

Abstract
Resistance to β-lactams in Acinetobacter baumannii involves various mechanisms. To decipher them, whole genome sequencing (WGS) and real-time quantitative polymerase chain reaction (RT-qPCR) were complemented by mass spectrometry (MS) in selected reaction monitoring mode (SRM) in 39 clinical isolates. The targeted label-free proteomic approach enabled, in one hour and using a single method, the quantitative detection of 16 proteins associated with antibiotic resistance: eight acquired β-lactamases (i.e. GES, NDM-1, OXA-23, OXA-24, OXA-58, PER, TEM-1, and VEB), two resident β-lactamases (i.e. ADC and OXA-51-like) and six components of the two major efflux systems (i.e. AdeABC and AdeIJK). Results were normalized using “bacterial quantotypic peptides,” i.e. peptide markers of the bacterial quantity, to obtain precise protein quantitation (on average 8.93% coefficient of variation for three biological replicates). This allowed to correlate the levels of resistance to β-lactam with those of the production of acquired as well as resident β-lactamases or of efflux systems. SRM detected enhanced ADC or OXA-51-like production and absence or increased efflux pump production. Precise protein quantitation was particularly valuable to detect resistance mechanisms mediated by regulated genes or by overexpression of chromosomal genes. Combination of WGS and MS, two orthogonal and complementary techniques, allows thereby interpretation of the resistance phenotypes at the molecular level.
The accurate description of cellular events remains a difficult task in post-genomic days when multiomics approaches are required to obtain integrative information (1). This is particularly the case for the elucidation of multifactorial antibiotic resistance in Acinetobacter baumannii. This Gram-negative opportunistic pathogen has the remarkable ability to become resistant to numerous antibiotics. β-lactams, together with quinolones, aminoglycosides, and polymixins, are the major drug classes used for therapy infections because of A. baumannii. There are two major mechanisms of resistance to β-lactams in A. baumannii: i) enzymatic modification of the drug by overexpression of the chromosomally encoded β-lactamases, ADC (Acinetobacter-derived Cephalosporinase, an AmpC-type β-lactamase) and OXA-51-like (2–3) or acquisition of genes for β-lactamases with either narrow (e.g. TEM-1, CARB, SCO, OXA-10, 20, and -21) or extended-spectrum substrate (e.g. TEM-type or SHV-type ESBL variants, VEB, GES, PER, CTX-M, and RTG-4), or metallo-β-lactamases (IMP-, VIM-, SIM- and NDM-type enzymes) or serine-carbapenemases (OXA-23, -24/40, -58); ii) overexpression of resistance-nodulation-cell division (RND)1 efflux systems (4). To date, three Acinetobacter Drug Efflux (Ade) RND pumps, AdeABC (5), AdeFGH (6), and AdeIJK (7) have been characterized in A. baumannii. Overexpression of adeABC plays a major role in multidrug resistance in clinical settings (8–9) whereas that of adeFGH (4, 10) has been responsible for a regional epidemic (11), and constitutively expressed adeIJK is associated with intrinsic resistance (4, 7). Expression of each pump is tightly regulated, AdeABC by a two-component regulatory system AdeRS (8), AdeFGH by the LysR-type transcriptional regulator AdeL (10), and AdeIJK by the TetR transcriptional regulator AdeN (12). Efflux pump overproduction contributes synergistically with β-lactamases to the level of resistance of the host (4, 13).
The resistance proteins must therefore be determined qualitatively and quantitatively in order to analyze, at the molecular level, the multifactorial resistance phenotype of A. baumannii and make predictions as to the outcome of antibiotic therapy. Quantitative mass spectrometry (MS) appears particularly attractive to achieve this goal (14). MS technologies are increasingly used to characterize A. baumannii at the protein level using both untargeted (4, 15–16) or targeted approaches (17). Untargeted MS has been extensively used in proteomics study for a long time because it allows stochastic identification of hundreds to thousands of proteins with a coarse quantitation. On the contrary, targeted MS provides a robust and accurate quantification but at the cost of the detection of a lower number of proteins in a single multiplex. We reported the adequacy of a targeted approach to assess resistance in Pseudomonas aeruginosa (18) and Staphylococcus aureus (19), using a liquid chromatography hyphenated to an electrospray triple quadrupole mass spectrometer (LC-ESI-QqQ-MS) in Selected Reaction Monitoring mode (SRM). This technology is compatible with analytical requirements of accurate, sensitive, and selective assays for quantitation of dozens of resistance traits. We thus applied SRM, in combination with genomic data, to study the more complex question of the biochemistry of multidrug resistance in A. baumannii.
A set of 39 previously characterized clinical isolates of A. baumannii (4, 10, 20–30) was used. These strains are known to produce resident (i.e. ADC and OXA-51-like) as well as acquired (i.e. GES, NDM-1, OXA-23, 24, and 58, PER, TEM-1, and VEB) β-lactamases or various levels of AdeABC, AdeFGH, and AdeIJK (4, 10). The presence of these resistance mechanisms was assessed by both whole genome sequencing (WGS) and quantitative label-free SRM. After normalization using A. baumannii quantotypic peptides (31, 32), SRM enabled to study the relative resistance protein levels which turned out to be in good agreement with the resistance phenotypes of the host bacteria.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Antibiotic Resistance
The clinical strains used in this study are described in Table I. They were grown on Columbia agar supplemented with 5% sheep blood (COS) (bioMérieux, Marcy l'Etoile, France) for WGS and SRM and in brain heart infusion, or Luria-Bertani broth for RT-qPCR. The minimal inhibitory concentrations (MICs) of antibiotics were determined by microdilution according to the guidelines of the Clinical and Laboratory Standards Institute (33), in cation adjusted Mueller-Hinton (CA-MH) broth.
Table I. Strains, antibiotic resistance phenotypes and β-lactamase genes identified by whole genome sequencing. CAZ, Ceftazidime; CTX, Cefotaxime; FEP, Cefepime; IPM, Imipenem; MEM, Meropenem; TIC, Ticarcillin.
| Name | Strain |
MICa (μg/ml) of: |
β-lactamase genes determined by WGS |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Other designation | Reference | Penicillins | Cephalosporins |
Carbapenems |
Resident | Acquired | ||||
| TIC | CAZ | CTX | FEP | IPM | MEM | |||||
| Group 1 | ||||||||||
| BM4587 | (10) | 8 | 4 | 8 | 1 | 0,12 | 0.5 | blaOXA-104, blaADC | ||
| CIP 70.34T | ATCC19606 | (20) | 16 | 16 | 32 | 16 | 0,5 | 2 | blaOXA-98, blaADC | |
| NIPH 146 | (23, 26) | 8 | 4 | 16 | 1 | 0,12 | 0,25 | blaOXA-64, blaADC | ||
| NIPH 190 | (23, 26) | 4 | 1 | 8 | 0.5 | 0,12 | 0,12 | blaOXA-65, blaADC | ||
| NIPH 201 | (23, 26) | 8 | 2 | 8 | 1 | 0,12 | 0,25 | blaOXA-51-like, blaADC | ||
| NIPH 329 | (23, 26) | 8 | 4 | 16 | 2 | 0,12 | 0,25 | blaOXA-51-like, blaADC | ||
| NIPH 335 | (23, 26) | 16 | 8 | 32 | 8 | 0,25 | 0.5 | blaOXA-128, blaADC | ||
| NIPH 410 | (23, 26) | 4 | 2 | 4 | 0.5 | 0,12 | 0,12 | blaOXA-88, blaADC | ||
| NIPH 60 | (23, 26) | 8 | 2 | 16 | 1 | 0,25 | 0,25 | blaOXA-69, blaADC | ||
| NIPH 601 | (23, 26) | 16 | 2 | 8 | 0,5 | 0,12 | 0,12 | blaOXA-69, blaADC | ||
| NIPH 615 | (23, 26) | 4 | 2 | 4 | 1 | 0,12 | 0,25 | blaOXA-120, blaADC | ||
| NIPH 67 | (23, 26) | 16 | 8 | 16 | 4 | 0,25 | 0,5 | blaOXA-51-like, blaADC | ||
| NIPH 70 | (23, 26) | 16 | 4 | 8 | 2 | 0,25 | 0,25 | blaOXA-256, blaADC | ||
| NIPH 80 | (23, 26) | 32 | 16 | 32 | 4 | 0,12 | 0,5 | blaOXA-78, blaADC | ||
| NIPH 1669 | (26) | 64 | 32 | 128 | 16 | 0,5 | 4 | blaOXA-71, blaADC** | ||
| BM4704 | 3220 | (29) | 64 | 128 | 512 | 32 | 1 | 2 | blaOXA-132*, blaADC* | |
| NIPH 2061 | (30) | 64 | 128 | 512 | 32 | 8 | 32 | blaOXA-109*, blaADC* | ||
| Group 2 | ||||||||||
| NIPH 24 | (25) | >1024 | 16 | 16 | 16 | 2 | 2 | blaOXA-66, blaADC | blaTEM-1 | |
| NIPH 290 | (23) | >1024 | 64 | 32 | 256 | 0,5 | 2 | blaOXA-69, blaADC | blaTEM-1 | |
| NIPH 527 | RUH 875 | (21, 26) | >1024 | 16 | 16 | 8 | 0,5 | 1 | blaOXA-69, blaADC | blaTEM-1 |
| NIPH 528 | RUH 134 | (21, 26) | >1024 | 8 | 16 | 8 | 0,25 | 1 | blaOXA-66, blaADC | blaTEM-1 |
| BM4701 | AB8 | (29) | 512 | 256 | 64 | 16 | 64 | 32 | blaOXA-64, blaADC | blaGES-14 |
| BM4702 | AB25 | (29) | >1024 | >1024 | >512 | >256 | 16 | 16 | blaOXA-64, blaADC | blaGES-11 |
| BM4703 | 3171 | (29) | >1024 | 4 | 16 | 2 | 16 | 8 | blaOXA-64, blaADC | blaOXA-58 |
| NIPH 1734 | (23) | >1024 | 16 | 32 | 4 | 64 | 32 | blaOXA-132, blaADC | blaOXA-58 | |
| BM4714 | 3142 | (29) | >1024 | 4 | 32 | 8 | 128 | >64 | blaOXA-51, blaADC | blaOXA-24 |
| NIPH 1362 | (23, 25) | >1024 | >1024 | 512 | 64 | 16 | 16 | blaOXA-51-like*, blaADC* | blaOXA-58 | |
| BM4713 | AB4 | (29) | 512 | 128 | >512 | 16 | 64 | >64 | blaOXA-66, blaADC* | blaOXA-72 |
| BM4706 | 3185 | (29) | >1024 | 1024 | >512 | 32 | 32 | 32 | blaOXA-94, blaADC* | blaOXA-23 |
| BM4710 | 3073 | (29) | >1024 | 1024 | 512 | 64 | 64 | 64 | blaOXA-69, blaADC* | blaOXA-23 |
| MRSN 3405 | (28) | >1024 | 256 | 512 | 64 | 64 | >64 | blaOXA-69, blaADC* | blaOXA-23 | |
| AYE | (24) | >1024 | >1024 | >512 | >256 | 1 | 1 | blaOXA-69, blaADC* | blaVEB-1 | |
| Group 3 | ||||||||||
| BM4705 | 3228 | (29) | >1024 | 256 | 128 | 4 | 32 | 64 | blaOXA-132, blaADC* | blaOXA-24, blaTEM-1 |
| BM4709 | 3048 | (29) | >1024 | 1024 | 512 | 64 | 32 | 64 | blaOXA-69, blaADC* | blaOXA-23, blaTEM-1 |
| BM4712 | 3162 | (29) | >1024 | 512 | >512 | 256 | 128 | >64 | blaOXA-66, blaADC* | blaOXA-23, blaTEM-1 |
| ANC 4097 | (27) | >1024 | >1024 | >512 | >256 | 128 | >64 | blaOXA-69, blaADC | blaOXA-23, blaTEM-1, blaNDM-1 | |
| BM4708 | AB20 | (29) | >1024 | >1024 | 512 | 256 | 32 | 32 | blaOXA-69, blaADC | blaOXA-23, blaPER-1 |
| BM4711 | AB18 | (29) | >1024 | >1024 | >512 | 256 | 32 | 32 | blaOXA-69, blaADC | blaOXA-23, blaPER-1 |
| BM4707 | 3221 | (29) | >1024 | 32 | 512 | 64 | 64 | 64 | blaOXA-65, blaADC* | blaOXA-23, blaOXA-58 |
* ISAba1 upstream from the gene.
** ISAba125 upstream from the gene.
a MICs were determined by microdilution.
DNA Sequencing
Scaffolds of strains ANC 4097, AYE, CIP 70.34T, MRSN 3405, and NIPH were downloaded from the Acinetobacter initiative, Broad Institute (broadinstitute.org). The BM4587 genome was previously reported (4). Illumina whole genome sequencing was performed on the remaining 14 strains (supplemental Table S1). UltraClean® Microbial DNA Isolation Kit (MO BIO, Carlsbad, CA) was used for DNA extraction. Library preparation was performed from 1 ng of DNA extracted with the Nextera® XT DNA sample preparation kit for 96 samples (Illumina, San Diego, CA) and Nextera® XT index kit (Illumina). Library validation was performed on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) to control the distribution of tagmented DNA. The samples were multiplexed in compliance with the Illumina optimal specifications and 1% (volume/volume) PhiX CONTROL V3 Kit (Illumina). A sequencing run of paired-end 2 × 200 cycles was performed with the MiSeq Reagent kit v3 (600 cycles) (Illumina) on a MiSeq instrument (Illumina) and controlled using Sequencing Analysis Viewer software provided by MiSeq instrument. De novo assembly was performed using A5-miseq pipeline steps 1 and 2 (34). BLASTn or tBLASTn of proteins or nucleotide sequences of interest on scaffolds of the strain(s) was achieved using NCBI program (35).
RT-qPCR
A. baumannii total RNA was extracted from exponentially grown bacteria (OD600 0.8–0.9) using RNeasy mini kit (Qiagen, Hilden, Germany). Gene expression of was quantified by RT-qPCR as described (10) using LightCycler® RNA Amplification Kit SYBR Green I (Roche Diagnostic, Basel, Switzerland) with the following cycle profile: 1 cycle at 95 °C for 30 s, followed by 45 cycles at 95 °C for 5 s, 56 °C for 10 s, and 72 °C for 20 s. The expression level of the rpoB gene was used for normalization. Each experiment was performed in duplicate at least twice independently.
Chemical Reagents for SRM Analysis
Methanol was from Merck Millipore (Billerica, MA). LC-MS grade water and acetonitrile were provided by Fisher Scientific (Illkirch, France) and all the other chemicals by Sigma-Aldrich (Lyon, France). Peptides were synthesized using Fmoc chemistry on a MultiPep RS peptide synthesizer from Intavis Bioanalytical Instruments AG (Koeln, Germany).
Bacterial Lysis, Digestion, and LC-ESI-QqQ-MS Analysis in SRM Mode
Bacterial digests were prepared as described (18) with few modifications. Briefly, bacterial colonies were suspended in 3 ml of suspension medium (bioMérieux), the bacterial concentration was adjusted to 4 McFarland (36), manually mixed twice and 1 ml was removed in two 1.5 ml tubes. Samples were centrifuged at 10,000 × g for 10 min and the supernatant removed. After 5 min of ultrasonic disruption with glass beads in 50 mm ammonium carbonate (150 μl) and 5 mm dithiotreitol, the pellets were carbamidomethylated using iodoacetamide (5 min) and digested 15 min with trypsin (10 μg) at 50 °C. The digestion was stopped by adding 0.5 μl formic acid (FA). The two digests were desalted using Oasis HLB, 3 cm3 (Waters, Milford, MA), pooled, and concentrated with a TurboVap (Biotage, Uppsala, Sweden) to 50 μl. The final volume was adjusted to 250 μl with LC-MS grade water containing 0.1% FA and 2% of acetonitrile. Targeted SRM analysis was carried out on a Nexera HPLC (Shimadzu, Kyoto, Japan) hyphenated to an ESI-QqQ-MS 5500 QTRAP (Sciex, Framingham, MA) in positive ionization mode according to (18). Concisely, LC separation was carried out at 300 μl/min on an XBridge BEH C18 column, 100 mm × 2.1 mm, particle size 3.5 μm, porosity 130 Å (Waters). Elution was performed with 0.1% FA in water (solvent A) or in acetonitrile (solvent B) using a 22-min linear gradient from 2% to 35% of solvent B.
SRM Assay Construction
Proteotypic peptides for resistance, i.e. peptides that are specific for the resistance proteins inferred from WGS, were selected in silico as described (18, 19, 37). Briefly, proteins of interest from consensual UniProtKB sequences were digested in silico by trypsin using Skyline software (38). Missed-cleavage sites and cleavage among Lysine, or Arginine, and Proline were not allowed. The resulting peptides were synthetized and separately injected in a 5500 QTRAP mass spectrometer. Reference spectra of peptide ion fragments were acquired using Skyline prediction equation for declustering potential, entrance potential, collision energy, and collision cell exit potential. For each peptide, the three most intense characteristic fragment ions were selected. This selection was verified, using strains overproducing (BM4689, BM4690, and BM4666) or deleted (BM4717, BM4718, and BM4719) for the Ade efflux pumps (4) as well as non-A. baumannii strains for β-lactamase production (data not shown). Data-dependent analyses were performed on a 5600 TripleTOF (Sciex) to select A. baumannii-specific peptide candidates for bacterial quantification. Specificity for A. baumannii was evaluated both experimentally and by BLASTp analysis (35) with UniProtKB database (data not shown). These peptides were named “bacterial quantotypic peptides.” Nonspecific peptides were discarded as well as peptides containing methionine or missed-cleavage sites. Peptides from β-lactamases, Ade efflux pumps and for bacterial quantity evaluation were combined in a single Scheduled SRM experiment composed of 558 transitions representing 186 peptides with a detection window of 155 s (supplemental Table S2).
SRM Data Analysis and Protein Quantitation
Data analysis by MultiQuantTM 2.1 software (Sciex) with the integration algorithm Summation for peak integration was applied to three replicates per strain obtained from three independent experiments. Peak integration parameters were set as follows: Gaussian smooth width of 1.0 point, summation window of 15 s and 40% noise level for baseline (Fig. 1, step I). Automatic integrations were visually checked. The simultaneity of the 3 transitions from the same peptide was checked by ensuring a standard deviation of their retention time (min) lower than 0.03. Areas under 3000 Sciex arbitrary unit (a.u.) were set to null. For a peptide, if at least one of the transitions was null, then all the transitions were set to null. Peptides not detected in any strain (supplemental Table S2) Were Discarded (Fig. 1, step II).
Fig. 1.

SRM data analysis and protein quantitation. Step I: peak integration with verification of transition coelution when appertaining to the same peptide. Step II: suppression of peptides with at least one transition signal below 3,000 a.u. cut-off. Step III: iterative deletion of non quantotypic peptides and transitions until reaching Pearson's product-moment coefficient r > 0.95 for all proteins in all strains. Step IV: minimization of the measurement error via backcalculation of all the transitions areas, including the missed and the discarded values, using linear regression equations from step III. Step V: quantity estimations by summing the two most intense transitions (TopTra2) of the three most intense peptides (TopPep3) per protein and protein quantity normalization by the bacterial quantity using “bacterial quantotypic transitions.”
Bona fide proteotypic peptides (i.e. peptides unique to the protein of interest) were experimentally selected according to Charretier et al. (19). Briefly, for a given peptide, transition areas were normalized using Equation 1:
| (1) |
where T1, T2, and T3 represent each of the three transitions and Ti one of the 3. We selected peptides with coefficients of variation (CVs) of all the three normalized transition areas below 20%, across all the data set after removal of outliers.
Quantotypic peptides and transitions (i.e. accurately representing the level of the proteins) (31, 32) were further selected using correlation criteria. For each protein, the Pearson's product-moment coefficient (r) was calculated between the nonzero transition areas of each strain and reference transition areas estimated by averaging all the nonzero transition areas from the whole data set (Fig. 1, step III). To reduce quantitation distortions by noisy transitions or nonquantotypic peptides, those accounting for poor regressions were iteratively discarded sample per sample until reaching r > 0.95 for all proteins in all strains (Fig. 1, step III). Bacterial quantotypic peptides were selected using the same process by considering their transitions as belonging to a unique artificial protein. We then took advantage of the observed strong correlation to minimize the measurement error via the least squares approach. All the transitions areas, including the missed and the discarded values, were back-calculated using linear regression models with null y-intercepts. These models were built for each protein and each SRM experiment (Fig. 1, step IV).
Quantity estimations were made according to Ludwig et al. (39) by summing the two most intense transitions (TopTra2) of the three most intense peptides per protein (TopPep3), except for NDM-1 (Fig. 1, step V). For this protein, a single peptide respecting all the previous criteria was detected. In this case, the three transitions areas for this unique peptide were summed. Quantifications were done using the same subset of TopPep3 peptides and TopTra2 transitions for all the three biological replicates (supplemental Table S2). As reference gene(s) is(are) used to normalize RT-qPCR data, “bacterial quantotypic transitions” were eventually used to normalize the SRM estimation of the relative quantity of protein per bacteria cell, following Equation 2:
| (2) |
The nonzero quantifications of the three biological replicates were then used to calculate averages, standard deviations (SDs), and CVs. For comparison, protein quantities were indicated by average numbers with a dash followed by CVs in percentage (Table II).
Table II. β-lactamase quantitation using multiplexed SRM (normalized relative quantity/coefficient of variation, %.
| Strain | β-lactamase: relative protein quantities determined by SRM |
Efflux pump: relative protein quantities determined by SRM |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acquired β-lactamase |
Resident β-lactamase |
AdeA | AdeB | AdeC | AdeI | AdeJ | AdeK | |||||||||
| GES | NDM-1 | OXA-23 | OXA-24 | OXA-58 | PER | TEM-1 | VEB | AmpC | OXA-51 | |||||||
| Group 1 | ||||||||||||||||
| BM4587 | 0.58/6.3% | 0.17/21.8% | 2.7/10.3% | 1.6/10.5% | 2.4/2.5% | |||||||||||
| CIP 70.34T | 3.6/NA | 7.4/10.9% | 0.08/10.5% | 11.4/9.3% | 4.1/4.3% | 3.3/10.1% | 2.2/7.7% | 4.2/12.1% | ||||||||
| NIPH 146 | 0.61/4.6% | 0.12/NA | 2.4/1.2% | 1.5/9.8% | 2.7/9.1% | |||||||||||
| NIPH 190 | 0.3/9.4% | 0.3/7.9% | 4.6/6.4% | 2.8/6% | 5.6/14.4% | |||||||||||
| NIPH 201 | 0.63/4.9% | 0.18/0.9% | 2.3/4% | 1.3/6.7% | 2.6/5.4% | |||||||||||
| NIPH 329 | 0.81/4.6% | 0.11/6% | 2.4/5.9% | 1.4/9.2% | 3/4.7% | |||||||||||
| NIPH 335 | 0.68/3.3% | 0.05/15.2% | 5.7/1.8% | 3.2/9.1% | 5.5/8.4% | |||||||||||
| NIPH 410 | 0.54/6.7% | 0.17/18.8% | 1.7/2.2% | 1.2/2.6% | 2.6/7.9% | |||||||||||
| NIPH 60 | 0.87/11.8% | 0.25/4.2% | 2.4/1.9% | 1.5/8.7% | 3/6.4% | |||||||||||
| NIPH 601 | 0.68/17.9% | 1.8/2.1% | 1.4/3.6% | 2.1/10.6% | ||||||||||||
| NIPH 615 | 0.55/9.9% | 0.22/13.8% | 2.4/3.5% | 1.5/12% | 2.8/5.6% | |||||||||||
| NIPH 67 | 1.1/3% | 3.1/7.1% | 2.1/8.3% | 3.5/9.9% | ||||||||||||
| NIPH 70 | 0.72/9.6% | 0.41/1% | 2.7/4.6% | 1.8/6.3% | 3.1/4.8% | |||||||||||
| NIPH 80 | 0.54/12.5% | 0.13/6% | 5.8/7.9% | 3.3/3.9% | 5.6/10.6% | |||||||||||
| NIPH 1669 | 9.7/13.7% | 0.24/15.9% | 1.9/4.4% | 1.2/4.1% | 0.05/0.6% | 6.5/1.5% | 4.4/7.6% | 7.2/3.6% | ||||||||
| BM4704 | 92.7/11.6% | 8.3/5.7% | 4.8/5.9% | 1.9/5% | 6/9.5% | 4.1/2.1% | 7.2/2.5% | |||||||||
| NIPH 2061 | 53.3/2% | 20.7/10.5% | 1.8/3.6% | 1/10.3% | 0.06/NA | 2.4/4.6% | 1.4/5.1% | 3/7.4% | ||||||||
| Group 2 | ||||||||||||||||
| NIPH 24 | 0.34/18.6% | 0.58/11% | 0.22/10.6% | 2.3/4.6% | 0.81/1.5% | 2.1/6.1% | 1.4/4.8% | 2.8/3.3% | ||||||||
| NIPH 290 | 0.23/19% | 0.86/8.1% | 0.12/13.3% | 23.3/4.9% | 10.4/39.9% | 3.5/30% | 2.8/2.6% | 1.8/7.8% | 3.9/9.5% | |||||||
| NIPH 527 | 0.25/30.7% | 0.94/2.8% | 0.16/8.4% | 2.2/7.5% | 0.92/10.8% | 0.09/17.3% | 2.8/7.3% | 1.8/12.5% | 3.3/6.3% | |||||||
| NIPH 528 | 0.33/32.1% | 0.78/2% | 0.28/9.5% | 3/3.2% | 1/2% | 2.8/3.7% | 1.5/9.1% | 3.4/0.6% | ||||||||
| BM4701 | 8.1/89.8% | 0.27/4.6% | 0.04/14% | 3.8/17% | 1.2/9.3% | 5.3/9.8% | 2.3/8.8% | 4.9/12.1% | ||||||||
| BM4702 | 12.6/84.2% | 0.56/5.7% | 0.13/11.6% | 2.1/9% | 1/6.7% | 5.7/7.8% | 2.8/2.9% | 4.8/7.3% | ||||||||
| BM4703 | 16.1/5.7% | 0.65/4.7% | 0.1/22.1% | 0.13/NA | 2.7/8.5% | 1.7/7.3% | 2.9/4.8% | |||||||||
| NIPH 1734 | 51.3/6.1% | 2.9/1% | 0.17/14.1% | 3.8/6.9% | 1.4/6% | 2.8/6.7% | 2.1/3% | 3.9/10.3% | ||||||||
| BM4714 | 0.47/NA | 13.5/10.2% | 1.1/10.2% | 0.23/21.1% | 5.3/4.5% | 3.4/7.7% | 5.2/5.1% | |||||||||
| NIPH 1362 | 15.1/10% | 26.6/23.9% | 10.4/12.9% | 2.2/6.7% | 1/9.2% | 5.9/2.6% | 4.1/12.5% | 5.7/8.5% | ||||||||
| BM4713 | 5.3/9.6% | 51.9/6.3% | 0.19/1.1% | 3.5/5.4% | 2.1/6.7% | 3.3/11% | ||||||||||
| BM4706 | 85.1/8.2% | 56.4/3.1% | 0.16/20% | 7.1/3.1% | 4.5/4.1% | 6.5/6.4% | ||||||||||
| BM4710 | 46.5/8.1% | 76.2/8.9% | 0.2/12.9% | 1.4/2.8% | 1/5.4% | 0.1/12% | 3.1/6.9% | 2/10.7% | 3.1/11.2% | |||||||
| MRSN 3405 | 73/5% | 72.8/7.2% | 0.2/7.7% | 1.5/16.2% | 1.1/9.1% | 0.1/3.8% | 5.4/7.6% | 3.5/5.8% | 5.6/5.1% | |||||||
| AYE | 0.76/18.8% | 94.4/7.7% | 0.17/18.3% | 1.3/12.3% | 0.87/3.8% | 0.09/14.2% | 2/4.6% | 1.3/7.2% | 2.8/6.1% | |||||||
| Group 3 | ||||||||||||||||
| BM4705 | 9.1/10% | 0.23/24.7% | 63.5/9.7% | 0.08/12.6% | 2.8/22.2% | 1.1/7.9% | 3.2/4.3% | 1.6/12.3% | ||||||||
| BM4709 | 76.3/11.7% | 0.47/24.9% | 12.3/11.6% | 0.12/12.2% | 1.8/9.4% | 0.82/10.5% | 0.07/7% | 3.1/8% | 2/9.6% | 2.9/8.9% | ||||||
| BM4712 | 72/6.9% | 0.3/5.7% | 64/5.3% | 0.32/4.2% | 2.3/1.9% | 0.85/6.5% | 2.1/6.7% | 1.3/8.1% | 2.9/1.8% | |||||||
| ANC 4097 | 0.04/31.9% | 56.1/2.7% | 0.49/24.3% | 0.81/2.8% | 0.17/8.2% | 1.5/7.2% | 0.6/5.8% | 0.16/4% | 3.7/5.3% | 2.4/4.9% | 4.7/9.2% | |||||
| BM4708 | 44.3/1.3% | 13.3/11.1% | 0.91/7.2% | 0.18/15.2% | 1/9.6% | 0.72/7.5% | 0.19/3.7% | 3.2/3.9% | 2.2/6.4% | 4.3/3.5% | ||||||
| BM4711 | 50.7/2.7% | 13.4/6.6% | 0.92/11.8% | 0.19/6.4% | 1.5/7.7% | 0.73/6.2% | 0.21/7.2% | 3.1/4.9% | 2.1/3% | 4.3/3.4% | ||||||
| BM4707 | 75.5/9.6% | 3/3.8% | 87/3.7% | 0.16/5.4% | 4/11.1% | 1.8/9% | 6.7/3% | 4.4/7.8% | 7.4/6.1% | |||||||
Evaluation of Bacterial Quantotypic Transitions
A. baumannii NIPH 527, ANC 4097, and BM4587, were diluted in series to obtain three independent concentration ranges of five points each, with a total of 15 points. For each point, enumeration was performed on three independent Petri dishes. In parallel, the 15 points were analyzed thrice by MS using the lysis, digestion, and SRM method described earlier. Bacterial numbers were expressed as Colony Forming Unit per ml (CFU/ml) using the Petri dish reference method. The numbers were corrected by counting bacterial clusters in suspensions under microscope to obtain cell numbers per ml (cell/ml). The resulting curve, cell/ml versus SRM a.u., enabled the calibration of bacterial load for the normalization of protein quantities per cell in samples.
RESULTS
Characterization of Antibiotic Resistance by Phenotype and WGS
The antibiotic resistance phenotypes determined by microdilution and the β-lactamase genes identified by WGS are listed in Table I. The 39 strains were grouped based on the presence of acquired β-lactamases: 17 strains were devoid of any acquired enzyme, 15 had one, and the remaining 7 produced multiple enzymes.
In the first group, most were susceptible to ticarcillin, cephalosporins, and carbapenems with six exceptions. Strain CIP 70.34T had reduced susceptibility to cephalosporins and carbapenems, NIPH 335 to ticarcillin and cefotaxime, NIPH 80 to ticarcillin, ceftazidime, and cefotaxime, and NIPH 1669, BM4704, and NIPH 2061 to all the tested β-lactams. Regulatory mutations, leading to overexpression of intrinsic β-lactamases and efflux pumps, were searched for in these strains. Five Insertion Sequences (IS) were detected upstream from a resident β-lactamase gene (blaOXA-51-like and/or blaADC) which can result in overexpression of the enzyme (2, 40). ISAba1 was found upstream from both blaOXA-51-like and blaADC in strains BM4704 and NIPH 2061. Overexpression of these genes was consistent with high resistance to both cephalosporins and carbapenems but was unable to account for a 4-fold difference in carbapenem MICs. In NIPH 1669, ISAba125 was identified upstream from blaADC consistent with resistance to cephalosporins. Substitutions T153A and D167N, in the dimerization and histidine-containing phosphotransfer domain of AdeS, were found in CIP 70.34T and NIPH 1669, respectively, putatively responsible for AdeABC overproduction and decreased susceptibility to ticarcillin and cephalosporins (8–9). In NIPH 80, a mutation in the DNA binding region of AdeN leading to overexpression of the AdeIJK pump (12) was found and could explain lower susceptibility to cephalosporins and carbapenems (4). However, this type of WGS-based analysis is fastidious and not yet comprehensive: presence of mutations can reflect polymorphisms and mutational alterations in regulatory mechanisms are still unknown. Therefore, the production levels of the intrinsic β-lactamases and of the efflux systems must be assessed quantitatively.
The second group comprised isolates having a single acquired β-lactamase (Table I). Strains NIPH 24, NIPH 290, NIPH 527, and NIPH 528 possessed a blaTEM-1 gene, for a narrow-spectrum class A β-lactamase, which confers high level resistance to ticarcillin. However, strain NIPH 290 also exhibited high level resistance to oxyiminocephalosporins, indicating that at least another mechanism was involved. Strains BM4701 and BM4702, producing a GES-type enzyme, were resistant to all β-lactams tested. Differences in the levels of resistance to cephalosporins and carbapenems were consistent with the functional properties of the two GES-type variants identified: GES-11 being a more efficient cephalosporinase but a less efficient carbapenemase than GES-14 (41). The presence of blaOXA-58 in strains BM4703 and NIPH 1734 could confer resistance to ticarcillin and carbapenems but not reduced susceptibility to ceftazidime and cefepime in NIPH 1734. Similarly, BM4714 having blaOXA-24 was resistant to ticarcillin and carbapenems but its reduced susceptibility to cefotaxime could not be explained by the presence of blaOXA-24 gene. The NIPH 1362 isolate had an acquired blaOXA-58 gene in addition to overproduction of both intrinsic blaADC and blaOXA-51, because of IS insertions, resulting in increased ticarcillin and carbapenems MICs. However, high-level resistance to cephalosporins, not efficiently hydrolyzed by OXA-58, suggests other mechanisms. Overproduction of intrinsic ADC-type cephalosporinase because of an ISAba1 copy and the carbapenemase genes blaOXA-23, blaOXA-24, or blaOXA-58 conferred resistance to all β-lactams in strains BM4713, BM4706, BM4710, and MRSN3405. The presence of blaVEB together with blaADC overexpression in strain AYE led to high-level resistance to ticarcillin and cephalosporins, and reduced susceptibility to the carbapenems. In the last group of strains with multiple β-lactam resistance genes, a synergistic effect of the mechanisms was generally observed resulting in resistance to all antibiotics. However, despite blaOXA-24, blaTEM-1, and insertion of ISAba1 upstream from blaADC, BM4705 was less resistant to cefepime than other ADC producers and less resistant to carbapenems than BM4714 only harboring the acquired blaOXA-24. A TetR transcriptional regulator AdeN, having only 90% identity with AdeN from other strains was found in this isolate, which could affect AdeIJK regulation. Unexpectedly, BM4709 and BM4712 exhibited different cefepime and carbapenem susceptibilities, despite analogous genotypes (i.e. ISAba1 upstream from blaADC and both blaOXA-23 and blaTEM-1). The presence of blaNDM-1, a metallo-carbapenemase gene, together with that of blaOXA-23 and blaTEM-1 in ANC 4097 conferred extremely high level of resistance levels to all β-lactams. Similarly, co-production of OXA-23 carbapenemase and PER-1 extended-spectrum β-lactamase in BM4708 and BM4711 also conferred resistance to all β-lactams. These data confirm that analysis of the β-lactam resistance phenotype in strains with multiple resistance determinants is often complex and may involve other mechanisms in addition to the production of one or more β-lactamases. Although the WGS data can provide information on the acquired determinants and the presence of insertion sequences, it has limitations in the understanding on how additional mechanisms which act synergistically with β-lactamases, such as efflux, contribute to the resistance phenotype.
Quality Assessment of Relative Protein Quantification by SRM
Despite adjustment at 4 McF, the CVs of bacterial quantities across all the experiments, estimated using bacterial quantotypic peptides, was 29.90% by SRM. This poor reproducibility prompted us to estimate the bacterial load directly via SRM to correct more accurately for sample preparation fluctuation. The correlation between bacterial quantotypic peptide summations and Petri dish enumeration led initially to parallel right curves. The reason for this observation was elucidated by microscopic examination of A. baumannii suspensions: single cells or double-cells were observed. Using three independent fields per strain, the numbers of cells per cluster were on the average 1.46, 1.75, and 1.1 for strains NIPH 527, ANC 4097, and BM4587, respectively. Assuming that every cluster leads to a single colony after growth on solid medium, these numbers were used to convert the measured CFU/ml to cells/ml. Cells/ml versus SRM signals of bacterial quantotypic transitions points were distributed on a single right curve (Fig. 2). The correlation was highly significant (p value 3.4 × 10−8 and squared Pearson's product-moment 0.914) demonstrating the ability of bacterial quantotypic transitions to accurately estimate bacterial counts and thus validating the normalization strategy adopted to assess the relative protein quantity per cell. After correction of this bacterial load disparity, mean CVs for protein quantitation were systematically reduced, e.g. 17.8% versus 7.1% and 18.6% versus 7.7% for AdeJ and ADC quantitation, respectively.
Fig. 2.
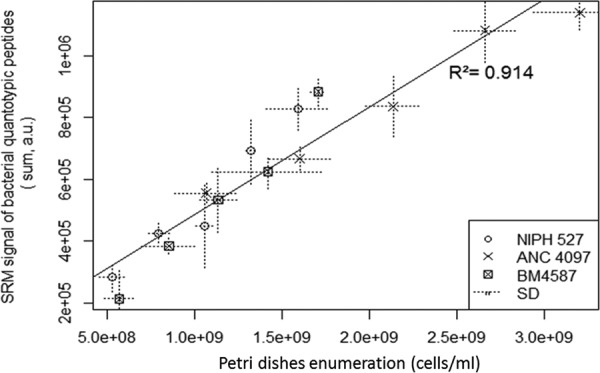
Calibration curve of SRM signals from bacterial quantotypic transitions versus cells/ml from Petri dish enumeration corrected by cell number per colony.
As in previous studies (18, 19), the quality of the protein measurements was assessed by verifying the preservation of the ratio among transitions belonging to the same protein. The quantities of quantotypic peptides from a given protein should be equimolar if they are not impaired by posttranslational modifications, stochastic digestion, or affected by contaminants. After selection of the peptides and transitions operated according to the Experimental Procedures, the correlations were rather high, Pearson's r average was 0.9845, the lower quartile was 0.9776 and the smallest observed r value was 0.9510 (r numbers are displayed in supplemental Table S3, r distributions in supplemental Fig. S1 and examples of typical correlation curves in supplemental Figs. S2). As a result, the CVs obtained for the three independent biological replicates (Table II) were 7.3% in median and 8.9% in average. Seventy-five percent of measurements had CVs below 10.4% and only 15/275 quantifications (i.e. 5.4%) had CVs higher than 20% (supplemental Fig. S3A). CVs, displayed in supplemental Fig. S3B, were uniformly distributed according to signal intensities, except for GES-type β-lactamases. In strains BM4701 and BM4702 producing this β-lactamase, we observed a clear difference among biological replicates (supplemental Fig. S2B) and, as a result, biological CVs were huge: 89.8% and 84.2%, respectively. The SRM quantitation seemed robust with measurements of 18 transitions from 6 peptides and excellent correlations (supplemental Fig. S2B). Of note, for both strains the bias was the same among the 3 biological replicates : replicates 1 had the lowest concentrations (0.19 and 0.24, respectively), replicates 2 the highest (1.1 and 1.6, respectively), and replicates 3 had intermediate concentrations (0.3 and 0.7, respectively). This trend was not observed for other proteins. For instance, ADC was quantified in BM4701 and BM4702 with 4.6% and 5.7% CVs, respectively. These results suggest potential growth-related differences in the blaGES expression levels, rather than analytical discrepancies. If GES was excluded, the mean CV dropped to 8.4% and 95.3% of the biological replicates had CVs below 20%.
Detection of β-lactam Resistance Determinants by SRM
All the targeted resistance mechanisms: acquired β-lactamases (i.e. GES, NDM-1, OXA-23, OXA-24, OXA-58, PER, TEM-1, and VEB), resident β-lactamases (i.e. ADC and OXA-51-like) and efflux pumps (i.e. AdeABC and AdeIJK) were quantified using multiplex SRM (supplemental Table S4). The various subgroups of OXA-type enzymes were distinguished by specific peptides (supplemental Table S5). Examples of characteristic SRM peaks are displayed in Fig. 3 and in supplemental Fig. S4.
Fig. 3.

SRM peaks. From left to right, up to bottom; A, AdeK in NIPH 2061 (3.0/7.4%); B, ADC in NIPH 1669 (9.7/13.7%); C, OXA-24 in BM4713 (5.3/9.6%); D, PER in BM4711 (13.4/6.6%).
Detection of Acquired β-lactamases
The GES, PER, TEM-1, VEB, and OXA-24 enzymes were detected by SRM as predicted by WGS: GES in BM4701 and BM4702; PER in BM4708 and BM4711; TEM-1 in BM4705, BM4709, BM4712, ANC 4097, NIPH 24, NIPH 290, NIPH 527, and NIPH 528; VEB in AYE; and OXA-24 in BM4705, BM4713, and BM4714.
SRM detected unambiguously the OXA-23 group in BM4706, BM4707, BM4708, BM4709, BM4710, BM4711, BM4712, ANC 4097, and MRSN 3405. Surprisingly, OXA-23 was also detected in one of the three biological replicates from strains BM4714 and CIP 70.34T. It was characterized by 18 and 21 transitions corresponding to 6 and 7 peptides, respectively. The detection in a single replicate of both strains was therefore significant but the relative protein amount was much lower, 0.47/NA and 3.62/NA, respectively, whereas an average of 64.4 was measured in the 9 other positive strains. False positive results because of HPLC memory effects were ruled out because complete recovery of columns was found in blank injections before and after the sample run. False negative results were also ruled out by the correct detection of bacterial quantotypic peptides and of peptides from ADC and OXA-51-like in the two strains. Interestingly, OXA-23 was initially detected in BM4714 using a multiplex PCR (30), but only OXA-24 was found in the present WGS analysis. A new PCR confirmed OXA-24 production but was unable to detect OXA-23. These discrepant results using both genomics and proteomics tools could be interpreted as plasmid instability. Several colonies are mixed to perform both techniques and SRM replicates come from three independent cultures. This particular case was not investigated further because OXA-23 and OXA-24 confer similar resistance phenotypes. As opposed to BM4714, CIP 70.34T was susceptible to most antibiotics (Table I). Thus, detection of OXA-23 in this strain was not studied further. New WGS or SRM analyses, after culture in the presence of antibiotics, could have been helpful to investigate these two cases.
The OXA-58 group was detected by SRM in BM4703, BM4707, NIPH 1362, and NIPH 1734. It was expected in BM4703, BM4707, and in NIPH 1734 but not in NIPH 1362, based on publicly available WGS (Acinetobacter initiative, Broad Institute). A new WGS of NIPH 1362 identified a gene for OXA-58 (supplemental Fig. S5).
As expected, NDM-1 was detected in ANC 4097. Of note, a single peptide (FGDLVFR) was detected with 3 transitions for NDM-1, 2 other peptides (AAITHTAR, AFGAAFPK) were detectable with 2 transitions each but were not considered. This β-lactamase was therefore the protein detected with the lower number of transitions but the level of confidence was still acceptable (see peak signals in supplemental Fig. S4).
Overproduction of Resident β-lactamases
The resident A. baumannii OXA-51-like carbapenemase is produced at low levels but can be overproduced when upstream insertion of an IS element provides a strong promoter (2). OXA-51-like enzymes were targeted by 12 transitions (i.e. 4 peptides) enabling co-detection of 51-like variants: 64, 65, 66, 69, 71, 78, 88, 98, 109, and 128. Characteristic transitions were detected in at least one of three biological replicates in all the strains except NIPH 601 and NIPH 67. A 2.0 cut-off was set to significantly differentiate between basal level and overproduction (Fig. 4, right part, p value < 0.0001, t test). Using this criterion, overproduction was found in BM4704, NIPH 1362, and NIPH 2061 (as expected according to the presence of ISAba1 upstream from the corresponding structural gene in the three strains). Basal expression levels were close to the sensitivity limit thus, for some strains, OXA-51-like proteins were not detected in all 3 replicates. In these samples, OXA-51-like peptides seemed to be present with transition areas just under the 3000 a.u. cut off.
Fig. 4.

Differentiation between ADC (A) and OXA-51-like (B) accumulation for natural and overproduced protein, t test p value <0.0001 for both ADC and OXA51.
The blaADC gene is also transcribed at low levels in A. baumannii (3) but SRM was sensitive enough to detect low levels of ADC production in the wild-type strains. Using a cut off of 2.0, as for OXA-51, SRM significantly distinguished the strains based on the level of ADC production (Fig. 4, left part, p value < 0.0001, t test). In 12 strains, the presence of an ISAba1 element upstream from blaADC, found by WGS (Table I), was systematically associated with high level ADC production: the average relative quantity was 62.6 compared with 0.71 for strains with basal level in the absence of IS. Insertion element ISAba125 (42) known to cause ADC overproduction was found in NIPH 1669. This strain displayed an intermediate accumulation level of 9.7/13.7%. Two other strains had also relative intermediate amounts: CIP 70.34T and NIPH 1734 with 7.4/10.9% and 2.9/1.0%, respectively. Being ten times higher than the basal level, accumulation of ADC conferred clearly resistance in CIP 70.34T. The clinical relevance of ADC concentration in NIPH 1734 may be questionable but low-level resistance was observed to ceftazidime and cefotaxime. In these strains neither ISAba1 nor ISAba125 were found but still unknown mechanisms of regulation modulation could be present.
Ade Efflux Systems
The AdeABC efflux system was detected with 30, 42, and 10 transitions corresponding to 10, 14, and 4 peptides for AdeA, AdeB, and AdeC, respectively. The average CVs of SRM biological replicates were 8.3%, 8.2%, and 10.0% for AdeA, AdeB, and AdeC, respectively. Quantifications were contrasted for this three-component efflux system. The entire system or only one or two proteins were detected in 11 and 12 strains, respectively, and none of these proteins were detected in the remaining 16 strains. AdeABC was not detected in the susceptible strain BM4587. Absence of detection corresponds to a basal level of protein accumulation below the sensitivity of SRM of this tightly regulated pump. On the contrary, AdeA, AdeB, and AdeC were highly produced in NIPH 290 raising 23.3/4.9%, 10.4/39.9% and 3.5/30.0%, respectively. In this strain AdeA was reproducibly quantitated but, surprisingly, we observed poor CVs for both AdeB and AdeC, with proportionally the same lower quantities in the third replicate. AdeABC were also moderately accumulated in CIP 70.34T with protein quantities of 11.4/9.3% and 4.1/4.3% for AdeA and AdeB, respectively but, surprisingly, AdeC was not detected. A single peptide showed three transitions above the threshold in one of the 3 biological replicates but was excluded because incompatible with an r > 0.95 in NIPH 290 and an absence in other strains. Excluding these up-regulated two cases, the level of accumulation was almost equivalent in the strains when the proteins were detected; levels of accumulation (SDs in curly brackets) were: 2.25 {1.11}, 1.05 {0.33}, and 0.11 {0.05} for AdeA, AdeB, and AdeC, respectively.
On the contrary, the AdeFGH complex was only detected in the engineered strain BM4690 (4), which overproduces AdeFGH (data not shown) but not in the 39 other isolates. Indeed, fold changes measured by RT-qPCR in BM4690 compared with BM4587 were 509, 402, and 82 for AdeF, AdeG, and AdeH, respectively, whereas the average fold change for AdeG was 2.4 with the other strains.
The AdeIJK complex was found in all the strains using 15, 33, and 30 transitions, corresponding to 5, 11, and 10 peptides for AdeI, AdeJ, and AdeK, respectively. This large number of peptides per protein ensured an accurate detection across samples. Average CVs of SRM quantifications were 5.3%, 7.1%, and 7.7% for AdeI, AdeJ, and AdeK, respectively. Complex accumulation was mostly constant over strains; levels of accumulation with {SDs} across samples were 3.65 {1.57}, 2.29 {1.00}, and 4.01 {1.51} for AdeI, AdeJ, and AdeK, respectively. These observations agree with constitutive basal production of AdeIJK and a contribution of the pump to the broad intrinsic resistance of A. baumannii (4). Interestingly, despite the presence of the corresponding gene, AdeJ was not detected in BM4705. The RT-qPCR adeJ expression was also very low: 0.3/66.7%. SRM and RT-qPCR were therefore in agreement with the loss of AdeJ production and possibly the presence of a non-functional AdeIJK pump.
Comparison of the quantitation of the three AdeIJK proteins indicated that the correlation was significant. Indeed, p values of Pearson's product moment correlation were 2.2 × 10−16 for AdeI/AdeK and inferior to 2.2 × 10−16 for AdeI/AdeJ and AdeJ/AdeK, respectively (Fig. 5). Besides, the slopes of linear regressions were 0.60, 0.88, and 1.41 for AdeI/AdeJ, AdeI/AdeK, and AdeK/AdeJ, respectively. The amounts of the three proteins of the pumps were compatible with a 1/1/1 stoichiometry (39), as expected from co-transcription of the three corresponding genes (7).
Fig. 5.

Correlation of protein expression for AdeI and AdeJ; AdeI and AdeK; AdeK and AdeJ (from left to right). Linear regression equation fitted using least squares approaches are indicated in top left corner. p values of Pearson's product moment correlation test were < 2.2 × 10–16, 2.2 × 10–16 and < 2.2 × 10–16 for AdeI and AdeJ; AdeI and AdeK; AdeK and AdeJ, respectively.
Deciphering Phenotypic Resistance by Genomics and Proteomics
As expected, in the first group composed of strains devoid of acquired β-lactamase determinants, the predicted overexpression of blaOXA-51-like, blaADC, adeABC, and adeIJK genes was confirmed by SRM. Moreover, SRM quantitation brought a new information, the 4-fold difference in carbapenem MICs between BM4704 and NIPH 2061 could be explained by the 2.5-fold difference of OXA-51 amounts. In CIP 70.34T, ADC overproduction was not anticipated from the genomic analysis but could be responsible for lower susceptibility to ticarcillin and cephalosporins, associated with predicted adeABC overexpression. However, abundant AdeA and AdeB without detectable AdeC, suggests the recruitment of another porin to achieve RND pump functionality. Overexpression of adeIJK was correctly predicted in NIPH 80 and similar AdeIJK levels in NIPH 335 could explain resistance to cephalosprins in both strains (4). A single amino acid substitution in AdeN, G530A to G177E, was found in NIPH 335 but its consequences were not studied.
In the second group of strains with a single acquired β-lactamase gene, the 2- to 4-fold carbapenem MIC differences between BM4703 and NIPH 1734 could be because of the 3.2 times more elevated production of OXA-58 in NIPH 1734. Among the four strains producing similar amounts of the TEM-1 enzyme, NIPH 290 had a 2- to 16-fold higher cefepime MIC without genomic explanation. This resistance level was probably because of the extremely high quantity of AdeABC production. Similarly, the high AdeIJK amounts in BM4714 and NIPH 1362, appeared associated with high-level resistance to cephalosporins not explained by the β-lactamase genes. The BM4706, BM4710, and MRSN3405 isolates harboring blaOXA-23 together with overexpression of blaADC presented similar levels of enzyme production resulting in similar β-lactam resistance phenotypes. The additive effect of AdeIJK overproduction in BM4706 and MRSN was not significant and was probably masked by the high levels of ADC and OXA-23 production. In the case of BM4701 and BM4702, almost the same amounts of GES enzyme and Ade systems production were observed, and the MIC differences reflected the functional properties of the GES-type variants (higher cephalosporinase but lower carbapenemase activity of GES-11 when compared with GES-14) (41). Low level of ADC production in NIPH 1734 was not predicted by genomics but could contribute to a moderate loss of susceptibility to ceftazidime and cefotaxime.
Knowledge of the level of protein amounts allowed to partially clarify the more complex third group composed of strains having multiple β-lactam-specific resistance determinants. BM4705 was less resistant to cefepime and carbapenems than expected despite the detection of ADC, OXA-24, and TEM-1 but absence of AdeJ, possibly because of an unusual adeN sequence, suggests a non-functional AdeIJK pump that could explain a more susceptible phenotype. The differences in cefepime and carbapenem MICs with strains BM4709 and BM4712 remain not entirely elucidated. These isolates had equivalent efflux pumps, OXA-23, and TEM-1 amounts but a 5.2-fold elevated production of ADC was detected in BM4712. ADC could contribute to cefepime resistance but is less likely to impact on carbapenems. Other resistance determinants not considered here, such as porin-mediated alterations of outer membrane permeability, could be involved (45). However, the CarO porin conferring resistance to imipenem was not detected by WGS, neither in BM4709 nor in BM4712. Strains BM4708 and BM4711 having the same resistance genotype, and similar production levels of enzymes and of RND efflux systems, had very similar β-lactams MICs.
DISCUSSION
Quantitative Protein Measurement Using Targeted MS
Normalization of bacterial counts is a prerequisite for accurate estimation of protein production. The initial normalization of bacterial numbers was by optical density adjustment of bacterial suspensions to 4 McF. This appeared to lack precision for bacterial quantity estimations and adjustment for sample preparation fluctuations. Thus, bacterial numbers were estimated using normalization by bacterial quantotypic peptides (46). The peptides, chosen essentially from ribosomal and structural proteins, play a normalization role equivalent to that of housekeeping genes in transcriptomic.
Similarly to a previous work studying resistance in P. aeruginosa by label free SRM (18), relative quantitation was sufficient in this study and the use of internal standards (47–49) appeared unnecessary. The addition of artificial molecules, synthetized with heavy isotopes, would allow assessing the absolute protein quantity and the exact protein numbers per cell. Such an approach was recently used by Lawless et al. (50) for absolute quantification of over 1800 yeast proteins via SRM. However, we demonstrate accurate relative quantitation without the need for heavy internal standards. Spiking specific synthetic molecules increases the cost of the test depending on the number of proteins and bacterial species studied. With bacterial quantotypic peptides, relative quantitation was obtained at no additional cost and without pipetting errors because of calibrant addition. This strategy eases the use of SRM to answer biological questions in domains where relative quantitation is required (46). Reaching analytical reproducibility, specificity, and sensitivity close to ELISA, SRM is now an attractive alternative when multiplex protein assays should be developed in a short time.
A special attention was paid to a putative quantitation bias generated by mutated peptides. Mutations are frequent in bacteria and were detected by WGS in the genes encoding ADC, OXA-51, OXA-58, TEM, AdeA, AdeB, AdeC, AdeJ, and AdeK proteins. Although resulting in single amino acid changes, the mutations accounted for modifications of the peptide mass (except Isoleucine/Leucine substitutions), the retention time, the ionization efficiency, and the fragmentation pattern. Alterations of these physical properties prevent the mutated peptide to be detected using the SRM transitions from the wild peptide. Both wild and mutated peptides could be easily multiplexed in the same SRM method but this was considered inappropriate to perform accurate relative quantitation. Ionization and fragmentation intensities depending on peptide sequences, substitution of signal integrations from a wild peptide by its corresponding mutated counterpart may therefore account for quantity discrepancies. We thus estimated missing peptide signals using the information provided by other peptides from the same protein.
The TopTra2/TopPep3 strategy proposed initially by Ludwig et al. (39), could be a good compromise between simplicity and accuracy. It favors the most intense transitions and, therefore, minimizes the risk of overestimation by co-detection of the biological noise. Ludwig et al. demonstrated a linear relationship between label-free SRM intensities and protein copies per cell, with a squared Pearson coefficient R2 = 0.88. However, we could not use directly Ludwig's equation because our ESI-QqQ-MS instrument was different. We thus preferred to normalize SRM with bacterial quantotypic peptides and to provide relative abundances rather than absolute protein molecules per cell. Nonetheless, it is possible to transform our simple approach into an absolute quantitation method via appropriate calibration using heavy internal standards.
Here, using a conventional bore chromatography and a short 30-min SRM gradient we were able to quantify 186 peptides representing twenty bacterial proteins (including eleven bacterial quantotypic peptides from four proteins). If needed, more targeted proteins could be easily monitored; the method was very far from the multiplexing capabilities of SRM. Moreover, the robustness of SRM hyphenated with standard chromatography was shown to be compatible with clinical applications (19, 51). A targeted strategy seems therefore a good compromise to accurately measure a reasonable number of proteins and to address biological questions at the protein level.
As expected SRM enabled highly specific protein detection in agreement with WGS predictions. Using, in quadrupoles Q1 and Q3, a resolution of 0.7 ± 0.1 atomic mass unit full width at half maximum (18–19), a single amino acid difference, accounting for a mass shift above 1 Da, should impair a peptide detection. Similarly, a peptide becomes undetectable if a sequence alteration modifies the fragmentation yields or moves the chromatographic retention time out of the 155 s window. Consequently, the probability to confuse two different sequences is very low, except may be in the case of an I/L mutation. The measurement of several peptides per protein strengthens the confidence in each protein detection and explains the high specificity of SRM assays.
Quantitative Measurement of Resistance Gene Expression by MS Versus RT-qPCR
In the past decades, when the development of large protein multiplexes at an affordable cost was a true challenge, transcriptomics was seen as an alternative solution. However, today MS has reached performances which question this approach. Protein quantities provide an information closer to the phenotype than mRNA and CV comparisons of SRM versus RT-qPCR are in favor of SRM. MS has the advantage to measure protein accumulation in cells rather than a transcription flux. It allows evaluating enzymes amounts for antibiotic degradation kinetics, efflux pump densities for assessment of antibiotic export and amounts of target for resistance by sequestration of the drug (52). SRM has moreover a reasonable time to results (around 1 h per sample), a high multiplexing capability (up to several hundred of peptides), and does not require expensive or specific reagents (i.e. no specific probes). This makes assay developments particularly straightforward when multiplexing is required. The analytical reproducibility of SRM enabled CVs below 20% (Table II) which is better than RT-qPCR reproducibility (supplemental Fig. S6). Observed correlations between mRNA and protein measurements were poor as already demonstrated (43–44). Protein abundance is not only determined by the rate of transcription but also by translational regulatory mechanisms and protein turnover. The latter is difficult to predict or measure, could be differently regulated among the strains, and could explain the observed discrepancy between transcription flow and protein accumulation. This is supported by the statement of Liu and Aebersold (44): “the relative contribution of mRNA- versus protein-level regulation seems to be dependent on the temporal scale, on the complexity level of the biological system, and on the type of perturbation applied”. This is an additional argument in favor of direct quantitation of proteins when phenotypes depend on their abundance.
Assuming that Ludwig's strategy (39) could apply to A. baumannii, SRM enabled the comparison of various enzyme amounts in various strains, with the enzyme kinetic parameters for β-lactam hydrolysis. For example, TEM-1 hydrolyzes ampicillin (kcat/Km, 3.8 × 107 m−1.s−1) (53) 10-fold more efficiently than OXA-23 (kcat/Km, 5.6 × 106 m−1.s−1) (54). This difference in catalytic properties of the enzymes seems to be counterbalanced by a level of accumulation 100 times more important for OXA-23 than for TEM-1. The accumulation of acquired enzymes was in the same order of magnitude among strains for some β-lactamases. CV of protein relative quantities across strains was 31.2%, 0.80%, 31.3%, 53.9%, 44.3%, and 97.5% for GES, PER, TEM-1, OXA-23, OXA-24, and OXA-58, respectively. On the contrary, for a given strain the level of accumulation of the various enzymes varied notably. For example, in BM4709, TEM-1 relative quantity was 0.49/24.3% but 56.1/2.7% for OXA-23. Globally, the accumulation of OXA-23 seemed to be very high (mean accumulation 53.1 compared with TEM-1 or VEB (mean accumulation 0.33 and 0.76, respectively). For OXA-24, GES, PER, and OXA-58 acquired β-lactamases, the mean accumulation level seemed similar: 9.3, 10.4, 13.3, and 21.4, respectively.
Resistance Phenotypes Versus Molecular Information
In an attempt to account for the resistance phenotypes by a molecular approach and in a comprehensive way, we tried to infer antibiotic resistance from both genomics and proteomics data. The presence of enzymes was deduced from WGS and protein quantities from SRM. DNA sequencing therefore was preferred to achieve full characterization of protein variants, MS leading only to partial sequence coverage. SRM confirmed or detected unexpected enzymes and was pivotal in providing quantitative information. Both technologies are therefore complementary and strengthen the molecular analysis of resistance phenotypes. A deep sequence coverage determination and quantitation of the β-lactamase abundance were necessary to correctly interpret the enzymatic behavior. On one hand, substitutions in β-lactamases may affect the functional properties of the enzyme, as illustrated by BM4701 and BM4702, in which production of GES-14 or GES-11 variants led to different phenotypes. On the other hand, the β-lactamase amount is directly proportional to the β-lactam hydrolysis rate (55). Quantitative differences in acquired OXA-58 as well as intrinsic ADC and OXA-51 were observed and associated with consistent variations of the MICs.
Expression of most resistance genes is tightly regulated. Unfortunately, gene expression levels cannot always be predicted by genomics because mutations in regulatory regions could be polymorphisms or not yet reported functional alterations. Protein overproduction was correctly predicted when ISs were inserted upstream from blaOXA-51-like or blaADC genes but moderate levels of ADC production were not explained in two isolates (CIP 70.34T and NIPH 1734). However, protein levels accounted for the resistance phenotypes.
CONCLUSIONS
SRM analysis, requiring a turnaround time of one hour, was able to decipher multifactorial resistance in A. baumannii via quantitation of antibiotic resistance determinants. The production levels of sixteen resistance proteins was measured with a single SRM method targeting 186 peptides: eight acquired β-lactamases (i.e. GES, NDM-1, OXA-23, OXA-24, OXA-58, PER, TEM-1, and VEB), two resident β-lactamases (i.e. ADC and OXA-51-like), and two efflux tripartite systems (i.e. AdeABC and AdeIJK). After normalization by “bacterial quantotypic peptides,” these proteins were accurately quantified with CVs of 8.93% on average over three biological replicates. This relative quantification was particularly relevant to differentiate basal level and overproduction of proteins leading to susceptible or resistant phenotypes, respectively. The ability of SRM to easily generate accurate multiplexed quantitation makes mass spectrometry the favored choice for direct quantitation of gene products. By combining WGS and MS, molecular characterization of resistance determinants is obtained using two orthogonal and complementary technologies. This paves the way to the understanding of the molecular mechanisms responsible for gene regulation and resistance phenotypes.
Data availability
The mass spectrometry data were deposited in Peptide Atlas (PASS01051) and the scaffolds of the whole genome sequencing to EMBL Nucleotide Sequence Submissions (http://www.ebi.ac.uk/).
Supplementary Material
Acknowledgments
We thank P. Mollon and B. Muller for DNA sequencing and E. Santiago-Allexant for assistance in interpretation.
Footnotes
* T.C., Y.C., and C.Ba. were funded by the Association Nationale de la Recherche et de la Technologie (ANRT), E.J.Y., P.C, and C.G.C. by an unrestricted grant from Reckitt-Benckiser.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- RND
- resistance-nodulation-cell division
- CAZ
- Ceftazidime
- CFU
- Colony Forming Unit
- CLSI
- Clinical & Laboratory Standards Institute
- CTX
- Cefotaxime
- CV
- Coefficient of Variation
- FEP
- Cefepime
- IPM
- Imipenem
- MEM
- Meropenem
- MIC
- Minimal Inhibitory Concentration
- RT-qPCR
- Real-Time quantitative Polymerase Chain Reaction
- SD
- Standard Deviation
- SRM
- Selected Reaction Monitoring
- TIC
- Ticarcillin
- WGS
- Whole Genome Sequencing.
REFERENCES
- 1. Low T. Y., and Heck A. J. (2015) Reconciling proteomics with next generation sequencing. Curr. Opin. Chem. Biol. 30, 14–20 [DOI] [PubMed] [Google Scholar]
- 2. Turton J. F., Ward M. E., Woodford N., Kaufmann M. E., Pike R., Livermore D. M., and Pitt T. L. (2006) The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol. Lett. 258, 72–77 [DOI] [PubMed] [Google Scholar]
- 3. Poirel L., and Nordmann P. (2006) Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clin. Microbiol. Infection 12, 826–836 [DOI] [PubMed] [Google Scholar]
- 4. Yoon E. J., Chabane Y. N., Goussard S., Snesrud E., Courvalin P., De E., and Grillot-Courvalin C. (2015) Contribution of resistance-nodulation-cell division efflux systems to antibiotic resistance and biofilm formation in Acinetobacter baumannii. mBio 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Magnet S., Courvalin P., and Lambert T. (2001) Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 45, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coyne S., Rosenfeld N., Lambert T., Courvalin P., and Perichon B. (2010) Overexpression of resistance-nodulation-cell division pump AdeFGH confers multidrug resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 54, 4389–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Damier-Piolle L., Magnet S., Bremont S., Lambert T., and Courvalin P. (2008) AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob. Agents Chemother. 52, 557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marchand I., Damier-Piolle L., Courvalin P., and Lambert T. (2004) Expression of the RND-type efflux pump AdeABC in Acinetobacter baumannii is regulated by the AdeRS two-component system. Antimicrob. Agents Chemother. 48, 3298–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoon E. J., Courvalin P., and Grillot-Courvalin C. (2013) RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: major role for AdeABC overexpression and AdeRS mutations. Antimicrob. Agents Chemother. 57, 2989–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coyne S., Guigon G., Courvalin P., and Perichon B. (2010) Screening and quantification of the expression of antibiotic resistance genes in Acinetobacter baumannii with a microarray. Antimicrob. Agents Chemother. 54, 333–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernando D., Zhanel G., and Kumar A. (2013) Antibiotic resistance and expression of resistance-nodulation-division pump- and outer membrane porin-encoding genes in Acinetobacter species isolated from Canadian hospitals. Can. J. Infectious Dis. Med. Microbiol. 24, 17–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenfeld N., Bouchier C., Courvalin P., and Perichon B. (2012) Expression of the resistance-nodulation-cell division pump AdeIJK in Acinetobacter baumannii is regulated by AdeN, a TetR-type regulator. Antimicrob. Agents Chemother. 56, 2504–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heritier C., Poirel L., Lambert T., and Nordmann P. (2005) Contribution of acquired carbapenem-hydrolyzing oxacillinases to carbapenem resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 49, 3198–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tiwari V., and Tiwari M. (2014) Quantitative proteomics to study carbapenem resistance in Acinetobacter baumannii. Front. Microbiol. 5, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trip H., Mende K., Majchrzykiewicz-Koehorst J. A., Sedee N. J., Hulst A. G., Jansen H. J., Murray C. K., and Paauw A. (2015) Simultaneous identification of multiple beta-lactamases in Acinetobacter baumannii in relation to carbapenem and ceftazidime resistance, using liquid chromatography-tandem mass spectrometry. J. Clin. Microbiol. 53, 1927–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee S. Y., Yun S. H., Lee Y. G., Choi C. W., Leem S. H., Park E. C., Kim G. H., Lee J. C., and Kim S. I. (2014) Proteogenomic characterization of antimicrobial resistance in extensively drug-resistant Acinetobacter baumannii DU202. J. Antimicrob. Chemother. 69, 1483–1491 [DOI] [PubMed] [Google Scholar]
- 17. Wang H., Drake S. K., Yong C., Gucek M., Tropea M., Rosenberg A. Z., Dekker J. P., and Suffredini A. F. (2016) A novel peptidomic approach to strain typing of clinical Acinetobacter baumannii isolates using mass spectrometry. Clin. Chem. 62, 866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Charretier Y., Kohler T., Cecchini T., Bardet C., Cherkaoui A., Llanes C., Bogaerts P., Chatellier S., Charrier J. P., and Schrenzel J. (2015) Label-free SRM-based relative quantification of antibiotic resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Front. Microbiol. 6, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charretier Y., Dauwalder O., Franceschi C., Degout-Charmette E., Zambardi G., Cecchini T., Bardet C., Lacoux X., Dufour P., Veron L., Rostaing H., Lanet V., Fortin T., Beaulieu C., Perrot N., Dechaume D., Pons S., Girard V., Salvador A., Durand G., Mallard F., Theretz A., Broyer P., Chatellier S., Gervasi G., Van N. M., Ann R. C., Van B. A., Lemoine J., Vandenesch F., and Charrier J. P. (2015) Rapid bacterial identification, resistance, virulence and type profiling using selected reaction monitoring mass spectrometry. Scientific Reports 5, 13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouvet P., and Grimont P. (1986) Taxonomy of the genus Acinetobacter with the recognition of Acinetobacter baumannii sp. nov., Acinetobacter haemolyticus sp. nov. Acinetobacter johnsonii sp. nov., and Acinetobacter junii sp. nov., and emended descriptions of Acinetobacter calcoaceticus and Acinetobacter lwoffii. Int. J. Syst. Evolutionary Microbiol. 36, 228–240 [Google Scholar]
- 21. Dijkshoorn L., Aucken H., Gerner-Smidt P., Janssen P., Kaufmann M. E., Garaizar J., Ursing J., and Pitt T. L. (1996) Comparison of outbreak and nonoutbreak Acinetobacter baumannii strains by genotypic and phenotypic methods. J. Clin. Microbiol. 34, 1519–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nemec A., Janda L., Melter O., and Dijkshoorn L. (1999) Genotypic and phenotypic similarity of multiresistant Acinetobacter baumannii isolates in the Czech Republic. J. Med. Microbiol. 48, 287–296 [DOI] [PubMed] [Google Scholar]
- 23. Nemec A., Dijkshoorn L., and van der Reijden T. J. (2004) Long-term predominance of two pan-European clones among multi-resistant Acinetobacter baumannii strains in the Czech Republic. J. Med. Microbiol. 53, 147–153 [DOI] [PubMed] [Google Scholar]
- 24. Fournier P. E., Vallenet D., Barbe V., Audic S., Ogata H., Poirel L., Richet H., Robert C., Mangenot S., Abergel C., Nordmann P., Weissenbach J., Raoult D., and Claverie J. M. (2006) Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genetics 2, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diancourt L., Passet V., Nemec A., Dijkshoorn L., and Brisse S. (2010) The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PloS ONE 5, e10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nemec A., Krizova L., Maixnerova M., van der Reijden T. J., Deschaght P., Passet V., Vaneechoutte M., Brisse S., and Dijkshoorn L. (2011) Genotypic and phenotypic characterization of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex with the proposal of Acinetobacter pittii sp. nov. (formerly Acinetobacter genomic species 3) and Acinetobacter nosocomialis sp. nov. (formerly Acinetobacter genomic species 13TU). Res. Microbiol. 162, 393–404 [DOI] [PubMed] [Google Scholar]
- 27. Krizova L., Bonnin R. A., Nordmann P., Nemec A., and Poirel L. (2012) Characterization of a multidrug-resistant Acinetobacter baumannii strain carrying the blaNDM-1 and blaOXA-23 carbapenemase genes from the Czech Republic. J. Antimicrob. Chemother. 67, 1550–1552 [DOI] [PubMed] [Google Scholar]
- 28. Lesho E., Yoon E. J., McGann P., Snesrud E., Kwak Y., Milillo M., Onmus-Leone F., Preston L., St C. K., Nikolich M., Viscount H., Wortmann G., Zapor M., Grillot-Courvalin C., Courvalin P., Clifford R., and Waterman P. E. (2013) Emergence of colistin-resistance in extremely drug-resistant Acinetobacter baumannii containing a novel pmrCAB operon during colistin therapy of wound infections. J. Infectious Dis. 208, 1142–1151 [DOI] [PubMed] [Google Scholar]
- 29. Jeannot K., Diancourt L., Vaux S., Thouverez M., Ribeiro A., Coignard B., Courvalin P., and Brisse S. (2014) Molecular epidemiology of carbapenem non-susceptible Acinetobacter baumannii in France. PloS ONE 9, e115452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perichon B., Goussard S., Walewski V., Krizova L., Cerqueira G., Murphy C., Feldgarden M., Wortman J., Clermont D., Nemec A., and Courvalin P. (2014) Identification of 50 class D beta-lactamases and 65 Acinetobacter-derived cephalosporinases in Acinetobacter spp. Antimicrob. Agents Chemother. 58, 936–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Worboys J. D., Sinclair J., Yuan Y., and Jorgensen C. (2014) Systematic evaluation of quantotypic peptides for targeted analysis of the human kinome. Nat. Methods 11, 1041–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brownridge P., Holman S. W., Gaskell S. J., Grant C. M., Harman V. M., Hubbard S. J., Lanthaler K., Lawless C., O'Cualain R., Sims P., Watkins R., and Beynon R. J. (2011) Global absolute quantification of a proteome: Challenges in the deployment of a QconCAT strategy. Proteomics 11, 2957–2970 [DOI] [PubMed] [Google Scholar]
- 33. Clinical and laboratory Standards Institute. (2015) Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fith Informational Supplement. M100-S25, In: Clinical and Laboratory Standards Institute (ed), Wayne, PA [Google Scholar]
- 34. Coil D., Jospin G., and Darling A. E. (2015) A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589 [DOI] [PubMed] [Google Scholar]
- 35. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 36. McFarland J. (1907) An instrument for estimating the number of bacteria in suspensions used for calculating the opsonic index and for vaccines. JAMA 49, 1176–1178 [Google Scholar]
- 37. Bereman M. S., MacLean B., Tomazela D. M., Liebler D. C., and MacCoss M. J. (2012) The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 12, 1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics (Oxford, England) 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ludwig C., Claassen M., Schmidt A., and Aebersold R. (2012) Estimation of absolute protein quantities of unlabeled samples by selected reaction monitoring mass spectrometry. Mol. Cell. Proteomics 11, M111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heritier C., Poirel L., and Nordmann P. (2006) Cephalosporinase over-expression resulting from insertion of ISAba1 in Acinetobacter baumannii. Clin. Microbiol. Infection 12, 123–130 [DOI] [PubMed] [Google Scholar]
- 41. Delbruck H., Bogaerts P., Kupper M. B., Rezende de, Bennink C. R. S., Glupczynski Y., Galleni M., Hoffmann K. M., and Bebrone C. (2012) Kinetic and crystallographic studies of extended-spectrum GES-11, GES-12, and GES-14 beta-lactamases. Antimicrob. Agents Chemother. 56, 5618–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hamidian M., Nigro S. J., and Hall R. M. (2012) Variants of the gentamicin and tobramycin resistance plasmid pRAY are widely distributed in Acinetobacter. J. Antimicrob. Chemother. 67, 2833–2836 [DOI] [PubMed] [Google Scholar]
- 43. Anderson L., and Seilhamer J. (1997) A comparison of selected mRNA and protein abundances in human liver. Electrophoresis 18, 533–537 [DOI] [PubMed] [Google Scholar]
- 44. Liu Y., Beyer A., and Aebersold R. (2016) On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550 [DOI] [PubMed] [Google Scholar]
- 45. Perez F., Hujer A. M., Hujer K. M., Decker B. K., Rather P. N., and Bonomo R. A. (2007) Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 51, 3471–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bluemlein K., and Ralser M. (2011) Monitoring protein expression in whole-cell extracts by targeted label- and standard-free LC-MS/MS. Nat. Protoc. 6, 859–869 [DOI] [PubMed] [Google Scholar]
- 47. Gerber S. A., Rush J., Stemman O., Kirschner M. W., and Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Picard G., Lebert D., Louwagie M., Adrait A., Huillet C., Vandenesch F., Bruley C., Garin J., Jaquinod M., and Brun V. (2012) PSAQ standards for accurate MS-based quantification of proteins: from the concept to biomedical applications. J. Mass Spectrometry 47, 1353–1363 [DOI] [PubMed] [Google Scholar]
- 49. Pratt J. M., Simpson D. M., Doherty M. K., Rivers J., Gaskell S. J., and Beynon R. J. (2006) Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protocols 1, 1029–1043 [DOI] [PubMed] [Google Scholar]
- 50. Lawless C., Holman S. W., Brownridge P., Lanthaler K., Harman V. M., Watkins R., Hammond D. E., Miller R. L., Sims P. F., Grant C. M., Eyers C. E., Beynon R. J., and Hubbard S. J. (2016) Direct and absolute quantification of over 1800 yeast proteins via selected reaction monitoring. Mol. Cell, Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fortin T., Salvador A., Charrier J. P., Lenz C., Lacoux X., Morla A., Choquet-Kastylevsky G., and Lemoine J. (2009) Clinical quantitation of prostate-specific antigen biomarker in the low nanogram/milliliter range by conventional bore liquid chromatography-tandem mass spectrometry (multiple reaction monitoring) coupling and correlation with ELISA tests. Mol. Cell. Proteomics 8, 1006–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McGann P., Courvalin P., Snesrud E., Clifford R. J., Yoon E. J., Onmus-Leone F., Ong A. C., Kwak Y. I., Grillot-Courvalin C., Lesho E., and Waterman P. E. (2014) Amplification of aminoglycoside resistance gene aphA1 in Acinetobacter baumannii results in tobramycin therapy failure. mBio 5, e00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lenfant F., Petit A., Labia R., Maveyraud L., Samama J. P., and Masson J. M. (1993) Site-directed mutagenesis of beta-lactamase TEM-1. Investigating the potential role of specific residues on the activity of Pseudomonas-specific enzymes. Eur. J. Biochem. 217, 939–946 [DOI] [PubMed] [Google Scholar]
- 54. Kaitany K. C., Klinger N. V., June C. M., Ramey M. E., Bonomo R. A., Powers R. A., and Leonard D. A. (2013) Structures of the class D Carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob. Agents Chemother. 57, 4848–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Page M. G. (2008) Extended-spectrum beta-lactamases: structure and kinetic mechanism. Clin. Microbiol. Infection 14, 63–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data were deposited in Peptide Atlas (PASS01051) and the scaffolds of the whole genome sequencing to EMBL Nucleotide Sequence Submissions (http://www.ebi.ac.uk/).