

Abstract
The Developmental Origins of Health and Disease hypothesis predicts that early-life environmental exposures can be detrimental to later-life health and that mismatch between the pre- and post-natal environment may contribute to the growing non-communicable disease epidemic. Within this is an increasingly recognized role for epigenetic mechanisms; for example, epigenetic modifications can be influenced by nutrition and can alter gene expression in mothers and offspring. Currently, there are few whole-genome transcriptional studies of response to nutritional alteration. Thus, we sought to explore how nutrition affects the expression of genes involved in epigenetic processes in Drosophila melanogaster. We manipulated Drosophila food macronutrient composition at the F0 generation, mismatched F1 offspring back to a standard diet and analysed the transcriptome of the F0–F3 generations by RNA sequencing. At F0, the altered (high-protein, low-carbohydrate) diet increased expression of genes classified as having roles in epigenetic processes, with co-ordinated down-regulation of genes involved in immunity, neurotransmission and neurodevelopment, oxidative stress and metabolism. Upon reversion to standard nutrition, mismatched F1 and F2 generations displayed multigenerational inheritance of altered gene expression. By the F3 generation, gene expression had reverted to F0 (matched) levels. These nutritionally induced gene expression changes demonstrate that dietary alterations can up-regulate epigenetic genes, which may influence the expression of genes with broad biological functions. Furthermore, the multigenerational inheritance of the gene expression changes in F1 and F2 mismatched generations suggests a predictive adaptive response to maternal nutrition, aiding the understanding of the interaction between maternal diet and offspring health, with direct implications for the current non-communicable disease epidemic.
Keywords: environment, nutrition, Drosophila, epigenetics, transriptomics, mismatch
Background
Exposure to aberrant or harmful environments during development and early life can be detrimental to later life health. From these observations is derived the Developmental Origins of Health and Disease (DOHaD) hypothesis [1–4], which seeks to explain why the period from conception to birth and the first few years of life is critical for determining life-long susceptibility to non-communicable diseases (NCDs). Many NCD phenotypes are thought to be caused by developmental perturbations that are a consequence of altered epigenetic marks [2, 5], induced by environmental exposure during critical periods of development [6]. Alteration to the epigenome regulates gene expression through DNA methylation, histone and chromatin modifications [7, 8] providing plasticity to the genome. Consequently, phenotypes under epigenetic regulation provide a pathway through which the genome can interact with the environment [9]. If the epigenetic modifications occur at a time during which they are able to affect the germ line, such modifications may also influence development of the offspring [6].
The interaction between the environment and the epigenome and the resulting phenotypic adaptations, coupled with the growing NCD epidemic, has led to the predictive adaptive response (PAR) hypothesis [10]. This hypothesis states that nature of the PAR is determined by the degree of mismatch between the foetal pre-natal and its ultimate post-natal environments. This mismatch results from the information that the foetus receives on environmental conditions while in utero, to which it will respond adaptively by programming its biology to expect that environment. If the actual post-natal environment matches the prenatal prediction, then the PARs are appropriate and disease risk is low; if they do not match then the PAR is inappropriate, and disease risk is increased. For instance, obesity has a distinct epigenetic profile. This pattern could be established in early life as a response to the maternal, foetal and/or early post-natal environment, and later-life nutritional mismatch could mean that the individual has been programmed inappropriately, leading to an increased risk of obesity and associated diseases later in life [11]. This implies that the epigenetic hallmarks of early-life exposures may be able to be maintained or ‘stored’ in such a way as to produce long-lasting effects [12].
Along with stress, drugs and environmental toxicants, one of the main factors that can cause epigenetic perturbation is nutrition; evidence from humans, supported by experiments in rodents, suggest that early-life nutrition can affect the long-term health not only of the individual but also of their offspring [13–15], potentially through epigenetic mechanisms [16–20]. Consistent with the DOHaD hypothesis, strong links exist between both maternal and early-life nutrition and cardiovascular disease [21], diabetes and obesity [22] along with asthma and allergy, autoimmune disease, cancer and mental health [23–26]. Inappropriate maternal nutrition in rodents has been linked to incorrect epigenetic ‘priming’ during foetal or post-natal life [27, 28]; in particular, experiments conducted in Drosophila show that diets high in carbohydrate content (sugar) have been shown to programme metabolic status and diabetes [29, 30].
Many nutrition-related phenotypes have been attributed to changes in epigenetic processes and a classic example of this is that of methyl supplementation, which influences coat colour in agouti mice [31]. Nutrition influences are separated into direct effects (on the mother/father themselves) and also indirect (on the offspring or maternal provisioning) effects. Direct effects can be exemplified by high-fat diets inducing obesity and metabolic syndrome, due to the methylation pattern of particular genes and promoters involved in body weight and adipocyte differentiation such as leptin [32] and peroxisome proliferator-activated receptor gamma (PPARγ) [33] and the differential DNA methylation detected in metabolic syndrome in both humans and rodents [34, 35]. Indirect effects on offspring metabolic phenotype can occur via maternal diet. For example, rodents exposed to high-fat diets in utero have altered epigenetic patterns and methylation status of particular genes, for example those expressed in and secreted by adipose tissue such as adiponectin and leptin genes [36]. Further, a high-fat diet during pregnancy and lactation can induce epigenetic modifications and differential expression of the μ-opioid receptor (involved in drug metabolism), and corresponding hypomethylation of the promoter regions of the gene, in mouse offspring [37]. Additionally, maternal protein restriction in rodents can cause hypomethylation of particular genes involved in metabolic processes in foetus and offspring such as those that regulate metabolism in the liver [38, 39], those that contribute to cholesterol and fatty acid metabolism [40] and those that regulate metabolic pathways that control lipid metabolism between the liver and adipose tissue [41] and can also affect methylation in the developing placenta [42]. Such epigenetic perturbation is not just limited to foetal and early life; the post-natal period is also susceptible to the epigenetic effects of nutrition. For example, hypermethylation of the promoter region of the anorexigenic neurohormone proopipmelanocortin occurs in overfed rats [43], and post-natal folic acid supplementation can lead to hypermethylation of peroxisome proliferator-activated receptor alpha (PPARα), a nuclear transcription factor gene [44]. This sensitivity of the epigenome to the effects of the environment (nutrition) also extends into adulthood, where epigenetic changes in response to nutritional changes have been observed in rats [45–47].
In addition to metabolic genes, altered nutrition appears to have broader genomic consequences. For instance, a study in dairy cattle showed that nutrition can also alter markers of inflammation and oxidative stress [48]; in rats, a protein-restricted diet in pregnancy leads to an increased susceptibility to oxidative stress in offspring [49], while in humans, a high carbohydrate diet increases the oxidative stress response [50]. A high-fat, high-carbohydrate meal can induce oxidative and inflammatory stress as reflected by increased reactive oxygen species (ROS) generation in both normal weight [51] and in obese people [52], suggesting that oxidative stress and inflammation are major mechanisms involved in metabolic disorders associated with obesity [53] and can also induce epigenetic changes [35]. Environmental stress can induce DNA and histone-modified changes in gene expression in organisms ranging from plants [54] to humans [55]. In terms of applicability to health, we know that low doses of ROS, from calorie-restricted or high-carbohydrate diets, promote health and lifespan in numerous species [56]. Thus, considering the above, along with the PAR hypothesis, it is likely that such biological responses to nutrition reflect the idea that induced epigenetic changes that underpin physiological change and aid in the adaptation of an individual, and potentially its offspring, to an adverse environment [57].
Thus, considering that gene-specific studies of altered nutrition have demonstrated broad and diverse genetic and epigenetic consequences, it is pertinent to apply this concept to the whole genome. Nutrition is commonly investigated as an environmental factor that is expected to influence the epigenetic landscape, and there are several examples in the literature of response to altered nutrition being inherited multigenerationally [27, 58–60] and transgenerationally (F3 and beyond, [31]). As such, altered gene expression, via epigenetic marks in response to nutrition, coupled with the PAR hypothesis, could be the key to understanding the prevalence of obesity and metabolic syndrome. Here, we explore the PAR hypothesis and the ability of nutrition to affect gene expression at a whole-genome level by manipulating the diet of the fruitfly, Drosophila melanogaster, to investigate the extent to which gene expression is changed by differing levels of macronutrients. Previous research has shown that a high-sugar maternal diet can alter the body composition of larval Drosophila offspring for at least two generations [29], as well as demonstrating that nutrition is able to influence traits relative to metabolic syndrome, longevity and the immune response [30, 61, 62]. Further, evidence also exists of a multigenerational response to the maternal condition (immune challenge, maternal age, Nystrand and Dowling, 2014 [63]). Considering such traits and responses are often under epigenetic control, we predict that dietary manipulation will have broad consequences for the expression of genes involved in epigenetic processes.
Methods
Fly Husbandry
Drosophila melanogaster stocks used in this study were wild-type Canton-S flies from the Bloomington Drosophila Stock Center at Indiana University. Drosophila were cultured in a dedicated invertebrate laboratory using standard techniques. Briefly, flies were maintained in laboratory incubators at 25°C in a P Selecta HOTCOLD-C incubator. Larvae were reared on either a standard low-protein high-carbohydrate (LPHC, standard laboratory fly food) or a high-protein low-carbohydrate (HPLC) diet. These differential diets consisted of standard brewer’s yeast (Health2000, New Zealand), sugar (New Zealand Sugar Company, Auckland, New Zealand) and cornmeal (Health 2000) in varying ratios (Table 1). Agar (A7002, Sigma-Aldrich, St Louis, MO, USA), propionic acid (Thermo Fisher, New Zealand, AJA693) and Nipagin (47889, Sigma-Aldrich, 10% w/v in 100% ethanol) were added in equal amounts. Gross energy (kJ/g) of both the LPHC and HPLC food types was determined by bomb calorimetry, and total protein content (%) was calculated using the total combustion method (Table 1) by the Institute of Food, Nutrition and Human Health at Massey University, Palmerston North, New Zealand.
Table 1:
Fly diet components and content information
| Component | High protein | Low protein |
|---|---|---|
| Agar (g) | 9 | 9 |
| Cornmeal (g) | 66.7 | 66.7 |
| Sugar (g) | 31.24 | 46.7 |
| Yeast (g) | 148.76 | 16.7 |
| Propionic acid (ml) | 6.6 | 6.6 |
| Nipagen (ml) | 5 | 5 |
| Water (l) | 1 | 1 |
| Protein (%) | 8 | 5.3 |
| Gross energy (kJ/g) | 0.9 | 2.1 |
| Yeast:sugar ratio | 1:0.2 | 1:2.8 |
Nutrition Experiments
Drosophila were manipulated under anaesthesia (CO2) and initially raised on LPHC (low-protein, high-carbohydrate, standard fly) food. To enable mating, 50 female flies were segregated within 4 h of eclosion and incubated with 10–15 male flies, on LPHC food, for 24 h. Female flies were separated and incubated on either LPHC or HPLC diet. Female flies laid eggs in their specified food and the F1 offspring from the HPLC diet were either maintained on HPLC, or mismatched onto LPHC diet (Fig. 1). The further offspring then remained on those matched or mismatched diets, relative to the F0 generation. Thus, the biological mismatch relates to flies from an HPLC (non-standard) dietary background that are mismatched onto an LPHC background.
Figure 1:
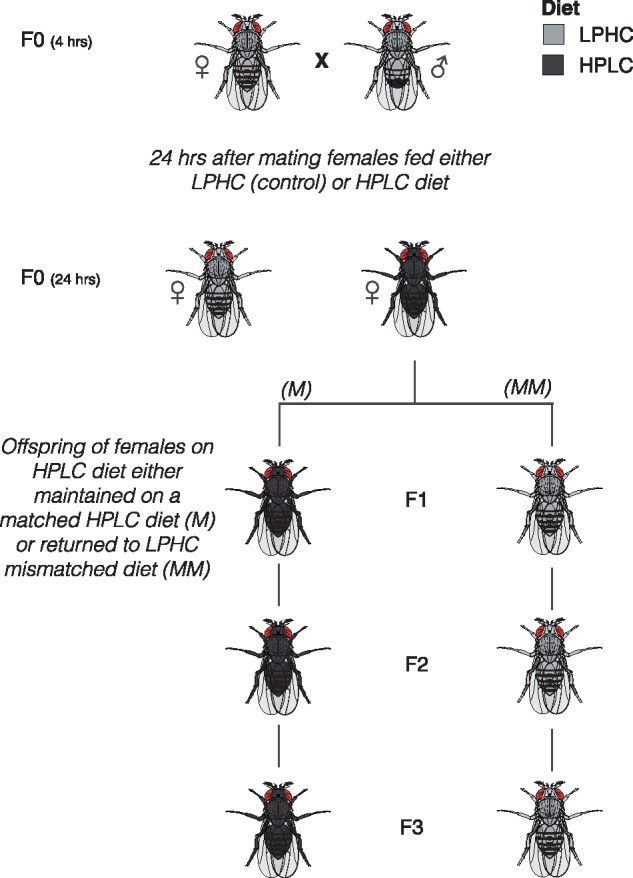
Fly diet experiments. LPHC, low-protein, high-carbohydrate (standard) diet; HPLC, high-protein low-carbohydrate diet; F1(M), F2(M) and F3(M), flies maintained on LPHC diet for three generations; F1(MM), F2(MM) and F3(MM), flies that were raised on HPLC in the F0 generation, and mismatched back to LPHC at eclosion in the F1 generation and were maintained for two more generations on the mismatched (LPHC) diet. Two replicates of each condition were used for transcriptomic experiments.
At each generation, RNA was extracted from female Drosophila at 5 days post-eclosion using a modified kit protocol (Supplementary File 1). Following the extraction process, RNA was stored at −80 °C until needed.
Transcriptomic Experiments
Two F0 generation replicates from each of the LPHC and HPLC diets and two replicates from the F1, F2 and F3 generations with matched and mismatched diets were prepared, resulting in 16 total RNA samples submitted to the Otago Genomics and Bioinformatics Facility at the University of Otago (Dunedin, New Zealand) under contract to the New Zealand Genomics Limited for library construction and sequencing. The libraries were prepared using TruSeq stranded mRNA sample preparation kit according to the manufacturer’s protocol (Illumina). All libraries were normalised, pooled and pair-end sequenced on 2 lanes of high-output flowcell HiSeq 2500, V3 chemistry (Illumina), generating 100 bp reads. Libraries had an average insert size of ∼208 bp.
Transcriptomic output was analysed in CLC Genomics Workbench Version 8.5.1. Reads were aligned to the D.melanogaster reference genome (BDGP6) as implemented in CLC Genomics Workbench, and differential gene expression (EDGE test) was calculated between samples using an absolute fold change value of >1.5, and an false discovery rate-corrected P-value of <0.001. These values were selected so that the fold change was of high enough magnitude that it could be validated in the lab by Nanostring, and the P-value was stringent to reduce false positives. Transcriptomic data were validated by Nanostring: samples were submitted to the Otago Genomics and Bioinformatics Facility at the University of Otago (Dunedin, New Zealand) under contract to the New Zealand Genomics Limited for nCounter Custom Gene Expression assays (Nanostring). Total RNA (100 ng) in a total volume of 5 µL was processed using the standard nCounter XT Total RNA protocol [64]. Raw data were exported and QC-checked using Nanostring’s nSolver data analysis tool (www.nanostring.com). Per the Nanostring CodeSet design criteria, 25 candidate genes for validation were chosen, including two housekeeping genes incorporated (Mnf and Rpl32, Table 2). Raw data were normalized to the geometric mean of both the positive controls (included in the hybridisation steps) and the nominated housekeeping genes. Normalized Nanostring data were compared with transcriptomic data, and the Pearson’s correlation coefficient was calculated in R [65].
Table 2:
Codeset design for Nanostring
| Gene | Accession | Position | NSID |
|---|---|---|---|
| asf1 | NM_079439.2 | 601–700 | NM_079439.2: 600 |
| Caf1-105 | NM_136745.3 | 1231–1330 | NM_136745.3: 1230 |
| Def | NM_078948.2 | 8–107 | NM_078948.2: 7 |
| E(bx) | NM_167819.2 | 4681–4780 | NM_167819.2: 4680 |
| E(Pc) | NM_078974.2 | 2551–2650 | NM_078974.2: 2550 |
| E(spl)m4-BFM | NM_079786.1 | 241–340 | NM_079786.1: 240 |
| E(spl)m5-HLH | NM_079787.2 | 396–495 | NM_079787.2: 395 |
| Fbp1 | NM_079341.1 | 3001–3100 | NM_079341.1: 3000 |
| Mnf | NM_168444.1 | 866–965 | NM_168444.1: 865 |
| Hml | NM_079336.2 | 7236–7335 | NM_079336.2: 7235 |
| Hmt4-20 | NM_130497.2 | 3201–3300 | NM_130497.2: 3200 |
| Inos | NM_058057.4 | 1026–1125 | NM_058057.4: 1025 |
| Lip3 | NM_057983.3 | 946–1045 | NM_057983.3: 945 |
| Lsd-1 | NM_170092.2 | 836–935 | NM_170092.2: 835 |
| Lsp1beta | NM_057276.3 | 1351–1450 | NM_057276.3: 1350 |
| Ocho | NM_080514.1 | 416–515 | NM_080514.1: 415 |
| Pc | NM_079475.2 | 991–1090 | NM_079475.2: 990 |
| Pgm | NM_079936.2 | 841–940 | NM_079936.2: 840 |
| Rpl32 | NM_170461.1 | 342–441 | NM_170461.1: 341 |
| Sap30 | NM_132934.2 | 216–315 | NM_132934.2: 215 |
| Su(var)3-3 | NM_140937.2 | 1781–1880 | NM_140937.2: 1780 |
| Su(var)3-7 | NM_079618.2 | 3516–3615 | NM_079618.2: 3515 |
| Top2 | NM_057412.3 | 3111–3210 | NM_057412.3: 3110 |
| Tps1 | NM_134983.2 | 2056–2155 | NM_134983.2: 2055 |
| Ubx | NM_206497.1 | 1321–1420 | NM_206497.1: 1320 |
Gene, gene symbol based on FlyBase nomenclature; NSID, Nanostring internal identifier; Position, region in the target mRNA being probed.
Gene Ontology Analyses
Functional annotation clustering (FAC) was undertaken in the Database for Annotation, Visualization and Integration of Discovery (DAVID) v6.8 [66, 67] with the following categories: COG_ONTOLOGY, GOTERM_BP_DIRECT, GOTERM_CC_DIRECT, GOTERM_MF_DIRECT, KEGG_PATHWAY and INTERPRO. Up- and down-regulated genes from the F0 generation were submitted separately to DAVID for FAC and analysed against a background of genes that were expressed and detectable in this dataset, to identify gene oncology (GO) terms that were significantly enriched between the HPLC and LPHC F0 generation.
Statistical Analyses
Significant differences in mean gene expression between different dietary conditions and generations were calculated via analysis of variance (ANOVA) and Tukey’s posthoc testing, as implemented in R [65].
Results
RNAseq Data
Summary statistics for transcriptomic work is shown in Supplementary File 2. Briefly, each sample yielded between 3164 Mbases and 4286 Mbases (average 4001 Mbases), with an average number of reads of 32 006 648 reads (range 25 310 758–34 288 584 reads). The mean quality score was an average of 36 Q (range 35.4–35.72 Q).
Differential Gene Expression
Of 17 490 genes annotated in the Drosophila genome and contained within the CLC reference database, 12 424 were expressed and detected across generations in these transcriptomic experiments. At F0, of these 12 424 genes, 2946 were differentially expressed between HPLC and LPHC [1074 (8.6%) down-regulated and 1872 (15.1%) up-regulated (Supplementary File 3)], with an absolute fold change of >1.5, and an FDR-corrected P-value of <0.001, as determined by EDGE test as implemented in CLC table.
Gene Ontology
DAVID uses a clustering approach to reduce redundancy; GO terms that are similar, are clustered together. Each term within the cluster is given a P-value, and the cluster itself is given an enrichment score (the geometric mean in –log scale of the individual GO term P-values). An enrichment score of 1.3 is equivalent to a non-log P-value of 0.05.
Functional annotation clustering of genes that are up-regulated in HPLC vs. LPHC indicated that the data set was highly enriched for genes that are involved in epigenetic processes such as chromatin binding [enrichment score (ES) 10.64], DNA replication (ES 4.64), chromatin regulation (ES 1.62), histone binding and phosphorylation (ES 1.60) (Table 3). Conversely, clustering of genes that are down-regulated in HPLC vs. LPHC indicated that the data set was highly enriched for genes that are involved in immunity (ES 11.41), fatty acid metabolism (ES 3.49), neurotransmission (ES 3.35), cellular metabolic processes (ES 2.49) and oxidative stress pathways (ES 2.19, Table 4). Table 5 lists the GO term classes and protein classes that are significantly up- or down-regulated in the F0 (HPLC vs. LPHC) generations, followed by those GO terms and protein classes that are up- or down-regulated in MM vs. M flies at each of the F1, F2 and F3 generations.
Table 3:
Functional annotation clustering (FAC) as performed in DAVID
| Annotation category | Term | Enrichment score | Gene | FlyBase gene ID | Function | P-value of ANOVA | Significance of ANOVA |
|---|---|---|---|---|---|---|---|
| Interpro | Zinc finger | 10.64 | Bre1 | FBgn0086694 | Ubiquitinates histone H2B on lysine 120 at most RNA Polymerase II transcribed genes | 0.0304 | * |
| pygo | FBgn0043900 | Binds His3 methylated tail to associate with arm as part of Wnt-induced transcription | 0.0030 | ** | |||
| e(y)3 | FBgn0087008 | PBAF complex, chromatin binding and gene silenecing | 0.0596 | . | |||
| nej | FBgn0261617 | Acetylates histone proteins and regulated gene expression | 0.1420 | NS | |||
| E(bx) | FBgn0000541 | Nucleosome remodelling, chromatin organization | 0.0809 | NS | |||
| Pcl | FBgn0003044 | Chromatin binding, co-localizes with the ESC/E(Z) complex | 0.0439 | * | |||
| Interpro/GOTERM_MF_DIRECT | Zinc finger, DNA binding | 4.64 | E(var)3-9 | FBgn0260243 | Chromatin maintenance | 0.0014 | ** |
| Br140 | FBgn0033155 | Enhancer of polycomb-like | 0.0027 | ** | |||
| MTA1-like | FBgn0027951 | Chromatin binding, chromosome condensation | 0.0665 | . | |||
| spn-E | FBgn0003483 | Chromatin binding, chromosome condensation | 0.0153 | * | |||
| hang | FBgn0026575 | Response to oxidative stress | 0.0031 | ** | |||
| phol | FBgn0035997 | Polycomb group protein recruitment to polycomb response elements | 0.0006 | *** | |||
| Chd1 | FBgn0250786 | Remodelling and assembly of chromatin | 0.0865 | . | |||
| Cp190 | FBgn0000283 | Chromatin binding | 0.0165 | * | |||
| cg | FBgn0000289 | Binds to polycomb response elements | 0.0047 | ** | |||
| su(Hw) | FBgn0003567 | Negative regulation of chromatin silencing | 0.0058 | ** | |||
| Chd3 | FBgn0023395 | Chromatin assembly or disassembly, nucleosome remodelling | 0.0027 | ** | |||
| GOTERM_BP/MF/CC_DIRECT, INTERPRO | DNA replication, MCM complex | 4.42 | Orc1 | FBgn0022772 | origin recognition complex, DNA replication, chromatin binding | 0.0013 | ** |
| Orc4 | FBgn0023181 | Initiation of DNA replication | 0.0136 | * | |||
| Orc2 | FBgn0015270 | DNA replication, chromatin binding | 0.0004 | *** | |||
| Mcm5 | FBgn0017577 | Chromatin binding, chromosome condensation | 0.0008 | *** | |||
| RecQ4 | FBgn0040290 | DNA stability, DNA rewinding | 0.0047 | ** | |||
| Mcm10 | FBgn0032929 | Heterochromatin organisation, chromatin silencing | 0.0008 | *** | |||
| polybromo | FBgn0039227 | Chromatin binding | 0.0104 | * | |||
| GOTERM_BP/MF/CC_DIRECT, INTERPRO | Microtublues, kinesin | 4.05 | cid | FBgn0040477 | Histone H3 variant, epigenetic mark for centromere identity | 0.0000 | *** |
| GOTERM_MF_DIRECT, INTERPRO | Helicase | 3.10 | XNP | FBgn0039338 | Heterochromatin organization, chromatin silencing | 0.0924 | . |
| me31B | FBgn0004419 | Gene silencing by miRNA | 0.0001 | *** | |||
| Mi-2 | FBgn0262519 | Chromatin binding, nucleosome binding | 0.0261 | * | |||
| INTERPRO | Structural maintenance of chromosomes | 2.61 | SMC2 | FBgn0027783 | Chromatin binding, chromosome condensation | 0.0020 | ** |
| glu | FBgn0015391 | Chromatin binding, chromosome condensation | 0.0146 | * | |||
| SMC1 | FBgn0040283 | Chromatin binding | 0.0263 | * | |||
| SMC3 | FBgn0015615 | Chromatin binding | 0.0129 | * | |||
| GOTERM_CC/BP_DIRECT | Rb-E2F complex, Myb complex | 2.32 | mor | FBgn0002783 | Brahma associated proteins complex, PBAF complex | 0.0100 | * |
| INTERPRO, GOTERM_MF_DIRECT | Chromo domain | 1.99 | Chro | FBgn0044324 | Histone binding, chromosome organization | 0.0098 | ** |
| Chd1 | FBgn0250786 | Remodelling and assembly of chromatin | 0.0865 | . | |||
| Mi-2 | FBgn0262519 | Chromatin binding, nucleosome binding | 0.0261 | * | |||
| msl-3 | FBgn0002775 | Methylated histone binding, chromatin binding | 0.1760 | NS | |||
| Chd3 | FBgn0023395 | Chromatin assembly or disassembly, nucleosome remodelling | 0.0027 | ** | |||
| GOTERM_MF/BP_DIRECT | Nucleosome mobilisation, nucleosome binding | 1.76 | dre4 | FBgn0002183 | Chromatin binding, nucleosome binding | 0.0021 | ** |
| E(bx) | FBgn0000541 | Histone binding, chromatin organization, chromatin remodelling | 0.0809 | . | |||
| Su(z)12 | FBgn0020887 | Polycomb repressive complex 2, histone methyltransferase activity' | 0.0391 | * | |||
| INTERPRO | WD40 repeat | 1.67 | Hira | FBgn0022786 | Chromatin binding | 0.0000 | *** |
| Caf1-105 | FBgn0033526 | Chromatin assembly factor, histone binding | 0.0001 | *** | |||
| wds | FBgn0040066 | Histone acetyltransferase activity, chromatin remodelling, Trx complex | 0.0032 | ** | |||
| ebi | FBgn0263933 | Chromatin binding | 0.0070 | ** | |||
| GOTERM_BP/CC_DIRECT | Histone phosphorylation | 1.6 | borr | FBgn0032105 | Histone phosphorylation, chromatin binding | 0.0006 | *** |
| ball | FBgn0027889 | Histone threonine kinase activity, histone phosphorylation | 0.0009 | *** | |||
| aurB | FBgn0024227 | Chromatin organization | 0.0004 | *** |
Genes that were significantly up-regulated in these data in HPLC compared to LPHC were compared to a background list of genes that were expressed and detected in these data. Analyses of variance were carried out on expression between all samples in this study (F0-F3 matched and mismatched) with P-values and statistical significances listed.
Table 4:
Functional annotation clustering (FAC) as performed in DAVID
| Annotation category | Term | Enrichment score | Gene | FlyBase gene ID | Function | P-value of ANOVA | Significance of ANOVA |
|---|---|---|---|---|---|---|---|
| INTERPRO | Immunoglobulin-like domain/fold | 11.42 | mesh | FBgn0051004 | Ig fold | 0.000611 | *** |
| Ppn | FBgn0003137 | Ig domain, extracellular matrix structural constituent | 0.0101 | * | |||
| INTERPRO/GO_TERM_BP_DIRECT/GOTERM_MF_DIRECT | Proteolysis, peptidase | 6.16 | Jon65Ai | FBgn0035667 | Serine-type endopeptidase, proteolysis | 0.00293 | ** |
| MP1 | FBgn0027930 | Serine-type endopeptidase, proteolysis | 0.00187 | ** | |||
| thetaTry | FBgn0011555 | Digestive enzyme with serine-type peptidase activity | 1.68E-08 | *** | |||
| zetaTry | FBgn0011556 | Digestive enzyme with serine-type peptidase activity | 0.000488 | *** | |||
| Decay | FBgn0028381 | Cysteine-type endopeptidase activity, caspase | 0.00188 | ** | |||
| Damm | FBgn0033659 | Cysteine-type endopeptidase activity, caspase | 6.79E-06 | *** | |||
| alphaTry | FBgn0003863 | Serine-type endopeptidase, proteolysis | 5.69E-05 | *** | |||
| Ser7 | FBgn0019929 | Serine-type endopeptidase, proteolysis | 1.51E-12 | *** | |||
| Ser6 | FBgn0011834 | Serine-type endopeptidase, proteolysis | 5.17E-03 | ** | |||
| Swim | FBgn0034709 | Polysaccharide binding, cysteine-type peptidase activity, proteolysis | 1.35E-02 | * | |||
| Jon74E | FBgn0023197 | Serine-type endopeptidase, proteolysis | 1.58E-03 | ** | |||
| psh | FBgn0030926 | Peptidase and serine-type endopeptidase activity, defense response | 4.41E-08 | *** | |||
| Jon65Aiv | FBgn0250815 | Serine-type endopeptidase, proteolysis | 1.74E-04 | *** | |||
| Jon65Aiii | FBgn0035665 | Serine-type endopeptidase, proteolysis | 6.77E-05 | *** | |||
| Ance-4 | FBgn0033366 | Peptidase | 6.97E-04 | *** | |||
| Ance | FBgn0012037 | peptidyl-dipeptidase activity | 6.97E-04 | *** | |||
| Jon25Bii | FBgn0031654 | Serine-type endopeptidase, proteolysis | 8.62E-04 | *** | |||
| GOERM_BP_DIRECT/INTERPRO/GOTERM_MF_DIRECT | G-protein-coupled receptor signalling, rhodopsin-like | 6.02 | Rh3 | FBgn0003249 | G-protein coupled photoreceptor activity | 3.73E-03 | ** |
| AkhR | FBgn0025595 | Ccarbohydrate, lipid and tryglyceride homeostasis | 1.88E-09 | *** | |||
| Rh2 | FBgn0003248 | G-protein coupled photoreceptor activity | 4.65E-02 | * | |||
| Galphas | FBgn0001123 | Signal transduction, chemical synaptic transmission | 1.07E-11 | *** | |||
| ninaE | FBgn0002940 | G-protein coupled photoreceptor activity, rhodopsin-like | 2.80E-05 | *** | |||
| Ggamma1 | FBgn0004921 | G-protein, gamma subunit, signal transducer activity | 4.16E-05 | *** | |||
| Gbeta76C | FBgn0004623 | G-protein coupled photoreceptor activity | 2.71E-02 | * | |||
| Rh4 | FBgn0003250 | G-protein coupled photoreceptor activity | 1.83E-03 | ** | |||
| GOTERM_MF_DIRECT/GOTERM_BP_DIRECT | Fatty acid elongation | 3.49 | eloF | FBgn0037762 | Fatty acid elongase | 4.60E-05 | *** |
| GOTERM_MF_DIRECT | Neuropeptide hormone activity/binding | 3.35 | Nplp2 | FBgn0040813 | Neuropeptide signaling, humoral immune response | 1.39E-02 | * |
| Nplp3 | FBgn0042201 | Neuropeptide hormone activity, neuropeptide signaling | 3.45E-01 | NS | |||
| Neb-cGP | FBgn0083167 | Hormone activity, regulation of growth | 3.72E-02 | * | |||
| GOTERM_MF/CC/BP_DIRECT | Calcium-dependent phospholipid binding | 2.72 | AnxB9 | FBgn0000083 | calcium ion binding, calcium-dependent phospholipid binding | 7.69E-03 | ** |
| GPTERM_MP_DIRECT, INTERPRO, COG_ONTOLOGY | Calcium ion binding, signal transduction, EF-hand domain | 2.5 | Cals | FBgn0039928 | Calcium ion binding, chemical synaptic transmission | 2.29E-02 | * |
| Gel | FBgn0010225 | Actin binding, calcium ion binding | 1.52E-03 | ** | |||
| Scp2 | FBgn0020907 | Calcium ion binding | 3.43E-02 | * | |||
| Scp1 | FBgn0020908 | Calcium ion binding | 8.74E-02 | . | |||
| Mlc2 | FBgn0002773 | Calcium ion binding | 3.33E-04 | *** | |||
| Zasp52 | FBgn0265991 | Muscle development | 3.91E-08 | *** | |||
| LpR1 | FBgn0066101 | Uptake of neutral lipids from circulation, calcium ion binding | 4.93E-01 | NS | |||
| Mlc1 | FBgn0002772 | Calcium ion binding, myosin | 3.91E-04 | *** | |||
| up | FBgn0004169 | Calcium ion binding, calcium ion homeostatis, muscle morphogenesis | 3.26E-04 | *** | |||
| TpnC73F | FBgn0010424 | Calcium ion binding | 3.01E-10 | *** | |||
| Gpo-1 | FBgn0022160 | Glycerol-3-phosphate dehydrogenase activity, calcium ion binding | 0.000209 | *** | |||
| INTERPRO, GOTERM_MF/BP/CC_DIRECT | Fatty acyl-CoA reductase, peroxisome | 2.47 | wat | FBgn0039620 | Long-chain-fatty-acyl-CoA reductase activity, oxidation-reduction process | 1.21E-06 | *** |
| Spat | FBgn0014031 | Amino transferase activity, glyoxylate catabolic process, proxisome | 2.02E-02 | * | |||
| Cat | FBgn0000261 | Catalase, antioxidant activity and ROS metabolic process | 1.15E-02 | * | |||
| Uro | FBgn0003961 | Urate oxidase activity, oxidation-reduction process, peroxisome | 1.96E-02 | * | |||
| Acox57D-d | FBgn0034629 | acyl-CoA dehydrogenase activity, fatty acid beta-oxidation, peroxisome | 4.63E-05 | *** | |||
| GOTERM_MF/BP/CC_DIRECT, INTERPRO | Oxidation−reduction, haeme binding, iron ion binding, Cytochrome P450 | 2.2 | Men | FBgn0002719 | Malic oxidoreductase, determination of adult lifespan | 5.16E-04 | *** |
| Cyp6a23 | FBgn0033978 | Cytochrome P450, heme and iron ion binding, oxidation-reduction process | 4.93E-01 | NS | |||
| Cyp4p1 | FBgn0015037 | Cytochrome P450, heme and iron ion binding, oxidation-reduction process | 7.98E-02 | . | |||
| Cpr | FBgn0015623 | NADPH-hemoprotein reductase activity, oxidation-reduction process | 2.27E-04 | *** | |||
| Cyt-b5-r | FBgn0000406 | Heme binding, oxidoreductase activity, lipid metabolic process | 2.69E-04 | *** | |||
| antdh | FBgn0026268 | Oxidoreductase activity, oxidation-reduction process | 4.85E-02 | * | |||
| AOX1 | FBgn0267408 | Aldehyde oxidase, pyridoxal oxidase activity | 5.82E-03 | ** | |||
| Cyt-b5 | FBgn0264294 | Cytocrome b5-like heme-binding site | 6.47E-04 | *** | |||
| Cyp313a1 | FBgn0038236 | Cytochrome P450, heme and iron ion binding, oxidation-reduction process | 1.45E-02 | * | |||
| mt: CoI | FBgn0013674 | Mitochondrial Cytochrome C oxidase, heme and iron binding | 7.39E-02 | . | |||
| Aldh | FBgn0012036 | Aldehyde dehydrogenase, oxidation-reduction | 9.39E-03 | ** | |||
| Cyp4e3 | FBgn0015035 | Cytochrome P450, heme and iron ion binding, oxidation-reduction process | 1.27E-05 | *** | |||
| hgo | FBgn0040211 | Homogentisate 1, 2-dioxygenase activity, oxidation-reduction process | 4.08E-03 | ** | |||
| Pdh | FBgn0011693 | Retinol and alcohol dehydrogenase activity, oxidation-reduction process | 4.75E-03 | ** | |||
| Desat1 | FBgn0086687 | Fatty acid desaturase, oxidation-reduction process | 2.55E-05 | *** | |||
| Plod | FBgn0036147 | Iron ion binding, oxidoreductase activity, oxidation-reduction process | 6.75E-04 | *** | |||
| Fad2 | FBgn0029172 | Fatty acid desaturase, oxidation-reduction process, lipid metabolic process | 1.15E-03 | ** | |||
| Gpdh | FBgn0001128 | Carbohydrate metabolic process, oxidation-reduction process | 6.13E-04 | *** | |||
| GOTERM_BP/CC/MF_DIRECT | Calcium ion channel, transmembrane transport | 2.13 | trpl | FBgn0005614 | Calcium channel activity, calcium ion transport | 2.13E-02 | * |
| trp | FBgn0003861 | Plasma membrane cation channel, calcium channel | 3.06E-09 | *** | |||
| GOTERM_BP/CC/MF_DIRECT | Voltage-gated potassium channel activity | 2.03 | Irk3 | FBgn0032706 | Potassium channel activity, potassium ion transport | 3.88E-02 | * |
| KEGG_PATHWAY, INTERPRO, GOTERM_BP/CC/MF_DIRECT | Fatty acid metabolism/biosynthetic process | 1.93 | ACC | FBgn0033246 | Acetyl-CoA carboxylase activity, fatty acid biosybthetic process | 3.44E-03 | ** |
| Cyt-b5-r | FBgn0000406 | Cytochrome b5-like heme binding, lipid metabolic process | 6.47E-04 | *** | |||
| Desat1 | FBgn0086687 | Fatty acid desaturase, oxidation-reduction process | 2.55E-05 | *** | |||
| Fad2 | FBgn0029172 | Fatty acid desaturase, oxidation-reduction process, lipid metabolic process | 1.15E-03 | ** | |||
| Acsl | FBgn0263120 | Long-chain fatty acid-CoA ligase activity | 1.03E-04 | *** | |||
| INTERPRO, GOTERM_MF_DIRECT | Carbohydrate binding | 1.91 | Clect27 | FBgn0031629 | Carbohydrate binding | 4.40E-11 | *** |
| lectin-37Db | FBgn0053533 | Galactose binding | 2.68E-04 | *** | |||
| INTERPRO, GOTERM_MF_DIRECT | Potassium ion transport, oxalate transmembrane transporter activity | 1.65 | Irk3 | FBgn0032706 | Potassium channel activity, potassium ion transport | 3.88E-02 | * |
| Irk2 | FBgn0039081 | Potassium channel activity, potassium ion transport | 1.96E-08 | *** | |||
| Dic1 | FBgn0027610 | Inorganic phosphate transmembrane transporter activity | 3.33E-05 | *** | |||
| GOTERM_MF/BP/CC_DIRECT, INTERPRO | Neurotransmitter-gated ion-channel ligand binding | 1.45 | Cals | FBgn0039928 | Calcium ion binding | 2.29E-02 | * |
| GOTERM_MF/BP/CC_DIRECT, INTERPRO, COG_ONTOLOGY | Receptor activity, carboxylesterase, lipid metabolism | 1.35 | alpha-Est7 | FBgn0015575 | Carboxylic ester hydrolase activity, lipid storage, determination of lifespan | 2.49E-04 | *** |
| ACC | FBgn0033246 | Acetyl-CoA carboxylase activity, fatty acid biosybthetic process | 3.44E-03 | ** | |||
| Gs2 | FBgn0001145 | Glutamate catabolic process, neurotransmitter receptor metabolic process | 1.46E-03 | ** | |||
| Est-6 | FBgn0000592 | Carboxylic ester hydrolase activity | 2.93E-02 | * | |||
| AcCoAS | FBgn0012034 | Acetyl-CoA ligase activity | 3.35E-04 | *** | |||
| GOTERM_MF/BP/CC_DIRECT, INTERPRO | Neurotransmitter transporter | 1.32 | NAAT1 | FBgn0029762 | Neurotransmitter transporter activity | 2.06E-08 | *** |
| Eaat1 | FBgn0026439 | Glutamate: sodium symporter activity, determination of adult lifespan | 2.06E-02 | * |
Genes that were significantly down-regulated in these data in HPLC compared to LPHC were compared to a background list of genes that were expressed and detected in these data. Analyses of variance were carried out on expression between all samples in this study (F0−F3 matched and mismatched) with P-values and statistical significances listed.
Table 5:
A functional annotation comparison of up and downregulated genes at each generation
| F0 |
F1 |
F2 |
F3 |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GO category | GO term |
n = 1074 |
n = 1867 |
n = 541 |
n = 1684 |
n = 17 |
n = 22 |
n = 1893 |
n = 2720 |
||||||||
| ↑ expression in HPLC | ↓ expressison in HPLC | ↓ expression in MM | ↑ expression in MM | ↓ expression in MM | ↑ expression in MM | ↓ expression in MM | ↑ expression in MM | ||||||||||
| Biological process | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | |
| Biological adhesion | GO: 0022610 | 3 | 0.30 | 34 | 1.90 | 1 | 0.20 | 29 | 1.90 | 1 | 7.10 | 7 | 0.40 | 35 | 1.50 | ||
| Biological regulation | GO: 0065007 | 80 | 7.60 | 127 | 7.00 | 25 | 5.10 | 109 | 7.20 | 1 | 4.50 | 143 | 7.50 | 175 | 7.40 | ||
| Cellular component organization or biogenesis | GO: 0071840 | 93 | 8.90 | 63 | 3.50 | 36 | 7.30 | 51 | 3.40 | 1 | 4.50 | 176 | 9.20 | 98 | 4.10 | ||
| Cellular process | GO: 0009987 | 370 | 35.30 | 475 | 26.30 | 164 | 33.50 | 383 | 25.50 | 2 | 14.30 | 4 | 18.20 | 696 | 36.30 | 602 | 25.50 |
| Developmental process | GO: 0032502 | 37 | 3.50 | 104 | 5.80 | 11 | 2.20 | 89 | 5.90 | 2 | 9.10 | 70 | 3.60 | 147 | 6.20 | ||
| Immune system process | GO: 0002376 | 10 | 1.00 | 26 | 1.40 | 5 | 1.00 | 25 | 1.70 | 16 | 0.80 | 33 | 1.40 | ||||
| Localization | GO: 0051179 | 78 | 7.40 | 139 | 7.70 | 25 | 5.10 | 130 | 8.60 | 2 | 14.30 | 2 | 9.10 | 151 | 7.90 | 194 | 8.20 |
| Locomotion | GO: 0040011 | 4 | 0.40 | 1 | 0.10 | 2 | 0.40 | 1 | 0.10 | 6 | 0.30 | 1 | 0.00 | ||||
| Metabolic process | GO: 0008152 | 383 | 36.50 | 393 | 21.80 | 179 | 36.50 | 328 | 21.80 | 5 | 35.70 | 5 | 22.70 | 708 | 36.90 | 508 | 21.50 |
| Multicellular organismal process | GO: 0032501 | 18 | 1.70 | 127 | 7.00 | 6 | 1.20 | 90 | 6.00 | 2 | 9.10 | 28 | 1.50 | 162 | 6.90 | ||
| Reproduction | GO: 0000003 | 16 | 1.50 | 30 | 1.70 | 9 | 1.80 | 24 | 1.60 | 1 | 4.50 | 28 | 1.50 | 36 | 1.50 | ||
| Response to stimulus | GO: 0050896 | 75 | 7.20 | 124 | 6.90 | 33 | 6.70 | 106 | 7.00 | 1 | 4.50 | 140 | 7.30 | 166 | 7.00 | ||
| Rhythmic process | GO: 0048511 | 14 | 0.80 | 1 | 0.20 | 13 | 0.90 | 1 | 4.50 | 1 | 0.10 | 14 | 0.60 | ||||
| Protein class | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | |
| Calcium-binding protein | PC00060 | 4 | 0.40 | 27 | 1.50 | 1 | 0.20 | 14 | 0.90 | 17 | 0.90 | 35 | 1.50 | ||||
| Cell adhesion molecule | PC00069 | 2 | 0.20 | 24 | 1.30 | 16 | 1.10 | 5 | 0.30 | 30 | 1.30 | ||||||
| Cell junction protein | PC00070 | 3 | 0.30% | 8 | 0.40 | 10 | 0.70 | 5 | 0.30 | 14 | 0.60 | ||||||
| Chaperone | PC00072 | 8 | 0.80% | 3 | 0.20 | 2 | 0.40% | 3 | 0.20 | 14 | 0.70 | 7 | 0.30 | ||||
| Cytoskeletal protein | PC00085 | 41 | 3.90% | 44 | 2.40 | 17 | 3.50% | 38 | 2.50 | 59 | 3.10 | 59 | 2.50 | ||||
| Defense/immunity protein | PC00090 | 2 | 0.20% | 8 | 0.40 | 1 | 0.20% | 7 | 0.50 | 7 | 0.40 | 10 | 0.40 | ||||
| Enzyme modulator | PC00095 | 42 | 4.00% | 59 | 3.30 | 14 | 2.90% | 51 | 3.40 | 1 | 4.50% | 101 | 5.30 | 72 | 3.00 | ||
| Extracellular matrix protein | PC00102 | 22 | 1.20 | 16 | 1.10 | 3 | 0.20 | 26 | 1.10 | ||||||||
| Hydrolase | PC00121 | 73 | 7.00% | 144 | 8.00 | 30 | 6.10% | 131 | 8.70 | 4 | 28.60% | 2 | 9.10% | 142 | 7.40 | 181 | 7.70 |
| Isomerase | PC00135 | 5 | 0.50% | 5 | 0.30 | 8 | 0.50 | 13 | 0.70 | 7 | 0.30 | ||||||
| Ligase | PC00142 | 24 | 2.30% | 20 | 1.10 | 8 | 1.60% | 15 | 1.00 | 1 | 7.10% | 1 | 4.50% | 48 | 2.50 | 25 | 1.10 |
| Lyase | PC00144 | 4 | 0.40% | 28 | 1.60 | 20 | 1.30 | 15 | 0.80 | 25 | 1.10 | ||||||
| Membrane traffic protein | PC00150 | 6 | 0.60% | 16 | 0.90 | 15 | 1.00 | 14 | 0.70 | 21 | 0.90 | ||||||
| Nucleic acid binding | PC00171 | 164 | 15.60% | 72 | 4.00 | 88 | 18.00% | 60 | 4.00 | 296 | 15.40 | 134 | 5.70 | ||||
| Oxidoreductase | PC00176 | 21 | 2.00% | 94 | 5.20 | 12 | 2.40% | 67 | 4.50 | 43 | 2.20 | 96 | 4.10 | ||||
| Receptor | PC00197 | 12 | 1.10% | 81 | 4.50 | 5 | 1.00% | 64 | 4.30 | 1 | 7.10% | 28 | 1.50 | 107 | 4.50 | ||
| Signalling molecule | PC00207 | 12 | 1.10% | 56 | 3.10 | 3 | 0.60% | 49 | 3.30 | 1 | 4.50% | 24 | 1.30 | 72 | 3.00 | ||
| Storage protein | PC00210 | 3 | 0.30% | 7 | 0.40 | 2 | 0.40% | 4 | 0.30 | 5 | 0.30 | 8 | 0.30 | ||||
| Structural protein | PC00211 | 2 | 0.20 | 2 | 0.10 | 1 | 0.20 | 3 | 0.20 | 7 | 0.40 | 3 | 0.10 | ||||
| Transcription factor | PC00218 | 81 | 7.70 | 68 | 3.80 | 33 | 6.70% | 59 | 3.90 | 130 | 6.80 | 117 | 5.00 | ||||
| Transfer/carrier protein | PC00219 | 13 | 1.20% | 36 | 2.00 | 1 | 0.20% | 31 | 2.10 | 28 | 1.50 | 51 | 2.20 | ||||
| Transferase | PC00220 | 62 | 5.90% | 56 | 3.10 | 25 | 5.10% | 49 | 3.30 | 1 | 4.50% | 131 | 6.80 | 66 | 2.80 | ||
| Transmembrane receptor regulatory/adaptor protein | PC00226 | 1 | 0.10% | 6 | 0.30 | 8 | 1.60% | 4 | 0.30 | 3 | 0.20 | 5 | 0.20 | ||||
| Transporter | PC00227 | 28 | 2.70% | 140 | 7.80 | 108 | 7.20 | 47 | 2.50 | 168 | 7.10 | ||||||
| Viral protein | PC00237 | 1 | 0.10 | ||||||||||||||
| Molecular function | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | # genes | % of total | |
| Antioxidant activity | GO: 0016209 | 2 | 0.20 | 9 | 0.50 | 3 | 0.60 | 7 | 0.50 | 5 | 0.30 | 12 | 0.50 | ||||
| Binding | GO: 0005488 | 258 | 24.60 | 240 | 13.30 | 117 | 23.90 | 201 | 13.40 | 1 | 7.10% | 1 | 4.50% | 455 | 23.70 | 349 | 14.80 |
| Catalytic activity | GO: 0003824 | 265 | 25.30 | 412 | 22.80 | 123 | 25.10 | 352 | 23.40% | 5 | 35.70% | 4 | 18.20% | 524 | 27.30 | 483 | 20.40 |
| Channel regulator activity | GO: 0016247 | 1 | 0.00 | ||||||||||||||
| Receptor activity | GO: 0004872 | 8 | 0.80 | 54 | 3.00 | 5 | 1.00 | 47 | 3.10 | 1 | 7.10 | 18 | 0.90 | 74 | 3.10 | ||
| Signal transducer activity | GO: 0004871 | 3 | 0.30 | 12 | 0.70 | 1 | 0.20 | 8 | 0.50 | 7 | 0.40 | 19 | 0.80 | ||||
| Structural molecule activity | GO: 0005198 | 22 | 2.10 | 64 | 3.50 | 6 | 1.20 | 44 | 2.90 | 45 | 2.30 | 87 | 3.70 | ||||
| Translation regulator activity | GO: 0045182 | 2 | 0.20 | 1 | 0.10 | 1 | 0.20 | 115 | 7.60 | 9 | 0.50 | 3 | 0.10 | ||||
| Transporter activity | GO: 0005215 | 34 | 3.20 | 149 | 8.30 | 13 | 2.70 | 1 | 7.10 | 1 | 4.50 | 58 | 3.00 | 177 | 7.50 | ||
Up- and down-regulated genes at each generation were submitted to PANTHER for an analysis of their annotated function. Genes were put into gene ontology (GO) classes, and the number of genes from each of the up- and down-regulated gene list in each class is reported. F0 compares the number of genes in each GO class that are up-regulated or down-regulated in HPLC vs. LPHC. For each subsequent generation (F1−F3), the number of genes in each class is reported as those that are up- or down-regulated in mismatched (MM) flies vs. matched (M) flies, at each generation. The percentages reported are the percentage of gene hits against the total number of genes identified in each list submitted for gene functional classification.
Validation
Based on the genes that were up-regulated in this study, we selected a panel for 20 genes for transcriptomic data validation. All transcriptomic samples were validated by Nanostring, per-sample r value of 0.81–0.95 and whole data set correlation r of 0.86 (Supplementary File 4).
Multigenerational Gene Expression of Genes of Interest
Alteration of diet appears to lead to a characteristic suite of gene expression changes. To determine whether the suite of up-regulated genes observed in HPLC is maintained across mismatched generations when the HPLC diet is removed, we analysed the expression levels of particular groups of genes classed as having roles in epigenetic processes in F1, F2 and F3 matched (M) and mismatched (MM) flies. We observed a specific pattern of expression for every gene; the intermediate-level maintenance of the up-regulation of the genes in the F1 and F2 generation, followed by a reversion to F0 (matched) gene expression levels by the F3 generation. Fig. 2 displays a selection of indicative graphs which display this effect, with ANOVA significance data listed in Table 3 (described in Supplementary File 5) and significant pairwise comparisons as determined by Tukey’s posthoc testing (Supplementary File 6) indicated by solid and dashed lines. There is no significant difference between the expression of epigenetic genes when comparing the F0 LPHC diet and the F3MM flies, despite an intermediate and significant difference between F1 and F2 flies mismatched onto LPHC diets. For genes that were significantly down-regulated in F0 HPLC flies, we observe the same pattern of gene expression in the opposite direction; genes that are down-regulated in response to diet remain at a low level in the F1 and F2 mismatched cohorts (F1MM and F2MM) but by F3, their gene expression has regained the same level as the LPHC (F0) generation (Fig. 3 and Supplementary Files 5 and 7). This effect is genomewide, and applies to every gene tested from the lists generated by DAVID.
Figure 2:

Indicative graphs of gene expression of genes up-regulated in the F0 generation, with significances as determined by ANOVA, between F0 and F3 generations on matched and mismatched diets. Pairwise comparisons by Tukey’s post hoc testing indicated by solid and dashed lines, as described in the key. Y axis denotes mean expression level from transcriptomic experiments, and X axis denotes the dietary condition as per Figure. 1. Note differing Y axis scales. Gene names are stated as per their FlyBase gene symbol IDs.
Figure 3:

Indicative graphs of gene expression of genes down-regulated in the F0 generation, with significances as determined by ANOVA, between F0 and F3 generations on matched and mismatched diets. Pairwise comparisons by Tukey’s post hoc testing indicated by solid and dashed lines, as described in the key. Y axis denotes mean expression level from transcriptomic experiments, and X axis denotes the dietary condition as per Figure. 1. Note differing Y axis scales. Gene names are stated as per their FlyBase gene symbol IDs.
Discussion
The primary goal of this study was to investigate the effect of nutrition on the expression of genes involved in epigenetic processes. We have demonstrated that an HPLC diet results in genomewide up-regulation of epigenetic genes in the F0 generation compared to that observed in a standard Drosophila LPHC diet; this effect was so strong that the overwhelming majority of genes that were up-regulated in response to diet had GO terms categorized as being involved in epigenetic processes, with very few other classes of genes categorized as significantly up-regulated. Classes of genes that were down-regulated in response to the HPLC diet vs. LPHC were broader in scope; these included genes that have GO terms with roles in the immune response, cell signalling, oxidative stress, carbohydrate and fatty acid metabolism and neurotransmission. Thus, this study shows that altering an LPHC diet to an HPLC results in genomewide up-regulation of genes classified as having roles in epigenetic processes, with a co-ordinated down-regulation of genes classified as having broad physiological functions. The co-ordinated nature of the gene expression data we observed imply, firstly, that genes with GO terms categorized as having roles in the processes of neurotransmission, oxidative stress, metabolism and immunity appear to be under epigenetic control, and, secondly, that this epigenetic control, and altered biological response, is influenced by nutrition.
In response to the genomic and epigenomic changes observed in the F0 generation, we further questioned whether dietary alteration resulted in developmental programming of the biological response to diet; specifically, whether the changes induced by the HPLC diet in the F0 generation persisted beyond F0, upon removal of the HPLC diet. The genomic changes induced in the F0 generation persisted at intermediate levels in mismatched F1 and F2 generations in the absence of the HPLC diet. By the F3 generation, gene expression in the mismatched flies had reverted back to the level observed in the F0 matched generation. We hypothesize, firstly, that this multigenerational inheritance of gene expression, followed by a reversion to matched F0 levels by the time the F3 generation is reached, indicates moderate epigenetic programming in the form of a predictive adaptive response (PAR); the gene expression changes induced by the dietary environment experienced by the F0 female flies may be maintained by her offspring. This intermediate maintenance may be considered either as an adaptive response to an environment that the F1-F3MM offspring are ultimately not experiencing or maintenance could be considered a form of bet-hedging, to mean that the offspring are primed to equally respond if their environment changes. Given that these data display a reversion to the unchallenged nutritional state after three generations, we further hypothesize that the ‘correction’ of the induced genomic changes by F3 implies that dietary reversion to match the F0 generation may be able to correct an altered genomic landscape, effectively reversing an altered nutrition-induced phenotype.
The environment is able to interact with genes through epigenetic mechanisms [9] particularly during development [2–4]. Crucially, this could also lead to the alteration of the epigenome of the germ cells [6]. Any permanent alteration to the germ cell epigenome [68] may then be transmitted through the germ line, with adverse phenotypic consequences for offspring [1, 69]. For example: adult-onset diseases can be induced through embryonic exposure to environmental toxins, primarily endocrine disruptors [5, 70–72]; toxic stress can modify Drosophila development by the suppression of Polycomb group genes, with epigenetic inheritance of developmental alterations by unchallenged offspring [73] and; mutations in chaperone proteins such as Hsp90 can induce a heritably altered chromatin state in Drosophila, suggesting a transgenerational epigenetic phenotype [74]. Thus, if epigenetic modifications do become permanent, these modifications can be inherited by future generations and affect disease susceptibility [58, 75].
A large number of studies report transgenerational inheritance in a range of eukaryotes (reviewed in [76]). Many of these studies, particularly in mammals, report inheritance of the acquired trait over two or three generations. Concordant with the work by Jirtle and Skinner [58], we agree that these effects should not be defined as truly transgenerational, because, mechanistically, exposure of an F0 gestating female to an environmental stimulus (nutrition, toxicants or stress) also exposes the F1 embryo (Fig. 1, [77]). Furthermore, for species that develop in utero, parental exposure also exposes the germ cells that will form the F2 generation. Thus, traits present in the F2 generation should be considered as multigenerational, rather than transgenerational, as they could have been induced by direct environmental exposure through the foetus and the germ line. This concept is equally applicable to Drosophila because, while they do not gestate, they do harbour ovarioles in their ovaries which contain developing follicles or egg chambers [78], thus their F1 eggs are exposed to the maternal environment. Further, F1 larvae, which in these experiments were raised on an HPLC diet before being mismatched back to LPHC, contain ovaries and germ cells for the F2 generation. Thus, to reflect this direct exposure, some studies searching for evidence of true transgenerational inheritance are declining to assay the F1 generation entirely [79] due to the fact that transmission to the F1 generation can be indicative of both parental effects and programming [80]. Thus, here we describe our findings from the F1 and F2 generations as multigenerational inheritance, because the gene expression changes induced by the environment in the parental generation revert to F0 levels after the F2 generation.
The current NCD epidemic is aetiologically very complex, but it is thought to be mediated, in part, by developmental aberrations arising from the inheritance of altered epigenetic marks [2, 5]. Many metabolic phenotypes and gene expression differences are linked to differential epigenetic marks that are nutritionally induced. For example, a protein-restricted diet during pregnancy causes hypomethylation of the hepatic PPARα and glucocorticoid receptor genes in rats and promotes the same hypomethylation in the F1 and F2 offspring of F0 rats fed a protein-restricted diet during pregnancy, despite the nutritional challenge being only in the F0 generation [27]. Others have reported evidence of embryonic environmental exposure influencing the phenotype of the F1 generation in species as diverse as humans, rats, chordate fish, Daphnia and isopods [59, 60, 81–84] as well as, specifically, maternal nutrition exerting effects on the F1 phenotype [59, 60]. Such research strongly implies that epigenetic effects could be the key to understanding the current epidemic of overweight and obese, and associated metabolic syndromes, particularly if nutrition in the F0 generation can induce a PAR to nutrition, as we hypothesize is occurring here. Interestingly, comprehensive studies using animal models that investigated the effect of both protein restricted and energy-rich diets during pregnancy on the phenotype of the offspring showed that offspring born to dams fed these different diets exhibited persistent metabolic changes, similar to those observed in human metabolic disease such as obesity, insulin resistance and hypertension [15], indicating an element of developmental programming and a possible PAR. These findings imply that both famine (protein restriction) and energy-rich diets, when mismatched back to adequate nutrition, are similarly detrimental to the metabolic health of offspring and that it is possibly the mismatch itself between inadequate nutrition and proper nutrition which is leading to metabolic disease phenotypes. This highlights the fact that epigenetic mechanisms play a highly complex role in human obesity and metabolic pathways [15, 85–87].
In addition to the striking expression level changes observed in genes that have GO terms classified as having roles in epigenetic processes, one major source of change that we observed in these data are genes involved in oxidative stress. Malnutrition or excess of particular nutrients can cause oxidative damage [88]. For example, hyperglycaemia, which is an excess of sugar in the blood and one of the hallmarks of diabetes, is linked to a diet that is rich in carbohydrates and fat [89, 90]. continuedAn accumulation of sugar can lead to tissue damage, and this can be maintained because of metabolic memory [35], which itself may induce epigenetic changes and altered gene expression [35, 91, 92]. Given that the increased carbohydrate intake can induce oxidative stress as reflected by increased ROS generation [51, 52], our results are consistent with the observation that high-carbohydrate diets are implicated in increased oxidative and metabolic stress [e.g. [49] and that the genome may be responding adaptively to dietary stressors]. We know that oxidative stress responses are often under epigenetic control [55] and also, that maternal nutritional deficiency in pregnancy can lead to altered methylation and increased oxidative DNA damage in the brains of adult offspring [93], which, as well as being directly influenced by nutrition, may predispose to neurological disorders in later life.
Consistent with, and leading on from this observation, these data display a decrease in the expression of genes involved in neurotransmission and neurodevelopment when exposed to an HPLC diet. There is strong evidence linking oxidative stress to neurodegeneration and neurodegenerative disease, such as Alzheimer’s disease [94]. In addition, it is also clear that an increase in the production of ROS, induced by environmental factors, can increase the risk of a multitude of neurodegenerative diseases [95]. Thus, it stands to reason that, in these data, nutrition may be impacting on the production of ROS and the expression of genes involved in ROS pathways and those involved in neurotransmission and neurodegeneration.
In addition to the DOHaD hypothesis, there are also free radical early-life theories, which link environmental agents (e.g. diet and heavy metals) with perturbations of gene regulation and expression (e.g. in the APP gene) and the onset of, for example Alzheimer’s disease [96]. Free radical early-life theories also link the necessity for oxygen in histone demethylase action to epigenetic processes in development [97]. These theories are supported by the observation that nutrition during pregnancy can induce epigenetic changes that result in altered nervous system development [98] and also offspring cerebral function [99]. Further, nutrient availability during the pre and postnatal periods can lead to long-lasting changes in neuron development [100] as well as influence the development of psychopathological behaviour [101]. This is because nutritional deficit may lead to altered brain development [102], possibly via epigenetic factors that can lead to changes in brain structure and function [103]. Micronutrient availability can heavily influence neurotransmission, due to the fact that the function of the brain is inherently related to its metabolism of nutrients [103] in the form of vitamins and minerals that function as co-enzymes in neurotransmission and neurotransmitter metabolism. Given that the gene expression data presented here display significant changes in gene expression in pathways relevant to neurotransmission, these data are supportive of these linkages.
Thus, through our data, we hypothesize that the genomewide changes we observe in genes involved in epigenetic pathways could be responsible for the gene expression changes in other, broad, biological process seen in response to diet. The intermediate maintenance of these gene expression changes, even when the HPLC diet is removed, suggests a PAR to diet; the biology of the mismatched flies is programmed to respond to a particular diet, and is responding adaptively, with altered gene expression in the absence of HPLC, albeit at a slightly lower level. The complete reversion of this in the F3 generation suggests an element of phenotypic rescue, implying that altered nutrition did not affect the germ line and that the gene expression changes are not fixed transgenerationally and thus may have the capacity to be corrected over time.
To date, while the effects of dietary manipulation on fecundity and lifespan in Drosophila have been reported [30, 104–106], none of the studies have assayed the whole-genome gene expression in response to diet. This study contributes to our understanding of the myriad ways in which nutrition can influence gene expression, phenotypes and future health outcomes, with relevance to the DOHaD hypothesis and the current NCD epidemic. While our study demonstrates multigenerational inheritance of gene expression values, rather than transgenerational, it is worth noting that the phenotypic effects of gene expression changes, rather than the gene expression itself, can persist and show multigenerational, and potentially transgenerational, inheritance. For example, a low-protein diet given to Drosophila can increase H3K27me3 through up-regulation of the Enhancer of zeste (E(z)) protein, a protein that is a catalytic component of the Polycomb Repressive Complex 2 methyltransferase. Interestingly, while the up-regulation of the (E)z protein was not detected in the F2 generation, the associated increase in methylation H3K27me3 (a specific chemical modification – trimethylation − of histone H3 at the lysine 27 residue) was in fact detected in the F2 generation [79] and the co-ordinated effect on longevity was also present through to the F2 generation. This suggests that while the gene expression and protein level is not inherited per se, the effects and/or functions of those genes possibly could be. It is possible that a phenomenon such as this may be present in these data; a permissive state may be achieved whereby we might not detect gene expression changes inherited to F3 and beyond, but we may see associated genomic conformational or phenotypic changes in F3 and beyond. Further functional studies based on dietary manipulation are required to confirm this. In particular, it will be pertinent to prove causality between an epigenetic alteration and a change in regulation of genes involved in the traits we observe. To do so, we suggest a combination of phenotypic measures such as assessing lifespan, oxidative stress resistance, and immunity, as well as exploiting mutant Drosophila strains for genes of interest, to assess the effect of epigenetic alterations on downstream gene expression and associated phenotypes. Further functional studies are especially pertinent due to the wealth of evidence demonstrating that high-protein diets in Drosophila can alter Drosophila development and fitness, particularly ovary development and reproductive output [107–110], heat stress tolerance [110, 111], body composition [109] and egg-to-adult viability [111]. The different caloric content between the HPLC and LPHC diets cannot be discounted as a confounding factor; however, the wealth of evidence pertaining to the epigenetic effects of high-protein diets that support these data implies that the gene expression changes detected in these data are driven by the protein/carbohydrate content of the food, rather than a caloric difference.
Thus, here we have identified characteristic suites of genes that are responsive to altered nutrition and the maintenance of altered gene expression upon removal of the nutritional challenge in multiple generations. This has broad implications for our understanding of DOHaD, the PAR hypothesis and the NCD epidemic and will be vital in directing future functional research in this area.
Supplementary Material
Acknowledgements
Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. Under New Zealand law, Drosophila research is waived from requiring ethical approval, therefore none were needed for this study. Drosophila melanogaster stocks were imported under the HSNO Approval number GMC001092. All Drosophila work was carried out in accordance with the regulations of the HSNO Act 1996, as described in Morgan, 2012. We are grateful to Professor Sir Peter Gluckman for his input into, and discussions around, this subject; Matthew Walters for technical assistance; and three anonymous referees, whose comments improved this manuscript. A.J.O. would like to thank Professor Martin Kennedy for hosting the latter stages of this work.
Data Availability
All data supporting this work is included in the supplementary material.
Funding
This work was supported by a Gravida grant (MP04) to P.K.D.
Supplementary data
Supplementary data are available at EnvEpig online.
Conflict of interest statement. None declared.
References
- 1. Barker DJ. The origins of the developmental origins theory. J Intern Med 2007;261:412–7.http://dx.doi.org/10.1111/j.1365-2796.2007.01809.x [DOI] [PubMed] [Google Scholar]
- 2. Gluckman PD, Hanson MA.. Developmental origins of disease paradigm: a mechanistic and evolutionary perspective. Pediatr Res 2004;56:311–7.http://dx.doi.org/10.1203/01.PDR.0000135998.08025.FB [DOI] [PubMed] [Google Scholar]
- 3. Gluckman PD, Hanson MA, Mitchell MD.. Developmental origins of health and disease: reducing the burden of chronic disease in the next generation. Genome Med 2010;2:14..http://dx.doi.org/10.1186/gm135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruce KD, Hanson MA.. The developmental origins, mechanisms, and implications of metabolic syndrome. J Nutr 2010;140:648–52.http://dx.doi.org/10.3945/jn.109.111179 [DOI] [PubMed] [Google Scholar]
- 5. Heindel JJ. Role of exposure to environmental chemicals in the developmental basis of reproductive disease and dysfunction. Semin Reprod Med 2006;24:168–77.http://dx.doi.org/10.1055/s-2006-944423 [DOI] [PubMed] [Google Scholar]
- 6. Skinner MK, Haque CG-BM, Nilsson E, Bhandari R, McCarrey JR, Cooney AJ.. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. PLoS One 2013;8:e66318.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Callinan PA, Feinberg AP.. The emerging science of epigenomics. Hum Mol Genet 2006;15:R95–R101. [DOI] [PubMed] [Google Scholar]
- 8. Peaston AE, Whitelaw E.. Epigenetics and phenotypic variation in mammals. Mamm Genome 2006;17:365–74.http://dx.doi.org/10.1007/s00335-005-0180-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holliday R, Pugh JE.. DNA modificaiton mechanisms and gene activity during development. Science 1975;187:226–32.http://dx.doi.org/10.1126/science.1111098 [PubMed] [Google Scholar]
- 10. Gluckman PD, Hanson MA.. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab 2004;15:183–7.http://dx.doi.org/10.1016/j.tem.2004.03.002 [DOI] [PubMed] [Google Scholar]
- 11. Cordero P, Li JW, Oben JA.. Epigenetics of obesity: beyond the genome sequence. Curr Opin Clin Nutr Metab Care 2015;18:361–6.http://dx.doi.org/10.1097/MCO.0000000000000179 [DOI] [PubMed] [Google Scholar]
- 12. Lee HS. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 2015;7:9492–507.http://dx.doi.org/10.3390/nu7115467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tarry-Adkins JL, Ozanne SE.. Mechanisms of early life programming: current knowledge and future directions. Am J Clin Nutr 2011;94:1765S–71S. [DOI] [PubMed] [Google Scholar]
- 14. Langley‐Evans S. Nutrition in early life and the programming of adult disease: a review. J Hum Nutr Diet 2015;28:1–14. [DOI] [PubMed] [Google Scholar]
- 15. Lillycrop KA, Burdge GC.. Maternal diet as a modifier of offspring epigenetics. J Dev Orig Health Dis 2015;6:88–95.http://dx.doi.org/10.1017/S2040174415000124 [DOI] [PubMed] [Google Scholar]
- 16. Aiken CE, Ozanne SE.. Transgenerational developmental programming. Hum Reprod Update 2014;20:dmt043.. [DOI] [PubMed] [Google Scholar]
- 17. Haggarty P. Epigenetic consequences of a changing human diet. Proc Nutr Soc 2013;72:363–71.http://dx.doi.org/10.1017/S0029665113003376 [DOI] [PubMed] [Google Scholar]
- 18. Vickers M. Developmental programming and transgenerational transmission of obesity. Ann Nutr Metab 2014;64:26–34.http://dx.doi.org/10.1159/000360506 [DOI] [PubMed] [Google Scholar]
- 19. Vickers MH. Early life nutrition, epigenetics and programming of later life disease. Nutrients 2014;6:2165–78.http://dx.doi.org/10.3390/nu6062165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaiserman A. Early-life nutritional programming of longevity. J Dev Orig Health Dis 2014;5:325–38.http://dx.doi.org/10.1017/S2040174414000294 [DOI] [PubMed] [Google Scholar]
- 21. Barker DJP. Maternal nutrition, fetal nutrition, and disease in later life. Nutrition 1997;13:807–13.http://dx.doi.org/10.1016/S0899-9007(97)00193-7 [DOI] [PubMed] [Google Scholar]
- 22. Uauy R, Kain J, Corvalan C.. How can the Developmental Origins of Health and Disease (DOHaD) hypothesis contribute to improving health in developing countries? Am J Clin Nutr 2011;94:1759S–64S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gluckman PD, Hanson MA, Low FM.. The role of developmental plasticity and epigenetics in human health. Birth Defects Res C Embryo Today 2011;93:12–8.http://dx.doi.org/10.1002/bdrc.20198 [DOI] [PubMed] [Google Scholar]
- 24. Barouki R, Gluckman P, Grandjean P, Hanson M, Heindel JJ.. Developmental origins of non-communicable disease: implications for research and public health. Environ Health 2012;11:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanson M, Gluckman P.. Developmental origins of health and disease—global public health implications. Best Pract Res Clin Obstet Gynaecol 2015;29:24–31. [DOI] [PubMed] [Google Scholar]
- 26. Balbus JM, Barouki R, Birnbaum LS, Etzel RA, Gluckman PD, Grandjean P, Hancock C, Hanson MA, Heindel JJ, Hoffman K. et al. Early-life prevention of non-communicable diseases. Lancet 2013;381:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burdge GC, Slater-Jefferies JL, Hanson MA, Lillycrop KA.. Differences in protein and folic acid intake in pregnant rats induce sex-specific changes to the epigenetic regulation of hepatic genes in the adult offspring. Early Hum Dev 2007;83:S97. [Google Scholar]
- 28. Brudasca I, Cucuianu M.. Abnormal lipid metabolism in metabolic syndrome: an epigenetic perspective. Rev Romana Med Lab 2016;24:153–60. [Google Scholar]
- 29. Buescher JL, Musselman LP, Wilson CA, Lang T, Keleher M, Baranski TJ, Duncan JG.. Evidence for transgenerational metabolic programming in Drosophila. Dis Model Mech 2013;6:1123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Musselman LP, Fink JL, Narzinski K, Ramachandran PV, Hathiramani SS, Cagan RL, Baranski TJ.. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis Model Mech 2011;4:842–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waterland RA, Jirtle RL.. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 2003;23:5293–300.http://dx.doi.org/10.1128/MCB.23.15.5293-5300.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milagro F, Campion J, Garcia-Diaz D, Goyenechea E, Paternain L, Martinez J.. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem 2009;65:1–9. [DOI] [PubMed] [Google Scholar]
- 33. Fujiki K, Kano F, Shiota K, Murata M.. Expression of the peroxisome proliferator activated receptor γ gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol 2009;7:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luttmer R, Spijkerman AM, Kok RM, Jakobs C, Blom HJ, Serne EH, Dekker JM, Smulders YM.. Metabolic syndrome components are associated with DNA hypomethylation. Obes Res Clin Pract 2013;7:e106–15. [DOI] [PubMed] [Google Scholar]
- 35. Sánchez I, Reynoso-Camacho R, Salgado LM.. The diet-induced metabolic syndrome is accompanied by whole-genome epigenetic changes. Genes Nutr 2015;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masuyama H, Mitsui T, Eguchi T, Tamada S, Hiramatsu Y.. The effects of paternal high-fat diet exposure on offspring metabolism with epigenetic changes in the mouse adiponectin and leptin gene promoters. Am J Physiol Endocrinol Metab 2016;311: E236–45. [DOI] [PubMed] [Google Scholar]
- 37. Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM.. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 2010;151:4756–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lillycrop KA, Slater-Jefferies JL, Hanson M, Godfrey K, Jackson AA, Burdge GC.. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr 2007;97:1064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA.. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br J Nutr 2007;97:435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Straten EME, Bloks VW, Huijkman NCA, Baller JF, van Meer W, Lutjohann H, Kuipers DF, Plosch T.. The liver x-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am J Physiol Regul Integr Comp Physiol 2010;298:R275–82. [DOI] [PubMed] [Google Scholar]
- 41. Burdge GC, Phillips ES, Dunn RL, Jackson AA, Lillycrop KA.. Effect of reduced maternal protein consumption during pregnancy in the rat on plasma lipid concentrations and expression of peroxisomal proliferator-activated receptors in the liver and adipose tissue of the offspring. Nutr Res 2004;24:639–46. [Google Scholar]
- 42. Reamon-Buettner SM, Buschmann J, Lewin G.. Identifying placental epigenetic alterations in an intrauterine growth restriction (IUGR) rat model induced by gestational protein deficiency. Reprod Toxicol 2014;45:117–24.http://dx.doi.org/10.1016/j.reprotox.2014.02.009 [DOI] [PubMed] [Google Scholar]
- 43. Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K.. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol 2009;587:4963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burdge GC, Lillycrop KA, Phillips ES, Slater-Jefferies JL, Jackson AA, Hanson MA.. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr 2009;139:1054–60. [DOI] [PubMed] [Google Scholar]
- 45. Christman JK, Sheikhnejad G, Dizik M, Abileah S, Wainfan E.. Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis 1993;14:551–7. [DOI] [PubMed] [Google Scholar]
- 46. Waterland RA, Lin J-R, Smith CA, Jirtle RL.. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum Mol Genet 2006;15:705–16.http://dx.doi.org/10.1093/hmg/ddi484 [DOI] [PubMed] [Google Scholar]
- 47. Hoile SP, Irvine NA, Kelsall CJ, Sibbons C, Feunteun A, Collister A, Torrens C, Calder PC, Hanson MA, Lillycrop KA.. Maternal fat intake in rats alters 20:4n-6 and 22: 6n-3 status and the epigenetic regulation of Fads2 in offspring liver. J Nutr Biochem 2013;24:1213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jacometo CB, Osorio JS, Socha M, Correa MN, Piccioli-Cappelli F, Trevisi E, Loor JJ.. Maternal consumption of organic trace minerals alters calf systemic and neutrophil mRNA and microRNA indicators of inflammation and oxidative stress. J Dairy Sci 2015;98:7717–29. [DOI] [PubMed] [Google Scholar]
- 49. Langley SC, Seakins M, Grimble RF, Jackson AA.. The acute phase response of adult rats is altered by in utero exposure to maternal low protein diets. J Nutr 1994;124:1588–96. [DOI] [PubMed] [Google Scholar]
- 50. Gregersen S, Samocha-Bonet D, Heilbronn L, Campbell L.. Inflammatory and oxidative stress responses to high-carbohydrate and high-fat meals in healthy humans. J Nutr Metab 2012;2012:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aljada A, Mohanty P, Ghanim H, Abdo T, Tripathy D, Chaudhuri A, Dandona P.. Increase in intranuclear nuclear factor κB and decrease in inhibitor κB in mononuclear cells after a mixed meal: evidence for a proinflammatory effect. Am J Clin Nutr 2004;79:682–90. [DOI] [PubMed] [Google Scholar]
- 52. Patel C, Ghanim H, Ravishankar S, Sia CL, Viswanathan P, Mohanty P, Dandona P.. Prolonged reactive oxygen species generation and nuclear factor-κB activation after a high-fat, high-carbohydrate meal in the obese. J Clin Endocrinol Metab 2007;92:4476–9. [DOI] [PubMed] [Google Scholar]
- 53. Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González Á, Esquivel-Chirino C, Durante-Montiel I, Sánchez-Rivera G, Valadez-Vega C, Morales-González JA.. Inflammation, oxidative stress, and obesity. Int J Mol Sci 2011;12:3117–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chinnusamy V, Zhu J-K.. Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol 2009;12:133–9.http://dx.doi.org/10.1016/j.pbi.2008.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cencioni C, Spallotta F, Martelli S, Valente A, Mai A, Zeiher AM, Gaetano C.. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int J Mol Sci 2013;14:17643–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ristow M, Schmeisser S.. Extending life span by increasing oxidative stress. Free Radic Biol Med 2011;51:327–36.http://dx.doi.org/10.1016/j.freeradbiomed.2011.05.010 [DOI] [PubMed] [Google Scholar]
- 57. Gluckman P, Hanson M, Spencer HG.. Predictive adaptive responses and human evolution. Trends Ecol Evol 2005;20:527–33.http://dx.doi.org/10.1016/j.tree.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 58. Jirtle RL, Skinner MK.. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007;8:253–62.http://dx.doi.org/10.1038/nrg2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carter MJ, Lardies MA, Nespolo RF, Bozinovic F.. Heritability of progeny size in a terrestrial isopod: transgenerational environmental effects on a life history trait. Heredity 2004;93:455–9. [DOI] [PubMed] [Google Scholar]
- 60. Golding J. The Avon Longitudinal Study of Parents and Children (ALSPAC)–study design and collaborative opportunities. Eur J Endocrinol 2004;151:U119–23. [DOI] [PubMed] [Google Scholar]
- 61. Morgan SM. Nutrition Forced Extension of Lifespan in Drosophila: A Whole-Genome Investigation. Dunedin, New Zealand, University of Otago, 2012. [Google Scholar]
- 62. Roussou IG, Savakis C, Tavernarakis N, Metaxakis A.. Stage dependent nutritional regulation of transgenerational longevity. Nutr Healthy Aging 2016;4:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nystand M, Dowling DK. Transgenerational interactions involving parental age and immune status affect female reproductive success in Drosophila melanogaster. Proc R Soc B 2014;281:20141241.http://dx.doi.org/10.1038/nprot.2008.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T. et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26:317.. [DOI] [PubMed] [Google Scholar]
- 65. Team RC. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2008. [Google Scholar]
- 66. Huang DW, Sherman BT, Lempicki RA.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008;4:44–57.http://dx.doi.org/10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 67. Huang DW, Sherman BT, Lempicki RA.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37:1–13.http://dx.doi.org/10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guerrero-Bosagna C, Settles M, Lucker B, Skinner MK, Gaetano C.. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One 2010;5:e13100.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Skinner MK. Role of epigenetics in developmental biology and transgenerational inheritance. Birth Defects Res C Embryo Today 2011;93:51–5.http://dx.doi.org/10.1002/bdrc.20199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Anway MD, Leathers C, Skinner MK.. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 2006;147:5515–23.http://dx.doi.org/10.1210/en.2006-0640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Newbold RR, Padilla-Banks E, Jefferson WN.. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology 2006;147:S11–7. [DOI] [PubMed] [Google Scholar]
- 72. Anway MD, Skinner MK.. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 2006;147:S43–9. [DOI] [PubMed] [Google Scholar]
- 73. Stern S, Fridmann-Sirkis Y, Braun E, Soen Y.. Epigenetically heritable alteration of fly development in response to toxic challenge. Cell Rep 2012;1:528–42.http://dx.doi.org/10.1016/j.celrep.2012.03.012 [DOI] [PubMed] [Google Scholar]
- 74. Sollars V, Lu X, Li X, Wang MD, Garfinkel DM, Ruden DM.. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat Genet 2003;33:70. [DOI] [PubMed] [Google Scholar]
- 75. Anway MD, Cupp AS, Uzumcu M, Skinner MK.. Epigenetic transgenerational actions of endocrine disruptors and mate fertility. Science 2005;308:1466–9.http://dx.doi.org/10.1126/science.1108190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jablonka E, Raz G.. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol 2009;84:131–76.http://dx.doi.org/10.1086/598822 [DOI] [PubMed] [Google Scholar]
- 77. Osborne AJ, Duncan EJ, Cridge AG, Dearden PK.. Epigenetics and the maternal germline In: Tollefsbol T. (ed.), Transgenerational Epigenetics: Evidence and Debate. Waltham, MA: Elsevier, 2013. [Google Scholar]
- 78. McCall K. Eggs over easy: cell death in the Drosophila ovary. Dev Biol 2004;274:3–14.http://dx.doi.org/10.1016/j.ydbio.2004.07.017 [DOI] [PubMed] [Google Scholar]
- 79. Xia B, Gerstin E, Schones DE, Huang W, Steven de Belle J.. Transgenerational programming of longevity through E(z)-mediated histone H3K27 trimethylation in Drosophila. Aging 2016;8:2988.http://dx.doi.org/10.18632/aging.101107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xia B, de Belle S.. Transgenerational programming of longevity and reproduction by post-eclosion dietary manipulation in Drosophila. Aging 2016;8:1115..http://dx.doi.org/10.18632/aging.100932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kang IJ, Yokota H, Oshima Y, Tsuruda Y, Oe T, Imada N, Tadokoro H, Honjo T.. Effects of bisphenol A on the reproduction of Japanese medaka (Oryzias latipes). Environ Toxicol Chem 2002;21:2394–400. [PubMed] [Google Scholar]
- 82. Klip H, Verloop J, van Gool JD, Koster M, Burger CW, van Leeuwen FE; OMEGA Project Group. Hypospadias in sons of women exposed to diethylstilbestrol in utero: a cohort study. Lancet 2002;359:1102–7.http://dx.doi.org/10.1016/S0140-6736(02)08152-7 [DOI] [PubMed] [Google Scholar]
- 83. Portha B. Programmed disorders of beta-cell development and function as one cause for type 2 diabetes?—The GK rat paradigm. Diabetes Metab Res Rev 2005;21:495–504. [DOI] [PubMed] [Google Scholar]
- 84. Tsui MTK, Wang WX.. Maternal transfer efficiency and transgenerational toxicity of methylmercury in Daphnia magna. Environ Toxicol Chem 2004;23:1504–11.http://dx.doi.org/10.1897/03-310 [DOI] [PubMed] [Google Scholar]
- 85. Xue J, Ideraabdullah FY.. An assessment of molecular pathways of obesity susceptible to nutrient, toxicant and genetically induced epigenetic perturbation. J Nutr Biochem 2016;30:1–13.http://dx.doi.org/10.1016/j.jnutbio.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lillycrop KA, Burdge GC.. The effect of nutrition during early life on the epigenetic regulation of transcription and implications for human diseases. J Nutrigenet Nutrigenomics 2011;4:248–60.http://dx.doi.org/10.1159/000334857 [DOI] [PubMed] [Google Scholar]
- 87. Li CCY, Maloney CA, Cropley JE, Suter JE.. Epigenetic programming by maternal nutrition: shaping future generations. Epigenomics 2010;2:539–49.http://dx.doi.org/10.2217/epi.10.33 [DOI] [PubMed] [Google Scholar]
- 88. Fang Y-Z, Yang S, Wu G.. Free radicals, antioxidants, and nutrition. Nutrition 2002;18:872–9.http://dx.doi.org/10.1016/S0899-9007(02)00916-4 [DOI] [PubMed] [Google Scholar]
- 89. Gupta N, Shah P, Nayyar S, Misra A.. Childhood obesity and the metabolic syndrome in developing countries. Indian J Pediatr 2013;80:28–37.http://dx.doi.org/10.1007/s12098-012-0923-5 [DOI] [PubMed] [Google Scholar]
- 90. Yang Z-H, Miyahara H, Takeo J, Katayama J.. Diet high in fat and sucrose induces rapid onset of obesity-related metabolic syndrome partly through rapid response of genes involved in lipogenesis, insulin signalling and inflammation in mice. Diabetol Metab Syndr 2012;4:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ceriello A. Hyperglycaemia and the vessel wall: the pathophysiological aspects on the atherosclerotic burden in patients with diabetes. Eur J Cardiovasc Prev Rehab 2010;17:s15–9. [DOI] [PubMed] [Google Scholar]
- 92. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M.. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008;205:2409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Langie SA, Achterfeldt S, Gorniak RW, Halley-Hogg J, Oxley D, van Schooten FJ, Godschalk RW, McKay JA, Mathers JC.. Maternal folate depletion and high-fat feeding from weaning affects DNA methylation and DNA repair in brain of adult offspring. FASEB J 2013;27:3323–34. [DOI] [PubMed] [Google Scholar]
- 94. Tabaton M, Tamagno E.. The molecular link between [beta]-and [gamma]-secretase activity on the amyloid [beta] precursor protein. Cell Mol Life Sci 2007;64:2211..http://dx.doi.org/10.1007/s00018-007-7219-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Migliore L, Coppedè F.. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mut Res/Genet Toxicol Environ Mutag 2009;674:73–84. [DOI] [PubMed] [Google Scholar]
- 96. Lahiri DK, Maloney B, Riyaz Basha M, Wen Ge Y, Zawia NH.. How and when environmental agents and dietary factors affect the course of Alzheimer’s disease: the “LEARn” model (latent early-life associated regulation) may explain the triggering of AD. Curr Alzheimer Res 2007;4:219–28. [DOI] [PubMed] [Google Scholar]
- 97. Hitchler MJ, Domann FE.. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med 2007;43:1023–36.http://dx.doi.org/10.1016/j.freeradbiomed.2007.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. McGowan PO, Meaney MJ, Szyf M.. Diet and the epigenetic (re) programming of phenotypic differences in behavior. Brain Res 2008;1237:12–24.http://dx.doi.org/10.1016/j.brainres.2008.07.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gallagher E, Newman J, Green L, Hanson M.. The effect of low protein diet in pregnancy on the development of brain metabolism in rat offspring. J Physiol (Lond) 2005;568:553–8.http://dx.doi.org/10.1113/jphysiol.2005.092825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Niculescu MD, Yamamuro Y, Zeisel SH.. Choline availability modulates human neuroblastoma cell proliferation and alters the methylation of the promoter region of the cyclin‐dependent kinase inhibitor 3 gene. J Neurochem 2004;89:1252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Neugebauer R, Hoek HW, Susser E.. Prenatal exposure to wartime famine and development of antisocial personality disorder in early adulthood. JAMA 1999;282:455–62.http://dx.doi.org/10.1001/jama.282.5.455 [DOI] [PubMed] [Google Scholar]
- 102. Liu J, Raine A.. The effect of childhood malnutrition on externalizing behavior. Curr Opin Pediatr 2006;18:565–70.http://dx.doi.org/10.1097/01.mop.0000245360.13949.91 [DOI] [PubMed] [Google Scholar]
- 103. Liu J, Zhao SR, Reyes T.. Neurological and epigenetic implications of nutritional deficiencies on psychopathology: conceptualization and review of evidence. Int J Mol Sci 2015;16:18129–48.http://dx.doi.org/10.3390/ijms160818129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Grandison RC, Piper MD, Partridge L.. Amino acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 2009;462:1061.http://dx.doi.org/10.1038/nature08619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lee KP, Simpson SJ, Clissold FJ, Brooks R, Ballard JWO, Taylor PW, Soran N, Raubenheimer D.. Lifespan and reproduction in Drosophila: new insights from nutritional geometry. Proc Natl Acad Sci U S A 2008;105:2498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ayres JS, Schneider DS.. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol 2009;7:e1000150.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Morris SNS, Coogan K, Chamseddin K, Fernandez-Kim SO, Kolli S, Keller JN, Bauer JH.. Development of diet-induced insulin resistance in adult Drosophila melanogaster. Biochim Biophys Acta 2012;1822:1230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Skorupa DA, Dervisefendic A, Zwiener J, Pletcher SD.. Dietary composition specifies consumption, obesity, and lifespan in Drosophila melanogaster. Aging Cell 2008;7:478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Matzkin LM, Johnson S, Paight C, Bozinovic G, Markow TA.. Dietary protein and sugar differentially affect development and metabolic pools in ecologically diverse Drosophila. J Nutr 2011;141:1127–33. [DOI] [PubMed] [Google Scholar]
- 110. Sisodia S, Singh BN.. Experimental evidence for nutrition regulated stress resistance in Drosophila ananassae. PLoS One 2012;7:e46131.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Andersen LH, Kristensen TN, Loeschcke V, Toft S, Mayntz D.. Protein and carbohydrate composition of larval food affects tolerance to thermal stress and desiccation in adult Drosophila melanogaster. J Insect Physiol 2010;56:336–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting this work is included in the supplementary material.