distortions and influencing how the β-sheets pack together. The effects seen here with A30P may be specific to the fibril polymorph formed at pH 5, which may not be observable under the different conditions previously studied. Of the four mutants studied, the vibrational spectrum of G51D is most distinct. Collectively, the spectral differences suggest a unique conformation unlike the other fibrils. Based on our proteolysis study on WT fibril degradation (35), Gly-51 is likely involved in a β-turn, and thus, a dramatic mutation to aspartic acid would have a strong impact on the β-sheet organization and fibril stability.
To conclude, our work demonstrates that Raman spectroscopy is sensitive to subtle structural differences in α-syn fibrils that are important not only for aggregation and amyloid formation but also for disease-related mutations. Within a cellular environment, α-syn aggregation could be initiated in different compartments that would be modulated by their characteristic pH such as the neutral cytosol or the acidic lysosome. Our results suggest that fibrils formed in different cellular compartments could indeed be structurally unique because of a pH effect and potentially result in distinct PD phenotypes. α-Syn fibril polymorphs have been shown to have significant physiological implications (52), and therefore the ability to see structural differences through noninvasive, cellular-compatible measurements, like Raman spectroscopy, is valuable. Because amyloid fibrils provide a direct structural link of α-syn to PD, this work provides an important step in establishing Raman spectroscopy as useful for detecting fibril structural features. Ultimately, understanding the relationship between amyloid structure and cell health would offer valuable insight for designing therapeutics that could treat disease or avoid its onset.
Experimental procedures
Chemicals
Sodium chloride (≥99.5%), sodium acetate trihydrate (≥99.5%), sodium phosphate monobasic (≥99.0%), sodium phosphate dibasic (≥99.0%), EDTA (≥99%), hydrochloric acid (36.5–38%), cyclohexane (anhydrous, 99.8%), PMSF (≥98.5%), and ThT were purchased from Sigma–Aldrich and used as received. Tris and guanidine hydrochloride were purchased from MP Biomedicals.
Recombinant protein expression, purification, and preparation
Recombinant human WT and mutant (A30P, E46K, G51D, and A53T) α-syn were expressed in BL21(DE3) pLysS cells (Invitrogen) and purified as previously described (53) with slight modifications. Cell pellets from a 3-liter culture were first homogenized in 60 ml of pH 8 buffer (100 mm Tris, 300 mm NaCl, 1 mm EDTA, and 100 μm PMSF), stirred under N2 atmosphere for 10 min, and followed by heat treatment in 100 °C water bath for 15 min. The soluble fraction was then separated from cell debris by centrifugation (30 min, Sorvall, SS-34 rotor, 18,000 rpm, 4 °C), titrated to pH 3.5 using 1 m HCl, and stirred at 4 °C for 10 min before a final centrifugation step (30 min, Beckman Coulter, 45Ti rotor, 30,000 rpm, 4 °C). The resulting supernatant was dialyzed overnight into low-salt buffer (20 mm Tris, 0.5 mm EDTA, 0.5 mm PMSF, pH 8) and then applied onto a HiPrep 16/10 DEAE FF anionic exchange column (GE Healthcare) and eluted with a linear gradient from 100–300 mm NaCl in 20 mm Tris buffer (pH 8). All dialysis and chromatography steps were performed at 4 °C. Fractions containing protein were determined using UV-visible spectroscopy (ϵ280 nm = 5120 m−1 cm−1) and SDS-PAGE on a Pharmacia Phastsystem (Amersham Biosciences) visualized by silver-staining methods. Protein fractions were again dialyzed overnight into low-salt buffer (20 mm Tris, 0.5 mm EDTA, 0.5 mm PMSF, pH 8) and then applied onto a Mono-Q HR 16/10 column (Amersham Biosciences). Protein-containing fractions were determined by UV-visible spectroscopy, and protein purity was confirmed using SDS-PAGE/silver-staining methods and mass spectrometry (NHLBI Biochemistry Core Facility). All purified proteins were concentrated to ∼200 μm using Amicon stirred ultrafiltration cells (molecular mass cutoff, 3 kDa; Millipore), flash frozen in liquid N2, and stored at −80 °C until use.
To form fibrils, protein solutions were exchanged into either pH 5 buffer (10 mm NaOAc, 0 mm NaCl, or 100 mm NaCl) or pH 7.4 buffer (10 mm NaPi, 0 mm NaCl, or 100 mm NaCl) using a PD-10 column (GE Healthcare) and filtered (YM100 membranes; Millipore) immediately prior to aggregation to remove any preformed aggregates. α-Syn was incubated ([α-syn] = 70–100 μm) in 1.5-ml Eppendorf tubes at 37 °C and shaken at 600 rpm (Mini-Micro 980140 shaker; VWR) for 3–10 days.
For aggregation kinetics, α-syn was prepared as described above and incubated in sealed, polypropylene 96-well microplates (655261, Greiner Bio-One, [α-syn] = 100 μm, [ThT] = 10 μm) or polystyrene 384-well microplates (781185, Greiner Bio-One, [α-syn] = 40 μm), with continuous orbital shaking (1.0 mm) at 37 °C using a microplate reader (Tecan Infinite M200 Pro). For ThT fluorescence, the excitation and emission wavelengths were 410 and 480 nm, respectively. For turbidity, the measurement wavelength was 400 nm. All buffers were filtered (0.22 μm; Millipore) prior to use.
Raman spectroscopy
Aggregated α-syn fibrils were deposited on to the surface of a well in an 8-well #1 coverglass LabTek chamber in a 10–20-μl droplet. All data were collected from fibrils in buffer solution using a home-built Raman microscope at room temperature. Briefly, the 514-nm line of an argon-ion laser (CVI Melles Griot, 35-MAP-431-200) was passed through a clean-up filter (Semrock, LL01-514-25) and then directed into a modified inverted microscope (Olympus IX71). Excitation light (∼65 milliwatt at the sample) was directed to the sample using a dichroic mirror (Semrock, LPD01-514RU-25 × 36–1.1) and a 60× water-immersion objective (Olympus, UPLSAPO60XW). Spontaneous Raman Stokes scattering was collected through the same objective, filtered (Semrock, LP02-514RE-25) to remove any residual excitation light or Rayleigh scattering and then directed into a 320-mm focal length (f/4.1 aperture) imaging spectrometer (Horiba Scientific, iHR 320) through a 50-μm slit, dispersed using a 1200 g/mm grating, and collected for 3–5-s acquisitions (×25) with high gain enabled on a liquid-nitrogen–cooled, back-illuminated, deep-depletion CCD array (Horiba Scientific, Symphony II, 1024 × 256 px, 26.6 mm × 6.6 mm, 1 MHz repetition rate). Daily calibration of imaging spectrometer was done using neat cyclohexane (20 μl in a sealed capillary tube). Bandpass and accuracy were found to be <12 cm−1 and ±1 cm−1, respectively. All Raman spectra were corrected by subtracting the buffer background collected under identical conditions, followed by applying a baseline polynomial fit (Lab Spec 5 software). The presented data have not been smoothed or averaged and have been normalized to the Phe band at 1003 cm−1. This band represents the breathing mode of the aromatic ring and is not sensitive to conformational changes. Second derivative analysis was performed using IGOR Pro 7 (Wavemetrics).
TEM
Aggregated α-syn samples (10 μl) were incubated on TEM grids (400-mesh Formvar carbon-coated copper; Electron Microscopy Sciences) for 1–2 min. Sample solution was wicked with filter paper, and the grid was quickly washed with water (10 μl) to remove any excess material and improve background contrast. The grids were then incubated with 1% (w/v) aqueous uranyl acetate solution (10 μl) for 30–60 s. Excess uranyl acetate was wicked away with filter paper, and the grids were air-dried. TEM images were collected using a JOEL JEM 1200EX TEM (accelerating voltage, 80 keV) equipped with an AMT XR-60 digital camera (NHLBI EM Core Facility).
Determination of protease-resistant amyloid core
In glass vials, mutant (A30P, E46K, G51D, and A53T) and WT α-syn (500 μl, 30 μm) in pH 5 buffer were incubated at 37 °C and agitated at 600 rpm for 3 days (Mini-Micro 980140 shaker). Resulting fibrils were treated with either 150, 300, 450, or 600 nm CtsL (Sigma–Aldrich, C6854-25UG) at 37 °C. Reactions (50 μl) were taken at 24 h and terminated with 2 m guanidine hydrochloride (final concentration). Samples (20 μl) were separated using a ZORBAX 300SB-C18 reverse-phase column (2.1 × 50 mm, 3.5 μm) on an Agilent 1100 series HPLC (NHLBI Biochemistry Core Facility) coupled to an Agilent G1946D mass selective detector equipped with an electrospray ionization interface (Agilent Technologies). Mass spectra were obtained using positive ion mode. Data were analyzed using LC/mass selective detector ChemStation software (Rev. A.10.02, Agilent Technologies).
Author contributions
J. D. F. built the Raman microscope, expressed/purified proteins, conducted Raman experiments, analyzed the results, collected some of the aggregation and TEM data, and wrote the paper. R. P. M. conducted limited proteolysis, LC-MS analysis, and some of the aggregation and TEM experiments. R. L. W. expressed and purified PD–related mutants. J. C. L. conceived the idea for the project and wrote the paper with J. D. F. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
Parts of this research was performed on instruments maintained by the NHLBI Electron Microscopy (TEM) and Biochemistry (LC-MS) Core Facilities.
This work was supported by the Intramural Research Program at the National Institutes of Health, NHLBI. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S3.
- PD
- Parkinson's disease
- α-syn
- α-synuclein
- ThT
- thioflavin T
- TEM
- transmission electron microscopy
- CtsL
- cathepsin L.
References
- 1. Lees A. J., Hardy J., and Revesz T. (2009) Parkinson's disease. Lancet 373, 2055–2066 10.1016/S0140-6736(09)60492-X [DOI] [PubMed] [Google Scholar]
- 2. Lotharius J., and Brundin P. (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 3, 932–942 10.1038/nrn983 [DOI] [PubMed] [Google Scholar]
- 3. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 4. Petrucci S., Ginevrino M., and Valente E. M. (2016) Phenotypic spectrum of α-synuclein mutations: new insights from patients and cellular models. Parkinsonism Relat. Disord. 22, S16–S20 10.1016/j.parkreldis.2015.08.015 [DOI] [PubMed] [Google Scholar]
- 5. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson, et al. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- 6. Deng H., and Yuan L. (2014) Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res. Rev. 15, 161–176 10.1016/j.arr.2014.04.002 [DOI] [PubMed] [Google Scholar]
- 7. Conway K. A., Harper J. D., and Lansbury P. T. (2000) Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry 39, 2552–2563 10.1021/bi991447r [DOI] [PubMed] [Google Scholar]
- 8. Narhi L., Wood S. J., Steavenson S., Jiang Y., Wu G. M., Anafi D., Kaufman S. A., Martin F., Sitney K., Denis P., Louis J. C., Wypych J., Biere A. L., and Citron M. (1999) Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 274, 9843–9846 10.1074/jbc.274.14.9843 [DOI] [PubMed] [Google Scholar]
- 9. Conway K. A., Harper J. D., and Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 10.1038/3311 [DOI] [PubMed] [Google Scholar]
- 10. Ghosh D., Mondal M., Mohite G. M., Singh P. K., Ranjan P., Anoop A., Ghosh S., Jha N. N., Kumar A., and Maji S. K. (2013) The Parkinson's disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 52, 6925–6927 10.1021/bi400999d [DOI] [PubMed] [Google Scholar]
- 11. Li J., Uversky V. N., and Fink A. L. (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 40, 11604–11613 10.1021/bi010616g [DOI] [PubMed] [Google Scholar]
- 12. Greenbaum E. A., Graves C. L., Mishizen-Eberz A. J., Lupoli M. A., Lynch D. R., Englander S. W., Axelsen P. H., and Giasson B. I. (2005) The E46K mutation in α-synuclein increases amyloid fibril formation. J. Biol. Chem. 280, 7800–7807 10.1074/jbc.M411638200 [DOI] [PubMed] [Google Scholar]
- 13. Lesage S., Anheim M., Letournel F., Bousset L., Honoré A., Rozas N., Pieri L., Madiona K., Dürr A., Melki R., Verny C., Brice A., and French Parkinson's Disease Genetics Study Group (2013) G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471 10.1002/ana.23894 [DOI] [PubMed] [Google Scholar]
- 14. Ranjan P., and Kumar A. (2017) Perturbation in long-range contacts modulates kinetics of amyloid formation in α-synuclein familial mutants. ACS Chem. Neurosci. 8, 2235–2246 10.1021/acschemneuro.7b00149 [DOI] [PubMed] [Google Scholar]
- 15. Burré J., Vivona S., Diao J., Sharma M., Brunger A. T., and Südhof T. C. (2013) Properties of native brain α-synuclein. Nature 498, E4–E7 10.1038/nature12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burré J., Sharma M., and Südhof T. C. (2014) α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283 10.1073/pnas.1416598111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dettmer U., Newman A. J., Luth E. S., Bartels T., and Selkoe D. (2013) In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 288, 6371–6385 10.1074/jbc.M112.403311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stefanis L. (2014) α-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 4, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pieri L., Madiona K., and Melki R. (2016) Structural and functional properties of prefibrillar α-synuclein oligomers. Sci. Rep. 6, 24526 10.1038/srep24526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lashuel H. A., Overk C. R., Oueslati A., and Masliah E. (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fauvet B., Mbefo M. K., Fares M.-B., Desobry C., Michael S., Ardah M. T., Tsika E., Coune P., Prudent M., Lion N., Eliezer D., Moore D. J., Schneider B., Aebischer P., El-Agnaf O. M., et al. (2012) α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364 10.1074/jbc.M111.318949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crowther R. A., Daniel S. E., and Goedert M. (2000) Characterisation of isolated α-synuclein filaments from substantia nigra of Parkinson's disease brain. Neurosci. Lett. 292, 128–130 10.1016/S0304-3940(00)01440-3 [DOI] [PubMed] [Google Scholar]
- 23. Tuttle M. D., Comellas G., Nieuwkoop A. J., Covell D. J., Berthold D. A., Kloepper K. D., Courtney J. M., Kim J. K., Barclay A. M., Kendall A., Wan W., Stubbs G., Schwieters C. D., Lee V. M., George J. M., et al. (2016) Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415 10.1038/nsmb.3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Serpell L. C., Berriman J., Jakes R., Goedert M., and Crowther R. A. (2000) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. U.S.A. 97, 4897–4902 10.1073/pnas.97.9.4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen M., Margittai M., Chen J., and Langen R. (2007) Investigation of α-synuclein fibril structure by site-directed spin labeling. J. Biol. Chem. 282, 24970–24979 10.1074/jbc.M700368200 [DOI] [PubMed] [Google Scholar]
- 26. Rodriguez J. A., Ivanova M. I., Sawaya M. R., Cascio D., Reyes F. E., Shi D., Sangwan S., Guenther E. L., Johnson L. M., Zhang M., Jiang L., Arbing M. A., Nannenga B. L., Hattne J., Whitelegge J., et al. (2015) Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490 10.1038/nature15368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dearborn A. D., Wall J. S., Cheng N., Heymann J. B., Kajava A. V., Varkey J., Langen R., and Steven A. C. (2016) α-Synuclein amyloid fibrils with two entwined, asymmetrically associated protofibrils. J. Biol. Chem. 291, 2310–2318 10.1074/jbc.M115.698787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vilar M., Chou H.-T., Lührs T., Maji S. K., Riek-Loher D., Verel R., Manning G., Stahlberg H., and Riek R. (2008) The fold of α-synuclein fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 8637–8642 10.1073/pnas.0712179105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heise H., Hoyer W., Becker S., Andronesi O. C., Riedel D., and Baldus M. (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 102, 15871–15876 10.1073/pnas.0506109102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peelaerts W., Bousset L., Van der Perren A., Moskalyuk A., Pulizzi R., Giugliano M., Van den Haute C., Melki R., and Baekelandt V. (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- 31. Khurana R., Uversky V. N., Nielsen L., and Fink A. L. (2001) Is Congo Red an amyloid-specific dye? J. Biol. Chem. 276, 22715–22721 10.1074/jbc.M011499200 [DOI] [PubMed] [Google Scholar]
- 32. Levine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfefferkorn C. M., Jiang Z., and Lee J. C. (2012) Biophysics of α-synuclein membrane interactions. Biochim. Biophys. Acta 1818, 162–171 10.1016/j.bbamem.2011.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McGlinchey R. P., and Lee J. C. (2015) Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 112, 9322–9327 10.1073/pnas.1500937112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McGlinchey R. P., Dominah G. A., and Lee J. C. (2017) Taking a bite out of amyloid: Mechanistic insights into α-synuclein degradation by cathepsin L. Biochemistry 56, 3881–3884 10.1021/acs.biochem.7b00360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kloepper K. D., Woods W. S., Winter K. A., George J. M., and Rienstra C. M. (2006) Preparation of α-synuclein fibrils for solid-state NMR: expression, purification, and incubation of wild-type and mutant forms. Protein Expr. Purif. 48, 112–117 10.1016/j.pep.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 37. Carey P. R. (2006) Raman crystallography and other biochemical applications of Raman microscopy. Annu. Rev. Phys. Chem. 57, 527–554 10.1146/annurev.physchem.57.032905.104521 [DOI] [PubMed] [Google Scholar]
- 38. Rygula A., Majzner K., Marzec K. M., Kaczor A., Pilarczyk M., and Baranska M. (2013) Raman spectroscopy of proteins: a review. J. Raman Spectrosc. 44, 1061–1076 10.1002/jrs.4335 [DOI] [Google Scholar]
- 39. Kurouski D., Van Duyne R. P., and Lednev I. (2015) Exploring structure and formation mechanism of amyloid fibrils by Raman spectroscopy. Analyst 140, 4967–4980 10.1039/C5AN00342C [DOI] [PubMed] [Google Scholar]
- 40. Louros N. N., Tsiolaki P. L., Baltoumas F. A., Chryssikos G. D., Gionis V., Hamodrakas S. J., and Iconomidou V. A. (2017) Tracking the amyloidogenic core of IAPP amyloid fibrils: insights from micro-Raman spectroscopy. J. Struct. Biol. 199, 140–152 10.1016/j.jsb.2017.06.002 [DOI] [PubMed] [Google Scholar]
- 41. Maiti N. C., Apetri M. M., Zagorski M. G., Carey P. R., and Anderson V. E. (2004) Raman spectroscopic characterization of secondary structure in natively unfolded proteins: α-synuclein. J. Am. Chem. Soc. 126, 2399–2408 10.1021/ja0356176 [DOI] [PubMed] [Google Scholar]
- 42. Mensch C., Konijnenberg A., Van Elzen R., Lambeir A.-M., Sobott F., and Johannessen C. (2017) Raman optical activity of human α-synuclein in intrinsically disordered, micelle-bound α-helical, molten globule and oligomeric β-sheet state. J. Raman Spectrosc. 48, 910–918 10.1002/jrs.5149 [DOI] [Google Scholar]
- 43. Apetri M. M., Maiti N. C., Zagorski M. G., Carey P. R., and Anderson V. E. (2006) Secondary structure of α-synuclein oligomers: characterization by Raman and atomic force microscopy. J. Mol. Biol. 355, 63–71 10.1016/j.jmb.2005.10.071 [DOI] [PubMed] [Google Scholar]
- 44. Naiki H., Higuchi K., Hosokawa M., and Takeda T. (1989) Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T. Anal. Biochem. 177, 244–249 10.1016/0003-2697(89)90046-8 [DOI] [PubMed] [Google Scholar]
- 45. LeVine H., 3rd (1999) Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284 10.1016/S0076-6879(99)09020-5 [DOI] [PubMed] [Google Scholar]
- 46. Uversky V. N., Li J., and Fink A. L. (2001) Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 276, 10737–10744 10.1074/jbc.M010907200 [DOI] [PubMed] [Google Scholar]
- 47. Chan J. W., Taylor D. S., Zwerdling T., Lane S. M., Ihara K., and Huser T. (2006) micro-Raman spectroscopy detects individual neoplastic and normal hematopoietic cells. Biophys. J. 90, 648–656 10.1529/biophysj.105.066761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Movasaghi Z., Rehman S., and Rehman I. U. (2007) Raman spectroscopy of biological tissues. Appl. Spectrosc. Rev. 42, 493–541 10.1080/05704920701551530 [DOI] [Google Scholar]
- 49. Ghosh D., Singh P. K., Sahay S., Jha N. N., Jacob R. S., Sen S., Kumar A., Riek R., and Maji S. K. (2015) Structure based aggregation studies reveal the presence of helix-rich intermediate during α-synuclein aggregation. Sci. Rep. 5, 9228 10.1038/srep09228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McColl I. H., Blanch E. W., Gill A. C., Rhie A. G., Ritchie M. A., Hecht L., Nielsen K., and Barron L. D. (2003) A new perspective on β-sheet structures using vibrational Raman optical activity: from poly(l-lysine) to the prion protein. J. Am. Chem. Soc. 125, 10019–10026 10.1021/ja021464v [DOI] [PubMed] [Google Scholar]
- 51. Comellas G., Lemkau L. R., Nieuwkoop A. J., Kloepper K. D., Ladror D. T., Ebisu R., Woods W. S., Lipton A. S., George J. M., and Rienstra C. M. (2011) Structured regions of α-synuclein fibrils include the early-onset Parkinson's disease mutation sites. J. Mol. Biol. 411, 881–895 10.1016/j.jmb.2011.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pfefferkorn C. M., and Lee J. C. (2010) Tryptophan probes at the α-synuclein and membrane interface. J. Phys. Chem. B. 114, 4615–4622 10.1021/jp908092e [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.