

ABSTRACT
Morphine is one of the most effective analgesics in medicine. However, its use is associated with the development of tolerance and dependence. Recent studies demonstrating epigenetic changes in the brain after exposure to opiates have provided insight into mechanisms possibly underlying addiction. In this study, we sought to identify epigenetic changes in ten regions of the rat brain following acute and chronic morphine exposure. We analyzed DNA methylation of six nuclear-encoded genes implicated in brain function (Bdnf, Comt, Il1b, Il6, Nr3c1, and Tnf) and three mitochondrially-encoded genes (Mtco1, Mtco2, and Mtco3), and measured global 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5 hmC) levels. We observed differential methylation of Bdnf and Il6 in the pons, Nr3c1 in the cerebellum, and Il1b in the hippocampus in response to acute morphine exposure (all P value < 0.05). Chronic exposure was associated with differential methylation of Bdnf and Comt in the pons, Nr3c1 in the hippocampus and Il1b in the medulla oblongata (all P value < 0.05). Global 5mC levels significantly decreased in the superior colliculus following both acute and chronic morphine exposure, and increased in the hypothalamus following chronic exposure. Chronic exposure was also associated with significantly increased global 5hmC levels in the cerebral cortex, hippocampus, and hypothalamus, but significantly decreased in the midbrain. Our results demonstrate, for the first time, highly localized epigenetic changes in the rat brain following acute and chronic morphine exposure. Further work is required to elucidate the potential role of these changes in the formation of tolerance and dependence.
KEYWORDS: morphine, addiction, DNA methylation, 5-methylcytosine, 5-hydroxymethylcytosine, mitochondrial epigenetics, BDNF, opiates
Introduction
Morphine is one of the most effective analgesic medications. Morphine, along with other opioids, relieves acute and chronic pain by acting at mu opioid receptors found throughout the central nervous system. Alongside its therapeutic effects, morphine use is also associated with adverse effects such as tolerance and dependence.
Morphine tolerance may originate in part through neuronal processes via activation of mu opioid receptors and subsequent neuroexcitation [1]. Preclinical gene expression profiling studies have demonstrated widespread changes in the transcriptome of the nucleus accumbens and striatum following chronic morphine exposure [2,3], including in genes associated with circadian rhythms, neurotransmitter release, and glucocorticoid receptor signaling. There is also increasing evidence for a role of microglia in tolerance. Microglia are macrophages present within the central nervous system that produce pro-inflammatory cytokines, such as interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNF-α), following their activation by morphine [4]. IL-1β, IL-6, and TNF-α have been shown to reduce morphine analgesia within five minutes of administration [4], and blockade of IL-1β signaling prolongs morphine-induced analgesia [5], thereby potentially implicating these three cytokines in morphine tolerance. Microglial activation is further implicated in the development of tolerance through increased expression of brain derived neurotrophic factor (BDNF) in the ventral tegmental area (VTA) that facilitates the switch to the dopamine-dependent reward system present in addiction [6]. Notably, Bdnf−/− knockout mice do not develop tolerance to morphine [7]. Other genes implicated in morphine tolerance include the catechol-O-methyltransferase (COMT) gene through its role in dopamine elimination, whose activity therefore regulates morphine response in mice [8], and the glucocorticoid receptor gene NR3C1, which is epigenetically silenced in the HPA axis following chronic, but not acute, morphine exposure [9].
Exposure to opiates is also known to induce mitochondrial dysfunction in neuronal and glioma cells [10,11], and chronic exposure is associated with reduced mitochondrial DNA copy number in the rat hippocampus [12]. Furthermore, morphine may induce oxidative stress in the brain [13], and this is known to induce the expression of mitochondrial DNA methyltransferase 1 (mtDNMT1), which regulates the mitochondrial epigenome [14].
Morphine exposure has been demonstrated to affect the epigenetic regulation of genes implicated in addiction and tolerance both in human studies and in animal models. Differential methylation of the BDNF [15,16] and opioid receptor Mu 1 (OPRM1) [17,18] gene promoters have been reported in the blood of opiate-dependent individuals. Epigenetic changes in the brain may be highly localized, as studies utilizing whole brain tissue have often reported no significant change in global DNA methylation levels following morphine exposure [19,20]. Targeted approaches have proven more insightful, identifying changes in regions of the brain associated with addiction, such as the hippocampus, medial prefrontal cortex, and VTA, in a rat model of heroin self-administration and reward devaluation [21] and post-mortem orbitofrontal cortex brain tissue from former heroin users [22].
However, a significant knowledge gap remains regarding the spatial and temporal epigenetic regulation in the brain of genes that are implicated in tolerance and dependence. In this study, we analyzed epigenetic changes in response to acute and chronic morphine induction in 10 regions of the rat brain reported to be implicated in response to opiates and the formation of addiction: the midbrain contains the VTA, which has been consistently implicated in addiction [23,24]; the pons contains the locus coeruleus, which is key in the integration of opioid and stress signaling and which is served by innervation from the paraventricular nucleus of the hypothalamus [25]; the inferior and superior colliculus regulate response to opiate withdrawal through reduced activation of mu-opioid receptor signalling [26,27]; the cerebral cortex (containing the dorsomedial prefrontal cortex), hippocampus, and cerebellum are implicated in reinforcement of drug-seeking beheaviors [28–30]; the medulla oblongata is crucial in pain modulation and opiate withdrawal behaviors [31]; and the thalamus, whose function is disrupted in opiate dependence [32,33]. We measured global 5-methylcytosine and 5-hydroxymethylcytosine levels and analyzed the DNA methylation of genes previously identified as implicated in morphine tolerance (Bdnf, Comt, Il1b, Il6, Nr3c1, and Tnf) and mitochondrially-encoded genes potentially implicated in mitochondrial dysfunction (Mt-co1, Mt-co2, and Mt-co3).
Results
Study overview
An overview of the study is provided in Figure 1. Male rats were assigned to control, acute challenge, and chronic induction groups (all n = 5). After 10 days, brain tissue was dissected and DNA extracted for epigenetic analysis.
Figure 1.
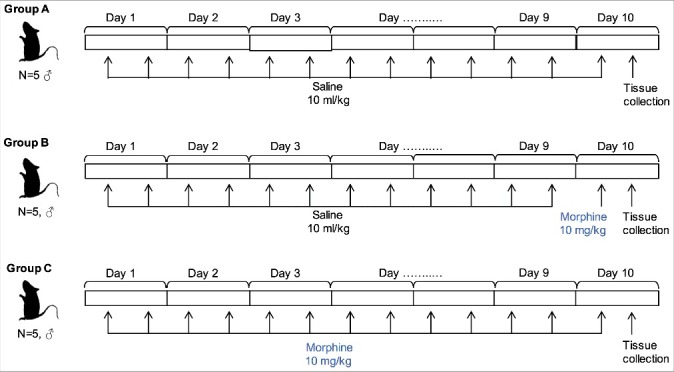
Overview of experimental approach.
Rats were assigned to control (A), acute morphine challenge (10 mg/kg one hour post injection, B) and chronic morphine induction (10 mg/kg/day bid for 10 days, C) groups. Saline or morphine was administered twice daily until tissue collection at Day 10.
Gene-specific DNA methylation variability by region of the rat brain
The nuclear-encoded genes Bdnf, Comt, Il1b, Il6, Nr3c1, and Tnf displayed differential variability by region of the brain (Figure 2A-F). Bdnf and Tnf displayed highly conserved methylation levels between the regions, with mean Bdnf methylation levels ranging between 3.0 and 4.6% (Figure 2A) and Tnf methylation between 54.0 and 62.5% (Figure 2F). The greatest variability was observed in Il6 methylation, which displayed low levels (<6.5%) in the cerebellum, cerebral cortex, and hippocampus, but markedly higher methylation (>20%) in the hypothalamus, inferior colliculus, superior colliculus, and the thalamus (Figure 2D). Global DNA methylation levels, estimated using LINE-1 as a surrogate marker (L1-5utr and L1-orf), were highly consistent between brain regions (Figure 2G-H).
Figure 2.
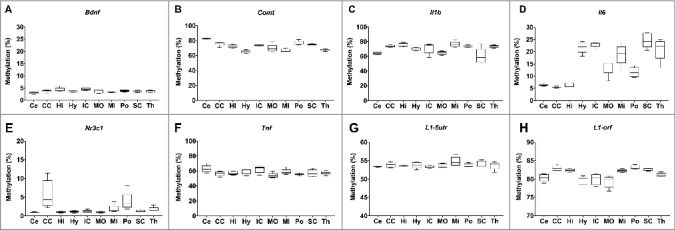
Variation in DNA methylation by region of the rat brain. A-F: DNA methylation of Bdnf, Comt, Il1b, Il6, Nrc31, and Tnf in the cerebellum (Ce), cerebral cortex (CE), hippocampus (Hi), hypothalamus (Hy), inferior colliculus (IC), medulla oblongata (MO), midbrain (Mi), pons (Po), superior colliculus (SC), and thalamus (Th). G-H: global DNA methylation estimated by measurement of L1-5utr and L1-orf elements in the same 10 regions of the brain.
Morphine induction is associated with gene-specific and global DNA methylation changes in the rat brain
Acute morphine exposure (10 mg/kg, one hour post injection) was associated with significantly increased methylation of Il6 in the pons (11.6 vs. 14.3%, P value < 0.05) and Nr3c1 in the cerebellum (1.0 vs. 1.2%, P value = 0.03), and significantly decreased methylation of Bdnf in the pons (3.9 vs. 3.3%, P value = 0.03) and Il1b in the hippocampus (76.5 vs. 71.8%, P value = 0.02) (Table 1, Figure 3). Chronic morphine exposure (10 mg/kg/day bid for 10 days) was associated with significantly increased methylation of Il1b in the medulla oblongata (64.8 vs. 68.4%, P value = 0.03) and Nr3c1 in the hippocampus (1.0 vs. 1.6%, P value = 0.02), and significantly decreased methylation of Bdnf (3.9 vs. 3.1, P value = 0.02) and Comt in the pons (77.3 vs. 66.9%, P value < 0.0001).
Table 1.
Nuclear DNA methylation in the rat brain following morphine induction.
| Region | Treatment | Bdnf | Comt | Il1b | Il6-1 | Nr3c1 | Tnf | L1-5utr | L1-orf | 5-hmc |
|---|---|---|---|---|---|---|---|---|---|---|
| Cerebellum | Control | 3.0 (0.2) | 82.5 (0.3) | 64.2 (0.7) | 6.3 (0.2) | 1.0 (0.0) | 62.5 (2.2) | 53.3 (0.1) | 80.4 (0.5) | 0.48 (0.07) |
| Acute | 3.0 (0.2) | 83.6 (0.5) | 66.2 (1.3) | 5.9 (0.3) | 1.2 (0.1) | 63.2 (2.8) | 53.8 (0.3) | 82.9 (0.3) | 0.51 (0.06) | |
| Chronic | 3.5 (0.4) | 82.4 (0.3) | 64.8 (0.4) | 6.0 (0.2) | 1.1 (0.2) | 65.3 (0.9) | 53.5 (0.1) | 82.4 (0.2) | 0.63 (0.03) | |
| Cerebral cortex | Control | 3.8 (0.2) | 75.8 (1.3) | 74.1 (0.7) | 5.5 (0.2) | 5.7 (1.7) | 56.4 (1.3) | 53.7 (0.4) | 79.7 (0.5) | 0.50 (0.06) |
| Acute | 4.1 (0.4) | 74.8 (0.9) | 76.1 (1.0) | 8.2 (1.8) | 3.1 (1.4) | 55.3 (1.4) | 53.3 (0.2) | 80.0 (0.7) | 0.54 (0.03) | |
| Chronic | 3.8 (0.4) | 77.5 (0.5) | 74.6 (2.2) | 5.5 (0.5) | 4.7 (0.9) | 52.8 (1.2) | 53.6 (0.2) | 79.3 (0.7) | 0.78 (0.03) | |
| Hippocampus | Control | 4.3 (0.4) | 72.4 (1.0) | 76.5 (1.3) | 6.5 (0.4) | 1.0 (0.1) | 57.1 (1.0) | 54.8 (0.6) | 82.2 (0.2) | 0.34 (0.02) |
| Acute | 3.5 (0.2) | 70.0 (1.1) | 71.8 (1.0) | 5.7 (0.5) | 1.2 (0.1) | 57.5 (1.8) | 53.7 (0.2) | 83.2 (0.2) | 0.35 (0.01) | |
| Chronic | 3.4 (0.2) | 71.9 (0.2) | 74.8 (0.7) | 6.4 (0.3) | 1.6 (0.2) | 58.9 (1.0) | 54.0 (0.4) | 82.5 (0.2) | 0.43 (0.01) | |
| Hypothalamus | Control | 3.6 (0.1) | 65.1 (0.8) | 70.1 (0.9) | 21.8 (1.1) | 1.1 (0.1) | 59.4 (1.5) | 53.6 (0.5) | 81.1 (0.2) | 0.40 (0.02) |
| Acute | 3.8 (0.2) | 65.2 (1.5) | 68.9 (2.1) | 16.4 (2.7) | 1.3 (0.2) | 55.9 (1.4) | 54.0 (0.6) | 81.5 (0.4) | 0.46 (0.02) | |
| Chronic | 3.7 (0.3) | 65.0 (0.8) | 68.0 (1.2) | 20.4 (0.9) | 1.1 (0.1) | 58.9 (1.1) | 54.3 (0.7) | 83.4 (0.3) | 0.54 (0.03) | |
| Inferior colliculus | Control | 4.6 (0.3) | 73.4 (0.5) | 71.1 (3.4) | 23.0 (0.5) | 1.2 (0.2) | 62.0 (2.1) | 54.5 (0.5) | 82.6 (0.1) | 0.57 (0.1) |
| Acute | 3.9 (0.4) | 72.6 (0.9) | 72.4 (3.8) | 22.5 (1.2) | 1.3 (0.1) | 62.5 (1.7) | 54.2 (0.4) | 80.0 (0.5) | 0.62 (0.07) | |
| Chronic | 4.0 (0.4) | 72.7 (0.5) | 76.1 (0.7) | 24.0 (0.5) | 1.3 (0.1) | 63.7 (1.3) | 53.6 (0.6) | 77.5 (2.2) | 0.79 (0.04) | |
| Medulla oblongata | Control | 3.7 (0.4) | 69.8 (2.3) | 64.8 (1.1) | 13.7 (1.4) | 1.0 (0.1) | 54.0 (1.5) | 53.4 (0.3) | 80.1 (0.5) | 0.51 (0.05) |
| Acute | 3.4 (0.3) | 69.1 (1.1) | 65.7 (3.6) | 13.2 (0.9) | 1.1 (0.1) | 53.0 (1.5) | 54.5 (0.6) | 81.8 (0.2) | 0.56 (0.07) | |
| Chronic | 3.8 (0.2) | 67.3 (0.8) | 68.4 (0.9) | 14.6 (0.3) | 0.9 (0.1) | 56.5 (0.4) | 53.9 (0.4) | 82.7 (0.1) | 0.66 (0.08) | |
| Midbrain | Control | 3.3 (0.1) | 66.8 (0.9) | 76.6 (1.7) | 18.8 (1.8) | 1.8 (0.5) | 58.8 (1.7) | 55.4 (0.6) | 81.9 (0.2) | 0.79 (0.05) |
| Acute | 3.7 (0.4) | 68.1 (1.1) | 72.0 (1.8) | 19.3 (0.9) | 1.9 (0.4) | 57.2 (1.8) | 53.3 (0.4) | 81.8 (0.4) | 0.72 (0.03) | |
| Chronic | 3.2 (0.3) | 68.2 (1.0) | 73.6 (2.0) | 21.3 (1.2) | 1.6 (0.3) | 60.6 (2.1) | 54.6 (0.7) | 80.7 (0.1) | 0.36 (0.01) | |
| Pons | Control | 3.9 (0.2) | 77.3 (1.1) | 74.3 (0.7) | 11.6 (0.9) | 3.9 (1.1) | 55.5 (0.6) | 53.6 (0.4) | 83.1 (0.3) | 0.69 (0.09) |
| Acute | 3.3 (0.1) | 71.6 (2.7) | 75.0 (1.4) | 14.3 (0.7) | 3.6 (0.8) | 54.3 (1.1) | 53.8 (0.3) | 82.3 (0.1) | 0.58 (0.05) | |
| Chronic | 3.1 (0.2) | 66.9 (0.8) | 72.5 (1.6) | 12.8 (1.4) | 3.7 (1.1) | 56.6 (2.5) | 54.9 (0.3) | 78.5 (0.2) | 0.57 (0.06) | |
| Superior colliculus | Control | 3.7 (0.2) | 74.8 (0.4) | 60.3 (4.7) | 24.6 (1.4) | 1.2 (0.2) | 57.0 (2.1) | 53.9 (0.4) | 79.7 (0.3) | 0.54 (0.04) |
| Acute | 3.3 (0.3) | 73.4 (1.8) | 59.4 (3.6) | 21.4 (1.8) | 1.8 (0.5) | 57.9 (1.6) | 53.9 (0.3) | 79.0 (0.2) | 0.53 (0.03) | |
| Chronic | 3.2 (0.3) | 74.1 (0.7) | 58.4 (2.2) | 25.1 (0.6) | 1.2 (0.1) | 60.8 (1.1) | 53.6 (0.0) | 81.9 (0.2) | 0.51 (0.03) | |
| Thalamus | Control | 3.6 (0.2) | 67.7 (0.6) | 73.5 (0.8) | 21.0 (2.0) | 1.7 (0.3) | 57.7 (1.1) | 54.4 (0.3) | 82.8 (0.1) | 0.37 (0.01) |
| Acute | 3.4 (0.1) | 67.1 (1.0) | 72.9 (2.4) | 20.1 (2.2) | 1.8 (0.4) | 57.9 (2.4) | 54.7 (0.1) | 81.3 (0.2) | 0.36 (<0.01) | |
| Chronic | 3.6 (0.3) | 67.9 (0.8) | 72.2 (1.0) | 22.2 (0.8) | 2.0 (0.4) | 60.1 (1.3) | 53.5 (0.2) | 81.7 (0.4) | 0.37 (<0.01) |
Mean DNA methylation levels of six genes (Bdnf, Comt, Il1b, Il6, Nr3c1, and Tnf), global DNA methylcytosine (L1-5utr and L1-orf) and global 5-hydroxymethylcytosine (5hmC) are provided for the 10 regions of the rat brain that were analyzed, with standard error of the mean in brackets.
Figure 3.
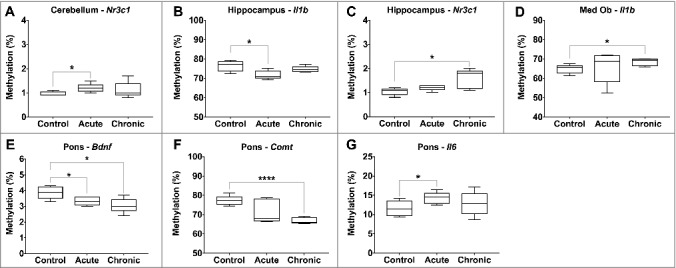
Differential gene-specific DNA methylation in response to morphine exposure. Gene-specific changes in DNA methylation by morphine exposure (control, acute, and chronic) are illustrated, with significant differences indicated (P value * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
Global DNA methylation levels, estimated through measurement of Line1 methylation, were significantly lower in the superior colliculus following both acute (L1-orf, 82.5 vs. 81.9%, P value < 0.05) and chronic morphine exposures (L1-orf, 82.5 vs. 81.3%, P value = 0.003), and higher in the hypothalamus following chronic exposure (L1-5utr, 53.7 vs. 54.9%, P value = 0.04) (Figure 4).
Figure 4.
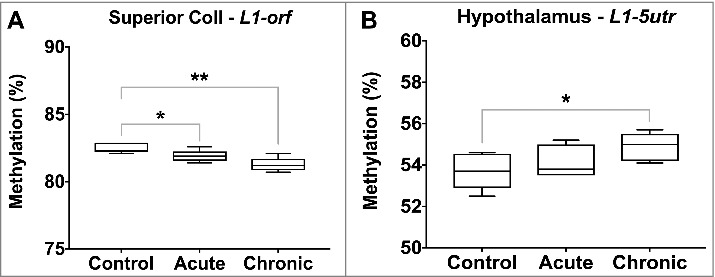
Global DNA methylation levels following acute and chronic morphine exposure. Global DNA methylation levels were estimated through measurement of L1-5utr and L1-orf elements in 10 regions of the rat brain. Regions displaying significant changes are illustrated (P value * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
Nuclear 5-hydroxymethylcytosine levels are altered following morphine induction
We measured global 5-hydroxymethylcytosine (5hmC) levels in each of the ten regions of the brain following acute and chronic morphine induction. In the control rats, 5hmC levels varied by brain region (Table 1). The lowest levels of 5hmC were observed in the hippocampus (0.34%) and thalamus (0.37%), with the highest in the midbrain (0.79%) and pons (0.69%). Significant changes in 5 hmC levels were identified following chronic morphine induction, but none following acute exposure. The levels of 5 hmC increased in the cerebral cortex (0.50% vs. 0.78%, P value = 0.002), hippocampus (0.34% vs. 0.43%, P value = 0.01) and hypothalamus (0.40% vs. 0.54%, P value = 0.008), while they decreased in the midbrain (0.79% vs. 0.36%, P value < 0.0001) (Figure 5).
Figure 5.
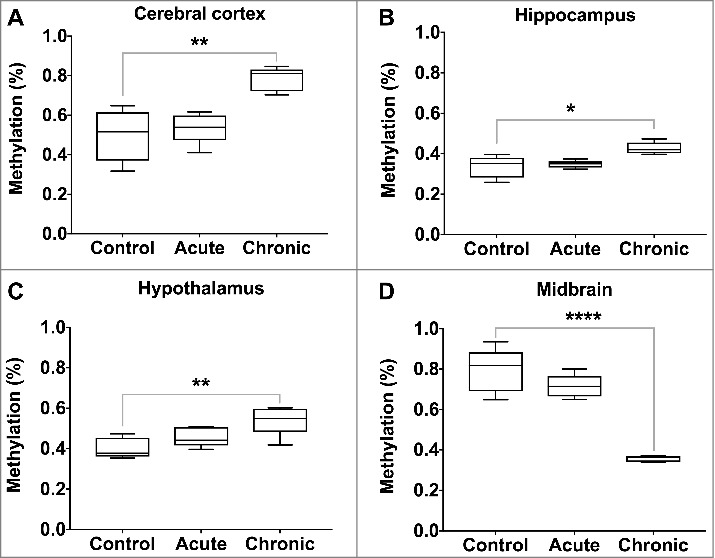
Global 5-hydroxymethylcytosine levels following acute and chronic morphine exposure. Global 5 hmC levels were measured by ELISA in 10 regions of the rat brain, with significant changes illustrated (P value * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
Morphine induction is not associated with alterations in DNA methylation of mitochondrially-encoded genes
We measured DNA methylation of three mitochondrially-encoded genes (Mtco1, Mtco2, and Mtco3) following morphine exposure. Methylation levels of the three genes were uniformly low across the 10 brain regions, with mean levels ranging between 0.8 (Mtco2, cerebral cortex and pons) and 2.7% (Mtco1, cerebral cortex) (Table 2). No significant effect of acute or chronic morphine induction was observed on methylation of the three genes in any of the brain regions. The most substantial changes in methylation were observed for Mtco1 in the cerebral cortex following acute (2.7 vs. 1.8%, P value = 0.23) and chronic (2.7 vs. 1.9%, P value = 0.29) morphine induction.
Table 2.
DNA methylation of mitochondrial-encoded genes in the rat brain following morphine induction.
| Region | Treatment | Mtco1 | Mtco2 | Mtco3 |
|---|---|---|---|---|
| Cerebellum | Control | 2.1 (0.1) | 0.9 (0.1) | 1.1 (0.1) |
| Acute | 2.0 (0.2) | 1.0 (0.1) | 1.2 (0.1) | |
| Chronic | 2.1 (0.1) | 0.9 (0.1) | 1.1 (0.1) | |
| Cerebral cortex | Control | 2.7 (0.6) | 0.8 (0.1) | 1.1 (0.1) |
| Acute | 1.8 (0.3) | 0.9 (0.2) | 1.1 (<0.01) | |
| Chronic | 1.9 (0.4) | 0.8 (0.1) | 1 (0.1) | |
| Hippocampus | Control | 1.9 (0.1) | 1.0 (0.1) | 1.2 (0.1) |
| Acute | 1.9 (0.2) | 0.8 (0.1) | 1.3 (0.1) | |
| Chronic | 1.8 (0.1) | 0.8 (0.1) | 1.1 (0.1) | |
| Hypothalamus | Control | 2.0 (0.1) | 0.9 (0.1) | 1.1 (0.1) |
| Acute | 2.1 (0.2) | 0.9 (0.1) | 1.1 (0.1) | |
| Chronic | 1.9 (0.2) | 0.7 (0.1) | 1.0 (0.1) | |
| Inferior colliculus | Control | 2.2 (0.1) | 1.0 (0.1) | 1.3 (0.2) |
| Acute | 2.2 (0.1) | 0.9 (0.1) | 1.2 (0.1) | |
| Chronic | 2.0 (0.1) | 0.8 (0.1) | 1.1 (0.1) | |
| Medulla oblongata | Control | 2.4 (0.3) | 1.0 (0.1) | 1.1 (0.1) |
| Acute | 2.0 (0.2) | 0.8 (0.1) | 1.1 (0.1) | |
| Chronic | 2.0 (0.2) | 0.8 (0.1) | 1.1 (0.1) | |
| Midbrain | Control | 1.9 (0.2) | 0.9 (0.1) | 1.2 (0.2) |
| Acute | 2.2 (0.4) | 0.9 (0.1) | 1.1 (0.1) | |
| Chronic | 2.0 (0.2) | 1.0 (0.1) | 1.1 (0.1) | |
| Pons | Control | 1.9 (0.3) | 0.8 (0.1) | 1.0 (0.2) |
| Acute | 2.3 (0.4) | 0.8 (0.1) | 1.6 (0.5) | |
| Chronic | 2.6 (1.0) | 0.8 (0.1) | 1.1 (0.1) | |
| Superior colliculus | Control | 2.1 (0.1) | 1.0 (0.1) | 1.3 (0.1) |
| Acute | 2.4 (0.2) | 0.8 (0.1) | 1.3 (<0.01) | |
| Chronic | 2.1 (0.2) | 0.9 (0.1) | 1.3 (0.1) | |
| Thalamus | Control | 2.1 (0.1) | 1.0 (0.1) | 1.3 (0.2) |
| Acute | 1.9 (0.1) | 0.9 (0.1) | 1.1 (<0.01) | |
| Chronic | 1.8 (0.2) | 1.0 (0.3) | 0.9 (0.1) |
Mean DNA methylation levels of three genes (Mtco1, Mtco2, and Mtco3) are provided, with standard error of the mean in brackets.
Discussion
In this study, we investigated the effect of acute and chronic morphine exposures upon nuclear and mitochondrial DNA methylation in ten regions of the rat brain known to be affected by opiates. Brain region-specific changes were identified in global 5-methylcytosine and 5-hydroxymethylcytosine content, and the Bdnf, Comt, Il1b, Il6, and Nr3c1 genes in response to morphine exposure. We did not observe differential methylation within the mitochondrial genome. To our knowledge, our study is the first to identify region-specific epigenetic changes in the brain in response to morphine exposure.
Morphine induces the dopaminergic mesolimbic pathway in the ventral tegmental area (VTA) within the midbrain. This region of the brain has a high concentration of dopaminergic neurons and is considered to be crucial in addiction. We observed significantly decreased global DNA methylation in the superior colliculus, estimated through analysis of Line1, and a highly significant decrease in 5 hmC content in the midbrain. Together, these observations demonstrate substantial epigenetic changes in this important region of the brain with morphine exposure.
The 5hmC modification is approximately 10-fold more abundant in the brain than other tissues [34], increasing during neuronal development but also implicated in age-related neurodegeneration [35]. While 5hmC is an intermediate in active DNA demethylation, 5mC and 5hmC levels are not inherently correlated [36] and therefore our observations of decreased 5mC and 5hmC are not contradictory. Furthermore, our results could also be the product of the direct role of 5hmC in RNA splicing [37] and gene expression through inhibition of chromatin remodelling [38], which may offer an alternative explanation for our observations. TET1 has been demonstrated to be downregulated in response to cocaine administration, leading to locus-specific decreases in 5hmC [39]. Whilst other studies have reported no change in global 5hmC content in response to heroin and cocaine exposure [19,20,39], they have focused upon a single region (nucleus accumbens) or utilized homogenized brain tissue and therefore could not exclude the possibility of localized changes in 5hmC in specific regions of the brain. The limitations of such approaches were addressed in the current study. Indeed, a particular strength of this work is the analysis of ten different regions of the brain that has facilitated the identification of region-specific changes in DNA methylation. Our findings demonstrated significantly increased 5hmC levels in the cerebral cortex, hippocampus, and hypothalamus, in contrast to observations elsewhere of increased hippocampal 5hmC levels following the cessation of cocaine [40]. Further work is required to elucidate the functional implications of these observations.
We identified differential methylation of three genes (Bdnf, Comt, and Il6) in the pons following morphine exposure. This region, located inferior to the midbrain, regulates respiration and sleep, which are commonly disturbed with morphine use. Animal studies have demonstrated a morphine-induced reduction in acetylcholine in the pons and medulla oblongata [41], thereby further implicating this region in response to the drug. BDNF expression facilitates opiate-associated neural plasticity [42] and the dopamine-dependent response to opiates [6]. Chronic exposure to opiates is associated with increased BDNF levels in the VTA [23], and it has been demonstrated that this increase begins rapidly after initial usage [43]. Other studies have reported increased expression of BDNF in the VTA following morphine withdrawal [24]. Our results provide further evidence for the role of BDNF in the acute response to morphine exposure and suggest that this may not be limited to the VTA. Catechol-O-methyltransferase (COMT) is implicated in dopamine elimination and, subsequently, COMT activity reduces response to morphine [8], while genetic variants in the COMT gene are associated with differential response to morphine as an analgesic [44]. Our observation of decreased Comt methylation with chronic exposure may be in response to increased dopamine signaling and a role in its catabolism. The changes seen in Il6 in the pons may be in accordance with the modulating role that opioids have in inflammatory pathways [45]. Our observation of increased Il6 promoter methylation is in contrast to reports of increased secretion in acute response to morphine in the spinal cord [4] and plasma [46,47]. Notably, however, we also identified regional-specificity in Il6 methylation levels and, therefore, the difference in measured methylation levels may represent alterations in cell composition, such as increased microglial infiltration. As IL-1β and TNF-α are also secreted by activated macrophages in response to morphine [48], but were not differentially methylated in the pons, it is unclear how IL-6 alone may be implicated in response to morphine.
Morphine administration can induce the generation of reactive oxygen species (ROS) by increasing the metabolism of substrates such as dopamine and xanthine oxidase [49]. Elevated ROS levels are known to induce site-specific alterations in DNA methylation by mechanisms such as modulation of the expression of DNA methyltransferases (DNMTs) [50]. While the observed gene-specific changes in DNA methylation are likely to be directly induced by morphine administration, global changes in 5mC and 5hmC content could in part be non-specific effects through increased ROS production.
The study of the mitochondrial epigenome is an emerging field of interest. Alterations in mitochondrial DNA methylation have been demonstrated in cardiovascular disease [51] and in response to environmental exposures [52,53]. It has been speculated that such epigenetic changes may give rise to the altered mitochondrial function observed in drug addiction [14]. However, our study did not identify differential methylation of mitochondrial genes in response to morphine induction. We cannot exclude the possibility that other regions of the mitochondrial epigenome may be differentially methylated in response to exposure, and therefore more comprehensive analysis is required.
Our study contains certain limitations that restrict the inferences that can be made from our observations. In particular, the absence of rat behavioral studies and the lack of RNA and protein samples to analyze gene expression have precluded the study of the functional impact of the observed epigenetic changes. The number of cells obtained from some of the dissected tissue specimens was only sufficient to extract DNA from and, therefore, no analysis of gene expression was possible. Gene-specific analysis of 5 hmC content would also have proven highly insightful, but it is currently prohibitively expensive. Nonetheless, our study offers several important strengths. Firstly, the study of ten different regions of the brain has enabled us to identify region-specific epigenetic changes in response to drug exposure. Furthermore, we have performed a range of epigenetic analyses (nuclear and mitochondrial gene-specific DNA methylation and global 5mC and 5hmC content) that have facilitated a broader study of the epigenome in response to morphine exposure. Finally, our study design has enabled the delineation of epigenetic changes associated with acute and chronic morphine exposure.
In conclusion, we have identified gene-specific and global changes in DNA methylation in response to acute and chronic morphine exposure, which were highly localized to specific regions of the brain. Of especial interest, we identified significant changes in 5 hmC content in four of the ten regions, suggestive of wider epigenetic remodeling in response to morphine exposure. We hope that our findings will help inform further studies in the treatment of dependence, including elucidation of the functional consequences of the observed epigenetic changes and how they relate to long-term response to opiates. Additionally, our findings may help facilitate the production of biomarkers of drug tolerance. Given the increase in deaths from opiate overdose within the past decade [54,55], there is urgent clinical need for tolerance biomarkers that could help reduce thousands of avoidable deaths.
Materials and methods
Animals and housing conditions
Male Wistar rats (180–200 g) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). Following shipment, rats were housed in a temperature (22–24°C) and humidity (60%) controlled environment on a 12-hour light/dark cycle (lights on at 7:00 AM) for three days to allow for acclimatization prior to experimental studies. The animals were given free access to food and water ad libitum. All efforts were made to reduce the number of animals used and minimize their suffering. Experimental study groups were assigned randomly. All procedures were approved by the Laboratory Animal Use Committee of Shanghai Jiao Tong University School of Pharmacy.
Drugs
Morphine hydrochloride was purchased from Shenyang First Pharmaceutical Co., Ltd. (Shenyang, Liaoning, China) and Maybridge Chemicals (Cornwall, UK). CBIO was freshly dissolved in sterile normal saline solution (Sinopharm Group Chemical Reagent Co., Ltd.) with the pH adjusted to 7.3–7.5 with 1 M NaOH solution.
Morphine induction
Male rats were assigned into three groups (each n = 5) receiving alternative treatment regimens: saline/saline (Group A, control); saline/morphine (Group B, acute challenge, 10 mg/kg); and morphine/morphine (Group C, chronic induction). For morphine chronic induction, the first treatment period consisted of subcutaneous injection of saline (Groups A and B, 10 ml/kg) or morphine (Group C, 10 mg/kg) twice daily at 12-hour intervals for nine consecutive days [56]. On Day 10, the second treatment was performed by a single subcutaneous injection of saline (Group A) or morphine (Groups B and C, 10 mg/kg) (Figure 1). Previous work has demonstrated tolerance to morphine is developed within 7–10 days, with epigenetic changes observable in the rodent brain 5–7 days following first treatment [57,58].
Sample preparation and DNA extraction
The rats were decapitated on Day 10, one hour after the last administration. Brains were removed from the skull and ten brain regions (cerebellum, cerebral cortex, hippocampus, hypothalamus, inferior colliculus, midbrain, pons, medulla oblongata, superior colliculus, and thalamus) were rapidly dissected, frozen on dry ice, and kept at −80°C until processed. DNA was extracted from the tissues using the Wizard Genomic DNA purification kit (Promega, Madison, WI) according to the manufacturer's instructions. Purified DNA was stored at −20°C until analysis.
DNA methylation analysis
Gene-specific and global 5mC DNA methylation were analyzed by pyrosequencing. Global methylation levels were estimated using two assays to interrogate LINE1 methylation (L1-utr and L1-orf) that have been widely utilized for this purpose since their first development [59] and which correlate well with measures of global 5mC content by high-performance liquid chromatography [60]. Gene-specific assays were designed to interrogate promoter regions for the Bdnf, Comt, Il1b, Il6, Nr3c1, and Tnf genes. Specifically, the assays interrogated Bdnf promoter IV, which influences response to levomethadone [15]; the Comt P1 promoter, which regulates expression of the shorter tissue-specific S-COMT isoform, which influences dopamine metabolism in the mouse brain [61]; and the Nr3c1 GR110 promoter, which we have previously demonstrated to be differentially methylated in the rat brain in response to environmental exposures [62].
Bisulfite conversion was performed using 1 µg of genomic DNA and the EZ-96 DNA Methylation-Gold Kit (Zymo Research, Orange, CA) according to the manufacturer's protocol. M-Elution Buffer (30 µl) was used for the elution of bisulfite-converted DNA. Following amplification of target regions by PCR, DNA methylation was analyzed by pyrosequencing. Details of the primers and thermocycling conditions are shown in Table 3. In brief, a 30 µl-PCR was carried out using 15 µl GoTaq Hot Start Green Master Mix (Promega), 10 pmol forward primer, 10 pmol reverse primer, 1 µl bisulfite-treated DNA, and water. Pyrosequencing was performed using the PyroMark Q96 MD Pyrosequencing System (QIAGEN, Germantown, MD). The percentage of methylated cytosines was quantified at three CpG sites for Bdnf, two for Comt, two for Il1b, one for Il6, six for Nr3c1, one for Tnf, two for L1-utr, and one for L1-orf. For the mitochondrially-encoded genes, the percentage-methylation was measured at three CpG sites for each of Mtco1, Mtco2, and Mtco3. Pyrosequencing reactions were performed in duplicate and the mean of the replicates taken forward for analysis. The correlation coefficient between replicate pyrosequencing runs ranged from 0.77 (L1-orf) to 0.97 (Comt).
Table 3.
PCR and pyrosequencing primer sequences.
| Gene name | Gene region | Primer | Sequence |
|---|---|---|---|
| Bdnf | Promoter IV | Forward primer (5' to 3') | TTTTTAGTTTTTGTTTAGATTAAATGGAGT |
| Reverse biotin primer (5' to 3') | CAACAAAAAAATTAAATTATTAATAATAAA | ||
| Sequencing primer (5' to 3') | GTTTTTTATTGAAGG | ||
| Sequence analyzed | C/TGTGC/TGAGTATTATTTTC/TGTTATG | ||
| Comt | Promoter P1 | Forward primer (5' to 3') | GTTTGTTGTAGTTTGGAGTTAGGT |
| Reverse biotin primer (5' to 3') | CTCATCCTCCCCCATTACCT | ||
| Sequencing primer (5' to 3') | TATGGTTTAGTGTGG | ||
| Sequence analyzed | C/TGGTTGTGGGGTGTAGGGGGC/TGGGGGGGT | ||
| Nr3c1 | GR110 | Forward primer (5' to 3') | GGTTGGTAAAAGTTTGTTAAGTT |
| Reverse biotin primer (5' to 3') | CTAAAAACTCTCCCCTCCCC | ||
| Sequencing primer (5' to 3') | GTTTATTTTAGTATT | ||
| Sequence analyzed | C/TGC/TGC/TGTTC/TGTTC/TGTTC/TGC | ||
| Il1b | Promoter | Forward primer (5' to 3') | TGTATAAGGAAGTTTGATTGGAGAG |
| Reverse biotin primer (5' to 3') | ATAAAATCAATTAACCCAAAAAAAA | ||
| Sequencing primer (5' to 3') | ATGTTTTGAATTATT | ||
| Sequence analyzed | C/TGGGGTTTGCTGTCCACTAGTTTTCTCTCCCTC/TGTTTTA | ||
| Il6-1 | Promoter | Forward primer (5' to 3') | TTGTGATTTTTTGGATGTTAAATGA |
| Reverse biotin primer (5' to 3') | CAAACATCCCCAATCTCATATTTAT | ||
| Sequencing primer (5' to 3') | TTGTGATTTTTTGGATGTTAAATGA | ||
| Sequence analyzed | C/TGTTATAT | ||
| Il6-2 | Promoter | Forward primer (5' to 3') | TTGTGATTTTTTGGATGTTAAATGA |
| Reverse biotin primer (5' to 3') | CAAACATCCCCAATCTCATATTTAT | ||
| Sequencing primer (5' to 3') | TTAAAAGTAGAGAGT | ||
| Sequence analyzed | C/TGATTTTTA | ||
| Tnf | Promoter | Forward primer (5' to 3') | GGATTGTTATAGAATTTTGGTGAGG |
| Reverse biotin primer (5' to 3') | ACTTCCTTAATAAAAAAAACCATAATCTC | ||
| Sequencing primer (5' to 3') | TTAAATTTTTGTTTT | ||
| Sequence analyzed | C/TGTATTGGAGAAGAAATTGA | ||
| MT-co1 | Exon | Forward primer (5' to 3') | AGTTGGAGTTGGAATAGGATGAATA |
| Reverse biotin primer (5' to 3') | TAACTCCTAAAATAAAAAACACCCC | ||
| Sequencing primer (5' to 3') | ATATTTTTTTTTAGT | ||
| Sequence analyzed | C/TGGAAATTTAGTTTATGTTGGGG/ATATTC/TGTAGATTTAA | ||
| MT-co2 | Exon | Forward primer (5' to 3') | TAATGATTTAAAATTAGGTGAATTT |
| Reverse biotin primer (5' to 3') | TAACCCTAATAAAAAAATAACTCATAAATA | ||
| Sequencing primer (5' to 3') | ATAGAATTTTTAATT | ||
| Sequence analyzed | C/TGTATATTAATTTTATTC/TGAAGAC/TGTTTTG | ||
| MT-co3 | Exon | Forward primer (5' to 3') | GTTATTATATTTTTATTGTATAAAAAGGTT |
| Reverse biotin primer (5' to 3') | AAATAATAAAATACTCAAAAAAATCC | ||
| Sequencing primer (5' to 3') | GTATAAAAAGGTTTT | ||
| Sequence analyzed | C/TGATAC/TGGAATAATTTTGTTTATTGTTTTC/TGAAGT | ||
| L1-utr | - | Forward primer (5' to 3') | GGTGTATAGGTTTTTTTGGTTGTTG |
| Reverse biotin primer (5' to 3') | AAATTCACCAAACAACTTTCTTACAA | ||
| Sequencing primer (5' to 3') | TTTTTTTGGTTGTTGT | ||
| Sequence analyzed | C/TGTTGTAGAGAGTTC/TGTGGTAGTATTTTA | ||
| L1-orf | - | Forward primer (5' to 3') | AGAAAGAATATTAAAAATAGTAAGGGAAAA |
| Reverse biotin primer (5' to 3') | ATCAATCCAAAATCTTCTAACCTTC | ||
| Sequencing primer (5' to 3') | TTATATTAGATTTTT | ||
| Sequence analyzed | C/TGTTAGAAATTAT |
Analysis of global 5-hydroxymethylcytosine levels
The 5hmC modification was measured in total DNA using the MethylFlash Global DNA Hydroxymethylation ELISA Easy Kit (Epigentek, Farmingdale, NY) according to the manufacturer's protocol. The correlation coefficient between replicates was 0.97.
Statistical analysis
Analysis was performed using GraphPad Prism version 7.0b. Differences in DNA methylation were determined by t-test, with significance defined as P value < 0.05.
Funding Statement
This work was supported by the Science Foundation of Tianjin Medical University under Grant 2GW033 and the National Natural Science Foundation of China under Grants 81602827 and 41601548.
Acknowledgments
We would like to thank the staff of the King's Lab, Shanghai Jiao Tong University School of Pharmacy, China for supplying the rats. This work was supported by the Science Foundation of Tianjin Medical University under Grant 2GW033 and the National Natural Science Foundation of China under Grants 81602827 and 41601548.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Hutchinson MR, Shavit Y, Grace PM, et al. . Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810; PMID:21752874; Available from: https://doi.org/10.1124/pr.110.004135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Albertson DN, Schmidt CJ, Kapatos G, et al. . Distinctive profiles of gene expression in the human nucleus accumbens associated with cocaine and heroin abuse. Neuropsychopharmacology. 2006;31:2304–2312; PMID:16710320; Available from: https://doi.org/10.1038/sj.npp.1301089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Piechota M, Korostynski M, Sikora M, et al. . Common transcriptional effects in the mouse striatum following chronic treatment with heroin and methamphetamine. Genes Brain Behav. 2012;11:404–414; PMID:22390687; Available from: https://doi.org/10.1111/j.1601-183X.2012.00777.x [DOI] [PubMed] [Google Scholar]
- [4].Hutchinson MR, Coats BD, Lewis SS, et al. . Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008;22:1178–1189; PMID:18599265; Available from: https://doi.org/10.1016/j.bbi.2008.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shavit Y, Wolf G, Goshen I, et al. . Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–59; PMID:15836969; Available from: https://doi.org/10.1016/j.pain.2005.02.003 [DOI] [PubMed] [Google Scholar]
- [6].Vargas-Perez H, Ting-A Kee R, Walton CH, et al. . Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science. 2009;324:1732–1734; PMID:19478142; Available from: https://doi.org/10.1126/science.1168501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Matsushita Y, Omotuyi IO, Mukae T, et al. . Microglia activation precedes the anti-opioid BDNF and NMDA receptor mechanisms underlying morphine analgesic tolerance. Curr Pharm Des. 2013;19:7355–7361; PMID:23448475; Available from: https://doi.org/10.2174/138161281942140105161733 [DOI] [PubMed] [Google Scholar]
- [8].Kambur O, Männistö PT, Viljakka K, et al. . Stress-induced analgesia and morphine responses are changed in catechol-O-methyltransferase-deficient male mice. Basic Clin Pharmacol Toxicol. 2008;103:367–373; PMID:18834357; Available from: https://doi.org/10.1111/j.1742-7843.2008.00289.x [DOI] [PubMed] [Google Scholar]
- [9].Zhu J, Zhu F, Zhao N, et al. . Methylation of glucocorticoid receptor gene promoter modulates morphine dependence and accompanied hypothalamus-pituitary-adrenal axis dysfunction. J Neurosci Res. 2017;95:1459–1473; PMID:27618384; Available from: https://doi.org/10.1002/jnr.23913 [DOI] [PubMed] [Google Scholar]
- [10].Cunha-Oliveira T, Rego AC, Garrido J, et al. . Street heroin induces mitochondrial dysfunction and apoptosis in rat cortical neurons. J Neurochem. 2007;101:543–554; PMID:17250679; Available from: https://doi.org/10.1111/j.1471-4159.2006.04406.x [DOI] [PubMed] [Google Scholar]
- [11].Mastronicola D, Arcuri E, Arese M, et al. . Morphine but not fentanyl and methadone affects mitochondrial membrane potential by inducing nitric oxide release in glioma cells. Cell Mol Life Sci. 2004;61:2991–2997; PMID:15583861; Available from: https://doi.org/10.1007/s00018-004-4371-x [DOI] [PubMed] [Google Scholar]
- [12].Feng YM, Jia YF, Su LY, et al. . Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy. 2013;9:1395–1406; PMID:23800874; Available from: https://doi.org/10.4161/auto.25468 [DOI] [PubMed] [Google Scholar]
- [13].Skrabalova J, Drastichova Z, Novotny J. Morphine as a potential oxidative stress-causing agent. Mini Rev Org Chem. 2013;10:367–372; PMID:24376392; Available from: https://doi.org/10.2174/1570193X113106660031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sadakierska-Chudy A, Frankowska M, Filip M. Mitoepigenetics and drug addiction. Pharmacol Ther. 2014;144:226–233; PMID:24956109; Available from: https://doi.org/10.1016/j.pharmthera.2014.06.002 [DOI] [PubMed] [Google Scholar]
- [15].Schuster R, Kleimann A, Rehme M-K, et al. . Elevated methylation and decreased serum concentrations of BDNF in patients in levomethadone compared to diamorphine maintenance treatment. Eur Arch Psychiatry Clin Neurosci. 2017;267:33–40; PMID:26801497; Available from: https://doi.org/10.1007/s00406-016-0668-7 [DOI] [PubMed] [Google Scholar]
- [16].Xu X, Ji H, Liu G, et al. . A significant association between BDNF promoter methylation and the risk of drug addiction. Gene. 2016;584:54–59; PMID:26976342; Available from: https://doi.org/10.1016/j.gene.2016.03.010 [DOI] [PubMed] [Google Scholar]
- [17].Marie-Claire C, Crettol S, Cagnard N, et al. . Variability of response to methadone: genome-wide DNA methylation analysis in two independent cohorts. Epigenomics. 2016;8:181–195; PMID:26792095; Available from: https://doi.org/10.2217/epi.15.110 [DOI] [PubMed] [Google Scholar]
- [18].Nielsen DA, Yuferov V, Hamon S, et al. . Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34:867–873; PMID:18650805; Available from: https://doi.org/10.1038/npp.2008.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chao M-R, Fragou D, Zanos P, et al. . Epigenetically modified nucleotides in chronic heroin and cocaine treated mice. Toxicol Lett. 2014;229:451–457; PMID:25064621; Available from: https://doi.org/10.1016/j.toxlet.2014.07.023 [DOI] [PubMed] [Google Scholar]
- [20].Fragou D, Zanos P, Kouidou S, et al. . Effect of chronic heroin and cocaine administration on global DNA methylation in brain and liver. Toxicol Lett. 2013;218:260–265; PMID:23454526; Available from: https://doi.org/10.1016/j.toxlet.2013.01.022 [DOI] [PubMed] [Google Scholar]
- [21].McFalls AJ, Imperio CG, Bixler G, et al. . Reward devaluation and heroin escalation is associated with differential expression of CRF signaling genes. Brain Res Bull. 2016;123:81–93; PMID:26655889; Available from: https://doi.org/10.1016/j.brainresbull.2015.11.009 [DOI] [PubMed] [Google Scholar]
- [22].Kozlenkov A, Jaffe AE, Timashpolsky A, et al. . DNA Methylation profiling of human prefrontal cortex neurons in heroin users shows significant difference between genomic contexts of hyper- and hypomethylation and a younger epigenetic age. Genes (Basel). 2017;PMID:828556790; Available from: https://doi.org/10.3390/genes8060152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bolaños CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. Neuromolecular Med. 2004;5:69–83; PMID:15001814; Available from: https://doi.org/10.1385/NMM:5:1:069 [DOI] [PubMed] [Google Scholar]
- [24].Mashayekhi FJ, Rasti M, Rahvar M, et al. . Expression levels of the BDNF gene and histone modifications around its promoters in the ventral tegmental area and locus ceruleus of rats during forced abstinence from morphine. Neurochem Res. 2012;37:1517–1523; PMID:22410736; Available from: https://doi.org/10.1007/s11064-012-0746-9 [DOI] [PubMed] [Google Scholar]
- [25].Commons KG. Neuronal pathways linking substance P to drug addiction and stress. Brain Res. 2010;1314:175–182; PMID:19913520; Available from: https://doi.org/10.1016/j.brainres.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].De Ross J, Avila MA, Ruggiero RN, et al. . The unconditioned fear produced by morphine withdrawal is regulated by mu- and kappa-opioid receptors in the midbrain tectum. Behav Brain Res. 2009;204:140–146; PMID:19520121; Available from: https://doi.org/10.1016/j.bbr.2009.05.033 [DOI] [PubMed] [Google Scholar]
- [27].Mihaly E, Legradi G, Fekete C, et al. . Efferent projections of ProTRH neurons in the ventrolateral periaqueductal gray. Brain Res. 2001;919:185–197; PMID:11701131; Available from: https://doi.org/10.1016/S0006-8993(01)02962-6 [DOI] [PubMed] [Google Scholar]
- [28].Alvandi MS, Bourmpoula M, Homberg JR, et al. . Association of contextual cues with morphine reward increases neural and synaptic plasticity in the ventral hippocampus of rats. Addict Biol. 2017;919:185–197; PMID:28940732; Available from: https://doi.org/10.1111/adb.12547 [DOI] [PubMed] [Google Scholar]
- [29].Fuchs RA, Evans KA, Ledford CC, et al. . The role of the dorsomedial prefrontal cortex, basolateral amygdala, and dorsal hippocampus in contextual reinstatement of cocaine seeking in rats. Neuropsychopharmacology. 2005;30:296–309; PMID:15483559; Available from: https://doi.org/10.1038/sj.npp.1300579 [DOI] [PubMed] [Google Scholar]
- [30].Taylor JA, Ivry RB. Cerebellar and prefrontal cortex contributions to adaptation, strategies, and reinforcement learning. Prog Brain Res. 2014;210:217–253; PMID:24916295; Available from: https://doi.org/10.1016/B978-0-444-63356-9.00009-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vera-Portocarrero LP, Ossipov MH, Lai J, et al. . Descending facilitatory pathways from the rostroventromedial medulla mediate naloxone-precipitated withdrawal in morphine-dependent rats. J Pain. 2011;12:667–676; PMID:21354865; Available from: https://doi.org/10.1016/j.jpain.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bora E, Yücel M, Fornito A, et al. . White matter microstructure in opiate addiction. Addict Biol. 2012;17:141–148; PMID:21070508; Available from: https://doi.org/10.1111/j.1369-1600.2010.00266.x [DOI] [PubMed] [Google Scholar]
- [33].Zhu Y, Wienecke CF, Nachtrab G, et al. . A thalamic input to the nucleus accumbens mediates opiate dependence. Nature. 2016;530:219–222; PMID:26840481; Available from: https://doi.org/10.1038/nature16954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930; PMID:19372393; Available from: https://doi.org/10.1126/science.1169786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Song C-X, Szulwach KE, Fu Y, et al. . Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72; PMID:21151123; Available from: https://doi.org/10.1038/nbt.1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nestor CE, Ottaviano R, Reddington J, et al. . Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–477; PMID:22106369; Available from: https://doi.org/10.1101/gr.126417.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Khare T, Pai S, Koncevicius K, et al. . 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. 2012;19:1037–1043; PMID:22961382; Available from: https://doi.org/10.1038/nsmb.2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jin S-G, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125; PMID:20371518; Available from: https://doi.org/10.1093/nar/gkq223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Feng J, Shao N, Szulwach KE, et al. . Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat Neurosci. 2015;18:536–544; PMID:25774451; Available from: https://doi.org/10.1038/nn.3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sadakierska-Chudy A, Frankowska M, Wydra K, et al. . Increased 5-hydroxymethylation levels in the hippocampus of rat extinguished from cocaine self-administration. Hippocampus. 2017;27:811–821; PMID:28422379; Available from: https://doi.org/10.1002/hipo.22733 [DOI] [PubMed] [Google Scholar]
- [41].Ge XQ, Xu PC, Bian CF. Relationship between morphine-induced respiratory depression and the cholinergic system of respiratory center. Yao Xue Xue Bao. 1990;25:566–572; 2082678 [PubMed] [Google Scholar]
- [42].Akbarian S, Rios M, Liu R-J, et al. . Brain-derived neurotrophic factor is essential for opiate-induced plasticity of noradrenergic neurons. J Neurosci. 2002;22:4153–4162; PMID:12019333; Available from: https://doi.org/20026381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Laviolette SR, van der Kooy D. GABA(A) receptors in the ventral tegmental area control bidirectional reward signalling between dopaminergic and non-dopaminergic neural motivational systems. Eur J Neurosci. 2001;13:1009–1015; PMID:11264674; Available from: https://doi.org/10.1046/j.1460-9568.2001.01458.x [DOI] [PubMed] [Google Scholar]
- [44].Rakvåg TT, Klepstad P, Baar C, et al. . The Val158Met polymorphism of the human catechol-O-methyltransferase (COMT) gene may influence morphine requirements in cancer pain patients. Pain. 2005;116:73–78; PMID:15927391; Available from: https://doi.org/10.1016/j.pain.2005.03.032 [DOI] [PubMed] [Google Scholar]
- [45].Ninković J, Roy S. Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids. 2013;45:9–24; PMID:22170499; Available from: https://doi.org/10.1007/s00726-011-1163-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Houghtling RA, Bayer BM. Rapid elevation of plasma interleukin-6 by morphine is dependent on autonomic stimulation of adrenal gland. J Pharmacol Exp Ther. 2002;300:213–219; PMID:11752119; Available from: https://doi.org/10.1124/jpet.300.1.213 [DOI] [PubMed] [Google Scholar]
- [47].Zubelewicz B, Muc-Wierzgoń M, Harbuz MS, et al. . Central single and chronic administration of morphine stimulates corticosterone and interleukin (IL)-6 in adjuvant-induced arthritis. J Physiol Pharmacol. 2000;51:897–906; PMID:11220497. [PubMed] [Google Scholar]
- [48].Roy S, Cain KJ, Chapin RB, et al. . Morphine modulates NF kappa B activation in macrophages. Biochem Biophys Res Commun. 1998;245:392–396; PMID:9571161; Available from: https://doi.org/10.1006/bbrc.1998.8415 [DOI] [PubMed] [Google Scholar]
- [49].Enrico P, Esposito G, Mura MA, et al. . Effects of allopurinol on striatal dopamine, ascorbate and uric acid during an acute morphine challenge: ex vivo and in vivo studies. Pharmacol Res. 1997;35:577–585; PMID:9356212; Available from: https://doi.org/10.1006/phrs.1997.0193 [DOI] [PubMed] [Google Scholar]
- [50].Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16:13–19; PMID:25585126; Available from: https://doi.org/10.2174/1389450116666150113121054 [DOI] [PubMed] [Google Scholar]
- [51].Baccarelli AA, Byun H-M. Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics. 2015;7:44; PMID:25901189; Available from: https://doi.org/10.1186/s13148-015-0078-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Byun H-M, Panni T, Motta V, et al. . Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 2013;10:18; PMID:23656717; Available from: https://doi.org/10.1186/1743-8977-10-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Byun H-M, Colicino E, Trevisi L, et al. . Effects of air pollution and blood mitochondrial DNA methylation on markers of heart rate variability. J Am Heart Assoc. 2016;5:e003218; PMID:27107129; Available from: https://doi.org/10.1161/JAHA.116.003218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Middleton J, McGrail S, Stringer K. Drug related deaths in England and Wales. BMJ. 2016;355:i5259; PMID:27754839; Available from: https://doi.org/10.1136/bmj.i5259 [DOI] [PubMed] [Google Scholar]
- [55].Rudd RA, Seth P, David F, et al. . Increases in Drug and Opioid-Involved Overdose Deaths – United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65:1445–1452; PMID:28033313; Available from: https://doi.org/10.15585/mmwr.mm655051e1 [DOI] [PubMed] [Google Scholar]
- [56].Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87; PMID:1824728; Available from: https://doi.org/10.1126/science.1824728 [DOI] [PubMed] [Google Scholar]
- [57].Chao YC, Xie F, Li X, et al. . Demethylation regulation of BDNF gene expression in dorsal root ganglion neurons is implicated in opioid-induced pain hypersensitivity in rats. Neurochem Int. 2016;97:91–98; PMID:26970395; Available from: https://doi.org/10.1016/j.neuint.2016.03.007 [DOI] [PubMed] [Google Scholar]
- [58].Sun H, Maze I, Dietz DM, et al. . Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci. 2012;32:17454–17464; PMID:23197736; Available from: https://doi.org/10.1523/JNEUROSCI.1357-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yang AS, Estécio MR, Doshi K, et al. . A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:38e; PMID:14973332; Available from: https://doi.org/10.1093/nar/gnh032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lisanti S, Omar WA, Tomaszewski B, et al. . Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS One. 2013;8:e79044; PMID:24260150; Available from: https://doi.org/10.1371/journal.pone.0079044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tammimäki A, Käenmäki M, Kambur O, et al. . Effect of S-COMT deficiency on behavior and extracellular brain dopamine concentrations in mice. Psychopharmacology (Berl). 2010;211:389–401; PMID:20617305; Available from: https://doi.org/10.1007/s00213-010-1944-2 [DOI] [PubMed] [Google Scholar]
- [62].Byun HM, Benachour N, Zalko D, et al. . Epigenetic effects of low perinatal doses of flame retardant BDE-47 on mitochondrial and nuclear genes in rat offspring. Toxicology. 2015;328:152–159; PMID:25533936; Available from: https://doi.org/10.1016/j.tox.2014.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]