

Abstract
There is intense interest in developing therapeutic strategies for RAS proteins, the most frequently mutated oncogene family in cancer. Development of effective anti-RAS therapies will be aided by the greater appreciation of RAS isoform-specific differences in signaling events that support neoplastic cell growth. Recognition that there are RAS mutation-specific differences has led to expectations that defining RAS mutation-selective vulnerabilities will lead to new therapies. However, critical issues remain that require resolution to facilitate the success of these efforts. In particular, the use of well-validated anti-RAS antibodies is essential for accurate interpretation of experimental data. We evaluated 22 commercially available RAS antibodies using a set of unique and innovative reagents and cell lines. We validated antibodies for each of the four RAS isoforms, and for G12D- or G12V-mutant RAS proteins, for Western blot but not for immunofluorescence (IF) or immunohistochemical (IHC) analyses. Our results may help ensure accurate interpretation of future RAS studies.
INTRODUCTION
The three RAS genes (HRAS, KRAS and NRAS) comprise the most frequently mutated oncogene family in cancer, with an overall 27% missense mutation frequency in all human cancers (COSMIC v80) (1–3). RAS mutation frequency is highly tissue context-dependent, with highest frequencies observed in lung, colorectal and pancreatic ductal adenocarcinoma (PDAC).
RAS genes encode four structurally and biochemically highly related 188–189 amino acid proteins (4). KRAS encodes two proteins, KRAS4A and KRAS4B, due to alternative fourth exon utilization (5–8). RAS proteins exhibit 82–90% overall amino acid sequence identity (9) and 93–99% sequence identity within the amino-terminal G domain (residues 1–164) (10). In sequences involved in interaction with the guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) that regulate GDP-GTP cycling, and in activation of downstream effectors (e.g., RAF), this region is invariant. In contrast, RAS proteins show only 8% sequence identity in the carboxyl-terminal hypervariable region (residues 165–184/185). Caax tetrapeptide motif-signaled posttranslational lipid modifications, an isoform-dependent process of prenylation and methylation, combined with the divergent hypervariable region, determine RAS intracellular trafficking and membrane interactions and contribute to RAS isoform-distinct biological functions. One issue now recognized, yet still poorly understood, is that the four RAS protein isoforms, despite their near-identical structure and biochemistry, serve distinct roles as cancer drivers (3, 9).
The three RAS genes are not mutated at comparable frequencies in human cancers. KRAS is by far the most commonly mutated (84%), followed by NRAS (12%), and HRAS (4%) (1). Additionally, there are tissue-specific preferences for the specific RAS gene mutated. For example, KRAS is exclusively the RAS isoform mutated in PDAC, whereas NRAS is the predominant (94%) isoform mutated in cutaneous melanoma, and HRAS mutations are favored (86%) in head and neck squamous cell carcinoma. Limited evidence that these frequencies reflect tissue-specific functions comes from mouse models of cancer. For example, KrasG12D but not NrasG12D promoted colon cancer development in Apc-deficient mice (11). Expression of activated KrasG12V and HrasG12V under control of the same regulatory sequences resulted in different types of tumors (12).
Cancer-associated RAS genes contain missense mutations that result in single amino acid substitutions; 99% are found at one of three mutational hotspots, glycine-12 (G12), glycine-13 (G13) or glutamine-61 (Q61) (1, 9, 13). G12 mutations comprise 83% of all KRAS mutations, followed by G13 mutations (14%), whereas Q61 mutations are rare (2%). In striking contrast, Q61 is the predominant mutational hotspot in NRAS (62%), followed by G12 (23%), and then G13 (12%). These frequencies suggest that G12 and Q61 mutations may have distinct consequences for different RAS isoforms. That NrasQ61R but not NrasG12D potenti caused melanoma development in Ink4a-deficient mice supports this possibility (14). Finally, although there are six possible single base missense mutations at each codon, the frequencies with which they occur are not uniform. It has also become evident that RAS mutant proteins exhibit distinct biochemical deficiencies (15, 16). An emerging premise in the field is that the different RAS mutations are not created equal and that the identification of mutation-selective differences will identify unique vulnerabilities that can then be exploited to develop mutation-selective therapies (17). The development of RAS inhibitors that are selective for one RAS mutation, G12C, represents this shift in the field (18, 19).
Antibodies are essential reagents in biomedical research, but poor quality antibodies are a major cause of the “reproducibility crisis” in understanding protein function and in the pathway to new drugs (20–22). This crisis, and the related need for better characterized and validated antibodies, is exemplified by a report that 47 of 53 landmark cancer research papers from the early 2000s could not be replicated (23). It has been estimated that fewer than half of the 6,000 commercially available antibodies recognize their advertised targets (21) and that $800 million annually is spent on poor quality antibodies ($350 million in the USA alone) (22). There are also numerous examples of antibody problems slowing medical progress. In 2009, a research group secured federal funding to stratify melanoma patient treatment regimens based on quantitative immunofluorescence staining of biopsy samples (24). However, they were forced to abandon this project because subsequent lots of antibodies from the same source no longer generated the same results on the same tumor samples (20). Further, another research group spent two years, thousands of patient samples, and $500,000 before realizing that the antibody they had been using for early detection of pancreatic cancer was recognizing the wrong protein (20), an astounding disappointment for pancreatic cancer patients, whose 5-year survival rate is an abysmal 8% (25). Nevertheless, despite extensive efforts of organizations such as the International Working Group on Antibody Validation, two International Antibody Validation Meetings, and crowd-funded scientific websites such as www.antybuddy.com (26, 27), the quality of commercially available antibodies and their degree of validation remain decidedly mixed.
With renewed interest in RAS and with new investigators entering the field (28, 29), we believe that now is an ideal time to revisit validation of RAS antibodies. To ensure the accuracy of our own studies and as a service to the field, we analyzed 22 commercially available RAS antibodies for their specificity and sensitivity in recognizing individual RAS isoforms. Our analyses demonstrate that the properties of many of these antibodies have not been accurately established. Our results support the likelihood that the conclusions from some RAS research have been compromised by misinterpretation of data due to faulty information regarding RAS antibody properties. Moving forward, we hope that our validation of antibodies for specific applications will facilitate greater reliability in future RAS studies.
RESULTS
Evaluation of RAS isoform selectivity against recombinant RAS proteins
We purified unprocessed wild-type full length human KRAS4B, HRAS, and NRAS proteins from E. coli. Additionally, both unprocessed and processed (farnesylated, proteolyzed and methylated) KRAS4B and KRAS4A were purified from insect cells as we described (30). Three concentrations of each purified RAS protein were resolved by SDS-PAGE to verify size and purity (Fig. 1A). KRAS bands appear slightly more intense across the blot, likely because of their lysine-rich C-terminal HVR sequences detected by the negatively charged Coomassie staining method. Notably, unprocessed KRAS4A protein from insect cells purified as two species, a minority of the full-length protein, and a major species that, based on mass spectrometry results, lacks the last five amino acids. Otherwise, RAS proteins were highly pure (>95%) and migrated at the expected molecular weight for full length proteins.
Fig. 1. Selectivity of RAS antibody detection of purified RAS proteins.
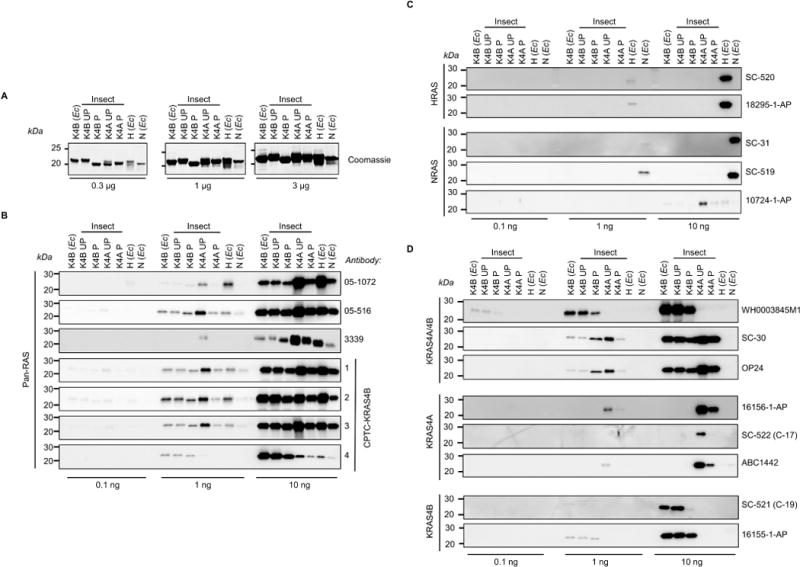
Three different concentrations of E. coli (Ec)- or insect cell-expressed unprocessed (UP) or processed (P) purified RAS proteins were resolved by SDS-PAGE and (A) stained with Coomassie blue to show protein loading, or subjected to western blot analysis using a panel of RAS antibodies: (B) pan-RAS, (C) HRAS- or NRAS-selective, and (D) “pan-KRAS” KRAS4A/4B-, or KRAS4A- or KRAS4B-selective.
We analyzed a panel of 22 commercially available RAS antibodies (Table 1) that includes those most widely cited in the literature. Also included in this panel were four monoclonal antibodies generated against human KRAS4B protein by the NCI RAS Initiative at the Frederick National Laboratory for Cancer Research (FNLCR) that have not yet been used in any published studies. We interrogated each antibody for RAS isoform selectivity and sensitivity, and asked whether Caax-signaled processing influenced recognition of KRAS proteins (Figs. 1, B–D). Most antibodies recognized their intended target, but there were some exceptions.
Table 1.
List of RAS Antibodies and Manufacturer Information
| Isoform | Cat # | Lot # | Vendor | Clonality | Clone # | Host | Species Reactivity1 | Applications2 | Immunogen/Epitope |
|---|---|---|---|---|---|---|---|---|---|
| H, K, N | 05–1072 | 2557627 | Millipore | mAb | 9A11.2 | Mouse | H, M, R | WB, IHC | Recombinant GST-FL HRAS |
| H, K, N | 05–516 | 2673037 | Millipore | mAb | Ras10 | Mouse | H, M, R | WB, ELISA, FC, ICC, IHC, IP | Recombinant RAS |
| H, K, N | 3339 | 5 | Cell Signaling | mAb | 27H5 | Rabbit | H, M, R | WB | N-terminal KRAS peptide |
|
| |||||||||
| K | CPTC-KRAS4B-13 | 1 | FNLCR/DSHB | mAb | 2D2 | Mouse | H | WB | Recombinant FL KRAS4B |
| K | CPTC-KRAS4B-23 | 1 | FNLCR/DSHB | mAb | 4E8 | Mouse | H | WB | Recombinant FL KRAS4B |
| K | CPTC-KRAS4B-33 | 1 | FNLCR/DSHB | mAb | 4H2 | Mouse | H | WB | Recombinant FL KRAS4B |
| K | CPTC-KRAS4B-43 | 1 | FNLCR/DSHB | mAb | 6C10 | Mouse | H | WB | Recombinant FL KRAS4B |
| K | WH0003845M1 | FA191-S2 | Sigma-Aldrich | mAb | 3B10–2F2 | Mouse | H | WB, indirect ELISA, IF | Recombinant GST-FL KRAS4B |
| K | SC-30 | K0315 | Santa Cruz Biol. | mAb | F234 | Mouse | H, M, R | WB, IP, IF | Recombinant rat KRAS4A (aa 54–189) |
| K | OP24 | D00175459 | Millipore | mAb | 234–4.2 | Mouse | H, M, R | WB, Frozen Sections, IP | Recombinant rat KRAS4A (aa 54–189) |
|
| |||||||||
| H | SC-520 | L3015 | Santa Cruz Biol. | polyclonal | n/a | Rabbit | H, M, R | WB, IP, IF, ELISA | Human HRAS C-terminal peptide |
| H | 18295–1-AP | 22367 | Proteintech | polyclonal | n/a | Rabbit | H, M | WB, ELISA, IF, IHC, FC | Peptide |
|
| |||||||||
| N | SC-31 | L0915 | Santa Cruz Biol. | mAb | F155 | Mouse | H, M, R | WB, IP, IF, IHC | Recombinant RAS |
| N | SC-519 (C-20) | E0515 | Santa Cruz Biol. | polyclonal | n/a | Rabbit | H, M, R | WB, IP, IF, IHC, ELISA | Human NRAS C-terminal epitope |
| N | 10724–1-AP | 17213 | Proteintech | polyclonal | n/a | Rabbit | H, M, R | WB, ELISA, IHC, FC, IF | Recombinant GST FL human NRAS protein |
|
| |||||||||
| K4A | 16156–1-AP | 26025 | Proteintech | polyclonal | n/a | Rabbit | H | WB, ELISA, IHC, IF, FC | Peptide |
| K4A | SC-522 (C-17) | H2815 | Santa Cruz Biol. | polyclonal | n/a | Rabbit | H, M, R | WB, IP, IF, ELISA | Human KRAS4A C-terminus epitope |
| K4A | ABC1442 | Q2737518 | Millipore | polyclonal | n/a | Rabbit | H | WB | KLH-KRAS4A peptide |
|
| |||||||||
| K4B | SC-521 (C-19) | I2315 | Santa Cruz Biol. | polyclonal | n/a | Rabbit | H, M, R | WB, IP, IF, IHC | Human KRAS4B C-terminal epitope |
| K4B | 16155–1-AP | 15812 | Proteintech | polyclonal | n/a | Rabbit | H, M | WB, ELISA, IHC, IF, FC | Peptide |
| Mutant-Specific Antibodies
| |||||||||
| G12D | 14429S | 1 | Cell Signaling | mAb | D8H7 | Rabbit | H | WB | RAS G12D mutant peptide |
| G12V | 14412S | 1 | Cell Signaling | mAb | D2H12 | Rabbit | H | WB | RAS G12V mutant peptide |
Species (incomplete listing) - H, human; M, mouse; R, rat;
Applications (incomplete listing): IF, immunofluorescence; IHC, immunohistochemistry; IP, immunoprecipitation
Hybridoma
No antibody described as having pan-RAS selectivity uniformly recognized all RAS isoforms. Ras10 (05–516) showed fairly equivalent recognition of all RAS isoforms and was not influenced by KRAS4B processing. However, another pan-RAS antibody (05–1072) was more selective for HRAS and for unprocessed KRAS4A than for other RAS proteins. Three of the four FNLCR-generated antibodies recognized all the RAS proteins, even though the immunogen used was recombinant full length KRAS4B (Fig. 1B). Notably, the FNLCR CPTC-KRAS4B-2 antibody showed pan-RAS sensitivity that was superior to any of the commercially available pan-RAS antibodies we tested.
Two HRAS-specific (SC-520 and 18295–1-AP) and two NRAS-specific (SC-31 and SC-519) antibodies showed strong selectivity for their intended targets, with no recognition of the other RAS isoforms (Fig. 1C). However, one antibody intended to be NRAS-specific (10724–1-AP) only weakly recognized NRAS at the highest protein abundance and instead more effectively recognized other RAS isoforms.
We evaluated three commercially available KRAS4A/4B antibodies (Fig. 1D). Two antibodies (SC-30 and OP24) recognized unprocessed and processed versions of both KRAS isoforms, whereas another (WH0003845M1) showed strong selectivity for KRAS4B with a higher affinity for unprocessed protein, and did not recognize KRAS4A. We evaluated three KRAS4A-specific antibodies [6156–1-AP, SC-522 (C-17) and ABC1422], none of which recognized KRAS4B (Fig. 1D). However, all three showed significantly stronger recognition of unprocessed than processed KRAS4A. This is not entirely surprising, given that the Caax-signaled posttranslational modifications are immediately adjacent to the major sequence differences between KRAS4A and KRAS4B and may therefore disrupt epitope recognition of antibodies generated against purified bacterial proteins that lack such modifications. The two KRAS4B-selective antibodies recognized purified KRAS4B but did not recognize KRAS4A. One of these (16155–1-AP) recognized both unprocessed and processed KRAS4B proteins with comparable signal strength (Fig. 1D). In contrast, the other (SC-521) recognized only unprocessed KRAS4B protein, even though the antibody was generated against the human KRAS4B carboxyl-terminus. Processed KRAS4B may not be recognized because the antibody epitope overlaps with the cleaved Caax fragment that is removed during processing.
Evaluation of RAS isoform selectivity in lysates of engineered mouse cells
We next investigated how well the antibodies performed when probing cell lysates from cells deficient in any Ras genes (henceforth called Rasless MEFs) and rescued by BRAFV600E or exogenous expression of a single RAS isoform. This approach took advantage of an innovative cell model developed by Barbacid and colleagues (31). Rasless MEFs are derived from Hras−/−/Nras−/− mice harboring a conditional Kras allele that is deleted upon ectopic expression of Cre recombinase (31). Upon Kras deletion, these MEFs lack all Ras isoforms, and become quiescent upon loss of all endogenous Ras activity. As was shown previously (31), we also determined that proliferation can be rescued by ectopic expression of activated BRAFV600E, or of wild-type versions of human KRAS4A, KRAS4B, HRAS, or NRAS. We established MEF cell lines using clonal populations from cultures infected with lentivirus expression vectors encoding each gene. Therefore, total RAS protein across the cell lines are comparable but most likely not equivalent (Fig. 2).
Fig. 2. RAS antibodies detect both RAS and also background bands and nonspecific 21-kDa bands in lysates of rescued Rasless MEFs.
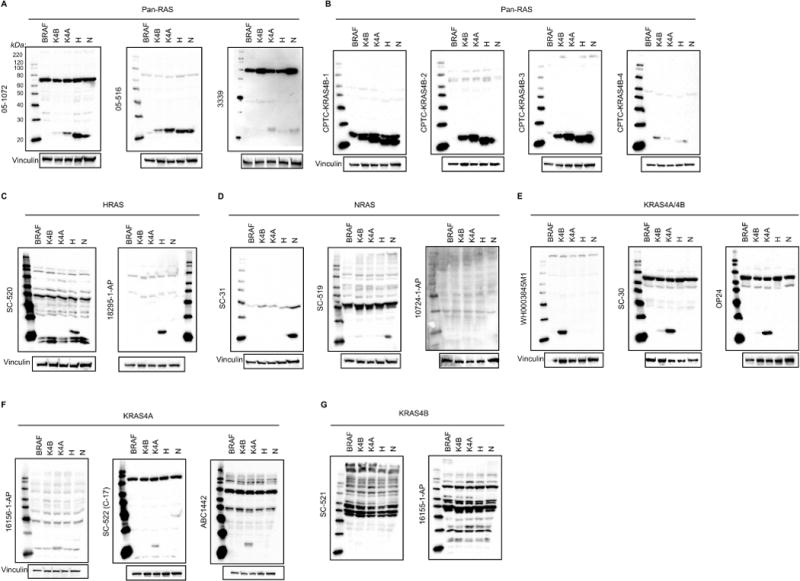
Western blot analyses were performed with (A,B) pan-RAS, (C) HRAS-, (D) NRAS-, (E) KRAS4A/B-, (F) KRAS4A- or (G) KRAS4B-selective antibodies on total cell lysates from Rasless MEFs ectopically expressing the indicated proteins. Vinculin (A-F) or background nonspecific bands (G) served as a loading control. Full (uncut) blots, including MagicMark XP size standards in the left lane of each panel, are shown to reveal degree of nonspecific protein recognition.
The results obtained with the three commercial pan-RAS antibodies in cell lines largely mirrored those using recombinant RAS proteins (Fig. 2A). Antibody 05–516 readily detected all RAS isoforms, albeit more weakly for KRAS4B. However, it also weakly detected a band in the Rasless MEFs expressing BRAFV600E that co-migrated with HRAS and NRAS, likely a RAS-related protein. As described above, three of the four FNLCR-generated anti-KRAS4B antibodies display pan-RAS specificity, with CPTC-KRAS4B-2 showing stronger and equivalent recognition of all RAS isoforms, no detection of any proteins in the ~21 kDa range in the Rasless MEFs expressing BRAFV600E, and faint recognition of other high MW cellular proteins (Fig. 2B). CPTC-KRAS4B-3 displayed similar pan-RAS reactivity and may better recognize NRAS. However, this antibody also weakly recognized a band in Rasless MEFs expressing BRAFV600E that migrates just above all the RAS isoforms; depending on the properties of the SDS-PAGE used to resolve cell lysates, this minor band may or may not be a concern in western blotting applications.
While both HRAS antibodies showed high specificity for the HRAS isoform, one (18295–1-AP) recognized few other cellular proteins whereas the second (SC-520) also recognized numerous others (Fig. 2C). The NRAS antibody SC-31 showed high specificity for NRAS and no recognition of any ~21 kDa bands in Rasless MEFs expressing BRAFV600E (Fig. 2D).
Of three putative KRAS4A/4B antibodies evaluated, both SC-30 and OP24 recognized both KRAS4A and KRAS4B and WH0003845M1 was highly selective for KRAS4B (Fig. 2E). However, based on the much stronger recognition of recombinant KRAS4A protein by SC-30 and OP24 (Fig. 1D), we suspect that neither antibody would provide an accurate determination of how expression of the two KRAS isoforms at the RNA level is related to isoform expression at the protein level.
One KRAS4A-specific antibody (16156–1-AP) appeared to recognize KRAS4A but additionally recognized a closely co-migrating band in Rasless MEFs expressing BRAFV600E (Fig. 2F). In contrast, two other KRAS4A-specific antibodies selectively recognized KRAS4A but not KRAS4B, with no detection of any ~21 kDa bands in Rasless MEFs expressing BRAFVs600E (Fig. 2F). While both KRAS4B antibodies specifically detected purified recombinant proteins (Fig. 1D), neither detected KRAS4B in cell lysates, but strongly recognized numerous nonspecific proteins (Fig. 2G). Instead, our data indicate that WH0003845M1, although described as detecting both KRAS4A and KRAS4B, is the best available reagent for selective detection of KRAS4B. We conclude that there are many experimental conditions under which even antibodies commonly used in the literature may lead to erroneous interpretations.
RAS isoform detection in a human cell line
Our analyses in Rasless MEFs assessed detection of exogenously expressed human RAS and also revealed highly variable nonspecific recognition of mouse proteins. To assess detection of endogenous human RAS proteins and to evaluate nonspecific reactivity in human cells, we also evaluated several antibodies using lysates of HEK293T cells (Fig. 3A). Two pan-RAS antibodies, 05–1072 and 05–516, recognized all RAS isoforms but also recognized other bands. The FNLCR antibody (CPTC-KRAS4B-2) that showed strong pan-RAS reactivity in Rasless MEFs expressing any RAS isoform without additional ~21 kDa bands in Rasless MEFs expressing BRAFV600E (Fig. 2B) also showed strong recognition of human RAS proteins and minimal recognition of nonspecific human proteins.
Fig. 3. RAS antibodies detect both endogenous RAS and nonspecific background bands in human cell lysates.
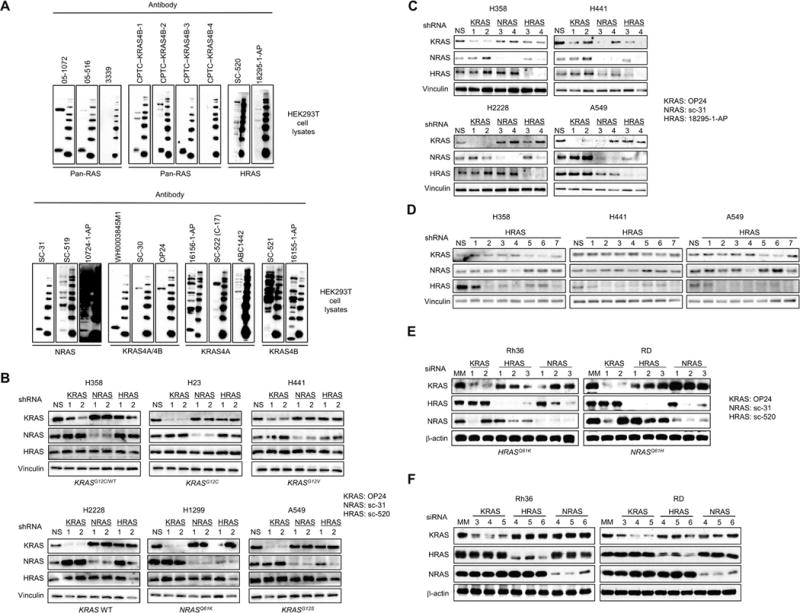
(A) HEK293T cell lysates were probed by western blotting with the indicated RAS antibodies. Full (uncut) blots, including molecular weight markers in the right lane of each panel, are shown to reveal the degree of nonspecific protein recognition. (B, C) To determine isoform selectivity of shRNAs, human NSCLC cell lines were infected with lentivirus vectors encoding shRNAs targeting the indicated RAS genes. Cell lysates were probed by western blotting with the indicated RAS antibodies. Vinculin served as a loading control. (D) To identify HRAS-selective shRNAs, 7 HRAS-targeting constructs were infected into NSCLC cells and probed with the indicated RAS antibodies. (E, F) To determine isoform selectivity of siRNAs, rhabdomyosarcoma cell lines were transfected with siRNAs targeting the indicated RAS genes. Cell lysates were probed by western blotting with the indicated RAS antibodies. β-actin served as a loading control.
Neither HRAS-specific antibody detected any ~21 kDa band in HEK293T cells, indicating that these cells do not express significant endogenous HRAS. The NRAS-selective antibody SC-31 strongly recognized endogenous NRAS, with essentially no nonspecific reactivity.
Collectively, our results using the three commercially available KRAS4A/4B antibodies in all three model systems enable us to conclude that HEK293T cells express only KRAS4B, and that WH0003845M1 is superior to the two commercial KRAS4B-selective antibodies in recognizing endogenous human KRAS4B. Similarly, despite the strong detection by 16156–1-AP of a ~21 kDa band in HEK293T cells, we conclude, based on our results using this antibody in Rasless MEFs, that this band(s) likely represents detection of RAS related proteins (e.g., RRAS isoforms). Finally, the lack of detection of any ~21 kDa band in HEK293T cells by the KRAS4A-specific antibodies SC-522 and ABC1442, coupled with the results from Rasless MEFs, validate both as excellent reagents for selective detection by western blotting of endogenous human KRAS4A but not KRAS4B protein. We suggest that such parallel analyses, using purified human RAS proteins, Rasless MEFs and human cells can more definitively identify the true specificity of RAS antibodies than any one system alone.
Use of RNA interference for antibody validation in human tumor cell lines
Other experimental strategies to assist in validation of the RAS isoform selectivity of antibodies employs genetic knockdown using shRNA and siRNA to selectively target each RAS gene. Currently, no shRNA has been reported that can selectively silence each KRAS splice variant. Using a panel of human non-small cell lung carcinoma (NSCLC) cell lines, we first stably infected each line with lentivirus vectors encoding a non-specific sequence (NS) or two distinct targeted sequences (Table S1) for human HRAS, NRAS or KRAS (Fig. 3B). Western blots on the resulting cell lysates with isoform-selective KRAS4A/4B and NRAS antibodies validated in Rasless MEFs expressing KRAS or NRAS (OP24 and SC-31, respectively). For comparison, we included an HRAS-selective antibody, SC-520, which failed in our validation experiments, to illustrate complications that may arise upon recognition of closely migrating nonspecific reactivity.
The protein levels detected by the KRAS4A/4B and NRAS antibodies were strongly reduced in cells stably expressing shRNA targeting KRAS and NRAS, respectively (Fig. 3B). In contrast, ~21 kDa bands were not reduced in cells stably expressing shRNA targeting HRAS and probed with the SC-520 antibody, suggesting that these HRAS-targeted shRNAs were not effective in silencing HRAS expression. However, the same analyses with a well-validated second HRAS-selective antibody, 18295–1-AP, did show strong suppression of HRAS protein levels (Fig. 3C–3D). Thus, the apparent detection of HRAS protein by SC-520 likely reflects instead its strong detection of non-RAS proteins that migrate near HRAS (Fig. 2C). Table 2 provides a list of the isoform-specific, pan-RAS, and pan-KRAS (KRAS4A/4B) antibodies that we have validated for western blot applications, based on the above experiments.
Table 2.
Validated RAS antibodies
| Isoform | Validated Antibodies | Comments |
|---|---|---|
| HRAS/KRAS/NRAS | CPTC-KRAS4B-2 | Nonspecific bands above 50 kDa, very sensitive |
| 05–1072 | Nonspecific bands above 30 kDa | |
|
| ||
| HRAS | 18295–1-AP | Nonspecific bands above 30 kDa |
|
| ||
| NRAS | SC-31 | May be useful for IF in overexpression models |
|
| ||
| KRAS4A/4B | OP24*/SC-30* | No IF |
|
| ||
| KRAS4B | WH0003845M1 | Very sensitive, no IF |
|
| ||
| KRAS4A | ABC1442 | Nonspecific bands above 40 kDa, may have difficulty detecting endogenous levels, no IF |
| SC-522 | Nonspecific bands above 30 kDa, may have difficulty detecting endogenous levels, no IF | |
same clone
With our validated antibodies, we could initially identify shRNAs that were selective for human KRAS or NRAS, without displaying off-target effects on other RAS isoforms. However, both of the original shRNAs targeting HRAS had isoform-specific off-target effects (Fig. 3C). To better identify an effective and selective HRAS shRNA, we further infected the panel of NSCLC lines with five additional HRAS shRNAs (Fig. 3D), and validated shRNAs that selectively suppressed HRAS. Our results using isoform-specific shRNAs are summarized in Supplementary Table S1.
Similarly, we evaluated isoform-specificity of siRNAs used to target different RAS isoforms in two RAS-mutant rhabdomyosarcoma cell lines. The protein levels detected by the specific antibodies were reduced in cells transiently transfected with the appropriate siRNAs (Fig. 3E). In this instance, HRAS knockdown is clearly visible with the SC-520 antibody, suggesting the nonspecific band(s) that co-migrate with HRAS in NSCLC lines are not expressed in rhabdomyosarcoma lines (Fig. 3E–F), stressing our antibody validations are context and cell-type dependent. Notably, NRAS protein expression is reduced with KRAS siRNA1, HRAS siRNA2 decreases NRAS expression, and NRAS siRNA2 and siRNA3 reduce HRAS expression. As done with the shRNAs, we further transfected these cells with additional KRAS, HRAS, and NRAS siRNAs and validated those that selectively suppress the correct isoform without targeting other RAS isoforms. Our results using isoform-specific siRNAs are summarized in Supplementary Table S2, and all siRNA and shRNA binding sites are shown in Fig. S1.
Analysis of RAS G12D and G12V mutation-specific antibodies
We also tested two commercially available G12 mutant-specific RAS antibodies for their ability to recognize ectopically expressed mutant human KRAS4B in rescued Rasless MEFs expressing different mutant KRAS proteins and endogenous mutant KRAS in human KRAS-mutant cell lines (Fig. 4). These antibodies were very specific for the indicated mutant proteins. No background bands were detected in any of the lysates, even at extended exposure times. Notably, our data indicate that KRAS protein levels in Rasless MEFs expressing different mutant KRAS proteins are comparable to endogenous levels in the established cancer cell lines. However, while very specific for their targets, these mutation-specific antibodies were considerably less sensitive than most of the isoform-specific antibodies. While we included only KRAS-mutant cell lines along with our controls in these experiments, these antibodies are also advertised to detect the G12D- and G12V-mutant forms of HRAS and NRAS.
Fig. 4. Mutation-specific RAS antibodies detect mutant KRAS proteins in MEFs and human cancer cell lines with high fidelity.
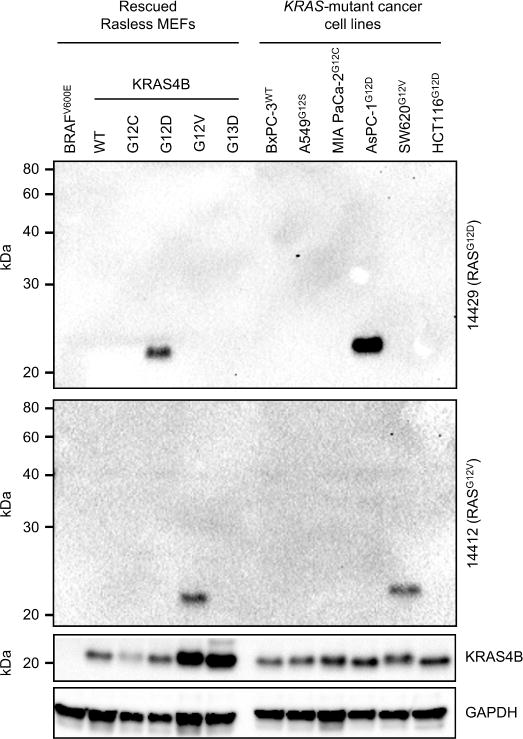
Western blotting was performed with G12D- (upper panel) and G12V- (lower panel) mutation-specific antibodies to probe (left) Rasless MEFs rescued with the indicated mutants of human KRAS (or WT RAS or BRAF controls), or (right) KRAS-mutant cancer cell lines. All cell lines except HCT116 are homozygous for mutant KRAS. Full (uncut) blots are shown to reveal the absence of nonspecific protein recognition. Blots were probed with KRAS4B antibody to show total KRAS protein. GAPDH served as a loading control.
RAS antibodies unsuitable for immunofluorescence of endogenous RAS proteins
We investigated whether any of the isoform- and mutant-specific RAS antibodies validated for use in western blotting could also be used for immunofluorescence (IF) detection of RAS proteins in Rasless MEFs (Fig. 5A). Our initial, baseline metric for suitability in IF was the lack of a significant signal in Rasless MEFs expressing BRAFV600E. Only one, the NRAS antibody SC-31, met this criterion (Fig. 5A). Notably, adherent Rasless MEFs expressing NRAS cover large surface areas when cultured on plastic or glass. Upon detaching these cells with trypsin, they are similar in size to Rasless MEFs expressing other RAS proteins in suspension. Moreover, the NRAS antibody selectively stained Rasless MEFs expressing NRAS, but not Rasless MEFs expressing KRAS4A, KRAS4B, or HRAS (Fig. 5B). The subcellular distribution of the signal was consistent with that of NRAS in our previous reports (32, 33). However, not all cells in each culture of Rasless MEFs expressing NRAS stained positively, presumably due to differing levels of NRAS expression.
Fig. 5. An immunofluorescence (IF) signal from isoform- or mutation-specific RAS antibodies does not reliably indicate the presence of the relevant RAS protein.
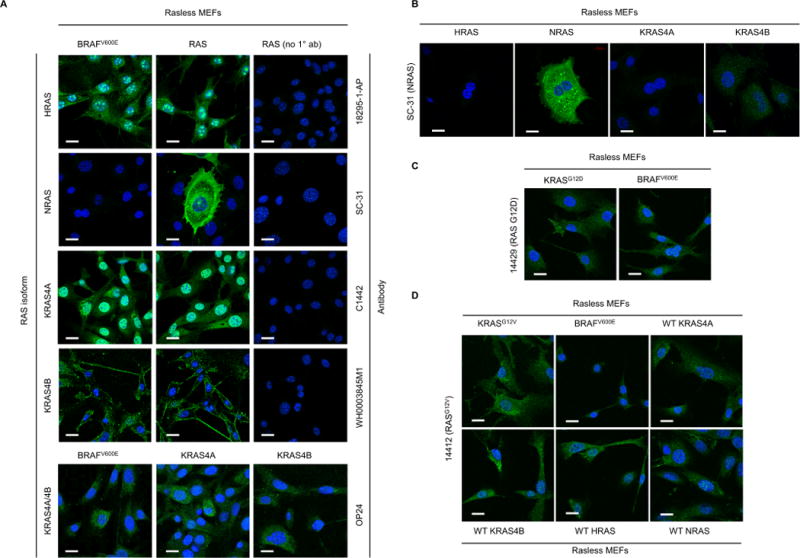
(A) The indicated RAS isoform-selective antibodies were used in IF analyses of Rasless MEFs expressing solely the relevant RAS isoform or BRAFV600E. (B) To determine the RAS isoform selectivity of the NRAS SC-31 signal shown in panel A, the same antibody was used to probe Rasless MEFs expressing each RAS isoform. (C, D) RASG12D- or RASG12V-mutation specific RAS antibodies were used for IF analyses of Rasless MEFs expressing the relevant KRAS mutant or BRAFV600E negative control (C, KRASG12D) or WT RAS isoforms (D, KRASG12V). Scale bars, 20 µm; blue, DAPI nuclear stain; green, goat anti-rabbit or anti-mouse AlexaFluor488, to detect each primary RAS antibody.
Despite their utility in western blotting, both RAS mutation-specific antibodies also failed to be suitable for immunofluorescence (IF, Fig. 5C). The G12D-specific antibody picked up substantial background in Rasless MEFs expressing BRAFV600E. While the G12V-specific antibody did not appreciably stain Rasless MEFs expressing BRAFV600E, it did stain Rasless MEFs expressing any other RAS isoform. We suspect that the use of these antibodies in IF may be limited by their low sensitivity, as shown in our western blots, making it difficult to distinguish endogenous levels of RAS from background signals in individual cells.
Similarly, our attempts to detect endogenous wild-type (WT) and mutant NRAS in human cancer cell lines by IF, using the same SC-31 antibody that selectively stained Rasless MEFs expressing NRAS, were unsuccessful. We first validated by western blotting that we could deplete the endogenous NRAS signal in cancer cells harboring wild type NRAS [A375 cells] or mutant NRAS (Q61K) [SBC12 cells]. Despite near-complete NRAS knockdown (Fig. 6A), the IF signal remained unchanged in each case (Fig. 6B).
Fig. 6. NRAS antibody can detect endogenous NRAS protein by western blot but not by immunofluorescence.
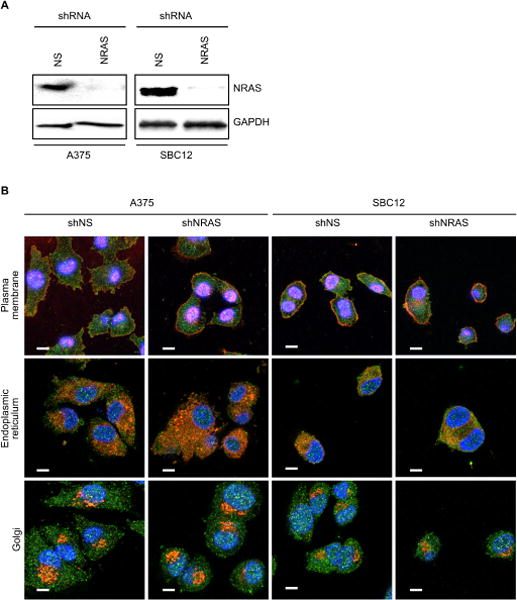
(A) Immunoblot showing shRNA knockdown of NRAS protein in A375WT and SBcl2Q61K melanoma cell lines. (B) Representative images of IF analyses performed as in Fig. 5, but in A375 and SBcl2 cells knocked-down with nonspecific (NS) or NRAS shRNA and co-stained with NRAS (SC-31) antibody and co-localization markers for plasma membrane (wheat germ agglutinin), endoplasmic reticulum (PDI), or Golgi (GM130). Scale bars, 10 µm; blue, DAPI nuclear stain; green, RAS; red, organelles.
It is widely appreciated that antibodies selected for utility in one application may not perform well in another (27). Importantly, western blots are performed under denaturing conditions whereas IF detects proteins in more native conformations. Thus, while disappointing, it may not be entirely surprising that we have been unable to identify RAS antibodies suitable for both western blotting and immunofluorescence.
DISCUSSION
There is now a greater appreciation that there are RAS isoform-distinct biological roles in normal and neoplastic cells and that there are mutation-distinct functional consequences of these differences for RAS function (9). Research that aims to successfully address these differences requires the use of antibody reagents that have been rigorously validated for specificity and for utility in designated experimental applications. We recognize that the degree of validation of commercially available RAS antibodies is highly variable, and that rigorous validation is not feasible for the majority of research laboratories to perform on their own. The NCI Ras Initiative was established in part to address such gaps in the field (29). We completed studies that critically assessed 22 commercially available RAS antibodies, including those most widely used in the literature. Our goals were to identify potential limitations of each antibody, to rigorously validate antibodies for use in the field in specific applications, and ultimately to establish how accurately it is possible to interpret data acquired using each antibody. A critical component of our analyses was the utilization of a unique panel of Rasless MEFs expressing BRAFV600E or other RAS isoforms. With this well-validated panel, which we established at the NCI RAS Initiative, coupled with our analyses of recombinant unprocessed and processed human KRAS4A/4B proteins and by using shRNA vectors targeting each RAS gene, we were able to very rigorously assess RAS specificity and isoform selectivity under different experimental conditions. We have identified antibodies that selectively recognize each of the four human RAS proteins in human cancer cells, thereby identifying a set of RAS antibodies that can be used with confidence in future studies employing western blot analyses. Our survey did not identify RAS antibodies useful for other experimental applications such as immunofluorescence or immunohistochemistry.
Consistent with perceptions of the antibody field in general (20–22), we found a broad spectrum of RAS antibody quality, from poor to excellent. Overall, the majority of these antibodies displayed their vendor-described specificities, but several vendors provided incomplete or overstated information that may lead to misinterpretation of data.
Given the high overall amino acid sequence identity of RAS proteins (9), many antibodies generated against one specific RAS isoform will also recognize other RAS isoforms. For example, while monoclonal antibody CPTC-KRAS4B-2 was generated against KRAS4B, the name is misleading in that it suggests KRAS4B selectivity. Instead, we found that CPTC-KRAS4B-2 was the most effective pan-RAS antibody available, and can provide a useful reagent to determine overall total RAS protein expression in cells. However, this antibody may also recognize other ~21 kDa small GTPases with sequence similarity to RAS. Since ectopic expression of other small GTPases cannot restore the growth of Rasless MEFs (31), we do not have reagents to address potential antibody selectivity for such proteins with similar rigor as done here for RAS. Conversely, one polyclonal antibody generated against recombinant NRAS poorly recognized recombinant NRAS, showed stronger recognition of other recombinant RAS proteins, and failed to detect NRAS in our panel of rescued Rasless MEFs expressing other RAS isoforms. Thus, while it did identify a ~21 kDa band in HEK293T cells, we suspect that the detected band is not NRAS. Finally, while the product information provided with this antibody showed that it recognizes only a single ~21 kDa band in HEK293 cell lysates, with no recognition of other cellular proteins, our analyses in both Rasless MEFs and HEK293T cells found very significant nonspecific reactivity. These and other examples emphasize the critical value of Rasless MEFs in evaluating RAS antibody selectivity and utility in different experimental applications.
Of the two KRAS splice variants, KRAS4B has been by far the most studied in the cancer field, due in large part to availability of reagents and a widely held perception that the KRAS4B splice variant is the predominant KRAS protein expressed in human tumor cells (5). However, in the absence of antibodies capable of distinguishing KRAS4A and KRAS4B, this perception was based on early studies of KRAS expression at the level of mRNA, not protein. Ironically, the very first discovery of KRAS as an oncogene was of the Kirsten murine sarcoma virus, which encodes a G12-mutated rat KRas4A protein (34). Furthermore, our studies found that 30 of 30 human cancer cell lines express both KRAS4A and KRAS4B mRNA, and that, in colorectal carcinoma tissues, KRAS4A and KRAS4B transcripts are similarly abundant (6). Our analyses here did identify useful antibodies for selective detection of KRAS4A and KRAS4B proteins. Regarding KRAS4B-selective antibodies, we found that the two commercially available antibodies, while clearly selective when evaluated using recombinant proteins, did not effectively recognize cell-expressed KRAS4B protein in Rasless MEFs. One antibody did detect ~21 kDa bands, but the equivalent detection of such bands in the Rasless MEFs expressing BRAFV600E demonstrate that this antibody does not detect cellular KRAS4B expression. Instead, we found that the best KRAS4B-selective antibody was one described as detecting both KRAS4A and KRAS4B. We did validate two useful KRAS4A-selective antibodies, with each having distinct nonspecific activities that do not compromise their application in western blots, but likely limit their usefulness in other applications. Currently, whether KRAS4A and KRAS4B serve distinct driver roles in cancer is not known. To date, the relative expression of the two KRAS splice variants has been determined only at the RNA level. Validated KRAS splice variant-selective antibodies will provide crucial reagents to accurately address this issue. Furthermore, using our validated antibodies, we identified shRNA constructs selective for each RAS isoform that can assist in determining the specificity of additional antibodies.
Finally, we validated two mutation-specific antibodies for western blot detection of RAS proteins with G12D or G12V mutations (which comprise 59% of all KRAS mutations), supporting the feasibility of developing additional antibodies selective for other common RAS mutations. Current assays to detect GTP-bound RAS are not amenable to immunohistochemical (IHC) evaluation of patient tumor tissue. Unfortunately, these mutation-selective antibodies were also not useful for IHC analyses of formalin-fixed, paraffin-embedded or frozen tissue from two cases harboring previously identified KRASG12D and KRASG12V mutations, even though western blot analyses of tissue from the same two cases confirmed expression of the G12D or G12V mutant RAS proteins (Fig. S2). Thus, these antibodies cannot selectively measure activated RAS in patient tissue.
In summary, we have performed comprehensive analyses of the selectivity of a large panel of RAS antibodies, and achieved our goal of validating antibodies that are useful for western blot detection of each of the four RAS proteins as well as of the two KRAS mutations most prevalent in cancer. Our studies also emphasize the need for well-validated RAS antibodies for other experimental applications, in particular for immunofluorescence analyses of endogenous RAS proteins, where our current knowledge of the spatial distribution of RAS proteins has relied exclusively on the study of exogenously expressed proteins. With the possible exception of a validated NRAS-selective antibody, we did not identify RAS antibodies useful for immunofluorescence analyses. Although many commercially available RAS antibodies are described as being useful for immunofluorescence (Table 1), our findings stress that more rigorous validation of antibody utility in specific applications, beyond simply detecting signal, needs to be done. We did not evaluate the usefulness of RAS antibodies for immunoprecipitation (IP) or (with the exception of G12 mutation-specific antibodies), for immunohistochemistry, although the inability of the antibodies we tested to perform well in IF suggests they may also not work well in IP applications. We did not evaluate all commercially available RAS antibodies. Nevertheless, our western blot results can serve as a resource and guide for further qualifying RAS antibodies for additional applications.
MATERIALS AND METHODS
E. coli expression and purification of human RAS proteins
Recombinant proteins were expressed as His6-MBP-tev-RAS fusion proteins in E. coli (BL21Star™ (DE3) pRare) and purified as we described (35). After proteolytic cleavage of the N-terminal tags, the final proteins generated were GlyGlySerGly-KRAS4B (2–188), Gly-HRAS (1–189), and Gly-NRAS (2–189). Protein concentrations were determined by UV280 using an extinction coefficient of 19865 M-1 cm-1 (11,920 from protein, 7945 from GDP) (36).
Insect cell expression and purification of KRAS4A and KRAS4B
Expression and purification of processed GlyGly-Hs.KRAS4A (2–189) and GlyGly-Hs.KRAS4B (2–188) were performed as we described previously, using a bacmid engineered to co-express FNTA and FNTB (35). Expression of non-processed GlyGly-Hs.KRAS4A and GlyGly-Hs.KRAS4B followed the same protocol except that the expression constructs were cloned into the standard bacmid. Purification of processed KRAS4B was performed as we described (35), whereas processed KRAS4A was purified using a modified protocol that added an anion exchange step after the initial capture from lysate. Non-processed KRAS4B was purified as we described previously (30).
Establishment of Rasless MEFs with isoform-specific RAS protein production
A clonal panel of MEF cell lines containing single transgene alleles was generated using the Rasless MEF system developed by Barbacid and colleagues (31). MEFs null for both Hras and Nras (provided by M. Barbacid) were treated with 600 nM 4-hydroxytamoxifen (4-OHT) for 9 to 11 d to remove the endogenous floxed KRAS gene. Cells lacking all endogenous Ras proteins arrest in G1, but resume proliferation upon lentiviral delivery of an ERK MAPK pathway activating gene (e.g., RAS or BRAF). Alternatively, cell line pools were generated through lentiviral transduction and antibiotic selection followed by treatment with 4-OHT and single-cell cloning. A single-cell clone was validated for each transgene. Confirmation of endogenous Kras gene removal was performed by western blotting and sequencing.
RAS antibody generation
Purified unprocessed full length wild type KRAS4B (30) was submitted for antibody generation at Precision Antibody Inc. Candidate clones were screened at the Antibody Characterization Lab (ACL; FNLCR) following procedures outlined on the antibody portal website (http://antibodies.cancer.gov). Four candidates were selected based on reactivity by western blot on a WES instrument (Protein Simple) and using the immunoprecipitation mass spectrometry method developed by the ACL. All antibodies and characterization data can be found on the ACL web portal, and antibodies and hybridomas are available through the Developmental Studies Hybridoma Bank (University of Iowa).
SDS-PAGE
Each purified RAS protein was diluted to 1X in NuPage LDS loading buffer (Invitrogen), containing 25 mM TCEP (Sigma-Aldrich) to final protein concentrations of 3, 1, and 0.3 µg per 10 µL and applied to 10–20% gradient Criterion 26-well Tris-HCl precast gels (Bio-Rad) in 1X Tris-Glycine-SDS running buffer. Benchmark™ protein ladder size standards (Thermo Fisher Scientific) flank each set of proteins. Gels were stained with SimplyBlue SafeStain (Thermo Fisher), then imaged in a Fujifilm LAS-3000 equipped with an LAS-4000 CCD camera.
Western blots
For purified protein, each RAS protein was diluted to 1X in NuPage LDS loading buffer containing 25 mM TCEP to final protein concentrations of 10, 1, and 0.1 ng per 10 µL and denatured by heating to 70°C for 10 min. Western blots were performed as described previously (37). Cells were lysed in 1% Triton detergent buffer and assayed for protein content using the BCA assay (Pierce Biotechnologies). Twenty µg of each lysate in NuPAGE LDS sample buffer were loaded into wells of 12% Criterion TGX Stain-Free Gels (Bio-Rad) to the right of a MagicMark™ XP protein ladder (Thermo Fisher Scientific). The resolved proteins were transferred to methanol-activated PVDF membranes for 90 min in 1X Transfer Buffer (25 mM Tris, 94 mM glycine, 20% methanol) at currents maintained between 350–450 mA. After blocking in 3% non-fat dry milk and washing in TBS-Tween, membranes were incubated overnight in 10 µg of primary antibody diluted in 10 ml of 3% dry milk in 1X TBS-T. Membranes were washed again and rocked at room temperature for 1 h in HRP-labeled secondary antibody (horse anti-mouse, Cell Signaling #7076 or goat anti-rabbit, Cell Signaling #7074) diluted 1:2000 in 3% dry milk in TBS-Tween. After washing, membranes were developed with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare) in a ChemiDoc Imaging System (Bio-Rad). Anti-vinculin (V9131) or anti-GAPDH (G8795) antibodies (1:5000, Sigma-Aldrich) were used as loading controls. Blots were not stripped prior to probing for loading controls. A full list of RAS antibodies can be found in Table 1.
Cell culture
Rescued Rasless MEFs, generated at FNLCR as described above, were maintained in DMEM (Corning, Inc.) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich). Established human cancer cell lines were obtained from the American Type Culture Collection (ATCC) and maintained in RPMI-1640 (Corning, Inc.; NSCLC cancer lines) or in DMEM (Corning; melanoma cell lines) supplemented with 10% FBS. Cell line identities were confirmed by STR analysis.
shRNA knockdowns of specific RAS isoforms
Lentivirus virus-like particles expressing shRNAs targeted against different RAS transcripts were produced in HEK293T cells by transfection of a 4:3:1 ratio of NRAS (or nonspecific) shRNA plasmid:psPAX2:pMD2.G and a 1:3 ratio of DNA:Fugene 6 (Promega). Sequences targeted by each shRNA are provided in Table S1.
Immunofluorescent laser scanning confocal microscopy
All rescued Rasless MEFs expressing BRAFV600E or RAS isoforms were plated at a density of 4 × 104 cells in 12-well No 1.5 Mattek plates with 14 mm coverslips (Mattek Corporation) and grown for 24 to 48 h before fixing in 2% paraformaldehyde for 15 min at room temperature, 4% paraformaldehyde for 5 minutes at room temperature, or 100% methanol for 20 min at −20°C. Paraformaldehyde-fixed cells were permeabilized with 0.1% Triton X-100 in 1X PBS for 15 min. All cells were then incubated in blocking buffer (Rockland) containing 0.1% Triton X-100 for 45 min at room temperature. Cells were incubated overnight in 1:100 dilutions of each RAS antibody. Cells were then incubated in 1:300 goat anti-mouse or goat anti-rabbit AlexaFluor488 (Invitrogen) in blocking buffer + 0.1% Triton X-100. Ten min prior to completion of secondary antibody incubation, a 1:100 dilution of 50 µg/ml DAPI (Thermo Fisher) was added to each well to stain nuclei. After washing, cells were imaged in a Zeiss LSM700 scanning confocal microscope with a 40X oil immersion lens. Images were analyzed using ZEN Blue software (Carl Zeiss). Changes in fixation method did not affect localization, except with anti-HRAS. Methanol fixation of Rasless MEFs expressing HRAS led to loss of nuclear staining in both Rasless MEFs expressing BRAFV600E or NRAS. Otherwise, the nonspecific signal was preserved. All images shown are from cells fixed in 2% paraformaldehyde, except for WH0003845M1 KRAS images, which were from methanol-fixed cells.
For co-localization experiments in melanoma lines, A375 and SBC12 cells were fixed in 2% paraformaldehyde for 15 min at room temperature. To visualize the plasma membrane, wheat germ agglutinin AlexaFluor647 conjugate (Thermo Fisher, W33466) labeling solution (5 µg/ml in 250 µl) was added to each well and incubated for 10 min at 37°C. For endoplasmic reticulum (ER) or Golgi labeling, anti-PDI (Invitrogen, MA3–019) or anti-GM130 (610823) (BD Biosciences) was incubated on the cells overnight (1:300), followed by goat anti-mouse Alexafluor568 for 45 min at room temperature. Cells were washed and imaged in a Zeiss LSM700 with a 63X oil immersion lens. Images were analyzed using ZEN Blue software.
Immunohistochemistry on formalin-fixed and frozen sections of human tumor tissues
Immunohistochemistry was performed on 4-μm-thick formalin-fixed, paraffin-embedded de-identified patient tumor sections using the Discovery XT automated system (Ventana Medical Systems). Sections were mounted on charged glass slides and baked at 60°C for 1 h before heat-induced antigen retrieval was performed using standard CC1 (Cell Conditioning 1 solution). Slides were incubated with G12D- (14429) or G12V- (14412) selective antibodies for either 2 h at room temperature (UltraMap protocol) or 1 h with heat (DABMap protocol). Chromogenic detection was done with the DABMap kit or the UltraMap DAB anti-Rb kit. Four-μm-thick frozen sections were fixed in neutral buffered formalin for 10 min before the assay was performed on the Discovery Ultra automated system (Ventana). Slides underwent 36 min of CC1 treatment before incubation with G12D or G12V antibodies, titrated at 1:100, for 60 min at 37°C. Detection was completed with the DABMap kit.
Supplementary Material
Fig. S1. RAS isoform siRNA and shRNA binding sites
Fig. S2. Immunohistochemistry analyses using mutation-specific RAS antibodies in tumor tissues harboring KRASG12D or KRASG12V
Table S1. RAS shRNA sequences
Table S2. RAS siRNA sequences
Acknowledgments
We thank P. Ariel and the Microscopy Services Laboratory at UNC-Chapel Hill and B. Papke and K. Bryant from the Der lab for technical assistance with confocal microscopy. We acknowledge the following personnel from the NCI RAS Initiative at the FNLCR for their technical expertise in cloning, expression, purification, and QC of materials: V. Wall, J. Mehalko, C. Grose, C. Bittner, T. Taylor, M. Sherekar, S. Messing, P. Frank, S. Perkins, R. Gapud, and Z. Meng. We thank D.G. Wong and A.N. Karnezis for assistance with the immunohistochemistry analyses.
Funding: This work was supported by grants (CA42978, CA179193, CA175747, CA199235 to C.J.D. and A.D.C. and CA165995 to P.J.H.) and training grants/fellowships (T32 CA009156 to A.V.V. and G.A.H., F32 CA200313 to G.A.H. and from the American Cancer Society to A.V.V.) from the NIH/NCI, and the Pancreatic Cancer Action Network-AACR. C.J.D. is also supported by a grant from the DoD and from the Lustgarten Pancreatic Cancer Foundation. A.M.W., W.K.G, L.G., N.F., G.R.W., R.B., F.M. and J.L.H. were funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Author contributions: A.M.W., A.D.C., C.J.D., J.L.H., A.V.V, G.A.H., B.T.-C. and D.G.H. designed the experiments; A.M.W., A.D.C., C.J.D., and J.L.H. wrote the manuscript; A.M.W., I.O-D., A.V.V., N.F. and B.T.-C. performed experiments; W.G., N.F, L.G., R.B., P.J.H. and G.W. generated tools, cell lines, and reagents for the experiments; M.R.P. and F.M. provided scientific guidance.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Cell lysates of Rasless MEFs expressing BRAFV600E or RAS proteins are available through the FNLCR. Antibodies generated by the Antibody Characterization Library at the FNLCR are available from the Developmental Studies Hybridoma Bank (DSHB) at the University of Iowa.
REFERENCES AND NOTES
- 1.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papke B, Der CJ. Drugging RAS: Know the enemy. Science. 2017;355:1158–1163. doi: 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- 3.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 4.Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004:Re13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capon DJ, Seeburg PH, McGrath JP, Hayflick JS, Edman U, Levinson AD, Goeddel DV. Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature. 1983;304:507–513. doi: 10.1038/304507a0. [DOI] [PubMed] [Google Scholar]
- 6.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112:779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011;2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oltean S, Bates D. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33:5311–5318. doi: 10.1038/onc.2013.533. [DOI] [PubMed] [Google Scholar]
- 9.Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drosten M, Simon-Carrasco L, Hernandez-Porras I, Lechuga CG, Blasco MT, Jacob HK, Fabbiano S, Potenza N, Bustelo XR, Guerra C, Barbacid M. H-Ras and K-Ras oncoproteins induce different tumor spectra when driven by the same regulatory sequences. Cancer Res. 2017;77:707–718. doi: 10.1158/0008-5472.CAN-16-2925. [DOI] [PubMed] [Google Scholar]
- 13.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, Fu K, Martin BL, Jeck WR, Souroullas GP, Darr DB, Zedek DC, Miley MJ, Baguley BC, Campbell SL, Sharpless NE. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014;4:1418–1429. doi: 10.1158/2159-8290.CD-14-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res. 2015;13:1325–1335. doi: 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- 16.Smith MJ, Neel BG, Ikura M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A. 2013;110:4574–4579. doi: 10.1073/pnas.1218173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montalvo SK, Li L, Westover KD. Rationale for RAS mutation-tailored therapies. Future Oncol. 2017;13:263–271. doi: 10.2217/fon-2016-0363. [DOI] [PubMed] [Google Scholar]
- 18.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Janne PA, Meyerson M, Marto JA, Engen JR, Gray NS. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker M. Reproducibility crisis: Blame it on the antibodies. Nature. 2015;521:274–276. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 21.Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, Persson A, Ottosson J, Wernerus H, Nilsson P, Lundberg E, Sivertsson A, Navani S, Wester K, Kampf C, Hober S, Ponten F, Uhlen M. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Bradbury A, Pluckthun A. Reproducibility: Standardize antibodies used in research. Nature. 2015;518:27–29. doi: 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- 23.Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature. 2012;483:531–533. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- 24.Gould Rothberg BE, Berger AJ, Molinaro AM, Subtil A, Krauthammer MO, Camp RL, Bradley WR, Ariyan S, Kluger HM, Rimm DL. Melanoma prognostic model using tissue microarrays and genetic algorithms. J Clin Oncol. 2009;27:5772–5780. doi: 10.1200/JCO.2009.22.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 26.Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J, Anagnostou V, Rimm D. Antibody validation. Biotechniques. 2010;48:197–209. doi: 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, Rimm DL, Rodriguez H, Hiltke T, Snyder M, Yamamoto T. A proposal for validation of antibodies. Nat Methods. 2016;13:823–827. doi: 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ledford H. Cancer: The Ras renaissance. Nature. 2015;520:278–280. doi: 10.1038/520278a. [DOI] [PubMed] [Google Scholar]
- 29.Thompson H. US National Cancer Institute’s new Ras project targets an old foe. Nat Med. 2013;19:949–950. doi: 10.1038/nm0813-949. [DOI] [PubMed] [Google Scholar]
- 30.Gillette WK, Esposito D, Abreu Blanco M, Alexander P, Bindu L, Bittner C, Chertov O, Frank PH, Grose C, Jones JE, Meng Z, Perkins S, Van Q, Ghirlando R, Fivash M, Nissley DV, McCormick F, Holderfield M, Stephen AG. Farnesylated and methylated KRAS4b: high yield production of protein suitable for biophysical studies of prenylated protein-lipid interactions. Sci Rep. 2015;5:15916. doi: 10.1038/srep15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. Embo j. 2010;29:1091–1104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 33.Zhou M, Wiener H, Su W, Zhou Y, Liot C, Ahearn I, Hancock JF, Philips MR. VPS35 binds farnesylated N-Ras in the cytosol to regulate N-Ras trafficking. The Journal of cell biology. 2016;214:445–458. doi: 10.1083/jcb.201604061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuchida N, Ryder T, Ohtsubo E. Nucleotide sequence of the oncogene encoding the p21 transforming protein of Kirsten murine sarcoma virus. Science. 1982;217:937–939. doi: 10.1126/science.6287573. [DOI] [PubMed] [Google Scholar]
- 35.Dharmaiah S, Bindu L, Tran TH, Gillette WK, Frank PH, Ghirlando R, Nissley DV, Esposito D, McCormick F, Stephen AG, Simanshu DK. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc Natl Acad Sci U S A. 2016;113:E6766–e6775. doi: 10.1073/pnas.1615316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith SJ, Rittinger K. Preparation of GTPases for structural and biophysical analysis. Methods Mol Biol. 2002;189:13–24. doi: 10.1385/1-59259-281-3:013. [DOI] [PubMed] [Google Scholar]
- 37.Waters AM, Bagni R, Portugal F, Hartley JL. Single synonymous mutations in KRAS cause transformed phenotypes in NIH3T3 cells. PLoS One. 2016;11:e0163272. doi: 10.1371/journal.pone.0163272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, Marayati R, Kher S, George SD, Xu M, Wang-Gillam A, Samatar AA, Maitra A, Wennerberg K, Petricoin EF, 3rd, Yin HH, Nelkin B, Cox AD, Yeh JJ, Der CJ. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29:75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. RAS isoform siRNA and shRNA binding sites
Fig. S2. Immunohistochemistry analyses using mutation-specific RAS antibodies in tumor tissues harboring KRASG12D or KRASG12V
Table S1. RAS shRNA sequences
Table S2. RAS siRNA sequences