

Mutations in the GBA gene, which cause Gaucher disease, are also risk factors for Parkinson’s disease. Leveraging sequencing data from a large cohort of patients and controls, Robak, Jansen et al. discover a significant burden of likely damaging variants among 54 lysosomal storage disorder genes in association with Parkinson’s disease risk.
Keywords: Parkinson’s disease, lysosomal storage disorders, genetics, whole exome sequencing
Abstract
Mutations in the glucocerebrosidase gene (GBA), which cause Gaucher disease, are also potent risk factors for Parkinson’s disease. We examined whether a genetic burden of variants in other lysosomal storage disorder genes is more broadly associated with Parkinson’s disease susceptibility. The sequence kernel association test was used to interrogate variant burden among 54 lysosomal storage disorder genes, leveraging whole exome sequencing data from 1156 Parkinson’s disease cases and 1679 control subjects. We discovered a significant burden of rare, likely damaging lysosomal storage disorder gene variants in association with Parkinson’s disease risk. The association signal was robust to the exclusion of GBA, and consistent results were obtained in two independent replication cohorts, including 436 cases and 169 controls with whole exome sequencing and an additional 6713 cases and 5964 controls with exome-wide genotyping. In secondary analyses designed to highlight the specific genes driving the aggregate signal, we confirmed associations at the GBA and SMPD1 loci and newly implicate CTSD, SLC17A5, and ASAH1 as candidate Parkinson’s disease susceptibility genes. In our discovery cohort, the majority of Parkinson’s disease cases (56%) have at least one putative damaging variant in a lysosomal storage disorder gene, and 21% carry multiple alleles. Our results highlight several promising new susceptibility loci and reinforce the importance of lysosomal mechanisms in Parkinson’s disease pathogenesis. We suggest that multiple genetic hits may act in combination to degrade lysosomal function, enhancing Parkinson’s disease susceptibility.
Introduction
Parkinson’s disease is a common neurodegenerative disorder with evidence for a substantial genetic aetiology (Kalia and Lang, 2015). Studies in families and large population-based cohorts have implicated more than 30 genes (Singleton et al., 2013; Bras et al., 2015; Verstraeten et al., 2015); however, the risk alleles identified to date explain only a fraction of Parkinson’s disease heritability estimates (Hamza and Paymi, 2010; Do et al., 2011; Keller et al., 2012), suggesting the involvement of additional loci. Beyond discovering the responsible genes, a major challenge remains to understand the mechanisms by which these factors alter disease onset and/or progression, including whether they act independently or interact within coherent biological pathways.
Substantial evidence highlights the importance of lysosomal mechanisms in Parkinson’s disease susceptibility and pathogenesis (Vekrellis et al., 2011; Kalia and Lang, 2015; Moors et al., 2016; Wong and Krainc, 2016). Prior to its discovery as a Parkinson’s disease risk locus, the glucocerebrosidase gene, GBA, was known to cause Gaucher disease, an autosomal recessive lysosomal storage disorder (LSD). Increased risk for Parkinson’s disease in heterozygous carriers of GBA loss-of-function alleles was first recognized in families of individuals with Gaucher disease (Tayebi et al., 2003; Goker-Alpan et al., 2004). Follow-up studies in large, case-control samples confirmed that heterozygous GBA variants confer at least a 5-fold increased risk of Parkinson’s disease (Aharon-Peretz et al., 2004; Sidransky et al., 2009). GBA variants may also modify Parkinson’s disease clinical manifestations (Clark et al., 2007; Winder-Rhodes et al., 2012; Brockmann et al., 2015; Davis et al., 2016), causing earlier age of onset, higher risk of cognitive impairment, and accelerated progression. LSDs—of which there are more than 50—are strictly Mendelian-inherited, metabolic disorders collectively caused by dysfunction in lysosomal biogenesis or function, and similarly characterized by the abnormal accumulation of non-degraded metabolites in the lysosome (Filocamo and Morrone, 2011; Boustany, 2013). The strong genetic evidence linking Gaucher disease and Parkinson’s disease risk leads to the intriguing, generalized hypothesis that LSDs and Parkinson’s disease may share a common genetic mechanism. Other LSD genes have therefore become attractive candidate risk factors for Parkinson’s disease (Shachar et al., 2011; Deng et al., 2015). Several studies have consistently supported a role for SMPD1 (Foo et al., 2013; Gan-Or et al., 2013, 2015; Wu et al., 2014; Clark et al., 2015), which causes Niemann-Pick disease, type A/B. Initial reports evaluating other LSD genes, including NPC1, NPC2, MCOLN1, NAGLU and ARSB, have either shown conflicting results or await further replication (Winder-Rhodes et al., 2012; Kluenemann et al., 2013; Zech et al., 2013; Clark et al., 2015; Jansen et al., 2017). LSDs are individually quite rare in populations of European ancestry, as are the known genetic variants established to cause these disorders (Filocamo and Morrone, 2011; Boustany, 2013). However, with the exception of GBA, most studies of LSD gene candidates have been small and therefore likely underpowered to detect the effects of rare alleles or those with more modest effect sizes. Genome-wide association studies in large Parkinson’s disease case-control cohorts have independently implicated more common risk alleles at two other LSD genes, SCARB2 and GALC (Do et al., 2011; Nalls et al., 2014; Chang et al., 2017). SCARB2 encodes a membrane protein required for correct targeting of glucocerebrosidase to the lysosome. GALC, encoding galactosylceramidase, participates in ceramide metabolism, similiar to GBA. Besides this growing genetic evidence, studies in cellular and animal models also implicate the lysosome in the clearance of α-synuclein (Cuervo et al., 2004; Lee, 2004; Vogiatzi et al., 2008), which aggregates to form Lewy body pathology in Parkinson’s disease. Reciprocally, α-synuclein disrupts neuronal vesicle trafficking and lysosomal function (Cooper et al., 2006; Mazzulli et al., 2011; Moors et al., 2016; Wong and Krainc, 2016).
In this study, we leverage the largest Parkinson’s disease whole exome sequencing (WES) dataset currently available to systematically examine the overlap between genes responsible for LSDs and Parkinson’s disease. Our results reveal an aggregate burden for genetic variants among 54 genes established to cause LSDs and suggest that many genes besides GBA likely contribute to susceptibility for Parkinson’s disease.
Materials and methods
Subjects
Clinical and demographic features for our study cohorts, which have also been described in other recent reports (Giri et al., 2017; Jansen et al., 2017), are shown in Supplementary Table 1. The International Parkinson’s Disease Genomics Consortium (IPDGC) WES discovery dataset used for this study consists of 2835 samples of Northern and Western European ancestry, including 1156 Parkinson’s disease cases and 1679 controls not known to have Parkinson’s disease. Subjects were recruited from academic medical centres across the USA and Europe. Cases were recruited at a mean age of 51.5 years [standard deviation (SD) = 11.5) and diagnosed with Parkinson’s disease at a mean age of 41.2 years (SD = 10.8); 40.4% reported a positive family history. Control subjects were on average 63.7 years of age (SD = 17.1). The majority of control exomes (n = 1201 of 1679) originated from the Rotterdam Study exome dataset version 1 (RSX-1) (Giri et al., 2017; van Rooij et al., 2017). The Rotterdam Study is a prospective population-based cohort study based in Rotterdam, The Netherlands. WES was performed on DNA from participants from the RSX-I subcohort, enrolled in 1990, with an average age at baseline of 68.6 years (SD = 8.6, 54.4% female) (Hofman et al., 2015). All IPDGC and RSX-1 subjects gave written informed consent for participation in genetic research, which was approved by relevant oversight committees and institutional review boards. Subjects with pathogenic variants in established Mendelian Parkinson’s disease genes (SNCA, LRRK2, VPS35, PARK2/parkin, PARK7/DJ-1, or PINK1) were excluded from analysis (Jansen et al., 2017). Following quality control filters, the Parkinson’s Progression Markers Initiative (PPMI) replication dataset (Parkinson Progression Marker Initiative, 2011) includes 436 cases and 169 controls of Northwest European descent. Cases were recruited at a mean age of 61.7 years (SD = 9.7) and diagnosed with Parkinson’s disease at an average age of 59.8 years (SD = 10.0); 27.1% reported a positive family history. PPMI controls were an average of 61.8 years of age (SD = 10.1) at the time of evaluation. Data used in the preparation of this article were obtained from the PPMI database (www.ppmi-info.org/data); up-to-date information on the study is available online (www.ppmi-info.org). Samples analysed for both the IPDGC and PPMI cohorts were derived from whole blood. The NeuroX cohort has also been previously described in detail (Nalls et al., 2015; Jansen et al., 2017). A minority of subjects overlapping with the IPDGC WES discovery sample were removed, such that the NeuroX replication cohort was a completely independent sample, including 6713 individuals with Parkinson’s disease and 5964 controls. NeuroX cases were diagnosed at an average age of 61.6 (SD = 12.4) and controls were evaluated at an average age of 64.1 (SD = 14.3).
Sequencing, genotyping and quality control
Data generation and detailed quality control procedures for the IPDGC and RSX-1 samples have recently been reported (Giri et al., 2017; Jansen et al., 2017; van Rooij et al., 2017). WES was performed using the Roche Nimblegen SeqCap v2 or Illumina exome capture kits to prepare sample libraries, followed by paired-end sequencing with Illumina HiSeq2000. The generation of the PPMI WES dataset are described elsewhere (www.ppmi-info.org). Although the datasets originate from different consortia, the same algorithms were used for read processing. The Burrows-Wheeler Aligner-MEM algorithm (Li and Durbin, 2010) was used for alignment of sequencing reads to the human reference genome (hg19). Using Picard tools (http://broadinstitute.github.io/picard), Binary Alignment/Map files were generated in a sorted and indexed manner. Alignments were Base-Quality score recalibrated and indels realigned using the Genome Analysis Toolkit (McKenna et al., 2010) v3.3-0, after which single nucleotide variants and small insertions/deletions were called with the HaplotypeCaller to one genomic Variant Call Format file per individual. The IPDGC and RSX-1 WES datasets (hereafter referred to as simply the IPDGC discovery dataset) were merged by joint variant calling from the individual genomic Variant Call Format files. Variants that were not assigned with the standard Genome Analysis Toolkit quality annotation ‘PASS’ were excluded for subsequent analyses. 94.4% and 98.0% of the IPDGC and PPMI exomes, respectively, achieved a minimum of 10× coverage.
As previously described (Giri et al., 2017; Jansen et al., 2017), for individual quality control, samples were excluded for ambiguous gender, deviating heterozygosity/genotype calls, low genotype call rates, or cryptic relatedness following identity-by-descent analyses. Population structure was further evaluated using multi-dimensional scaling component analysis based on linkage disequilibrium-pruned, genome-wide common variant markers. Prior to these calculations, our datasets were merged with available genotypes from 1000 Genomes Project (1000GP) ancestry-based population samples, including African (AFR), East Asian (EAS), European (EUR) and the Americas (AMR) (1000 Genomes Project Consortium, 2012). Using the European samples as a reference, population outliers were excluded, resulting in the removal of 39 or 9 individuals from the IPDGC and PPMI datasets, respectively. All remaining samples cluster tightly with European ancestry subjects on multi-dimensional scaling plots (Supplementary Fig. 1). Genotype and variant quality control was accomplished by removal of low-quality genotypes (Phred-scaled genotype quality score < 20, depth < 8) and variants with low call rates or departure from Hardy-Weinberg equilibrium. Furthermore, for the IPDGC discovery dataset, variants were only considered when located within the overlapping targeted regions of the applied library preparation capture kits. Post-quality control procedures, a total of 462 946 and 192 421 variants were called for the IPDGC and PPMI datasets, respectively.
Data generation and quality control for the NeuroX cohort has also previously been described in detail (Nalls et al., 2015; Jansen et al., 2017). NeuroX consists of 242 901 exonic variants from the Illumina Infinium HumanExome BeadChip and 24 706 custom variants related to neurological disease. For individual quality control, as above, samples were excluded for gender ambiguity, dubious heterozygosity/genotype calls, evidence of relatedness, or poor clustering on multi-dimensional scaling plots (Supplementary Fig. 1). We similarly excluded variants for low call rates, departure from Hardy-Weinberg equilibrium, or for significant differences in missingness rate between cases and controls. Post-quality control, we called 177 028 exonic variants from the NeuroX dataset.
Where allowable based on individual consents and institutional review board approval, the datasets used in this study, including WES and NeuroX data from the IPDGC, are publicly available. Data availability is detailed in Jansen et al. (2017) and at http://pdgenetics.org/resources. Data from PPMI is also available for download at http://www.ppmi-info.org/access-data-specimens/download-data/.
Variant selection
Our analyses initially considered 54 LSDs (Table 1), defined based on widely accepted clinical, pathological, and metabolic criteria (Filocamo and Morrone, 2011; Boustany, 2013; Amberger et al., 2015). All variants within the LSD gene set were extracted from the three datasets. For the IPDGC WES dataset, no variants in the genes CLN5 and NEU1 passed the prespecified maximum missingness criteria of 15%, yielding 1136 total exonic variants for consideration in these analyses. In addition, there were no non-synonymous variants identified in SUMF1. Variants were categorized in nested groups (Fig. 1) including (i) non-synonymous (n = 760 variants in 51 genes); (ii) likely damaging (n = 596 variants in 51 genes); or (iii) loss-of-function (n = 69 variants in 27 genes) (Table 1 and Supplementary Table 2). Loss-of-function variants included stop gain/loss, frameshift, and splicing mutations falling within two base pairs of exon-intron junctions. Predictions of variant pathogenicity were obtained from ANNOVAR (Wang et al., 2010), based on the Combined Annotation Dependent Depletion (CADD) algorithm (v1.3, http://cadd.gs.washington.edu) (Kircher et al., 2014). CADD integrates predictions from numerous bioinformatic algorithms into a single ‘C-score’ and ranks all possible nucleotide changes in the genome based on potential to disrupt gene/protein function. In accordance with prior work (Amendola et al., 2015), we selected a stringent CADD C-score ≥ 12.37, representing the top ∼2% most damaging of all possible nucleotide changes in the genome—this subset is enriched for known pathogenic alleles. For descriptive purposes, all putative damaging variants within the IPDGC discovery cohort were further cross-referenced with ClinVar (Landrum et al., 2016) to identify those previously established with pathogenicity for LSDs (Supplementary Table 3). For the PPMI cohort, no variants were called in DNAJC5, resulting in a dataset of 515 total exonic variants, of which 256 variants from 49 genes were non-synonymous and 187 variants in 47 genes met the CADD criteria for putative damaging changes (Supplementary Table 2). For the NeuroX cohort, all genes in the 54-gene set were represented, resulting in 467 non-synonymous variants, of which 348 were classified as likely damaging (Supplementary Table 2). Within these categories, variants were filtered based on two minor allele frequency (MAF) thresholds: (i) <1%; and (ii) <3% (Fig. 1). The latter, more relaxed frequency threshold is based on the population prevalence (de Lau and Breteler, 2006; Pringsheim et al., 2014) and known incomplete penetrance of Parkinson’s disease risk alleles (Anheim et al., 2012; Rana et al., 2013; Trinh et al., 2014; Marder et al., 2015). For a subset of individuals in the IPDGC (n = 572) and PPMI (n = 566) WES cohorts, array-based genotyping data were also available, allowing us to compute concordance rates for genotyping calls present in both datasets using two independent assays (Supplementary Table 4). We observe complete concordance for GBA variants as well as nearly perfect concordance (>>99%) for variant genotype calls in the full LSD gene set.
Table 1.
LSD genes and variants in the IPDGC cohort
| Disease | Gene | Variantsa |
|---|---|---|
| Aspartylglucosaminuria | AGA | 13 (10) |
| Metachromatic leukodystrophy | ARSA | 5 (5) |
| Maroteaux-Lamy disease | ARSB | 11 (10) |
| Farber Lipogranulomatosis | ASAH1 | 20 (17) |
| Kufor-Rakeb syndrome | ATP13A2 | 24 (18) |
| Neuronal ceroid lipofuscinosis (CLN3) | CLN3 | 18 (17) |
| Neuronal ceroid lipofuscinosis (CLN5) | CLN5 | - |
| Neuronal ceroid lipofuscinosis (CLN6) | CLN6 | 10 (7) |
| Neuronal ceroid lipofuscinosis (CLN8) | CLN8 | 9 (4) |
| Cystinosis | CTNS | 13 (12) |
| Galactosialidosis | CTSA | 14 (11) |
| Neuronal ceroid lipofuscinosis (CLN10) | CTSD | 7 (4) |
| Neuronal ceroid lipofuscinosis (CLN13) | CTSF | 11 (9) |
| Pycnodysostosis | CTSK | 6 (5) |
| Neuronal ceroid lipofuscinosis (CLN4B) | DNAJC5 | 5 (5) |
| Fucosidosis | FUCA1 | 15 (12) |
| Pompe disease | GAA | 15 (10) |
| Krabbe disease | GALC | 36 (30) |
| Morquio A disease | GALNS | 22 (14) |
| Gaucher disease | GBA | 39 (32) |
| Fabry disease | GLA | 9 (7) |
| GM1-gangliosidosis/Morquio B | GLB1 | 8 (4) |
| GM2-gangliosidosis | GM2A | 1 (1) |
| I-Cell disease | GNPTAB | 39 (31) |
| Sanfilippo D syndrome | GNS | 20 (11) |
| Neuronal ceroid lipofuscinosis (CLN11) | GRN | 19 (12) |
| Sly disease | GUSB | 17 (10) |
| Tay-Sachs disease | HEXA | 20 (18) |
| Sandhoff disease | HEXB | 8 (6) |
| Sanfilippo C syndrome | HGSNAT | 18 (15) |
| Mucopolysaccharidosis type IX | HYAL1 | 13 (9) |
| Hunter syndrome | IDS | 9 (8) |
| Hurler syndrome | IDUA | 8 (4) |
| Neuronal ceroid lipofuscinosis (CLN14) | KCTD7 | 4 (3) |
| Danon disease | LAMP2 | 9 (7) |
| Wolman disease | LIPA | 14 (10) |
| Alpha-mannosidosis | MAN2B1 | 12 (11) |
| Beta-mannosidosis | MANBA | 18 (15) |
| Mucolipidosis type IV | MCOLN1 | 19 (14) |
| Neuronal ceroid lipofuscinosis (CLN7) | MFSD8 | 18 (14) |
| Schindler disease/Kanzaki disease | NAGA | 9 (8) |
| Sanfilippo B syndrome | NAGLU | 10 (9) |
| Sialidosis | NEU1 | - |
| Niemann-Pick disease type C1 | NPC1 | 43 (35) |
| Niemann-Pick disease type C2 | NPC2 | 2 (2) |
| Neuronal ceroid lipofuscinosis (CLN1) | PPT1 | 9 (7) |
| Sphingolipid-activator deficiency | PSAP | 22 (16) |
| Action mycolonus-renal failure syndrome | SCARB2 | 10 (7) |
| Sanfilippo A syndrome | SGSH | 10 (8) |
| Salla disease | SLC17A5 | 18 (17) |
| Niemann-Pick disease type A/B | SMPD1 | 25 (21) |
| GM3-gangliosidosis | ST3GAL5 | 11 (11) |
| Multiple sulfatase deficiency | SUMF1 | - |
| Neuronal ceroid lipofuscinosis (CLN2) | TPP1 | 15 (13) |
aThe number of variants (MAF < 3%) in each LSD gene is shown for the IPDGC discovery cohort, including total number of non-synonymous variants and likely damaging variants based on CADD (in parentheses). Of the 54 LSD genes considered, no exonic variants in CLN5 or NEU1 passed quality control filters (see ‘Materials and methods’ section), and no non-synonymous variants were identified in SUMF1.
Figure 1.

Overall analytic strategy. (Left) Variant categories. Because the number, frequency, and effect sizes of Parkinson’s disease risk variants remains incompletely defined, our analyses considered three nested categories based on increasing variant pathogenicity: (1) all non-synonymous variants (Nonsyn); (2) likely damaging variants based on combined annotation dependent depletion (CADD) score; and (3) loss-of-function (LoF) variants. Based on the known prevalence of Parkinson’s disease and incomplete penetrance documented for many risk alleles, we also considered two frequency thresholds, including rare (MAF < 1%) and somewhat more common (MAF < 3%) variants. (Right) Analysis flow chart. The SKAT-O was initially performed for the complete LSD gene set in the IPDGC discovery cohort, considering each variant category separately. For those categories with a significant SKAT-O association in the full gene set, a secondary analysis was performed excluding all GBA variants in order to confirm the involvement of additional genes. This was repeated in each of the replication cohorts (PPMI and NeuroX). Lastly, to highlight those loci driving associations detected in the gene set, secondary analyses were performed in the IPDGC cohort using SKAT-O to evaluate variants in each LSD gene independently.
Statistical analysis
The sequence kernel association test – optimal (SKAT-O) (Lee et al., 2012, 2016) was implemented in R using SKAT v1.0.9 to determine the difference in the aggregate burden of rare LSD gene variants between Parkinson’s disease cases and controls. SKAT-O aggregates genetic information across defined genomic regions to test for associations. Covariates were included to adjust analyses for gender and WES coverage (pre-quality control missingness). Twenty multi-dimensional scaling components were also included to account for other possible confounding factors (four components for analyses of the NeuroX genotyping cohort). An empirical P-value (P) was derived from the distribution of null results based on 10 000 permutation trials in which case/control assignment was randomized. As shown in Fig. 1, SKAT-O analysis was initially performed for the complete LSD gene set, considering each class of variants defined based on frequency and functional characteristics. To adjust for multiple comparisons, we applied the Bonferroni-Holm stepwise procedure to control for the familywise error rate and establish a corrected statistical significance threshold and adjusted P-value (Padj) based on a significance level, α of 0.05 (Holm, 1979). For those categories with a significant SKAT-O association in the full gene set, a secondary analysis was performed excluding all GBA variants to confirm the involvement of additional genes. For example, in the IPDGC discovery cohort, we adjusted for k = 5 or k = 2 comparisons for the number of variant categories evaluated in the primary and secondary analyses, respectively. Due to the nested variant categories (Fig. 1) and the highly interdependent nature of the respective burden tests, we separately considered those results with an empirical SKAT-O P-value < 0.05, but not surviving the Bonferonni-Holm correction, as ‘suggestive’. Unadjusted, empiric SKAT-O P-values for all gene set analyses are included in Supplementary Table 5. Lastly, to highlight those loci driving associations detected in the gene set, secondary analyses were also performed using SKAT-O to evaluate variants in each LSD gene independently. For these per gene analyses, which we considered exploratory due to limited statistical power, we report all findings with an empirical unadjusted P-value < 0.05.
To estimate statistical power, we performed 1000 SKAT simulations of causal subregions within the discovery or replication datasets. We assumed a Parkinson’s disease prevalence of 0.0041 and 0.0017 for the IPDGC and PPMI datasets, respectively, based on their distinct ages of onset (Supplementary Table 1) (Pringsheim et al., 2014). For gene set simulations, subregion length was defined as the sum of individual LSD gene coding region lengths (169.5 kb and 170.4 in IPDGC and PPMI, respectively). For single gene simulations, the average gene length was used (3.5 kb and 3.2 kb, respectively). The MAF cut-off for causal variants was set to 0.00035 (based on the frequency of rare GBA loss-of-function alleles in the IPDGC dataset) or 0.03 for the rare or more common variant models, respectively, and penetrance was assumed to be either 100% or 10%. Because we predict that LSD gene variants associated with Parkinson’s disease will have a damaging effect, all causal variants were assumed to have a positive coefficient (risk rather than protective alleles).
Results
Variants were extracted from 54 genes responsible for LSDs, defined based on widely accepted criteria (Table 1), and filtered into nested categories based on two frequency thresholds and three tiers of functional criteria (Fig. 1A). Our overall analytic approach is illustrated in Fig. 1B. To test our hypothesis that an aggregate burden of variants in the LSD gene set contributes to Parkinson’s disease, we first implemented SKAT-O within the IPDGC WES discovery cohort (Table 2). Following adjustment for multiple comparisons (see ‘Materials and methods’ section), significant associations were detected for the LSD gene set considering either all non-synonymous variants (Category 1b, Padj = 0.014) or likely damaging variants (Category 2b, Padj = 0.0055), when using the more relaxed frequency threshold of MAF < 3%. When considering only the subset of rare (MAF < 1%) non-synonymous or likely damaging variants, the SKAT-O result was attenuated and no longer significant (Category 1a, Padj = 0.056 and category 2a, 0.066, respectively). No association was observed when considering only loss-of-function alleles (Category 3, Padj = 0.464), possibly due to the relative paucity of such variants limiting statistical power (Supplementary Table 2). We next repeated analyses with significant results, but excluding all GBA variants. As expected, the strength of the associations was attenuated; however, both SKAT-O results, including either all non-synonymous variants (MAF < 3%) or the subset of likely damaging variants, were robust to the exclusion of GBA and remained significant (Category 1b, Padj = 0.026 and Category 2b, Padj = 0.0198). Our results indicate that the association between variant burden and Parkinson’s disease risk in the IPDGC discovery cohort is mediated, at least in part, by the effects of LSD genes other than GBA, an established Parkinson’s disease susceptibility locus.
Table 2.
Analyses of LSD variant burden in Parkinson’s disease
| (a) MAF < 1% | (b) MAF < 3% | ||||||
|---|---|---|---|---|---|---|---|
| Cohort | Cases (n) | Controls (n) | Variantsa | nb | PLSDc | nb | PLSD (P-GBA)c |
| Discovery | |||||||
| IPDGC | 1167 | 1685 | (1) Nonsyn | 746 (709) | 0.056 | 760 (721) | 0.014 (0.026) |
| (2) CADD | 585 (555) | 0.066 | 596 (564) | 0.0055 (0.0198) | |||
| (3) LoF | 69 (65) | 0.464 | –d | – | |||
| Replication | |||||||
| PPMI | 436 | 169 | (1) Nonsyn | 243 (237) | 0.096 | 256 (248) | 0.320 |
| (2) CADD | 179 (174) | 0.294 | 187 (180) | 0.281 | |||
| NeuroX | 6713 | 5964 | (1) Nonsyn | 452 (443) | 0.068 | 467 (456) | 0.0004 (0.002) |
| (2) CADD | 338 (331) | 0.057 | 348 (339) | 0.0003 (0.020) | |||
aVariants were classified into nested categories (Fig. 1A) based on two frequency thresholds, (a) MAF < 1% or (b) MAF < 3%, and three functional filters, all non-synonymous (1), CADD likely damaging (2), and loss-of-function (3).
bn = total number of LSD variant (number of variants excluding GBA). In parentheses, the number of variants excluding those in GBA are shown.
cEmpirical SKAT-O P-values are based on 10 000 permutations following randomization of case/control status, and adjusted for multiple comparisons using the Bonferroni-Holm method (see ‘Materials and methods’ section). As shown in Fig. 1, primary analyses consider the variant burden among 54 LSD genes (PLSD). For significant SKAT-O results, secondary analyses were performed excluding all variants in GBA (P-GBA). Unadjusted P-values are reported in Supplementary Table 5.
dNo additional LoF variants met the relaxed frequency threshold (MAF < 3%).
LoF = loss of function variants; NeuroX = NeuroX exome array cohort; Nonsyn = non-synonymous variants.
To replicate our findings, we leveraged two independent cohorts, including an additional WES dataset from PPMI (436 Parkinson’s disease cases and 169 controls) (Parkinson Progression Marker Initiative, 2011) and the NeuroX exome-wide genotyping dataset from IPDGC (6713 Parkinson’s disease cases and 5964 controls) (Nalls et al., 2015). We again implemented SKAT-O to detect a potential variant burden in Parkinson’s disease cases versus controls. In the smaller PPMI replication cohort, we discovered suggestive evidence for an excessive LSD variant burden in Parkinson’s disease (Table 2 and Supplementary Table 5); however, this finding was not significant following adjustment for multiple comparisons (Category 1a, Padj = 0.096). The association signal—which appeared independent of GBA—was detected exclusively among rare alleles (MAF < 1%) and only when considering all non-synonymous variants. It is possible that SKAT-O is sensitive to cohort differences between PPMI and the IPDGC, including both sample size and pertinent demographic features (e.g. age of onset and family history; Supplementary Table 1). However, in the substantially larger NeuroX dataset, significant burden associations were detected for the same two variant categories implicated by SKAT-O in the IPDGC discovery cohort (Table 2), despite the less comprehensive genotyping coverage compared to WES. A major driver for the robust LSD gene set association in NeuroX (Category 1b, Padj = 0.0004 and Category 2b, Padj = 0.0003) appears to be the more common GBAE326K variant (FreqCases = 0.021, FreqControls = 0.011), which has been reported to be associated with Parkinson’s disease risk in several large studies (Duran et al., 2012; Pankratz et al., 2012). Importantly, consistent with our findings in the IPDGC discovery cohort, the LSD gene set burden association for both of these variant categories remained significant in NeuroX following exclusion of GBA (Category 1b, Padj = 0.002 and Category 2b, Padj = 0.020). When considering only the subset of rare (MAF < 1%) variants in the NeuroX dataset, the SKAT-O result for the LSD gene set was attenuated and no longer significant; although, the association in the non-synonymous variant group remained suggestive, and this association was independent of GBA (Supplementary Table 5). In sum, based on analyses in three independent Parkinson’s disease case-control datasets, we demonstrate a burden of variants in LSD genes associated with Parkinson’s disease risk, and this signal is at least partially independent of GBA.
To determine which additional LSD genes/variants may be responsible for the observed association with Parkinson’s disease risk, we performed exploratory analyses using SKAT-O to assess for potential contribution of variants within each gene considered independently. For these analyses, we returned to the IPDGC discovery dataset, and again focused on likely damaging variants, which showed the strongest association signal in our primary analysis (Category 2b). In these gene-based analyses, besides the expected result for GBA (P = 0.0001) and confirmation of SMPD1 (P = 0.029), we discover evidence of novel aggregate associations for variants in CTSD (P = 0.002), SLC17A5 (P = 0.005), and ASAH1 (P = 0.031). The specific variants implicated for each of these genes are included in Supplementary Table 3, along with all other putative damaging variants considered in our full LSD gene set analysis. While our datasets are underpowered to definitively assess the contributions of a particular rare variant in any single gene (see ‘Discussion’ section), these results identify the most likely specific loci driving the aggregate LSD gene set association signal detected in the IPDGC discovery sample.
Lastly, we examined the distribution of putative damaging LSD gene variants (MAF < 3%, Category 2b) within the IPDGC WES cohort (Fig. 2). Consistent with our finding of an excessive variant burden in Parkinson’s disease, the distribution of variants appeared modestly right-skewed in cases. The average variant burden among IPDGC cases was 0.9 alleles per individual, which was slightly higher than that seen in controls (0.8 alleles per individual). Given their commonality, the majority of IPDGC cases (56%) have at least one putative damaging variant in an LSD gene, and 21% carry multiple alleles. Notably, only 22 of 1156 total Parkinson’s disease cases are homo- or hemizygous for putative damaging LSD variants (Supplementary Table 6), suggesting that Mendelian recessive or X-linked inheritance may contribute minimally to the overall burden association. As discussed further below, our findings are consistent with a hypothetical model in which multiple LSD gene variants may interact to influence Parkinson’s disease risk.
Figure 2.
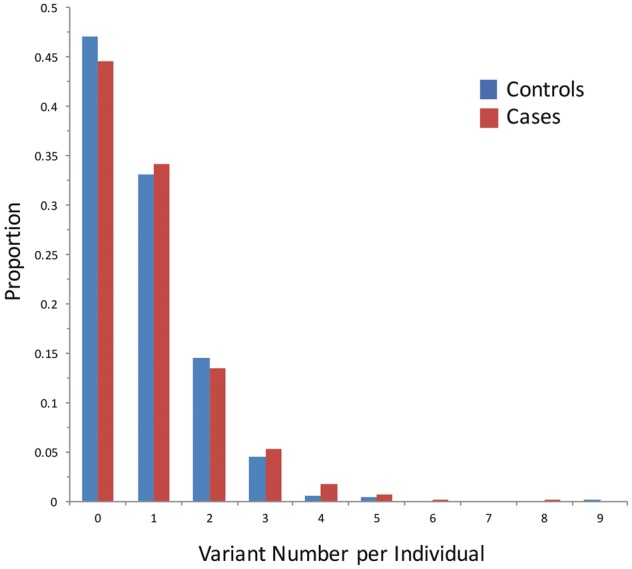
Distribution of LSD variants in the IPDGC cohort. The number of likely damaging LSD variants (MAF < 3%, CADD C-score ≥ 12.37) per individual is shown versus the proportional representation in the IPDGC discovery cohort. Cases (red) and controls (blue) are plotted separately. Many individuals harbour multiple LSD alleles, and the distribution is right-skewed among Parkinson’s disease cases. The analysis considers variants in all 54 LSD genes. Supplementary Fig. 2 shows a similar plot restricted to the five top driver genes.
Discussion
This study reveals an important connection between the genetic factors broadly responsible for LSDs, which are predominantly paediatric Mendelian disorders, and Parkinson’s disease, an adult-onset neurodegenerative disorder with complex genetic aetiology. Specifically, among 54 genes that cause LSDs, we find evidence for a burden of damaging alleles in association with Parkinson’s disease risk. This association persisted after excluding GBA, consistent with a contribution from additional LSD genes. More than half of Parkinson’s disease cases in our cohort harbour one or more putative damaging variants among the LSD genes. Thus, our results implicate several promising new Parkinson’s disease susceptibility loci and reinforce the importance of lysosomal mechanisms in Parkinson’s disease pathogenesis.
The strengths of this study include a large Parkinson’s disease case/control discovery cohort as well as two independent datasets for replication of our findings. The IPDGC WES discovery sample is characterized by younger-onset Parkinson’s disease cases (mean age ∼41 years) and those with a positive family history, thereby enriching for individuals with a potential genetic contribution. Recruitment of a substantially older IPDGC control group (mean age ∼64), reduces the possibility of latent, unrecognized Parkinson’s disease (i.e. with minimal or absent symptoms), likely further increasing power for genetic discovery. By contrast, our PPMI and NeuroX replication cohorts include older cases (mean age ∼62 years) and age-matched controls, making them more broadly representative of the older adult population commonly affected by Parkinson’s disease. Consistent findings of an excessive LSD variant burden across these three datasets, especially the large NeuroX sample (n ∼12 677), strongly enhances the generalizability of our conclusions. To minimize the possibility of population stratification, stringent quality control filters were implemented to ensure a homogeneous European ancestry sample in all study cohorts (Supplementary Fig. 1). Nevertheless, it will also be important to examine other ethnic populations in the future, especially those potentially enriched for LSD-causing variants due to genetic bottlenecks.
Since our understanding of the characteristics of causal alleles—including in both Parkinson’s disease and LSDs—is incomplete, our initial analyses systematically considered multiple variant classes binned into categories based on frequency and putative functional impact. In the IPDGC and PPMI cohorts, WES offers comprehensive characterization of LSD gene variants. By contrast, since the NeuroX data are restricted to those variants included on the genotyping array, it is possible that many potential pathogenic variants would be missed. Nevertheless, a total of 348 putative damaging variants were detected, including alleles for all LSD genes (Supplementary Table 2). Importantly, the selected analytic tool, SKAT-O, is robust to a wide frequency spectrum, including rare and more common alleles, and to variants with different magnitudes and directions of effect (Lee et al., 2012, 2016). Our results suggest that consideration of likely damaging alleles based on bioinformatic predictions, including more common LSD variants (MAF < 3%), appeared to offer optimal sensitivity for detection of a significant aggregate variant association. Many of these variants are known to be pathogenic for LSDs (Supplementary Table 3). For example, of the GBA variants considered in our analyses, 27% of those with annotations available in ClinVar (Landrum et al., 2016) are rated as likely or definitively pathogenic. Critically, the implementation of burden association tests for joint consideration of LSD genes significantly improves statistical power over single gene and variant tests (Zuk et al., 2014). In populations of European ancestry similar to our study cohorts, loss-of-function alleles, including those established to cause LSDs, are individually rare (Supplementary Table 2), and based on post hoc simulations (see ‘Materials and methods’ section), we estimate poor power for discovery of rare Parkinson’s disease risk alleles at single loci. For example, assuming a rare variant model (MAF = 0.035%, as for GBA loss-of-function alleles in our sample) and even assuming full penetrance, the IPDGC discovery cohort has only 30% power to discover an association for a single gene. However, a similar simulation considering the full set of 54 LSD genes was fully powered (100%). Our consideration of higher frequency variants further enhances power for both discovery and replication, especially when coupled with filtering based on potential pathogenicity. For example, allowing for more common variants (MAF < 3%) and assuming 10% of such alleles are causal, we estimate that the smaller PPMI cohort achieves 95% power for replication of a gene set association, whereas negligible power (1%) is available for interrogation of a single gene candidate. We anticipate that larger WES datasets will significantly improve power, including for per gene analyses.
We also performed analyses in the IPDGC cohort to pinpoint the specific drivers from the LSD gene set responsible for increasing Parkinson’s disease risk. Our results (i) recapitulate the established association with GBA; (ii) strengthen the emerging evidence in support of SMPD1; and (iii) newly implicate SLC17A5, ASAH1, and CTSD as candidate Parkinson’s disease susceptibility genes. Recessive mutations in SMPD1 cause Niemann-Pick type A/B disease and this locus has been independently implicated in Parkinson’s disease risk based on several published studies (Lee et al., 2012; Foo et al., 2013; Gan-Or et al., 2013, 2015; Clark et al., 2015). While our analysis identified 21 candidate, putative damaging SMPD1 risk alleles (Supplementary Table 3), most appear distinct from those reported in other studies of Parkinson’s disease. One notable exception, SMPD1 p.L304P, was previously implicated in a study of Ashkenazi Jewish subjects (Gan-Or et al., 2013). Another non-synonymous variant identified in the IPDGC sample, p.P332L, is at the same amino acid position as a different substitution, p.P332R, which was previously implicated in a Chinese Parkinson’s disease cohort (Foo et al., 2013). Among the novel candidate genes, SLC17A5, ASAH1, and CTSD, most of the implicated variants are rare (MAF < 1%). Only two of these variants (rs16883930 and rs141068211 in SLC17A5 and ASAH1, respectively) are present in the 1000 Genomes reference (The 1000 Genomes Project Consortium, 2012), having been previously examined in genome-wide scans, and both were non-associated with Parkinson’s disease risk (P > 0.05) based on available data (Lill et al., 2012). Mutations in SLC17A5, ASAH1, and CTSD cause the rare LSDs, Salla disease, Farber lipogranulomatosis, and neuronal ceroid lipofuscinosis (CLN10), respectively. Whereas sialin (the protein product of SLC17A5) is a lysosomal membrane transporter for sialic acid, acid ceramidase (ASAH1) participates in ceramide metabolism, similar to glucocerebrosidase and sphingomyelinase (SMPD1). In addition to promoting lysosomal stress, glucosylceramide, which accumulates in Gaucher disease, has been suggested to directly promote the aggregation of α-synuclein (Mazzulli et al., 2011; Moors et al., 2016). Interestingly, CTSD encodes a lysosomal aspartyl proteinase that has been independently implicated in α-synuclein degradation (Cullen et al., 2009; McGlinchey and Lee, 2015). In sum, the LSD genes and variants implicated by our studies are excellent candidates for further replication, including resequencing and/or genotyping in the largest available Parkinson’s disease case/control samples. Although we used standard quality control procedures for calling variants from WES and genotyping data, definitive confirmation of specific variants will require additional studies.
There is a growing recognition of the importance of lysosomal biology in Parkinson’s disease pathogenesis (Moors et al., 2016; Wong and Krainc, 2016). First, the lysosome is an important route for α-synuclein degradation (Cuervo et al., 2004; Lee, 2004; Vogiatzi et al., 2008). Genomic variants that elevate α-synuclein protein levels—such as rare locus multiplication (Singleton et al., 2003) or a common polymorphism that enhances promoter activity (Soldner et al., 2016)—also increase Parkinson’s disease risk. Knockdown of selected LSD genes, including GBA or SCARB2, in neuronal cells or in mouse models impairs α-synuclein clearance (Cooper et al., 2006; Sardi et al., 2011; Rothaug et al., 2014), whereas increasing glucocerebrosidase activity has the opposite effect (Sardi et al., 2011; Mazzulli et al., 2016b; Migdalska-Richards et al., 2016). Second, lysosomal autophagy plays a critical role in mitochondrial quality control, and substantial evidence, including from genetics, highlight mitochondrial dysfunction in Parkinson’s disease (Haelterman et al., 2014). Third, there is accumulating evidence from numerous experimental models that α-synuclein interferes with endoplasmic reticulum-to-Golgi vesicle trafficking, inducing reciprocal disruptions in lysosomal biogenesis (Cooper et al., 2006). Expression of α-synuclein impeded trafficking of multiple hydrolases linked to LSDs, including GBA, within human dopaminergic neurons (Mazzulli et al., 2016a). In one recent study, subjects with idiopathic Parkinson’s disease, in which GBA carriers were excluded, were found to have modest but significantly reduced glucocerebrosidase enzymatic activity based on peripheral blood testing (Alcalay et al., 2015). Fourth, besides GBA and the other LSD genes implicated in our study, genome-wide association studies of Parkinson's disease have recently identified common variants at the GALC locus, which causes Krabbe disease (Chang et al., 2017). Further, mutations in ATP13A2, a rare cause of recessive juvenile-onset parkinsonism and dementia, have been independently implicated to cause the LSD neuronal ceroid lipofuscinosis (Bras et al., 2012). Lastly, many other common and rare Parkinson’s disease risk alleles, including at RAB7L1, GAK, LRRK2, and VPS35 have strong functional links to vesicle trafficking, including for lysosomal biogenesis and function. Together, these findings support a model in which partial loss-of-function in genes regulating lysosomal activity, such as those that cause LSDs, may increase vulnerability to α-synuclein-mediated mechanisms in Parkinson’s disease.
While our analyses reveal a robust and replicable LSD variant burden in Parkinson’s disease cases, the overall magnitude of the difference between variant frequencies in cases and controls appears modest (Fig. 2). We speculate that this is probably an underestimate of the true difference due to several assumptions. Specifically, only a subset of the 54 LSD genes and 760 non-synonymous variants considered in our burden analyses are likely to be truly involved in Parkinson’s disease risk. Further, as noted above, while the CADD framework allowed us to prioritize 596 variants as putative damaging alleles, larger Parkinson’s disease exome datasets with improved statistical power will be required to resolve the specific LSD genes and variants that contribute to Parkinson’s disease risk. Lastly, similar to GBA (Anheim et al., 2012; Rana et al., 2013), we expect that many of the other LSD gene variants contributing to Parkinson’s disease risk may have individually modest and therefore incompletely penetrant effects, perhaps modified by alleles at other loci (Cooper et al., 2013). In sum, the likely (i) incomplete penetrance of many pathogenic variants; along with (ii) contamination of our analyses by many benign variants would be expected to inflate estimates for the LSD variant burden among controls and attenuate the overall SKAT-O association.
Parkinson’s disease heritability remains incompletely explained by the genes and variants identified to date (Hamza and Paymi, 2010; Do et al., 2011; Keller et al., 2012; Verstraeten et al., 2015). Besides the likelihood of yet undiscovered loci, alternative explanations for familial aggregation of disease include epigenetic changes due to shared environmental exposures or even false positive diagnoses due to phenocopies (Pihlstrøm and Toft, 2011; Mullin and Schapira, 2015). In complex genetic disorders such as Parkinson’s disease, the cumulative impact of common and rare variants at multiple genomic loci, as well as non-additive interactions among alleles, likely also play an important role (Lupski et al., 2011; Cooper et al., 2013). Polygenic modelling approaches have previously demonstrated how common risk alleles can cumulatively impact Parkinson’s disease risk and age-of-onset (Nalls et al., 2014; Escott-Price et al., 2015). In addition, a recently published analysis in the IPDGC WES and NeuroX cohorts identified evidence for oligogenic interactions underlying Parkinson’s disease risk, including alleles for GBA and those for established Mendelian Parkinson’s disease genes (Lubbe et al., 2016). In the IPDGC, WES reveals a substantial proportion of Parkinson’s disease cases (21%) carrying two or more likely damaging variants in LSD genes. Consistent with other reports (Clark et al., 2015), our observation suggests the possibility that LSD gene variants may interact in a multi-hit, combinatorial manner to degrade lysosomal function, causing the accumulation of α-synuclein and potentially other toxic substrates, and increasing susceptibility for Parkinson’s disease. Oligogenic interactions such as those proposed here may be an important source for ‘missing heritability’ in Parkinson’s disease (Pihlstrøm and Toft, 2011; Mullin and Schapira, 2015). Recent work has also implicated oligogenic inheritance in other neurological disorders, including amyotrophic lateral sclerosis (van Blitterswijk et al., 2012; Kenna et al., 2013; Cady et al., 2015) and idiopathic peripheral neuropathy (Gonzaga-Jauregui et al., 2015), and further reveals how pleiotropic genes causing early-onset, monogenic disorders may act in combination to additionally trigger late-onset, complex genetic disorders (Lupski et al., 2011; Cooper et al., 2013). Future studies, including even larger, case-control cohorts with WES and complementary experiments in Parkinson’s disease cellular or animal models, are needed to further investigate whether a variant burden in LSD genes, perhaps in combination with other susceptibility loci, underlies oligogenic risk and contributes substantially to Parkinson’s disease heritability.
Supplementary Material
Acknowledgements
We thank Drs Andrew Singleton, John Hardy, V. Reid Sutton, Mike Nalls, Manu Sharma, Zongqi Xia, Genevera Allen, Richard Gibbs, and Huda Zoghbi for helpful discussions related to the manuscript. We also thank Anamika Giri for assistance with data processing. We thank Mr Pascal Arp, Ms Mila Jhamai, and Mr Marijn Verkerk for their help in creating the RSX exome sequencing database. The authors are grateful to the Rotterdam study participants, staff, and the contributing general practitioners and pharmacists. The IPDGC would also like to thank all of the subjects who donated their time and biological samples to be a part of this study. We thank the French Parkinson’s Disease Genetics Study Group and the Drug Interaction with genes (DIGPD) study group: Y Agid, M Anheim, A-M Bonnet, M Borg, A Brice, E Broussolle, J-C Corvol, P Damier, A Destée, A Dürr, F Durif, A Elbaz, D Grabil, S Klebe, P. Krack, E Lohmann, L. Lacomblez, M Martinez, V Mesnage, P Pollak, O Rascol, F Tison, C Tranchant, M Vérin, F Viallet, and M Vidailhet. We also thank the members of the French 3C Consortium: A. Alpérovitch, C. Berr, C. Tzourio, and P. Amouyel for allowing us to use part of the 3C cohort, and D Zelenika for support in generating the genome-wide molecular data. We thank P Tienari (Molecular Neurology Programme, Biomedicum, University of Helsinki), T Peuralinna (Department of Neurology, Helsinki University Central Hospital), L Myllykangas (Folkhalsan Institute of Genetics and Department of Pathology, University of Helsinki), and R Sulkava (Department of Public Health and General Practice Division of Geriatrics, University of Eastern Finland) for the Finnish controls (Vantaa85+ GWAS data). This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD, (http://biowulf.nih.gov), and DNA panels, samples, and clinical data from the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository. People who contributed samples are acknowledged in descriptions of every panel on the repository website. As with previous IPDGC efforts, this study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
Funding
L.A.R. received support from the T32 GM07526-37, the Pfizer/ACMG Foundation Clinical Genetics Combined Residency for Translational Genomic Scholars, and the American Academy of Neurology Clinical Research Training Fellowship in Parkinson’s disease. J.M.S. is supported by NIH grants (R21NS089854, R01AG050631, R01AG053960, C06RR029965), Huffington Foundation, the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and a Career Award for Medical Scientists from the Burroughs Wellcome Fund. I.E.J. and P.H. receive funding from the Prinses Beatrix Spierfonds. P.H. is supported by the EU joint Program-Neurodegenerative Diseases (JPND):COURAGE-PD, and the Federal Ministry of Education and Research Germany (BMBF):MitoPD. The generation and management of the exome sequencing data for the Rotterdam Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Netherlands. The Exome Sequencing data set was funded by the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) sponsored Netherlands Consortium for Healthy Aging (NCHA; project nr. 050-060-810), by the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, and by a Complementation Project of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRI-NL; www.bbmri.nl; project number CP2010-41). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. PPMI – a public-private partnership – is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including Abbvie, Avid Radiopharmaceuticals, Biogen, Bristol-Myers Squibb, Covance, GE Health Care, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Priamal, Roche, Servier and UCB. The IPDGC was supported in part by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institute on Aging (NIA), and the National Institute of Environmental Health Sciences both part of the National Institutes of Health, Department of Health and Human Services; project numbers Z01-AG000949-02 and Z01-ES101986. In addition, this work was supported by the Department of Defense (award W81XWH-09-2-0128) and The Michael J Fox Foundation for Parkinson’s Research. This work was supported by National Institutes of Health grants R01NS037167, R01CA141668, P50NS071674, American Parkinson Disease Association (APDA); Barnes Jewish Hospital Foundation; Greater St Louis Chapter of the APDA; Hersenstichting Nederland; Neuroscience Campus Amsterdam; and the section of medical genomics, the Prinses Beatrix Fonds. The KORA (Cooperative Research in the Region of Augsburg) research platform was started and financed by the Forschungszentrum für Umwelt und Gesundheit, which is funded by the German Federal Ministry of Education, Science, Research, and Technology and by the State of Bavaria. This study was also funded by the German National Genome Network (NGFNplus number 01GS08134, German Ministry for Education and Research); by the German Federal Ministry of Education and Research (NGFN 01GR0468, PopGen); and 01EW0908 in the frame of ERA-NET NEURON and Helmholtz Alliance Mental Health in an Ageing Society (HA-215), which was funded by the Initiative and Networking Fund of the Helmholtz Association. The French GWAS work was supported by the French National Agency of Research (ANR-08-MNP-012). This study was also funded by France-Parkinson Association, the French program ‘Investissements d’avenir’ funding (ANR-10-IAIHU-06) and a grant from Assistance Publique-Hôpitaux de Paris (PHRC, AOR-08010) for the French clinical data. This study was also sponsored by the Landspitali University Hospital Research Fund (grant to S.Sv.); Icelandic Research Council (grant to S.Sv.); and European Community Framework Programme 7, People Programme, and IAPP on novel genetic and phenotypic markers of Parkinson’s disease and Essential Tremor (MarkMD), contract number PIAP-GA-2008-230596 MarkMD (to H.P. and J.Hu). This study was supported by the Medical Research Council and Wellcome Trust disease centre (grant WT089698/Z/09/Z to N.W., J.Ha., and A.Sc.). Funding for the project was provided by the Wellcome Trust under award 076113, 085475 and 090355. This study was also supported by Parkinson’s UK (grants 8047 and J-0804) and the Medical Research Council (G0700943). DNA extraction work that was done in the UK was undertaken at University College London Hospitals, University College London, who received a proportion of funding from the Department of Health’s National Institute for Health Research Biomedical Research Centres funding. This study was supported in part by the Wellcome Trust/Medical Research Council Joint Call in Neurodegeneration award (WT089698) to the Parkinson’s Disease Consortium (UKPDC), whose members are from the UCL Institute of Neurology, University of Sheffield, and the Medical Research Council Protein Phosphorylation Unit at the University of Dundee.
Supplementary material
Supplementary material is available at Brain online.
Appendix 1
See Supplementary material for details of authors and affiliations.
IPDGC Consortium members
Glossary
Abbreviations
- CADD
combined annotation dependent depletion
- IPDGC
International Parkinson’s Disease Genomics Consortium
- LSD
lysosomal storage disorder
- MAF
minor allele frequency
- PPMI
Parkinson’s Progression Markers Initiative
- RSX-1
Rotterdam Study exome dataset version 1
- SKAT-O
sequence kernel association test – optimized
- WES
whole exome sequencing
Contributor Information
International Parkinson’s Disease Genomics Consortium (IPDGC):
Mike A Nalls, Vincent Plagnol, Dena G Hernandez, Manu Sharma, Una-Marie Sheerin, Mohamad Saad, Javier Simón-Sánchez, Claudia Schulte, Suzanne Lesage, Sigurlaug Sveinbjörnsdóttir, Sampath Arepalli, Roger Barker, Yoav Ben-, Henk W Berendse, Daniela Berg, Kailash Bhatia, Rob M A de Bie, Alessandro Biffi, Bas Bloem, Zoltan Bochdanovits, Michael Bonin, Jose M Bras, Kathrin Brockmann, Janet Brooks, David J Burn, Elisa Majounie, Gavin Charlesworth, Codrin Lungu, Honglei Chen, Patrick F Chinnery, Sean Chong, Carl E Clarke, Mark R Cookson, J Mark Cooper, Jean Christophe Corvol, Carl Counsell, Philippe Damier, Jean-François Dartigues, Panos Deloukas, Günther Deuschl, David T Dexter, Karin D van Dijk, Allissa Dillman, Frank Durif, Alexandra Dürr, Sarah Edkins, Jonathan R Evans, Thomas Foltynie, Jing Dong, Michelle Gardner, J Raphael Gibbs, Alison Goate, Emma Gray, Rita Guerreiro, Clare Harris, Jacobus J van Hilten, Albert Hofman, Albert Hollenbeck, Janice Holton, Michele Hu, Xuemei Huang, Isabel Wurster, Walter Mätzler, Gavin Hudson, Sarah E Hunt, Johanna Huttenlocher, Thomas Illig, Pálmi V Jónsson, Jean-Charles Lambert, Cordelia Langford, Andrew Lees, Peter Lichtner, Patricia Limousin, Grisel Lopez, Delia Lorenz, Codrin Lungu, Alisdair McNeill, Catriona Moorby, Matthew Moore, Huw R Morris, Karen E Morrison, Valentina Escott-Price, Ese Mudanohwo, Sean S O’Sullivan, Justin Pearson, Joel S Perlmutter, Hjörvar Pétursson, Pierre Pollak, Bart Post, Simon Potter, Bernard Ravina, Tamas Revesz, Olaf Riess, Fernando Rivadeneira, Patrizia Rizzu, Mina Ryten, Stephen Sawcer, Anthony Schapira, Hans Scheffer, Karen Shaw, Ira Shoulson, Joshua Shulman, Ellen Sidransky, Colin Smith, Chris C A Spencer, Hreinn Stefánsson, Francesco Bettella, Joanna D Stockton, Amy Strange, Kevin Talbot, Carlie M Tanner, Avazeh Tashakkori-Ghanbaria, François Tison, Daniah Trabzuni, Bryan J Traynor, André G Uitterlinden, Daan Velseboer, Marie Vidailhet, Robert Walker, Bart van de Warrenburg, Mirdhu Wickremaratchi, Nigel Williams, Caroline H Williams-Gray, Sophie Winder-Rhodes, Kári Stefánsson, Maria Martinez, Nicholas W Wood, John Hardy, Peter Heutink, Alexis Brice, Thomas Gasser, and Andrew B Singleton
References
- Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004; 351: 1972–7. [DOI] [PubMed] [Google Scholar]
- Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo S-H, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain 2015; 138: 2648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res 2015; 43: D789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res 2015; 25: 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012; 78: 417–20. [DOI] [PubMed] [Google Scholar]
- Boustany R-MN. Lysosomal storage diseases—the horizon expands. Nat Rev Neurol 2013; 9: 583–98. [DOI] [PubMed] [Google Scholar]
- Bras J, Guerreiro R, Hardy J. SnapShot: genetics of Parkinson's disease. Cell 2015; 160: 570.e1. [DOI] [PubMed] [Google Scholar]
- Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet 2012; 21: 2646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann K, Srulijes K, Pflederer S, Hauser A-K, Schulte C, Maetzler W, et al. GBA-associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015; 30: 407–11. [DOI] [PubMed] [Google Scholar]
- Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 2015; 77: 100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 2017; 49: 1511–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Chan R, Cheng R, Liu X, Park N, Parmalee N, et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS One 2015; 10: e0125204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007; 69: 1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 2006; 313: 324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Humangenetik 2013; 132: 1077–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305: 1292–5. [DOI] [PubMed] [Google Scholar]
- Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009; 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016; 73: 1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol 2006; 5: 525–35. [DOI] [PubMed] [Google Scholar]
- Deng H, Xiu X, Jankovic J. Genetic convergence of Parkinson's disease and lysosomal storage disorders. Mol Neurobiol 2015; 51: 1554–68. [DOI] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson's disease. PLoS Genet 2011; 7: e1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran R, Mencacci NE, Angeli AV, Shoai M, Deas E, Houlden H, et al. The glucocerobrosidase E326K variant predisposes to Parkinson‘s disease, but does not cause Gaucher’s disease. Mov Disord 2012; 28: 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Nalls MA, Morris HR, Lubbe S, Brice A, et al. Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann Neurol 2015; 77: 582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filocamo M, Morrone A. Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics 2011; 5: 156–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo J-N, Liany H, Bei J-X, Yu X-Q, Liu J, Au W-L, et al. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson's disease. Neurobiol Aging 2013; 34: 2890.e13–5. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Orr-Urtreger A, Alcalay RN, Bressman S, Giladi N, Rouleau GA. The emerging role of SMPD1 mutations in Parkinson's disease: implications for future studies. Parkinsonism Relat Disord 2015; 21: 1294–5. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013; 80: 1606–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri A, Mok KY, Jansen I, Sharma M, Tesson C, Mangone G, et al. Lack of evidence for a role of genetic variation in TMEM230 in the risk for Parkinson's disease in the Caucasian population. Neurobiol. Aging 2017; 50: 167e11–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet 2004; 41: 937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Harel T, Gambin T, Kousi M, Griffin LB, Francescatto L, et al. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep 2015; 12: 1169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ. A mitocentric view of Parkinson's disease. Annu Rev Neurosci 2014; 37: 137–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza TH, Payami H. The heritability of risk and age at onset of Parkinson's disease after accounting for known genetic risk factors. J Hum Genet 2010; 55: 241–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman A, Brusselle GGO, Murad SD, van Duijn CM, Franco OH, Goedegebure A, et al. The Rotterdam Study: 2016 objectives and design update. Eur J Epidemiol 2015; 30: 661–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Statistics 1979; 6: 65–70. [Google Scholar]
- Jansen IE, Ye H, Heetveld S, Lechler MC, Michels H, Seinstra RI, et al. Discovery and functional prioritization of Parkinson's disease candidate genes from large-scale whole exome sequencing. Genome Biol 2017; 18: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Lang AE. Parkinson's disease. Lancet 2015; 386: 896–912. [DOI] [PubMed] [Google Scholar]
- Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simon-Sanchez J, et al. Using genome-wide complex trait analysis to quantify ‘missing heritability“ in Parkinson”s disease. Hum Mol Genet 2012; 21: 4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet 2013; 50: 776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluenemann HH, Nutt JG, Davis MY, Bird TD. Parkinsonism syndrome in heterozygotes for Niemann–Pick C1. J Neurol Sci 2013; 335: 219–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016; 44: D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ. Clearance of -synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004; 24: 1888–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Fuchsberger C, Kim S, Scott L. An efficient resampling method for calibrating single and gene-based rare variant association analysis in case-control studies. Biostatistics 2016; 17: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 2012; 13: 762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26: 589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide B-MM, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: the PDGene database. PLoS Genet 2012; 8: e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubbe SJ, Escott-Price V, Gibbs JR, Nalls MA, Bras J, Price TR, et al. Additional rare variant analysis in Parkinson’s disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum Mol Genet 2016: 25:5483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell 2011; 147: 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder K, Wang Y, Alcalay RN, Mejia-Santana H, Tang M-X, Lee A, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015; 85: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146: 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci USA 2016a; 113: 1931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J Neurosci 2016b; 36: 7693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci USA 2015; 112: 9322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska-Richards A, Daly L, Bezard E, Schapira AHV. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol 2016; 80: 766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moors T, Paciotti S, Chiasserini D, Calabresi P, Parnetti L, Beccari T, et al. Lysosomal dysfunction and α-synuclein aggregation in Parkinson's disease: diagnostic links. Mov Disord 2016; 31: 791–801. [DOI] [PubMed] [Google Scholar]
- Mullin S, Schapira A. The genetics of Parkinson's disease. Br Med Bull 2015; 114: 39–52. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, et al. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 2015; 36: 1605.e7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 2014; 46: 989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, et al. Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann Neurol 2012; 71: 370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson Progression Marker Initiative. The Parkinson progression marker initiative (PPMI). Prog Neurobiol 2011; 95: 629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlstrøm L, Toft M. Parkinson's disease: What remains of the “missing heritability"? Movement Disorders 2011; 26: 1971–3. [DOI] [PubMed] [Google Scholar]
- Pringsheim T, Jette N, Frolkis A, Steeves TDL. The prevalence of Parkinson's disease: a systematic review and meta-analysis. Mov Disord 2014; 29: 1583–90. [DOI] [PubMed] [Google Scholar]
- Rana HQ, Balwani M, Bier L, Alcalay RN. Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013; 15: 146–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lüllmann-Rauch R, et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci USA 2014; 111: 15573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci USA 2011; 108: 12101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar T, Bianco CL, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov Disord 2011; 26: 1593–604. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009; 361: 1651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003; 302: 841–1. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord 2013; 28: 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016; 533: 95–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 2003; 79: 104–9. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Guella I, Farrer MJ. Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol 2014; 71: 1535–9. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk M, van Es MA, Hennekam EAM, Dooijes D, van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012; 21: 3776–84. [DOI] [PubMed] [Google Scholar]
- van Rooij, Jhamai M, Arp PP, Nouwens SCA, Verkerk M, Hofman A, et al. Population-specific genetic variation in large sequencing datasets: why more data is still better. Eur J Hum Genet 2017; 25; 1173–5. doi: 10.1038/ejhg.2017.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol 2011; 10: 1015–25. [DOI] [PubMed] [Google Scholar]
- Verstraeten A, Theuns J, Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet 2015; 31: 140–9. [DOI] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 2008; 283: 23542–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder-Rhodes SE, Garcia-Reitböck P, Ban M, Evans JR, Jacques TS, Kemppinen A, et al. Genetic and pathological links between Parkinson's disease and the lysosomal disorder Sanfilippo syndrome. Mov Disord 2012; 27: 312–15. [DOI] [PubMed] [Google Scholar]
- Wong YC, Krainc D. Lysosomal trafficking defects link Parkinson‘s disease with Gaucher’s disease. Mov Disord 2016; 31: 1610–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R-M, Lin C-H, Lin H-I. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2014; 82: 283–3. [DOI] [PubMed] [Google Scholar]
- Zech M, Nübling G, Castrop F, Jochim A, Schulte EC, Mollenhauer B, et al. Niemann-Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS One 2013; 8: e82879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: Designing rare variant association studies. Proc Natl Acad Sci USA 2014; 111: E455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.