

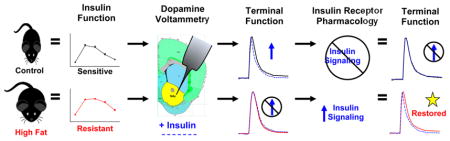
Abstract
Systemically released insulin crosses the blood-brain barrier and binds to insulin receptors on several neural cell types, including dopaminergic neurons. Insulin has been shown to decrease dopamine neuron firing in the ventral tegmental area (VTA), but potentiate release and reuptake at dopamine terminals in the nucleus accumbens (NAc). Here we show that prolonged consumption of a high fat diet blocks insulin’s effects in the NAc, but insulin’s effects are restored by inhibiting protein tyrosine phosphatase 1B, which supports insulin receptor signaling. Mice fed a high fat diet (60% kcals from fat) displayed significantly higher fasting blood glucose 160 mg/dL, compared to 101 mg/dL for control-diet-fed mice, and high-fat-diet-fed mice showed reduced blood glucose clearance after an intraperitoneal glucose tolerance test. Using fast scan cyclic voltammetry to measure electrically evoked dopamine in brain slices containing the NAc core, high-fat-diet-fed mice exhibited slower dopamine reuptake compared to control-diet-fed mice (2.2 ± 0.1 and 2.67 ± 0.15 μM/s, respectively). Moreover, glucose clearance rate was negatively correlated with Vmax. Insulin (10 nM to 1 μM) dose dependently increased reuptake rates in control-diet-fed mice compared with in the high-fat-diet group; however, the small molecule insulin receptor sensitizing agent, TCS 401 (300 nM), restored reuptake in high-fat-diet-fed mice to control-diet levels, and a small molecule inhibitor of the insulin receptor, BMS 536924 (300 nM), attenuated reuptake, similar to high-fat-diet-fed mice. These data show that a high-fat diet impairs dopamine reuptake by attenuating insulin signaling at dopamine terminals.
Keywords: Voltammetry, high fat diet, insulin resistance, obesity, dopamine, DAT
Graphical Abstract
INTRODUCTION
Prolonged consumption of a high-fat diet is known to cause obesity and type 2 diabetes, which are characterized by insulin resistance that impacts both the brain and peripheral tissues.1–3 An emerging body of evidence has revealed that insulin plays multiple roles in the brain, expanding on its well-known function to regulate glucose homeostasis and energy balance via the hypothalamus.4–6 Recent studies have shown that insulin modulates dopamine neurotransmission by altering the excitability of dopamine neuron cell bodies in the ventral tegmental area (VTA),7–9 in addition to augmenting dopamine release in striatal terminal fields.10 Behavioral outcomes associated with insulin’s effects on the dopamine system, however, differ based on the location of insulin infusion, whether into dopamine terminal fields in the striatum or cell bodies in the VTA. Insulin infused into the VTA reduced consumption of a sweetened high-fat diet8 and attenuated sucrose self-administration,11,12 whereas blocking insulin’s actions at dopamine terminal fields with microinjections of insulin antibodies decreased preference for a sucrose solution, suggesting that insulin in the nucleus accumbens (NAc) helps attribute flavor preference.10 Taking these findings into consideration, it appears that insulin signaling in the VTA presumably aids in the termination of food intake in concert with satiety mechanisms initiated by insulin and other glucose sensing neurons in the hypothalamus,13 while insulin signaling at dopamine terminals is postulated to contribute to the rewarding properties of food consumption.10 This scenario supports a dual hypothesis for insulin to aid in satiety, but also increase reward salience, possibly promoting future seeking of palatable food.
Insulin receptors are found throughout the brain, with the greatest density in the choroid plexus, followed by the hypothalamus, amygdala, and the NAc.14 Homeostatic regulation of blood glucose and food intake by insulin in the hypothalamus is well documented,4–6 but insulin may assume a nontraditional role in the NAc where it helps regulate the rewarding characteristics of food intake by acting on dopamine terminals.10 Interestingly, autoradiography shows binding of [125I]insulin to its receptors is nearly doubled in the NAc compared to the caudate putamen of rats.14 Dopamine signaling in the NAc helps encode drug reinforcement as well as incentive salience,15 and a high density of insulin receptors in the NAc suggests that insulin may play a role in the initiation of appetitive behaviors. Indeed, insulin has been shown to increase preference for a sucrose solution,10 and hypoinsulinemic conditions can reduce drug self-administration.16 However, food restriction, which lowers circulating insulin levels, has also been shown to increase drug self-administration.17–19 Mechanisms underlying both increased and decreased drug and food self-administration following hypoinsulinemia are poorly understood, and highlight the need to understand how insulin functions in discrete regions of the mesolimbic dopamine system. However, because administration of insulin antibodies enhances flavor preference when administered at dopamine terminals,10 the regulation of dopamine terminals by insulin needs to be further examined, specifically in the NAc.
A well-documented role of insulin on dopamine terminals is to increase dopamine reuptake by enhancing dopamine transporter (DAT) membrane levels and function.20–23 Insulin signaling at dopamine terminals has been investigated using streptozotocin (STZ) to produce hypoinsulinemic rats. Dopamine efflux in response to amphetamine is markedly reduced in the absence of insulin in these rats.22,24 Moreover, insulin has been shown to contribute to DAT function by increasing DAT recruitment to the plasma membrane, and enhancing dopamine clearance through downstream activation of Akt.25 Attenuated Akt activity following a high-fat diet reduced membrane DAT levels and function, but viral restoration of Akt normalized DAT activity and reduced hyperphagia.25 However, hypoinsulinemic conditions have been shown in separate studies to either attenuate26 or enhance16 [3H]-dopamine clearance in striatal synaptosomes.26 Discrepancies in these studies highlight a need for further work, and the use of more sophisticated measurements of dopamine neurotransmission, like fast scan cyclic voltammetry. Additionally, inhibition of downstream insulin signaling also blocked cocaine-induced [3H]-dopamine overflow from NAc brain slices.23 These findings indicate that insulin promotes DAT function by increasing reuptake and improving drug-induced dopamine efflux through the DAT. Impairments in DAT function have been observed after prolonged consumption of a high-fat diet, which leads to insulin resistance.27,28 Recent studies have shown that obesogenic weight gain cause by a high-fat diet reduces dopamine reuptake in rats29,30 and mice,31 but it is not known whether impaired dopamine uptake observed with obesity is directly caused by a reduction in insulin sensitivity. It is likely that changes in insulin receptor function contribute to dysregulated dopamine neurotransmission after the development of high-fat-diet-induced insulin resistance.
The purpose of this study was to further characterize how high-fat-diet-induced insulin resistance alters dopamine terminal function, to expand on previous reports,10 and to investigate whether the onset of systemic insulin resistance coincides with reduced neuronal insulin function. Additionally, we wanted to test if deficits in dopamine terminal function caused by high-fat-diet- induced insulin resistance could be reversed by improving insulin signaling. We show that mice fed a high-fat diet for six weeks were systemically insulin-resistant, which correlated with deficits in dopamine terminal function. A high-fat diet blocked insulin’s ability to facilitate dopamine release and reuptake in the NAc, and we show that restored insulin receptor signaling in high-fat diet-fed mice reversed deficits in dopamine terminal function.
RESULTS AND DISCUSSION
Six Week Access to a High-Fat Diet Increased Body Weight and Impaired Glucose Clearance
Clinical obesity is characterized by obesogenic weight gain that coincides with glucose intolerance, and ultimately the development of insulin resistance. To verify that our model replicates this clinical phenotype, we provided mice with a high-fat diet which led to obesogenic weight gain (Figure 1A, B), and then we conducted intraperitoneal glucose tolerance tests (IPGTT) to confirm the onset of insulin resistance (Figure 1A, C). IPGTTs offer an indirect measure of insulin receptor function that reliably correlates with insulin resistance.32 Mice with access to high fat gained ~2.5 times more weight than control-chow-fed littermates over the 6 weeks, replicating the weight gain observed in previous studies, which resulted in hyper-insulinemia, 33 hyperleptinemia,34 and elevated serum triglycerides. 31 Significant differences in body weight were detected as early as 3 weeks (p < 0.01), with final averaged body weights of 24.88 ± 1.16 and 31.36 ± 3.06 g for control and high-fat-fed mice, respectively (p < 0.001) (Figure 1B). Two-way analysis of variance (ANOVA) detected main effects of time (F18,324 = 127.36, p < 0.0001) and diet (F1,324 = 15.33, p = 0.001) with a time × diet interaction (F18,324 = 27.09, p < 0.0001). A significant elevation in fasting blood glucose was also observed in high fat mice (160.9 ± 8.0 mg/dL; n = 15) compared with controls (101.5 ± 5.1 mg/dL; n = 12) (p < 0.001) (Figure 1C), and high-fat-fed mice displayed significantly reduced glucose clearance compared to chow-fed littermates with main effects of time (F4,112 = 241.36, p < 0.0001), diet (F1,112 = 37.65, p < 0.0001), and a time × diet interaction (F4,112 = 6.53, p < 0.0001) (Figure 1D). Area under the curve (AUC) analysis showed significantly impaired glucose clearance in the high-fat-diet-fed group (p < 0.001) (Figure 1E). These data indicate the onset of systemic insulin resistance in mice after 6 week access to a high-fat diet, verifying an insulin-resistant phenotype in our preclinical model of diet-induced obesity.
Figure 1.

Increased body weight and impaired blood glucose clearance. (A) Experimental design showing mice with 6 week dietary exposure to high-fat diet (HF) or a control chow diet, followed by an i.p. glucose tolerance test (IPGTT) which measures glucose clearance to identify insulin sensitivity, and slice voltammetry (Volt) in the nucleus accumbens core (NAc). Access to a high-fat diet significantly increases body weight (B) and blood glucose levels following a 5 h fast (C), compared to controls. Blood glucose clearance was also significantly impaired in high-fat-diet-fed mice, compared to controls, following a bolus injection of glucose (2g/kg) (D), quantified by area under the curve (AUC) (E). (*p < 0.05, **p < 0.01, ***p < 0.001.)
Insulin-Resistant High-Fat-Diet-Fed Mice Have Reduced Dopamine Reuptake
After establishing a clinically obese phenotype, we next sought to determine if systemic insulin resistance coincided with impaired dopamine function in the brain. We used fast scan cyclic voltammetry in NAc containing slices to identify differences in dopamine release and reuptake between control chow-fed mice and high fat-fed insulin-resistant mice. No difference in dopamine release, evoked by a single pulse stimulations, was detected between groups (1.34 ± 0.12 μM, control (n = 10); 1.35 ± 0.13 μM, high-fat diet (n = 15) (Figure 2A); however, a significant positive correlation between dopamine release and glucose AUC was identified in control mice, (r2 = 0.629; p = 0.006) (Figure 2B) indicating that dopamine release increases with reduced glucose clearance. This correlation showed a relationship between control mice with high sensitivity that had lower dopamine release, but control mice with similar insulin sensitivity as high-fat-diet-fed mice had higher dopamine release, indicating that evoked dopamine release is lower in control mice with superior insulin sensitivity. Additionally, a significant reduction in the maximal rate of dopamine reuptake (Vmax) was observed in insulin-resistant high-fat-fed mice (2.20 ± 0.10 μM/s) versus controls (2.67 ± 0.15 μM/s) (p > 0.01) (Figure 2C). Moreover, Vmax was negatively correlated with AUC in high-fat-diet-fed mice (r2 = −0.461, p = 0.005), a correlation that was less robust in control mice (r2 = −0.26, p = 0.09) (Figure 2D), indicating that maximal dopamine clearance occurs with greater insulin sensitivity. These findings suggest that systemic measures of glucose clearance and overall insulin function may predict dopamine release and reuptake in the NAc, and under basal conditions, high-fat-fed insulin-resistant mice have reduced DAT function. Because the DAT clears extracellular dopamine throughout the rising phase of dopamine release, as well as rapidly reducing peak dopamine to basal levels, DAT function associated with insulin sensitivity can alter peak synaptic dopamine levels and dopamine reuptake. A correlation between lower evoked dopamine release and insulin sensitivity putatively corresponds with constitutive insulin signaling that supports the recruitment and maintenance of DATs at the plasma membrane of control insulin sensitive mice. Fasting brain levels of insulin have been reported to be between 20 and 90 pM,35,36 but postprandial systemic insulin reaches 60–100 nM.37 Central insulin levels shift corresponding with fasting and fed systemic insulin levels,35 but overall concentrations are ~25% of systemic circulation,35,38,39 suggesting that postprandial insulin can reach 15–30 nM in the brain. We did not measure circulating insulin, but can estimate brain insulin levels in our mice to be in the mid-picomolar to low-nanomolar range. This insulin range appears to be sufficient to recruit and maintain DATs at the plasma membrane in control insulin sensitive mice. However, since insulin resistance coincides with hyperinsulinemia33 and reduced insulin receptor signaling, insulin-resistant mice are predicted to have reduced DAT function, which corresponds with reports of reduced membrane DAT levels following exposure to high fat.25,31,40 This is consistent with our data showing reduced DAT function in high fat-fed insulin-resistant mice, and demonstrates that prolonged consumption of a high-fat diet interferes with dopamine reuptake and insulin enhanced dopamine release in the NAc.
Figure 2.

Dopamine release and reuptake. Dopamine (DA) release (A) was similar between dietary groups; however, greater DA release was correlated with reduced glucose clearance in chow-fed control mice, as indicated by a greater area under the curve (AUC) showing slower glucose clearance following an i.p. glucose tolerance test (IPGTT) (B). The maximal rate of DA reuptake (Vmax) was lower in mice fed a high-fat diet (C), and negatively correlated with glucose clearance (D). Insulin increased DA release in control mice, but not in slices from high-fat-fed mice, measured as percent change from baseline (BL) DA release (E), and visualized with representative line traces following 30 nM insulin (F). (*p < 0.05, ***p < 0.001.)
Given the relationship between insulin sensitivity and dopamine release and reuptake, along with reports that insulin increases dopamine release at dopamine terminals in the striatum,10 we bath applied insulin over NAc slices in a half-log stepwise fashion (10 nM to 1 μM) to examine if insulin-resistant high-fat-fed mice were unresponsive to insulin’s effect on dopamine release. We observed a significant group effect on dopamine release (F1,96 = 6.18, p = 0.02), with insulin increasing dopamine release in the chow-fed control mice only (n = 9) (Figure 2E), as depicted by representative line traces showing increase dopamine release in control, but not high-fat-fed (n = 15), mice after the application of 30 nM insulin (Figure 2F). These data corroborate previous reports showing that insulin augments dopamine release, potentially by acting on cholinergic interneurons in the striatum.10 Interestingly, we observed an effect of insulin on dopamine release up to 1 μM, which is consistent with studies showing that neuronal insulin receptors do not desensitize like systemic insulin receptors, even in the presence of more than 800 nM insulin,41,42 but contrasts with findings from Stouffer et al., showing diminishing effects above 30 nM. However, our data show that insulin functions at dopamine terminals to increase dopamine release, and that high-fat-fed insulin-resistant mice have reduced basal reuptake. Accordingly, high-fat-diet-induced insulin resistance blocks insulin’s actions on dopamine release, which compliments behavioral studies that show increased sensitivity to dopamine agonists,43–45 and suggests that high-fat-diet-induced insulin resistance is another mechanism that regulates dopamine signaling. Our findings also suggest that neuronal insulin resistance coincides with systemic markers of glucose intolerance providing a complementary measure of dopamine system function to behavioral pharmacology tests.
Blocking Downstream Insulin Signaling in Control Mice Reduced Dopamine Reuptake Resembling Mice Fed a High-Fat Diet
To further examine the results of insulin resistance at dopamine terminals, we used wortmannin, a PI3-kinase (PI3K) inhibitor, to test whether blocking downstream insulin signaling in chow-fed control mice recapitulates the dopaminergic phenotype of high-fat-fed insulin-resistant mice. The insulin receptor-activated intracellular signaling cascade includes activation of PI3K, which phosphorylates Akt, initiating numerous cellular responses.46,47 Importantly, PI3K activation promotes DAT recruitment to the plasma membrane. 48 Inhibition of PI3K causes DAT internalization and reduces amphetamine-induced dopamine efflux,48,49 which is achieved through reverse transport of dopamine through DATs at the plasma membrane. With respect to diet-induced obesity, a high-fat diet has been shown to reduce Akt phosphorylation leading to hyperphagia, presumably caused by insulin resistance.25 Moreover, chronoamperometry measurements showed reduced clearance of dopamine that was injected into the striatum of obese rats, indicating reduced DAT function.25 We tested whether similar DAT disruptions were present at dopamine terminals in NAc of obese high-fat-fed mice, and whether this was caused by impaired insulin signaling.
To determine the effect of insulin signaling on DAT function at dopamine terminals, we added 1 μM insulin, which produced the greatest effect on dopamine release (Figure 2E), to NAc containing slices from control (n = 9) and insulin-resistant high-fat-fed mice (n = 15). In chow-fed controls, 1 μM insulin increased Vmax by an average of 46 ± 6.8%, indicated by reduced AUC and a sharp increase in the downward slope of the top third of the representative line plot depicting dopamine release and reuptake (Figure 3A). The representative line plots are normalized to 100% peak height for better visual comparisons between groups. Mice fed a high-fat diet showed no change in Vmax after 1 μM insulin (Figure 3B). We then pretreated slices from control mice with wortmannin (n = 7) and blocked insulin-enhanced dopamine reuptake (Figure 3C). Moreover, wortmannin blocked the dose dependent increase in Vmax caused by insulin (Figure 3D), resembling the lack of response shown by slices from insulin-resistant high fat-fed mice. Overall, main effects of insulin dose (F5,100 = 15.93, p < 0.0001), diet (F1,100 = 12.98, p = 0.0018), and a dose × diet interaction (F5,100 = 10.89, p < 0.0001) were observed on Vmax after half-log additions of insulin, with Tukey’s posthoc analysis identifying significantly increased Vmax in control mice at 0.3 and 1 μM (p < 0.001) (Figure 3D). The inset to Figure 3D shows a main effect of diet on Vmax at physiological concentrations of insulin (F1,38 = 4.64, p = 0.044), with Tukey’s posthoc analysis identifying a significant Vmax increase with 30 nM insulin in control mice compared to mice fed a high-fat diet. Our findings show that insulin increases Vmax at physiological concentrations, with maximal effects observed at supraphysiological doses. Inhibiting downstream signaling of the insulin receptor with wortmannin, however, blocked insulin’s effect on dopamine reuptake. Interfering with insulin signaling at this step in the downstream cascade replicates ex vivo tissue studies from obese diabetic patients that presented impaired Akt activation, a direct result of PI3K activity.46 Ex vivo tissue studies from insulin-resistant patients, which better reflect our model, show that deficiencies in the insulin signaling pathway occur upstream to alterations in these broad signaling kinases, starting with phosphorylation of the insulin receptor substrates,46 which occur subsequent to insulin receptor activation50,51
Figure 3.

Insulin enhanced dopamine clearance is absent in high-fat diet-fed mice. Representative line traces and color plots depicting evoked DA release normalized to peak height show that insulin (1 μM), dashed lines (–––), increases Vmax compared to baseline, solid lines (—), in NAc slices from control chow-fed mice (A), but not in slices from high-fat (HF)-fed mice (B), or in slices from control mice pretreated with 100 nM wortmannin (Wort), a PI3-kinase inhibitor that blocks downstream insulin signaling (C). Below the representative line traces are corresponding color plots showing the oxidation and reductions peaks of dopamine at +0.6 and −0.4 V, respectively. (D) Slices from high fat-fed mice and slices from control mice pretreated with Wort were resistant to insulin’s effect on Vmax in slices from control mice, and insulin significantly increased Vmax at physiological concentrations in control, but not high-fat-fed (E, inset). (*p < 0.05, ***p < 0.001.)
Improving Insulin Receptor Signaling in High-Fat-Fed Mice Restored Insulin’s Effects on Dopamine Release and Reuptake
In order to confirm that deficits in dopamine terminal function are a direct result of insulin receptor activity, and test if high-fat diet-induced impairments can be reversed, we used two different approaches. First, in addition to blocking downstream insulin signaling in chow-fed control mice, which resembles an obese diabetic phenotype, we confirmed the role of insulin receptor activation on dopamine neurotransmission, which was lacking in high-fat-fed insulin-resistant mice, by using a small molecule inhibitor of the insulin receptor, BMS 536924, in controls. Second, to support the hypothesis that insulin resistance drives deficits in dopamine terminal function in the NAc, we used the protein tyrosine phosphatase 1B (PTP1B) inhibitor, TCS 401 (300 nM), to sensitize the insulin signaling pathway. PTP1B effectively removes the “brakes” on the insulin signaling pathway by preserving phosphorylation at tyrosine residues. Phosphorylation of tyrosine residues on insulin receptor substrates (IRS1 and 2) propagates downstream effects by activating PI3K and Akt,51 but dephosphorylation of tyrosine residues by PTP1B or increased phosphorylation of serine residues, as observed with obesity and other inflammatory disorders, effectively blocks insulin signaling, contributing to insulin resistance.51–53
Application of BMS 536924 (300 nM) to NAc containing slices from control mice (n = 6) did not significantly reduce dopamine release from baseline (92.3% ± 4.7%), but effectively blocked the significant increase in dopamine release observed with 500 nM insulin (125.2% ± 6.2%; p < 0.05, p < 0.01, and p < 0.01 compared to baseline, BMS, and BMS+Insulin, respectively) (Figure 4A). Similarly, BMS pretreatment blocked the significant increase in Vmax (137.3% ± 7.4%; p < 0.01 compared to all other groups) observed following 500 nM insulin (Figure 4C). These findings support previous reports of insulin augmenting dopamine release10 and enhancement of DAT function by increasing DAT membrane levels and dopamine reuptake.20,25,54 Additionally, we show that improving insulin signaling in high-fat insulin-resistant mice (n = 6) by inhibiting PTP1B restores dopamine terminal function. Application of TCS 401 (300 nM) increased dopamine release in response to 500 nM insulin (p = 0.09; overall one-way ANOVA model of dopamine release) (Figure 4B), and restored Vmax responsiveness to insulin (p < 0.05; compared to high fat baseline, high fat + insulin, and high fat + TCS groups) (Figure 4D). These changes are likely due to the DAT’s responsiveness to enhance insulin signaling in slices from these mice. High fat-fed mice have an increased ratio of cytosolic to membrane DAT, but greater overall DAT in tissue lysate from the NAc.31 Improving insulin signaling in high fat insulin-resistant mice putatively supports redistribution of the DAT to the plasma membrane where a functional increase in DAT activity can be measured with voltammetry. To rule out the possibility that high-fat mice simply had reduced insulin receptor content in the NAc, we used a quantitative ELISA to measure total insulin receptor protein in the NAc (n = 7, controls; n = 8, high fat). High-fat-fed mice showed no reduction in insulin receptor content (Figure 4E), indicating that our observations in the high-fat mice were not due to reduced receptor levels.
Figure 4.

Insulin receptor function, not receptor number, modulates dopamine reuptake. Pretreating slices from chow-fed control mice with BMS 536924 (300 nM), a small molecule inhibitor of insulin receptor autophosphorylation blocks insulin-induced increases on dopamine (DA) release (A) and Vmax (C), recreating the high-fat (HF)-fed phenotype. Conversely, insulin’s effect on Vmax and DA release can be restored in slices from high fat-fed mice by pretreating them with TCS 401, a protein tyrosine 1B inhibitor that sensitizes downstream insulin signaling (B, D). No difference in NAc insulin receptor protein content was found between controls and high fat mice (E). (*p < 0.05; **p < 0.01.)
Overall, our findings show that restoring insulin signaling at dopamine terminals of high fat-fed insulin-resistant mice improved deficits in dopamine terminal function. These deficits presumably stem from a reduction in insulin receptor function, consistent with a high fat insulin-resistant phenotype. Our study aligns well with previous reports of insulin signaling on dopamine neurons showing insulin-induced increases in dopamine terminal function, which coincided with enhanced palatable food preference.10 Together with studies that support a reduction in palatable food intake when insulin is applied to the VTA,8 it appears that insulin may have a dual role to aid in satiety, but also influence reward salience, possibly promoting future seeking of palatable food. It is difficult, however, to behaviorally delineate this dual role of insulin on dopaminergic neurotransmission in chronic feeding models that promote insulin resistance. Because postprandial insulin helps dampen dopaminergic output by the VTA, but enhances dopamine reuptake at terminals, insulin resistance would dysregulate the mesolimbic dopamine system preserving dopaminergic output in response to a meal, which would have normally been quenched by insulin. The fact that insulin signaling enhanced dopamine release in the NAc, putatively increasing reward salience of the meal, suggests that exposure to palatable food while insulin sensitive promotes future consumption of that food. Conversely, exposure to palatable food when insulin-resistant would prevent appetitive consumption, but associations with palatable food intake prior to the onset of insulin resistance would be preserved. To better understand the impact of insulin resistance on satiety, food seeking, and decreased palatability of novel foods (often observed in obese children55,56), future studies should focus on behavioral outcomes of insulin resistance on dopamine neurotransmission after the onset of obesity, and whether therapeutics targeted at sensitizing an insulin-resistant dopamine system have efficacy as obesity treatments.
METHODS
Animals and Diet
C57BL/6 mice originally purchased from Jackson Laboratories (Bar Harbor, ME) were bred in house to produce 6 week old male mice for all experiments. Mice were housed 2–3 per cage depending on litter size, maintained on a 12 h light/dark cycle with access to water, and fed either a control diet consisting of standard lab chow (3.46 kcal/g) (5P00, Prolab RMH 3000; LabDiet, St. Louis, MO) or a high-fat diet (5.24 kcal/g) (DIO, D12492; Research Diets, New Brunswick, NJ) containing 14% and 60% total kcal from fat, respectively. Mice remained on respective diets for 6 weeks prior to experimental tests (Figure 1A). All experiments were in compliance with the Wake Forest Animal Care and Use Committee.
Intraperitoneal Glucose Tolerance Test
At the end of the 6 week dietary paradigm, all mice underwent an intraperitoneal glucose tolerance test (IPGTT) to measure glucose clearance. Briefly, mice were placed in clean cages with access to water, but no food, for a 5 h fast. Blood glucose levels were then measured from the tail vein using a TRUEtrack glucometer and blood glucose test strips (Rite Aid Pharmacy, Camp Hill, PA) to establish fasting blood glucose levels. An i.p. bolus of glucose (2g/kg in 20% w/v saline) was delivered, and repeat blood glucose measurements were conducted 15, 30, 60, and 120 min thereafter. This procedure provides a reliable indirect measurement of insulin function by examining the disappearance of blood glucose over time following a single bolus of intraperitoneal i.p. glucose.32
Fast Scan Cyclic Voltammetry
Fast scan cyclic voltammetry was used to identify changes in dopamine signaling within the NAc core. Voltammetry experiments were conducted after 6 week access to the control or high-fat diets, and 5 days following the IPGTT to allow the mice to reacclimate to their respective diets. All voltammetry experiments began ~3 h into the light cycle, and were executed and modeled as previously described.57,58 Briefly, mice were rendered unconscious using 5% isoflurane, decapitated, and the brain swiftly removed. The brain was hemisected with one hemisphere submerged in ice cold artificial cerebral spinal fluid (aCSF) for slicing on a vibratome (Leica Microsystems Inc.; Buffalo Grove, IL), and the other was used to excise NAc tissue for insulin receptor quantification. Then 300 μm brain slices containing the NAc (from +1.34 to +0.74 from bregma) were transferred to the voltammetry chamber and allowed to equilibrate for 60 min at 32.5 °C while being bathed in oxygenated (95% O2/5% CO2) aCSF (NaCl 126 mM, NaHCO3 25 mM, D-glucose 11 mM, KCl 2.5 mM, CaCl2 2.4 mM, MgCl2 1.2 mM, NaH2PO4 1.2 mM, L-ascorbic acid 0.4 mM, pH adjusted to 7.4) at 1 mL/min. A triangular waveform was used for voltammetry, scanning from −0.4 to +1.2 to −0.4 V vs Ag/AgCl at 400 V/s every 100 ms. A glass capillary pulled carbon fiber electrode (100–150 μM length, 7 μM diameter, (Goodfellow, C005722, batch 4) penetrated the NAc core ~75 μM, and dopamine release was evoked with single 4 ms pulse stimulations (monophasic, 350 μA) from a bipolar stimulating electrode (Plastics One, Roanoke, VA, 8IMS3033SPCE) every 3 min. All recording electrode placements were grouped in the NAc core directly ventral to the anterior commissure (Figure 5). Dopamine signals were acquired and kinetically modeled using Demon Voltammetry Software, based on Michaelis–Menten kinetics, as previously described,58 holding the Km at 160 nM59 with the assumption that Km does not differ between our mouse model and the well-documented affinity of dopamine for the DAT in rats.59 Insulin has been shown to not affect Km;10 therefore, we assumed that consumption of a high-fat diet does not change the affinity of dopamine at the transporter in our Vmax calculations. Recordings were taken from single slices from each animal for baseline measures, and then drugs were washed over the slice with cumulative applications to obtain a dose response for insulin (10 nM to 1 μM) (Sigma-Aldrich, St. Louis, MO) at the same location as baseline collections. On additional slices, the PI3-kinase inhibitor, wortmannin (100 nM) (Sigma-Aldrich, St. Louis, MO), a small molecule inhibitor of the insulin receptor, BMS 536924 (300 nM) (Tocris Bioscience, Bristol, United Kingdom), and an insulin sensitizing agent, TCS 401 (Tocris Bioscience, Bristol, United Kingdom), were applied in order to characterize the insulin receptor function at dopamine terminals in control and high-fat-fed mice. Electrodes were calibrated after each experiment in a flow cell by adding 3 μM dopamine to the remaining experimental aCSF.
Figure 5.

Voltammetry recording locations. Mouse brain atlas pictures from Franklin and Paxinos (2008)60 show placement of the carbon fiber electrode for voltammetry recordings. All recordings were grouped ventral to the anterior commissure in the area contained by the yellow circles. Reproduced with permission from ref 60. Copyright 2008 Elsevier.
Insulin Receptor Protein Quantification
The contralateral hemisphere of the brain used for voltammetry was rapidly dissected in order to excise the ventral striatum, predominantly containing the NAc. Tissue was flash frozen using cold 2-methylbutane (Sigma- Aldrich, St. Louis, MO) and stored at −80 °C until use. An enzyme-linked immunosorbent assay (ELISA) kit (LifeSpan BioSciences Inc., Seattle, WA) was used to quantify insulin receptor protein content in frozen NAc tissue as per the manufacturer’s instructions. Briefly, NAc tissue was homogenized in phosphate buffered saline (0.02 mol/L pH7.2) and tissue was further lysed by ultrasonication. Homogenates were centrifuged at 5000g for 5 min, and the supernatant was used for BCA protein assay (ThermoScientific, Rockford, IL) and the insulin receptor ELISA kits. Insulin receptor was quantified in pg/mg protein and reported as percent control.
Statistics
Graph Pad Prism v.5 (La Jolla, CA) was used for all statistical analyses. Two-way repeated measures analysis of variance (ANOVA) was used to identify significant differences within and between treatment groups for body weight, IPGTT, and voltammetry dose response curves using Bonferroni’s or Tukey’s posthoc analysis to identify significant variations between groups at individual data points, when applicable. Student’s t-Tests were used to identify differences in fasting blood glucose, area under the curve analysis, and baseline dopamine release and reuptake data between control chow-fed and high fat-fed mice. Linear regression analysis was used to identify goodness of fit r2 values for correlational analysis between Vmax and IPGTT. One-way ANOVA tests were used to analyze between group differences in Vmax observed in the insulin receptor manipulation studies. Group data are presented ± SEM.
Acknowledgments
Funding
This work was funded by NIH Grants AA0014091, AA021099, DA021325, DA030161, and DA006634 (S.R.J.) and T32 AA007565 (S.C.F.)
ABBREVIATIONS
- HF
high fat
- DAT
dopamine transporter
- NAc
nucleus accumbens
- VTA
ventral tegmental area
- DA
dopamine
- IRS 1/2
insulin receptor subtrates 1 and 2
- aCSF
artificial cerebral spinal fluid
- PTP1B
protein tyrosine phosphatase 1B
- AUC
area under the curve
- BL
baseline
- IPGTT
intraperitoneal glucose tolerance test
- STZ
Streptozotocin
- ELISA
enzyme linked immunosorbent assay
- ANOVA
analysis of variance
Footnotes
Author Contributions
S.C.F. and S.R.J. developed a theoretical framework for the study and designed the experiments. S.C.F. executed the experiments. S.C.F. and S.R.J. completed data analysis, interpretation, and manuscript assembly.
Notes
The authors declare no competing financial interest.
References
- 1.Storlien LH, Pan DA, Kriketos AD, Baur LA. High fat diet-induced insulin resistance. Lessons and implications from animal studies. Ann N Y Acad Sci. 1993;683:82–90. doi: 10.1111/j.1749-6632.1993.tb35694.x. [DOI] [PubMed] [Google Scholar]
- 2.Freeman LR, Haley-Zitlin V, Rosenberger DS, Granholm AC. Damaging effects of a high-fat diet to the brain and cognition: a review of proposed mechanisms. Nutr Neurosci. 2014;17:241–51. doi: 10.1179/1476830513Y.0000000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinuesa A, Pomilio C, Menafra M, Bonaventura MM, Garay L, Mercogliano MF, Schillaci R, Lux Lantos V, Brites F, Beauquis J, Saravia F. Juvenile exposure to a high fat diet promotes behavioral and limbic alterations in the absence of obesity. Psychoneuroendocrinology. 2016;72:22–33. doi: 10.1016/j.psyneuen.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Heni M, Kullmann S, Preissl H, Fritsche A, Häring HU. Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol. 2015;11:701–11. doi: 10.1038/nrendo.2015.173. [DOI] [PubMed] [Google Scholar]
- 5.Williamson R, McNeilly A, Sutherland C. September 15) Insulin resistance in the brain: An old-age or newage problem? Biochem Pharmacol. 2012;84:737. doi: 10.1016/j.bcp.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Daws LC, Avison MJ, Robertson SD, Niswender KD, Galli A, Saunders C. Insulin signaling and addiction. Neuropharmacology. 2011;61:1123–1128. doi: 10.1016/j.neuropharm.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labouèbe G, Liu S, Dias C, Zou H, Wong JCY, Karunakaran S, Clee SM, Phillips AG, Boutrel B, Borgland SL. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci. 2013;16:300–8. doi: 10.1038/nn.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mebel DM, Wong JCY, Dong YJ, Borgland SL. Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur J Neurosci. 2012;36:2336–2346. doi: 10.1111/j.1460-9568.2012.08168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Könner AC, Hess S, Tovar S, Mesaros A, Sánchez-Lasheras C, Evers N, Verhagen LAW, Brönneke HS, Kleinridders A, Hampel B, Kloppenburg P, Brüning JC. Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab. 2011;13:720–728. doi: 10.1016/j.cmet.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 10.Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, Machold RP, Jones KT, de Vaca SC, Reith ME, Carr KD, Rice ME. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat Commun. 2015;6:8543. doi: 10.1038/ncomms9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Figlewicz DP, Bennett JL, Aliakbari S, Zavosh A, Sipols AJ. Insulin acts at different CNS sites to decrease acute sucrose intake and sucrose self-administration in rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R388–94. doi: 10.1152/ajpregu.90334.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figlewicz DP, Bennett JL, Naleid AM, Davis C, Grimm JW. Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol Behav. 2006;89:611–616. doi: 10.1016/j.physbeh.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Routh VH, Hao L, Santiago AM, Sheng Z, Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci. 2014;8:236. doi: 10.3389/fnsys.2014.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, Mendelsohn FAO. Localization and Characterization of Insulin Receptors in Rat Brain and Pituitary Gland Using in Vitro Autoradiography and Computerized Densitometry*. Endocrinology. 1987;121:1562–1570. doi: 10.1210/endo-121-4-1562. [DOI] [PubMed] [Google Scholar]
- 15.Carelli RM. The nucleus accumbens and reward: neurophysiological investigations in behaving animals. Behav Cogn Neurosci Rev. 2002;1:281–296. doi: 10.1177/1534582302238338. [DOI] [PubMed] [Google Scholar]
- 16.Galici R, Galli A, Jones DJ, Sanchez TA, Saunders C, Frazer A, Gould GG, Lin RZ, France CP. Selective Decreases in Amphetamine Self-Administration and Regulation of Dopamine Transporter Function in Diabetic Rats. Neuroendocrinology. 2003;77:132–140. doi: 10.1159/000068650. [DOI] [PubMed] [Google Scholar]
- 17.Branch SY, Goertz RB, Sharpe AL, Pierce J, Roy S, Ko D, Paladini CA, Beckstead MJ. Food Restriction Increases Glutamate Receptor-Mediated Burst Firing of Dopamine Neurons. J Neurosci. 2013;33:13861–13872. doi: 10.1523/JNEUROSCI.5099-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cabeza de Vaca S, Carr KD. Food restriction enhances the central rewarding effect of abused drugs. J Neurosci. 1998;18:7502–10. doi: 10.1523/JNEUROSCI.18-18-07502.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macenski MJ, Meisch RA. Cocaine self-administration under conditions of restricted and unrestricted food access. Exp Clin Psychopharmacol. 1999;7:324–37. doi: 10.1037//1064-1297.7.4.324. [DOI] [PubMed] [Google Scholar]
- 20.Garcia BG, Wei Y, Moron JA, Lin RZ, Javitch JA, Galli A. Akt Is Essential for Insulin Modulation of Amphetamine-Induced Human Dopamine Transporter Cell-Surface Redistribution. Mol Pharmacol. 2005;68:102–109. doi: 10.1124/mol.104.009092. [DOI] [PubMed] [Google Scholar]
- 21.Figlewicz DP, Szot P, Chavez M, Woods SC, Veith RC. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994;644:331–334. doi: 10.1016/0006-8993(94)91698-5. [DOI] [PubMed] [Google Scholar]
- 22.Owens WA, Williams JM, Saunders C, Avison MJ, Galli A, Daws LC. Rescue of Dopamine Transporter Function in Hypoinsulinemic Rats by a D2 Receptor-ERK-Dependent Mechanism. J Neurosci. 2012;32:2637–2647. doi: 10.1523/JNEUROSCI.3759-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoffelmeer ANM, Drukarch B, De Vries TJ, Schetters D, Pattij T. Insulin Modulates Cocaine-Sensitive Monoamine Transporter Function and Impulsive Behavior. J Neurosci. 2011;31:1284–1291. doi: 10.1523/JNEUROSCI.3779-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams JM, Owens WA, Turner GH, Saunders C, Dipace C, Blakely RD, France CP, Gore JC, Daws LC, Avison MJ, Galli A. Hypoinsulinemia regulates amphetamine-induced reverse transport of dopamine. PLoS Biol. 2007;5:e274. doi: 10.1371/journal.pbio.0050274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speed N, Saunders C, Davis AR, Owens WA, Matthies HJG, Saadat S, Kennedy JP, Vaughan RA, Neve RL, Lindsley CW, Russo SJ, Daws LC, Niswender KD, Galli A. Impaired striatal akt signaling disrupts dopamine homeostasis and increases feeding. PLoS One. 2011;6:e25169. doi: 10.1371/journal.pone.0025169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patterson TA, Brot MD, Zavosh A, Schenk JO, Szot P, Figlewicz DP. Food deprivation decreases mRNA and activity of the rat dopamine transporter. Neuroendocrinology. 1998;68:11–20. doi: 10.1159/000054345. [DOI] [PubMed] [Google Scholar]
- 27.Gupta N, Jensen MD. Clinical effects of high-fat meals and weight gain due to high-fat feeding. Int J Obes. 2012;2:S51–S55. doi: 10.1038/ijosup.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuang CC, Shen W, Chen H, Xie G, Jia W, Chung S, McIntosh MK. Differential effects of grape powder and its extract on glucose tolerance and chronic inflammation in high-fat-fed obese mice. J Agric Food Chem. 2012;60:12458–12468. doi: 10.1021/jf3028107. [DOI] [PubMed] [Google Scholar]
- 29.Narayanaswami V, Thompson AC, Cassis LA, Bardo MT, Dwoskin LP. Diet-induced obesity: dopamine transporter function, impulsivity and motivation. Int J Obes. 2013;37:1095–1103. doi: 10.1038/ijo.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baladi MG, Horton RE, Owens WA, Daws LC, France CP. Eating high fat chow decreases dopamine clearance in adolescent and adult male rats but selectively enhances the locomotor stimulating effects of cocaine in adolescents. Int J Neuropsychopharmacol. 2015;18:pyv024. doi: 10.1093/ijnp/pyv024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fordahl SC, Locke JL, Jones SR. High fat diet augments amphetamine sensitization in mice: Role of feeding pattern, obesity, and dopamine terminal changes. Neuropharmacology. 2016;109:170–182. doi: 10.1016/j.neuropharm.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Models & Mech. 2010;3:525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winzell MS, Ahren B. The High-Fat Diet-Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes. 2004;53:S215–219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 34.Ahrén B, Scheurink AJW. Marked hyper-leptinemia after high-fat diet associated with severe glucose intolerance in mice. Eur J Endocrinol. 1998;139:461–467. doi: 10.1530/eje.0.1390461. [DOI] [PubMed] [Google Scholar]
- 35.Strubbe JH, Porte D, Woods SC. Insulin responses and glucose levels in plasma and cerebrospinal fluid during fasting and refeeding in the rat. Physiol Behav. 1988;44:205–208. doi: 10.1016/0031-9384(88)90139-4. [DOI] [PubMed] [Google Scholar]
- 36.May AA, Liu M, Woods SC, Begg DP. CCK increases the transport of insulin into the brain. Physiol Behav. 2016;165:392. doi: 10.1016/j.physbeh.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Runchey SS, Pollak MN, Valsta LM, Coronado GD, Schwarz Y, Breymeyer KL, Wang CY, Wang CY, Lampe JW, Neuhouser ML. Glycemic load effect on fasting and post-prandial serum glucose, insulin, IGF-1 and IGFBP-3 in a randomized, controlled feeding study. Eur J Clin Nutr. 2012;66:1146–52. doi: 10.1038/ejcn.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function. Diabetes. 2014;63:2232–2243. doi: 10.2337/db14-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woods SC, Seeley RJ, Baskin DG, Schwartz MW. Insulin and the Blood-Brain Barrier. Curr Pharm Des. 2003;9:795–800. doi: 10.2174/1381612033455323. [DOI] [PubMed] [Google Scholar]
- 40.Hryhorczuk C, Florea M, Rodaros D, Poirier I, Daneault C, Des Rosiers C, Arvanitogiannis A, Alquier T, Fulton S. Dampened Mesolimbic Dopamine Function and Signaling by Saturated but not Monounsaturated Dietary Lipids. Neuropsychopharmacology. 2016;41:811. doi: 10.1038/npp.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zahniser NR, Goens MB, Hanaway PJ, Vinych JV. Characterization and Regulation of Insulin Receptors in Rat Brain. J Neurochem. 1984;42:1354–1362. doi: 10.1111/j.1471-4159.1984.tb02795.x. [DOI] [PubMed] [Google Scholar]
- 42.Boyd FT, Raizada MK. Effects of insulin and tunicamycin on neuronal insulin receptors in culture. Am J Physiol. 1983;245:C283–7. doi: 10.1152/ajpcell.1983.245.3.C283. [DOI] [PubMed] [Google Scholar]
- 43.Serafine KM, Bentley TA, Grenier AE, France CP. Eating high fat chow and the behavioral effects of direct-acting and indirect-acting dopamine receptor agonists in female rats. Behav Pharmacol. 2014;25:287–295. doi: 10.1097/FBP.0000000000000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baladi MG, Koek W, Aumann M, Velasco F, France CP. Eating high fat chow enhances the locomotor-stimulating effects of cocaine in adolescent and adult female rats. Psychopharmacology (Berl) 2012;222:447–457. doi: 10.1007/s00213-012-2663-7. [DOI] [PubMed] [Google Scholar]
- 45.McGuire BA, Baladi MG, France CP. Eating high-fat chow enhances sensitization to the effects of methamphetamine on locomotion in rats. Eur J Pharmacol. 2011;658:156–159. doi: 10.1016/j.ejphar.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fröjdö S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta, Mol Basis Dis. 2009;1792:83–92. doi: 10.1016/j.bbadis.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 47.Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: More than just a peripheral hormone. J Aging Res. 2012;2012:1. doi: 10.1155/2012/384017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carvelli L, Moron JA, Kahlig KM, Ferrer JV, Sen N, Lechleiter JD, Leeb-Lundberg LMF, Merrill G, Lafer EM, Ballou LM, Shippenberg TS, Javitch JA, Lin RZ, Galli A. PI 3-kinase regulation of dopamine uptake. J Neurochem. 2002;81:859–869. doi: 10.1046/j.1471-4159.2002.00892.x. [DOI] [PubMed] [Google Scholar]
- 49.Lute BJ, Khoshbouei H, Saunders C, Sen N, Lin RZ, Javitch JA, Galli A. PI3K signaling supports amphetamine-induced dopamine efflux. Biochem Biophys Res Commun. 2008;372:656–661. doi: 10.1016/j.bbrc.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9:753–762. doi: 10.1016/j.coph.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 52.Koren S, Fantus IG. Inhibition of the protein tyrosine phosphatase PTP1B: potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab. 2007;21:621–640. doi: 10.1016/j.beem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Goldstein BJ. Protein-tyrosine phosphatase 1B (PTP1B): a novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr Drug Targets: Immune, Endocr Metab Disord. 2001;1:265–275. doi: 10.2174/1568008013341163. [DOI] [PubMed] [Google Scholar]
- 54.Morris JK, Bomhoff GL, Gorres BK, Davis VA, Kim J, Lee PP, Brooks WM, Gerhardt GA, Geiger PC, Stanford JA. Insulin resistance impairs nigrostriatal dopamine function. Exp Neurol. 2011;231:171–180. doi: 10.1016/j.expneurol.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rigal N, Frelut ML, Monneuse MO, Hladik CM, Simmen B, Pasquet P. Food neophobia in the context of a varied diet induced by a weight reduction program in massively obese adolescents. Appetite. 2006;46:207–214. doi: 10.1016/j.appet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Fildes A, Mallan KM, Cooke L, van Jaarsveld CHM, Llewellyn CH, Fisher A, Daniels L. The relationship between appetite and food preferences in British and Australian children. Int J Behav Nutr Phys Act. 2015;12:116. doi: 10.1186/s12966-015-0275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calipari ES, Ferris MJ, Siciliano CA, Zimmer BA, Jones SR. Intermittent cocaine self-administration produces sensitization of stimulant effects at the dopamine transporter. J Pharmacol Exp Ther. 2014;349:192–8. doi: 10.1124/jpet.114.212993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yorgason JT, España RA, Jones SR. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods. 2011;202:158–64. doi: 10.1016/j.jneumeth.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu Q, Reith MEA, Wightman RM, Kawagoe KT, Garris PA. Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J Neurosci Methods. 2001;112:119–133. doi: 10.1016/s0165-0270(01)00459-9. [DOI] [PubMed] [Google Scholar]
- 60.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. 3. Elsevier; Amsterdam: 2008. [Google Scholar]