

Abstract
Cognitive deficits represent core symptoms in schizophrenia (SZ) and predict patient outcome; however, they remain poorly treated by current antipsychotic drugs. Elevated levels of the endogenous alpha7 nicotinic receptor negative allosteric modulator and NMDA receptor antagonist, kynurenic acid (KYNA), are commonly seen in post-mortem tissue and cerebrospinal fluid of patients with SZ. When acutely or chronically elevated in rodents, KYNA produces cognitive deficits similar to those seen in the disease, making down-regulation of KYNA, via inhibition of kynurenine aminotransferase II (KAT II), a potential treatment strategy. We determined, in adult Wistar rats, if the orally available KAT II inhibitor BFF816 a) prevents KYNA elevations in prefrontal cortex (PFC) after a systemic kynurenine injection and b) reverses the kynurenine-induced attenuation of evoked prefrontal glutamate release caused by stimulation of the nucleus accumbens shell (NAcSh). Systemic injection of kynurenine (25 or 100 mg/kg, i.p.) increased KYNA levels in PFC (532% and 1104% of baseline, respectively). NMDA infusions (0.15 μg/0.5 μL) into NAcSh raised prefrontal glutamate levels more than 30-fold above baseline. The two doses of kynurenine reduced evoked glutamate release in PFC (by 43% and 94%, respectively, compared to NMDA alone). Co-administration of BFF816 (30 or 100 mg/kg, p.o.) with kynurenine (25 mg/kg, i.p.) attenuated the neosynthesis of KYNA and dose-dependently restored NMDA-stimulated glutamate release in the PFC (16% and 69%, respectively). The ability to prevent KYNA neosynthesis and to normalize evoked glutamate release in PFC justifies further development of KAT II inhibitors for the treatment of cognitive deficits in SZ.
Keywords: glutamate, KAT II, kynurenic acid, microelectrode array, prefrontal cortex, schizophrenia
1.0 Introduction
Schizophrenia (SZ), a devastating psychiatric disorder that can be traced to a combination of genetic vulnerabilities and environmental causes, affects approximately 1% of the world’s population (Lewis and Lieberman, 2000). One symptom cluster of the disease, cognitive deficits, includes impairments in executive functions, such as attention, working memory, and cognitive flexibility (Keefe, 2007; Kerns et al., 2008). These are considered core symptoms (Elvevag and Goldberg, 2000) because they emerge during the prodromal period, can often be seen in first-degree relatives of patients, and correlate with the long-term functional outcome of the patient (Gold, 2004). Unfortunately, currently available antipsychotic drugs produce, at best, minor improvements in these cognitive impairments (Keefe et al., 2007a; Keefe et al., 2007b).
The cause(s) of the cognitive deficits seen in SZ patients remains unknown, but could be related to abnormally high levels of kynurenic acid (KYNA), a neuroinhibitory metabolite of the kynurenine pathway of tryptophan degradation (see Muller et al., 2011; Schwarcz et al., 2012 for review). KYNA is a potent negative modulator of the alpha7 nicotinic acetylcholine receptor (α7nAChR; Hilmas et al., 2001) and a competitive antagonist of the glycine co-agonist site of the N-methyl-D-aspartate receptor (NMDAR; Parsons et al., 1997), and its levels are elevated in post-mortem brain tissue and cerebrospinal fluid of patients with SZ (Erhardt et al., 2001; Schwarcz et al., 2001), irrespective of antipsychotic medication (Ceresoli-Borroni et al., 2006). In rodents, even relatively modest KYNA increases acutely reduce basal extracellular levels of glutamate (Konradsson-Geuken et al., 2010; Wu et al., 2010), GABA (Beggiato et al., 2014) and dopamine (Rassoulpour et al., 2005) in the prefrontal cortex (PFC) as well as in other brain areas (Rassoulpour et al., 2005; Pocivavsek et al., 2011; Beggiato et al., 2013). Notably, elevations in KYNA during sensitive periods of late gestation, have also been shown to reduce the gain of GABAergic transmission that occurs post-adolescence in the maturing PFC (Thomases et al., SFN 2014). In line with these neurochemical and electrophysiological effects, acute elevation of KYNA levels in adults, or chronic elevations of KYNA during sensitive periods of development, causes cognitive deficits that resemble those seen in persons with SZ, including diminished cognitive flexibility (Alexander et al., 2012; Alexander et al., 2013; Pershing et al., 2015), trace fear conditioning (Pershing et al., 2016), spatial working memory, and conditioned avoidance tasks (Pocivavsek et al., 2012).
In the mammalian brain, KYNA is formed mainly by the irreversible enzymatic transamination of its immediate bioprecursor kynurenine (Schwarcz et al., 2012). Of several kynurenine aminotransferases (KATs), KAT II is preferentially responsible for the neosynthesis of easily mobilizable KYNA (Guidetti et al., 2007) and has therefore been characterized in considerable detail (Han et al., 2008; Passera et al., 2011; Nematollahi et al., 2016). Inhibition of this enzyme provides a means to down-regulate cerebral KYNA production and may result in an attenuation of cognitive deficits (Schwarcz et al., 2012). Experimental proof-of-concept was provided in studies with KAT II knockout mice, which show improved performance in three hippocampus-related cognitive paradigms and a significant increase in hippocampal long-term potentiation (Potter et al., 2010). Additionally, the first-generation KAT II inhibitor S-ESBA demonstrates pro-cognitive effects following intracerebral infusion in intact rats (Pocivavsek et al., 2012). Second-generation KAT II inhibitors were recently developed that are active in brain following systemic administration (Tuttle et al., 2013; Dounay et al., 2013; Koshy Cherian et al., 2014; Kozak et al., 2014). One of these compounds, the orally available BFF816, was not only found to reduce extracellular KYNA and increase extracellular glutamate levels in the rat hippocampus, but also to improve the performance of intact rats in the Morris water maze (Wu et al., 2014).
In the present study, we take advantage of BFF816 to determine, for the first time, if the effect of increased KYNA levels to reduce evoked glutamate release in PFC can be reversed by inhibiting the synthesis of KYNA. To this end, we utilized an experimental paradigm in which the release of glutamate was evoked by an infusion of NMDA into the shell region of the nucleus accumbens (NAcSh; Bortz et al., 2014; Bortz et al., 2016). This procedure results in an increase in cortical acetylcholine (ACh) release from basal forebrain and, subsequently, a local α7nAChR-dependent increase in prefrontal glutamate levels (Bortz et al., 2016). Notably, stimulation of the NAcSh in this manner facilitates the filtering of distractors during a sustained attention task in rodents, indicating that prefrontal glutamate, evoked under these conditions, has a positive impact on cognitive performance (St Peters et al., 2011). Thus, the restoration of stimulated prefrontal glutamate levels in PFC by BFF816 would represent a proof of principle for the use of KAT II inhibitors for the treatment of cognitive dysfunctions produced, in part, by elevations in brain KYNA levels.
2.0 Materials and Methods
2.1 Animals
Male Wistar rats (65-90 days of age, 280-420 g) were maintained in a temperature- and humidity-controlled room on a 12:12-hour light:dark cycle (lights on at 06:00 a.m.), and housed in pairs (pre-surgery) in plastic cages lined with corn cob bedding (Harlan Teklad, Madison, WI, USA). After implantation of the microelectrode array (MEA), animals were singly housed with ad libitum access to food and water. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to consider alternatives to in vivo techniques. Procedures involving animals were approved by the Institutional Animal Care and Use Committees of The Ohio State University and the University of Maryland School of Medicine, in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
2.2 Reagents and test compounds
The following reagents were used to prepare and calibrate the glutamate-sensitive MEAs: m-phenylenediamine dihydrochloride (purchased from Acros Organics, NJ, USA), L-ascorbic acid, dopamine, L-glutamate monosodium salt, glutaraldehyde (25% solution in water), bovine serum albumin, and hydrogen peroxide (all obtained from Sigma Aldrich Corp., St. Louis, MO, USA) and L-glutamate oxidase (purchased from United States Biological; Salem, MA, USA).
For administration to animals in vivo, NMDA (Sigma Aldrich) was prepared in artificial cerebrospinal fluid (aCSF; pH 7.1-7.4; see Zmarowski et al., 2007), L-kynurenine sulfate (purity 94%; Sai Advantium, Hyderabad, India) was dissolved in physiological saline (pH 7.1-7.4), and BFF816 (provided by Mitsubishi-Tanabe, Yokohama, Japan) was suspended in a solution of hydroxyl-propyl-beta-cyclodextrin (HPBCD; Sigma Aldrich; 20%; pH 7.1-7.4).
2.3 Microdialysis studies
Rats were anesthetized (chloral hydrate, 360 mg/kg, i.p.) and placed in a stereotaxic frame. A guide cannula (0.65 mm o.d.; Carnegie Medicin, Stockholm, Sweden) was positioned unilaterally over the medial PFC (AP: 3.2 mm anterior to bregma, ML: ± 0.6 mm from midline, DV: 2.0 mm below dura; hemispheres counterbalanced) and secured to the skull with acrylic dental cement. On the next day, a microdialysis probe (CMA/10, membrane length: 3.0 mm, Carnegie Medicin) was inserted through the guide cannula and connected to a microperfusion pump set to a speed of 1 μL/min. The freely moving animals were perfused with Ringer solution containing (in mM): NaCl, 144; KCl, 4.8; MgSO4, 1.2; CaCl2, 1.7; pH 6.7. Microdialysis samples were collected every 30 min for a total of 8 hrs. Data were not corrected for recovery from the microdialysis probe.
2.3.1 Measurement of KYNA in dialysate
The content of KYNA in microdialysate samples was determined by high performance liquid chromatography. To this end, KYNA was isocratically eluted with a mobile phase containing zinc acetate (200 mM) and acetonitrile (5%; pH 6.2) and detected fluorimetrically (excitation wavelength: 344 nm; emission wavelength: 398 nm), as described (Wu et al., 2010).
2.4 Microdialysis studies: experimental procedures
2.4.1 Experiment 1: Effect of kynurenine on extracellular KYNA levels
Following the establishment of a stable baseline of KYNA (~2 hrs), rats received an i.p. injection of 25 mg/kg or 100 mg/kg kynurenine (n = 6/ group). These and subsequent i.p. and p.o. injections were delivered at a volume of 0.1% body weight. Dialysates were collected every 30 min for a total of 8 hrs.
2.4.2 Experiment 2: Effect of BFF816 on basal and kynurenine-induced elevations of KYNA
Five groups of rats (n = 6/ group) were tested following the establishment of a stable baseline of KYNA (~2 hrs). To assess the effects of KAT II inhibition on basal levels of KYNA, BFF816 was administered (p.o.) at 30 mg/kg (Group 1) or 100 mg/kg (Group 2). To assess the effects of BFF816 on elevated KYNA levels, three groups of animals received a systemic injection of kynurenine (25 mg/kg, i.p.) immediately following a p.o. administration of HPBCD (vehicle for BFF816; Group 3), 30 mg/kg BFF816 (Group 4) or 100 mg/kg BFF816 (Group 5). In all groups, dialysates were collected every 30 min for a total of 8 hrs.
2.5 Biosensor studies
2.5.1 Preparation of glutamate-sensitive MEAs
MEAs were composed of a ceramic paddle with a stainless steel tip bearing four 15 × 333 μm platinum recording sites and a region that interfaces with the preamplifier. Each pair of recording sites (Figure 1A) was designated to be either glutamate-sensitive (Gluox) or not (sentinel; see Rutherford et al., 2007 for further details on MEA assemblage). This coating design (Figure 1B) permits the isolation of the electrical signal driven solely by the oxidation of glutamate by subtracting the sentinel current from that from of the Gluox channel (i.e. self-referencing; Burmeister and Gerhardt, 2001; Rutherford et al., 2007, Konradsson-Geuken et al., 2009; Bortz et al., 2014). MEAs were calibrated in vitro using the FAST-16 MKII electrochemical recording system just prior to implantation (Figure 1C). Calibration criteria for each sensor were determined as previously described (Burmeister and Gerhardt, 2001; Rutherford et al., 2007 Konradsson-Geuken et al., 2009; Bortz et al., 2014), and all sensors used for analysis met these criteria.
Figure 1. MEA design, signal transduction scheme, in vitro calibration, and representative placement.
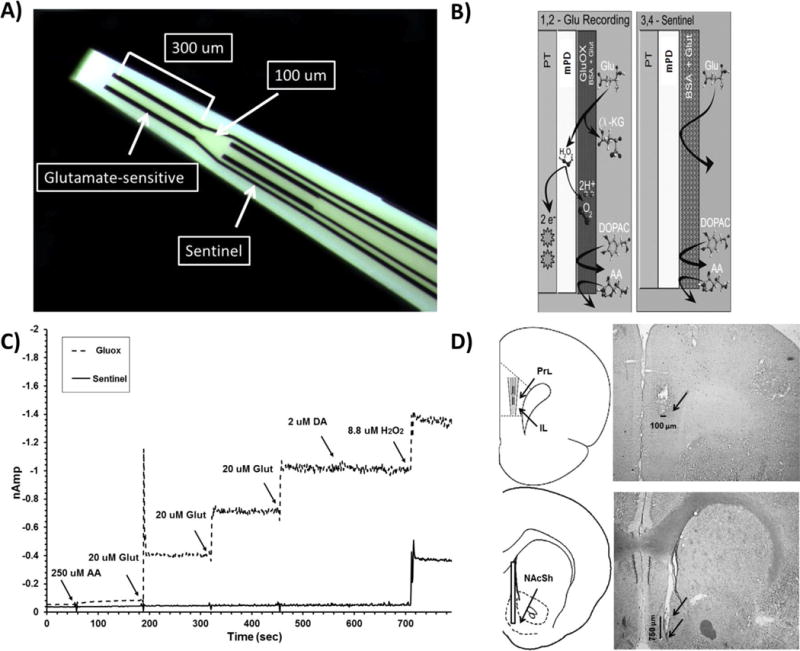
(A): Four platinum recording channels near the tip of the MEA allow for the detection and isolation of glutamate within a single brain sub-region (see Burmeister and Gerhardt, 2001 for further detail on self-referencing); (B): Glutamate is detected in vivo when a glutamate molecule (Glu) contacts the platinum surface (PT) coated with glutamate oxidase (Gluox; see Rutherford et al., 2007) and is oxidized to the reporting molecule hydrogen peroxide (H2O2). The oxidation of H2O2 generates a current that is proportional to the concentration of glutamate at the surface of the MEA; (C): In vitro calibrations of the MEA are conducted immediately before implantation into the PFC. The solid line tracing is from the glutamate-sensitive (Gluox) recording channel, and the dashed tracing is from the sentinel background channel. Arrows correspond to the addition of ascorbic acid (AA, 500 μL), glutamate (glut, 3 × 40 μL), dopamine (DA, 40 μL), and H2O2, (40 μL) into the stirred calibration beaker. Current (nAmp) is depicted along the y-axis, and time (sec) along the x-axis. The successive additions of glutamate (20 μM/aliquot) produce a linear increase on Gluox, but not sentinel channels. Important for self-referencing, the Gluox and sentinel channels respond equally to every addition, except glutamate; (D): The cartoon and the corresponding photomicrograph depict the desired target location for both the MEAs (between the prelimbic (PrI) and infralimbic (IL) cortices) and infusion cannulae (within the nucleus accumbens shell). Arrows indicate the ventral termination of the MEA, guide, and infusion cannula within the respective photomicrographs. NAcc: Nucleus accumbens core; CC: Corpus callosum; aca: anterior commissure; NAcSh: Nucleus accumbens shell.
2.5.2 Implantation of MEAs and infusion cannulae
Animals were anesthetized using isoflurane (2%, 0.8 L/min) and implanted with an MEA unilaterally in the PFC (in mm from bregma, midline and dura: AP: +2.7, ML: ±0.65, DV: –3.9; hemispheres counterbalanced), and a stainless steel guide cannula (Plastics One, Roanoke, VA) ipsilaterally in the NAcSh (at a 10° angle, in mm: AP: +0.4, ML: ±0.7, DV: –7.4; Figure 1D). The Ag/AgCl reference electrode was implanted in the contralateral side at a site distant from the recording area. All coordinates were determined using a stereotaxic atlas (Paxinos and Watson, 1998).
2.5.3 In vivo glutamate recordings
On the first day after MEA implantation, rats were placed in a wooden, electrically- shielded testing box (H 58 cm; W 342 cm; L 317 cm), without connecting them to the preamplifier, to allow habituation to the test environment prior to recording. On the second day, recordings of cortical glutamate began (3 consecutive test days). At the onset of each test day, the animals, allowed to move freely, were connected to the preamplifier and then remained undisturbed for 1–3 hours in order to obtain a stable baseline.
2.6 Biosensor studies: experimental procedures
2.6.1 Experiment 3: Effect of kynurenine on the evoked release of glutamate
Following the baseline period, animals in Group 1 (n = 6) received (counterbalanced sessions) an injection of either saline or kynurenine (25 mg/kg, i.p.). Two hrs later, NMDA (0.15 μg in 0.5 μL) was delivered over 2 sec into the NAcSh using an infusion cannula attached to a Hamilton PB600-1 manual dispenser (Hamilton Company, Reno, NV, USA), and phasic increases in extracellular glutamate levels were recorded. Animals in Group 2 (n = 6) underwent the same protocol but received saline or the higher dose of kynurenine (100 mg/kg, i.p.).
2.6.2 Experiment 4: Effect of BFF816 on evoked glutamate release
A single group of rats (n = 5) was tested in 3 counterbalanced test sessions. Following the baseline period, animals received an oral dose of vehicle for BFF816 (HPBCD), BFF816 (30 mg/kg), or BFF816 (100 mg/kg). Two hrs later, NMDA (0.15 μg) was infused into the NAcSh as described in Experiment 3, and phasic increases in extracellular glutamate levels were recorded.
2.6.3 Experiment 5: Effect of BFF816 + kynurenine on evoked glutamate release
Two groups of rats were tested in 3 counterbalanced sessions. Following the baseline period, animals in Group 1 (n = 6) received one of three drug combinations: a) saline (i.p.) and HPBCD vehicle (p.o.); b) kynurenine (25 mg/kg, i.p.) and HPBCD vehicle (p.o.); or c) kynurenine (25 mg/kg, i.p.) and BFF816 (30 mg/kg, p.o.). Rats in Group 2 (n = 6) were administered identical drug combinations but received a higher dose of BFF816 (100 mg/kg, p.o.). Two hrs later, on each testing day, rats in both groups received an intra-NAcSh infusion of NMDA (0.15 μg), and phasic increases in extracellular glutamate levels were recorded.
2.7 Histology
At the conclusion of each experiment, animals received a sub-lethal dose of sodium pentobarbital and were transcardially perfused with 200 ml of heparinized saline following by 100 ml of 10% formalin. Cryostat-sectioned coronal brain slices (70 μm) were mounted on gelatin-coated slides, stained using cresyl violet, and examined under a light microscope for verification of microelectrode and cannula placement (Figure 1D).
2.8 Data analysis
The microdialysis data from Experiments 1 & 2 were analyzed using an initial 2-way omnibus ANOVA with time-point as a within-subjects condition and drug combination or drug dose as a between-subjects condition. If significance was determined in any omnibus ANOVA, post-hoc multiple comparisons were performed using the Bonferroni-corrected t-test.
The glutamate signal (Experiments 3-5), initially measured in pA, was transformed to a concentration equivalent (μM) on the basis of each sensor’s individual calibration slopes, generated immediately before surgery. Measurements (taken after self-referencing) included: (i) basal glutamate levels (μM) in the PFC (calculated as the average of 10 sampling points prior to the marking of an event), (ii) maximal prefrontal glutamate increase (μM and % change from basal) following stimulation of the NAcSh, (iii) effect of kynurenine as measured by the % reduction of NMDA-evoked glutamate release; (iv) effect of BFF816 as measured by the % recovery of kynurenine-induced inhibition of evoked glutamate; (v) the latency (sec) between NMDA infusion and the onset in the rise of the glutamate peak; and (vi) time to clear 80 percent (T80) of the glutamate increase from its maximal amplitude (sec). Comparisons were performed using the IBM SPSS statistics program (version 22, IBM Corporations, Armonk, NY, USA). An initial, omnibus ANOVA was performed separately for each dependent measure in each experiment. If significance was determined, post-hoc multiple comparisons were performed using the Bonferroni-corrected t-test.
3.0 Results
3.1 MEA, cannula, and probe placements
All subjects included in this analysis had histologically confirmed microdialysis probe, MEA, and cannula placements (Figure 1D). Notably, animals that had errant cannula placements (for example in the NAc core) in the biosensor experiments did not show any stimulation-evoked glutamate increase in the PFC (data not shown).
3.2 Experiments 1 & 2: Systemically administered kynurenine increases KYNA levels in the PFC: attenuation by co-administration of BFF816
In line with previous reports (Konradsson-Geuken et al., 2010; Alexander et al., 2012), systemically administered kynurenine, but not its vehicle, increased extracellular KYNA levels in the PFC in a dose- and time-dependent fashion (dose × time interaction; F32,176 = 23.33, P < 0.001; Figure 2). Both doses significantly increased KYNA levels compared to vehicle (timepoints 2.5-8, P’s < 0.05). The higher dose of kynurenine (100 mg/kg) was more effective than the lower dose (25 mg/kg) at 2.5 - 6.5 hrs (all P’s < 0.05).
Figure 2. Systemic kynurenine administration increases extracellular KYNA levels in the PFC.
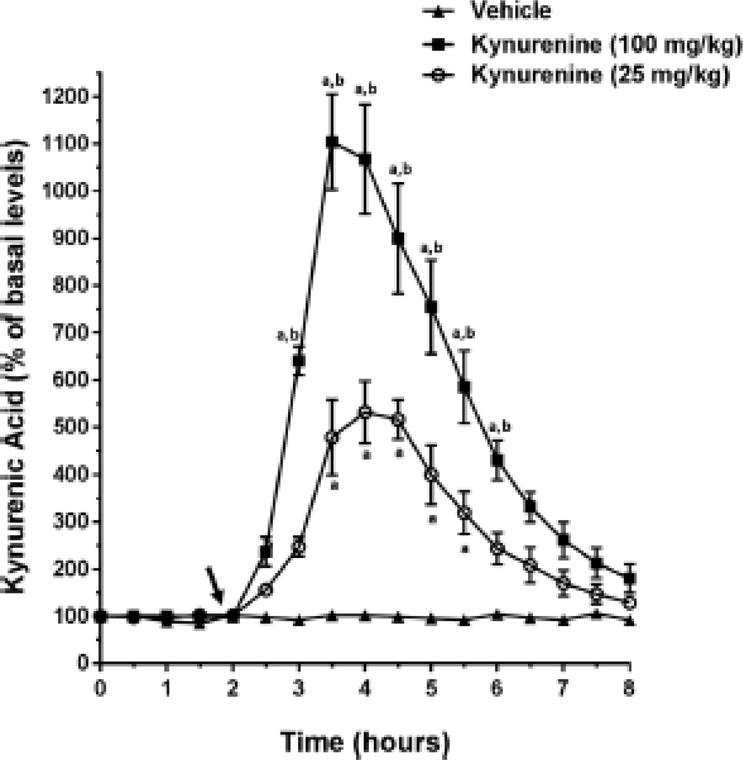
Microdialysis samples were collected every 30 min after subjects received one of two doses of kynurenine (25 or 100 mg/kg, i.p.) or its vehicle solution (saline). The arrow indicates the timing of the injection. Post-hoc multiple comparisons were conducted using a Bonferroni adjusted t-test. a P < 0.05 vs. baseline values; b P < 0.05 vs. 25 mg/kg kynurenine.
Oral administration of BFF816 (30 and 100 mg/kg), but not its vehicle (HPBCD), significantly reduced basal KYNA levels in the PFC (Figure 3; maximum decrease from baseline: 30 mg/kg: 22.8 ± 7.7%; 100 mg/kg: 29.1 ± 3.8%; dose × time interaction: F32,176 = 2.303, P = 0.004).
Figure 3. Oral administration of BFF816 reduces extracellular KYNA levels in the PFC.
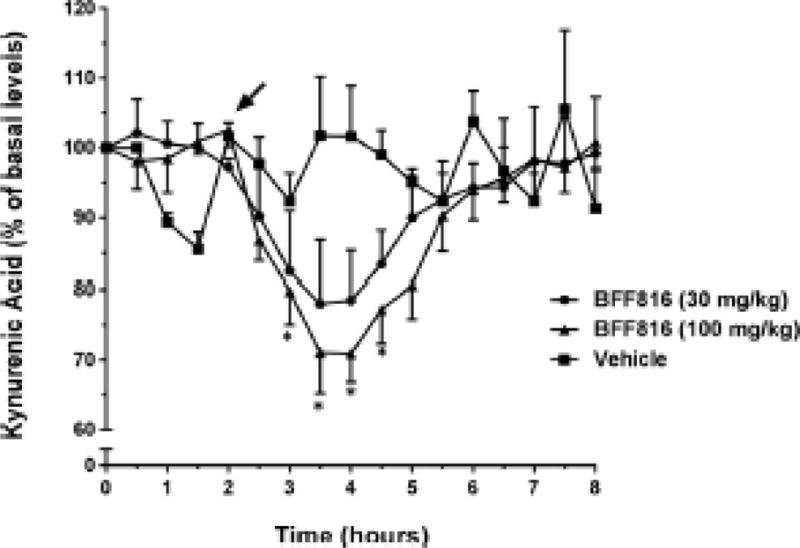
Microdialysis samples were collected every 30 min. The arrow indicates the timing of the oral administration of BFF816 (30 or 100 mg/kg, p.o.) or its vehicle solution HPBCD. * P < 0.05 vs. baseline values (Bonferroni adjusted t-test).
Both doses of BFF816 also reduced experimentally elevated levels of KYNA in the PFC. Compared to the effect of treatment with kynurenine + HPBCD (the vehicle for BFF816), BFF816 (30 mg/kg) attenuated kynurenine (25 mg/kg)-induced KYNA increases over an extended period of time (F(32,192) = 49.487, P < 0.001). There was a trend for the higher dose of BFF816 (100 mg/kg) to be more effective than the lower dose, particularly in the first hours after application (Figure 4), but there was no significant difference between the two doses overall (P > 0.05).
Figure 4. BFF816 attenuates the kynurenine-induced increase in extracellular KYNA levels in the PFC.
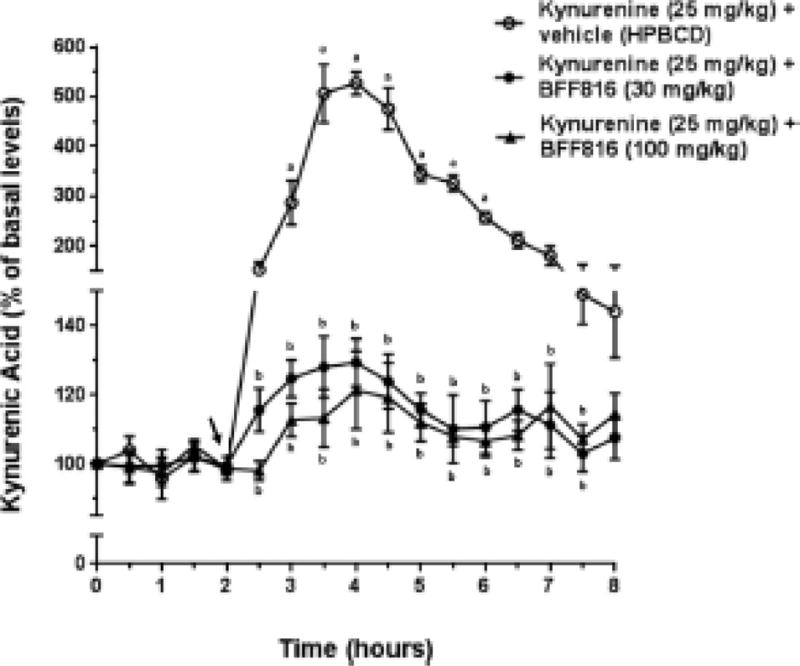
Microdialysis samples were collected every 30 min. The timing of the administration of vehicle or BFF816 (30 or 100 mg/kg, p.o.), followed immediately by kynurenine (25 mg/kg, i.p.), is indicated by the solid arrow. a P < 0.05 vs. baseline values; b P < 0.05 vs. kynurenine alone (Bonferroni adjusted t-test).
3.3 Experiment 3: Kynurenine attenuates evoked but not basal glutamate release in the PFC
Systemic administration of kynurenine dose-dependently reduced stimulated glutamate release in PFC following an infusion of NMDA into the NAcSh. Figures 5A–C illustrate the MEA tracings from a single representative animal. Figure 5A depicts current/time recordings from the individual Gluox and sentinel recording sites, whereas Figures 5B and 5C show the self-referenced current tracings that are due specifically to the oxidation of glutamate (following 25 and 100 mg/kg kynurenine, respectively). NAcSh stimulation with NMDA resulted in phasic increases in PFC glutamate (left section of traces in Figures 5B and 5C), which were similar in amplitude to those reported previously (Bortz et al., 2014). However, when NMDA was infused 2 hours after kynurenine (25 or 100 mg/kg; right section of traces in Figures 5B and 5C), the stimulated glutamate increases in the PFC were attenuated, with the higher dose of kynurenine producing a significantly greater degree of attenuation. These individual effects were replicated across the group data (Figure 5D; 25 mg/kg group: NMDA + saline = 6.22 ± 1.47 μM; NMDA + kynurenine = 3.32 ± 0.97 μM, 42.5 ± 10.1% decrease of evoked release; 100 mg/kg group: NMDA + saline = 3.77 ± 0.56 μM; NMDA + kynurenine = 0.24 ± 0.13 μM, 94.2 ± 4.4% decrease of evoked release; n = 6 each, 25 mg/kg vs. 100 mg/kg, F1,11 = 24.97, P = 0.001). Glutamate peak onset (overall mean = 53.2 ± 10.3 sec) and clearance (T80, overall mean = 7.5 ± 2.5 sec) after NMDA infusion were stable across all experiments and not affected by kynurenine or BFF816 pre-treatment (all P values > 0.05). Therefore, these values will not be discussed in subsequent experiments.
Figure 5. Systemic kynurenine decreases evoked glutamate levels in the PFC.
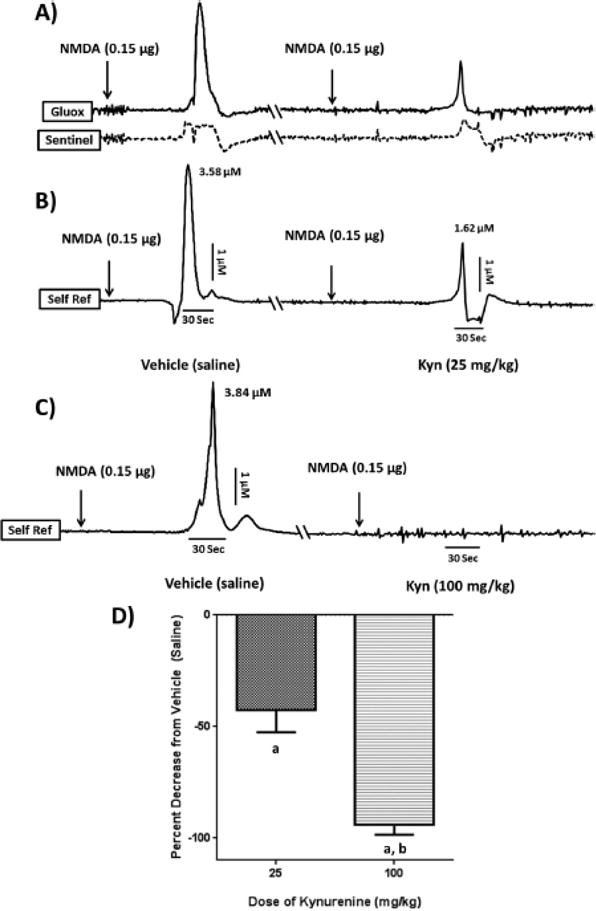
Each set of representative tracings (panels A–C) was recorded from the PFC of the same intact animal following an infusion of NMDA (0.15 μg in 0.5 μL) into the NAcSh 2 hrs after a systemic injection of either vehicle or kynurenine. Each tracing is separated by at least 24 hrs (cut \\ in the axis), and baselines are displayed uniformly for ease of comparison (see text for actual values). The vertical dimension represents changes in concentration (μM), whereas the horizontal dimension reflects the passage of time (sec). The timing of NMDA infusions relative to the glutamate peaks are indicated by arrows. (A): The solid-line tracing represents the signal from the glutamate-sensitive channel (Gluox), whereas the dashed tracing represents the signal from its adjacent sentinel channel; (B/C): Self-referenced (Self Ref) signals derived from subtracting the sentinel channel from the Gluox channel, thereby isolating the signal obtained exclusively from the oxidation of extracellular glutamate. NMDA causes a robust increase in PFC glutamate, which is attenuated after an injection of 25 mg/kg kynurenine (i.p.) (B) and essentially abolished after an injection of 100 mg/kg kynurenine (i.p.) (C); (D): Group data (n = 6/ group). a P < 0.05 vs. vehicle; b P < 0.05 vs. 25 mg/kg kynurenine.
In contrast to the effects on stimulated glutamate release, kynurenine treatment produced only a small attenuation of basal glutamate levels that did not reach statistical significance (25 mg/kg kynurenine group: saline = 1.67 ± 1.25 μM, kynurenine = 1.36 ± 0.98 μM; 100 mg/kg kynurenine group: saline = 0.80 ± 0.45 μM, kynurenine = 0.56 ± 0.25 μM, P > 0.05).
3.4 Experiment 4: Orally administered KAT II inhibitor does not affect basal or evoked glutamate release in the PFC under endogenous levels of KYNA
We next determined the impact of inhibition of endogenous KYNA synthesis via administration of BFF816 (30 and 100 mg/kg), on basal and stimulated glutamate release. BFF816 did not significantly affect basal glutamate levels (saline = 1.42 ± 1.09 μM; 30 BFF816 = 1.55 ± 1.11 μM; 100 BFF816 = 1.69 ± 1.26 μM; n = 5, F2,14 = 0.012, P = 0.988) in the absence of elevated kynurenine levels. Similarly, the decrease in endogenous KYNA levels produced by BFF816 (see Figure 3) did not significantly change NMDA-stimulated glutamate levels in the PFC (NMDA + saline = 5.11 ± 1.57 μM; NMDA + 30 BFF816 = 8.80 ± 1.64 μM; NMDA + 100 BFF816 = 7.53 ± 1.81 μM; n = 5, F2,14 = 1.249, P = 0.322).
3.5 Experiment 5: Effect of BFF816 + kynurenine on basal and evoked glutamate release in the PFC
The final set of experiments was designed to examine whether BFF816 could restore stimulated glutamate levels that had been attenuated following kynurenine administration (i.e. elevated levels of KYNA). Figures 6A–B illustrate the self-referenced recordings from individual animals. NMDA (0.15 μg) infusion into the NAcSh, 2 hours after i.p. saline (vehicle for kynurenine) + p.o. HPBCD (vehicle for BFF816), resulted in a phasic increase in PFC glutamate (Figure 6A) similar to that seen in Experiment 3. Again similar to Experiment 3, kynurenine (25 mg/kg), administered to the same animals in a separate test session, significantly attenuated the stimulated glutamate release (from 3.10 to 1.33 μM; Figure 6A middle section). 30 mg/kg BFF816 reduced the magnitude of the kynurenine-suppressed glutamate release by 14%, but this did not reach statistical significance (Figure 6A right section). In contrast, rats that received kynurenine and the higher dose of BFF816 (100 mg/kg) showed a partial, but significant, restoration of the evoked glutamate release relative to the kynurenine-induced suppression (by 41%; Figure 6B right section). These findings were replicated across a group of animals (Figure 6C), revealing a main effect of the two drug combinations (F2,20 = 31.505, P = 0.001) that interacted with the dose of BFF816 (F2,20 = 4.621, P = 0.022). Specifically, 30 mg/kg BFF816 partially prevented the suppressing effects of kynurenine on NMDA-evoked glutamate release (30 mg/kg BFF816 group: NMDA = 6.01 ± 1.57 μM; NMDA + kynurenine = 3.04 ± 1.36 μM, NMDA + kynurenine + BFF = 4.00 ± 0.87 μM, 15.6 ± 3.0% recovery from NMDA + kynurenine; F1,11 = 15.055, P = 0.003). Following the administration of kynurenine + 100 mg/kg BFF816, glutamate levels were significantly rescued (Figure 6C) and no longer significantly different from saline + HPBCD controls (100 mg/kg BFF816 group: NMDA = 4.76 ± 0.88 μM, NMDA + kynurenine = 0.79 ± 0.44 μM, NMDA + kynurenine + BFF = 4.01 ± 1.61 μM, 68.8 ± 19.4% rescue from NMDA + kynurenine; F(1,11) = 0.906, P = 0.364). Similar to previous experiments, basal glutamate levels were not significantly affected by kynurenine or BFF816 treatment (30 mg/kg BFF816 group: saline + HPBCD = 0.45 ± 0.18 μM, kynurenine + HPBCD = 0.31 ± 0.11 μM, kynurenine + BFF = 0.34 ± 0.08 μM; 100 mg/kg BFF816 group: saline + HPBCD = 0.44 ± 0.18 μM, kynurenine + HPBCD = 0.47 ± 0.15 μM, kynurenine + BFF = 0.50 ± 0.21 μM, P > 0.05).
Figure 6. BFF816 counteracts the attenuating effect of kynurenine on evoked glutamate levels in the PFC.
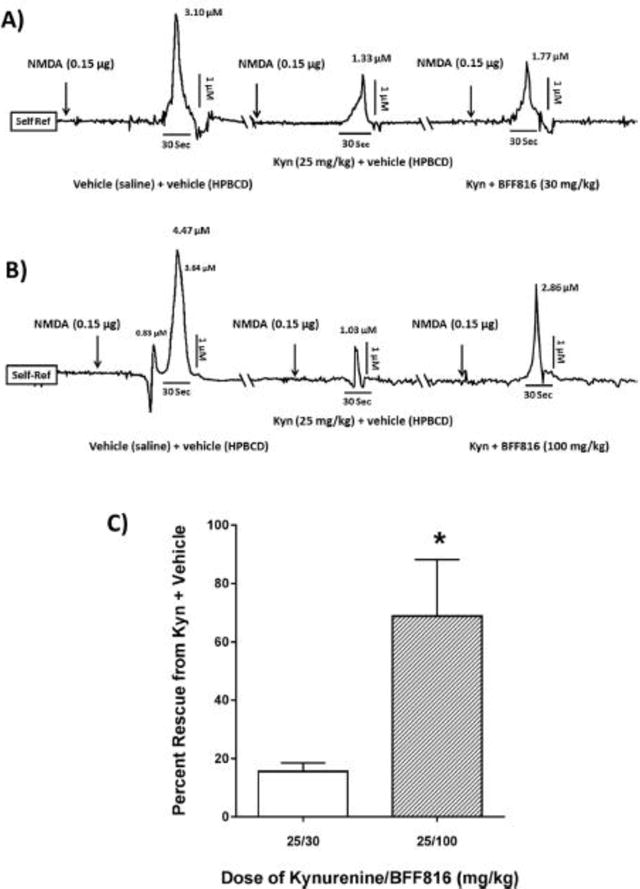
Two sets of self- referenced tracings (A,B) are taken from the same animal, separated by 24 hrs (cut \\ in the axis). (A): NMDA infused into the NAcSh after saline/HPBCD causes a robust increase in glutamate in the PFC (similar to Figure 5B). Administration of kynurenine (25 mg/kg, i.p.) + HPBCD 2 hrs prior to NMDA decreases stimulated glutamate release by 57%. Kynurenine + BFF816 (30 mg/kg, p.o.) modestly reduces this effect by 17%; (B): Using the same paradigm in a separate animal, a higher dose of BFF816 (100 mg/kg, p.o.) attenuates the kynurenine-induced reduction in evoked glutamate release by 41%; (C): Group data (n = 6/ group) illustrating percent rescue from kynurenine + vehicle. Percent rescue = BFF/kynurenine – HPBCD/kynurenine divided by HPBCD/saline. * P < 0.05 vs. the lower dose of BFF816 (Bonferroni adjusted t-test).
4.0 Discussion
The present experiments generated several novel results. First, systemic administration of kynurenine dose-dependently attenuates stimulated glutamate released in the PFC following an infusion of NMDA into the NAcSh. Second, BFF816, a selective KAT II inhibitor that can be administered orally and crosses the blood-brain barrier (Wu et al., 2014), substantively counteracts, in a dose-dependent manner, kynurenine-induced increases in extracellular KYNA levels. Finally, administration of BFF816 partially restores stimulated glutamate release in the PFC, and the magnitude of this effect is dependent upon the BFF816 dose used. Taken together, these data indicate that increases in cortical KYNA levels disrupt glutamate transmission in the PFC, and that both KYNA levels and glutamate transmission can be partially normalized via selective inhibition of KAT II activity. The following sections will address several issues raised by these experimental results and discuss the implications with respect to the etiology and therapeutics of SZ.
4.1 Intra-accumbens stimulation of prefrontal glutamate release
Infusions of NMDA into the NAcSh consistently elevate extracellular glutamate levels in the PFC, as demonstrated using rapid in situ electrochemistry (i.e. the MEA; Bortz et al., 2014; Bortz et al., 2016). The glutamate signal measured by this method has been referred to as phasic based on its high temporal resolution and faster kinetics (Burmeister & Gerhardt, 2001; Bortz et al., 2014; Parikh et al., 2008; Pocivavsek et al., SFN abstract 2014). It is not known, however, if this signal is caused by a single release event or by several signals that are captured under a single symmetrical wave form due to limited temporal resolution. In fact, the glutamate wave recorded in the present study was occasionally clearly split into what appeared to be multiple release events (Figure 6).
The extended latency (40-120 sec) between the NAcSh stimulation by NMDA and the increase in the glutamate signal in the PFC was unexpected, as trans-synaptic activation of the presumed neural circuit (NAcSh to basal forebrain to PFC), which is believed to mediate this effect (Zaborszky and Cullinan, 1992; Zmarowski et al., 2007; St Peters et al., 2011; Bortz et al., 2016), would take only msec to occur. The delayed release of prefrontal glutamate following intra-NAcSh NMDA therefore suggests a more complex mechanism. We are currently testing the hypothesis that protracted inhibitory processes initiated by interneurons within the NAcSh and/or the basal forebrain contribute to this phenomenon.
The increase in prefrontal glutamate levels following intra-accumbens infusions of NMDA has also been seen using microdialysis/HPLC methodology (Valentini et al., SFN abstract 2016). Measured by either MEA or microdialysis, this effect is secondary to an initial activation of local α7nAChRs (Bortz et al., 2016; Valentini et al., SFN abstract 2016), which are located on both neurons (Wang et al., 2006; Livingstone et al., 2010) and astrocytes (Duffy et al., 2011). Both cell types participate in the regulation of glutamatergic neurotransmission in the PFC (Wang et al., 2006; Vollbrecht et al., 2014), however, the relative roles of neuronal and astrocytic mechanisms remain to be determined. Notably, diverse cellular origins of glutamate may also account for the currently unexplained, different temporal profiles observed by microdialysis and MEA. Thus, glutamate levels measured by dialysis remained elevated for ~40 min after the infusion of NMDA into the NAcSh (Valentini et al., SFN abstract, 2014), whereas evoked glutamate levels determined by the sensor typically returned to baseline values in ~30-45 sec. Future studies will also need to elucidate why basal levels of glutamate are affected by kynurenine or BFF816 when measured by dialysis (Pocivavsek et al., 2016; Wu et al., 2010; Wu et al., 2014) but not by the MEA (Sections 3.3-3.5).
4.2 Effects of KAT II inhibition on KYNA and glutamate levels in the PFC
In line with our recent microdialysis study (Wu et al., 2014), BFF816 decreased basal extracellular KYNA levels in the PFC relatively modestly, to a nadir of 30%, following administration of the higher dose of the KAT II inhibitor (100 mg/kg). Interestingly, BFF816 was far more effective following an acute, kynurenine-induced elevation of KYNA levels, causing a reduction by almost 75% (Figures 3 and 4). Thus, as discussed in more detail recently (Pocivavsek et al., 2016), KAT II inhibition appears to preferentially affect a rapidly mobilizable pool of KYNA.
We demonstrated previously that the kynurenine-induced reduction of basal extracellular glutamate levels was secondary to the neosynthesis of KYNA (Konradsson-Geuken et al., 2010; Wu et al., 2010; Wu et al., 2014). Thus, we assumed that inhibition of KYNA production by BFF816 was responsible for the KAT II inhibitor’s ability to counteract the reduction of stimulated glutamate levels by kynurenine, as demonstrated in Experiment 5. However, Experiment 4 revealed that BFF816, when delivered alone, i.e. in the absence of exogenously supplied kynurenine, was insufficient to produce a significant increase in stimulated glutamate release, as assessed by MEA. This finding was unexpected, because both S-ESBA, perfused locally (Wu et al., 2010), and BFF816, delivered orally (Wu et al., 2014), increase basal glutamate levels in the PFC, as measured via microdialysis. We therefore conclude that endogenous KYNA does not regulate stimulated glutamate release unless KYNA levels are elevated. This notion is supported by another study where a systemically-administered KAT II inhibitor, PF-04859989, was shown to restore nicotine-induced glutamate transients in PFC after kynurenine injections, but had no effect in the absence of exogenous kynurenine (Koshy Cherian et al., 2014).
4.3 Translational implications
One of the key findings of the present study was that an acute elevation of KYNA inhibits NAcSh- mediated glutamate release in the PFC. As NAcSh stimulation via the same experimental protocol used here facilitates attentional processing in rodents (St Peters et al., 2011), the prefrontal glutamate release measured in these experiments may have cognitive relevance. Notably, glutamate transmission in the PFC is critical for several higher order cognitive functions that are deficient in SZ (Keefe, 2007; Kerns et al., 2008) such as cognitive flexibility (Stefani and Moghaddam, 2005), working memory (Aultman and Moghaddam, 2001), and sustained attention (Parikh et al., 2008). Thus, the present data provide further support to the notion, posited previously (see Muller et al., 2011; Schwarcz et al., 2012 for review), that the elevated KYNA levels seen in patients (Erhardt et al., 2001; Schwarcz et al., 2001) may play a role in the occurrence of cognitive deficits in SZ via its interference with cortical glutamate transmission.
If elevations in KYNA levels indeed contribute to the cognitive deficits in SZ, pharmacological interventions leading either to reduced uptake of kynurenine into the brain (Sekine et al., 2016) or to a reduction in KYNA neosynthesis within the brain may result in cognitive enhancement. Using the latter approach, pro-cognitive effects were indeed demonstrated with intracerebral application of the KAT II inhibitor S-ESBA (Pocivavsek et al., 2011), which does not cross the blood-brain barrier. Systemically active KAT II inhibitors such as BFF816 and PF-04859989 therefore represent excellent tools for hypothesis testing in pre-clinical studies (Dounay et al., 2015). BFF816 indeed improves performance in the Morris water maze after acute administration in intact rats (Wu et al., 2014). Moreover, preliminary results indicate that BFF816 is able to reverse cognitive deficits in a well-validated neurodevelopmental animal preparation that duplicates several cognitive impairments seen in SZ (Pocivavsek et al., SFN abstract 2014).
4.4 Conclusions
We showed here that acute elevation of KYNA dose-dependently inhibits stimulated glutamate release in the PFC. Co-administration of BFF816 reduced prefrontal KYNA levels and concurrently partially rescued glutamate release. The ability to both regulate KYNA production and attenuate the (often detrimental) effects of elevated KYNA in the PFC bodes well for the development of novel KAT II inhibitors (Lu et al., 2016), as well as other conceptually related, innovative approaches (Zakrocka et al., 2016), for the purpose of treating cognitive deficits in individuals with SZ.
Acknowledgments
This work was supported by USPHS grants RO1 MH083729 and P50 MH103222.
Abbreviations
- ACh
acetylcholine
- α7nAChR
alpha7 nicotinic acetylcholine receptor
- Gluox
glutamate-sensitive
- HPBCD
hydroxyl-propyl-beta-cyclodextrin
- KYNA
kynurenic acid
- KAT II
kynurenine aminotransferase-II
- MEA
microelectrode array
- NMDAR
N-methyl-D-aspartate receptor
- NAcSh
nucleus accumbens shell
- PFC
prefrontal cortex
- SZ
schizophrenia
References
- Alexander KS, Pocivavsek A, Wu HQ, Pershing ML, Schwarcz R, Bruno JP. Early developmental elevations of brain kynurenic acid impair cognitive flexibility in adults: reversal with galantamine. Neuroscience. 2013;238:19–28. doi: 10.1016/j.neuroscience.2013.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KS, Wu HQ, Schwarcz R, Bruno JP. Acute elevations of brain kynurenic acid impair cognitive flexibility: normalization by the alpha7 positive modulator galantamine. Psychopharmacology (Berl) 2012;220:627–637. doi: 10.1007/s00213-011-2539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology (Berl) 2001;153:353–364. doi: 10.1007/s002130000590. [DOI] [PubMed] [Google Scholar]
- Beggiato S, Antonelli T, Tomasini MC, Tanganelli S, Fuxe K, Schwarcz R, Ferraro L. Kynurenic acid, by targeting alpha7 nicotinic acetylcholine receptors, modulates extracellular GABA levels in the rat striatum in vivo. Eur J Neurosci. 2013;37:1470–1477. doi: 10.1111/ejn.12160. [DOI] [PubMed] [Google Scholar]
- Beggiato S, Tanganelli S, Fuxe K, Antonelli T, Schwarcz R, Ferraro L. Endogenous kynurenic acid regulates extracellular GABA levels in the rat prefrontal cortex. Neuropharmacology. 2014;82:11–18. doi: 10.1016/j.neuropharm.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Bortz DM, Jorgensen CV, Mikkelsen JD, Bruno JP. Transient inactivation of the ventral hippocampus in neonatal rats impairs the mesolimbic regulation of prefrontal glutamate release in adulthood. Neuropharmacology. 2014;84:19–30. doi: 10.1016/j.neuropharm.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Bortz DM, Upton BA, Mikkelsen JD, Bruno JP. Positive allosteric modulators of the alpha7 nicotinic acetylcholine receptor potentiate glutamate release in the prefrontal cortex of freely-moving rats. Neuropharmacology. 2016;111:78–91. doi: 10.1016/j.neuropharm.2016.08.033. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal Chem. 2001;73:1037–1042. doi: 10.1021/ac0010429. [DOI] [PubMed] [Google Scholar]
- Ceresoli-Borroni G, Rassoulpour A, Wu HQ, Guidetti P, Schwarcz R. Chronic neuroleptic treatment reduces endogenous kynurenic acid levels in rat brain. J Neural Transm (Vienna) 2006;113:1355–1365. doi: 10.1007/s00702-005-0432-z. [DOI] [PubMed] [Google Scholar]
- Dounay AB, Anderson M, Bechle BM, Evrard E, Gan X, Kim JY, McAllister LA, Pandit J, Rong S, Salafia MA, Tuttle JB, Zawadzke LE, Verhoest PR. PF-04859989 as a template for structure-based drug design: identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorg Med Chem Lett. 2013;23:1961–1966. doi: 10.1016/j.bmcl.2013.02.039. [DOI] [PubMed] [Google Scholar]
- Dounay AB, Tuttle JB, Verhoest PR. Challenges and Opportunities in the Discovery of New Therapeutics Targeting the Kynurenine Pathway. J Med Chem. 2015;58:8762–8782. doi: 10.1021/acs.jmedchem.5b00461. [DOI] [PubMed] [Google Scholar]
- Duffy AM, Fitzgerald ML, Chan J, Robinson DC, Milner TA, Mackie K, Pickel VM. Acetylcholine alpha7 nicotinic and dopamine D2 receptors are targeted to many of the same postsynaptic dendrites and astrocytes in the rodent prefrontal cortex. Synapse. 2011;65:1350–1367. doi: 10.1002/syn.20977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14:1–21. [PubMed] [Google Scholar]
- Erhardt S, Blennow K, Nordin C, Skogh E, Lindstrom LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. 2001;313:96–98. doi: 10.1016/s0304-3940(01)02242-x. [DOI] [PubMed] [Google Scholar]
- Gold JM. Cognitive deficits as treatment targets in schizophrenia. Schizophr Res. 2004;72:21–28. doi: 10.1016/j.schres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Amori L, Sapko MT, Okuno E, Schwarcz R. Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. J Neurochem. 2007;102:103–111. doi: 10.1111/j.1471-4159.2007.04556.x. [DOI] [PubMed] [Google Scholar]
- Han Q, Cai T, Tagle DA, Robinson H, Li J. Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci Rep. 2008;28:205–215. doi: 10.1042/BSR20080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe RS. Cognitive deficits in patients with schizophrenia: effects and treatment. J Clin Psychiatry. 2007;68(Suppl 14):8–13. [PubMed] [Google Scholar]
- Keefe RS, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, Meltzer HY, Green MF, Capuano G, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Davis CE, Hsiao JK, Lieberman JA, Investigators, C., Neurocognitive Working, G. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007a;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- Keefe RS, Sweeney JA, Gu H, Hamer RM, Perkins DO, McEvoy JP, Lieberman JA. Effects of olanzapine, quetiapine, and risperidone on neurocognitive function in early psychosis: a randomized, double-blind 52-week comparison. Am J Psychiatry. 2007b;164:1061–1071. doi: 10.1176/ajp.2007.164.7.1061. [DOI] [PubMed] [Google Scholar]
- Kerns JG, Nuechterlein KH, Braver TS, Barch DM. Executive functioning component mechanisms and schizophrenia. Biol Psychiatry. 2008;64:26–33. doi: 10.1016/j.biopsych.2008.04.027. [DOI] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Gash CR, Alexander K, Pomerleau F, Huettl P, Gerhardt GA, Bruno JP. Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats. Synapse. 2009;63:1069–1082. doi: 10.1002/syn.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Wu HQ, Gash CR, Alexander KS, Campbell A, Sozeri Y, Pellicciari R, Schwarcz R, Bruno JP. Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience. 2010;169:1848–1859. doi: 10.1016/j.neuroscience.2010.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy Cherian A, Gritton H, Johnson DE, Young D, Kozak R, Sarter M. A systemically-available kynurenine aminotransferase II (KAT II) inhibitor restores nicotine-evoked glutamatergic activity in the cortex of rats. Neuropharmacology. 2014;82:41–48. doi: 10.1016/j.neuropharm.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak R, Campbell BM, Strick CA, Horner W, Hoffmann WE, Kiss T, Chapin DS, McGinnis D, Abbott AL, Roberts BM, Fonseca K, Guanowsky V, Young DA, Seymour PA, Dounay A, Hajos M, Williams GV, Castner SA. Reduction of brain kynurenic acid improves cognitive function. J Neurosci. 2014;34:10592–10602. doi: 10.1523/JNEUROSCI.1107-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Livingstone PD, Dickinson JA, Srinivasan J, Kew JN, Wonnacott S. Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by Alpha7 nicotinic receptors and potentiated by PNU-120596. J Mol Neurosci. 2010;40:172–176. doi: 10.1007/s12031-009-9232-5. [DOI] [PubMed] [Google Scholar]
- Lu H, Kopcho L, Ghosh K, Witmer M, Parker M, Gupta S, Paul M, Krishnamurthy P, Laksmaiah B, Xie D, Tredup J, Zhang L, Abell LM. Development of a RapidFire mass spectrometry assay and a fluorescence assay for the discovery of kynurenine aminotransferase II inhibitors to treat central nervous system disorders. Anal Biochem. 2016;501:56–65. doi: 10.1016/j.ab.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Muller N, Myint AM, Schwarz MJ. Kynurenine pathway in schizophrenia: pathophysiological and therapeutic aspects. Curr Pharm Des. 2011;17:130–136. doi: 10.2174/138161211795049552. [DOI] [PubMed] [Google Scholar]
- Nematollahi A, Sun G, Harrop SJ, Hanrahan JR, Church WB. Structure of the PLP-Form of the Human Kynurenine Aminotransferase II in a Novel Spacegroup at 1.83 A Resolution. Int J Mol Sci. 2016;17:446. doi: 10.3390/ijms17040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Man K, Decker MW, Sarter M. Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex. J Neurosci. 2008;28:3769–3780. doi: 10.1523/JNEUROSCI.5251-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G, Hartmann S, Lorenz B, Wollenburg C, Baran L, Przegalinski E, Kostowski W, Krzascik P, Chizh B, Headley PM. Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther. 1997;283:1264–1275. [PubMed] [Google Scholar]
- Passera E, Campanini B, Rossi F, Casazza V, Rizzi M, Pellicciari R, Mozzarelli A. Human kynurenine aminotransferase II–reactivity with substrates and inhibitors. FEBS J. 2011;278:1882–1900. doi: 10.1111/j.1742-4658.2011.08106.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th. Academic Press; San Diego: 1998. [DOI] [PubMed] [Google Scholar]
- Pershing ML, Bortz DM, Pocivavsek A, Fredericks PJ, Jorgensen CV, Vunck SA, Leuner B, Schwarcz R, Bruno JP. Elevated levels of kynurenic acid during gestation produce neurochemical, morphological, and cognitive deficits in adulthood: implications for schizophrenia. Neuropharmacology. 2015;90:33–41. doi: 10.1016/j.neuropharm.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershing ML, Phenis D, Valentini V, Pocivavsek A, Lindquist DH, Schwarcz R, Bruno JP. Prenatal kynurenine exposure in rats: age-dependent changes in NMDA receptor expression and conditioned fear responding. Psychopharmacology (Berl) 2016;233:3725–3735. doi: 10.1007/s00213-016-4404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Notarangelo FM, Wu HQ, Bruno JP, Schwarcz R. Astrocytes as pharmacological targets in the treatment of schizophrenia: focus on kynurenic acid. In: Pletnikov M, Waddington J, editors. Modeling the Psychopathological Dimensions of Schizophrenia Handbooks of Behavioral Neuroscience. Vol. 23. Elsevier; 2016. pp. 423–443. [Google Scholar]
- Pocivavsek A, Wu HQ, Elmer GI, Bruno JP, Schwarcz R. Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur J Neurosci. 2012;35:1605–1612. doi: 10.1111/j.1460-9568.2012.08064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Wu HQ, Potter MC, Elmer GI, Pellicciari R, Schwarcz R. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology. 2011;36:2357–2367. doi: 10.1038/npp.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu HQ, Schwarcz R. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology. 2010;35:1734–1742. doi: 10.1038/npp.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassoulpour A, Wu HQ, Ferre S, Schwarcz R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J Neurochem. 2005;93:762–765. doi: 10.1111/j.1471-4159.2005.03134.x. [DOI] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J Neurochem. 2007;102:712–722. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13:465–477. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry. 2001;50:521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- Sekine A, Kuroki Y, Urata T, Mori N, Fukuwatari T. Inhibition of Large Neutral Amino Acid Transporters Suppresses Kynurenic Acid Production Via Inhibition of Kynurenine Uptake in Rodent Brain. Neurochem Res. 2016;41:2256–2266. doi: 10.1007/s11064-016-1940-y. [DOI] [PubMed] [Google Scholar]
- St Peters M, Demeter E, Lustig C, Bruno JP, Sarter M. Enhanced control of attention by stimulating mesolimbic-corticopetal cholinergic circuitry. J Neurosci. 2011;31:9760–9771. doi: 10.1523/JNEUROSCI.1902-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Transient N-methyl-D-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry. 2005;57:433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Tuttle JB, Anderson M, Bechle BM, Campbell BM, Chang C, Dounay AB, Evrard E, Fonseca KR, Gan X, Ghosh S, Horner W, James LC, Kim JY, McAllister LA, Pandit J, Parikh VD, Rago BJ, Salafia MA, Strick CA, Zawadzke LE, Verhoest PR. Structure-Based Design of Irreversible Human KAT II Inhibitors: Discovery of New Potency-Enhancing Interactions. ACS Med Chem Lett. 2013;4:37–40. doi: 10.1021/ml300237v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollbrecht PJ, Simmler LD, Blakely RD, Deutch AY. Dopamine denervation of the prefrontal cortex increases expression of the astrocytic glutamate transporter GLT-1. J Neurochem. 2014;130:109–114. doi: 10.1111/jnc.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BW, Liao WN, Chang CT, Wang SJ. Facilitation of glutamate release by nicotine involves the activation of a Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse. 2006;59:491–501. doi: 10.1002/syn.20267. [DOI] [PubMed] [Google Scholar]
- Wu HQ, Okuyama M, Kajii Y, Pocivavsek A, Bruno JP, Schwarcz R. Targeting kynurenine aminotransferase II in psychiatric diseases: promising effects of an orally active enzyme inhibitor. Schizophr Bull. 2014;40(Suppl 2):S152–158. doi: 10.1093/schbul/sbt157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R. The astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J Mol Neurosci. 2010;40:204–210. doi: 10.1007/s12031-009-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Cullinan WE. Projections from the nucleus accumbens to cholinergic neurons of the ventral pallidum: a correlated light and electron microscopic double-immunolabeling study in rat. Brain Res. 1992;570:92–101. doi: 10.1016/0006-8993(92)90568-t. [DOI] [PubMed] [Google Scholar]
- Zakrocka I, Turski WA, Kocki T. Angiotensin-converting enzyme inhibitors modulate kynurenic acid production in rat brain cortex in vitro. Eur J Pharmacol. 2016;789:308–312. doi: 10.1016/j.ejphar.2016.07.023. [DOI] [PubMed] [Google Scholar]
- Zmarowski A, Sarter M, Bruno JP. Glutamate receptors in nucleus accumbens mediate regionally selective increases in cortical acetylcholine release. Synapse. 2007;61:115–123. doi: 10.1002/syn.20354. [DOI] [PubMed] [Google Scholar]
Web References
- Thomases DR, Flores-Barrera E, Bruno JP, Schwarcz R, Tseng KY, Society for Neuroscience abstract Contribution of alpha-7 nAChR tone in sustaining the gain of GABAergic transmission in the adult prefrontal cortex. 2014 Available at http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=be19888d-ce89-477d-a053-e0afc808da6a&cKey=621d6c10-05ff-4ddb-9426-87e2c97b53f0&mKey=54c85d94-6d69-4b09-afaa-502c0e680ca7 (accessed 12.19.16)
- Pocivavsek A, Wu H-Q, Okuyama M, Kajii Y, Elmer GI, Bruno JP, Schwarcz R, Society for Neuroscience abstract The systemically active kynurenine aminotransferase II inhibitor BFF816 attenuates contextual memory deficit induced by chronic prenatal kynurenine elevation in rats. 2014 Available at http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=37d17ee9-b44a-43e9-ab3f-dd51ebb2c555&cKey=00fd86e0-6701-47f1-b455-39717485110e&mKey=%7b54C85D94-6D69-4B09-AFAA-502C0E680CA7%7d (accessed 12.19.16)
- Valentini V, Schumacher JD, Phenis D, Bortz DM, Bruno JP, Society for Neuroscience abstract Accumbens-prefrontal interactions in the regulation of multiple transmitter systems: implications for cognitive deficits in schizophrenia. 2016 Available at http://www.abstractsonline.com/pp8/index.html#!/4071/presentation/17935 (accessed 12.19.16)