

Abstract
The rise of antibacterial resistance among human pathogens represents a problem that could change the landscape of healthcare unless new antibiotics are developed. The methyl erythritol phosphate (MEP) pathway represents an attractive series of targets for novel antibiotic design, considering each enzyme of the pathway is both essential and has no human homologs. Here we describe a pilot scale high-throughput screening (HTS) campaign against the first and second committed steps in the pathway, catalyzed by DXP reductoisomerase (IspC) and MEP cytidylyltransferase (IspD), using compounds present in the commercially available LOPAC1280 library as well as in an in-house natural product extract library. Hit compounds were characterized to deduce their mechanism of inhibition; most function through aggregation. The HTS workflow outlined here is useful for quickly screening a chemical library, while effectively identifying false positive compounds associated with assay constraints and aggregation.
Keywords: MEP pathway, antimicrobial development, LOPAC1280, HTS assay development
1. Introduction
As emphasized in a recent report by the World Health Organization on global antibiotic resistance, we are heading towards a post-antibiotic era in which “common infections and minor injuries can kill.”[1] Highlighting acquired resistance in pathogens such as Escherichia coli, Neisseria gonnorrhae, Streptococcus pneumoniae, Mycobacterium tuberculosis, and Klebsiella pneumonia, the World Health Organization is among several groups raising concerns and awareness of the spread of antibiotic resistance and its consequences. Beyond the natural means by which microbes may acquire resistance, others have also noted that resistance can be intentionally introduced through deliberate bioengineering, making antibiotic resistance a potential bioterrorist threat.
In an effort to produce new antibiotics targeting novel cellular processes, there has been a rise in attention given to the methyl erythritol phosphate (MEP) pathway (Figure 1A). The MEP pathway is essential for the production of isoprenoids in many bacterial species, including human pathogens like Mycobacterium tuberculosis [2], Francisella tularensis [3], Escherichia coli [4], and Vibrio cholera [5]. All isoprenoid metabolites, of which there are over 25,000 identified, are produced by the condensation of two end products of the MEP pathway, isopentenyl pyrophosphate (IPP, compound 10 in Figure 1) and dimethylallyl pyrophosphate (DMAPP, compound 11) [6]. Essential metabolites derived from isoprenoids are involved in a number of vital cellular processes, including electron transport, membrane fluidity and cell signaling. Additionally, while mammals also use IPP and DMAPP for biosynthesis of isoprenoids, these two building blocks are produced in mammals through an alternative pathway called the mevalonic acid pathway (MVA, Figure 1B). The MVA pathway has little semblance to the MEP pathway, making the bacterial MEP pathway a very attractive target for the development of new antibiotics.
Figure 1. The MVA and MEP biosynthetic pathways.
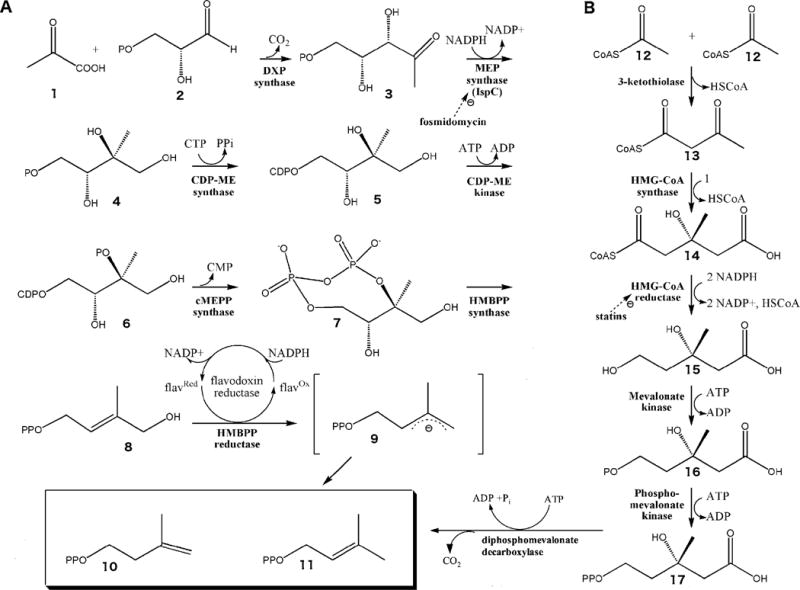
A) The MEP pathway is used by higher plants, the plastids of algae, apicomplexan protozoa, and many eubacteria, including numerous human pathogens. Pyruvate (1) is condensed with glyceraldehyde 3-phosphate (2) to yield 1-deoxy-D-xylulose 5-phosphate (DXP; (3)) [60], a branch point intermediate with a role in E. coli vitamin B1 and B6 biosynthesis ([61], [62], [63], [64]) as well as isoprene biosynthesis. In the first committed step of the E. coli MEP pathway, 1-Deoxy-D-xylulose 5-phosphate reductoisomerase (also called MEP synthase, Dxr or IspC) catalyzes the reduction and rearrangement of 3 to yield MEP (4) [65]. CDP-ME synthase then converts MEP into 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol (CDP-ME; (5)). CDP-ME kinase phosphorylates CDP-ME, which is subsequently cyclized (coupled with the loss of CMP) by cMEPP synthase to yield 2-C-methyl-D-erythritol 2,4-cyclodiphosphate (7) ([66], [67], [68], [69], [70]). A reductive ring opening of 7 produces 1-hydroxy-2-methyl-2-butenyl diphosphate (HMBPP; (8))([71], [72], [73], [74], [75]) which is then reduced to both IPP and DMAPP in a ~5:1 ratio ([76], [77], [78], [79], [80], [81], [82]). B) The MVA pathway is utilized by humans and other eukaryotes, archaebacteria, and certain eubacteria to produce IPP and DMAPP, the building blocks of isoprenoids. The pathway is initiated by the enzymatic condensation of 3 molecules of acetyl-CoA (12) to form 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) (14), which is then reduced to MVA (15) by HMG-CoA reductase ([83], [84]). Subsequent phosphorylation and decarboxylation yield IPP (10) ([85], [86], [87]) which is converted to DMAPP (11) by an isomerase [88].
The enzyme catalyzing the first committed step of the MEP pathway (called DXP reductoisomerase, or alternatively IspC or Dxr; Figure 1A) has received copious attention and is the only enzyme reported to have been targeted and tested with an inhibitor (fosmidomycin) in clinical trials [7]. MEP pathway inhibitors (collectively called MEPicides [8]) are most often rationally designed on the fosmidomycin scaffold, balancing target specificity with bioavailability ([9],[10],[11],[12]). High-throughput screening (HTS) of molecular libraries (containing a random array of structurally diverse molecules) affords the potential of identifying novel compounds in new chemical space, enabling the rational design of alternate MEPicides with a scaffold different than that of fosmidomycin. Additionally, the second committed step of the MEP pathway (called MEP cytidylyltransferase, or alternatively IspD) has received renewed attention. While the active site of IspD is particularly polar and traditionally difficult to target with substrate analogs[9], the identification of a potential allosteric site on IspD [13] has provided a feasible way to target IspD with more drug-like small molecules. Coupled with the discovery that IspD is the sole intercellular target of known antimalarial compound MMV008138 [14], part of the publically available Malaria Box [15] collection, IspD represents an excellent target for non-directed molecular screening in order to identify diverse structures potentially targeting its proposed allosteric site.
In this report, we describe a bench scale high throughput screening (HTS) campaign using purified, recombinant IspC and IspD, a commercially available 1280 compound molecular library, and a 150 sample, in-house prepared, natural product extract library [16]. We report a screening protocol for both IspC and IspD that minimizes the number of assays required while effectively identifying false positive compounds. Ultimately, this protocol can be used to screen diverse library collections in an effort to expand the chemical space of IspC and IspD inhibitors.
2. Materials and Methods
2.1 Expression and purification of enzymes
Recombinant Y. pestis IspC (YpIspC), M. tuberculosis IspC (MtbIspC), F. tularensis IspD (FtIspD), and E.coli IspD (EcIspD) were expressed and purified essentially as described in detail previously ([16],[17]). In general, a 10 mL overnight culture of E.coli BL21 Codon Plus (DE3)-RIL containing the protein-encoding plasmid was used to inoculate 1 L of Luria-Bertani media in a 2800 mL Fernbach shake flask at 37°C. Protein expression was induced with 0.5 mM isopropyl B-D-thiogalactopyranoside (IPTG) upon shake flask cultures reaching an OD600 of 1.2, and cells were incubated at 37°C for 18 hours. Cells were harvested by centrifugation at 4°C and 5000 rpm and stored at −80°C. Cell pellets were thawed and lysed with Lysis Buffer A, containing 100 mM Tris pH 8, 0.032% lysozyme at 3 mL per g cell pellet, then Lysis Buffer B, containing 0.1 M MgCl2, 0.1 M CaCl2, 0.020% DNase at 0.3 mL per g cell pellet. Total concentration of NaCl was brought to 0.1 M and clarified cell lysate was obtained by centrifugation (48,000 × g, 20 min, 4°C). His-tagged proteins were purified on a TALON metal affinity chromatography column (Clontech Laboratories, Mountain View, CA) by washing with 20 column volumes of 1X equilibrium buffer (50 mM HEPES pH 7.5, 300 mM NaCl), 10 column volumes of 1X wash buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM imidazole), and 15 column volumes of 2X wash buffer (100 mM HEPES pH 7.5, 600 mM NaCl, 20 mM imidazole). The protein was then eluted with 5 column volumes of 1X elution buffer (150 mM imidazole pH 7.0, 300 mM NaCl). Buffer was exchanged with 0.1 M Tris pH 7.8, 1 mM NaCl, 5 mM DTT during concentration by ultrafiltration. Protein concentration was determined using Advanced Protein Assay Reagent (Cytoskeleton, Denver CO) with γ-globulins (Sigma-Aldrich) as the standard. Purified protein was visualized via Coomassie-stained SDS-PAGE.
2.2 IspC Enzyme Assays
IspC activity was assayed by spectrophotometrically monitoring the enzyme-catalyzed oxidation of NADPH, as described previously.[18] The screening of a commercially available chemical library was conducted in three stages with the Library of Pharmacologically Active Compounds (LOPAC1280) purchased from Sigma-Aldrich[19]. The primary screen of the LOPAC1280 library consisted of 96-well plate based assays, with 0.5 μL of DMSO (for the uninhibited control) or the appropriate inhibitor (dissolved in DMSO) first added directly to the plate, to give a final inhibitor concentration of 100 μM in a total assay volume of 50 μL. Subsequently, 45 μL of assay master mix, containing 100 mM Tris pH 7.8, 25 mM MgCl2, 150 μM NADPH, and 0.89 μM YpIspC was added to each well and the plate was allowed to incubate at room temperature for 10 minutes to facilitate inhibitor binding. The reaction was initiated with the addition of the Km value of DXP (252 μM [16]), and was subsequently quenched after 1 minute with the addition of EDTA to a final concentration of 100 mM. Decrease in A340 as compared to wells without DXP was measured using a Beckman Coulter DTX800 plate reader.
The secondary screen consisted of cuvette-based assays with MtIspC. Enzymatic activity was monitored using 120 μL assay solutions containing 100 mM Tris pH 7.8, 25 mM MgCl2, 150 μM NADPH, 0.89 μM MtbIspC, and 100 μM inhibitor. Assay solutions were incubated at 37°C for 10 minutes to allow NADPH and the inhibitor to associate with the enzyme. Addition of the Km value of DXP (47 μM [20]) was then used to initiate the reaction. The enzymatic consumption of NADPH was continuously monitored (A340) using an Agilent 8453 spectrophotometer equipped with a multi-cuvette holder.
The tertiary screen consisted of a YpIspC-FtIspD coupled assay, where the 96-well plate assay used for the primary screen was adapted to assay IspD activity, as detailed elsewhere [21]. The assay master mix of the primary screen was modified to additionally contain 1.8 μM FtIspD, 200 μM CTP, and 100 mU/mL pyrophosphatase. The assay was quenched after 1 minute by the addition of 40 μL of an acidic malachite green assay solution (2.4 M sulfuric acid containing 1.5% ammonium molybdate, 1.6% Tween 20, and 9.5% malachite green) and 120 μL water. Sodium citrate was added to 3.4% after 10 minutes, and an increase in A660 as compared to wells without DXP was read using a Beckman Coulter DTX800 plate reader.
To screen the inhibitory activity of the natural product extracts, the YpIspC activity was monitored using 120 μL assay solutions containing 100 mM Tris pH 7.8, 25 mM MgCl2, 150 μM NADPH, 0.89 μM YpIspC, and 0.5% (1.98 μL) natural product extract. Assay solutions were incubated at 37°C for 10 minutes to allow NADPH and the inhibitor to associate with the enzyme. Addition of 252 μM DXP was used to initiate the reaction. The enzymatic consumption of NADPH was then continuously monitored (A340) using an Agilent 8453 spectrophotometer equipped with a multi-cuvette holder.
Where appropriate, half maximal inhibitory activity (IC50) was determined by assaying YpIspC in the presence of varying concentrations of inhibitor, ranging from 1 nM to 100 μM, then plotting fractional enzyme activity as a function of inhibitor concentration. GraphPad Prism (La Jolla, CA) was used for nonlinear curve fitting the assay results to a sigmoidal dose response curve.
Lineweaver Burk plots were used to determine the mechanism of enzyme inhibition. Accordingly, 120 μL assay solutions contained 100 mM Tris pH 7.8, 25 mM MgCl2, 0.89 μM YpIspC, and variable concentrations of an inhibitor. Each assay solution was incubated for 10 min at 37°C to facilitate inhibitor binding. NADPH was subsequently added, at 150 μM for DXP-dependent plots, and at variable concentrations for the NADPH-dependent plots (ranging from 20 to 100 μM), and the assays were incubated an additional 10 minutes to allow NADPH binding. Enzymatic reactions were then initiated with addition of 252 μM DXP for NADPH dependent plots, and at variable concentrations (from 50 to 300 μM) for the DXP-dependent plots. Lineweaver-Burk plots were fit by linear regression using GraphPad Prism.
2.3 IspD Enzyme Assays
IspD activity was monitored colorimetrically by quantifying production of inorganic phosphate using pyrophosphatase and malachite green dye as described previously[21]. The screening of a commercially available chemical library was conducted in three stages with the Library of Pharmacologically Active Compounds (LOPAC1280) purchased from Sigma-Aldrich[19]. The primary screen of the LOPAC1280 library consisted of 96-well plate based assays, with 0.5 μL of DMSO (for the uninhibited control) or the appropriate inhibitor (dissolved in DMSO) first added directly to the plate, to give a final inhibitor concentration of 100 μM in a total assay volume of 50 μL. Subsequently, 45 μL of assay master mix, containing 100 mM Tris pH 8.2, 1 mM MgCl2, 1mM DTT, 200 μM CTP, 100 mU/mL pyrophosphatase, and 1.8 μM FtIspD was added to each well and the plate was allowed to incubate at room temperature for 10 minutes to facilitate inhibitor binding. The assay was initiated with the addition of 200 μM MEP. The assay was quenched after 1 minute by the addition of 40 μL of an acidic malachite green assay solution (2.4 M sulfuric acid containing 1.5% ammonium molybdate, 1.6% Tween 20, and 9.5% malachite green) and 120 μL water. Sodium citrate was added to 3.4% after 10 minutes, and an increase in A660 as compared to wells without DXP was read using a Beckman Coulter DTX800 plate reader.
The secondary screen consisted of cuvette-based assays with EcIspD. Enzymatic activity was monitored using 250 μL assay solutions containing 100 mM Tris pH 8.2, 1 mM MgCl2, 1mM DTT, 200 μM CTP, 100 mU/mL pyrophosphatase, 1.8 μM EcIspC, and 100 μM inhibitor. Assay solutions were incubated at 37°C for 10 minutes to allow CTP and the inhibitor to associate with the enzyme. The addition of 200 μM MEP was used to initiate the reaction. Every 30 seconds, 40 μL aliquots were removed from the assay mixture and combined with 40 μL of the acidic malachite green assay solution and 120 μL water. The A660 of each solution was measured on an Agilent 8453 spectrophotometer equipped with a multi-cuvette holder.
The tertiary screen consisted of a pyrophosphatase assay. Enzymatic activity was monitored using 250 uL assay solutions containing 100mM Tris pH8.2, 1mM MgCl2, 1mM DTT, 100mU/mL pyrophosphatase, and 100μM inhibitor. Assay solutions were incubated for 10 minutes to allow the inhibitor to bind, then enzymatic activity was initiated with the addition of 20 μM pyrophosphate. Every 30 seconds, 40 μL aliquots were removed from the assay mixture and combined with 40 μL of the acidic malachite green assay solution and 120 μL water. The A660 of each solution was measured on an Agilent 8453 spectrophotometer.
To screen the inhibitory activity of the natural product extracts, FtIspD activity was monitored using 250 μL assay solutions containing 100 mM Tris pH 8.2, 1 mM MgCl2, 1mM DTT, 200 μM CTP, 100 mU/mL pyrophosphatase, 1.8 μM FtIspD, and 0.5% (1.25 μL) natural product extract. Assays were conducted as described above in the secondary IspD screen for the LOPAC library.
Where appropriate, half maximal inhibitory activity (IC50) was determined by assaying FtIspD in the presence of varying concentrations of inhibitor, ranging from 1 nM to 100 μM, then plotting fractional enzyme activity as a function of inhibitor concentration. GraphPad Prism (La Jolla, CA) was used for nonlinear curve fitting the assay results to a sigmoidal dose response curve.
2.4 Preparation of Natural Product Extracts
A natural product extract library was curated in house using specimens of plants and fungi indigenous to the northern Virginia area. One gram of each specimen was frozen with liquid nitrogen, ground with a mortar and pestle, and mixed with 15 mL of ethyl acetate. The mixture was vacuum-filtered using a Büchner funnel and subsequently collected and transferred into a round bottom flask. The flask was placed on a rotovaporator with a 44°C water bath to remove the solvent. Samples were then resuspended to 1.0 mL in ethyl acetate, transfered into a tared 1.5 mL microcentrifuge tube, then evaporated to dryness in a SpeedVac. Each residue was weighed and stored at 4°C. Working solutions were prepared by resuspending the dry extracts in DMSO to a concentration of 10 mg/mL.
2.5 Selective Binding and Isolation of the Active Inhibitor from the Natural Product Extract
IspC solutions (1.5 mL) were prepared with 50 mM Tris pH 7.8, 25 mM MgCl2, 150 μM NADPH, 1.7 μM YpIspC, and 3.3% wt/vol of either DMSO (negative control) or the natural product extract (e29). Samples were incubated at 37°C for 15 min, then transferred to a centrifugal filter concentrator with 30 kDa cutoff (Amicon Millipore). Additional wash buffer (500 μL) was added to each sample (50 mM Tris-HCL pH.7.8, 25 mM MgCl) before centrifugation at 35°C and 4000 × g to a final sample volume of 200 μL. The sample retentate was removed and combined with 200 μL of wash buffer in a microcentrifuge tube. The microfuge tube was placed at 65°C for 20 minutes to denature the protein (and release of the active inhibitor from the enzyme), then the denatured sample was transferred to a 10 kDa concentrator and centrifuged at 35°C, 4000 × g, for 45 min. The filtrate containing the inhibitor was retained at −80°C for LC-QToF analysis.
IspD solutions (1.5 mL) were prepared with 100 mM Tris pH 8.0, 1 mM MgCl2, 200 μM CTP, 3.4 μM FtIspD, 1 mM DTT and 2% wt/vol of either DMSO (negative control) or the natural product extract (e29). The inhibitor was extracted essentially as above, using a wash buffer of 100 mM Tris pH 8.0, 1 mM MgCl2, 1 mM DTT.
2.6 LC-QToF Analysis
Filtrate samples containing the active inhibitor were removed from the −80°C freezer, diluted 1:1 with LC-MS Grade acetonitrile (Fisher Scientific), filtered using a Supelco (54145-U) Iso-disc, N-4-2 nylon, 4 mm × 0.2 μm filter (Sigma-Aldrich), and transferred to high recovery amber vials (Agilent Technologies, Inc., Santa Clara, CA). Reverse-phase liquid chromatography was performed on the purified analyte using an Agilent 1290 Infinity Ultra High-Performance Liquid Chromatography system (UHPLC) coupled with an Agilent 6530 Quadrupole Time of Flight (QToF) detector. Mobile phase was delivered by a binary pump at a flow rate of 0.4 mL/min. Solvent A was composed of LCMS Grade water + 0.1% v/v formic acid (Proteochem, Loves Park, IL) and solvent B was composed of LCMS Grade acetonitrile + 0.1% v/v formic acid (Proteochem). The chromatography gradient used is as follows: 0–1 min, 5% solvent B; 10 min, 30% B; 15 min, 70% B; 22 min, 90% B; 24–25 min, 100% B; 27 min, 2% B; 30 min, 5% B. The autosampler was set with an injection volume of 5 μL. Flush port was set to clean the injection needle for 30 s intervals. A ZORBAX Rapid Resolution High Throughput (RRHT), 2.1 × 50 mm, 1.8 μm C18 column (Agilent Technologies, Inc.) was used for the chromatography. The column was maintained at an isothermal temperature of 38°C. The QToF was equipped with an electrospray ionization (ESI) source, and was set for detection of ions within a mass-to-charge ratio (m/z) ranging from 100 to 1000. A dedicated isocratic pump continuously infused reference standards of purine (Agilent Technologies, Inc.) and hexakis-H, 1H, 3H-tetrafluoropropoxy-phosphazine, or HP-921 (Agilent Technologies, Inc.) at flow rate of 0.5 mL/min to achieve accurate mass correction. The nebulizer pressure was set at 35 psig with a surrounding sheath gas temperature of 350°C and gas flow rate of 11 L/min. Drying gas temperature was set at 300°C with a flow rate of 10 L/min. Default settings were used to set voltage gradient for nozzle at 1000 V, skimmer at 65 V, capillary (VCap) at 3500 V, and fragmentor at 175 V. Each cycle of acquisition was performed at a constant collision energy that varied between 0 V, 10 V, 20 V, and 40 V for subsequent tandem MS analysis. Agilent MassHunter Acquisition SW Version, 6200 series TOF/6500 series Q-TOF B.05.01 (B5125.1) was used to record LCMS data. Agilent MassHunter Qualitative Analysis B.06.00 was used to analyze data and to generate the total ion chromatogram (TIC), extracted compound chromatogram (ECC) and mass spectra for analyte compounds. Tandem mass spectra were processed using the Find Compound by MSMS function to envelope product ions and related features (adducts, isotopes, and fragment ions that elute at the same retention time). Mass spectra of analytes were processed using the Find Compound by Options function set to consider factors including sodium (Na+) and hydrogen (H+) adducts and neutral loses of water (H2O) while utilizing METLIN Metabolite Personal Compound Database add-in for Agilent MassHunter Qualitative Analysis B.06.00. The initial step in the identification of metabolites relied on the METLIN Metabolite PCD match-scoring criteria filters that evaluated m/z, potential adducts, potential neutral loses, accurate mass and isotope effect to calculate and propose chemical formula matches for precursor ions [22]. Follow-on manual comparative analysis of raw tandem MS data was performed with online MS/MS spectra library references.
2.7 Antibacterial Assays
The following reagents were obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH: Yersinia pestis subsp. A1122 and Francisella tularensis subsp. novicida strain Utah 112. Both species were cultured at 37°C in tryptic soy broth with 0.1% cysteine (TSB-C) and constantly shaken at 250 rpm. Agar (1.5% wt/vol) was added to prepare solid media. For whole cell assays, dose-response plot of cell growth (OD600) was measured as a function of inhibitor concentration. An overnight culture of Y. pestis subsp. A1122 or F. tularensis subsp. novicida was grown in TSB-C and diluted to OD600 of 0.2. Aliquots of the culture (40 μL) were then dispensed into foam-capped 1.5 mL microcentrifuge tubes containing 360 μL of fresh TSB and the appropriate concentration of inhibitor. Bacterial growth was monitored until OD600 = 1.5 for uninhibited samples; approximately 22 hours for Y. pestis and 7 hours for F. tularensis. Each condition was evaluated in duplicate. Nonlinear regression fitting the resulting dose-response plot was achieved using GraphPad Prism version 4.00 for Windows (GraphPad software Inc, San Diego, CA) and the equation F = 1/(1+[I]/IC50) where F = fractional growth and [I] = inhibitor concentration.
3. Results
3.1 Overview of the Approach to High-Throughput Screening
In separate high-throughput screens, the LOPAC1280 library from Sigma Aldrich (containing 1280 purified pharmaceutically active compounds) was screened for inhibitory activity against purified recombinant IspC and IspD. In order to identify hit compounds from the LOPAC1280 library, a stepwise screen was performed in the manner illustrated Figure 1.
For the IspC screens (shown in teal in Figure 2), initial inhibitory activity was assayed in a primary screen using Yersinia pestis IspC (YpIspC), then compounds demonstrating ≥75% inhibition of native enzyme activity were taken into a secondary screen performed with the Mycobacterium tuberculosis IspC (MtbIspC). Compounds with ≥75% inhibition in the secondary screen were subsequently evaluated in a tertiary screen involving a coupled IspC-IspD assay. This tertiary screen was designed to identify and eliminate library molecules harboring chromophores with absorbance at 340 nm (which would appear as false hits in the primary and secondary screens). Compounds retained from the tertiary screen were evaluated for antimicrobial activity in a quaternary screen with F. tularensis subsp. novicida, and were considered leads for further characterization if the compound reduced microbial growth by 75% or greater at 100 μM. Further details on the development of the IspC screening protocol are detailed in the Supplementary Materials.
Figure 2. Tiered screening procedure utilized with the LOPAC1280 library screen.
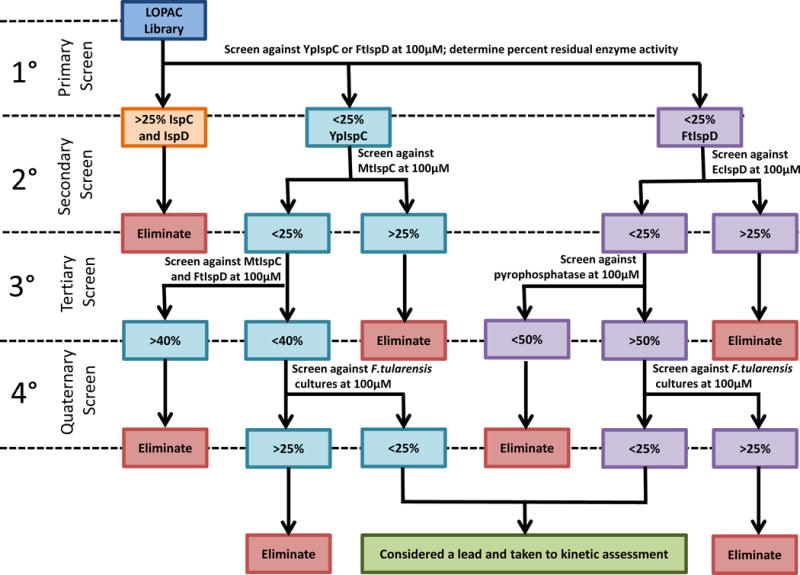
Compounds were passed through 4 tiers of screening for both IspC (teal) and IspD (purple). For IspC, these included a primary screen with YpIspC, a secondary screen with MtbIspC, a tertiary screen with MtbIspC coupled with FtIspD, and a quaternary antibacterial assay. For IspD, these included a primary screen with FtIspD, a secondary screen with EcIspD, a tertiary screen with pyrophosphatase, and a quaternary antibacterial screen. Cut-off values for each screen are indicated (% residual enzyme activity or % residual growth). Compounds retained through the quaternary screen for both IspC and IspD were considered lead compounds and were examined for mechanism of inhibition.
For the IspD screens (shown in purple in Figure 2), initial inhibitory activity was assayed in a primary screen using Francisella tularensis IspD (FtIspD), then compounds demonstrating ≥75% inhibition of native enzyme activity were taken into a secondary screen performed with Escherichia coli IspD (EcIspD). Compounds with ≥75% inhibition in the secondary screen were subsequently evaluated in a tertiary screen investigating inhibition of pyrophosphatase, a component of the IspD assay. This tertiary screen was designed to identify and eliminate library molecules which inhibited pyrophosphatase activity rather than IspD activity in the primary and secondary screens. As for the inhibitors identified with IspC, compounds retained from the tertiary screen with pyrophosphatase were evaluated in a quaternary screen with F. tularensis subsp. novicida to determine antimicrobial activity. Compounds reducing microbial growth by 75% or greater were retained and subsequently further characterized. Further details on the development of the IspD screening protocol are detailed in the Supplementary Materials.
3.2 The Screening of the LOPAC1280 Library — IspC
From the primary (Supplementary Figure 1A), secondary (Figure 3A), and tertiary screens (Figure 3B) with IspC, a total of seven compounds were identified as potential lead molecules and moved forward to a quaternary screen for antibacterial activity. The antibacterial activity of the seven highlighted library compounds was evaluated using a growth-inhibition assay with Francisella tularensis subsp. novicida Utah 112. All compounds were screened in duplicate at 100 μM. As shown in Figure 3C, four of the compounds significantly inhibit bacterial growth (structures of the compounds are provided in Figure 4D). These compounds, including 3-(3, 5-Dibromo-4-hydroxybenzylidine-5-iodo-1,3-dihydro-indol-2-one) or GW5074, 1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole or PPT, 3′-[(8-Cinnamoyl-5,7-dihy droxy-2,2-dimethyl-2H-1-benzopyran-6-yl)methyl]-2′,4′,6′-trihydroxy-5′-methylacetophenone or Rottlerin, and 13-Methyl-[1,3]benzodioxolo[5,6-c]-1,3-dioxolo[4,5-i] phenanthridinium chloride or Sanguinarine chloride, were thus considered lead molecules as inhibitors of IspC.
Figure 3. Screening of the LOPAC1280 library against IspC.
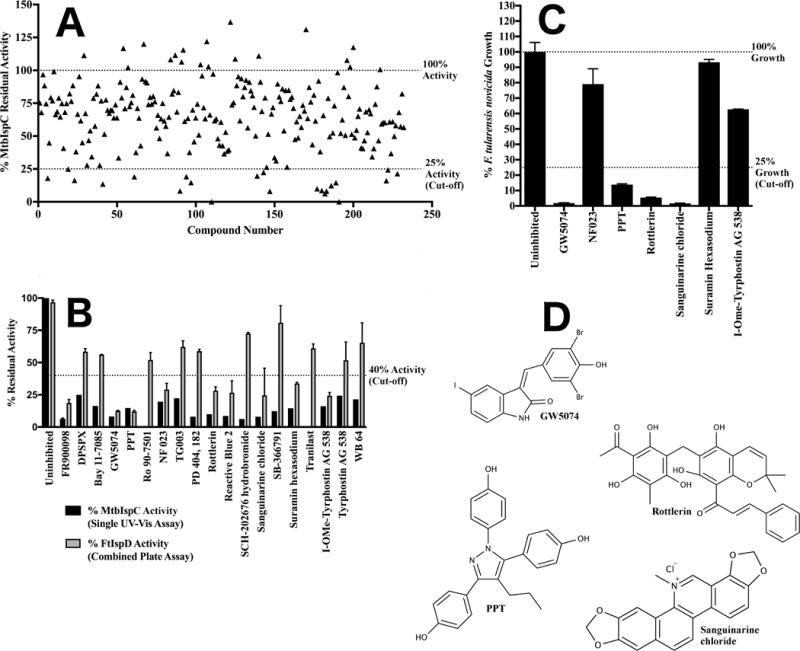
A) Secondary screening of the LOPAC1280 primary screen hits for IspC. Residual activity of MtbIspC when assayed in the presence of 100 μM of each of the LOPAC1280 library compounds identified in the primary screen. For clarity, each library molecule was assigned a numeric designation, ranging from 1–232 (the association of number, compound name, and residual enzyme activity is tabulated in Supplementary Figure 1B). Residual activity exceeding 100% reflects additional oxidation of NADPH over a well containing vehicle only. Library compounds demonstrating 75% or greater reduction in MtbIspC activity were selected for the tertiary screen. B) Tertiary screening of the LOPAC1280 library secondary screen hits for IspC via MtbIspC-FtIspD coupled assay. The residual activity of MtbIspC was measured with a coupled assay utilizing M. tuberculosis IspC and F. tularensis IspD (grey), and compared to the secondary screen values measuring MtbIspC activity using the A340 (black). Those compounds with less than 40% residual activity in the coupled assay were retained for quaternary antibacterial screening. Error bars are calculated as the deviation from the mean of two independent measurements. C) Quaternary screening of the LOPAC1280 tertiary screen hits for IspC against F. tularensis novicida. Antibacterial activity of LOPAC1280 inhibitors against F. tularensis subsp. novicida Utah 112 was measured in duplicate at 100 μM; compounds with less than 25% fractional bacterial growth were selected for EC50 determination. Fractional growth is calculated as the ratio of cell density (OD600) in the presence of 100 μM inhibitor to cell density in the presence of vehicle only. Error bars are calculated as the deviation from the mean of two independent measurements. D) Structures of lead compounds.
Figure 4. Screening of the LOPAC1280 library against IspD.
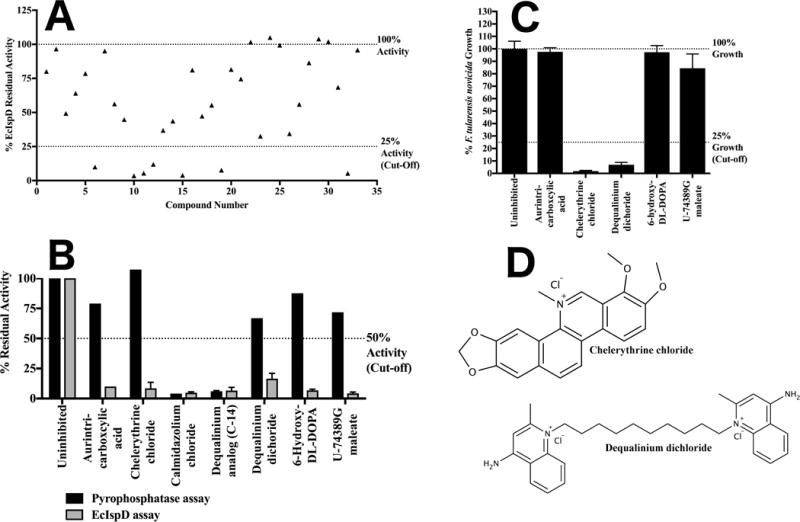
A) Secondary screening of the LOPAC1280 primary screen hits for IspD. Residual activity of EcIspD when assayed in the presence of 100 μM of each of the LOPAC1280 library compounds identified in the primary screen. For clarity, each library molecule was assigned a numeric designation, ranging from 1–33 (the association of number, compound name, and residual enzyme activity is tabulated in Supplementary Figure 1C). Library compounds demonstrating 75% or greater reduction in EcIspD activity were selected for the tertiary screen. B) Tertiary screening of the LOPAC1280 secondary screen hits for IspD against pyrophosphatase. Shown in black is the residual activity of pyrophosphatase versus the residual activity of EcIspD shown in grey in the presence of 100 μM inhibitor. Those compounds with greater than 50% residual activity in the pyrophosphatase assay were retained for quaternary antibacterial screening. Error bars are calculated as the deviation from the mean of two independent measurements. C) Quaternary screening of the LOPAC1280 tertiary screen hits for IspD against F. tularensis novicida. Antibacterial activity of LOPAC1280 inhibitors against F. tularensis subsp. novicida Utah 112 was measured in duplicate at 100 μM; compounds with less than 25% fractional bacterial growth were selected for EC50 determination. Fractional growth is calculated as the ratio of cell density (OD600) in the presence of 100 μM inhibitor to cell density in the presence of vehicle only. Error bars are calculated as the deviation from the mean of two independent measurements. D) Structures of lead compounds.
To quantify the potency of each of these molecules, EC50 values were derived from additional growth-inhibition assays performed with either F. tularensis subsp. novicida Utah 112 (presented in Supplementary Figure 2) or Yersinia pestis subsp. A1122 (presented in Supplementary Figure 3). As each of the four select compounds demonstrate low micromolar growth-inhibitory activity against F. tularensis and Y. pestis, we elected to further characterize each in follow-on enzyme assays performed with YpIspC and FtIspD (detailed following in Section 3.6) At the conclusion of primary through quaternary screening of the LOPAC1280 library with IspC, the 4 identified hit compounds from the initial library of 1280 gives a hit rate of 0.31%.
3.3 The Screening of the LOPAC1280 Library — IspD
From the primary (Supplementary Figure 1A), secondary (Figure 4A), and tertiary screens (Figure 4B) with IspD, a total of 5 molecules were identified as potential lead molecules and moved forward to a quaternary screen for antibacterial activity. As with IspC, the quaternary screen of IspD measured antibacterial activity of each of the potential lead molecules, evaluated using a growth-inhibition assay with Francisella tularensis subsp. novicida Utah 112. All compounds were screened in duplicate against the microbe at 100 μM as previously. Shown in Figure 4C, two of the compounds significantly inhibit bacterial growth (structures of the compounds are provided in Figure 4C). These compounds, including 1,2-Dimethoxy-N-methyl(1,3) benzodioxolo(5,6-c)phenanthridinium chloride or chelerythrine chloride and 1,1′-Decamethylenebis(4-aminoquinaldinium) dichloride hydrate or Dequalinium chloride, were thus considered lead molecules as inhibitors of IspD.
For the two lead compounds, EC50 values were determined from additional growth-inhibition assays performed with either F. tularensis subsp. novicida Utah 112 or Yersinia pestis subsp. A1122 (the dose-response plots for F. tularensis are presented in Supplementary Figure 2, and for Y. pestis are presented in Supplementary Figure 3). Both compounds demonstrate low micromolar growth-inhibitory activity against F. tularensis, but neither is active against Y. pestis. Given that the compounds were effective against F. tularensis, we elected to further characterize each in follow-on enzyme assays performed with YpIspC and FtIspD (detailed following in Section 3.6). At the conclusion of primary through quaternary screening of the LOPAC1280 library with IspD, the 2 identified hit compounds from the initial library of 1280 gives a hit rate of 0.15%.
3.4 Overview of the Approach to Natural Product Screening
In separate screens, a proprietary natural product library was screened for inhibitory activity against purified recombinant IspC and IspD. In order to identify hit compounds from the natural product library, a stepwise screen was performed in the manner illustrated in Figure 5.
Figure 5. Tiered Screening procedure utilized with the natural product library screen.
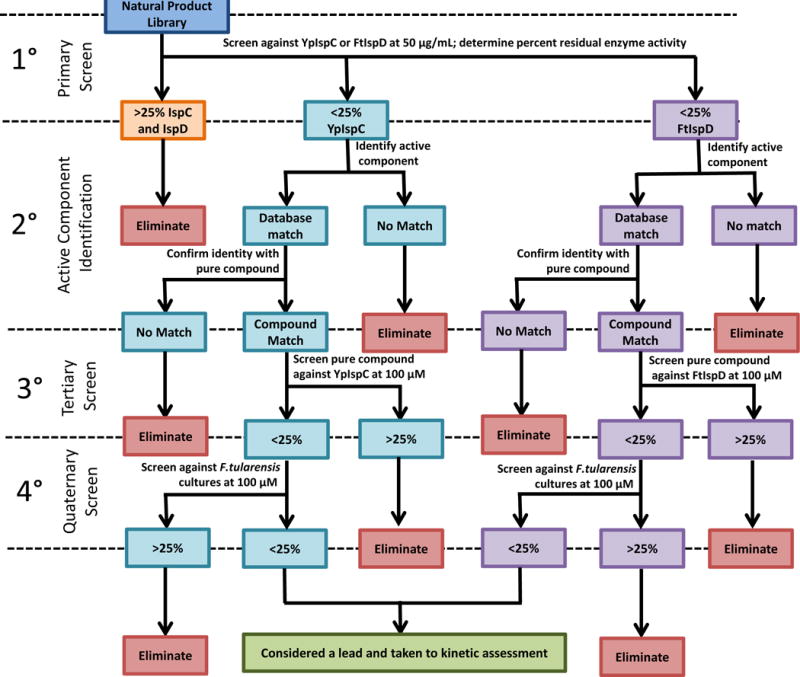
Compounds were passed through 4 tiers of screening for both IspC (teal) and IspD (purple). For both enzymes, these included a primary screen with the appropriate enzyme (YpIspC for IspC screen, and FtIspD for IspD screen), a secondary assessment to determine the active component in the extract, a tertiary screen to confirm the compound identification, and a quaternary screen for antibacterial activity. Cut-off values for each screen are indicated (% residual enzyme activity in primary and tertiary screens or % residual growth for quaternary antibacterial screen). Compounds retained through the quaternary screen for both IspC and IspD were considered lead compounds and were examined for mechanism of inhibition.
For the IspC screens (shown in blue in Figure 5) and the IspD screens (shown in purple in Figure 5), initial inhibitory activity was assessed in a primary screen with either YpIspC or FtIspD, respectively. Compounds demonstrating ≥75% inhibition of native enzyme activity were considered suitable compounds for active component identification, and select compounds were pursued based on percent inhibition of the enzyme and abundance of the starting material (the extract). Active components were identified using an affinity extraction method utilizing the enzyme as bait and subsequent matching of an MS/MS spectrum of the isolated compound to that of a standard in the Metlin metabolite database. Once a compound was initially identified using database matching, the pure compound was purchased and its MS/MS spectrum was derived then compared to the MS/MS spectrum of the compound isolated from the extract. If matched, then the compound was moved forward to a tertiary screen with the purified enzyme (YpIspC for the IspC screen or FtIspD for the IspD screen). Residual enzyme activity under 25% justified moving the compound to quaternary antibacterial screen with F. tularensis subsp. novicida. Compounds reducing microbial growth by at least 75% at 100 μM were retained for EC50 determination and subsequent further characterization.
3.5 The Screening of a Natural Product Extract Library
Run separate from the LOPAC1280 library screen, a natural product library was also evaluated for inhibitory activity utilizing purified recombinant YpIspC and FtIspD. This library was produced in-house and comprises 155 individual ethyl acetate extracts, derived from a wide variety of regional plants and fungi, each extract containing a diverse mixture of metabolites. The extracts were individually evaluated in the assay at 50 μg/mL final concentration, while 100 μM fosmidomycin and FR900098 were each used as positive controls for inhibition of IspC, and 100 μM anthracene blue was used as the positive control for IspD. Overall, nine different extracts were found to inhibit YpIspC activity by 75% or greater, and 11 different extracts were found to inhibit FtIspD activity by 75% or greater (Figure 6A). Most interestingly, e29 was found to inhibit the activity of both YpIspC and FtIspD by greater than 95%. In light of this, e29 was prioritized for subsequent active component identification.
Figure 6. Screening the natural product library against IspC and IspD.
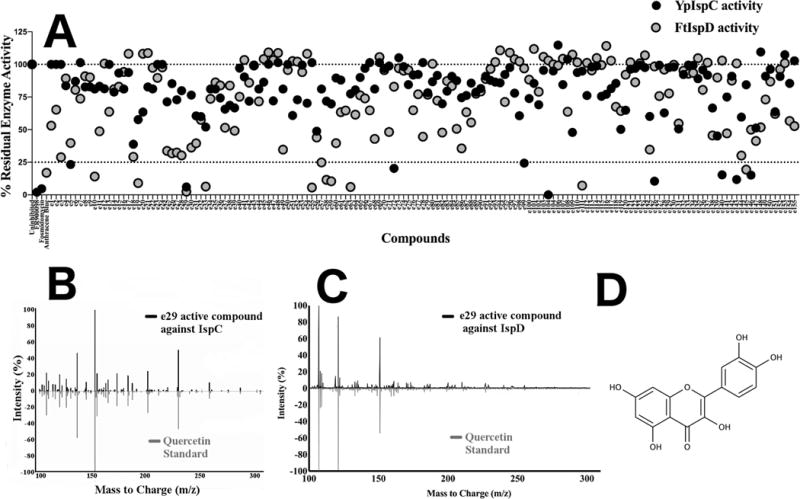
A) Primary natural product library screen for inhibitors of IspC or IspD. Each natural product extract is alphanumerically identified as e1 through e155. FR900098 and fosmidomycin (100 μM) serve as positive inhibition controls with YpIspC. Anthracene blue (100 μM) serves as a positive control for FtIspD. The residual enzyme activity for YpIspC is indicated in black, whereas the residual enzyme activity of FtIspD is indicated in grey. All residual activity values were calculated using an uninhibited assay with vehicle (DMSO) only. B) Secondary IspC screen for active compound identification of e29. Comparison of MS/MS spectrum of e29 active component with respect to YpIspC (black) with an MS/MS spectrum of a pure quercetin standard run in-house (green). Comparison of spectra shown at a collision energy of 40 V. Peaks at 302 m/z indicate the precursor ion. C) Secondary IspD screen for active compound identification of e29. Comparison of MS/MS spectrum of e29 active component with respect to FtIspD (black) with an MS/MS spectrum of a pure quercetin standard run in-house (grey). Comparison of spectra shown at a collision energy of 40 V. Peaks at 302 m/z indicate the precursor ion. D) Structure of quercetin.
The e29 extract was obtained from the plant Rumex crispus, commonly known as Curly Dock, an invasive weed species found throughout North America [23]. Although e29 inhibited both YpIspC and FtIspD, YpIspC was chosen first for active component identification via the use of YpIspC as an affinity bait. After incubation of the extract with the enzyme, the enzyme-inhibitor complex was separated from the extract by centrifugation through a 30 kDa ultrafiltration concentrator (the enzyme-inhibitor complex remains in the retentate) and washed. Subsequent heat denaturation of the enzyme-inhibitor complex released the associated inhibitor, which was immediately recovered by centrifugation through a second 30kDa ultrafiltration concentrator (the enzyme remains in the retentate whereas the inhibitor is now in the filtrate). The filtrate was then directly analyzed using an LC-QToF (LC-MS/MS). Mass spectra comparison of the inhibitor to the METLIN Metabolite and Tandem MS Database identified the molecule quercetin as the putative IspC inhibitor. Figure 6B shows a comparison of the MS/MS spectrum of the isolate from e29 and the MS/MS spectrum of a purchased quercetin standard analyzed on our LC-QToF at a collison energy of 40V.
To validate the inhibitory activity of quercetin, it and a small series of related flavonoids, including common glycosylated forms of quercetin and the structurally similar catechins, were purchased and screened at 100 μM against YpIspC in a tertiary screen. As reported in Table 1, quercetin effectively inhibits over 95% of the catalytic activity of YpIspC, while none of the glycosylated forms of quercetin nor the catechins showed any significant inhibitory activity.
Table 1.
Tertiary IspC Screen for inhibitory activity of commercially purchased quercetin and additional structurally related flavonoids when assayed with YpIspC.
| Compound | % Residual Activity |
|---|---|
| Quercetin | 4.83 |
| Quercitrin | 78.21 |
| Quercetin 3-β-D-glucoside | 76.25 |
| Quercetin 3-D-Galactoside | 76.779 |
| Catechin | 96.65 |
Quaternary bacterial growth-inhibition assays performed with quercetin and Francisella tularensis subsp. novicida Utah 112 at 100 μM showed less than 25% residual growth, justifying subsequent EC50 determination. Significant inhibitory activity associated with quercetin is observed against both Yersinia pestis subsp. A1122 and Francisella tularensis subsp. novicida Utah 112 (Supplementary Figure 4), with EC50 values under 50 μM for both microbes. Notably, the EC50 values with quercetin are comparable to those obtained in growth inhibition assays performed with FR900098 (Francisella tularensis: 36 μM with quercetin, 23 μM with FR900098[12]; Yersinia pestis: 29 μM with quercetin, 29 μM with FR900098 [16]).
Next, to identify the inhibitor of IspD present in e29, we used the same affinity bait approach described with YpIspC. LC-MS/MS reveals the active inhibitor of IspD also to be quercetin, as shown in Figure 4C with a comparison of the MS/MS spectrum of the isolate from e29 and the MS/MS spectrum of a purchased quercetin standard analyzed at a collison energy of 40V. To validate this discovery, we assayed the activity of FtIspD in the presence of 100 μM commercially purchased quercetin. The FtIspD activity was reduced to 4% of the uninhibited control, confirming that quercetin is indeed an inhibitor of IspD. Given the known antibacterial activity of quercetin against both F. tularensis and Y.pestis (as determined in the quaternary natural product screen for IspC inhibitors; see Supplementary Figure 4), quercetin was retained as a lead molecule for inhibition of IspD. The structure of quercetin is given in Figure 6D.
Literature records a striking number of biological activities associated with quercetin. The flavonoids as a class are noted for their antioxidant properties; quercetin is no exception, having been reported to inhibit the production of both reactive oxygen species (ROS) and nitric oxide (NO). [24] It has been reported as a direct competitive inhibitor of cytochrome p450 function, with a Ki of 15.4 μM against CYP34A [25], and as a noncompetitive inhibitor of lipoxygenase with an IC50 of 4.8 μM. [26] Quercetin has been described to have anticancer properties, interfering with signaling in the JAK-STAT pathway [27], PI3K-Akt-mTOr pathway (competitive inhibitor of PI3K, Kd = 0.28 μM [28]) and the MAPK-JNK pathway (inhibitor of JNK, IC50 = 1 μM [29]), and shows biological effects at physiologically relevant concentrations [30]. In addition, quercetin has noted antiviral properties, including effects on hepatitis C ([31], inhibitor of nonstructural protein 5A of HCV [32]) as well as influenza A (inhibitor of HA2 subunit [33]). Quercetin shows noted anti-MRSA activity by several groups ([34] [35] [36]), although reports conflict over its activity against E. coli ([37], [38]). Antibacterial activity has been observed in various flavonoid-containing plants used in folk medicine in places like Iran [39], Turkey [40], and Kenya [41]. The anti-inflammatory effects of quercetin [42] are well documented, including evidence that quercetin blocks mast cell cytokine release [43]. Despite the abundance of biological activities associated with quercetin, reports have become available implicating quercetin as a “Pan Assay Interference compound”, or PAIN, which nonspecifically interferes with a wide spectrum of assays ([44] [45]), leading to some concern over the association between target and biological effect. Further clinical trials have been called for to establish the clinical uses of quercetin. [46] Given the interest in this molecule from a large range of fields, we next elected to further characterize quercetin, along with the 6 top hits obtained from the LOPAC1280 library screen. This characterization is fully described in the subsequent Section 3.6.
3.6 Hit Characterization
To determine relative potency of the hit compounds from both the LOPAC1280 and natural product libraries, inhibitory dose-response curves for quercetin and each of the 6 LOPAC1280 hit compounds were determined using the enzyme assay with YpIspC or FtIspD, as appropriate, and are given in Supplementary Figure 5. Given the hydrophobicity of several of the inhibitors, as well as the steep dose response curves of several of the inhibitors, most specifically sanguinarine chloride and PPT, each inhibitor was examined for aggregation using a 0.01% Triton X-100 detergent screen, as recommended as a simple predictive test to identify aggregating inhibitors [47]. Neither YpIspC nor FtIspD showed significant changes in activity in the presence of this quantity of detergent. As shown in Figure 7A, each compound was determined to be significantly less effective in the presence of small amounts of detergent, defined as a 25% increase in residual activity in the presence of detergent, with the exception of sanguinarine chloride. Additional examination of sanguinarine chloride, given in Supplementary Figure 6, reveals that sanguinarine chloride is also an aggregating inhibitor, based on its mixed inhibition profile and its variable IC50, dependent on enzyme concentration. Based on these positive control tests, all compounds identified were determined to aggregate in assay buffer conditions and are unsuitable for further investigation. A literature search reveals reports of rottlerin, quercetin, and dequalinium chloride forming aggregates in solution, and we corroborate this observation. ([48], [49], [38], [50].)
Figure 7. Aggregation inhibition determination using detergent screening.
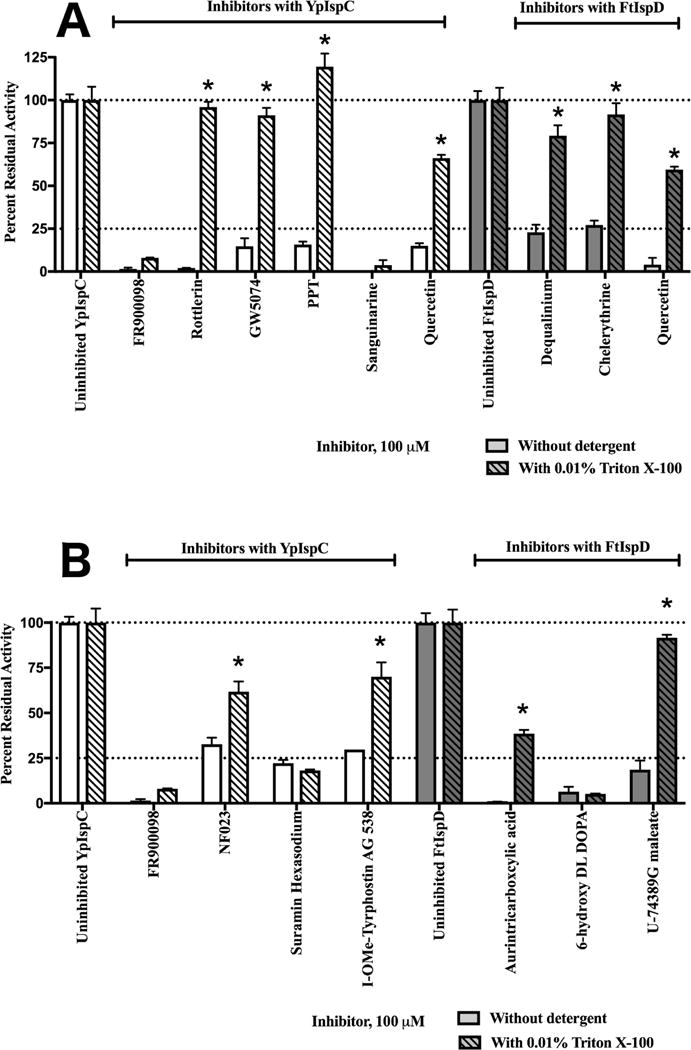
Residual activity of YpIspC (white) or FtIspD (dark grey) in the absence (open bars) and presence (hatched bars) of 0.01% Triton X-100. All samples are standardized to an uninhibited control containing the vehicle (DMSO) and 0.01% Triton X-100 if necessary. FR900098 was used as a positive control for specific inhibition. The cut-off for significant attenuation of inhibition was set as 25% or greater increase in residual enzyme activity; compounds with significantly attenuated activity in the presence of Triton X-100 are indicated with a star. Error bars are calculated as the deviation from the mean of two independent measurements. A) Inhibition is attenuated in the presence of Triton X-100 for all inhibitors except sanguinarine chloride, and inhibitor of IspC. B) Inhibition is attenuated in the presence of Triton X-100 for all inhibitors except suramin hexasodium, an inhibitor of IspC, and 6-hydroxy DL DOPA, an inhibitor of IspD.
Given the lack of suspected specific lead compounds identified in the LOPAC1280 and natural product screen, further investigation was considered for LOPAC1280 compounds that passed primary, secondary and tertiary screening, but did not show antibacterial activity. Both Francisella tularensis and Yersinia pestis are Gram negative microbes that may be limiting antimicrobial activity through reduced cell well penetration or efflux pumps. Therefore, it was hypothesized that select other compounds that were identified in the LOPAC1280 screening campaign still may function as novel scaffolds for IspC or IspD inhibitor development, albeit that these scaffolds would likely need to be optimized to improve cellular penetration. There were three inhibitors of IspC and of IspD identified in tertiary screening that failed to show significant antimicrobial activity in the quaternary screen (Figure 3 and Figure 4): NF023, Suramin Hexasodium, and I-OMe-Tyrphostin AG 538 with respect to IspC, and Aurintricarboxylic acid, 6-hydroxy-DL-DOPA, and U-74389G maleate with respect to IspD. These compounds were subsequently examined for their inhibition in the presence and absence of Triton X-100 to determine if any of these compounds were also functioning as aggregators. Figure 7B shows that addition of 0.01% Triton X-100 did not attenuate inhibition for suramin hexasodium, an identified IspC inhibitor, or 6-hydroxy-DL-DOPA, an identified IspD inhibitor. Because neither of these compounds were as likely to be aggregating inhibitors given the insensitivity to detergent, both were investigated for potency and mechanism of action. As shown in Supplementary Figure 7, both suramin and 6-hydroxy-DL-DOPA were confirmed as non-aggregating compounds, based on suramin’s consistent IC50 against variable YpIspC concentrations, and 6-hydroxy-DL-DOPA’s competitive mechanism with respect to CTP. However, given concerns over suramin’s specificity given it’s known side effects and wide range of other targets ([51], [52], [53], [54], [55], [56]), and 6-hydroxy-DL-DOPA’s known neurotoxicity and chemical instability ([57], [58]), both suramin and 6-hydroxy-DL-DOPA would be challenging lead molecules for further development as MEP pathway inhibitors.
4. Discussion
Each of the initial hits compounds identified from the LOPAC1280 and natural product libraries displaying antimicrobial activity showed evidence of aggregation, either through sensitivity of inhibition to detergent, or sensitivity of inhibition to enzyme concentration. Given the percentage of hits identified as aggregators in HTS campaigns, both in this work and others [59], not including such analysis of hits increases the likelihood of pursuing compounds that do not inhibit the expected target in a specific manner. Given aggregating inhibitors would be expected to inhibit diverse enzymes if assay conditions are similar, further investigation into the specificity of each cohort of LOPAC1280 inhibitors is warranted. For the aggregating inhibitors of IspD, investigation into the IspC primary and secondary screens reveals that both chelerythrine and dequalinium were identified as inhibitors of IspC in the primary screen with YpIspC. They were subsequently both eliminated following the secondary screen with MtbIspC, with chelerythrine reducing the residual activity of MtbIspC to 27%, only 2% over the set 25% cut-off for the secondary screen. It is likely that the identified aggregating inhibitors of IspD function as aggregating inhibitors of IspC, with decreases in potency just great enough to avoid being retained in the secondary screen of IspC. In the same way, investigation of the aggregating IspC inhibitors reveals that all showed some activity against IspD in the primary screen, with rottlerin being the least potent at 52% residual FtIspD activity in the primary screen. Sanguinarine reduced FtIspD residual activity to 27%, just missing the primary screen cut-off. Both GW5074 and PPT were identified as inhibitors of FtIspD in the primary screen, and were eliminated with 47% and 55% residual activity of EcIspD in the secondary screen, respectively. It is known that aggregating inhibitors are exceptionally sensitive to changes in protein concentration, as shown in the case of sanguinarine in Supplementary Figure 7. Because the IspD assays were performed with roughly twice the concentration of enzyme as the IspC assays (1.8 μM versus 0.89 μM), in addition to the pyrophosphatase, it is perhaps unsurprising that aggregating inhibitors in the IspC assays were in general less potent when examined in the IspD assays.
Based on these observations, it is apparent that HTS protocol design is crucial in identifying false positive compounds. In the protocol used here, we account for false positives due to assay constraints using the tertiary screens, and false positives due to aggregation phenomena using a combination of screens for detergent sensitivity, changes in potency dependent on enzyme concentration, and mechanism of action via Lineweaver Burk plots. The HTS protocol delineated provides a rational start to probing IspC and IspD for additional sites to expand the chemical space for inhibitors of these enzymes. Use of this HTS protocol for IspC and IspD with additional molecular libraries, including the appropriate control assays and investigation for aggregation, may unveil further compounds suitable as novel antibiotics.
Supplementary Material
Supplementary Figure 1. Primary and Secondary LOPAC1280 Screening Data. A) Primary Screen of the LOPAC1280 Library with YpIspC and FtIspD. B) Secondary Screen of MtbIspC with lead compounds from the primary screen of LOPAC1280. C) Secondary Screen of EcIspD with lead compounds from the primary screen of LOPAC1280
Supplementary Figure 2. Half-maximal effective concentration of LOPAC1280 hit compounds against F. tularensis subsp novicida Utah 112. Percent residual growth is calculated as the ratio of cell density (OD600) in the presence of inhibitor to cell density in the presence of vehicle (DMSO) only for inhibitors A) GW5074, B) PPT, C) Rottlerin, D) Sanguinarine chloride, E) Chelerythrine chloride and F) Dequalinium chloride. The EC50 values and R2 values for each plot are indicated. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 3: Half-maximal effective concentration of LOPAC1280 hit compounds against Y. pestis subsp A1122. Percent residual growth is calculated as the ratio of cell density (OD600) in the presence of inhibitor to cell density in the presence of vehicle (DMSO) only for inhibitors A) GW5074, B) PPT, C) Rottlerin, D) Sanguinarine, E) Chelerythrine chloride, and F) Dequalinium chloride. Neither chelerythrine chloride nor dequalinium chloride were active against Y.pestis up to 100 μM. The EC50 values and R2 values for each plot are indicated. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 4. Half-maximal effective concentration of quercetin against liquid cultures of A) Y. pestis subsp. A1122 and B) F. tularensis subsp. novicida Utah 112. Percent residual growth is calculated as the ratio of culture cell density (OD600) in the presence of quercetin to culture cell density in the presence of vehicle (DMSO) alone. The potency (EC50) and R2 values for each plot are indicated. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 5. Dose-response plots of select inhibitors of YpIspC or FtIspD. Assays were performed with each enzyme with various concentrations of inhibitors including A) GW5074 with YpIspC, B) PPT with YpIspC, C) Rottlerin with YpIspC, D) Sanguinarine with YpIspC, E) Chelerythrine chloride with FtIspD, F) Dequalinium chloride with FtIspD, G) Quercetin with YpIspC and H) Quercetin with FtIspD For each assay, after a ten minute incubation of the enzyme with inhibitor and cofactor, the substrate was added to initiate the reaction. The IC50 and goodness-of-fit (R2) value for each plot is indicated. The enzymatic activity is relative to an uninhibited control using vehicle (DMSO) alone. All assays were performed in duplicate. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 6. Mode of inhibition by sanguinarine. All assays were performed in duplicate using purified YpIspC. Error bars are calculated as the deviation from the mean of two independent measurements A) The Lineweaver–Burk plot indicates that sanguinarine is noncompetitive with respect to DXP. B) The Lineweaver-Burk plot indicates that sanguinarine is noncompetitive with respect to NADPH. C) Dependence of IC50 with sanguinarine on enzyme concentration. IC50 with sanguinarine decreases with a decrease in YpIspC concentration. Concentrations of YpIspC, IC50, and Hill slope are indicated. D) The dependence of IC50 value and Hill slope of each IC50 plot decreased linearly with decrease in YpIspC concentration.
Supplementary Figure 7. Characterization of suramin and 6-hydroxy-DL-DOPA. All assays were performed in duplicate. Error bars are calculated as the deviation from the mean of two independent measurements. A) IC50 of suramin with YpIspC. The IC50 with suramin does not significantly decreases with a decrease in YpIspC concentration, nor does the Hill slope decrease. B) Structure of suramin. C) The Lineweaver–Burk plot indicates that suramin is noncompetitive with respect to DXP. D) The Lineweaver-Burk plot indicates that suramin is noncompetitive with respect to NADPH. E) IC50 of 6-hydroxy-DL-DOPA with FtIspD. F) Structure of 6-hydroxy-DL-DOPA. C) The Lineweaver–Burk plot indicates that 6-hydroxy-DL-DOPA is competitive with respect to CTP. D) The Lineweaver-Burk plot indicates that 6-hydroxy-DL-DOPA is noncompetitive with respect to MEP.
Acknowledgments
This study was supported by George Mason University’s Department of Chemistry and Biochemistry, the U.S. Army Medical Research and Materiel Command (W23RYX1291N601), and the NIH (NIAID 1R01AI123433-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. Antimicrobial resistance: global report on surveillance. WHO; 2014. n.d. http://www.who.int/drugresistance/documents/surveillancereport/en/ accessed November 15, 2014. [Google Scholar]
- 2.Brown AC, Parish T. Dxr is essential in Mycobacterium tuberculosis and fosmidomycin resistance is due to a lack of uptake. BMC Microbiol. 2008;8:78. doi: 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallagher LA, Ramage E, Jacobs MA, Kaul R, Brittnacher M, Manoil C. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc Natl Acad Sci. 2007;104:1009–1014. doi: 10.1073/pnas.0606713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron DE, Urbach JM, Mekalanos JJ. A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proc Natl Acad Sci. 2008;105:8736–8741. doi: 10.1073/pnas.0803281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odom AR. Five Questions about Non-Mevalonate Isoprenoid Biosynthesis. PLoS Pathog. 2011;7:e1002323. doi: 10.1371/journal.ppat.1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandes JF, Lell B, Agnandji ST, Obiang RM, Bassat Q, Kremsner PG, Mordmüller B, Grobusch MP. Fosmidomycin as an antimalarial drug: a meta-analysis of clinical trials. Future Microbiol. 2015;10:1375–1390. doi: 10.2217/FMB.15.60. [DOI] [PubMed] [Google Scholar]
- 8.San Jose G, Jackson ER, Haymond A, Johny C, Edwards RL, Wang X, Brothers RC, Edelstein EK, Odom AR, Boshoff HIM, Couch RD, Dowd CS. Structure-Activity Relationships of the MEPicides: N-Acyl and O-linked Analogs of FR900098 as Inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis. ACS Infect Dis. 2016 doi: 10.1021/acsinfecdis.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masini T, Kroezen BS, Hirsch AKH. Druggability of the enzymes of the non-mevalonate-pathway. Drug Discov Today. 2013;18:1256–1262. doi: 10.1016/j.drudis.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Na-Bangchang K, Ruengweerayut R, Karbwang J, Chauemung A, Hutchinson D. Pharmacokinetics and pharmacodynamics of fosmidomycin monotherapy and combination therapy with clindamycin in the treatment of multidrug resistant falciparum malaria. Malar J. 2007;6:70. doi: 10.1186/1475-2875-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson ER, Dowd CS. Inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr): a review of the synthesis and biological evaluation of recent inhibitors. Curr Top Med Chem. 2012;12:706–728. doi: 10.2174/156802612799984599. [DOI] [PubMed] [Google Scholar]
- 12.McKenney ES, Sargent M, Khan H, Uh E, Jackson ER, San Jose G, Couch RD, Dowd CS, van Hoek ML. Lipophilic prodrugs of FR900098 are antimicrobial against Francisella novicida in vivo and in vitro and show GlpT independent efficacy. PloS One. 2012;7:e38167. doi: 10.1371/journal.pone.0038167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Witschel MC, Höffken HW, Seet M, Parra L, Mietzner T, Thater F, Niggeweg R, Röhl F, Illarionov B, Rohdich F, Kaiser J, Fischer M, Bacher A, Diederich F. Inhibitors of the herbicidal target IspD: allosteric site binding. Angew Chem Int Ed Engl. 2011;50:7931–7935. doi: 10.1002/anie.201102281. [DOI] [PubMed] [Google Scholar]
- 14.Imlay LS, Armstrong CM, Masters MC, Li T, Price KE, Edwards RL, Mann KM, Li LX, Stallings CL, Berry NG, O’Neill PM, Odom AR. Plasmodium IspD (2-C-Methyl-D-erythritol 4-Phosphate Cytidyltransferase), an Essential and Druggable Antimalarial Target. ACS Infect Dis. 2015;1:157–167. doi: 10.1021/id500047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. The Open Access Malaria Box: A Drug Discovery Catalyst for Neglected Diseases. PLoS ONE. 2013;8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haymond A, Johny C, Dowdy T, Schweibenz B, Villarroel K, Young R, Mantooth CJ, Patel T, Bases J, Jose GS, Jackson ER, Dowd CS, Couch RD. Kinetic Characterization and Allosteric Inhibition of the Yersinia pestis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (MEP Synthase) PLoS ONE. 2014;9:e106243. doi: 10.1371/journal.pone.0106243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsang A, Seidle H, Jawaid S, Zhou W, Smith C, Couch RD. Francisella tularensis 2-C-Methyl-D-Erythritol 4-Phosphate Cytidylyltransferase: Kinetic Characterization and Phosphoregulation. PLoS ONE. 2011;6:e20884. doi: 10.1371/journal.pone.0020884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi S, Kuzuyama T, Watanabe H, Seto H. A 1-deoxy-d-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-d-erythritol 4-phosphate in an alternative nonmevalonate pathway for terpenoid biosynthesis. Proc Natl Acad Sci. 1998;95:9879–9884. doi: 10.1073/pnas.95.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LOPAC1280 – Library of Pharmacologically Active Compounds, Sigma-Aldrich. n.d. http://www.sigmaaldrich.com/life-science/cell-biology/bioactive-small-molecules/lopac1280-navigator.html (accessed December 12, 2015)
- 20.Dhiman RK, Schaeffer ML, Bailey AM, Testa CA, Scherman H, Crick DC. 1-Deoxy-d-Xylulose 5-Phosphate Reductoisomerase (IspC) from Mycobacterium tuberculosis: towards Understanding Mycobacterial Resistance to Fosmidomycin. J Bacteriol. 2005;187:8395–8402. doi: 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernal C, Palacin C, Boronat A, Imperial S. A colorimetric assay for the determination of 4-diphosphocytidyl-2-C-methyl-D-erythritol 4-phosphate synthase activity. Anal Biochem. 2005;337:55–61. doi: 10.1016/j.ab.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Scripps Center For Metabolomics and Mass Spectrometry - METLIN. n.d. https://metlin.scripps.edu/index.php (accessed October 9, 2016)
- 23.Curly Dock (Rumex crispus) n.d. http://www.illinoiswildflowers.info/weeds/plants/curly_dock.htm (accessed April 5, 2017)
- 24.Zhang M, Swarts SG, Yin L, Liu C, Tian Y, Cao Y, Swarts M, Yang S, Zhang SB, Zhang K, Ju S, Olek DJ, Schwartz L, Keng PC, Howell R, Zhang L, Okunieff P. Antioxidant properties of quercetin. Adv Exp Med Biol. 2011;701:283–289. doi: 10.1007/978-1-4419-7756-4_38. [DOI] [PubMed] [Google Scholar]
- 25.Östlund J, Zlabek V, Zamaratskaia G. In vitro inhibition of human CYP2E1 and CYP3A by quercetin and myricetin in hepatic microsomes is not gender dependent. Toxicology. 2017;381:10–18. doi: 10.1016/j.tox.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Ha TJ, Shimizu K, Ogura T, Kubo I. Inhibition mode of soybean lipoxygenase-1 by quercetin. Chem Biodivers. 2010;7:1893–1903. doi: 10.1002/cbdv.200900249. [DOI] [PubMed] [Google Scholar]
- 27.Khan F, Niaz K, Maqbool F, Ismail Hassan F, Abdollahi M, Nagulapalli Venkata KC, Nabavi SM, Bishayee A. Molecular Targets Underlying the Anticancer Effects of Quercetin: An Update. Nutrients. 2016;8 doi: 10.3390/nu8090529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 29.Yoshizumi M, Tsuchiya K, Kirima K, Kyaw M, Suzaki Y, Tamaki T. Quercetin Inhibits Shc- and Phosphatidylinositol 3-Kinase-Mediated c-Jun N-Terminal Kinase Activation by Angiotensin II in Cultured Rat Aortic Smooth Muscle Cells. Mol Pharmacol. 2001;60:656–665. [PubMed] [Google Scholar]
- 30.Jeong JH, An JY, Kwon YT, Rhee JG, Lee YJ. Effects of low dose quercetin: Cancer cell-specific inhibition of cell cycle progression. J Cell Biochem. 2009;106:73–82. doi: 10.1002/jcb.21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khachatoorian R, Arumugaswami V, Raychaudhuri S, Yeh GK, Maloney EM, Wang J, Dasgupta A, French SW. Divergent antiviral effects of bioflavonoids on the hepatitis C virus life cycle. Virology. 2012;433:346–355. doi: 10.1016/j.virol.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattacharya D, Ansari IH, Mehle A, Striker R. Fluorescence Resonance Energy Transfer-Based Intracellular Assay for the Conformation of Hepatitis C Virus Drug Target NS5A. J Virol. 2012;86:8277–8286. doi: 10.1128/JVI.00645-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu W, Li R, Li X, He J, Jiang S, Liu S, Yang J. Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry. Viruses. 2015;8 doi: 10.3390/v8010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirai I, Okuno M, Katsuma R, Arita N, Tachibana M, Yamamoto Y. Characterisation of anti-Staphylococcus aureus activity of quercetin. Int J Food Sci Technol. 2010;45:1250–1254. doi: 10.1111/j.1365-2621.2010.02267.x. [DOI] [Google Scholar]
- 35.Betts JW, Sharili AS, Phee LM, Wareham DW. In Vitro Activity of Epigallocatechin Gallate and Quercetin Alone and in Combination versus Clinical Isolates of Methicillin-Resistant Staphylococcus aureus. J Nat Prod. 2015;78:2145–2148. doi: 10.1021/acs.jnatprod.5b00471. [DOI] [PubMed] [Google Scholar]
- 36.Amin MU, Khurram M, Khattak B, Khan J. Antibiotic additive and synergistic action of rutin, morin and quercetin against methicillin resistant Staphylococcus aureus. BMC Complement Altern Med. 2015;15:59. doi: 10.1186/s12906-015-0580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu T, He M, Zang X, Zhou Y, Qiu T, Pan S, Xu X. A structure–activity relationship study of flavonoids as inhibitors of E. coli by membrane interaction effect. Biochim Biophys Acta BBA - Biomembr. 2013;1828:2751–2756. doi: 10.1016/j.bbamem.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 38.Tritsch D, Zinglé C, Rohmer M, Grosdemange-Billiard C. Flavonoids: True or promiscuous inhibitors of enzyme? The case of deoxyxylulose phosphate reductoisomerase. Bioorganic Chem. 2015;59:140–144. doi: 10.1016/j.bioorg.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 39.Tofighi Z, Molazem M, Doostdar B, Taban P, Shahverdi AR, Samadi N, Yassa N. Antimicrobial Activities of Three Medicinal Plants and Investigation of Flavonoids of Tripleurospermum disciforme. Iran J Pharm Res IJPR. 2015;14:225–231. [PMC free article] [PubMed] [Google Scholar]
- 40.Orhan DD, Özçelik B, Özgen S, Ergun F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol Res. 2010;165:496–504. doi: 10.1016/j.micres.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Njeru SN, Obonyo MA, Nyambati SO, Ngari SM. Antimicrobial and cytotoxicity properties of the crude extracts and fractions of Premna resinosa (Hochst.) Schauer (Compositae): Kenyan traditional medicinal plant. BMC Complement Altern Med. 2015;15:295. doi: 10.1186/s12906-015-0811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Yao J, Han C, Yang J, Chaudhry MT, Wang S, Liu H, Yin Y. Quercetin, Inflammation and Immunity. Nutrients. 2016;8 doi: 10.3390/nu8030167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weng Z, Zhang B, Asadi S, Sismanopoulos N, Butcher A, Fu X, Katsarou-Katsari A, Antoniou C, Theoharides TC. Quercetin is more effective than cromolyn in blocking human mast cell cytokine release and inhibits contact dermatitis and photosensitivity in humans. PloS One. 2012;7:e33805. doi: 10.1371/journal.pone.0033805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pohjala L, Tammela P. Aggregating behavior of phenolic compounds–a source of false bioassay results? Mol Basel Switz. 2012;17:10774–10790. doi: 10.3390/molecules170910774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baell JB. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS) J Nat Prod. 2016;79:616–628. doi: 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- 46.Miles SL, McFarland M, Niles RM. Molecular and physiological actions of quercetin: need for clinical trials to assess its benefits in human disease. Nutr Rev. 2014;72:720–734. doi: 10.1111/nure.12152. [DOI] [PubMed] [Google Scholar]
- 47.McGovern SL, Helfand BT, Feng B, Shoichet BK. A Specific Mechanism of Nonspecific Inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 48.Coan KED, Maltby DA, Burlingame AL, Shoichet BK. Promiscuous Aggregate-Based Inhibitors Promote Enzyme Unfolding. J Med Chem. 2009;52:2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGovern SL, Shoichet BK. Kinase Inhibitors: Not Just for Kinases Anymore. J Med Chem. 2003;46:1478–1483. doi: 10.1021/jm020427b. [DOI] [PubMed] [Google Scholar]
- 50.Weissig V, Lasch J, Erdos G, Meyer HW, Rowe TC, Hughes J. DQAsomes: a novel potential drug and gene delivery system made from Dequalinium. Pharm Res. 1998;15:334–337. doi: 10.1023/a:1011991307631. [DOI] [PubMed] [Google Scholar]
- 51.pmhdev, Suramin (Injection route), PubMed Health. 2017 https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0045224/ accessed February 11, 2017.
- 52.Zhang YL, Keng YF, Zhao Y, Wu L, Zhang ZY. Suramin Is an Active Site-directed, Reversible, and Tight-binding Inhibitor of Protein-tyrosine Phosphatases. J Biol Chem. 1998;273:12281–12287. doi: 10.1074/jbc.273.20.12281. [DOI] [PubMed] [Google Scholar]
- 53.De Clercq E. Suramin in the treatment of AIDS: Mechanism of action. Antiviral Res. 1987;7:1–10. doi: 10.1016/0166-3542(87)90034-9. [DOI] [PubMed] [Google Scholar]
- 54.Zhou W, Wang Y, Xie J, Geraghty RJ. A fluorescence-based high-throughput assay to identify inhibitors of tyrosylprotein sulfotransferase activity. Biochem Biophys Res Commun. 2017;482:1207–1212. doi: 10.1016/j.bbrc.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 55.Krüger EA, Figg WD. Protein Binding Alters the Activity of Suramin, Carboxyamidotriazole, and UCN-01 in an ex Vivo Rat Aortic Ring Angiogenesis Assay. Clin Cancer Res. 2001;7:1867–1872. [PubMed] [Google Scholar]
- 56.Bruijn EAD, Pattyn G, Denis L, Mahler C, Oosterom ATV, Desmedt E. Therapeutic Drug Monitoring of Suramin and Protein Binding. J Liq Chromatogr. 1991;14:3719–3733. doi: 10.1080/01483919108049489. [DOI] [Google Scholar]
- 57.Cha JH, Dure LS, IV, Sakurai SY, Penney JB, Young AB. 2,4,5-Trihydroxyphenylalanine (6-hydroxy-dopa) displaces [3H]AMPA binding in rat striatum. Neurosci Lett. 1991;132:55–58. doi: 10.1016/0304-3940(91)90432-s. [DOI] [PubMed] [Google Scholar]
- 58.Rosenberg PA, Loring R, Xie Y, Zaleskas V, Aizenman E. 2,4,5-trihydroxyphenylalanine in solution forms a non-N-methyl-D-aspartate glutamatergic agonist and neurotoxin. Proc Natl Acad Sci U S A. 1991;88:4865–4869. doi: 10.1073/pnas.88.11.4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 60.Lange BM, Wildung MR, McCaskill D, Croteau R. A family of transketolases that directs isoprenoid biosynthesis via a mevalonate-independent pathway. Proc Natl Acad Sci U S A. 1998;95:2100–2104. doi: 10.1073/pnas.95.5.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Julliard JH, Douce R. Biosynthesis of the thiazole moiety of thiamin (vitamin B1) in higher plant chloroplasts. Proc Natl Acad Sci. 1991;88:2042–2045. doi: 10.1073/pnas.88.6.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Julliard JH. Biosynthesis of the pyridoxal ring (vitamin B6) in higher plant chloroplasts and its relationship with the biosynthesis of the thiazole ring (vitamin B1) Comptes Rendus Académie Sci Sér 3 Sci Vie. 1992;314:285–290. [Google Scholar]
- 63.Hill RE, Sayer BG, Spenser ID. Biosynthesis of vitamin B6: incorporation of D-1-deoxyxylulose. J Am Chem Soc. 1989;111:1916–1917. doi: 10.1021/ja00187a076. [DOI] [Google Scholar]
- 64.Hill RE, Himmeldirk K, Kennedy IA, Pauloski RM, Sayer BG, Wolf E, Spenser ID. The Biogenetic Anatomy of Vitamin B6 A 13CC NMR Investigation of the Biosynthesis of Pyridoxol in Escherichia coil. J Biol Chem. 1996;271:30426–30435. doi: 10.1074/jbc.271.48.30426. [DOI] [PubMed] [Google Scholar]
- 65.Andrew DTF, Koppisch T. E. coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry (Mosc) 2002;41:236–43. doi: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- 66.Rohdich F, Wungsintaweekul J, Fellermeier M, Sagner S, Herz S, Kis K, Eisenreich W, Bacher A, Zenk MH. Cytidine 5′-triphosphate-dependent biosynthesis of isoprenoids: YgbP protein of Escherichia coli catalyzes the formation of 4-diphosphocytidyl-2-C-methylerythritol. Proc Natl Acad Sci U S A. 1999;96:11758–11763. doi: 10.1073/pnas.96.21.11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuzuyama T, Takagi M, Kaneda K, Dairi T, Seto H. Formation of 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol from 2-C-methyl-d-erythritol 4-phosphate by 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase, a new enzyme in the nonmevalonate pathway. Tetrahedron Lett. 2000;41:703–706. doi: 10.1016/S0040-4039(99)02143-7. [DOI] [Google Scholar]
- 68.Kuzuyama T, Takagi M, Kaneda K, Watanabe H, Dairi T, Seto H. Studies on the nonmevalonate pathway: conversion of 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol to its 2-phospho derivative by 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol kinase. Tetrahedron Lett. 2000;41:2925–2928. doi: 10.1016/S0040-4039(00)00295-1. [DOI] [Google Scholar]
- 69.Lüttgen H, Rohdich F, Herz S, Wungsintaweekul J, Hecht S, Schuhr CA, Fellermeier M, Sagner S, Zenk MH, Bacher A, Eisenreich W. Biosynthesis of terpenoids: YchB protein of Escherichia coli phosphorylates the 2-hydroxy group of 4-diphosphocytidyl-2C-methyl-d-erythritol. Proc Natl Acad Sci. 2000;97:1062–1067. doi: 10.1073/pnas.97.3.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herz S, Wungsintaweekul J, Schuhr CA, Hecht S, Lüttgen H, Sagner S, Fellermeier M, Eisenreich W, Zenk MH, Bacher A, Rohdich F. Biosynthesis of terpenoids: YgbB protein converts 4-diphosphocytidyl-2C-methyl-d-erythritol 2-phosphate to 2C-methyl-d-erythritol 2,4-cyclodiphosphate. Proc Natl Acad Sci. 2000;97:2486–2490. doi: 10.1073/pnas.040554697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Altincicek B, Kollas AK, Eberl M, Wiesner J, Sanderbrand S, Hintz M, Beck E, Jomaa H. LytB, a novel gene of the 2-C-methyl-D-erythritol 4-phosphate pathway of isoprenoid biosynthesis in Escherichia coli. FEBS Lett. 2001;499:37–40. doi: 10.1016/S0014-5793-(01)02516-9. [DOI] [PubMed] [Google Scholar]
- 72.Campos N, Rodr໑guez-Concepción M, Seemann M, Rohmer M, Boronat A. Identification of gcpE as a novel gene of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis in Escherichia coli. FEBS Lett. 2001;488:170–173. doi: 10.1016/S0014-5793(00)02420-0. [DOI] [PubMed] [Google Scholar]
- 73.Hecht S, Eisenreich W, Adam P, Amslinger S, Kis K, Bacher A, Arigoni D, Rohdich F. Studies on the nonmevalonate pathway to terpenes: The role of the GcpE (IspG) protein. Proc Natl Acad Sci. 2001;98:14837–14842. doi: 10.1073/pnas.201399298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kollas AK, Duin EC, Eberl M, Altincicek B, Hintz M, Reichenberg A, Henschker D, Henne A, Steinbrecher I, Ostrovsky DN, Hedderich R, Beck E, Jomaa H, Wiesner J. Functional characterization of GcpE, an essential enzyme of the non-mevalonate pathway of isoprenoid biosynthesis. FEBS Lett. 2002;532:432–436. doi: 10.1016/S0014-5793(02)03725-0. [DOI] [PubMed] [Google Scholar]
- 75.Rohdich F, Zepeck F, Adam P, Hecht S, Kaiser J, Laupitz R, Gräwert T, Amslinger S, Eisenreich W, Bacher A, Arigoni D. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: Studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proc Natl Acad Sci. 2003;100:1586–1591. doi: 10.1073/pnas.0337742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rohdich F, Bacher A, Eisenreich W. Isoprenoid biosynthetic pathways as anti-infective drug targets. Biochem Soc Trans. 2005;33:785. doi: 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- 77.Altincicek B, Kollas AK, Sanderbrand S, Wiesner J, Hintz M, Beck E, Jomaa H. GcpE Is Involved in the 2-C-Methyl-d-Erythritol 4-Phosphate Pathway of Isoprenoid Biosynthesis in Escherichia coli. J Bacteriol. 2001;183:2411–2416. doi: 10.1128/JB.183.8.2411-2416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cunningham FX, Lafond TP, Gantt E. Evidence of a Role for LytB in the Nonmevalonate Pathway of Isoprenoid Biosynthesis. J Bacteriol. 2000;182:5841–5848. doi: 10.1128/JB.182.20.5841-5848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altincicek B, Duin EC, Reichenberg A, Hedderich R, Kollas AK, Hintz M, Wagner S, Wiesner J, Beck E, Jomaa H. LytB protein catalyzes the terminal step of the 2-C-methyl-D-erythritol-4-phosphate pathway of isoprenoid biosynthesis. FEBS Lett. 2002;532:437–440. doi: 10.1016/S0014-5793(02)03726-2. [DOI] [PubMed] [Google Scholar]
- 80.McAteer S, Coulson A, McLennan N, Masters M. The lytB Gene of Escherichia coli Is Essential and Specifies a Product Needed for Isoprenoid Biosynthesis. J Bacteriol. 2001;183:7403–7407. doi: 10.1128/JB.183.24.7403-7407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rohdich F, Hecht S, Gärtner K, Adam P, Krieger C, Amslinger S, Arigoni D, Bacher A, Eisenreich W. Studies on the nonmevalonate terpene biosynthetic pathway: Metabolic role of IspH (LytB) protein. Proc Natl Acad Sci. 2002;99:1158–1163. doi: 10.1073/pnas.032658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adam P, Hecht S, Eisenreich W, Kaiser J, Gräwert T, Arigoni D, Bacher A, Rohdich F. Biosynthesis of terpenes: Studies on 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase. Proc Natl Acad Sci. 2002;99:12108–12113. doi: 10.1073/pnas.182412599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.J PM, G PN. Mechanism of action of beta-oxoacyl-CoA thiolase from rat liver cytosol. Direct evidence for the order of addition of the two acetyl-CoA molecules during the formation of acetoacetyl-CoA. 1983 doi: 10.1042/bj2130153. http://www.biochemj.org/bj/213/bj2130153.htm accessed August 15, 2013. [DOI] [PMC free article] [PubMed]
- 84.Miziorko HM, Lane MD. 3-Hydroxy-3-methylgutaryl-CoA synthase. Participation of acetyl-S-enzyme and enzyme-S-hydroxymethylgutaryl-SCoA intermediates in the reaction. J Biol Chem. 1977;252:1414–1420. [PubMed] [Google Scholar]
- 85.Amdur BH, Rilling H, Bloch K. The enzymatic conversion of mevalonic acid to squalene. J Am Chem Soc. 1957;79:2646–2647. doi: 10.1021/ja01567a077. [DOI] [Google Scholar]
- 86.Bloch K, Chaykin S, Phillips AH, De Waard A. Mevalonic acid pyrophosphate and isopentenylpyrophosphate. J Biol Chem. 1959;234:2595–2604. [PubMed] [Google Scholar]
- 87.Dhe-Paganon S, Magrath J, Abeles RH. Mechanism of Mevalonate Pyrophosphate Decarboxylase: Evidence for a Carbocationic Transition State. Biochemistry (Mosc) 1994;33:13355–13362. doi: 10.1021/bi00249a023. [DOI] [PubMed] [Google Scholar]
- 88.Agranoff BW, Eggerer H, Henning U, Lynen F. Isopentenol pyrophosphate isomerase. J Am Chem Soc. 1959;81:1254–1255. doi: 10.1021/ja01514a059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Primary and Secondary LOPAC1280 Screening Data. A) Primary Screen of the LOPAC1280 Library with YpIspC and FtIspD. B) Secondary Screen of MtbIspC with lead compounds from the primary screen of LOPAC1280. C) Secondary Screen of EcIspD with lead compounds from the primary screen of LOPAC1280
Supplementary Figure 2. Half-maximal effective concentration of LOPAC1280 hit compounds against F. tularensis subsp novicida Utah 112. Percent residual growth is calculated as the ratio of cell density (OD600) in the presence of inhibitor to cell density in the presence of vehicle (DMSO) only for inhibitors A) GW5074, B) PPT, C) Rottlerin, D) Sanguinarine chloride, E) Chelerythrine chloride and F) Dequalinium chloride. The EC50 values and R2 values for each plot are indicated. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 3: Half-maximal effective concentration of LOPAC1280 hit compounds against Y. pestis subsp A1122. Percent residual growth is calculated as the ratio of cell density (OD600) in the presence of inhibitor to cell density in the presence of vehicle (DMSO) only for inhibitors A) GW5074, B) PPT, C) Rottlerin, D) Sanguinarine, E) Chelerythrine chloride, and F) Dequalinium chloride. Neither chelerythrine chloride nor dequalinium chloride were active against Y.pestis up to 100 μM. The EC50 values and R2 values for each plot are indicated. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 4. Half-maximal effective concentration of quercetin against liquid cultures of A) Y. pestis subsp. A1122 and B) F. tularensis subsp. novicida Utah 112. Percent residual growth is calculated as the ratio of culture cell density (OD600) in the presence of quercetin to culture cell density in the presence of vehicle (DMSO) alone. The potency (EC50) and R2 values for each plot are indicated. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 5. Dose-response plots of select inhibitors of YpIspC or FtIspD. Assays were performed with each enzyme with various concentrations of inhibitors including A) GW5074 with YpIspC, B) PPT with YpIspC, C) Rottlerin with YpIspC, D) Sanguinarine with YpIspC, E) Chelerythrine chloride with FtIspD, F) Dequalinium chloride with FtIspD, G) Quercetin with YpIspC and H) Quercetin with FtIspD For each assay, after a ten minute incubation of the enzyme with inhibitor and cofactor, the substrate was added to initiate the reaction. The IC50 and goodness-of-fit (R2) value for each plot is indicated. The enzymatic activity is relative to an uninhibited control using vehicle (DMSO) alone. All assays were performed in duplicate. Error bars are calculated as the deviation from the mean of two independent measurements.
Supplementary Figure 6. Mode of inhibition by sanguinarine. All assays were performed in duplicate using purified YpIspC. Error bars are calculated as the deviation from the mean of two independent measurements A) The Lineweaver–Burk plot indicates that sanguinarine is noncompetitive with respect to DXP. B) The Lineweaver-Burk plot indicates that sanguinarine is noncompetitive with respect to NADPH. C) Dependence of IC50 with sanguinarine on enzyme concentration. IC50 with sanguinarine decreases with a decrease in YpIspC concentration. Concentrations of YpIspC, IC50, and Hill slope are indicated. D) The dependence of IC50 value and Hill slope of each IC50 plot decreased linearly with decrease in YpIspC concentration.
Supplementary Figure 7. Characterization of suramin and 6-hydroxy-DL-DOPA. All assays were performed in duplicate. Error bars are calculated as the deviation from the mean of two independent measurements. A) IC50 of suramin with YpIspC. The IC50 with suramin does not significantly decreases with a decrease in YpIspC concentration, nor does the Hill slope decrease. B) Structure of suramin. C) The Lineweaver–Burk plot indicates that suramin is noncompetitive with respect to DXP. D) The Lineweaver-Burk plot indicates that suramin is noncompetitive with respect to NADPH. E) IC50 of 6-hydroxy-DL-DOPA with FtIspD. F) Structure of 6-hydroxy-DL-DOPA. C) The Lineweaver–Burk plot indicates that 6-hydroxy-DL-DOPA is competitive with respect to CTP. D) The Lineweaver-Burk plot indicates that 6-hydroxy-DL-DOPA is noncompetitive with respect to MEP.