

Abstract
Population stratification is a useful approach towards a better understanding of complex biological problems in human health and well-being. The proposal that such stratification applies to the human gut microbiome, in the form of distinct community composition types, termed “enterotypes”, was met with both excitement and controversy. In view of accumulated data and re-analyses since the original work, we revisit the enterotype concept, discuss different methods of dividing up the landscape of possible microbiome configurations, and put these concepts into a functional, ecological and medical context. As enterotypes are of use in describing the gut microbial community landscape and may become relevant in clinical practice, we aim to reconcile differing views and encourage a balanced application of the concept.
The human body is colonized by trillions of microbes that contribute to our health and well-being. Different communities of microbes inhabit various anatomical regions (Fig. 1). Inter-individual variation at each of these body sites is considerable, but the separation among sites within individuals remains apparent1 (Fig. 1). The most densely populated habitat is the gut, with an estimated 0.15 kg of microbial biomass2. The gut harbors hundreds of bacterial and archaeal species, with Firmicutes and Bacteroidetes as dominant phyla1,3–5. Considerable variation in microbiota composition has been described among individuals, for example in the US NIH Human Microbiome Project (HMP)1, the European Metagenomics of the Human Intestinal Tract project (MetaHIT)3,6 and multiple other population studies7,8. The gut microbial ecosystem shows a succession of different microbiota stages: community composition changes rapidly in early childhood, stabilizes in adults and deteriorates in old age8,9. There is nosimple description of this complex landscape across large populations and geographies, in part because some tax a vary monotonically among individuals while most others show bimodal or more complex distributions10 (Fig. 2A). Given the importance and complexity of the gut ecosystem, there is great interest in identifying compositional patterns and their underlying rules, as they may help us understand human health and disease states. A classification based on compositional patterns would potentiate microbiota-based diagnostics, therapies or prevention of disease, with implications for personalized treatment through nutritional, microbial, and pharmaceutical interventions. Such patterns of microbial composition could be used to stratify populations, similar to the molecular subtyping commonly used in cancer research, where, for example, breast cancer subclasses based on gene expression patterns are clinically relevant11,12. However, in other cases, like colorectal cancer, the determination and usefulness of such classifications remains unclear13, highlighting the fact that molecular stratification is notactionable in all situations.
Figure 1. The microbiota of distinct body locations within the healthy human is separable at the genus level.
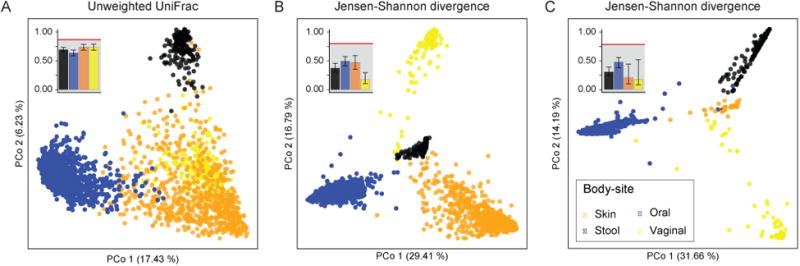
Using 2381 HMP samples profiled with 16S rRNA, we illustrate the degree of separation between body-sites using different distance measures and taxonomic resolutions: (A) unweighted UniFrac on OTU level, (B) Jensen-Shannon divergence on genus level (OTUs belonging to the same genus are added up together) and (C) Jensen-Shannon divergence on OTU level. Shown are the first two principal coordinates of a PCoA analysis for each, as well as a summary of the within and between body-site distances in the top left. Median inter-sample distances (error bars ranging from the 25th to the 75th quantile) compared to the median between all body-sites (red line) illustrate the ability to capture similarities and differences between these biomes, albeit with different effectiveness. We note that the Silhouette Index (a measure of clustering strength) in the case of unweighted UniFrac suggests a clustering into only three types, with an absolute value of ∼0.2 (Suppl. Fig.4).
Figure 2. Stratification of the microbial composition landscape of the human gut microbiome.
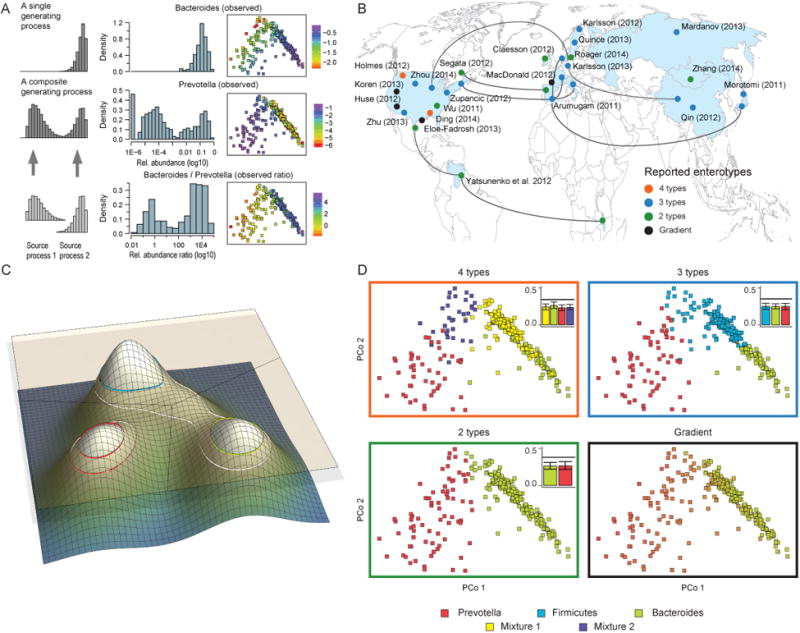
(A) Abundance distributions of prevalent microbial genera of the human gut are often complex. Theoretical beta distributions (left panels) were compared with observed distributions (middle panel) and the observed abundance plotted in enterotype space (right panel) of key enterotype tax a or ratios thereof, based on 278 MetaHIT samples6. While Bacteroides abundance distribution is close to log-normal in the three large-scale datasets studied, that of Prevotella is bimodal, suggesting that the observed values are perhaps better explained by a mixture of two distributions, generated by two distinct processes, one of which corresponds to a dominating role in the community, while the other to a low abundance state.
(B) Geographical distribution of studies that report enterotypes (Suppl. Table 1), colored according to the number of microbial clusters reported. Map locations indicate the country from which samples were collected. Links between locations represent samples belonging to a single study. Overrepresentation of “Western” countries is a well-known bias and probably misses a portion of variation in other human societies.
(C) Schematic representation of the simulated microbial composition landscape with three density peaks, modeled as multivariate normal distributions, each representing an enterotype and drawn out of scale to make the concept more accessible. This figure illustrates how segmentation of this space by clustering with different parameters would result indifferent numbers of clusters (three and two here) and in differential coverage of individuals (represented by intersecting planes). Top-most overlay presents the discretizing segmentation, which splits the space into three zones.
(D) Projection onto a set of 278 Danish samples6 of the three most frequent enterotype classification schemes based on different methods, including the Prevotella/Bacteroides gradient. This shows a split into a gradient and two, three (distance based clustering) or four enterotypes (Dirichlet multinomial mixture models). The local structure is preserved regardless of the method applied, and Prevotella (ET P) remains separated, suggesting the methods mostly differ in dividing the area between ET B and ET F. Additionally, the top right of each PCoA with a number of clusters greater than or equal to two shows the distance within a cluster (colored accordingly) compared to the median distance between the clusters (black line), showing that for all cases the distances within are smaller than between; bar height is the median distance and the whiskers represent the 25th and 75th quantile. It should be noted that a “horseshoe effect” can occur in ordinations, in particular if samples contain non-overlapping compositions84, which is not the case in the datasets analyzed here.
Reproducible patterns of variation in the microbiota, like the proportions of major tax a such as Bacteroides and Prevotella, have been observed in the adult human gut (Fig. 2A, Suppl. Fig. 1). When separated into clusters, they have been termed “enterotypes” 14 and proposed as a useful method to stratify human gut microbiomes. Later, other studies found stratification in other ecosystem types, such as the vagina15and other body sites16–18. However, due to the nature of the clustering in the gut, the number, or even existence of different community types has been a topic of heated debate after the publication of the original approach14.
Here we assess gut microbial community composition and test the different hypotheses using three of the largest available metagenomic datasets, which include data from three continents (from HMP, MetaHIT and a Chinese type II diabetes study)1,6,19. We perform a refined meta-analysis and propose a modified concept of enterotypes, with the goal of reconciling divergent viewpoints. The results illustrate the advantages and disadvantages of clustering and other stratification approaches. We find that the gut microbial composition is structured and that clustering can provide useful insights in to some microbiome datasets, even when not strongly supported statistically. This approach does not diminish the need to pursue other analyses and avenues for interpretation, since broad community-wide stratification captures only some of the dimensions of microbiota complexity.
Recurrent microbial compositional patterns in the gut microbiome
From the survey of the three large datasets mentioned above, it can be seen that groups of samples tend towards preferred genus level composition (Suppl. Fig. 2), as was also reported in the original study14. That is, some configurations of relative microbial abundance occur more frequently than others. This can be observed by calculating distances between samples and investigating the resulting clustering, as well as by directly observing the complex abundance distributions of some gut microbial tax a (Fig. 2). This preference for specific microbial community profiles is modest, resulting in higher sample density around the preferred constellations, but with a considerable proportion of samples falling between them. This makes it hard to describe these preferential microbial compositions mathematically or determine the number of such densely populated areas, prompting alternative description of this space as consisting of gradients16. However, it is important to characterize these local optima of community composition to understand the mechanisms responsible for these ecological constraints and community properties.
In 2011, clustering human fecal metagenomic samples from three continents, using three sequencing technologies (Illumina, 454, Sanger), as well as 16S rRNA gene profiling data, based on their taxonomic composition, resulted in the proposal of three enterotypes. They were described as being “densely populated areas in a multidimensional space of community composition”, and were independent of age, gender, cultural background and geography14.An investigation of the properties of each enterotype found networks of co-occurring microbes centered around one indicator (driver) tax on, that is the tax on correlating best to that given enterotype: enterotype 1, here denoted ET B for clarity, has Bacteroides as its best indicator; enterotype 2, here ET P, is driven by Prevotella, a genus whose abundance is inversely correlated with Bacteroides; and enterotype 3, here ET F, is distinguished by an overrepresentation of Firmicutes, most prominently Ruminococcus14. Analyses were performed at genus level, where microbial ecological niches are hypothesized to be most clearly reflected20, notwithstanding functional heterogeneity of some genera (e.g. streptococci, grouping deadly pathogens with common commensals and useful food fermenting species). Species- and strain-level variations are neglected, although they can contribute to functional differences between individuals that are important in a clinical context21,22.
Although much of the discussion emphasized the existence of three enterotypes, the original definition had made clear that they are not discrete, and that clustering is just one way to define them and stratify samples to reduce complexity (see Fig. 3A comparing clustering and genera abundances). There are limitations to this operational definition, and although the resulting stratification only partially reflects the more complex structure within the population space, the definition has been used to demonstrate that such stratification can be useful in analyzing microbiome data.
Figure 3. Microbiota of human fecal samples has local substructure.
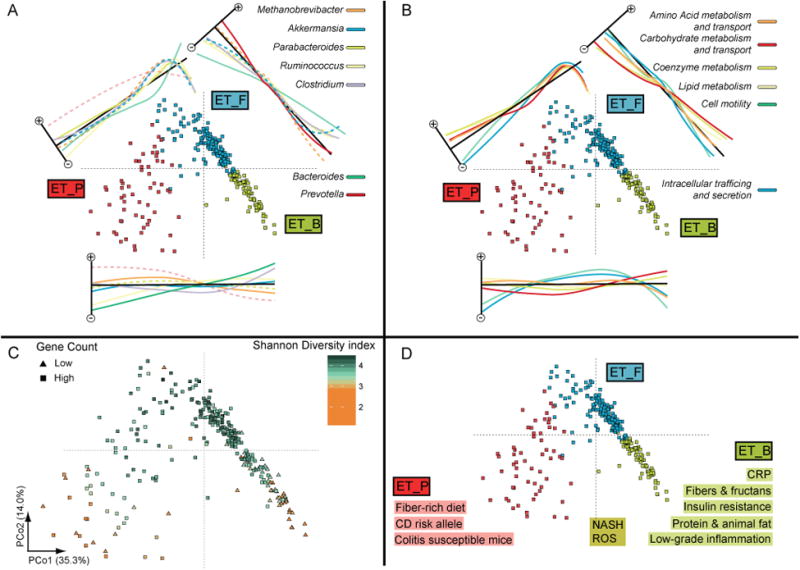
Ordination of 278 MetaHIT6 samples on Jensen-Shannon distance transformed space. For orientation, a three-enterotype model is illustrated by color in A, B and D. (A) The log-transformed relative abundance of the most significantly differing genera. On the adjacent axis, the projected abundance changes between the respective community types are shown. Bimodal abundance profiles (dotted lines, dip test p-value < 0.05) as well as gradual abundance changes (solid lines) can be identified, supporting a gradient or cluster model, respectively. (B) Abundance changes of selected COG categories were projected onto the ordination, illustrating that functional composition differs between enterotypes. (C) mOTU level Shannon diversity index and gene richness (low gene count is considered for subjects with less than 480k genes according to 6; all other subjects have high gene count) are significantly different between enterotypes (Suppl. Fig. 8), mostly following gradual changes over the whole enterotype space. (D) Summary of the diseases and dietary constituents that have been associated with Prevotella, Firmicutes or Bacteroides enrichedgut communities (Suppl. Table 5).Acronyms: CD: Crohn's disease, CRP: C-reactive protein, NASH: Nonalcoholic Steatohepatitis, ROS: reactive oxygen species.
Some later studies replicate dentero types in new datasets to different extents, both in the numbers of enterotypes and the strength of the statistical support; others reported finding no structure (Fig. 2, Suppl. Table 1). For example, a large scale, diet-focused study in a U.S. cohort23reported support for two attractors, one of which shared similar dominant tax a with ET P, while the other was a merge of ETs F and B. Analysis of the HMP 16S rRNA data18, a meta-analysis of four metagenomics datasets24 and a population-wide Flemish study7 showed a preference for three enterotypes, similar to the originally proposed ones. A study of individuals from Venezuelan and Malawian rural areas and US metropolitan areas emphasized the importance of Prevotella and Bacteroides as driving tax a, as well as a strikingly different composition in infants, with their communities mostly containing Bifidobacteria and Proteobacteria8. The establishment of an enterotype-like structure has been estimated to occur between month 9 and 36 in humans25, highlighting the need for caution when extrapolating overall community patterns from a limited sampling of the world population at different ages.
Departing from the clustering approach, Holmes et al. propose an alternate approach to identify structure26. Their method identifies a generative model for each possible state and determines how each explains the observed data, focusing on the actual genera abundances rather than the distances. Using this approach (i.e. Dirichlet multinomial mixture models; DMMs) they reported that the data from the original study most likely results from four generative processes (loosely referred to here as clusters). Two of the “clusters” resembled ET B and P, while a third showed an increased prevalence of Ruminococcus and other Firmicutes genera, which are usually low lyabundant in the gut microbiome. The last cluster had a high fraction of unidentified tax a. DMMs have also been used to identify three optimal clusters in a healthy Swedish cohort, again showing compositions similar to ET B and ET P, with one additional cluster dominated by unknown taxa27. A further study that applied the same method to the HMP 16S rRNA data, found that the gut microbiome is best approximated with four similar models17. When applying DMMs to the Meta HIT metagenomics dataset, we identified four groups. Two of these are overlapping with enterotypes B and P, while the other two are a more complex mixture (Fig. 2, Suppl. Fig.3). While DMMs represent a statistically more rigorous approach, further research is needed to determine if the distributional assumptions of generative models hold on microbiome data.
The three dominant gut tax a that contribute to enterotype clustering (Prevotella, Bacteroides and Ruminococcaceae) have been shown to have the largest variance in terms or relative abundance, despite being core taxa7. Therefore, it is not surprising that an ensemble-based network approach recovered them as hubs of three co-occurrence network clusters and showed that their abundances are mutually negatively correlated (Ref. 1 and Suppl. Fig. 5 therein). This negative correlation was also shown with qPCR data of 35 signature taxa28. Three distinct networks were found in adult Amish individuals29, with the dominant genera in these networks largely overlapping with the driver tax a of the original enterotypes. Similarly, six species co-abundance groups (CAGs) were reported in a dataset consisting of Irish adults and elderly individuals, with healthy hosts mostly possessing networks that correspond to the original three enterotypes9,30. Thus, independent of clustering and modeling approaches, bacterial co-abundance networks provide a species network that may underline the fundamental properties of these preferred community profiles. Theoretical studies show that enterotype-like structures can be an emerging feature of communities over a wide range of species interaction strengths31.
Enterotype-like structures have also been reported in several animal studies, although their gut composition is distinct from that of humans. In mouse gut microbiomes, obtained from hosts living under controlled experimental conditions32, clustering showed a clear compositional stratification while in animals living in the wild (mice33, primates34,35 and pigs36,37) clustering was considerably weaker. This is suggestive of preferred community states emerging more clearly when no external factors influence the microbiome. Thus the enterotype concept is not anthropocentric and can be defined in animals as well38, which has led to the speculation that enterotypes have existed before the pan-human split39. As gut commensals are mostly evolving in competition with each other, under the restraints of the host organism40, enterotypes might thus represent optimized states of symbiont compositions, that represent local optima in community effectiveness, that are still compatible with the restraints imposed by the host.
Challenges in defining microbial community types
Assessing clustering in fecal microbiota profiles is non-trivial, given that demonstration of alternate states is debated in disciplines from ecology to philosophy16,41. Given the nature of enterotype clustering, and additional factors including multiple choices for taxonomic levels, distance metrics, clustering algorithms and cluster optimality scores, it is not surprising that analysis can yield different numbers of clusters (Suppl. Fig. 4 and Supplementary Material), even on the same dataset (e.g. 16). Thus some have argued that there is little support for enterotypes in the data8,16,23,29,42,43. However, separating samples by body-sites(skin, stool, vaginal, and oral)using the same methods also has little statistical support (Suppl. Fig. 4), even though this separation is widely accepted in the scientific community.
Regardless of clustering support or modelling assumptions (Supplementary information), an analysis of the largest three public datasets, backed by reports from the literature (Suppl. Table 1), reveals that the local substructure is always similar, i.e., a three-cluster model finds Bacteroides, Prevotella and Firmicutes-dominated clusters, and a two-cluster model separates Prevotella-driven samples from the rest. Partitioning of the gut microbiota is thus stable in the sense that related cluster compositions are recovered, reconciling many studies and supporting the existence of preferred community compositions.
There is certainly agreement that there are distinct areas within the complex microbial composition landscape in which the respective gut communities show biological differences44. The concept of enterotypes can help capture such differences, although defining meaningful and robust boundaries remains a challenge. This is analogous to clustering of macro-biomes, which faces similar problems despite the recognition of separate types of environments. For example, Treeless, Savannah, and Forest ecosystems in sub-Saharan Africa could equally be represented as a gradient in response to mean precipitation45 or as contrasting stable states46.
Given the practical challenges in accurately determining gut community structure, such as overcoming batch effects, considering confounders (Suppl. Fig. 5) and accounting for temporal variation, an objective number of stable states is difficult to determine. Still, in the (mostly Western) subjects studied cross-sectionally, Bacteroides and Prevotella act as the driving tax a that explain inter-individual differences, and delineate the main sources of variation regardless of the technique employed. The extremes of the enterotypes pace are substantially different in microbial composition and diversity, and these are discussed in the following sections in terms of their function, ecology and disease. While three enterotypes may not always be the best explanation of the data, it is the model that has been used most and that provides a framework that we use below.
Functional and ecological context of enterotypes
Differences in taxonomic composition suggest that enterotypes may differ in functional and ecological properties. Analysis of the three large datasets revealed significant functional variation associated with microbial composition (Fig. 3B).Indeed, when gene types are considered, most KEGG Orthologs (KOs)47 and Non-supervised Orthologous Groups (NOGs)48 differ in abundance among the three enterotypes (64% and 77%, respectively, FDR<0.1). The same is true for egg NOG functional categories, where 23 out of 25 are significantly different (Suppl. Fig. 6). Other models choosing two or four enterotypes show similar broad functional differences (Suppl. Table 3), with some differences highly relevant to gut metabolism. For example, it has been shown on several occasions that either ET P23,29,49,50 or Prevotella (when no enterotype was reported) was enriched in individuals with non-Western and/or fiber-rich diets8,51–53. This association can be better understood in light of functional differences, as Prevotella hydrolases are specialized in the degradation of plant fibers54 and an overall decreased lipolytic and proteolytic fermentation potential has been reported for the whole ET P community44. Conversely, ET B has been associated with diets enriched in animal proteins and saturated fats23,53, in line with a large proportion of Bacteroides-specific CAZyme's (50%)55 being specialized on animal carbohydrates (Suppl. Table 2). Further, we find enzymes specific to carbohydrate metabolism overrepresented in ET B (Suppl. Table 3), corroborating recent research showing an increased saccharolytic as well as proteolytic potential44. While some of the functional differences between enterotypes can be attributed to the driver genera, others emerge only after imposing structure on the variation space.
The observed functional differences between enterotypes support the notion that they have varying community properties, such as richness, diversity and temporal stability. Such characteristics are relevant from an ecological perspective, where theory predicts a higher diversity in dynamic systems like the gut, with nutrient availability and type fluctuating over time56. Using 16S amplicon sequencing, richness differences between three enterotypes were first shown in an Amish population29, with a cluster similar to ET B having the lowest richness, as has been recently confirmed in a large population-wide study7. Our analysis of the three large datasets used here replicated these differences, with ET B having the lowest and ET F having the highest taxonomic as well as functional richness (Suppl. Fig.7 and 8). Community diversity, as measured by the Shannon diversity index, is also highest in ET F in all datasets, while ET B and ET P are similarly decreased in diversity (Suppl. Fig. 8). Moreover, such differences go hand in hand with differences in stool consistency57 and/or transit time58– with slow transit associated ETs also showing a higher relative ratio of proteolytic over saccharolytic potential44 and proteolisis-derived metabolites58.
Gut community composition in healthy adults in many studiesdoes not change substantially over long time periods23,29,59, indicative of agenerally stable ecosystem and enterotype stability. There are, however, important exceptions. Our analysis of the HMP metagenomic time-series dataset, containing individuals sampled more than 6 months apart, reveals significant stability in all three enterotypes, though 16% of individuals switched putative enterotypes between visits (Suppl. Fig. 9). This suggests that, at least for some individuals, gut microbial types are relatively fluid and do not have discrete boundaries (Suppl. Fig. 9). These observations could be explained through alternate models of gut community dynamics: a) the existence of preferred community compositions (i.e., enterotypes, Suppl. Fig. 10A) or b) individual-specific attractors that exist mostly due to temporal autocorrelation of that individual's gut community60 (Suppl. Fig. 10B). To disentangle these models requires information about the response of the microbial community to different perturbations there by allowing us to determine if individuals are more likely to maintain/return to their original composition or maintain/return to their enterotype. Unfortunately, only limited data on gut community perturbations are available—e.g., antibiotics, fecal microbiota transplantation (FMT) and diet—many of which were not considered in the enterotype frame work, making it difficult to draw conclusions about which steady-state model is correct. Short-term therapeutic antibiotic treatment was shown to induce substantial, partially recoverable shifts in the gut microbiota of humans61,62, suggesting little resistance to such a dramatic disruption. Indeed, antibiotics treatment canlead to a complete deterioration of the community and subsequent pathogen invasion (e.g., C. difficile63), effectively resulting in failure to recover the original community state.
Dietary interventions, which cause considerably less perturbation to the microbial ecosystem of the human gut, may thus be better suited for investigating community resilience. Effect of such interventions, with significant compositional changes, have been observed within four days and could cause an enterotype shift23,64. However, after about ten days, enterotypes appeared to be stable23, suggesting a tendency of recovering the original state. Stability was also observed in a 6-month intervention, using the ratio of Prevotella to Bacteroides (obtained by qPCR) as a proxy for enterotype assignments28. These results suggest that there are limitations on how much an individual's microbiome may be perturbed by short-term dietary interventions and support enterotype resilience. In contrast, long-term perturbations have a more profound effect, with dietary modulation over the period of a year having a strong impact on the Bacteroidetes/Firmicutes ratio23,65, potentially leading to enterotype switches. As enterotypes were generally stable over time and no follow-up studies exist for the long-term interventions, no approximation of their resilience either in terms of overall community resemblance or enterotype assignmentcan be derived from the available data. There are however indications that enterotypes may vary in their recovery after intervention, with ET F estimated to have the lowest overall bacterial growth rate44, possibly resulting in a delayed return to equilibrium.
Although it is not yet possible to predict how particular perturbations will modify the microbiota, it is possible that different microbiome configurations, including those stratified as enterotypes, might allow stratified treatment and diet recommendations in the future. Modulation of the gut microbiome is particularly relevant for diseases, where the challenge is to shift the microbiome back to a healthy pre-disease state in a given individual.
Clinicalrelevance of enterotypes
A simple classification scheme of gut community structure by enterotypes has the potential to be clinically useful. First, it can help in diagnosis, contributing to the identification of a disease state in an individual. Second, it can serve as an indicator of the risk or susceptibility of developing certain conditions. Third, the stratification may be a useful biomarker for changes that occur upon disease progression. Fourth, given that the gut microbiota influences xenobiotic metabolism, it may be that different enterotypes are associated with different pharmacokinetics and dynamics of drug metabolism66–69. Thus, enterotyping may guide treatment options and help in understanding different treatment responses.
Several associations between enterotypes, or their main taxonomic drivers, and human disease phenotypes have been reported (Fig. 3D). For example, an increase of Bacteroides or ET B itself, which tends towards lower overall diversity (Suppl. Fig. 7&8), has been linked to NASH70, colorectal cancer49,71,72, celiac disease73, immune-senescence and constant low-grade inflammation6,30. Reanalysis of the MetaHIT dataset found lymphocyte counts and C-reactive protein to be significantly increased in ET B compared to ET F (FDR<0.1), with ET F samples on average lower in insulin resistance index (HOMA IR) and insulin levels, (FDR=0.107 for both) (Suppl. Table 4). Increased Prevotella abundance has been linked to long-term antibiotic usage74, rheumatoid arthritis75, type IIdiabetes76 as well as HIV77, although the latter enriched in one of the risk groups that is in men who have sex with men78, which might confound the reported association. Lastly, ET F has been linked to high microbiota diversity and decreased host inflammatory status, and has only been associated with an increased risk of atherosclerosis79. Given the multitude of associations to different disease phenotypes, an enterotype classification by itself may not be sufficiently specific as a stand-alone diagnostic marker of any disease80, but may be able to indicate an increased risk of some. Enterotype associations within groups of healthy individuals at risk of certain conditions are rare, and it remains unclear if enterotype classifications might be useful as prognostics for disease development. In one example, increased prevalence of ET P had been reported in healthy individuals who had the heterozygous form of a Crohn's Disease (CD) risk allele27, while in the MetaHIT cohort, there is a significant enrichment of CD patients in ET B, implying inflammation shifting community states to these two enterotypes and thus indicating an increased risk for IBD.
Finally, it is possible that some diseases will have different etiologies, depending on enterotype. Stratification could allow discovery of these underlying signals, thereby eliminating part of the large variation observed in microbial communities between individuals that may be irrelevant to the disease itself. In one mouse study, for example, such stratification allowed discovery of genotype-microbiome and cage-microbiome associations32. Similarly, stratifying human patients into 8 microbial clusters helped identify medical parameters that correlated with microbial composition30, and microbial stratification significantly improved accuracy in classifying C. difficile-associated diarrhea81. Although there are currently no long-term data, responses to diet and drugs as well as the impact of intestinal physiology and lifestyle are also likely to differ depending on the position of an individual in the compositional landscape, and thus stratification represents an entry point into various clinically relevant areas. It can be implemented largely independently of a gradient- or cluster-centric view, analogous to the body mass index (BMI) where defined cutoffs are an important guide to patient disease risk82.
Towards guidelines for rational enterotyping
For enterotyping to be useful, standardization is essential. In addition to the technical challenges mentioned above, an inherent property of clustering is that assignments of single samples depend on which other samples are analyzed at the same time. An enterotype defined this way makes comparisons across studies difficult. For example, if by chance the majority of samples in a single study are ET B or ET F and only a few are ET P, the optimal cluster score might indicate two or even only one cluster(s). Nevertheless, one might identify these few ET P samples based on the knowledge that similar samples have been clustered in other data sets. Combining data from multiple studies is often challenging, because differences in DNA extraction methods, sample handling, sequencing technology, primer choice (for 16S rRNA gene amplification) and data processing (e.g. 16S rRNA clustering, copy number correction and chimera reduction) influence the proportions of bacteria detected and lead to biases in detecting enterotype clusters83. Extreme rigor is needed in standardizing these steps, perhaps in conjunction with artificial “mock” communities that span a large proportion of the phylogenetic spectrum of microbes found in the gut, and enable comparability between standard and clinical samples. Furthermore, there is a need for more longitudinal studies involving larger population cohorts across multiple continents to identify additional confounding factors. Indeed, several consortia such as IHMS41, MBQC42 and GSC43 are already trying to set standards for metagenomics and identify sources of variation.
We propose a classification procedure that both circumvents many of the problems outlined above as well as providing more comparable results (Fig. 4). While we do not want to limit other explorations of the data or novel analysis options, alternative schemes should at least be compared with the results from the procedure described here. Based on the MetaHIT data6 set, we have trained a classifier at genus level on taxonomic and functional features that recovers putative clustering observed in the Chinese type II diabetes study19 and in the HMP1 data set (Suppl. Fig.11). The classifier is available at [http://enterotypes.org]. If the results of a denovo clustering differ from the classifier results, we recommend caution in directly comparing the stratification outcome to the enterotypes described in this meta-analysis. Moreover, this approach also defines an enterotyping space, by determining which samples are compositionally similar to a reference set. This could be used to define the boundaries of ‘normal’ gut communities and identify individuals outside of them, serving as a health indicator. Unusual disease states have been previously reported: for example, by using a model of six-species communities, networks resembling the three enterotypes were most strongly overrepresented in healthy patients, whereas two new states were overrepresented in frail, elderly patients9. Another case reported in new enterotype H, enriched in Enterobacteriaceae70. The above classifier would consider samples from this “enterotype” compositionally dissimilar to those present in large datasets, and they would thus be labeled as being outside the enterotyping space. The individuals with this unusual composition frequently suffered from obesity, NASH, high blood ethanol as well as reactive oxygen species (ROS) levels70, suggesting this unusual compositional state to be dysbiotic.
Figure 4. Determination of Enterotype structure.
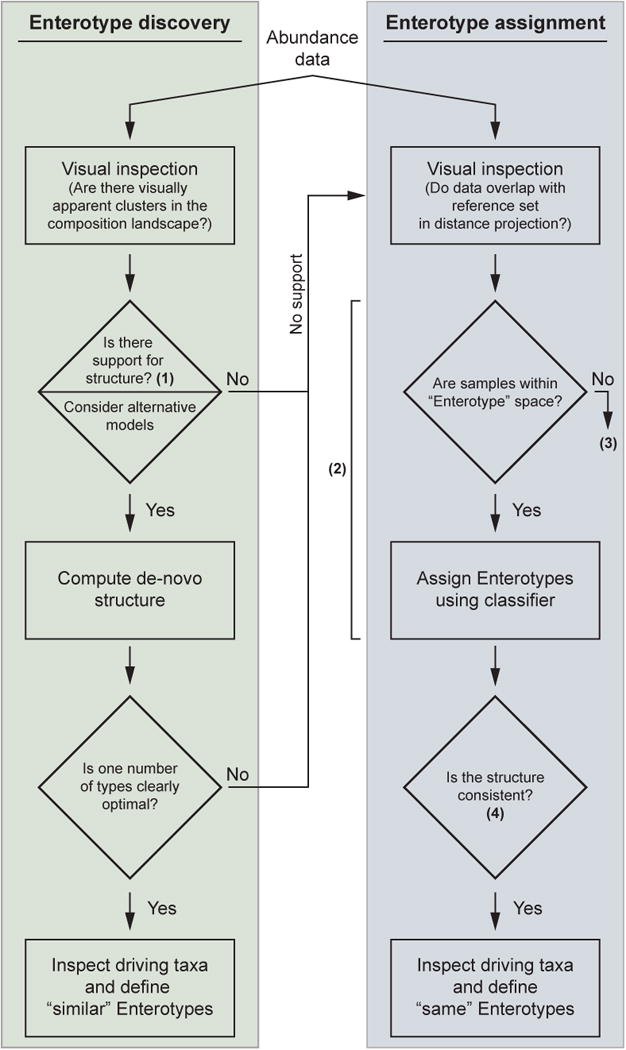
Flow diagram of recommended steps for determining enterotype assignment based on microbial abundance data. Two main routes to obtain enterotype assignments are depicted: denovo identification (enterotype discovery) and enterotype assignment based on a reference dataset. The suitability of existing models imposed on the data to describe the composition landscape (1) can be assessed by either determining the existence of cluster structure, using one of the proposed clustering strength measures (Suppl. Fig. 4) or by using a DMM modeling framework26. Other models might also be useful in capturing the structure in the data, although an exact implementation is not yet available. Determining whether samples are within the enterotype space (2) is based on similarity incomposition to adult human stool samples from the HMP1 and MetaHIT6 studies. This suitability check and a respective classifier are available at [http://enterotypes.org]. There are many explanations for the different compositional structures (3); for example, they may come from non-western individuals, or from infants. Technical issues such as DNA extraction, PCR primers, and/or bioinformatics preprocessing, may skew the analysis. The consistency of the separation (4) obtained from the classifier may be determined using a Silhouette index.
Whether used for disease state identification, prospective stratification, or the flagging of technical issues, standardized enterotyping will ensure comparability across a wide range of studies and will facilitate our understanding of the role and importance of enterotypes.
Conclusions
Identification and characterization of the major patterns related to human gut microbiota configurations remains challenging. Given an array of available approaches, each with their advantages and caveats, the number of recovered enterotype states and their statistical support can vary. With more standardization, control of sample processing and data analysis, increased concordance among different studies can be expected. Enterotype attribution can be further refined by the addition of a wider range of samples and contextual information, extending beyond the industrialized world to better represent the global human population. For now, however, we here propose a way of restricting the enterotyping space, allowing for the detection of samples that are outside of it.
Independent of the many difficulties outlined above, multiple studies have reported enterotypes with similar compositional properties albeit with varying statistical support (Fig. 2). While clearly not discrete and confounded by various factors, they differ in taxonomic, functional and ecological properties, and can be accurately recovered across large datasets (Suppl. Fig.11).They represent a way of capturing preferred microbial compositions in the human gut and thus appear to be useful stratifiers in many settings.
Relying solely on enterotypes classifications, can obscure potentially important microbial variation, and therefore should not replace direct clinical associations and expert statistical analysis with microbial species and functions, where possible. However, enterotypes may still be relevant in various clinical settings, ranging from direct disease associations to prospective study stratification or even personalized dietary interventions or other gut modulation treatments. We believe, despite our still limited knowledge, that enterotypes can be a useful tool for studying the human microbial community landscape.
Supplementary Material
Acknowledgments
The authors are grateful to the members of the Bork group at EMBL for discussions and assistance. The research leading to these results has received funding from EMBL, the VIB, the Rega institute for Medical Research, the European Research Council via the Cancer Biome project (project reference 268985), Microbes Inside (250172) and the European Community's Seventh Framework Programme via the MetaHIT (HEALTH-F4-2007-201052), the METACARDIS project (FP7-HEALTH-2012-INNOVATION-I-305312), the European Union's Horizon 2020 research and innovation programme (Marie Sklodowska-Curie grant 600375), Metagenopolis grant ANR-11-DPBS-0001 and the IHMS project (FP7-HEALTH-2010-single-stage-261376.
Footnotes
Author Contributions: PB, RK and JR conceived the review. PIC, FH and GZ performed data analysis. FH, PIC, JR and PB performed literature research, with input from all coauthors. PIC, FH, SS, RK, JR and PB wrote the manuscript with contributions from MA, FB, MJB, FDB, WMdV, SDE, CmF, MH, CH, IBJ, DK, JDL, REL, HO, PWO, CQ, DAR, FS, JW, GMW, GDW, GZ and LZ.
Author Information: The authors declare no competing financial interests.
References
- 1.Huttenhower C, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLOS Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faith JJ, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 7.Falony G, et al. Population-level analysis of gut microbiome variation. Science (80-) 2016;352:560–564. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 8.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–7. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeffery IB, Claesson MJ, O'Toole PW, Shanahan F. Categorization of the gut microbiota: enterotypes or gradients? Nat Rev Microbiol. 2012;10:591–592. doi: 10.1038/nrmicro2859. [DOI] [PubMed] [Google Scholar]
- 10.Lahti L, Salojärvi J, Salonen A, Scheffer M, de Vos WM. Tipping elements in the human intestinal ecosystem. Nat Commun. 2014;5:4344. doi: 10.1038/ncomms5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sørlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sorlie T, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamauchi M, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61:847–54. doi: 10.1136/gutjnl-2011-300865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravel J, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koren O, et al. A Guide to Enterotypes across the Human Body: Meta-Analysis of Microbial Community Structures in Human Microbiome Datasets. PLoS Comput Biol. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014 doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, et al. Exploration of bacterial community classes in major human habitats. Genome Biol. 2014;15:R66. doi: 10.1186/gb-2014-15-5-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 20.Dethlefsen L, Eckburg PB, Bik EM, Relman Da. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21:517–23. doi: 10.1016/j.tree.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Schloissnig S, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu A, Sunagawa S, Mende DR, Bork P. Inter-individual differences in the gene content of human gut bacterial species. Genome Biol. 2015;16:82. doi: 10.1186/s13059-015-0646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlsson FH, Nookaew I, Nielsen J. Metagenomic Data Utilization and Analysis (MEDUSA) and Construction of a Global Gut Microbial Gene Catalogue. PLoS Comput Biol. 2014;10:e1003706. doi: 10.1371/journal.pcbi.1003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergstrom A, et al. Establishment of Intestinal Microbiota during Early Life: a Longitudinal, Explorative Study of a Large Cohort of Danish Infants. Appl Environ Microbiol. 2014;80:2889–2900. doi: 10.1128/AEM.00342-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmes I, Harris K, Quince C. Dirichlet Multinomial Mixtures: Generative Models for Microbial Metagenomics. PLoS One. 2012;7:e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quince C, et al. The impact of Crohn's disease genes on healthy human gut microbiota: a pilot study. Gut. 2013;62:952–4. doi: 10.1136/gutjnl-2012-304214. [DOI] [PubMed] [Google Scholar]
- 28.Roager HM, Licht TR, Poulsen SK, Larsen TM, Bahl MI. Microbial enterotypes, inferred by the prevotella-to-bacteroides ratio, remained stable during a 6-month randomized controlled diet intervention with the new nordic diet. Appl Environ Microbiol. 2014;80:1142–9. doi: 10.1128/AEM.03549-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zupancic ML, et al. Analysis of the Gut Microbiota in the Old Order Amish and Its Relation to the Metabolic Syndrome. PLoS One. 2012;7:e43052. doi: 10.1371/journal.pone.0043052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claesson MJ, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–84. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 31.Gibson TE, Bashan A, Cao HT, Weiss ST, Liu Y-Y. On the Origins and Control of Community Types in the Human Microbiome. arXiv. 2015 doi: 10.1371/journal.pcbi.1004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hildebrand F, et al. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013;14:R4. doi: 10.1186/gb-2013-14-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, et al. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc Natl Acad Sci U S A. 2014;111:E2703–10. doi: 10.1073/pnas.1402342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moeller AH, et al. Chimpanzees and humans harbour compositionally similar gut enterotypes. Nat Commun. 2012;3:1179. doi: 10.1038/ncomms2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moeller AH, et al. Stability of the gorilla microbiome despite simian immunodeficiency virus infection. Mol Ecol. 2015;24:690–697. doi: 10.1111/mec.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mach N, et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ Microbiol Rep. 2015;7:554–569. doi: 10.1111/1758-2229.12285. [DOI] [PubMed] [Google Scholar]
- 37.Ramayo-Caldas Y, et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 2016:1–5. doi: 10.1038/ismej.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, et al. Two gut community enterotypes recur in diverse bumblebee species. Curr Biol. 2015;25:R652–R653. doi: 10.1016/j.cub.2015.06.031. [DOI] [PubMed] [Google Scholar]
- 39.Moeller aH, et al. Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci. 2014:1–5. doi: 10.1073/pnas.1419136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S. The evolution of the host microbiome as an ecosystem on a leash. Nature. 2017;548:43–51. doi: 10.1038/nature23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheffer M, Carpenter SR. Catastrophic regime shifts in ecosystems: linking theory to observation. Trends Ecol Evol. 2003;18:648–656. [Google Scholar]
- 42.Huse SM, Ye Y, Zhou Y, Fodor Aa. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One. 2012;7:e34242. doi: 10.1371/journal.pone.0034242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorvitovskaia A, Holmes SP, Huse SM, Interpreting Prevotella. Bacteroides as biomarkers of diet and lifestyle. Microbiome. 2016;4:15. doi: 10.1186/s40168-016-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vieira-Silva S, et al. Species–function relationships shape ecological properties of the human gut microbiome. Nat Microbiol. 2016;1:16088. doi: 10.1038/nmicrobiol.2016.88. [DOI] [PubMed] [Google Scholar]
- 45.Sankaran M, et al. Determinants of woody cover in African savannas. Nature. 2005;438:846–9. doi: 10.1038/nature04070. [DOI] [PubMed] [Google Scholar]
- 46.Staver aC, Archibald S, Levin S. Tree cover in sub-Saharan Africa: rainfall and fire constrain forest and savanna as alternative stable states. Ecology. 2011;92:1063–72. doi: 10.1890/10-1684.1. [DOI] [PubMed] [Google Scholar]
- 47.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell S, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–9. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ou J, et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr. 2013;98:111–20. doi: 10.3945/ajcn.112.056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakayama J, et al. Diversity in gut bacterial community of school-age children in Asia. Sci Rep. 2015;5:8397. doi: 10.1038/srep08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith MI, et al. Gut Microbiomes of Malawian Twin Pairs Discordant for Kwashiorkor. Science (80-) 2013;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107:14691–6. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2013 doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purushe J, et al. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol. 2010;60:721–9. doi: 10.1007/s00248-010-9692-8. [DOI] [PubMed] [Google Scholar]
- 55.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pereira FC, Berry D. Microbial nutrient niches in the gut. Environ Microbiol. 2017;19:1366–1378. doi: 10.1111/1462-2920.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vandeputte D, et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65:57–62. doi: 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roager HM, et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat Microbiol. 2016;1:16093. doi: 10.1038/nmicrobiol.2016.93. [DOI] [PubMed] [Google Scholar]
- 59.Caporaso JG, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gibbons SM, Kearney SM, Smillie CS, Alm EJ. Two dynamic regimes in the human gut microbiome. PLOS Comput Biol. 2017;13:e1005364. doi: 10.1371/journal.pcbi.1005364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Voigt AY, et al. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015;16:73. doi: 10.1186/s13059-015-0639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl):4554–61. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Nood E, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–15. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 64.Kovatcheva-Datchary P, et al. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015;22:971–982. doi: 10.1016/j.cmet.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 65.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature. 2006;444:1022–3. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 66.Haiser HJ, et al. Predicting and Manipulating Cardiac Drug Inactivation by the Human Gut Bacterium Eggerthella lenta. Science (80-) 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liang X, et al. Bidirectional interactions between indomethacin and the murine intestinal microbiota. Elife. 2015;4:e08973. doi: 10.7554/eLife.08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol. 2016;14:273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Forslund K, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu L, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–9. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- 71.Zeller G, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sobhani I, et al. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Palma G, et al. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010;10:63. doi: 10.1186/1471-2180-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology. 2010;156:3216–23. doi: 10.1099/mic.0.040618-0. [DOI] [PubMed] [Google Scholar]
- 75.Scher JU, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202–e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Larsen N, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lozupone Ca, et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14:329–39. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noguera-Julian M, et al. Gut Microbiota Linked to Sexual Preference and HIV Infection. EBioMedicine. 2016;5:135–146. doi: 10.1016/j.ebiom.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karlsson FH, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Knights D, et al. Rethinking ‘Enterotypes’. Cell Host Microbe. 2014;16:433–437. doi: 10.1016/j.chom.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schubert AM, et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio. 2014;5:e01021–14. doi: 10.1128/mBio.01021-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA. 2013;309:71–82. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu GD, et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morton JT, et al. Uncovering the Horseshoe Effect in Microbial Analyses. mSystems. 2017;2:e00166–16. doi: 10.1128/mSystems.00166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.