

SUMMARY
Toxoplasma gondii is an obligate intracellular apicomplexan parasite with high seroprevalence in humans. Repeated lytic cycles of invasion, replication and egress drive both the propagation and the virulence of this parasite. Key steps in this cycle, including invasion and egress, depend on tightly regulated calcium fluxes and, while many of the calcium-dependent effectors have been identified, the factors that detect and regulate the calcium fluxes are mostly unknown. To address this knowledge gap, we used a forward genetic approach to isolate mutants resistant to extracellular exposure to the calcium ionophore A23187. Through whole genome sequencing and complementation we have determined that a nonsense mutation in a previously uncharacterized protein is responsible for the ionophore resistance of one of the mutants. The complete loss of this protein recapitulates the resistance phenotype and importantly shows defects in calcium regulation and in the timing of egress. The affected protein, GRA41, localizes to the dense granules and is secreted into the parasitophorous vacuole where it associates with the tubulovesicular network (TVN). Our findings support a connection between the TVN and ion homeostasis within the parasite, and thus a novel role for the vacuole of this important pathogen.
Keywords: Toxoplasma gondii, calcium, dense granule, tubulovesicular network, egress, GRA41
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite of the apicomplexan phylum that causes widespread infection among many vertebrates, including humans (San Miguel et al., 2016). It is estimated that approximately a third of the world’s human population is infected with this opportunistic pathogen (Pappas et al., 2009). Humans become infected congenitally or by ingestion of either environmental oocysts, which are shed in the feces of cats, or tissue cysts, found in the undercooked meat of infected animals. Though immunocompetent individuals will not generally experience symptoms during infection, Toxoplasma can be particularly devastating in immunocompromised individuals and those infected congenitally (Mazzillo et al., 2013, Oray et al., 2015). During the acute stage of the infection, Toxoplasma propagates through repeated lytic cycles of host cell invasion, growth and egress as a rapidly dividing form known as the tachyzoite (Black et al., 2000b). The tissue damage elicited by parasite propagation is normally limited by the immune response, which induces conversion to the cyst-forming bradyzoite form and the establishment of a chronic infection. In immunocompromised individuals and lymphoma patients, new infections or rupture of pre-existing cysts can lead to life-threatening toxoplasmic encephalitis (Luft et al., 1992, Israelski et al., 1993, Slavin et al., 1994). Additionally, in congenital infections, toxoplasmosis can lead to blindness, severe neurological problems, or even death, given the immature nature of the fetal immune system (Wilson et al., 1980).
The lytic cycle of Toxoplasma, which is at the center of its propagation and pathogenesis, begins with the active invasion of parasites into host cells, a process that is dependent on the secretion of proteins from specialized secretory organelles known as the micronemes and rhoptries (Carruthers et al., 1997). Additionally, invasion initiates the formation of a parasitophorous vacuole (PV) through the invagination of the host cell membrane (Suss-Toby et al., 1996). Following parasite growth within the PV, the cycle is reinitiated after the parasites actively egress the host cell and invade neighboring host cells. Many of the events in Toxoplasma’s lytic cycle such as egress, motility, invasion and micronemal protein secretion are accompanied by and dependent on calcium fluxes within both the parasite and the host cell (Arrizabalaga et al., 2004a). Calcium levels increase in the host cell, the PV and the parasite cytoplasm just prior to the initiation of parasite egress from its host cell (Borges-Pereira et al., 2015). Oscillations in parasite calcium, which are enhanced in the presence of extracellular calcium, have been observed during periods of parasite motility using both chemical and genetically encoded calcium indicators, (Lovett et al., 2003, Borges-Pereira et al., 2015). Secretion of the proteins from the micronemes, which are required for parasite attachment to a host cell, can be stimulated by artificially inducing calcium fluxes (Carruthers et al., 1999b).
The parasite can access the calcium required for these signaling events from both intraparasite calcium stores and the extracellular milieu once it has egressed from its host cell (Moreno et al., 2011). Toxoplasma has been shown by transmission electron microscopy of precipitated calcium to store intracellular calcium within the perinuclear endoplasmic reticulum as well large cytoplasmic vacuoles, which likely are what has been referred to as plant like vacuoles (PLV), (Miranda et al., 2010), and within the flattened sacs of the inner membrane complex which lie just beneath the parasite plasma membrane (Bonhomme et al., 1993). Release of calcium from intra-parasitic compartments such as the ER can induce invasion related events such as protein secretion and cytoskeletal rearrangement of the apical end of the parasite (Moreno et al., 1996, Carruthers et al., 1999b). Invasion can also be enhanced by extracellular calcium which is likely taken up by a nifedipine-sensitive calcium channel in the parasite plasma membrane, though the channel responsible for this uptake has not been identified (Pace et al., 2014). Once inside a cell the parasite divides within the PV, where it is presumed to have access to the host cell calcium through the presence of a nonselective pore in the parasitophorous vacuole membrane (PVM), though the exact molecular mechanism for calcium import from the PV lumen across the parasite plasma membrane has yet to be elucidated (Gold et al., 2015). Interestingly, electron microscopy analysis of intracellular tachyzoites suggests that calcium is concentrated within the tubulovesicular network (TVN), a network of membranous tubules found throughout the PV (Bonhomme et al., 1993). Whether this accumulation of TVN is an active process or if it plays a role in the parasite’s biology is not known.
Though many of the key molecular mechanisms and factors that respond to calcium during invasion and egress have been identified, how the parasite detects and regulates the calcium fluxes is not completely understood. In this context, ionophores such as ionomycin, A23187, and nigericin have been instrumental in studying calcium signaling in Toxoplasma (Mondragon et al., 1996, Pingret et al., 1996, Stommel et al., 1997, Black et al., 2000a, Arrizabalaga et al., 2004b, Fruth et al., 2007, Caldas et al., 2010). Brief (<2 minute) treatment of intracellular parasites with the calcium ionophore A23187 results in rapid exit from the host cell, a process known as ionophore induced egress (iiEgress), while the same length of treatment of extracellular parasites induces micronemal secretion and parasite motility (Carruthers et al., 1999b). When this treatment is prolonged, the parasites lose their ability to attach and invade host cells, resulting in parasite death (ionophore induced death, iiDeath) (Mondragon et al., 1996, Black et al., 2000a). In an effort to identify the proteins that allow Toxoplasma to respond to calcium, we have exploited these calcium ionophore induced phenomena to isolate mutants with altered sensitivity to A23187. From a series of selections and screens we have isolated six independent mutants that fall into three phenotypic categories: delay in iiEgress and resistance to iiDeath, delay in iiEgress but normal sensitivity to iiDeath, and resistance to iiDeath but normal iiEgress (Black et al., 2000a, Lavine et al., 2007). We have previously reported that all mutants that are both delayed in iiEgress and resistant to extracellular ionophore death have causative mutations in a calcium dependent protein kinase, TgCDPK3, which regulates egress by phosphorylating the major motor driving Toxoplasma motility (Garrison et al., 2012, Gaji et al., 2015).
To understand how Toxoplasma responds to calcium fluxes we have now focused our attention to mutant strain MBD2.1, which is able to survive prolonged exposure to the ionophore while extracellular, but has no delay in iiEgress. Besides ionophore resistance, this mutant strain also exhibited hypersensitivity to treatment of extracellular parasites with the intracellular calcium chelator BAPTA AM, suggesting that it has altered calcium homeostasis or sensitivity (Black et al., 2000a). Here we describe how these phenotypes are due to a nonsense mutation in a previously uncharacterized protein, GRA41, which localizes to the parasites’ secretory organelles known as dense granules and is secreted into the PV, where it associates with the TVN. Importantly, we also show that GRA41 is critical for calcium homeostasis and the timing of natural non-induced egress. In conjunction, our findings suggest a connection between the TVN and ion homeostasis within parasite, and thus a novel role for the vacuole of this important pathogen.
RESULTS
Nonsense mutation in novel gene is responsible for iiDeath− phenotype of MBD2.1
To identify the causative mutation in mutant strain MBD2.1, whole genome sequencing was undertaken of both the mutant and its parental strain, RhΔhxgprt. Single nucleotide variants (SNVs) between the two were mapped against the Toxoplasma genomic database (ToxoDB). In total 18 SNVs occurred between mutant and parental; 5 were in intergenic regions, 11 within introns and 2 in exons. Of the two mutations within exons, which were both confirmed by PCR and sequencing, one results in a missense mutation in the hypothetical protein TGGT1_306020 and the other in a nonsense mutation in the hypothetical protein TGGT1_069070 (ToxoDB v7.2). Although both transcriptomic and proteomic evidence could be found for TGGT1_069070 in version 7.2 of the Toxoplasma genomic database, this predicted gene has not been annotated as a gene in subsequent genome versions. Analysis of a cDNA library using 5′ and 3′ RACE confirmed the gene model of TGGT1_069070 shown in ToxoDBv7.2 (Supplemental Fig. 1). TGGT1_069070 encodes a putative 179 amino acid (aa) protein with an N terminal signal sequence but no known functional domains were identified in the rest of the sequence. Though homologs of this gene are not annotated in any genomes present in the EuPathDB database, a tblastn search against the parasite genomes available in EuPathDB identifies potential homologs in the closely related parasites Hammondia hammondi (KL544038:723,066..723,587, 77% identity with TGGT1_069070) and Neospora caninum (FR823391:5,147,783..5,148,286, 47% identity).
The nonsense mutation in TGGT1_069070 detected in the mutant strain is a single C to G transversion, which results in the conversion of the serine at position 91 to a premature stop codon (Fig. 1A). To investigate whether the mutation in this gene was responsible for the observed phenotype of MBD2.1, we transfected a cosmid containing a parental copy of the gene into the mutant and generated stable clones by limiting dilution. Two independent clones (MBD2.1 Comp Clone 1 and MBD2.1 Comp Clone 7) were established and the incorporation of the parental allele was confirmed by sequencing of a PCR fragment spanning the TGGT1_069070 coding sequence. The sequence chromatogram from Clone 1 shows a mixed peak at the position of interest (Fig. 1B) indicating that this clone carries both a mutant and a parental copy of the gene as expected for random integration of the cosmid. On the other hand, Clone 7 contains only the wild-type copy of the gene (Fig. 1B), suggesting that the cosmid recombined by homologous recombination with the endogenous locus resulting in an allelic replacement event. We confirmed that Clone 7 still carries the missense mutation in TGGT1_306020 corroborating that it is indeed a derivative of the mutant strain MBD2.1 (data not shown). Importantly, regardless of whether by random integration or by allelic replacement, introduction of wildtype TGGT1_069070 restored ionophore sensitivity to MBD2.1 (Fig. 1C). Treatment of extracellular parasites with A23187 followed by plaque assay to determine number of viable parasites showed that, while mutant MBD2.1 has increased survivability (54%±9%, p<0.05) as compared to a parental strain (18%±11%), both MBD2.1 Comp Clone 1 and MBD2.1 Comp Clone 7, were statistically as sensitive to iiDeath as the parental strain (28%±5% and 26%±8% respectively, Fig. 1C). Thus, presence of a wildtype copy of TGGT1_069070 restores ionophore sensitivity to mutant MBD2.1. Based on this result and those described below we have focused our studies on the protein product of TGGT1_069070 and have not as of yet explored the role of the mutation found in TGGT1_306020 as it is unlikely to influence iiDeath.
Figure 1.

The iiDeath− phenotype of MBD2.1 is due to the introduction of a premature stop codon in TGGT1_069070. (A) Diagrams of the protein encoded at the TGGT1_069070 locus in the parental strain (top) and the putative truncated protein encoded in MBD2.1 as a result of the nonsense mutation in TGGT1_069070 (bottom) are shown. Gray rectangle represents predicted signal peptide and asterisks indicate relative positions of known phosphorylation sites (ToxoDB proteomic databases). (B) Chromatograms from sequencing of PCR fragment spanning mutated region for the parental, MBD2.1 mutant, and two MBD2.1 clones complemented with cosmid TOXO119 (Comp clones 1 and 7) are shown. In clone 7 the mutation has been repaired to wild type base indicating allelic replacement, while in clone 1 both a mutant and wild type allele are present as observed by mixed peak for base of interest (arrow). (C) Extracellular parasites of the parental, mutant (MBD2.1) and two complemented strains were exposed to 1 μM A23187 for 1 hour and allowed to form plaques to determine survival rate which was calculated based on survival of untreated parasites. Bars are average of three independent experiments and error bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05.
TGGT1_069070 encodes a novel dense granule protein, GRA41
In order to confirm the expression of TGGT1_069070 at the protein level and to determine its localization, sequences encoding a carboxy-terminal (C-terminal) triple-hemagglutinin (HA) tag were introduced into the endogenous locus in the RhΔKu80Δhxgprt strain just before the stop codon, using a previously described approach (Huynh et al., 2009). The resulting strain grows normally in culture and does not exhibit resistance to ionophore induced death (data not shown), indicating that the tag does not interfere with the protein’s function. Western blot analysis of protein extract from the endogenously tagged strain revealed a doublet migrating at approximately 30 kDa, which is higher than the predicted molecular weight of the tagged protein, 24 KDa (Fig. 2A). The doublet pattern was consistently observed in extracts from both intracellular and extracellular parasites (Fig. 2A). At present we do not know the reason for the migration pattern although the protein is predicted to be phosphorylated, which could account for size differences as it does for other Toxoplasma proteins such as GRA7 (Dunn et al., 2008). To explore the localization of TGGT1_069070, we performed an immunofluorescence analysis (IFA) and found that the encoded protein appears to localize within the PV, where it colocalizes with the PV localized protein, GRA7 (Fischer et al., 1998, Jacobs et al., 1998) (Fig. 2B). IFA of the engineered parasites with antibodies against the parasite surface antigen SAG1 (Kasper et al., 1984) confirmed that the majority of the HA-tagged protein is present within PV and not within the parasites (Fig. 2C). The majority of proteins in the PV are secreted by intracellular parasites from the dense granules, whose contents are more easily detected in extracellular parasites. To determine if TGGT1_069070 is also a dense granule protein, we performed an IFA of extracellular parasites with anti HA antibodies to detect TGGT1_069070 and antibodies to dense granule marker GRA7, which shows co-localization of both signals within intracellular vesicles consistent with dense granules (Fig. 2D). To confirm and refine the results from immunofluorescence analysis, we performed immunoelectron microscopy using anti-HA antibodies to detect the tagged protein (Fig. 2E). Within the parasite cytoplasm, the signal is detected predominantly within the membrane bound electron dense structures whose morphology and location are consistent with that of dense granules, confirming our observations from IFA. Within the PV the protein is exclusively found associated with the vesicles and tubules of the TVN and not free in the vacuole lumen or membrane (Fig. 2E). Consistent with this membranous localization, partitioning with TX-114 showed the tagged protein partitions almost exclusively with the detergent phase, consistent with a membrane-associated protein (Supplemental Fig. S3). Based on these results, we concluded that the protein encoded at the TGGT1_069070 locus is a dense granule derived protein that is secreted from the parasite into the PV where it associates with the TVN. According to the currently published literature, the dense granule proteins have been named from GRA1 to 40 so here we designate TGGT1_069070 as GRA41.
Figure 2.
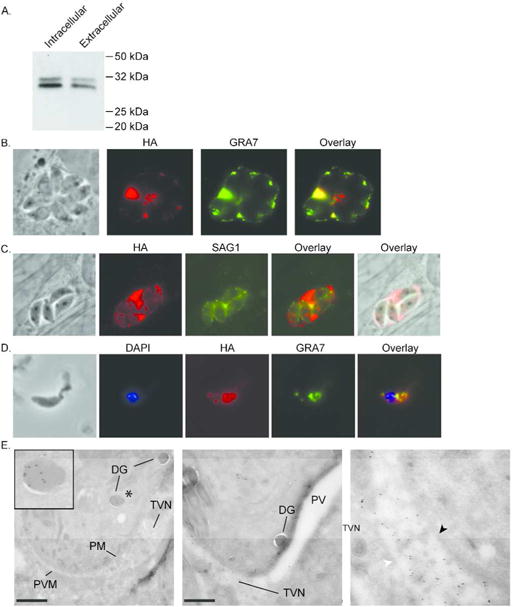
TGGT1_069070 encodes a novel dense granule protein, GRA41. A strain in which a 3xHA tag is expressed at the C terminus of the endogenous TGGT1_069070 protein was generated and analyzed for protein expression and localization. (A) Expression of HA tagged TGGT1_069070 was confirmed by Western blot in both intracellular and extracellular parasites. TGGT1_069070 migrates as a protein doublet of approximately 30 kDa (predicted molecular weight ~ 24 kDa). (B–C) Intracellular parasites were analyzed by Immunofluorescence Assay (IFA) using anti-HA antibodies to detect TGGT1_069070 (in red) and antibodies against either the dense granule/parasitophorous marker GRA7 (B, in green) or the surface antigen SAG1 (C, in green). (D) Extracellular parasites were analyzed by IFA using HA antibodies to detect TGGT1_069070 within the parasite and with antibodies against the dense granule marker GRA7 (in green). DAPI stain was used to detect the nucleus. (E) Immunoelectron microscopy was used to confirm the localization of TGGT1_069070, which we designated GRA41, to the dense granule and parasitophorous vacuole. Antibodies directed against HA conjugated to gold particles were used to detect protein. Inset in left panel is magnification of the dense granule marked with asterisk in image. Middle panel focuses on vacuolar space and shows that staining is confined to regions where tubulovesicular network (TVN) is present. Right panel is magnification of region of the middle panel where TVN is present. Black arrow head points at parasite membrane, white arrow head at parasitophorous vacuole membrane. DG = dense granule, PM = parasite membrane, PVM = parasitophorous vacuole membrane, TVN = tubulovesicular network. Scale bars, 500 nm.
Complete knockout of GRA41 recapitulates iiDeath− phenotype
To further study the role of GRA41 in the parasite’s life cycle, we generated a knockout of the gene by replacing the entire coding sequence with the selectable marker hypoxanthine guanine phosphoribosyltransferase (HXGPRT) through double homologous recombination into the RhΔKu80Δhxgprt strain, hereafter referred to as RhΔKu80 (Huynh et al., 2009) (Fig. 3A). Parasites transfected with the knockout vector (pGRA41KO) and stably expressing HXGPRT were selected by resistance to mycophenolic acid and xanthine and cloned by limiting dilution. Through two separate attempts to generate knockout strains, we established two independent clones (RhΔKu80Δgra41-1, aka KO1 and RhΔKu80Δgra41-2, aka KO2) that were shown to have correct integration of the construct by PCR (Fig. 3B). To confirm that the genetic disruption of the locus resulted in a functional knockout of GRA41, we measured relative transcript abundance by quantitative PCR (qPCR), which fail to detect any GRA41 message in either of the knockout clones (Fig. 3C). In addition, through qPCR, we determined that the genetic disruption did not affect transcript levels of the genes immediately upstream and downstream of GRA41 (Fig. 3C). Based on the nature of the genetic disruption expected from double recombination of the knockout construct (pGRA41KO, Fig. 3A) and the lack of detectable transcript (Fig. 3C), the two established clones represent functional knockouts and are unlikely to produce any GRA41 protein. We then assessed whether or not the complete knockout of GRA41 recapitulated the iiDeath− phenotype seen in GRA41 truncation mutant MBD 2.1. Both RhΔKu80Δgra41-1 (45% ± 16% versus 11% ± 12% for the parental strain, p<0.05, Fig. 3D) and RhΔKu80Δgra41-2 (25% ± 5% versus 7% ± 5% for the parental strain, p<0.05, Fig. 3D) showed statistically significant increases in survival as compared to the parental strain after 60 minutes of treatment with A23187. Thus, the recapitulation of the phenotype seen in the mutant MBD2.1 by the complete lack of GRA41 suggests that the iiDeath phenotype of MBD2.1 is likely due to the absence of GRA41 product and not a dominant negative effect imparted by a possible truncated protein.
Figure 3.

Complete knockout of GRA41 recapitulates the iiDeath− phenotype seen in MBD2.1 mutant. (A) Diagram shows strategy used to knockout GRA41 by double homologous recombination replacing the GRA41 locus with the HXGPRT selectable marker. Colored arrow heads indicate beginning and end of genomic regions used to drive homologous recombination of pGRA41 KO vector with target locus in chromosome X (ChrX). Arrows indicate relative direction of transcription of the three genes depicted and black arrow heads represent primers p1, 2 and 3 used to corroborate integration. (B) PCR analysis of DNA from parental strain (Par) and two independent putative knockouts KO1 (top panels) and KO2 (bottom panels) using either a primer pair expected to produce an amplicon in both parental and knock strains (p1-p2, left panels) or a test primer pair that would produce a product only if the GRA41 gene were replaced (p2-p3, right panels). (C) qPCR analysis was used to monitor transcript levels of GRA41 and the two flanking genes TGGT1_236870 and TGGT1_236860 in the parental (Par) and two knockout strains (KO1 and KO2). Data is expressed as ΔΔCt of each relative to that of parental and represents average of 3 independent experiments (D) Extracellular parasites of the parental and KO1 (left) and KO2 (right) mutant (MBD2.1 were exposed to 1 μM A23187 for 45 or 60 minutes and survival rate was determined as in Fig. 1. Bars are average of three independent experiments and error bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05.
Complete knockout of GRA41 results in decreased plaquing efficiency
While performing the iiDeath assays described above we noted that, although we used the same number of untreated parasites for all strains, the knockout parasites consistently established fewer plaques as compared to the parental. To determine whether the knockout strains truly had reduced plaquing efficiency, which would suggest a defect in the lytic cycle of the parasite, we infected cells with an equal number of parasites of the parental and knockout strains and allowed them to infect for two hours before removing extracellular parasites. Intracellular parasites were allowed to form plaques, which were quantitated on the sixth day post infection. Both clones showed a statistically significant decrease in the number of plaques as compared to the parental without any noticeable differences in plaque size (18%±12% of parental efficiency for RhΔku80Δgra41-1, 63%±9% for RhΔku80Δgra41-2, p<0.05), although the phenotype of the second independently generated clone was attenuated compared to the first (Fig. 4A). Consistent with this apparently variable phenotype, we observed that continuous passage of the RhΔku80Δgra41-1 in in vitro culture results in a relative loss of phenotype over the course of a few weeks. Plaque assays conducted over the course of two weeks showed the gradual increase in plaquing efficiency from 11% to 56% for clone RhΔKu80Δgra41-1 (Fig. 4B). Continued passage of adapted parasites in culture resulted in a strain that grows robustly, exhibiting more and larger-sized plaques as compared to the parental strain (data not shown). Thus, the loss of GRA41 resulted in reduced plaquing efficiency of tachyzoites, and the parasites adapted to this phenotype during continuous passage in culture.
Figure 4.

Complete knockout of GRA41 results in reduced plaquing efficiency which is rescued by complementation. (A) Extracellular parasites of the parental and knockout strains were allowed to form plaques to assess plaquing efficiency, which was calculated by dividing the number of plaques formed for each knockout strain by that formed by the parental strain. (B) Extracellular parasites of RhΔku80Δgra41-1, which had been maintained in culture for different amounts of time, were allowed to form plaques as in (A) to determine if knockout parasites were able to adapt to loss of GRA41 in respect to plaquing efficiency. (C) Diagram (left) shows the strategy for complementation of gra41 knockout by double homologous recombination of cosmid TOXO119 into the target locus. Parasites transformed with cosmid were exposed to 6-thioxanthine to select for loss of the HXGPRT cassette. Flow chart (right) shows the isolation of resulting clones. Indicated below each clone is the relative transcript level of gra41 (as described in Figure 3) and the presence/absence of the HXGPRT cassette, which was determine by sensitivity to MPA. (D) Extracellular parasites of the parental, knockout and complemented clones were allowed to grow for twenty-four hours before fixing and counting the number of vacuoles per field of view (vacuole formation). Bars are average of three independent experiments and errors bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05.
Complementation of GRA41 knockout with the parental gene rescues both iiDeath sensitivity and lytic cycle defects
While the fact that two independent knockout strains exhibited the same plaquing phenotype support the idea that the lack of GRA41 affects parasite propagation, there is always the possibility that secondary mutations are responsible for the observed phenotype. The introduction of a wild-type allele of GRA41, as we did for MBD2.1, would address this issue however, the rapidity with which this strain adapts to in vitro culture would complicate the interpretation of phenotypic complementation. A return to wild type levels of plaquing by a complemented strain could be due to either the addition of the wild type allele or simply to adaptation during the complementation process, which can take several weeks. Thus, we devised a complementation strategy that could control for adaptation, and that was based on repairing the disrupted GRA41 locus by homologous recombination using the same cosmid utilized to complement mutant MBD2.1 (Fig 4C). Our approach took advantage of the fact that the HXGPRT marker, which was used to replace GRA41 in the knockout strains (Fig 3A), can be selected against with 6-thioxanthine (Donald et al., 1996). Because 6-thioxanthine’s effect is static and not cidal (Pfefferkorn et al., 2001) we were able to transfect the knockout strain with the cosmid containing GRA41, select for lack of HXGPRT for 2 weeks, clone by limiting dilution and obtain two types of randomly selected clones: those with detectable levels of GRA41 transcript by qPCR and still HXGPRT+ as demonstrated by resistance to MPA/XAN treatment (Δgra41A and B in Fig. 4C), and those with measurable levels of GRA41 transcript and now HXGPRT− (Δgra41C and D in Fig. 4C). Since all clones, both the ones with GRA41 and those without, had gone through the same manipulations and been in culture for the same amount of time, direct comparison of phenotypes could be performed with some control over possible adaptation. Though the knockout strain also differs from the parental and the complemented strain by the presence of HXGPRT, previous work has shown that the loss of HXGPRT does not play a role in iiEgress, iiDeath or parasite propagation (Arrizabalaga et al., 2004b), supporting the conclusion that any differences seen are due to the loss of GRA41. The ability of these parasites to grow efficiently in in vitro culture was assessed by allowing parasites to infect a confluent monolayer of host cells for two hours, then allowing them to grow for an additional 22 hours before fixing them and counting the number of vacuoles per field of view. The number of vacuoles formed by the KO clones over that by the parental strain was used to convey the relative efficiency of vacuole formation. While both clones still lacking GRA41 (Δgra41a and Δgra41b) showed a significantly decreased frequency of vacuole formation (49%±7% and 50%±33% respectively), the two clones in which GRA41 expression was restored (Δgra41c and Δgra41d) had significantly enhanced ability to establish vacuoles (125%±20% and 121%±37% respectively, Fig. 5C). Thus, GRA41 plays a role in the efficiency of the parasite’s propagation cycle in addition to sensitivity to iiDeath treatment.
Figure 5.

Complete knockout of GRA41 leads to premature egress of parasites. (A) Extracellular parasites of the parental, knockout and complemented strains were allowed to invade for two hours before removing parasites that remained outside. Invaded parasites were allowed to grow for a total of 12, 24 or 30 hours before fixing and counting the number of parasites per vacuole for a minimum of 100 vacuoles per strain. (B) The average number of parasites per vacuole for the data in (A) was calculated for each strain and graphed as a function of time. (C) The average number of vacuoles per field of view for the data in (A) was calculated for each strain/time point and normalized to the twelve-hour time point. (D) Percentage egress was calculated for each strain following 5, 10 or 20 minute treatment with 5 mM DTT. All data is the average of at least three independent experiments and errors bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05.
Complete knockout of GRA41 affects timing of natural egress
To better define the phenotype of GRA41 loss of function we probed the ability of RhΔku80Δgra41-1 to complete the various steps of the lytic cycle. We first performed a 30 minute post-invasion assay as described previously (Carruthers et al., 1999a) to measure the parasite’s ability to attach and invade host cells. A slight but statistically significant reduction in the percentage of invaded parasites was seen from 63%±5% in the parental strain to 45%±11% in RhΔku80Δgra41-1, though no difference was seen in attachment efficiency of the GRA41 knockout strain (Supplemental Fig. S2). Thus, loss of GRA41 leads to a small reduction in invasion efficiency.
Since the level of reduction in invasion efficiency is not sufficient to explain the 50 to 80% reduction plaquing efficiency, we looked at post-invasion lytic cycle steps to determine why parasites were inefficient in forming plaques after successful initial invasion. From the vacuole formation assay used to assess successful complementation, we knew that there was a decrease in number of vacuoles present as early as 24 hours post-infection (hpi), so we chose to see what was occurring before and after this time point during parasite replication. Intracellular parasites from the parental, knockout and complemented strains were manually extracted from host cell, harvested, counted and allowed to invade confluent HFFs in multiple twenty-four well plates for two hours. Each plate was fixed at twelve, twenty-four or thirty hours post-infection and the number of parasites per vacuoles were determined for at least 100 vacuoles per sample (Fig. 5). No statistically significant difference in the average number of parasites per vacuole was seen at twelve or twenty-four hours, suggesting that the knockout parasites were replicating at a normal rate (Figs. 5A and B). However, the knockout parasites showed a more than two-fold decrease in the average number of parasites per vacuole at thirty hpi (from 15±1 parasites/vacuole for parental to 4±0.3 parasites/vacuole for knockout, p < 0.001, Fig. 5B), while the complemented strain is not significantly different from the parental (14±3 parasites/vacuole, p>0.05). This decrease in the average number of parasite per vacuole from 24 to 30 hpi for the knockout strain is due to a decrease in the proportion of large vacuoles, which coincides with an increase in the number of vacuoles containing 1–2 parasites (Fig. 5A). This pattern, (i.e. decrease in large vacuoles and increase in small ones) is consistent with egress and reinvasion, and suggests that natural egress for the GRA41 knockout strain is occurring between 24 and 30 hours. This contrasts with what is observed with both the parental and complemented parasite strain, which continue to divide within the original vacuoles between 24 and 30 hours (Fig. 5A).
In addition to quantifying the average number of parasites per vacuole, the average number of vacuoles per field of view was also assessed for each strain to see if vacuoles were being lost (due to egress without reinvasion or death) or gained (due to egress with reinvasion). The average number of vacuoles per field of view for both the parental and the complemented strains stayed relatively constant overtime, while the knockout parasites first appeared to lose vacuoles between 12 and 24 hpi (48±13% of the vacuoles twelve hours, p<0.05, Fig. 5C) and then gain vacuoles between 24 and 30 hpi (195±48% of the vacuoles at twelve hours, p<0.05, Fig. 5C). The reduction in number of vacuoles between 12 and 24 hpi is consistent with our previous data looking at parasites either shortly after invasion or at twenty-four hours after infection. When looking shortly after invasion, there was only a modest decrease in invasion efficiency for the knockout parasites (Supplemental Figure S3). However, there is a pronounced difference in the number of vacuoles present at 24 hpi between the strains, which would be explained by losing vacuoles between 12 and 24 hpi (Fig. 4D). In conjunction, these results are consistent with an early egress phenotype in the knockout parasites, with the parasites egressing between 12 and 24 hpi failing to reinvade and the parasites egressing between 24 and 30 hpi reinvading successfully. Thus, analysis of the lytic cycle indicates that the loss of GRA41 does not impact division rate of the parasites, but does lead to early egress of parasites, which would account for a majority of the decrease in plaquing efficiency reported above.
Interestingly, the original MBD2.1 mutant (Black et al., 2000a) as well as the GRA41 knockout strain (data not shown) appear to also undergo egress at a faster rate than their parental strain after induction with the ionophore A23187. Nonetheless, because of the high efficiency and fast rate of iiEgress the effect is very slight and difficult to statistically validate. Accordingly, we tested the sensitivity of the GRA41 knockout to a different inducer of egress, the redox reagent DTT, which typically requires that parasites be infected for approximately 36 hours before induction and induces egress with slower kinetics than A23187 (Stommel et al., 2001). For this purpose, we allowed parasites to infect host cells for 24 hours before inducing egress with 5 mM DTT. The GRA41 knockout strain does show evidence of significant enhanced egress as compared to the wild type and complemented strains at both 10 and 20 minutes post-induction (62%±19% versus 2±2% and 15±13% at 10 minutes, 85%±14% versus 5%±9% and 14%±13% at 20 minutes, Fig 5D, p<0.05), indicating that the GRA41 knockout strain is sensitive to induction of egress by DTT earlier in the replication phase of the lytic cycle as compared to the parental and complemented strains.
Complete knockout of GRA41 leads to dysregulation of parasite calcium
Because GRA41 was initially identified as a result of a forward genetic screen involving altered sensitivity to treatment with calcium ionophore and its complete knockout led to defects in events known to depend on calcium homeostasis and fluxes, we compared calcium levels in knockout and complemented strains with the parental (Fig. 6). For this purpose, extracellular parasites of the parental, GRA41 knockout and complemented strains were loaded with fluorescent calcium indicator Fura-2 AM and fluorescence measurements were made before and after adding calcium to the suspension buffer. Compared to the parental strain, the knockout has elevated cytosolic calcium under calcium-free buffer conditions, which is not rescued by complementation with wild-type GRA41 (Fig. 6). This suggests that the elevated cytosolic calcium might not be a direct consequence of the loss of GRA41. Instead it could be either part of the way the parasites adapt to its loss or an unrelated event. However, when the ability of parasites to uptake extracellular calcium was measured by adding 1mM CaCl2 to the buffer, the knockout strain showed a 2.45-fold increase in uptake of extracellular calcium over both the parental and complemented strains (Fig. 6, p<0.05). Thus, these results suggest that the loss of GRA41 leads to a dysregulation of parasite calcium, which likely has wide-reaching impacts on calcium-dependent events in the lytic cycle.
Figure 6.
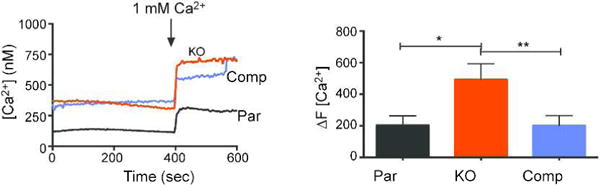
GRA41 knockout parasites exhibit enhanced uptake of extracellular calcium Extracellular parasites from the parental, knockout and complemented strains were loaded with Fura-2 AM to monitor calcium levels. The arrows represent the time at which 1 mM Ca2+ was spiked in to the medium. Increase in calcium levels after addition of extracellular calcium over basal levels was quantitated for at least three independent replicates. Error bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05, double asterisks denotes p value of <0.01.
Complete knockout of GRA41 disrupts vacuolar morphology
During the lytic cycle analysis described above, we noticed consistent differences in the organization of parasites within the vacuole and the gross morphology of the vacuole membrane between the knockout and parental strain, suggesting a role of GRA41 to maintain normal vacuolar morphology. To investigate the possibility that the loss of GRA41 disrupted vacuolar morphology, we visualized the parasites under high power magnification and noted that many of the vacuoles in the knockout strain appeared to be collapsed around the parasites within (Fig. 7A). Quantitation of this “collapsed vacuole” phenotype showed it was significantly more common in the GRA41 knockout strain as compared to the parental and complemented strains (43±1% versus 10±6% and 10±4%, respectively, p<0.05 Fig. 7B). Furthermore, we noted that the vacuoles in the knockout strain were more likely to have non-typical numbers of parasites. Typically, all parasites within the vacuole divide simultaneously into two daughter parasites leading to a doubling of parasite number and a geometric sequence of parasite number within each vacuole (i.e. 2, 4, 8, 16, etc.). Nonetheless, 35±2% of the knockout vacuoles contained a number of parasites that did not follow the normal pattern. This was significantly higher than both the parental and complemented strains, with 12±3% and 7±2% respectively (Fig. 7C, p<0.0001). To investigate how this might be occurring, we stained both parental and GRA41 knockout parasites for acetyl tubulin. The acetylation of tubulin occurs during parasite replication and can be used to visualize the daughter parasites as they are dividing (Varberg et al., 2016). Using this method, we were able to image GRA41 knockout parasites that showed evidence of more than two daughters forming per mother parasites, which would explain the abnormal number of parasites in some vacuoles (Fig. 7D). The formation of triplet and even quadruplet parasites has been observed in wild-type parasites at low levels (Hu et al., 2002), suggesting that the knockout of GRA41 has increased the frequency with which this happens.
Figure 7.

GRA41 knockout parasites have altered vacuole morphology and division pattern. (A) Intracellular parasites of the parental, knockout and complemented strains were fixed and imaged at 24 hours post infection. White arrowheads indicate the collapsed appearance of a vacuole around the parasites in parasites of the knockout strain. (B) Vacuoles from the parental, knockout and complemented strains containing four parasites each were assessed for the “collapsed vacuole” phenotype. Bars are average of three independent experiments and errors bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05. (C) Vacuoles from the parental, knockout and complemented strains were fixed at twenty-four hours post infection. Vacuoles with an abnormal number of parasites (i.e. not 2, 4, 8 or 16) were quantified for each strain. Bars are average of three independent experiments and errors bars represent standard deviation. Asterisk denotes statistical significance based on the results of an unpaired Student’s t-test with a p-value of <0.05. (D) Intracellular parasites of the parental and knockout strains were stained with an antibody against acetylated tubulin to visualize dividing parasites. Arrows show three daughter parasites for a single mother parasite in the knockout strain.
Complete knockout of GRA41 leads to altered structure of the tubulovesicular network
The “collapsed vacuole” and abnormal cell division are both similar to the abnormal vacuole morphology and parasite organization associated with the genetic disruption of GRA2, another secreted protein that localizes to the TVN and that is critical for its formation (Mercier et al., 2002). Accordingly, we examined the structure of the TVN in the GRA41 knockout strain by transmission electron microscopy (Fig. 8 and Supplemental Figure S4). Host cells were infected with freshly egressed tachyzoites for twenty-four hours prior to fixing and preparing samples for thin sectioning. The parental, knockout and complemented strains all showed normal morphology of the PV membrane, but the knockout strain appeared to have fewer tubules within its network and an increase in the small vesicles from which the tubules form (Fig. 8, black arrowhead). Additionally, the tubules that do form in the TVN of the knockout strain appeared to be shorter than those of the parental and complemented strains (Fig 8 and Supplemental Figure S4.)
Figure 8.

GRA41 knockout parasites show altered morphology and size of tubulovesicular network (TVN) as compared to the parental and complemented strains. Intracellular parasites were fixed at 24 hours post infection and prepared for imaging. Red arrowheads in left panels point at regions that were imaged at higher magnification in the respective right panels. Additional representative images can be seen in supplemental figure S3.
Since the GRA41 knockout showed similar disruption of the tubulovesicular network with the GRA2 knockout, we tested to see whether the GRA2 knockout would mirror the resistance to iiDeath treatment of the GRA41 knockout. In contrast to the GRA41 knockout, the GRA2 knockout did not show any increased survival over its parental and complemented strains at either 45 or 60 minutes of treatment (32±10% versus 26±4% and 30±13% at 45 minutes, respectively and 17±17% versus 12±7% and 18±14% at 60 minutes, respectively p>0.05, Supplemental Fig. S5), suggesting that this phenomenon is restricted to the disruption of GRA41 and not a general feature of parasites with disrupted tubulovesicular networks.
DISCUSSION
Calcium-dependent signaling is central to the propagation of parasites of the phylum Apicomplexa, including the causative agent of malaria, Plasmodium falciparum, and the opportunistic pathogen Toxoplasma gondii. Calcium-dependent events underlie the motility, invasion, and egress of these parasites and thus calcium homeostasis and fluxes are tightly controlled. While many of the effectors of calcium that drive events of the lytic cycle in both Plasmodium and Toxoplasma have been identified, much remains unknown about how these calcium fluxes are regulated. In an effort to address this knowledge gap we used a forward genetics approach to identify genes that influence Toxoplasma’s sensitivity to calcium fluxes, specifically those induced by the calcium ionophore A23187. Surprisingly, we have discovered that a secreted protein, which resides within the parasitophorous vacuole during parasite intracellular growth, influences several calcium related events in both intra and extracellular parasites. Our results suggest a role for PV proteins in the regulation of calcium homeostasis, timing of egress, and parasite division.
We first identified the novel protein GRA41 through the sequencing of a mutant resistant to extracellular exposure to the ionophore A23187. Normally, exposure to this calcium ionophore for over thirty minutes renders extracellular parasites non-invasive, which, for this obligate intracellular parasite, results in death. While this phenomenon, known as ionophore induced death (iiDeath), has been known and exploited for decades (Mondragon et al., 1996, Black et al., 2000a), the underlying mechanism is not well understood. It has been proposed that prolonged exposure to elevated calcium, which normally induces secretion of adhesive molecules required for invasion, exhausts the contents of the secretory organelles that harbor such proteins. Other plausible mechanisms include toxic effects from elevated cytoplasmic calcium levels, and changes to the calcium sensitivity of the molecules and machinery that regulate secretion. We have actually detected that following a one-hour ionophore exposure, the levels of micronemal proteins do not change compared to untreated parasites but the ability to elicit micronemal secretion upon subsequent ionophore exposure is significantly affected (Supplemental Fig. S6). This observation would indicate that iiDeath is not merely the consequence of the exhaustion of proteins to be secreted, but of an alteration in the sensitivity to calcium, which would lead to a reduction in active secretion. In this context, we hypothesize that the resistance to iiDeath of the parasites lacking GRA41 is due to an altered sensitivity to the ionophore, brought upon by the clear changes in calcium regulation we detect in the GRA41 knock out strain. Indeed, the gra41 mutant strain (MBD2.1) is over-sensitive to the effects of calcium chelation by BAPTA-AM (Mondragon et al., 1996, Black et al., 2000a). We do not know whether the altered calcium regulation exhibited by the GRA41 knockout strain is due to a direct effect of lack of GRA41 or an indirect consequence of how the parasites adapts to the loss of GRA41. The latter explanation is supported by our observation that the mutant strain has an elevated level of cytosolic calcium that is not corrected by genetic complementation. Nonetheless, lack of GRA41 does seem affect calcium regulation as the mutant parasites exhibited dysregulated calcium uptake, a phenotype that is reverted by complementation. Though it is difficult to determine the specific function of GRA41 the calcium uptake defect of extracellular parasites suggests that GRA41 is involved in regulating calcium homeostasis. Higher levels of cytosolic calcium and increased rates of calcium uptake by extracellular parasites shows how the parasites responds to the loss of GRA41.
How a protein that resides outside of the parasite but within the parasitophorous vacuole might affect calcium regulation within the cytoplasm of the parasite is puzzling, especially considering that some of the phenotypes related to its deletion, such as iiDeath resistance, manifest only once the parasites are extracellular. It is plausible that the protein, which is in the dense granules before secreted, has a role within the organelle that influences calcium homeostasis in extracellular parasites. Nonetheless, there is no evidence that dense granules accumulate significant amounts of calcium (Bonhomme et al., 1993) or are involved in the regulation of calcium homeostasis or fluxes. It is also conceivable that GRA41, while in the PV, directly or indirectly influences the physiology of the vacuole, which in turns affects that of the parasite. In such a model, the absence of GRA41 would alter the physiology of the intracellular parasite and, once outside, these parasites would react differently to external stimuli such as the calcium ionophore.
Though the PV lumen has long been considered physiologically at equilibrium with the host cell through a nonselective pore (Schwab, 1994), whose components were recently identified (Gold, 2015), several studies indicate that ionic concentration of the PV can vary in respect to that of the host cytoplasm during intracellular growth. For instance, measurements of calcium levels using non-ratiometric indicators in the PV have shown that calcium is concentrated in the PV relative to the host cell cytoplasm and the frequency of this calcium sequestration increases as egress approaches (Pingret, 1996). Interestingly, one of the most abundant PV proteins, GRA1, has been shown to bind calcium (Cesbron-Delauw, 1989), which suggests a potential mechanism for this calcium sequestration; however, GRA1 localizes to the vacuolar space and is not associated with the TVN. Interestingly, the calcium concentration of the Plasmodium PV has been determined through the use of ratiometric indicators to be approximately 40 μM, many fold higher than the 20 nM that would be expected if the PV calcium levels were in equilibrium with the host red blood cell’s cytosol (Murphy et al., 1987, Gazarini et al., 2003). Additionally, the same study showed that depletion of calcium in the Plasmodium PV leads to a depletion of intracellular calcium stores, which suggests that the parasite depends on the sequestration of calcium in the PV to maintain its intracellular pools of calcium (Gazarini et al., 2003). Interestingly, calcium is not the only ion which the parasite regulates the concentration of within the PV; it has recently been shown that the pH of the vacuole decreases as egress approaches and this increased acidification is associated with enhanced parasite motility and protein secretion (Roiko et al., 2014). While it is possible that one or more dense granule proteins that Toxoplasma secretes into its vacuole are responsible for the sequestration of ions as egress approaches, it is unlikely that this is occurring in Plasmodium species, since the dense granule proteins described to date are restricted to the tissue-cyst forming coccidia and are not found in other apicomplexans such as Plasmodium (Mercier et al., 2015). Therefore, it is possible that these two related organisms have evolved different strategies to utilize calcium signaling within the context of the calcium-poor cytosol of their host cells.
It is unlikely that GRA41 binds calcium directly, since bioinformatic analysis fails to identify any conserved calcium binding domains (SMART, NCBI, Prosite, Interpro) and recombinant GRA41 protein fails to bind calcium in vitro (Supplemental Fig. S7). Rather, we hypothesize that GRA41 might influence physiological calcium homeostasis by its localization and functional role within the tubulovesicular network (TVN). Loss of GRA41 leads to altered morphology of the tubulovesicular network, which interestingly has been proposed to play a role in calcium storage based on electron microscopy studies (Bonhomme et al., 1993). Nonetheless, other dense granule proteins that traffic exclusively to the TVN of the parasitophorous vacuole are also critical for its formation (Mercier et al., 2002), but do not seem to appear to affect the same of processes influenced by GRA41. The loss of the tubulovesicular network in parasites lacking GRA2 leads to a variety of phenotypes, including the formation of vacuoles containing poorly organized parasites that egress erratically (Muniz-Hernandez et al., 2011), altered presentation of antigens to the host immune system (Lopez et al., 2015), decreased ingestion of host cytosolic proteins (Dou et al., 2014) and decreased virulence in mice (Mercier et al., 1998b). However, we have shown that the loss of GRA2 does not phenocopy the iiDeath resistance of the GRA41 knockout and forward genetic mutant, suggesting that the disruption of the tubulovesicular network might not be the underlying reason for the iiDeath resistance of GRA41. Rather, it appears that the iiDeath resistance of the GRA41 knockout strain is a direct effect of GRA41 by a novel mechanism we have yet to completely discern. Nonetheless, we need to consider the possibility that although the TVN is structurally affected in both the GRA41 and GRA2 knockout strains, the nature, level and the functional consequences of that effect might be different between the two mutant strains.
At the current time, it is unclear which of the pleiotropic effects of GRA41 loss is the primary impact of its loss and which are simply downstream defects. However, one of the most severe and distinct phenotypes of the GRA41 knockout strain is a premature egress from the host cell. Natural egress from host cell can be an active process dependent on calcium fluxes (Moudy et al., 2001). Two alternative hypotheses to explain the premature egress are 1) that the altered TVN, which might normally act as a mechanical barrier, allows for early exit and 2) that the dysregulation of calcium homeostasis results in changes in the timing of egress. The fact that other mutant parasites lacking a normal TVN, such as the GRA2 knockout, don’t have a premature egress phenotype would argue against a model in which the altered TVN, caused by gra41 disruption, allows for early egress. Additionally, the only other dense granule protein whose loss leads to an early egress phenotype similar to the GRA41 knockout is GRA22. However, the GRA22 knockout does not exhibit any obvious defect in TVN formation (Okada et al., 2013), suggesting that GRA41 plays a structural role within the TVN that is independent from its role in egress and, by inference, calcium-dependent signaling processes within the parasite. It remains unclear whether or not these two proteins are interacting together directly or within the same pathway to influence the timing of egress. Future studies to identify interactors of GRA41 and to create a strain lacking both GRA22 and GRA41 would address some of these questions. Considering that tight control of calcium levels is needed for egress, it is plausible that the early egress phenotype of the GRA41 knockout is related to the dysregulation of calcium we see in this strain. Interestingly, the MBD2.1 mutant, as well as the GRA41 knockout, show a slight but consistent increase in the rate of calcium dependent iiEgress ((Black et al., 2000a) and unpublished data, LaFavers and Arrizabalaga). Additional support for a connection between early egress and calcium dysregulation in the mutants, is the fact that the GRA41 knockout is much more sensitive to DTT induced egress than the parental strain. The redox reagent DTT is proposed to induce egress by activating NTPases located in the PV since it is capable of activating isolated enzyme (Bermudes et al., 1994). Treatment with DTT has also been shown to lead to calcium fluxes within the parasite, resulting in induced egress, which can be blocked by chelating calcium, indicating that DTT-induced egress is also calcium dependent (Stommel et al., 1997, Borges-Pereira et al., 2015). Unlike calcium ionophores such as A23187, DTT is not capable of inducing egress at early stages of parasite vacuole formation and requires longer growth times (~36 hours) for vacuoles to consistently egress. This suggests that DTT-induced egress might be dependent on processes preceding natural egress and is therefore a more accurate model for what is happening in natural uninduced egress (Stommel et al., 2001). In this context, the ability of the GRA41 knockout strain to undergo DTT-induced egress at 24 hours is consistent with its early egress phenotype. The formation of more than two daughter parasites in the dividing GRA41 knockout strain could also occur as a result of calcium dysregulation rather than simply being an additional function of GRA41. Knockdown of the calcium-dependent kinase CDPK7 has previously been shown to disrupt normal division (Morlon-Guyot et al., 2014). Additionally, experiments to identify novel proteins in the inner membrane complex (IMC) of the parasite, which serves as the scaffold for daughter cell formation (Harding et al., 2014), identified a protein (ISC6, TgGT1_267620, ToxoDB.org), that localizes to the IMC and contains a C2 calcium binding domain (Chen et al., 2017), which could suggest a link between calcium and daughter parasite formation in T. gondii.
Of note is the fact that GRA41 is not annotated as a protein encoding gene in the latest version of the Toxoplasma genome database. This locus was denoted as a gene in earlier annotations with both transcriptomic and proteomic corroborating evidence. As we have shown there is clearly a protein encoding locus in that genomic region; underscoring the limitation of genome annotations. One of the drawbacks of GRA41 not being denoted as a protein in the current annotated genome is that it is not considered when analyzing datasets that rely on the annotation. In the many gene expression studies, mappings of post-translational modifications, and sequencing of mutant strains that have been undertaken in the last few years, GRA41 has not been included, significantly reducing our ability to understand its function. Indeed, we were very fortunate that this gene was still listed in the annotated genome we used to map the missense mutations present in the mutant strain MBD21. Had we used a later version of annotated genome, we would have not identified GRA41 as central to the phenotypes of our mutant strain. Thus, constant monitoring, validation, and curation of genome annotations is needed to fulfill the promise of genome sequencing. For example, the presence of certain post-translational modifications could provide clues to how GRA41 is associated with the TVN membrane. Though GRA41 lacks an amphipathic alpha helix (Helical Wheel Predictions, RZ Lab) like those found in other TVN-localized proteins such as GRA2 (Mercier et al., 1998a), it does contain a predicted palmitoylation site at Cys26 (CSS-Palm, Cuckoo Workgroup) that could explain membrane association. Though the palmitoylome for T. gondii has been published (Foe et al., 2015), GRA41 was not included in the database at the time and any potential palmitoylation modifications were not mapped. As a result, future directions for the study of GRA41 would include mapping post-translational modifications and determining how they impact protein localization and function by mutational analysis.
The discovery of GRA41 as a novel dense granule protein with a role in calcium-dependent events helps to both answer and create new questions about the role of the PV in these signaling processes. This work demonstrates that the parasite is dependent on a dense granule protein for normal calcium homeostasis, but it is still unclear how and if the parasite might regulate PV calcium levels independent of the host cell cytosol in the presence of a nonselective PVM pore (Schwab et al., 1994). The explanation for this might be the existence of additional calcium binding proteins that, in addition to GRA1 (Cesbron Delauw et al., 1989), could bind and sequester calcium within the PV. Future efforts to directly measure calcium concentration in the PV of GRA41 mutant strains and identifying proteins that interact with GRA41 might shed light on the specific function of this protein. The localization of GRA41 to the tubulovesicular network suggests that this membranous structure may also play a direct or indirect role in the physiological regulation of the parasite, pointing to a novel function for this poorly understood element of intracellular parasite development. Thus, in conjunction with previous knowledge, our results underscore the critical importance of the parasitophorous vacuole and its contents in the life cycle of Toxoplasma and, likely, of related parasites.
EXPERIMENTAL PROCEDURES
Parasite propagation
Toxoplasma gondii tachyzoites were propagated by passaging in human foreskin fibroblasts (HFFs, purchased from the American Tissue Culture Collection, ATCC) in a humidified incubator maintained at a temperature of 37°C and 5% CO2 concentration. Normal growth medium used was Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum, 2 mM L-gluatamine and 50 μg/mL penicillin streptomycin.
Genome sequencing and complementation
Extracellular parasites from strains MBD2.1 and RHΔhxgprt (the parental strain) were purified through a 3 μm filter to eliminate human cell contamination. Genomic DNA from both strains was isolated using the DNeasy Blood and Tissue Kit (Qiagen). Sample preparation, sequencing, genome assembly, and annotation was performed at the University of Idaho IBEST Genomics Resources Core facility. Genomic DNA libraries were constructed using the Illumina TruSeq library kit and quantified with rtPCR using the Kapa Illumina library quantification kit. 100bp paired-end Illumina sequencing was used to an estimated > 100× coverage per genome. Mapping of Illumina sequence was performed using GMAP to the TGGT1 reference sequence from ToxoDB (Gajria et al., 2008) with output to SAM format files for further processing. Genomic variants in the mutant strains in comparison to the reference sequence were detected and extracted from the mapped data using the Broad Institutes GATK toolkit. Data was exported as a variant call format (VCF) file, which listed each genomic variant, its position in the genome, and the quality of sequence data for that particular region. In total we detected 14 SNVs between the mutant and parental strain. The two SNVs that resulted in missense or nonsense mutations in MBD2.1 were confirmed by sequencing fragments of genomic DNA amplified by PCR from both the parental and mutant strains and that spanned regions with putative mutations.
To determine which of the identified variants in the mutant was responsible for the phenotype, a cosmid-based complementation approach was used. Cosmids generated from the Rh (Type I strain) containing the genomic regions of interest were identified on ToxoDB.org and were graciously provided by Dr. David Sibley at Washington University, St. Louis. For TGGT1_069070 we utilized cosmid TOXO119, which was linearized by digestion with NotI (TOXO119), purified and electroporated into Toxoplasma tachyzoites of the MBD 2.1 mutant according to established protocols (Kim et al., 1993, Soldati et al., 1993). Transfected parasites were maintained in the presence of 1 μM pyrimethamine prior to cloning by limiting dilution to select for stable transformants.
iiDeath survival assay
The iiDeath survival assay was performed as described previously (Black et al., 2000a). In brief, intracellular parasites were harvested by passage through a 27 gauge needle three times before dilution and treatment with either 1 μM A23187 or DMSO solvent control for 45 or 60 minutes in a humidified incubator at 37°C and 5% CO2 concentration. At each time point, 500 parasites were removed from the treatment and allowed to infect a confluent monolayer of HFFs in a twelve well plate format for two hours before changing the media to remove non-invading parasites. Parasites were allowed to grow and form plaques for 6 days before the cultures were fixed and scored. Each combination of treatment and time point was the average of a minimum of three technical replicates per experiment and the experiments were performed a minimum of three times for statistical analysis. The percent survival for each strain and time point was calculated as a ratio of the number of plaques scored in the wells infected with treated parasites as compared to the wells infected with untreated (DMSO solvent control) parasites.
Generation of endogenously tagged GRA41 line
For the expression of GRA41 tagged with hemagglutinin at the carboxyl terminal of the encoded protein, an 800 base pair fragment of genomic DNA was amplified by PCR with specific primers GRA41 Tag.FOR and GRA41 Tag.REV (see Table 1 for sequence of all primers used in this study) and directionally cloned into the PacI site of the 3xHA.Lic.DHFR-TS plasmid using In-Fusion Cloning (Clontech). The 3xHA.LIC.DHFR-TS plasmid is a derivative of the YFP.LIC.DHFR-TS plasmid (Huynh et al., 2009) with the YFP coding sequence replaced by a triple hemagglutinin tag. The resulting construct was verified by restriction digestion and sequencing. The plasmid construct was linearized with the restriction enzyme XcmI, which cuts within the region containing the insert and allows for integration of the construct by single homologous recombination when transfected into the RhΔKu80 strain (Huynh et al., 2009). Toxoplasma tachyzoites were transfected with the linearized vector by electroporation according to established protocols (Soldati et al., 1993). Transfected parasites were maintained in the presence of 1 μM pyrimethamine prior to cloning by limiting dilution to select for stable transformants.
Table 1.
Name and sequence of primers used in this study. All sequences are 5′ to 3′.
| Primer Name | Sequence |
|---|---|
| Gra41 Tag.FOR | TACTTCCAATCCAATTTAATGCAATAGAGACGCAGCACCCTTG |
| Gra41 Tag.REV | TCCTCCACTTCCAATTTTAGCTTCGGGTAGGTGAAGTTTAG |
| Gra41 qPCR.FOR | ACGGCAGCTTGAGATACCAC |
| Gra41 qPCR.REV | GCAGCCCTGGTATTGTATCG |
| Gra41 KO 5′Flank.FOR | CGGTATCGATAAGCTTTTTGGTGTGGGCCTGTCGAAT |
| Gra41 KO 5′Flank.REV | CGTGCTGATCAAGCTTCCACGCGTGGATTGCAGAAAA |
| Gra41 KO 3′ Flank.FOR | AGTTCTAGAGCGGCCGCTTTTGGTACCTCCATGGGCAAGG |
| Gra41 KO 3′ Flank.REV | ACCGCGGTGGCGGCCGCATACAAAGGCGAGGTCATTGCTGTG |
| GraX 41 3′UTR Mid.FOR | GCGGATTTGTGGCTACAATACTGC |
| Gra41 Downstream.REV | CTGCTCTATGTCCTCTGTTCGATGT |
| DHFR 3′UTR End.FOR | GGCCATTCATGCCAGTCAGT |
| TgGT1_236870 qPCR.FOR | TTGATGACGACGAGGATGAA |
| TgGT1_236870 qPCR.REV | GTATGCGGGTAACGACGAGT |
| TgGT1_236860 qPCR.FOR | TCTCGCGACAACATTTCAAG |
| TgGT1_236860 qPCR.REV | GCTTTGTCTTCGCTTCCATC |
| Gra41 qPCR.FOR | ACGGCAGCTTGAGATACCAC |
| Gra41 qPCR.REV | GCAGCCCTGGTATTGTATCG |
| RPL29 qPCR.FOR | GGCGAAATCAAAGAACCACAC |
| RPL29 qPCR.REV | TCGGACTTCATCATGCCTCTC |
| GRA41 pet28a.FOR | CGCGCGGCAGCCATATGGACTGTATTCCTTCGCATCAA |
| GRA41 pet28a.REV | GTCATGCTAGCCATATGTTATTCGGGTAGGTGAAGTTTAGA |
Immunofluorescence assays
For immunofluorescence assays, HFFs infected 18–24 hours prior were fixed with 3.5% methanol-free formaldehyde in PBS, blocked in PBS/3% BSA and permeabilized in PBS/3% BSA/0.2% TX-100. Coverslips were then incubated in primary antibodies (Rabbit anti-HA, Cell Signaling Technology, mouse anti-Gra1, Biotem) diluted in PBS/3% BSA/0.2% TX-100, washed and then incubated in secondary antibodies (goat anti-mouse/rabbit Alexafluor 488/594 conjugated, Invitrogen) diluted in PBS/3% BSA. Coverslips were mounted onto microscope slides with Vectashield mounting media that included DAPI (Vector Laboratories). IFAs were inspected using a Nikon Eclipse E100080i microscope and images captured with a Hamamatsu C4742-95 charge-coupled device camera using NIS elements software.
Western Blot Analysis
To examine protein expression by Western Blot, parasites were lysed in 150 Mm NaCl, 50 mM Tris-Cl, pH 7.5, 0.1% NP-40 for a minimum of twenty minutes. Samples were centrifuged at 14,000 × g’s for 20 minutes to remove insoluble proteins before combining with an equal amount of 2X Laemmli Sample Buffer (Bio-Rad) with freshly added 5% beta-mercaptoethanol (Thermo Scientific) and heating at 95°C for 5 minutes. Samples were separated on a 4–20% gradient SDS-PAGE gel (Bio-Rad) before transferring to a nitrocellulose membrane using a Trans-Blot semi-dry transfer cell (Bio-Rad). Western Blots were probed with rabbit anti-HA at a dilution of 1:5000 (Cell Signaling Technologies), mouse anti-ROP1 at a dilution of 1:5000 (Schwartzman et al., 1989) or mouse anti-SAG1 at a dilution of 1:5000 (Genway).
Triton-X 114 Membrane Partitioning
Membrane partitioning of lysates with Triton-X 114 (Sigma-Aldrich) was performed as described previously (Rome et al., 2008). Briefly, parasites were lysed in 10% Triton-X 114, 10 mM Tris, pH 7.4, 5 mM NaCl on ice for 30 minutes, before centrifuging the samples at 2500 × g’s for five minutes before removing the supernatant to a new tube for partitioning. Lysates were incubated at 30°C for five minutes, followed by centrifugation at 3,000 × g’s for 5 minutes to separate samples into the aqueous and detergent phases. The aqueous phase was re-extracted by incubating with 10% Triton-X 114, 10 mM Tris, pH 7.4, 5 mM NaCl on ice for 5 minutes, then at 30°C for five minutes, followed by centrifugation as described above. The detergent phase was re-extracted by incubating with PBS on ice for 5 minutes, then at 30°C for five minutes, followed by centrifugation. Proteins were precipitated from the final aqueous and detergent phases by adding two volumes of ice cold acetone to each sample and incubating at −20°C for a minimum of two hours, followed by centrifugation at 15,000 × g’s for ten minutes. The resulting pellets were washed once more with ice cold acetone before they were resuspended in 2X Laemlli Sample Buffer (Bio-Rad) with freshly added 5% beta-mercaptoethanol (Thermo Scientific) and processed for Western Blot analysis as described above.
Electron microscopy
For transmission electron microscopy, confluent monolayers of HFFs were infected with parasites of the strain of interest 24 hours prior to sample preparation. Infected cells were washed four times with PBS before fixing for one hour in freshly prepared 2.5% glutaraldehyde in 100 mM sodium cacodylate, pH 7.4 buffer in the dark. Fixative was then removed and samples were washed three times with PBS before being harvested by gentle scraping followed by centrifugation at 3,000 × g’s for 10 minutes. The pellets were then incubated in 1% osmium tetroxide in 100 mM sodium cacodylate pH 7.4 buffer that had been reduced by adding solid potassium ferrocyanide to a final concentration of 1.5% (W/V) and incubated in the dark at 4°C for one hour. The pellets were washed three times with 100 mM sodium cacodylate, pH 7.4 buffer before dehydration in a series of ethanol washes. Pellets were dehydrated with five minute incubations in 30%, 50%, 70%, 90% and 95% ethanol followed by four five minute washes of 100% ethanol. The samples were infiltrated with Embed-812/Araldite 502 resin (Electron Microscopy Sciences), at a dilution of 1:1 resin with 100% ethanol and in 3 changes of 100% resin. The formulation of resin used was 12.5 ml Embed-812, 7.5 ml Araldite-502, 27.5 ml Dodecenyl Succinic Anhydride, and 0.95 ml DMP-30. All resin incubations were for at least 2 hours and were done at room temperature in a rotator at low speed. After the final change the inserts were polymerized at 65°C. Thin sections were obtained using a diamond knife, mounted on copper grids and stained with uranyl acetate and lead citrate before imaging on a JEOL 1010 transmission electron microscope. Images were captured with a 1k × 1k Gatan CCD camera (MegaScan model 794) and a tungsten filament was used as an electron source.
For immunoelectron microscopy, HFFs infected with parasites for 24–36 hours were washed three times with PBS before fixing for one hour in freshly prepared 4% paraformaldehyde (EM grade, VWR) in 250 mM HEPES pH 7.4. Fixative was then removed and replaced with a solution of 8% paraformaldehyde in 250 mM HEPES pH 7.4 for overnight incubation at 4°C. Samples were harvested by gentle scraping to detach sheets of cells followed by centrifugation at 2,000 rpm for 10 minutes. Infected cells were pelleted in 10% fish skin gelatin and the gelatin-embedded pellets were infiltrated overnight with 2.3 M sucrose at 4°C and frozen in liquid nitrogen. Ultrathin cryosections were incubated in PBS and 1% fish skin gelatin containing mouse anti-HA antibody at 1/250 dilution, and then exposed to the secondary antibody that were revealed with 10 nm protein A-gold conjugates. Sections were observed and images were recorded with a Philips CM120 Electron Microscope (Eindhoven, the Netherlands) under 80 kV.
Generation of gra41 knockout strain
The GRA41 knockout strain, RhΔku80Δgra41, was generated by replacing the entire GRA41 coding sequence with the HXGPRT selectable marker by double homologous recombination in the RhΔKu80 parental strain (Huynh et al., 2009). The pGRA41KO vector was generated by cloning fragments approximately 1.5 kbp in length directly upstream (primers GRA41 KO 5′ Flank.FOR and GRA41 KO 5′ Flank.REV, Table 1) and downstream (primers GRA41 KO 3′ Flank.FOR and GRA41 KO 3′ Flank.REV, Table 1) of the GRA41 coding sequence into the HindIII and NotI sites of the pmini vector (Donald et al., 1996) by In-Fusion cloning (Clontech). The resulting construct was verified by restriction digestion and sequencing. Toxoplasma tachyzoites were transfected with purified linearized vector by electroporation according to established protocols (Soldati et al., 1993). Transfected parasites were maintained in the presence of 50 μg/ml mycophenolic acid and 50 μg/ml xanthine prior to cloning by limiting dilution to select for stable transformants. Individual clones were screened by PCR to verify correct replacement of the GRA41 locus with the HXGPRT selectable marker.
To confirm the correct insertion, primers complementary to the 3′ end of HXGPRT selectable marker and downstream of the insertion site were used to perform a PCR analysis of genomic DNA isolated from GRA41 knockout clones. A primer within the 3′ homology region of the pGRA41 KO vector (p1, Fig. 3, GRA41 3′UTR Mid.FOR, Table 1) and one downstream of the insertion site (p3, Fig. 3, GRA41 Downstream.REV, Table 1) were used as a positive control, while a primer within the selectable marker of the pGRA41KO vector (p2, Fig. 3, DHFR 3′UTR End.FOR, Table 1) was combined with the same downstream primer (p3) to identify clones with the desired incorporation of the pGRA41KO vector. Loss of GRA41 mRNA transcript was verified by qPCR analysis (primers GRA41 qPCR.FOR and GRA41 qPCR.REV, Table 1) using the SYBR Green detection reagent (Fast Syber Green Master Mix, Applied Biosystems). Relative gene expression was calculated by normalizing to levels of the housekeeping gene ribosomal protein 29 (TGGT1_2345500, Primers RPL29 qPCR.FOR and RPL29 qPCR.REV, Table 1) Relative transcript abundance of the genes directly flanking GRA41 (TGGT1_236860 using primers TGGT1_236860 qPCR.FOR and TGGT1_236860 qPCR.REV and TGGT1_236870 using primers TGGT1_236870 qPCR.FOR and TGGT1_236860 qPCR.REV, Table 1) was also measured by qPCR analysis to ensure that their levels did not differ significantly from the parental strain.
Generation of GRA41 knockout complemented clones
The GRA41 knockout strain, RhΔku80Δgra41 was complemented by transfecting with the cosmid TOXO119. Complemented clones were selected for integration of the gene back into its original locus by selecting against the presence of the HXGPRT gene using 80 μg/ml 6-thioxanthine. Following limited dilution cloning, individual clones were subjected to treatment with 50 μg/ml mycophenolic acid and 50 μg/ml xanthine and qPCR analysis of GRA41 transcript levels to identify clones which were either HXGPRT+GRA41− or HXGPRT−GRA41+. Transcript levels of GRA41 were normalized to the levels of ribosomal protein RPL29 as described above.
Parasite growth assays
Growth of all parasite strains was assessed by determining plaquing efficiency and growth rate as follows. Intracellular parasites were harvested from a confluent monolayer of HFFs by passage through a 27-gauge needle 2–3 times. Parasites were then diluted in normal growth medium and allowed to infect a confluent monolayer of HFFs and returned to a humidified incubator for two hours before changing the media to remove any extracellular parasites. For plaque assays, parasites were allowed to form plaques for six days before the monolayer was fixed and stained with Crystal Violet (ACROS Organics) to visualize plaques. For doubling assays, parasites were allowed to grow for twelve, twenty-four and or thirty hours before they were fixed and stained with Hema3 Manual Staining System (Fisher) to count the number of parasites per vacuole for a minimum of 50 vacuoles per sample.
Parasite egress assays
Egress in response to DTT treatment was assessed as follows. Parasites were allowed to invade host cells for 24 hours prior to the start of the assay. Infected monolayers were washed twice with 1X PBS and once with 1X HBSS (Invitrogen) before incubating in either 5 mM DTT in 1X HBSS or buffer only control for the required time. Monolayers were then fixed with methanol and stained with Hema3 Manual Staining System (Fisher) to assess parasite egress.
Parasite Invasion Assays
The efficiency of attachment and invasion for various parasite strains was measured as follows. Intracellular parasites were harvested as for plaque assays. Parasites were then diluted in normal growth medium and allowed to infect a confluent monolayer of HFFs for two hours. Unattached parasites were removed by washing with phosphate buffered saline (PBS, pH 7.4) three times. Cells were then fixed in 3.5% methanol-free formaldehyde before blocking in PBS/3% BSA. Prior to permeabilization, extracellular parasites were labeled with mouse anti-P30 antibody (Genway) for one hour in PBS/3% BSA. Cells were then washed to eliminate unbound antibody and permeabilized in PBS/3% BSA/0.2% TX-100 and incubated with rabbit anti-Toxoplasma (MyBioSource) for one hour. Cells were washed and then incubated with goat anti-mouse alexa fluor 488 and goat anti-mouse alexa flour 594 (Invitrogen) for one hour before washing and mounting in Vectashield mounting media with DAPI (Vector Labs). Intracellular and extracellular parasites were distinguished between by the presence of dual-color staining (extracellular parasites) or single-color staining (intracellular parasites, which only stain after permeabilization). The efficiency of attachment was calculated by counting the total number of parasites for ten random fields of view and normalizing to 100% attachment for the parental strain. The efficiency of invasion was calculated as the percentage of invaded (intracellular) parasites out of the total number of parasites.
Calcium Measurements
Loading of parasites with Fura-2-AM was performed as previously described (Moreno and Zhong, 1996). Briefly, freshly egressed parasites were washed and then centrifuged twice at 500×g for 10 minutes at room temperature in buffer A (BAG) (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM d-glucose and 50 mM HEPES, pH 7.4). Then, parasites were resuspended to a final density of 1×109 parasites/ml in Ringer buffer (155 mM NaCl, 3 mM KCl, 1 mM MgCl2, 3 mM NaH2PO4H2O, 10 mM HEPES, pH 7.3, and 5 mM glucose; plus 1.5% sucrose and 5 μM of Fura 2-AM). The suspension was incubated for 26 minutes in a 26°C water bath with mild agitation. Subsequently, the parasites were washed twice with Ringer’s buffer to remove extracellular dye. Parasites were resuspended to a final density of 1×109 parasites/ml in Ringer’s buffer and kept on ice. Parasites are known to be viable for a few hours under these conditions (Pace et al., 2014). For fluorescence measurements, 2×107 parasites/mL were added to a cuvette with 2.5 mL of Ringer’s buffer. Cuvette was placed in a thermostatically controlled Hitachi F-7000 fluorescence spectrophotometer, using the conditions for Fura-2-AM excitation (340 and 380nm) and emission (510nm). The Fura-2-AM fluorescence response to intracellular Ca2+ concentration ([Ca2+]i) was calibrated from the ratio of 340/380 nm fluorescence values after subtraction of the background fluorescence of the cells at 340 and 380 nm as previously described (Grynkiewicz et al., 1985). Changes in [Ca2+]i (ΔF [Ca2+]) were measured by subtracting the highest peak of calcium in the first 50 seconds after addition of calcium minus the baseline.
Recombinant Protein Production
The GRA41 recombinant expression vector was generated by cloning the GRA41 coding sequence without the signal peptide into the pet28a (+) vector (EMD Biosciences) using In-fusion Cloning (Clontech). The GRA41 coding sequence was amplified using primers GRA41 pet28a.FOR and GRA41 pet28a.REV (Table 1) and inserted into the NdeI sites of the pet28a (+) vector. The resulting construct was verified by restriction digestion and sequencing. Rosetta (DE3) pLysS E. coli BL21 competent cells (Novagen) were transformed with purified vector according to the manufacturer’s protocol. Transformed cells were then grown at 37°C in LB media to an O.D. of 0.6 before inducing with 1 mM IPTG for 3 hours. After induction, cells were harvested by centrifugation at 10,000 × g’s for 20 minutes at 4°C. Pellets were stored at −80°C. Pellets were resuspended in ~ 100 mL of lysis buffer (50 mM Tris-HCl, pH 7.4, 10 mM MgSO4, 1 mM beta-mercaptoethanol, 0.1% Triton-X 100, 5% glycerol) before adding lysozyme to a final concentration of 50 μg/mL and incubating for ten minutes on ice. Samples were then sonicated before removing the insoluble portion by centrifugation at 14,000 × g’s for 30 minutes at room temperature. The supernatant was filtered through a 0.45 μm filter before purifying on a 5 ml HisTrap HP Nickel column (GE) on a NGC Quest FPLC (Bio-Rad). Fractions were collected and analyzed by SDS-PAGE and Western Blot to determine purity. Fractions positive for the his-tagged protein were combined and further purified using a HiLoad 26/600 Superdex 200 size exclusion column (GE).
Calcium Thermal Shift Assays
Differential scanning fluorimetry was utilized to measure the ability of calcium ions to bind to and stabilize the secondary structures of recombinant proteins as described in (Niesen et al., 2007) using recombinant CDPK3 (Gaji et al., 2015) as a positive control. N-terminally His tagged GRA41 (final concentration of 8 μM) or CDPK3 (final concentration of 3.5 μM) in 100 mM HEPES, pH 7.0, 150 mM NaCl was incubated with 4X Sypro Orange (Sigma Aldrich) in and CaCl2 in a two-fold dilution series ranging from 0–250 μM. Melt curve analysis was performed from 25 to 99°C with 1°C steps on a StepOne Plus Real-Time PCR System (Applied Biosystems). Data were analyzed in Prism (GraphPad) to determine melting temperatures for each condition.
Supplementary Material
Acknowledgments
We would like to thank Dr. Vern Carruthers for sharing the RhΔKu80 strain used for endogenous tagging and generation of the gra41 knockout as well as the gra2 knockout strains along with its parental and complemented strains and the MIC2 antibody, Dr. Peter Bradley for the GRA7 and ROP1 antibodies, Dr. Barry Stein for valuable training and advice in obtaining the transmission electron microscopy of the gra41 parental, knockout and complemented strains and Dr. Sanofar Abdeen for training and advice in recombinant protein purification. Also, we appreciate the intellectual input of Drs. Bill Sullivan, Rajshekhar Gaji and Stacey Gilk into our work. We also like to thank all members of Arrizabalaga lab for critical reading of the manuscript. This research has been funded by NIH grants R21AI119516, R01AI123457, and RO3AI101624 to GA. KAL has been funded by an NIH training grant AI060519, and a fellowship from the American Heart Association 16PRE27260042.
Footnotes
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: KAL KMMN SNJM GA. Performed the experiments: KAL KMMN IC. Analyzed the data: KAL KMMN IC GA. Contributed reagents/materials/analysis tools: KAL KMMN IC SNJM GA. Wrote the paper: KAL KMMN IC SNJM GA. The authors have no conflicts of interest to declare.
References
- Arrizabalaga G, Boothroyd JC. Role of calcium during Toxoplasma gondii invasion and egress. International journal for parasitology. 2004a;34:361–368. doi: 10.1016/j.ijpara.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Arrizabalaga G, Ruiz F, Moreno S, Boothroyd JC. Ionophore-resistant mutant of Toxoplasma gondii reveals involvement of a sodium/hydrogen exchanger in calcium regulation. J Cell Biol. 2004b;165:653–662. doi: 10.1083/jcb.200309097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudes D, Peck KR, Afifi MA, Beckers CJ, Joiner KA. Tandemly repeated genes encode nucleoside triphosphate hydrolase isoforms secreted into the parasitophorous vacuole of Toxoplasma gondii. The Journal of biological chemistry. 1994;269:29252–29260. [PubMed] [Google Scholar]
- Black MW, Arrizabalaga G, Boothroyd JC. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Molecular and cellular biology. 2000a;20:9399–9408. doi: 10.1128/mcb.20.24.9399-9408.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MW, Boothroyd JC. Lytic cycle of Toxoplasma gondii. Microbiology and molecular biology reviews : MMBR. 2000b;64:607–623. doi: 10.1128/mmbr.64.3.607-623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhomme A, Pingret L, Bonhomme P, Michel J, Balossier G, Lhotel M, et al. Subcellular calcium localization in Toxoplasma gondii by electron microscopy and by X-ray and electron energy loss spectroscopies. Microscopy research and technique. 1993;25:276–285. doi: 10.1002/jemt.1070250403. [DOI] [PubMed] [Google Scholar]
- Borges-Pereira L, Budu A, McKnight CA, Moore CA, Vella SA, Hortua Triana MA, et al. Calcium Signaling throughout the Toxoplasma gondii Lytic Cycle: A STUDY USING GENETICALLY ENCODED CALCIUM INDICATORS. The Journal of biological chemistry. 2015;290:26914–26926. doi: 10.1074/jbc.M115.652511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas LA, de Souza W, Attias M. Microscopic analysis of calcium ionophore activated egress of Toxoplasma gondii from the host cell. Veterinary parasitology. 2010;167:8–18. doi: 10.1016/j.vetpar.2009.09.051. [DOI] [PubMed] [Google Scholar]
- Carruthers VB, Giddings OK, Sibley LD. Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cellular microbiology. 1999a;1:225–235. doi: 10.1046/j.1462-5822.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- Carruthers VB, Sibley LD. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur J Cell Biol. 1997;73:114–123. [PubMed] [Google Scholar]
- Carruthers VB, Sibley LD. Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Molecular microbiology. 1999b;31:421–428. doi: 10.1046/j.1365-2958.1999.01174.x. [DOI] [PubMed] [Google Scholar]
- Cesbron-Delauw MF, Guy B, Torpier G, Pierce RJ, Lenzen G, Cesbron JY, et al. Molecular characterization of a 23-kilodalton major antigen secreted by Toxoplasma gondii. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:7537–7541. doi: 10.1073/pnas.86.19.7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AL, Moon AS, Bell HN, Huang AS, Vashisht AA, Toh JY, et al. Novel insights into the composition and function of the Toxoplasma IMC sutures. Cellular microbiology. 2017;19 doi: 10.1111/cmi.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, Carter D, Ullman B, Roos DS. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. The Journal of biological chemistry. 1996;271:14010–14019. doi: 10.1074/jbc.271.24.14010. [DOI] [PubMed] [Google Scholar]
- Dou Z, McGovern OL, Di Cristina M, Carruthers VB. Toxoplasma gondii ingests and digests host cytosolic proteins. mBio. 2014;5:e01188–01114. doi: 10.1128/mBio.01188-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JD, Ravindran S, Kim SK, Boothroyd JC. The Toxoplasma gondii dense granule protein GRA7 is phosphorylated upon invasion and forms an unexpected association with the rhoptry proteins ROP2 and ROP4. Infection and immunity. 2008;76:5853–5861. doi: 10.1128/IAI.01667-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer HG, Stachelhaus S, Sahm M, Meyer HE, Reichmann G. GRA7, an excretory 29 kDa Toxoplasma gondii dense granule antigen released by infected host cells. Molecular and biochemical parasitology. 1998;91:251–262. doi: 10.1016/s0166-6851(97)00227-2. [DOI] [PubMed] [Google Scholar]
- Foe IT, Child MA, Majmudar JD, Krishnamurthy S, van der Linden WA, Ward GE, et al. Global Analysis of Palmitoylated Proteins in Toxoplasma gondii. Cell Host Microbe. 2015;18:501–511. doi: 10.1016/j.chom.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruth IA, Arrizabalaga G. Toxoplasma gondii: induction of egress by the potassium ionophore nigericin. International journal for parasitology. 2007;37:1559–1567. doi: 10.1016/j.ijpara.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaji RY, Johnson DE, Treeck M, Wang M, Hudmon A, Arrizabalaga G. Phosphorylation of a Myosin Motor by TgCDPK3 Facilitates Rapid Initiation of Motility during Toxoplasma gondii egress. PLoS Pathog. 2015;11:e1005268. doi: 10.1371/journal.ppat.1005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, et al. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic acids research. 2008;36:D553–556. doi: 10.1093/nar/gkm981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Treeck M, Ehret E, Butz H, Garbuz T, Oswald BP, et al. A forward genetic screen reveals that calcium-dependent protein kinase 3 regulates egress in Toxoplasma. PLoS pathogens. 2012;8:e1003049. doi: 10.1371/journal.ppat.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazarini ML, Thomas AP, Pozzan T, Garcia CR. Calcium signaling in a low calcium environment: how the intracellular malaria parasite solves the problem. The Journal of cell biology. 2003;161:103–110. doi: 10.1083/jcb.200212130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold DA, Kaplan AD, Lis A, Bett GC, Rosowski EE, Cirelli KM, et al. The Toxoplasma Dense Granule Proteins GRA17 and GRA23 Mediate the Movement of Small Molecules between the Host and the Parasitophorous Vacuole. Cell host & microbe. 2015;17:642–652. doi: 10.1016/j.chom.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding CR, Meissner M. The inner membrane complex through development of Toxoplasma gondii and Plasmodium. Cellular microbiology. 2014;16:632–641. doi: 10.1111/cmi.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Mann T, Striepen B, Beckers CJ, Roos DS, Murray JM. Daughter cell assembly in the protozoan parasite Toxoplasma gondii. Molecular biology of the cell. 2002;13:593–606. doi: 10.1091/mbc.01-06-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh MH, Carruthers VB. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryotic cell. 2009;8:530–539. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelski DM, Remington JS. Toxoplasmosis in patients with cancer. Clin Infect Dis. 1993;17(Suppl 2):S423–435. doi: 10.1093/clinids/17.supplement_2.s423. [DOI] [PubMed] [Google Scholar]
- Jacobs D, Dubremetz JF, Loyens A, Bosman F, Saman E. Identification and heterologous expression of a new dense granule protein (GRA7) from Toxoplasma gondii. Molecular and biochemical parasitology. 1998;91:237–249. doi: 10.1016/s0166-6851(97)00204-1. [DOI] [PubMed] [Google Scholar]
- Kasper LH, Bradley MS, Pfefferkorn ER. Identification of stage-specific sporozoite antigens of Toxoplasma gondii by monoclonal antibodies. Journal of immunology. 1984;132:443–449. [PubMed] [Google Scholar]
- Kim K, Soldati D, Boothroyd JC. Gene replacement in Toxoplasma gondii with chloramphenicol acetyltransferase as selectable marker. Science. 1993;262:911–914. doi: 10.1126/science.8235614. [DOI] [PubMed] [Google Scholar]
- Lavine MD, Knoll LJ, Rooney PJ, Arrizabalaga G. A Toxoplasma gondii mutant defective in responding to calcium fluxes shows reduced in vivo pathogenicity. Molecular and biochemical parasitology. 2007;155:113–122. doi: 10.1016/j.molbiopara.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J, Bittame A, Massera C, Vasseur V, Effantin G, Valat A, et al. Intravacuolar Membranes Regulate CD8 T Cell Recognition of Membrane-Bound Toxoplasma gondii Protective Antigen. Cell reports. 2015;13:2273–2286. doi: 10.1016/j.celrep.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Lovett JL, Sibley LD. Intracellular calcium stores in Toxoplasma gondii govern invasion of host cells. Journal of cell science. 2003;116:3009–3016. doi: 10.1242/jcs.00596. [DOI] [PubMed] [Google Scholar]
- Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clin Infect Dis. 1992;15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- Mazzillo FF, Shapiro K, Silver MW. A new pathogen transmission mechanism in the ocean: the case of sea otter exposure to the land-parasite Toxoplasma gondii. PloS one. 2013;8:e82477. doi: 10.1371/journal.pone.0082477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier C, Cesbron-Delauw MF. Toxoplasma secretory granules: one population or more? Trends in parasitology. 2015;31:60–71. doi: 10.1016/j.pt.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Mercier C, Cesbron-Delauw MF, Sibley LD. The amphipathic alpha helices of the toxoplasma protein GRA2 mediate post-secretory membrane association. J Cell Sci. 1998a;111(Pt 15):2171–2180. doi: 10.1242/jcs.111.15.2171. [DOI] [PubMed] [Google Scholar]
- Mercier C, Dubremetz JF, Rauscher B, Lecordier L, Sibley LD, Cesbron-Delauw MF. Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Molecular biology of the cell. 2002;13:2397–2409. doi: 10.1091/mbc.E02-01-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier C, Howe DK, Mordue D, Lingnau M, Sibley LD. Targeted disruption of the GRA2 locus in Toxoplasma gondii decreases acute virulence in mice. Infection and immunity. 1998b;66:4176–4182. doi: 10.1128/iai.66.9.4176-4182.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda K, Pace DA, Cintron R, Rodrigues JC, Fang J, Smith A, et al. Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole. Molecular microbiology. 2010;76:1358–1375. doi: 10.1111/j.1365-2958.2010.07165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon R, Frixione E. Ca(2+)-dependence of conoid extrusion in Toxoplasma gondii tachyzoites. The Journal of eukaryotic microbiology. 1996;43:120–127. doi: 10.1111/j.1550-7408.1996.tb04491.x. [DOI] [PubMed] [Google Scholar]
- Moreno SN, Ayong L, Pace DA. Calcium storage and function in apicomplexan parasites. Essays in biochemistry. 2011;51:97–110. doi: 10.1042/bse0510097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno SN, Zhong L. Acidocalcisomes in Toxoplasma gondii tachyzoites. Biochem J. 1996;313(Pt 2):655–659. doi: 10.1042/bj3130655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlon-Guyot J, Berry L, Chen CT, Gubbels MJ, Lebrun M, Daher W. The Toxoplasma gondii calcium-dependent protein kinase 7 is involved in early steps of parasite division and is crucial for parasite survival. Cellular microbiology. 2014;16:95–114. doi: 10.1111/cmi.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy R, Manning TJ, Beckers CJ. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. The Journal of biological chemistry. 2001;276:41492–41501. doi: 10.1074/jbc.M106154200. [DOI] [PubMed] [Google Scholar]
- Muniz-Hernandez S, Carmen MG, Mondragon M, Mercier C, Cesbron MF, Mondragon-Gonzalez SL, et al. Contribution of the residual body in the spatial organization of Toxoplasma gondii tachyzoites within the parasitophorous vacuole. J Biomed Biotechnol. 2011;2011:473983. doi: 10.1155/2011/473983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Berkowitz LR, Orringer E, Levy L, Gabel SA, London RE. Cytosolic free calcium levels in sickle red blood cells. Blood. 1987;69:1469–1474. [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Okada T, Marmansari D, Li ZM, Adilbish A, Canko S, Ueno A, et al. A novel dense granule protein, GRA22, is involved in regulating parasite egress in Toxoplasma gondii. Molecular and biochemical parasitology. 2013;189:5–13. doi: 10.1016/j.molbiopara.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Oray M, Ozdal PC, Cebeci Z, Kir N, Tugal-Tutkun I. Fulminant Ocular Toxoplasmosis: The Hazards of Corticosteroid Monotherapy. Ocul Immunol Inflamm. 2015:1–10. doi: 10.3109/09273948.2015.1057599. [DOI] [PubMed] [Google Scholar]
- Pace DA, McKnight CA, Liu J, Jimenez V, Moreno SN. Calcium entry in Toxoplasma gondii and its enhancing effect of invasion-linked traits. The Journal of biological chemistry. 2014;289:19637–19647. doi: 10.1074/jbc.M114.565390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas G, Roussos N, Falagas ME. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. International journal for parasitology. 2009;39:1385–1394. doi: 10.1016/j.ijpara.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn ER, Bzik DJ, Honsinger CP. Toxoplasma gondii: mechanism of the parasitostatic action of 6-thioxanthine. Experimental parasitology. 2001;99:235–243. doi: 10.1006/expr.2001.4673. [DOI] [PubMed] [Google Scholar]
- Pingret L, Millot JM, Sharonov S, Bonhomme A, Manfait M, Pinon JM. Relationship between intracellular free calcium concentrations and the intracellular development of Toxoplasma gondii. J Histochem Cytochem. 1996;44:1123–1129. doi: 10.1177/44.10.8813077. [DOI] [PubMed] [Google Scholar]
- Roiko MS, Svezhova N, Carruthers VB. Acidification Activates Toxoplasma gondii Motility and Egress by Enhancing Protein Secretion and Cytolytic Activity. PLoS pathogens. 2014;10:e1004488. doi: 10.1371/journal.ppat.1004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome ME, Beck JR, Turetzky JM, Webster P, Bradley PJ. Intervacuolar transport and unique topology of GRA14, a novel dense granule protein in Toxoplasma gondii. Infection and immunity. 2008;76:4865–4875. doi: 10.1128/IAI.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Miguel JM, Gutierrez-Exposito D, Aguado-Martinez A, Gonzalez-Zotes E, Pereira-Bueno J, Gomez-Bautista M, et al. Effect of Different Ecosystems and Management Practices on Toxoplasma gondii and Neospora Caninum Infections in Wild Ruminants in Spain. J Wildl Dis. 2016;52:293–300. doi: 10.7589/2015-07-176. [DOI] [PubMed] [Google Scholar]
- Schwab JC, Beckers CJ, Joiner KA. The parasitophorous vacuole membrane surrounding intracellular Toxoplasma gondii functions as a molecular sieve. Proc Natl Acad Sci U S A. 1994;91:509–513. doi: 10.1073/pnas.91.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzman JD, Krug EC. Toxoplasma gondii: characterization of monoclonal antibodies that recognize rhoptries. Experimental parasitology. 1989;68:74–82. doi: 10.1016/0014-4894(89)90010-6. [DOI] [PubMed] [Google Scholar]
- Slavin MA, Meyers JD, Remington JS, Hackman RC. Toxoplasma gondii infection in marrow transplant recipients: a 20 year experience. Bone Marrow Transplant. 1994;13:549–557. [PubMed] [Google Scholar]
- Soldati D, Boothroyd JC. Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science. 1993;260:349–352. doi: 10.1126/science.8469986. [DOI] [PubMed] [Google Scholar]
- Stommel EW, Cho E, Steide JA, Seguin R, Barchowsky A, Schwartzman JD, Kasper LH. Identification and role of thiols in Toxoplasma gondii egress. Experimental biology and medicine. 2001;226:229–236. doi: 10.1177/153537020122600311. [DOI] [PubMed] [Google Scholar]
- Stommel EW, Ely KH, Schwartzman JD, Kasper LH. Toxoplasma gondii: dithiol-induced Ca2+ flux causes egress of parasites from the parasitophorous vacuole. Experimental parasitology. 1997;87:88–97. doi: 10.1006/expr.1997.4187. [DOI] [PubMed] [Google Scholar]
- Suss-Toby E, Zimmerberg J, Ward GE. Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc Natl Acad Sci U S A. 1996;93:8413–8418. doi: 10.1073/pnas.93.16.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varberg JM, Padgett LR, Arrizabalaga G, Sullivan WJ., Jr TgATAT-Mediated alpha-Tubulin Acetylation Is Required for Division of the Protozoan Parasite Toxoplasma gondii. mSphere. 2016;1 doi: 10.1128/mSphere.00088-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, Remington JS, Stagno S, Reynolds DW. Development of adverse sequelae in children born with subclinical congenital Toxoplasma infection. Pediatrics. 1980;66:767–774. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.