

Abstract
Phosphoenolpyruvate carboxykinase (Pck1) is a metabolic enzyme that is integral to the gluconeogenic and glyceroneogenic pathways. However, Pck1's role in macrophage metabolism and function is unknown. Using stable isotopomer MS analysis in a mouse model with a myeloid cell–specific Pck1 deletion, we show here that this deletion increases the proinflammatory phenotype in macrophages. Incubation of LPS-stimulated bone marrow–derived macrophages (BMDM) with [U-13C]glucose revealed reduced 13C labeling of citrate and malate and increased 13C labeling of lactate in Pck1-deleted bone marrow–derived macrophages. We also found that the Pck1 deletion in the myeloid cells increases reactive oxygen species (ROS). Of note, this altered macrophage metabolism increased expression of the M1 cytokines TNFα, IL-1β, and IL-6. We therefore conclude that Pck1 contributes to M1 polarization in macrophages. Our findings provide important insights into the factors determining the macrophage inflammatory response and indicate that Pck1 activity contributes to metabolic reprogramming and polarization in macrophages.
Keywords: calcium, glycolysis, inflammation, macrophage, tricarboxylic acid cycle (TCA cycle) (Krebs cycle), PEP, Pck1, ROS, SERCA, polarization, phosphoenolpyruvate carboxykinase, isotopic tracer, glucose metabolism, chemokine, lipogenesis
Introduction
Macrophages are versatile cells that adapt themselves to the microenvironment in tissues. Stimulation of macrophage with either Th1 cytokines, with interferon-γ (IFN-γ), or lipopolysaccharide (LPS) promotes the inflammatory phenotype with increased secretion of tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), interleukin-6 (IL-6) and increased production of reactive oxygen species (ROS)2 and nitric oxides (NOs). To meet energetic and biosynthetic demands, inflammatory macrophages are subjected to metabolic reprogramming (1, 2). This reprogramming increases glycolytic rates and lactate production. The mechanism is thought to occur through repression of pyruvate dehydrogenase (PDH) by activating pyruvate dehydrogenase kinase (PDHK), thus limiting carbon flux into the citric acid cycle (CAC). However, some pyruvate is still oxidized through PDH, which is essential for full LPS activation in the macrophage (3).
The CAC is central to carbon metabolism coordinating the metabolism of glucose, glutamine, and other biosynthetic pathways such as lipogenesis and nucleic acid synthesis. Upon activation, macrophages increase their uptake of glutamine to sustain these cataplerotic reactions (4). Glutamate generated from glutamine is converted to α-ketoglutarate via glutamate dehydrogenase and replenishes the CAC. It has been suggested that metabolites from CAC impact macrophage inflammatory phenotypes. For example, increased succinate stabilizes hypoxia-inducible factor 1α (HIF-1α). The increased HIF-1α positively regulates proinflammatory cytokine IL-1β expression and other HIF-dependent genes, including some of the glycolytic enzymes (5). Another CAC metabolite, citrate, also modulates macrophage function. Citrate exported by citrate carrier (CIC) from mitochondria to the cytosol is cleaved by ATP-citrate lyase (ACLY) into acetyl-CoA and oxaloacetate (OAA). Acetyl-CoA provides carbons for fatty acid synthesis to meet the demand for lipids in the proinflammatory state (6). The second product of citrate metabolism, OAA, is reduced by NADH to form malate, and malate is decarboxylated and oxidized to form pyruvate by the malic enzyme. This produces NADPH, which is used to generate reactive oxygen species by NADPH oxidase (7, 8).
Pck1 is an integral metabolic enzyme for gluconeogenic and glyceroneogenic pathways (9, 10). It catalyzes the conversion of OAA into phosphoenolpyruvate (PEP). Although the production of PEP from OAA is a cataplerotic reaction during gluconeogenesis, Pck1 has also been found to play an important role in anaplerosis of the CAC. First when Pck1 is deleted in the liver, it results in ∼90% decreased CAC flux, which in turn impairs fatty acid oxidation and ultimately contributes to the development of hepatic steatosis (11). In the small intestine, it has been proposed that PEP produced from Pck1 can be converted into pyruvate and can re-enter the mitochondria through PDH (12). Recently, in tumor cells that exhibit the Warburg effect, Pck1 was discovered to play a key role for promoting anabolic metabolism, including synthesis of fatty acids and nucleic acids (13).
In this study, we found Pck1 expression is induced in LPS-activated macrophages. We determined the metabolic role of Pck1 in the macrophage and show that Pck1 has regulatory effects that promote the proinflammatory phenotype. Using Pck1-deleted macrophage we show that upon LPS stimulation, the macrophages exhibited less carbon entry into the CAC and had reduced lipid synthesis. We provide important insights into the macrophage inflammatory response and show that Pck1 contributes to metabolic reprogramming in the macrophage.
Results
Pck1 regulates inflammatory phenotype and redox state of macrophages
To establish whether Pck1 expression and activity in the macrophage have biological significance, we generated myeloid-specific deletion of Pck1 (Pck1MC-KO) (Fig. 1A). To confirm that Pck1 was successfully deleted, differentiated thioglycollate elicited peritoneal macrophages and bone marrow–derived macrophages (BMDM) were isolated and treated with vehicle or LPS for 4 h. The expression of the Pck1 mRNA in macrophages isolated from Pck1MC-KO mice was greatly reduced (Fig. 1B). To determine whether Pck1 expression in other tissues was altered, we isolated skeletal muscle, kidney, brown adipose tissue, white adipose tissue, and muscle from fasted Pck1flox/flox and Pck1MC-KO mice. Protein was isolated and analyzed by Western blotting (Fig. S1). There were no differences in Pck1 protein expression between Pck1flox/flox and Pck1MC-KO mice for the tissues analyzed except for skeletal muscle which showed a reduction of Pck1 in both parental strains (Pck1flox/flox and Pck1MC-KO) compared with C57BL/6J (B6) muscle.
Figure 1.

Generation of myeloid Pck1-deficient mice. A, the schematic represents the generation of LysM-specific Pck1 knock-out mice (Pck1MC-KO) by crossing Pck1flox/flox mice with LysM-Cre+/− transgenic mice expressing Cre-recombinase under control of the LysM promoter. B and C, to confirm that the deletion of Pck1 in macrophage cells, BMDM (B) and IPDM (C) from Pck1flox/flox and Pck1MC-KO mice were isolated and treated with LPS (100 ng/ml). Pck1 mRNA expression was measured by RT-PCR. The values are the mean ± S.E. for 4–6 mice per group normalized with 18S rRNA and expressed as -fold difference over Pck1flox/flox.
Role of Pck1 in the macrophage
To investigate the role of Pck1 in the macrophage, we performed stable isotope analysis of BMDM isolated and differentiated from Pck1MC-KO and control Pck1flox/flox mice. Glucose uptake in BMDM was measured by administration of 2-[14C] deoxyglucose (2-DG) to these cells with or without LPS stimulation. No differences for glucose uptake were found between Pck1flox/fox and Pck1MC-KO BMDMs in the basal or stimulated states (Fig. 2A). To determine the impact of Pck1 on glucose metabolism, we first measured expression of glycolysis-related genes in macrophages after LPS treatment. Only lactate dehydrogenase (LDH) expression was altered with 23% less expression in Pck1MC-KO cells compared with Pck1flox/flox (Fig. 2B). As lactate is the major end product of the glycolytic pathway, we measured lactate concentration in cells by GC-MS. The lactate concentration in Pck1MC-KO cell was 2.5-fold higher than Pck1flox/flox cells (Fig. 2C).
Figure 2.

Effects of Pck1 on LPS-induced glycolytic pathway. BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice were incubated with LPS for 4 h. A, 2-[U-14C]deoxyglucose uptake was measured and normalized by protein concentration. Values represent the mean ± S.E. for n = 6 per group. *, p < 0.05 compared with vehicle group with the same genotype. B, mRNA expression of glycolytic enzymes was measured by RT-PCR. The values are the mean ± S.E. for n = 6–8 per group normalized with 18S rRNA and expressed as -fold difference over Pck1flox/flox. C, lactate concentrations from cell lysates were determined by GC-MS. Values represent the mean ± S.E. for n = 6 per group. *, p < 0.05 compared with Pck1flox/flox group. GLUT1, glucose transporter 1; PFK2, phosphofructokinase 2; LDH, lactate dehydrogenase.
Utilization of glucose and glutamine in the macrophage for CAC
To monitor intracellular glucose utilization in macrophages under LPS stimulation, we incubated BMDMs for 16 h in the presence of uniformly 13C-labeled glucose, [U-13C]glucose. Cells were collected and analyzed for the total concentrations of intermediates (unlabeled plus 13C-labeled) and 13C labeling patterns (isotopomers) using gas chromatography mass spectrometry methods (14). The abundances (mole percent enrichments (MPE%)) from [13C]glucose incorporation into the intermediates included the measurements of the isotopomers as calculated by MPE% = [(13C-labeled M1 thru M + for each of the CAC intermediates) × (13C-labeled + unlabeled) −1] × 100.
The conversion of [13C]glucose toward lactate was measured by the MPE% of M3 lactate. M3 is a mass isotopomer with a mass shift of 3 m/z from 12C (unlabeled). Pck1MC-KO cells had 1.8-fold more M3 lactate measured compared with Pck1flox/flox (Fig. 3A). Pyruvate generated in the glycolytic pathway can also enter into CAC. Therefore, we investigated the first turn of the CAC by measuring the total concentrations and abundances of 13C-isotopomers of the CAC intermediates. No differences were found between Pck1flox/flox and Pck1MC-KO cells in total concentrations of citrate, succinate, and malate (Fig. 3B). However the abundances of M2 citrate and M2 malate were 5 and 40% less in Pck1MC-KO cells (Fig. 3C and Fig. S2), respectively. M2 is a mass isotopomer with a mass shift of 2 m/z from 12C (unlabeled). The data suggest decreased pyruvate flux through PDH in Pck1MC-KO cells.
Figure 3.

Contribution of glucose to central metabolism of LPS-activated macrophages. A–C, BMDMs isolated from Pck1flox/flox and Pck1MC-KO mice were incubated with [U-13C]glucose in the presence of LPS for 16 h and GC-MS was performed for absolute concentration of M3 lactate (A), total concentrations of CAC (B), and contribution of 13C to M2-CAC (C). D and E, BMDMs isolated from Pck1flox/flox and Pck1MC-KO mice were incubated with [U-13C]glutamine in the presence of LPS for 16 h and GC-MS was performed for total CAC concentrations (D) and contribution of 13C label to M4-CAC (E). Values represent the mean ± S.E. for n = 6 per group. *, p < 0.05 compared with Pck1flox/flox group.
Upon LPS stimulation, glutamine serves as the main substrate for anaplerosis of CAC intermediates (5). To determine whether the contribution of glutamine to CAC intermediates was altered in Pck1MC-KO macrophages, [U-13C]glutamine was incubated with LPS-activated BMDMs for 16 h. The total concentrations and abundances of 13C-isotopomers of the CAC intermediates (citrate, succinate, and malate) were measured and were not different between Pck1flox/flox and Pck1MC-KO cells (Fig. 3, D and E, and Fig. S3). Glutamine enters the TCA cycle as α-ketoglutarate, which is further converted to succinate and releases CO2. Therefore, M4 CAC intermediates were examined and similar abundance was found in both Pck1flox/flox and Pck1MC-KO cells. M4 is a mass isotopomer with a mass shift of 4 m/z from 12C (unlabeled). These data suggest that anaplerosis of the CAC by glutamine was not affected in Pck1MC-KO cells.
Pck1MC-KO macrophages have increased glutamine concentration
Because glutamine is an important amino acid in growing active cells (4), we measured the concentration of glutamine and glutamate as well as the production of glutamine and glutamate from 13C-labeled precursors. Using [U-13C]glucose administration to cells, the total concentrations of glutamine and the 13C label incorporation to glutamine were measured (Fig. 4, A and B). The concentration of glutamine was 1.5-fold greater in Pck1MC-KO cells. However the incorporation of labeled glucose to glutamine was not different between genotypes. To measure glutamine uptake in the cells, we used uniformly [U-13C]glutamine administrated to LPS-activated BMDMs for 16 h. The abundance of total glutamine in the cells was measured. There was a 1.7-fold increase in glutamine concentration in Pck1MC-KO cells compared with Pck1flox/flox cells (Fig. 4C). There were no differences in M2 or M4 13C-labeled glutamine (Fig. 4D). We also measured total glutamate concentration (Fig. 4E) and the incorporation of [13C]glutamine into glutamate (Fig. 4F). There were no differences found between Pck1MC-KO and Pck1flox/flox cells. These data suggest that the Pck1MC-KO cells have increased glutamine concentration that may reflect increased uptake but the synthesis of glutamate from glutamine was the same.
Figure 4.

Contribution of glutamine to central metabolism of LPS-activated macrophages. BMDMs were isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice. They were incubated with [U-13C]glucose in the presence of LPS for 16 h. A and B, GC-MS was performed for total glutamine concentration (A) and contribution of 13C label to glutamine (B). BMDMs isolated from Pck1flox/flox and Pck1MC-KO mice were incubated with [U-13C]glutamine in the presence of LPS for 16 h. C–F, GC-MS was performed for total glutamine concentrations (C), contribution of 13C label to M2 and M4 glutamine (D), total glutamate concentrations (E), and contribution of 13C label to M2 and M4 glutamate (F). Values represent the mean ± S.E. for n = 6 per group. *, p < 0.05 compared with Pck1flox/flox group.
Pck1 needed for palmitate synthesis
Upon activation by LPS, macrophages have increased demand of lipids for membrane formation. To meet this demand, acetyl-CoA in activated macrophages is utilized for the synthesis of large amounts of fatty acids (15). To determine whether decreased PDH flux in Pck1MC-KO cells impacts fatty acid synthesis, we measured de novo lipogenesis using deuterium-labeled water method (16). Deuterium incorporation into triacylglycerol (TG) and TG-palmitate is used for the quantification of newly synthesized glycerol and palmitate in macrophages. We found similar synthesis of new glycerol in Pck1MC-KO and Pck1flox/flox cells, but the Pck1MC-KO cells synthesized 40% less palmitate than Pck1flox/flox cells (Fig. 5, A and B).
Figure 5.

De novo lipogenesis in LPS-activated macrophages. A–E, BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice were incubated with [2H2O] (A and B) or [3-13C]lactate (D and E) in the presence of LPS for 16 h. GC-MS was performed for lipid-bound glycerol and palmitate. The values are expressed as newly synthesized or abundance (MPE%) as indicated. Values represent the mean ± S.E. for n = 3 per group. C, BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice were incubated with LPS for 4 h. mRNA expression of lipid metabolism genes were measured by RT-PCR. F, BMDMs isolated from Pck1flox/flox and Pck1MC-KO mice were incubated with LPS for 4 h and total triglyceride concentration in the cells was measured. The values are the mean ± S.E. for n = 6–8 per group normalized with 18S rRNA and expressed as -fold difference over Pck1flox/flox. *, p < 0.05. FASN, fatty acid synthase; ACC, acetyl-CoA carboxylase; CD36, cluster of differentiation 36.
To further determine whether the decrease of fatty acid synthesis is directly impacted by deletion of Pck1 in myeloid cells, we analyzed the synthesis of lipids using a known substrate of gluconeogenesis and glyceroneogenesis, [3-13C]lactate. The [13C]lactate was administered to LPS-activated BMDMs for 16 h. The abundance of 13C labeling into TG-glycerol and TG-palmitate was calculated. Consistent with our de novo lipogenesis study using deuterium-labeled water, we found that the Pck1MC-KO cells synthesized less palmitate than Pck1flox/flox cells, and very little glycerol was synthesized from the lactate precursor (Fig. 5, D and E). The glycerol-3-phosphate would be generated from glucose in the media. This suggests that Pck1 in the macrophage is not required for glycerol-3-phosphate synthesis or glyceroneogenesis. In addition, expression of genes associated with lipid metabolism was measured in macrophages. We found no differences in acetyl-CoA carboxylase and CD36 mRNA expression, but fatty acid synthase expression was reduced in Pck1MC-KO cells (Fig. 5C). The total concentration of triglyceride in the BMDM was not different between genotypes (Fig. 5F). As Pck1 is known for its gluconeogenic role, the abundance of 13C-labeled glucose from [13C]lactate and [13C]glutamine were also measured in these cells. However, we did not detect 13C-labeled glucose in either Pck1flox/flox or Pck1MC-KO cells for either substrate, suggesting Pck1 does not participate in gluconeogenesis in macrophages (data not shown).
Lack of Pck1 in macrophages results in increased M1 cytokines
We investigated the impact of the deletion of Pck1 in macrophages on macrophage polarization using quantitative RT-PCR analysis. We measured expression of macrophage phenotype markers and cytokines, iNOS, ARG1, TNFα, IL-1β, IL-6, and IL-10. We found significantly increased expression of TNFα, IL-1β, and IL-6 in the Pck1MC-KO cells, suggesting the cells have more of an M1 proinflammatory phenotype (Fig. 6A). To investigate the importance of Pck1 on expression of M1 markers, we overexpressed Pck1 in RAW 264.7 cells and analyzed proinflammatory markers upon LPS stimulation (Fig. 6B). We found that overexpression of Pck1 dramatically reduced TNFα, IL-1β, IL-6, and IL-10, suggesting that Pck1 may be a key regulator of macrophage metabolism and thus macrophage polarity.
Figure 6.

Effects of Pck1 on the regulation of LPS-induced inflammatory phenotypes. A and B, LPS was administered to (A) BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice (n = 8) and (B) RAW 264.7 cells transfected with a construct without Pck1 overexpression (control CMV promoter only) or with Pck1 overexpressed (Pck1 o/e) (n = 3). The cells were stimulated with LPS (100 ng/ml). mRNA expression of macrophage parameters was measured by RT-PCR. The values are the mean ± S.E. normalized with 18S rRNA and expressed as -fold difference over Pck1flox/flox. C, BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice (n = 5–6 mice). The cells were stimulated with LPS (100 μg/ml). The concentration of PEP was measured using the PEP Assay Kit (BioVision, Milpitas, CA). D, BMDMs isolated from Pck1flox/flox and Pck1MC-KO mice were incubated with LPS for 4 h (n = 6). Total ROS production was assessed with CM-H2DCFDA and the intensity of fluorescence was measured by flow cytometry. The values are expressed as -fold difference over Pck1flox/flox. Values represent the mean ± S.E. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 compared with Pck1flox/flox group.
To determine whether the altered cytokine mRNA expression in Pck1MC-KO cells influences inflammatory response, we measured ROS production in BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice. To examine ROS production directly, Pck1flox/flox and Pck1MC-KO BMDMs were incubated with cell-permeant reagent 2′,7′-dichlorofluorescin diacetate (DCFDA), a fluorogenic dye that measures hydroxyl, peroxyl, and other ROS activity within the cell in the presence of LPS for 4 h. The fluorescent intensity measured in Pck1MC-KO cells was significantly higher than Pck1flox/flox cells (Fig. 6D).
Because the glycolytic metabolite PEP has been shown to play a role in sustaining T cell receptor–mediated Ca+2-NFAT signaling and effector functions by repressing sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity (17), we measured the PEP concentration in our BMDM (Fig. 6C). We found that the Pck1MC-KO cells had 37% more PEP than Pck1flox/flox BMDM after 4 h of incubation with LPS (100 ng/ml). The Pck1MC-KO cells also have increased ROS (Fig. 6D), which suggests that these cells may have endoplasmic reticulum (ER) stress. Therefore, we analyzed total PK RNA-like ER kinase (PERK) protein concentrations in BMDM from Pck1MC-KO and Pck1flox/flox cells with LPS for 4 h (Fig. 7). The Pck1MC-KO cells had lower levels of PERK compared with Pck1flox/flox cells. Because ATF4 is downstream of PERK activation, we measured the expression levels of ATF4 mRNA in BMDM from Pck1MC-KO and Pck1flox/flox cells with and without LPS for 4 h and found that Pck1MC-KO has increased ATF4 mRNA expression (Fig. S4). However the protein levels of ATF4 were not increased. We also measured BiP protein levels but they were not increased in the Pck1MC-KO cells (Fig. S4) either. These data suggest that even though there are low levels of PERK in Pck1MC-KO cells, the Pck1MC-KO cells do not have increased ER stress.
Figure 7.
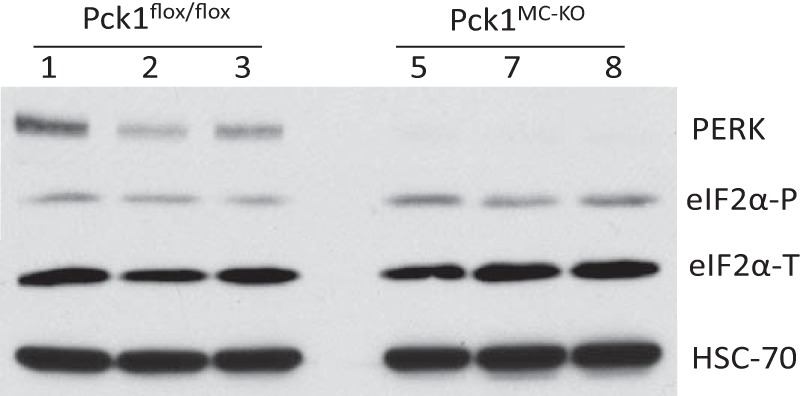
PERK deficiency in Pck1MC-KO cells. BMDMs isolated and differentiated from Pck1flox/flox and Pck1MC-KO mice (n = 8). A, after treatment with LPS (100 ng/ml) whole cell lysates were made and immunoblotted for the indicated proteins. The values are the mean ± S.E. normalized with 18S rRNA and expressed as -fold difference over Pck1flox/flox. PERK, total PKR-like ER kinase; eIF2α-T, total eIF2α; eIF2α-P, phospho-eIF2α (Ser-51); HSC-70, heat shock cognate 70.
Discussion
Using stable isotope techniques we have investigated the role of Pck1 in macrophages. The data from the [U-13C]glucose tracer studies indicated that glucose uptake was not altered in Pck1MC-KO mice but the flux of glucose to acetyl-CoA and citrate was reduced. The consequences were increased lactate concentration and reduced palmitate synthesis. We also found reduced M2 labeling from glucose. This suggests an anaplerotic role for Pck1 in the macrophage. When Pck1 is deleted, the conversion of PEP to OAA to malate to CAC would be inhibited. It is possible that unlabeled aspartate could be converted to OAA to malate to CAC. This would reduce labeling of M2 by glucose and support an important role for Pck1 in the macrophage. From our studies it is evident that Pck1 did not participate in gluconeogenesis or glyceroneogenesis because these pathways were not detectable or not altered, respectively, in the Pck1MC-KO cells. More importantly Pck1 is required for a balance of the M1/M2 macrophage phenotype. When Pck1 was overexpressed it reduced the M1 phenotype, whereas deletion of Pck1 promoted the proinflammatory phenotype. Our data also suggested that Pck1 was required for lipid metabolism in the macrophage because of reduced palmitate synthesis in the Pck1MC-KO macrophages.
In response to infection or during states of chronic stress, macrophages require extensive reprogramming metabolically and phenotypically to amplify proinflammatory responses. The macrophages develop a Warburg-like phenotype, similar to tumor cells, by increasing glycolysis generation of lactate. However, little is known about how macrophage metabolism regulates its polarization. It has been demonstrated that Pck1 in tumor cells promotes the utilization of glucose and glutamine for anabolic metabolism, including synthesis of fatty acids and nucleic acids (18). In the current study, we demonstrate that deletion of Pck1 in the myeloid cells impacts CAC concentrations, and increased cytokine expression of TNFα, IL-1β, and IL-6.
When Pck1 was knocked down in macrophages, we found the same level of glucose uptake into the cells, yet we found elevated lactate production. Concurrently, carbon flux through PDH reaction was reduced when Pck1 was knocked down, especially impacting on citrate and malate generation. These data suggest that the role of Pck1 may be to promote carbon flux from pyruvate toward CAC instead of lactate production. CAC in activated macrophage is important to provide the substrates for the biosynthesis of ribose, fatty acids, and nonessential amino acids (19). To sustain the central pathways, increased uptake of glucose and glutamine promotes anaplerotic reactions. Recently it has been demonstrated that PDH flux is essential to sustain LPS activation (3). This reaction provides carbons for citrate synthesis, which are then transported out of mitochondrion into the cytosol for lipogenesis (3). Our study supports this because Pck1-deleted macrophages have less palmitate synthesized. This may be because of the limited acetyl-CoA derived from pyruvate. When PDH flux is repressed, evidence shows that citrate is increasingly generated by reductive carboxylation of α-ketoglutarate, which is mostly originated from glutamine (20). Although total glutamine concentrations were elevated in Pck1MC-KO cells, the first-phase contribution of glutamine to citrate was not significantly different compared with Pck1flox/flox cells, as indicated by the abundance of 13C-labeled M5 (data not shown). This suggests the high demand of citrate in LPS-stimulated macrophages is mostly supplied from glucose via PDH.
Elevated glutamine concentration in Pck1MC-KO cells may promote a proinflammatory state with increased production of proinflammatory cytokines and mediators. We observed that deletion of Pck1 in macrophages with enhanced lactate production resulted in promoting the proinflammatory phenotypes. Tannahill et al. (5) have demonstrated that LPS-induced IL-1β release is dependent on glycolytic pathways with increased succinate level for the stabilization of HIF-1α signaling. Specifically, they showed that chronic LPS activation in macrophages causes an increase in intracellular succinate via glutamine-dependent anaplerosis. Succinate stabilizes HIF-α by inhibiting prolyl hydroxylase domain (PHD) enzyme activity. Stabilized HIF-α binds to HIF response elements (HREs) in target genes, including those encoding glycolytic enzymes and IL-1β (21). However, alteration in succinate abundance was not detected in our cell system, suggesting a novel and alternate mechanism linking reduced malate and citrate to inflammation.
When Pck1 was knocked down, the levels of TNFα, IL-1β, and IL-6 were increased whereas the overexpression of Pck1 in RAW 264.7 cells reduced the levels of TNFα, IL-1β, and IL-6. These data suggest that Pck1 plays an integral role in the macrophage polarization.
Previous studies have shown that PEP inhibits SERCA activity in T cells and reduces ER Ca+2 concentrations (17). When calcium concentration in the ER is chronically decreased, the function of the chaperones becomes disturbed and unfolded proteins accumulate, resulting in ER stress. As a consequence, ER-stress sensors become devoid of GRP78/BiP and become activated causing an early adaptive response or late responses promoting apoptosis (22). Ca+2 has also been shown to impact IL-1β cytokine expression. Reduced ER Ca+2 stores can activate the NLRP3 inflammasome and increase IL-1β (23). In our study we have a modest increase of PEP which did not increase ER stress in our Pck1MC-KO cells, but did alter the total concentration of PERK. The reason we find reduced total PERK protein in our Pck1MC-KO macrophages is not fully understood. However, other studies have shown that a deficiency of PERK in cancer cells have blunted Ca+2 signaling, increased eIF2α phosphorylation, and improved cell viability (24).
In our study we find elevated ROS production in the Pck1MC-KO cells. We hypothesize that deletion of Pck1 may enhance the malic enzyme activity. Thus when malate is decarboxylated and oxidized to form pyruvate by the malic enzyme, there would be an increased production of NADPH by NADPH oxidase resulting in increased proinflammatory cytokines.
ROS production from the glycolytic pathway has been exhibited to enhance inflammation and production of cytokines because of mitochondrial ROS generation (25). Upon LPS stimulation, NADPH oxidase activity is significantly enhanced and generates large amounts of ROS to destroy pathogens (26). ROS production has been implicated in regulating proinflammatory cytokines IL-1β and IL-18 production (27). The activation by a second stimulus such as ATP will trigger ROS generation, followed by caspase-1 and inflammasome activation and cytokine production.
In conclusion, in this study we show that deletion of Pck1 in macrophages promotes a more M1-like phenotype with increased expression of proinflammatory cytokines. In our model, we found that loss of Pck1 represses the anaplerotic pathway and enhances lactate production. This is accomplished by 1) the decreased flux through PDH and 2) the increased production of ROS in the Pck1MC-KO cells (Fig. 8). It is now recognized that chronic inflammation has a crucial role in the pathogenesis of obesity-related diseases (28). Elevated levels of proinflammatory markers including TNFα, IL-1β, and IL-6 are predictive of type 2 diabetes (28–30). Thus by understanding the regulation of macrophage polarization we may identify novel therapeutic approaches for type 2 diabetes or other inflammatory diseases.
Figure 8.
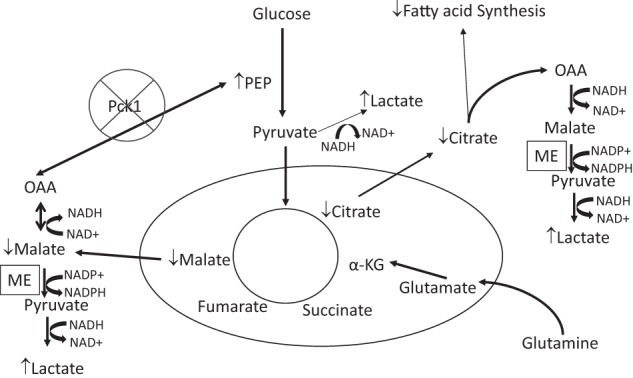
Deletion of Pck1 in macrophage cells. The deletion of Pck1 results in increased lactate concentrations in the cell. The flux through pyruvate dihydrogen complex is reduced resulting in decreased citrate levels and reduced fatty acid synthesis. The deletion of Pck1 prevents the decarboxylation of OAA to PEP. Thus, OAA would be reduced by NADH to form malate; malate would be decarboxylated and oxidized to form pyruvate by the malic enzyme. This produces NADPH, which is used to generate ROS by NADPH oxidase. Generation of ROS serves as defense against invading microbes and is viewed as the assurance of monocyte/macrophage activation to M1-like phenotype.
Materials and methods
Chemicals
For the Pck1 construct, mouse Pck1 cDNA cloned into the pCMV6–entry vector was purchased (OriGene Technologies). RAW 264.7 cells were transfected with either the control pCMV6-entry vector or pCMV6-entry vector containing the mouse Pck1 cDNA using FuGENE 6 transfection agent (Roche). Lipopolysaccharide (Sigma-Aldrich) or recombinant murine interleukin-4 (Peprotech, Rocky Hill, NJ) was prepared as a working stock of 25 μg/ml in sterile 1× phosphate buffered saline (PBS) and added into the wells of the culture dish for a final concentration of 100 ng/ml (LPS) or 20 ng/ml (IL-4).
Cell culture
RAW 264.7 cells from ATCC (ATCC* TIB-71, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 6 mm glutamine in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. For experimental incubations, cells in log-phase growth were plated at a density of 1–5 × 105 cells/ml and allowed to attach overnight. Cells at 80% confluency were then treated with 100 ng/ml LPS or 20 ng/ml IL-4 for 4 h. The media were removed and the cells were collected for analysis. Experimental incubations were terminated by directly lysing cells in the appropriate lysis buffer.
Animals and bone marrow–derived macrophages
All animals were housed in a temperature-controlled facility with a 12-h light/dark cycle in compliance with the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University. LysM-specific Pck1 knock-out mice (Pck1MC-KO mice) were generated by crossing Pck1flox/flox mice with LysM-Cre+/− transgenic mice expressing Cre-recombinase under control of the LysM promoter. These mice display no alterations in immune cell frequencies. Pck1flox/flox mice (designated as controls) were used as control animals, and LysM-Pck1 mice (designated as Pck1MC-KO) were used as experimental animals. Pck1foxl/flox mice were generated as described previously and maintained in C57BL/6J background (31). LysM-Cre transgenic mice were obtained from The Jackson Laboratory (Bar Harbor, ME).
BMDMs were generated as described previously (32, 33). Briefly, mononuclear phagocyte progenitor cells derived from femur and tibia bones of male Pck1flox/flox and Pck1MC-KO mice were cultured in cell-culture medium supplemented with mouse recombinant macrophage–colony stimulating factor (M-CSF) (Peprotech, Rocky Hill, NJ) for 7 days. Peritoneal macrophages were elicited and cultured 3 days after the induction of peritonitis by 4% thioglycollate broth administered to male Pck1flox/flox and Pck1MC-KO mice as described previously (34). Peritoneal macrophages and BMDMs were left untreated or polarized using 100 ng/ml LPS or 20 ng/ml IL-4 for 4 h.
Glucose uptake assay
BMDM cells were washed twice with 0.5 ml of 37 °C 1× PBS and then incubated for 10 min at 37 °C in 0.5 ml/well DMEM (10 mm glucose, 5 mm glutamine, 136 mm NaCl, 4.7 mm KCl, 1.25 mm CaCl2, 1.25 mm MgSO4, 10 mm HEPES, pH 7.4) with 0.5 μCi/ml of 2-[U-14C]deoxy-d-glucose (PerkinElmer Life Sciences). Cells were washed twice with 0.5 ml of cold 1× PBS with 20 mm d-glucose and then lysed in 0.5 ml of lysis buffer (0.025% SDS, 1% Triton X-100 in 1× PBS). Lysates were centrifuged at 20,000 × g for 5 min at 4 °C, and the protein concentrations were determined with a protein quantification kit (Bradford Assay, Abcam, Cambridge, MA) according to manufacturer's directions. Remaining lysates were counted using a scintillation counter. The data are expressed as disintegrations per minute per milligram of protein (35).
Mass spectrometric measurement of 13C incorporation into central metabolism
Cells were incubated in DMEM with 10% FBS and 1% penicillin/streptomycin that contained U-13C-labeled substrates and LPS (100 ng/ml): Condition 1, 10 mm [U-13C]glucose (99 atom %) with 5 mm glutamine; condition 2, 10 mm glucose with 5 mm [U-13C]glutamine (99 atom %); and condition 3, 10 mm glucose, 5 mm glutamine, and 1 mm [3-13C]lactate (99 atom %). 13C label incorporation into cells was at 80–90% confluence. 13C label incorporation into cells at 80–90% confluence was measured in medium containing 100% of one of the 13C-labeled substrates outlined in the above conditions, and cultured at 37 °C for 16 h. The media were then removed and saved for measurements of concentrations and MPE of the 13C-labeled precursors and 13C-labeled respective products. The isolated cells were stored at −80 °C and then analyzed for total concentrations and MPEs (13C-labeled isotopomers).
The fractional contribution of the precursor to the product was based on the MPE-derived from the 13C label incorporation into the total pool of each of the products. Mass isotopomer analysis enables the measurements of unlabeled analyte (MO) to the labeled analyte (M+1, 2, or 3, etc.). The measured mass isotopomer distribution was corrected for natural enrichment of each mass. All stable isotopomer analysis was completed at the Case Mouse Metabolic Phenotyping Center. The 13C-labeled CAC intermediates were determined by using gas chromatography–mass spectrometry (14, 36).
Mass spectrometric measurement of deuterium incorporation into palmitate and triglyceride
To measure stable isotope incorporation, cells at 80–90% confluence were switched into fresh medium containing 10% 2H2O and cultured at 37 °C for 16 h. Medium was removed and saved for percent enrichment measurements and the cells were isolated and stored at −80 °C until analysis. Triglyceride concentrations and de novo lipogenesis were measured by mass spectrometry by the Case Mouse Metabolic Phenotyping Center. The [2H] label on triglyceride-derived glycerol and palmitate was measured as the amount of newly synthesized triglyceride and palmitate and calculated as described by Bederman et al. (16).
Extraction of intracellular metabolites and gas chromatography–mass spectrometry
Cells homogenized with reference compounds were extracted with 1 ml of chloroform-methanol 2:1. The slurry was centrifuged at 11,000 × g for 20 min at 4 °C. The upper methanol-water phase was collected and evaporated in a Savant vacuum centrifuge. To determine glucose and triglyceride-bound glycerol, the residue was reacted with 50 μl of acetic anhydride solvent at 70 °C for 1 h. To determine CAC intermediates, the residue was reacted with 70 μl bis(trimethylsilyl)trifluroacetamide with 10% trimethylchlorosilane at 70 °C for 1 h. The chloroform phase was evaporated and used to determine triglyceride-bound palmitate (14, 36). The trimethylsilyl-derivatized samples were injected into an SSL injector at 270 °C in splitless mode. GC-MS analysis was performed using an Agilent 5973N-MSD equipped with an Agilent 6890 GC system and a DB-17 MS capillary column; helium was used as carrier gas. For each analyte the signals at the nominal m/z were monitored. All analytes were corrected for natural abundances (above background) and the MPEs were calculated.
Real-time RT-PCR analysis
Total RNA was isolated with an NucleoSpin® RNA kit (Macherey-Nagel Inc., Bethlehem, PA) and synthesized to single-strand cDNA from 500 ng of total RNA using random hexamer primers and murine mammary tumor virus (MMTV) reverse transcriptase (Applied Biosystems, Foster City, CA). Real-time qRT-PCR analysis was performed using Bullseye EvaGreen SYBR qPCR reagent (MidSci, St. Louis, MO) on a Chromo4 Cycler (MJ Research/Bio-Rad, Hercules, CA) with specific primer sequences (see Table S1). Data were normalized using the comparative threshold cycle (Ct) method with load variations normalized to 18S rRNA as described (37).
Intracellular reactive oxygen species production
The 5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate, acetyl ester (CM-H2DCFDA) were obtained from Life Technologies. BMDM cells were seeded on the 6-well plates at a confluence of 70%. After full attachment, cells were loaded with 5 μm CM-H2DCFDA dye following the protocol provided by Life Technologies and incubated at 37 °C for 30 min. The intensity of fluorescence was determined by flow cytometry.
Measurement of PEP in BMDM
BMDM cells were isolated from femur and tibia bones of male Pck1flox/flox and Pck1−/− mice were cultured in cell-culture medium supplemented with mouse recombinant M-CSF (Peprotech) for 7 days. Cells at 80% confluency were then treated as with 100 ng/ml LPS for 4 h. The medium was removed and the cells were collected for analysis. We used the PEP Assay Kit (BioVision, Milpitas, CA) to measure PEP concentrations in the cells according to manufacturer's directions. In the assay, PEP is converted to ATP and pyruvate. The generated pyruvate is quantified by colorimetric (λmax = 570 nm) or fluorometric methods (Ex/Em 535/587 nm). The concentrations were normalized by total protein.
Western blot analysis
Pck1flox/flox and Pck1MC-KO mice were euthanized and tissue samples were collected, protein was isolated and analyzed by Western blot analysis as previously described (34). The membranes were incubated with antibodies to Pck1 (anti-PCK1 antibody, ab70358, rabbit Ab, Abcam, Cambridge, MA). BMDM cells were isolated from femur and tibia bones of male Pck1flox/flox and Pck1MC-KO mice were cultured in cell-culture medium supplemented with mouse recombinant M-CSF (Peprotech) for 7 days. Cells at 80% confluency were then treated as with 100 ng/ml LPS for 4 h. C57BL6/J mouse embryonic fibroblasts (MEFs) were grown in high glucose DMEM supplemented with 10% defined FBS (Invitrogen), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. ER stress was induced with 400 nm thapsigargin (Sigma-Aldrich) for 12 h. Cells were collected, protein was isolated and analyzed by Western blot analysis as previously described (34). The membranes were incubated with antibodies to total PERK (PERK (C33E10) rabbit mAb), total eIF2α (eIF2α (L57A5) mouse mAb), phospho-eIF2α (Ser-51) (phospho-eIF2α (Ser-51) 9721 rabbit), BiP (BiP (C50B12) 3177 rabbit mAb), ATF-4 (ATF-4 11815 rabbit mAb) (Cell Signaling Technology). The negative controls (with no primary antibodies added) showed no staining.
The immunoreactive proteins were detected using the SuperSignal West Pico Chemiluminescent Substrate Kit (Thermo Scientific) and the density of the immunoreactive bands was measured by scanning densitometry (UN-SCAN-IT, Silk Scientific, Orem, UT). The membranes were normalized for loading differences using heat shock cognate-70 (HSC70) protein (HSC 70 Antibody (1B5), sc-59560 rat mAb, Santa Cruz Biotechnology, Santa Cruz, CA) as previously described (38).
Statistical analysis
The values reported are mean ± S.E. Data were analyzed with Student's t-test using GraphPad Prism 4 (GraphPad Software, San Diego CA). p value < 0.05 was considered significant.
Author contributions
C.-W. K. had substantial contributions to the conception and design, acquisition of data, analysis and interpretation of the data and writing of the manuscript. D. C. and J. W. performed the experiments for ER stress in the BMDM. M. H. designed and interpreted the ER stress experiments. M. A. P. had substantial contributions to the design, analysis of the data, and editing the manuscript for the isotope studies. C. M. C. had substantial contributions to the conception and design, analysis and interpretation of the data, and writing of the manuscript.
Supplementary Material
The Case Mouse Metabolic Phenotyping Center (MMPC) was supported by National Institutes of Health Grant U24 DK76174. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Table S1.
- ROS
- reactive oxygen species
- PDH
- pyruvate dehydrogenase
- PDHK
- pyruvate dehydrogenase kinase
- CAC
- citric acid cycle
- OAA
- oxaloacetate
- PEP
- phosphoenolpyruvate
- BMDM
- bone marrow–derived macrophages
- MPE%
- mole percent enrichments
- TCA
- tricarboxylic acid
- TG
- triacylglycerol
- DCFDA
- 2′,7′-dichlorofluorescin diacetate
- ER
- endoplasmic reticulum
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- M-CSF
- macrophage–colony stimulating factor.
References
- 1. Altenberg B., and Greulich K. O. (2004) Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 84, 1014–1020 10.1016/j.ygeno.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 2. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 3. Meiser J., Krämer L., Sapcariu S. C., Battello N., Ghelfi J., D'Herouel A. F., Skupin A., and Hiller K. (2016) Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J. Biol. Chem. 291, 3932–3946 10.1074/jbc.M115.676817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He L., Weber K. J., and Schilling J. D. (2016) Glutamine modulates macrophage lipotoxicity. Nutrients 8, 215 10.3390/nu8040215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., et al. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 10.1038/nature11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Infantino V., Convertini P., Cucci L., Panaro M. A., Di Noia M. A., Calvello R., Palmieri F., and Iacobazzi V. (2011) The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 438, 433–436 10.1042/BJ20111275 [DOI] [PubMed] [Google Scholar]
- 7. Geeraerts X., Bolli E., Fendt S. M., and Van Ginderachter J. A. (2017) Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front. Immunol. 8, 289 10.3389/fimmu.2017.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Infantino V., Iacobazzi V., Palmieri F., and Menga A. (2013) ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 440, 105–111 10.1016/j.bbrc.2013.09.037 [DOI] [PubMed] [Google Scholar]
- 9. Beale E. G., Harvey B. J., and Forest C. (2007) PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem. Biophys. 48, 89–95 10.1007/s12013-007-0025-6 [DOI] [PubMed] [Google Scholar]
- 10. Franckhauser S., Muñoz S., Pujol A., Casellas A., Riu E., Otaegui P., Su B., and Bosch F. (2002) Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes 51, 624–630 10.2337/diabetes.51.3.624 [DOI] [PubMed] [Google Scholar]
- 11. Burgess S. C., Hausler N., Merritt M., Jeffrey F. M., Storey C., Milde A., Koshy S., Lindner J., Magnuson M. A., Malloy C. R., and Sherry A. D. (2004) Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J. Biol. Chem. 279, 48941–48949 10.1074/jbc.M407120200 [DOI] [PubMed] [Google Scholar]
- 12. Yang J., Kalhan S. C., and Hanson R. W. (2009) What is the metabolic role of phosphoenolpyruvate carboxykinase? J. Biol. Chem. 284, 27025–27029 10.1074/jbc.R109.040543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Owen O. E., Kalhan S. C., and Hanson R. W. (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412 10.1074/jbc.R200006200 [DOI] [PubMed] [Google Scholar]
- 14. Kombu R. S., Brunengraber H., and Puchowicz M. A. (2011) Analysis of the citric acid cycle intermediates using gas chromatography-mass spectrometry. Methods Mol. Biol. 708, 147–157 10.1007/978-1-61737-985-7_8 [DOI] [PubMed] [Google Scholar]
- 15. O'Neill L. A. (2015) A broken Krebs cycle in macrophages. Immunity 42, 393–394 10.1016/j.immuni.2015.02.017 [DOI] [PubMed] [Google Scholar]
- 16. Bederman I. R., Foy S., Chandramouli V., Alexander J. C., and Previs S. F. (2009) Triglyceride synthesis in epididymal adipose tissue: Contribution of glucose and non-glucose carbon sources. J. Biol. Chem. 284, 6101–6108 10.1074/jbc.M808668200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ho P. C., Bihuniak J. D., Macintyre A. N., Staron M., Liu X., Amezquita R., Tsui Y. C., Cui G., Micevic G., Perales J. C., Kleinstein S. H., Abel E. D., Insogna K. L., Feske S., Locasale J. W., Bosenberg M. W., Rathmell J. C., and Kaech S. M. (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 10.1016/j.cell.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Montal E. D., Dewi R., Bhalla K., Ou L., Hwang B. J., Ropell A. E., Gordon C., Liu W. J., DeBerardinis R. J., Sudderth J., Twaddel W., Boros L. G., Shroyer K. R., Duraisamy S., Drapkin R., Powers R. S., Rohde J. M., Boxer M. B., Wong K. K., and Girnun G. D. (2015) PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol. Cell 60, 571–583 10.1016/j.molcel.2015.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ganeshan K., and Chawla A. (2014) Metabolic regulation of immune responses. Annu. Rev. Immunol. 32, 609–634 10.1146/annurev-immunol-032713-120236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wise D. R., and Thompson C. B. (2010) Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433 10.1016/j.tibs.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mills E., and O'Neill L. A. (2014) Succinate: A metabolic signal in inflammation. Trends Cell Biol. 24, 313–320 10.1016/j.tcb.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 22. Mekahli D., Bultynck G., Parys J. B., De Smedt H., and Missiaen L. (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3 10.1101/cshperspect.a004317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sano R., and Reed J. C. (2013) ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470 10.1016/j.bbamcr.2013.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verfaillie T., Rubio N., Garg A. D., Bultynck G., Rizzuto R., Decuypere J. P., Piette J., Linehan C., Gupta S., Samali A., and Agostinis P. (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891 10.1038/cdd.2012.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liemburg-Apers D. C., Willems P. H., Koopman W. J., and Grefte S. (2015) Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 89, 1209–1226 10.1007/s00204-015-1520-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sheppard F. R., Kelher M. R., Moore E. E., McLaughlin N. J., Banerjee A., and Silliman C. C. (2005) Structural organization of the neutrophil NADPH oxidase: Phosphorylation and translocation during priming and activation. J. Leukocyte Biol. 78, 1025–1042 10.1189/jlb.0804442 [DOI] [PubMed] [Google Scholar]
- 27. Strowig T., Henao-Mejia J., Elinav E., and Flavell R. (2012) Inflammasomes in health and disease. Nature 481, 278–286 10.1038/nature10759 [DOI] [PubMed] [Google Scholar]
- 28. Ouchi N., Parker J. L., Lugus J. J., and Walsh K. (2011) Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97 10.1038/nri2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hotamisligil G. S., Shargill N. S., and Spiegelman B. M. (1993) Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 259, 87–91 10.1126/science.7678183 [DOI] [PubMed] [Google Scholar]
- 30. Pradhan A. D., Manson J. E., Rifai N., Buring J. E., and Ridker P. M. (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286, 327–334 10.1001/jama.286.3.327 [DOI] [PubMed] [Google Scholar]
- 31. She P., Shiota M., Shelton K. D., Chalkley R., Postic C., and Magnuson M. A. (2000) Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mole. Cell. Biol. 20, 6508–6517 10.1128/MCB.20.17.6508-6517.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weischenfeldt J., and Porse B. (2008) Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc. 2008, pdb.prot5080 10.1101/pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- 33. Zhang X., Goncalves R., and Mosser D. M. (2008) The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 83, 14.1.1–14.1.14 10.1002/0471142735.im1401s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeSantis D. A., Ko C. W., Liu Y., Liu X., Hise A. G., Nunez G., and Croniger C. M. (2013) Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediators Inflamm. 2013, 751374 10.1155/2013/751374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Freemerman A. J., Johnson A. R., Sacks G. N., Milner J. J., Kirk E. L., Troester M. A., Macintyre A. N., Goraksha-Hicks P., Rathmell J. C., and Makowski L. (2014) Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289, 7884–7896 10.1074/jbc.M113.522037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang L., Kombu R. S., Kasumov T., Zhu S. H., Cendrowski A. V., David F., Anderson V. E., Kelleher J. K., and Brunengraber H. (2008) Metabolomic and mass isotopomer analysis of liver gluconeogenesis and citric acid cycle. I. Interrelation between gluconeogenesis and cataplerosis; formation of methoxamates from aminooxyacetate and ketoacids. J. Biol. Chem. 283, 21978–21987 10.1074/jbc.M803454200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan J. S., Reed A., Chen F., and Stewart C. N. Jr. (2006) Statistical analysis of real-time PCR data. BMC Bioinformatics 7, 85 10.1186/1471-2105-7-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Millward C. A., Desantis D., Hsieh C. W., Heaney J. D., Pisano S., Olswang Y., Reshef L., Beidelschies M., Puchowicz M., and Croniger C. M. (2010) Phosphoenolpyruvate carboxykinase (Pck1) helps regulate the triglyceride/fatty acid cycle and development of insulin resistance in mice. J. Lipid Res. 51, 1452–1463 10.1194/jlr.M005363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.