

Abstract
Motile and non-motile (primary) cilia are nearly ubiquitous cellular organelles. The dysfunction of cilia causes diseases known as ciliopathies. The number of reported ciliopathies (currently 35) is increasing, as is the number of established (187) and candidate (241) ciliopathy-associated genes. The characterization of ciliopathy-associated proteins and phenotypes has improved our knowledge of ciliary functions. In particular, investigating ciliopathies has helped us to understand the molecular mechanisms by which the cilium-associated basal body functions in early ciliogenesis, as well as how the transition zone functions in ciliary gating, and how intraflagellar transport enables cargo trafficking and signalling. Both basic biological and clinical studies are uncovering novel ciliopathies and the ciliary proteins involved. The assignment of these proteins to different ciliary structures, processes and ciliopathy subclasses (first order and second order) provides insights into how this versatile organelle is built, compartmentalized and functions in diverse ways that are essential for human health.
Many people are introduced to cilia together with microscopy. A compound microscope — or, increasingly, a smartphone fitted with a ball lens — allows young schoolchildren to see protists such as Paramecium using cilia (also known as flagella) to swim. Antonie van Leeuwenhoek first observed these thin ‘nimbly moving feet’, and even early microscopists appreciated that cilia can exist as either solitary or multiple structures on a single eukaryotic cell, and that they can be either motile or immotile (FIG. 1); these criteria are still used to discriminate between different types of cilia.
Figure 1. Structures and functions of motile and non-motile cilia.

All cilia extend from a basal body that typically consists of triplet microtubules, and subdistal and distal appendages. Distal appendages (also known as transition fibres) tether the basal body to the base of the ciliary membrane. Immediately distal to the basal body is the transition zone, which contains doublet microtubules that are connected to the ciliary membrane via Y-shaped structures. Axonemes (the ciliary backbone) are composed of doublet microtubules. In motile cilia, axonemes usually contain associated structures and proteins (for example, the central pair and axonemal dyneins) that are required for ciliary motility. Nodal cilia are an exception as they are motile but lack a central pair of microtubules. Cilia may contain additional subdomains, including singlet microtubules at the distal end, and regions with specific protein compositions or functions (for example, the inversin domain (INV; involved in signalling). Key cell signalling functions and roles in motility are summarized. PKD, polycystin.
Like protists, most vertebrate cells have either a single non-motile (‘primary’) cilium or multiple cilia, as found in kidney and olfactory epithelial cells, respectively. Cilia can be either actively motile, as observed at the embryonic node or in sperm, or immotile, as in photoreceptor cells or olfactory neurons. Immotile cilia function in transducing signals from the environment or from other cells, whereas motile cilia propel cells (such as sperm cells) or move extracellular fluids (for example, to clear mucus and debris from the lung) (FIG. 1).
The dysfunction of motile or immotile cilia is associated with a wide range of human diseases that are known as ciliopathies (FIG. 2). As the physiological consequences of defects in motile and immotile cilia are different, we discuss these two classes of ciliopathies separately (BOX 1). In addition, we distinguish between first-order ciliopathies, which are caused by the disruption of ciliary proteins, and second-order ciliopathies which result from the disruption of non-ciliary proteins that are required for ciliary function (BOX 1).
Figure 2. Dysfunctions in motile and/or non-motile cilia cause ciliopathies that encompass most human organ systems.

The figure shows the different organ systems or tissues that are affected in diverse ciliopathies, and the principle phenotypic manifestations of the disease in each organ. Ciliopathies that are caused primarily by defects in motile cilia are shown in orange, those that result from defects in non-motile (primary) cilia are shown in blue and those associated with defects in both types of cilia are shown in green. NPHP, nephronophthisis; PKD, polycystic kidney disease.
Box 1 | Proposed classification scheme for ciliopathies.
The term ciliopathy, which was first used in 1984 (REF. 183) and popularized in the 21st century184–186, describes human disorders that are caused by ciliary dysfunction. Dysfunction of basal body and ciliary proteins can affect both motile cilia and non-motile primary cilia, separately or together. Non-ciliary proteins can also contribute to ciliopathies, and ciliary proteins can have extraciliary functions that, when impaired, cause phenotypes that are unrelated to ciliopathies.
We propose a flexible classification system to describe the various ways in which ciliary and non-ciliary proteins relate to ciliopathies (see the figure).
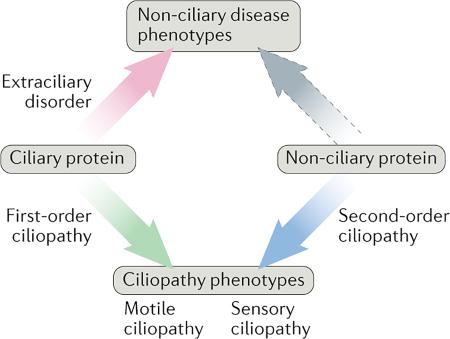
First-order and second-order ciliopathies
First-order ciliopathies: diseases that are caused by the dysfunction of a protein that principally localizes to, and functions within, the basal body and/or the ciliary compartment. For example, the disruption of intraflagellar transport (IFT) components, which are involved in protein transport to and within cilia, can result in the first-order ciliopathy Jeune asphyxiating thoracic dystrophy (JATD).
Second-order ciliopathies: diseases that are caused by mutations in proteins that are not localized within cilia but that have a role in cilium formation or function. Examples include the cytoplasmic assembly factors for outer arm dyneins that are involved in primary ciliary dyskinesia (PCD). Ciliary defects that are not caused by mutations in protein-coding genes can also be classified as second-order ciliopathies. For example, multicilin (MCIDAS) is a transcription factor that regulates genes that are required for ciliary motility63. mlR-34-449, which is a micro RNA that regulates the levels of basal body proteins that are involved in motile ciliogenesis187, also has a second-order ciliary function, although it has not yet been linked to a ciliopathy).
Motile and sensory ciliopathies
Motile ciliopathies: disorders, such as PCD, which result from impairment of ciliary motility.
Sensory ciliopathies: diseases that result from defects in the sensory and/or signalling functions of cilia. Examples include polycystic kidney disease (PKD) and Joubert syndrome (JBTS).
Using this classification scheme, first-order and second-order ciliopathies can be motile or sensory. For example, PCD that is caused by mutations in DNAAF4 is a second-order motile ciliopathy, whereas PKD is a first-order sensory ciliopathy.
Ciliary proteins with non-ciliary functions that are relevant to disease
Extraciliary disorder: disruption of a protein with both ciliary and non-ciliary functions causes phenotypes that are unrelated to ciliary function. For example, the role of IFT20 in collagen trafficking, which was discovered in a mouse model166 of a craniofacial skeletal development disorder, may be ultimately linked to an extraciliary disorder.
Many ciliary proteins are known to have essential roles in human physiology, signalling and development, and their importance is striking when we consider their collective contribution to the ciliopathy disease spectrum. Supplementary information S1 (table) lists 187 genes that have been implicated in 35 established ciliopathies, and at least another 241 genes that have been associated with ciliary structures and/or functions that could potentially result in known or novel ciliopathies if disrupted in humans.
In this Review, we briefly describe the structural features of motile and non-motile cilia, and how they are assembled to form discrete, compartmentalized organelles. We then summarize the functions of these two classes of cilia, and elaborate on how the impairment of different cilium-associated processes and structures — namely, signalling, ciliogenesis, compartmentalization (or gating) and dynamic trafficking — results in ciliopathies with distinct phenotypes. We also discuss evidence that nonciliary proteins influence ciliary functions, and that some ciliary proteins can have non-ciliary roles. Finally, we conclude by describing approaches for identifying the full complement of ciliary and ciliopathy-associated proteins.
Conserved ciliary structures
The first details of ciliary structure were described in the mid-20th century, using transmission electron microscopy (TEM). The ability to resolve subcellular structures led to the realization that cilia from diverse organisms and tissues can look different and can have different accessory substructures, but can also have many structural commonalities.
Basal body and axoneme
The most stereotypical features of cilia are the basal body and the axoneme1,2 (FIG. 1). The basal body describes the mother centriole when it is associated with a cilium. Most basal bodies comprise a barrel of nine triplet microtubules, subdistal appendages and nine strut-like structures, known as distal appendages or transition fibres, which connect to the membrane at the base of the cilium. The skeleton of the ciliary shaft, or axoneme, consists of doublet microtubules that originate from the basal body. Additional structures that are often found in motile cilia include a central pair of microtubules and axonemal inner and outer dynein arms that power ciliary movement (FIG. 1). Given their intimate relationship1,2, the basal body and cilium are considered in this Review as a functional unit.
Transition zone: compartmentalisation of signalling and motility functions
To move and/or to function as a signalling device, cilia must contain the motility and/or signal transduction machineries, and must have a composition that is distinct from the rest of the cell3–5 (FIGS 1,3). Unlike most other organelles, the ciliary membrane is contiguous with the plasma membrane. To achieve such compartmentalization and to thus maintain the distinct composition of the cilium, the proximal-most region of the axoneme consists of a transition zone (FIG. 1) that controls which proteins can enter and leave the cilium4,5. The transition zone features prominent Y-shaped structures that connect the ciliary membrane to the underlying axoneme and that are thought to form or to organize a diffusion barrier for membrane-associated soluble proteins6–10.
Figure 3. Structural and functional features of motile and sensory cilia are associated with ciliopathies.

a | The major structures of motile and non-motile cilia (also see FIG. 1). b | Major sites of action for ciliopathy-associated proteins that are components of motile cilia (motility apparatus or transcription factors required for the generation of motile cilia) and sensory cilia (axonemal and signalling proteins, ciliary tip proteins or inversin (INV) compartment proteins). The asterisks indicate proteins that are also localized to other ciliary regions during ciliogenesis (shown in FIG. 4) or ciliary trafficking (shown in FIG. 5). Circled numbers indicate one or more ciliopathies that result from defects in the different ciliary compartments and proteins. c | Ciliopathies grouped into major categories that are associated with the proteins and ciliary regions shown in part b.
Control of selective entry into cilia probably involves other structural and functional features. For example, septins may form part of the membrane diffusion barrier11, and the basal body distal appendages may prevent the inappropriate entry of vesicles into the cilia. Furthermore, a sieve-like functionality at the base of the cilium — potentially within the transition zone — controls the access of soluble proteins, restricting the entry of large (~70 kDa) proteins into the ciliary compartment6,12–14. It is unclear whether the different gate components have partially overlapping functions or how they help to regulate the entry of distinct ciliary components.
Key stages of ciliogenesis
As early as 1959, a study of vertebrate ciliary photoreceptor biogenesis described the key steps in ciliogenesis15. In brief, the mother centriole is modified to become a basal body and attaches to the plasma membrane through its distal appendages; the transition zone (known as the connecting cilium in photoreceptor cells (FIG. 4)) then forms, and finally, the rest of the axoneme extends out from the cell body (FIGS 4,5).
Figure 4. Ciliogenesis and ciliary compartmentalization are associated with ciliopathies.

a | The early steps of ciliogenesis. A mother centriole matures into a basal body and migrates towards the plasma membrane. The basal body distal appendages interact either directly with the plasma membrane, or via an intermediary ciliary vesicle (as shown), and the basal body-associated membrane becomes the incipient ciliary membrane. The transition zone is the first ultrastructure of the cilium to form. Centriolar satellites have a role in ciliogenesis, potentially as an intermediate storage compartment for ciliogenic proteins. b | Ciliopathy proteins associated with different sub-compartments of the basal body, the centriolar satellites or the ciliary apparatus during and/or after ciliogenesis. Circled numbers indicate which ciliopathies (listed in part c) result from defects in these sub-compartments, as well as the organs, tissues or physiological functions that are affected. The asterisks indicate proteins that are also localized to other ciliary regions during ciliogenesis or ciliary trafficking (shown in FIG. 5). c | Ciliopathies grouped into major categories that are associated with the proteins and ciliary compartments shown in part b.
Figure 5. Links between ciliary trafficking and ciliopathies.

a | The functional components of two ciliary trafficking pathways: intraflagellar transport (IFT) and lipidated protein intraflagellar targeting (LIFT). Ciliary proteins are trafficked from the Golgi or cytosol to the base of the cilium, after which they are transported into the ciliary compartment. IFT modules that mediate trafficking include anterograde (kinesin-2) and retrograde (dynein-2) motors, IFT subcomplexes A and B, and an accessory module that contains Bardet–Biedl syndrome (BBS) proteins (the BBSome). b | Ciliopathy proteins that constitute, or are regulators of, the IFT and LIFT trafficking systems. Circled numbers indicate which ciliopathies (listed in part c) result from defects in these ciliary trafficking components. The asterisks indicate proteins that are also localized to other ciliary regions during ciliogenesis (shown in FIG. 4) or ciliary trafficking. c | Ciliopathies that result from defects in ciliary trafficking grouped into categories according to the tissues affected.
Therefore, the separation of the ciliary compartment from the cell body by the transition zone occurs early during ciliogenesis4,10 (FIG. 4a). The axoneme is then constructed and maintained by the intraflagellar transport (IFT) machinery16 (FIG. 5a). The IFT machinery comprises several subcomplexes, including heterotrimeric kinesin-2, which moves anterogradely from the transition fibres to the ciliary tip, and dynein-2, which moves retrogradely to return the IFT complex to the base. The IFT complex that binds to the motors consists of two core subcomplexes, IFT-A and IFT-B, and an accessory module that contains Bardet–Biedl syndrome (BBS) proteins (known as the BBSome) (FIG. 5a); together, these IFT components traffic various ciliary proteins, including α-/β-tubulin ‘building blocks’ of the axoneme and signalling proteins17,18. There is some evidence that the transition zone proteins work together with the IFT trafficking machinery to dynamically deliver or to remove ciliary components19, which warrants further investigation.
Motile ciliopathies
Motile cilia on different cell types have different waveforms and functions20. Similar to choanoflagellates, sperm use a specialized cilium (flagellum) to locomote. Other motile cilia do not propel cells but instead move the overlying fluid. Such cilia are found on airway epithelial cells, oviduct cells, ependymal cells that line the brain ventricles, and on the node, which is a transient developmental structure that is crucial for left–right axis determination21.
Primary Ciliary Dyskinesia (PCD)
Impaired ciliary motility almost exclusively results in a motile ciliopathy (BOX 1) that is known as primary ciliary dyskinesia (PCD)20 (FIG. 3). PCD results in situs inversus (a left–right patterning anomaly), chronic bronchitis, sinusitis and atelectasis (attributable to defective clearance of lung mucus) and male infertility (owing to defective sperm locomotion)20. Less common manifestations of PCD include decreased female fertility (resulting from improper oocyte transport through the oviducts) and a disposition to headaches and hydrocephalus (presumably resulting from impaired cerebrospinal fluid movement by ependymal cilia).
PCD is an inherited autosomal recessive disease. It is usually caused by the impaired formation or function of the inner or outer dynein arms, dynein regulatory complex or central pair, all of which are required for ciliary motility20, and does not normally affect ciliary signalling (FIG. 1). As in many ciliopathies, PCD displays considerable genetic heterogeneity, and mutations in at least 37 separate loci have been linked to the syndrome (FIG. 3; see Supplementary information S1 (table)). Such heterogeneity probably reflects the complexity of the structures required to generate ciliary motility.
First-order and second-order PCD ciliopathies
Most forms of PCD are linked to genes that encode ciliary components that are directly required for motility, and thus can be considered first-order ciliopathies (BOX 1). However, not all PCD-associated genes encode ciliary proteins. For example, mutations in DNAAF2, DNAAF3 and DYX1C1 cause PCD but encode cytoplasmic proteins that are involved in the pre-assembly of axonemal dynein complexes before their import into cilia22–24. PCDs that are a result of the loss of these non-ciliary components are examples of second-order ciliopathies (BOX 1).
Sensory ciliopathies
Sensory ciliopathies result specifically from defects in the sensory and/or signalling functions of cilia, and are primarily caused by defects in non-motile cilia (although either motile or non-motile cilia can be involved) (FIG. 3).
Defects in primary cilium structure or signalling cause sensory ciliopathies
Although motile cilia also have sensory capabilities, such as sensing noxious chemicals in the respiratory airway25, the phenotypic presentations of PCD are distinct from those exhibited when nonmotile primary cilia functions are disrupted. Metazoan non-motile primary cilia have evolved different sensory modalities for environmental cues and intercellular cues. Therefore, defects in primary cilia function lead to more varied sensory, physiological and developmental anomalies than do defects in motile cilia (FIGS 1,3). Sensory ciliopathies have several possible molecular aetiologies, including impaired cilium formation or maintenance; abrogation of ciliary signal transduction pathway components; or trafficking defects that prevent the signalling machinery from being localized to, or removed from, cilia.
Sensory ciliopathies can impair the perception of environmental cues
Different cell types make use of different forms of ciliary signalling. The determinants of this ciliary signalling specificity are only beginning to be understood, but are probably determined by the nature of the signal transduction machinery that is expressed by different cell types.
For example, in the human retina, photoreceptor cells that are responsible for vision express proteins that are specialized for phototransduction. In the photoreceptor outer segment, which is part of a modified cilium26, light interacts with opsin to activate cGMP-specific phosphodiesterase via the G protein transducin; this reduces intracellular levels of cGMP and leads to the closing of ion channels on the cell membrane. Consequent decreases in the intracellular calcium concentration outside the outer segment then promote neurotransmitter release. Therefore, for vision, the cilium is the site of both signal reception and initial transduction, with the subsequent transmission of the information to the cell body being required for interpretation and communication with other cells.
Similarly, in olfaction27, an odorant is detected by its G protein-coupled receptor (GPCR) on the ciliary membrane. However, instead of being transmitted via cGMP, the activation of the olfactory receptor stimulates an adenylate cyclase (ADCY3) to generate a different second messenger, cAMP. In further contrast to vision, olfaction uses the G protein Golf and opens ion channels to trigger an action potential. Thus, although the machinery is different, sight and smell rely on conceptually similar mechanisms for signal reception and transduction.
Defects in ciliary signalling through opsins and olfactory receptors are linked to sensory ciliopathies such as retinal degeneration and anosmia (olfaction impairment), respectively26–28. Three different molecular aetiologies for a sensory ciliopathy can be observed in retinal degeneration (retinitis pigmentosa (RP)): cilium formation or length control may be impaired29; an enzyme that lowers ciliary cGMP concentration may be disrupted30; or mislocalization of opsin or other phototransduction components may occur31,32. In these cases, the ciliary defects promote apoptotic cell death through a mechanism that is unknown but may involve the accumulation of opsin in the endoplasmic reticulum and the subsequent activation of the unfolded protein response33.
Sensory defects can also result from anomalies in the structure or signalling functions of olfactory epithelial cell cilia34. In olfactory cells, mistrafficking of ciliary proteins does not cause apoptosis but impairs their function, resulting in anosmia. Consistently, anosmia is one hallmark of ciliopathies, such as BBS35 or those caused by transition zone dysfunction36.
Impaired ciliary signalling impacts development
Sensory ciliopathies extend beyond defects in interpreting environmental cues. Primary cilia also regulate intercellular signalling pathways (FIG. 1) that, when impaired, result in defects that affect physiology (for example, weight control) or the function of various organs, including the heart, kidney, skeleton and brain (FIG. 2).
Although cilia participate in multiple intercellular signalling pathways (FIG. 1), Hedgehog signalling is one of the pathways that has been most strongly linked to ciliary function. Hedgehog is a lipoprotein morphogen that participates in the developmental patterning of many vertebrate tissues, including the neural tube and limb buds37. We focus below on the Hedgehog signal transduction pathway, as its relationship to cilia function may elucidate general principles by which other signalling systems use cilia. Many components of the Hedgehog signal transduction pathway, including the Hedgehog receptor PTCH1, localize to cilia38. Binding of Hedgehog to PTCH1 allows the downstream seven-pass transmembrane protein SMO to accumulate inside cilia, where it converts its transcriptional effectors, the GLI proteins, from repressors to activators39. GLI proteins regulate transcription in the nucleus, but also localize to cilia to function in signalling40,41. As discussed below, both IFT (trafficking) and the transition zone (gating) have important roles in dynamically modulating the localization of Hedgehog (and other) signalling components to the cilia. Therefore, many developmental abnormalities that are associated with syndromic ciliopathies, such as polydactyly in BBS and the neural tube defects of Meckel syndrome (MKS), may result from compromised ciliary Hedgehog signalling42.
Other ciliary proteins help to modulate the output of Hedgehog signalling. These include the GPCR GPR161, disruption of which can cause pituitary stalk interruption syndrome, and the EVC–EVC2 complex, dysfunction of which can cause two ciliopathies that are associated with skeletal malformations: Ellis–van Creveld syndrome (EVC) and Weyers acrofacial dysostosis43–45 (FIG. 3).
Hedgehog signalling in Drosophila melanogaster imaginal discs and cuticle does not require cilia, indicating that at least some organisms have evolved ciliumindependent mechanisms for Hedgehog signalling37. Cilia form on some sensory neurons in D. melanogaster, including those involved in olfaction, and these neurons use cilium-dependent Hedgehog signalling46. Thus, within a single organism, a signal transduction pathway can be deployed in both ciliary-dependent and ciliary-independent manners. In vitro evidence suggests that mammals may also be able to interpret Hedgehog signals through a cilium-independent mechanism, with different outputs from those of cilium-dependent signalling47.
Early ciliogenis is linked to ciliopathies
Defects in specific ciliary signal transduction components, such as those required for olfaction, phototransduction and Hedgehog signalling, can result in ciliopathies without impairing cilium structure. However, many ciliopathies are caused by the disruption of a specific aspect of the ciliogenic programme, such as the transcriptional regulation of ciliogenesis, basal body formation and the early ciliogenesis pathway, the formation of the transition zone (ciliary gate), and the trafficking machinery responsible for building the ciliary axoneme.
Initiation of the ciliogenic programme
In metazoans, ciliogenesis is initiated by a transcriptional cascade that involves one or more RFX transcription factors, namely, RFX2, RFX3, RFX4 and RFX7 in vertebrates, DAF-19 in Caenorhabditis elegans and Rfx in D. melanogaster48–55. These transcription factors bind to X box regulatory motifs to activate the transcription of many genes that are required to build cilia52,55,56. RFX targets include genes that encode components of the transition zone and the IFT–BBSome system. The formation of specialized forms of cilia also requires other transcriptional regulators that may cooperate with RFX transcription factors48. Examples include forkhead box protein J1 (FOXJ1) for generating motile cilia, GEMC1 (also known as GMNC) and MCIDAS for producing multiciliated cells, the homeobox transcription factor NOTO for generating nodal cilia, and CRX for producing photoreceptors57–60. CRX is associated with two retinal ciliopathies: Leber congenital amaurosis (LCA) and cone–rod dystrophy (CRD)61. Recently, the TAp73 isoform of TP73 was found to function upstream of RFX and FOXJ1 to contribute to motile multiciliogenesis62. At least two proteins that are needed for multiciliated cell differentiation, MCIDAS and the regulator of centriole duplication cyclin O (CCNO), underlie motile ciliopathies63,64 (FIG. 3; see Supplementary information S1 (table)). Additional regulators of ciliogenesis probably await discovery, and it is possible that mutations in gene-regulatory elements or in non-coding genes (BOX 1) may also cause ciliopathies.
Basal body and initiation of ciliogenesis
Ciliogenesis has long been known to involve basal body docking to the incipient ciliary membrane, after which the transition zone forms and IFT extends the axoneme (FIGS 4,5). Further mechanistic details have described, for example, how binding of a ciliary vesicle to distal appendages may be a prerequisite for basal body migration to the cell surface65. More recent analyses of ciliogenesis have uncovered proteins that functionally connect basal bodies via their distal appendages to membranes, including small GTPases that regulate vesicular trafficking (such as RAB8 and RAB11)66–68, proteins that shape membranes (such as EHD1 and EHD3)69 and proteins that promote membrane fusion (for example, the exocyst complex)70,71. Consistent with their essential roles in an early step in ciliogenesis, five distal appendage components (CEP164, CEP89, CEP83 (also known as CCDC41), FBF1 and SCLT1) are required for ciliogenesis72–74.
Mutations in genes encoding distal appendage components can cause a variety of ciliopathies. For example, the disruption of CEP164 or CEP83 causes nephronophthisis (NPHP)75,76, a cystic kidney disease, and mutations in SCLT1 may result in orofaciodigital syndrome (OFD)77, which is characterized by polydactyly and craniofacial abnormalities (FIG. 4).
Other ciliopathy-associated proteins localize to the distal basal body region and are essential for distal appendage formation or function (FIG. 4). One such protein, HYLS1, is associated with hydrolethalus syndrome, which is a perinatal lethal syndrome that is characterized by hydrocephalus and brain malformation, or the milder ciliopathy Joubert syndrome (JBTS)78–80. Other proteins include OFD1 (associated with JBTS, RP, OFD and Simpson–Golabi–Behmel syndrome81–83) and C2CD3 (which is linked to OFD84). Yet another distal basal body component, TALPID3 (also known as KIAA0586), supports ciliogenesis and underlies some cases of JBTS, hydrolethalus syndrome and short-rib polydactyly syndrome85.
How mutations in genes encoding either distal appendage components or distal basal body components engender such a pleiotropic range of human syndromes is unclear. Hydrolethalus syndrome may result from strong loss-of-function alleles, as the absence of distal appendages in mice severely impairs ciliogenesis and is incompatible with life85. Other non-lethal ciliopathies are likely to be caused by hypomorphic alleles or mosaicism (BOX 2). For example, null alleles of mouse Ofd1, an X-linked gene, are lethal in males but recapitulate many human OFD phenotypes in heterozygous females, which are epigenetic mosaics owing to X inactivation81.
Box 2 | The complexity of ciliopathies: multigenicity, allelism, cell type specificity, redundancy and modifiers.
As the number of ciliopathy-associated genes grows and the range and overlap between ciliopathy phenotypes increase, it is clear that the relationship between a ciliary gene and a ciliopathy is often more complex than a deterministic, Mendelian one-gene-to-one-phenotype relationship.
For example, a gene can be implicated in multiple ciliopathies with no, or limited, phenotypic overlap. A single gene can be linked to multiple phenotypes if the alleles are of differing strength. For example, presumed nonsense mutations in CC2D2A may cause Meckel syndrome (MKS) (MKS6 subtype), whereas missense mutations in the same gene lead to Joubert syndrome (JBTS) (JBTS9 subtype)188, suggesting that MKS and JBTS are caused by an allelic series that affects the same essential ciliary function. Similarly, different alleles of TMEM231 are associated with MKS, orofaciodigital syndrome (OFD) and JBTS, even within one family101,111,189.
Different missense mutations in the same gene can also result in ciliopathies that are associated with distinct ciliary functions. For example, hypomorphic mutations that affect the core IFT-B protein IFT172 result in a skeletal ciliopathy, whereas other mutations cause retinitis pigmentosa (RP) or Bardet–Biedl syndrome (BBS)190. An intriguing hypothesis to explain how IFT172 can give rise to disparate ciliopathies is that certain mutations do not impair core IFT-B functions but specifically disrupt the association of IFT172 with the BBSome and thus cause BBS. Mutations in CEP290 provide another example, as they are associated with JBTS, BBS, Leber congenital amaurosis (LCA), MKS and Senior–Løken syndrome (SLSN)186,191–193 (Supplementary information S1 (table)). As it is not clear whether these different ciliopathy-associated mutations form an allelic series, it is possible that they affect distinct functions of CEP290 at the transition zone172,194 and centriolar satellites88,195 (FIGS 4,5).
Another way in which different mutations in the same gene can result in distinct phenotypes is by affecting protein isoforms that have different functions. For example, disruption of the BBSome-associated protein ARL6 (also known as BBS3) causes typical BBS phenotypes, whereas a longer isoform (BBS3L) is specifically required for photoreceptor maintenance in mice and zebrafish196.
Additionally, genetic modifiers influence the clinical manifestation of mutations in ciliopathy-associated genes. Such modifiers help to identify genes with overlapping or antagonistic functions. For example, mutations in RPGRIP1L, which encodes a transition zone component, are associated with MKS, JBTS and COACH (cerebellar vermis hypo/aplasia, oligophrenia (mental retardation), ataxia, ocular coloboma, and hepatic fibrosis) syndrome197,198. Mutations in a paralogue, RPGRIP1, cause isolated retinal phenotypes (cone–rod dystrophy (CRD) and LCA)199,200. Caenorhabditis elegans has only one orthologue of RPGRIP1L and RPGRIP1, which is crucial for transition zone assembly10,99. In mammals, RPGRIP1 and RPGRIP1L might have overlapping functions, and non-pathogenic alleles may modify the phenotypes that are caused by the pathogenic alleles. Indeed, components of different complexes (transition zone and BBS) have overlapping functions in cilium formation in C. elegans and mice10,99,106. These findings in model organisms indicate that the type of alleles, the modifiers present in different genetic backgrounds, overlapping protein functions and cell-type specificity can all influence the phenotypic outcome, suggesting that similar genetic complexities underlie human ciliopathies.
Centriolar satellites
In addition to the distal centriole, OFD1, C2CD3 and TALPID3 also localize to centriolar satellites86–88 (FIG. 4). Various centrosomal and ciliary proteins partially localize to these regions, which are found near centrosomes or basal bodies89. Like OFD1 and C2CD3, many centriolar satellite proteins are essential for cilium formation89–91. However, it remains unclear whether such proteins simply localize within the centriolar satellites before their transit to other locations that are directly relevant to ciliogenesis. Some proteins that are involved in centriole duplication and microcephaly, a disorder that is mainly associated with centrosomal dysfunction and with possible ramifications for ciliary signalling92,93, also require centriolar satellites94. Thus, centriolar satellites have roles that are both relevant to, and potentially independent of, ciliary function.
In summary, basal body-associated defects can compromise cilium formation or function, resulting in diverse ciliopathies that are often characterized by developmental abnormalities (FIG. 4). Numerous basal body proteins that are relevant to cilia function have been identified95–98, and we anticipate that some of these — as well as centriolar satellite proteins — will be implicated in other ciliopathies (Supplementary information S1 (table)).
Transition zone is a hotspot for ciliopathies
Once the basal body has docked to a membrane, the nascent cilium becomes a separate compartment that is separated from the cytosol by the transition zone4,5. Like the distal basal body, components of the transition zone have been extensively implicated in ciliopathies (FIG. 4; see Supplementary information S1 (table)). Transition zone-associated ciliopathies have effects that are generally restricted to single organs, such as effects in NPHP, but can also have pleiotropic effects, as in MKS4,5.
The transition zone influences ciliary composition
A large network of proteins that are present at the transition zone modulates the composition of the cilium in diverse organisms, including vertebrates, C. elegans, D. melanogaster and Chlamydomonas reinhardtii6–10,99. The MKS complex and the NPHP complex are the two main functional modules of the transition zone, which are associated with, respectively, MKS and NPHP ciliopathies10,100. In mammals, the disruption of the MKS complex reduces the ciliary abundance of membraneassociated ciliary proteins, including ARL13B, INPP5E, ADCY3 and the central Hedgehog signal transduction component SMO8,9,101,102. As Hedgehog signalling is crucial for specifying digit number and central nervous system development37, compromised SMO localization to cilia may well be sufficient to account for several of the ciliopathy-associated developmental defects.
Transition zone proteins are also crucial for the ciliary localization of polycystin 2 (PKD2), which is a transmembrane protein that interacts with PKD1 (REF. 9). As mutations in either PKD1 or PKD2 cause autosomal dominant polycystic kidney disease (ADPKD) in humans (FIG. 3; see Supplementary information S1 (table)), decreased ciliary localization of PKD2 may account for the kidney cysts in MKS103. Therefore, although it is possible to build cilia without a transition zone9,99, transition zone-associated ciliopathies are probably the result of the altered distribution of one or more ciliary signalling proteins.
Transition zone proteins are also critical for the ciliary localisation of PKD2, a transmembrane protein that interacts with PKD19. As mutations in either PKD1 or PKD2 cause autosomal dominant polycystic kidney disease (ADPKD) in humans (Fig. 3; Table S1), decreased ciliary localisation of PKD2 may account for the kidney cysts in MKS103. Hence, although it is possible to build cilia without a transition zone9,99, transition zone-associated ciliopathies are probably due to the altered distribution of one or more ciliary signalling proteins.
Transition zone involvement in ciliopathies
In addition to MKS, mutations in genes that encode components of the MKS complex are associated with JBTS and COACH (cerebellar vermis hypo/aplasia, oligophrenia (mental retardation), ataxia, ocular coloboma, and hepatic fibrosis) syndromes, whereas mutations that affect the NPHP complex also cause Senior–Løken syndrome (SLSN), which is characterized by NPHP with RP (FIG. 4; see Supplementary information S1 (table)). Some mutations may bridge these complexes, as exemplified by the minority of JBTS-affected individuals who have NPHP and RP in addition to the pathognomonic cerebellar defects104. Similar to the basal body-associated ciliopathies discussed above, the phenotypic diversity that is caused by transition zone dysfunction may result from alleles of different strengths (BOX 2). Perhaps MKSassociated alleles compromise MKS complex function to such an extent that ciliogenesis is compromised, whereas JBTS-associated alleles spare ciliogenesis but alter ciliary membrane composition (and thus ciliary signalling).
Another non-exclusive possibility to explain the pleiotropy of ciliopathies is the alteration of phenotypic outcomes of Mendelian-inherited ciliopathies by modifiers (BOX 2). For example, the BBSome, NPHP and MKS complexes may have partially overlapping roles in promoting the ciliary localization of membrane proteins, as exemplified by the finding that BBS can be caused by mutations in core transition zone proteins such as MKS1 (BBS13, a subtype of BBS) and CEP290 (BBS14)105 (FIGS 4,5; see Supplementary information S1 (table)). Thus, modest effects on one complex may phenocopy the consequences caused by the disruption of another complex. In C. elegans and mice, disruption of both the MKS complex and the NPHP complex, or both the MKS complex and the BBS complex, has a synthetic (synergistic) effect on the phenotypes10,102,106. Whether similar genetic interactions affect the manifestations of human ciliopathies will require careful phenotyping of large pedigrees.
With their moderate level of allelism and easily quantifiable discrete phenotypic outcomes, ciliopathies may represent a particularly tractable ‘sweet spot’ between strictly Mendelian and complex polygenic disorders. For example, genome-wide association studies (GWAS) cannot account for more than a small proportion of the estimated heritability of polygenic traits, leading to searches for the ‘missing heritability’ (REF. 107). At least in ciliopathies, specific genetic interactions between distinct functional complexes, such as the MKS and NPHP complexes, which GWAS fail to detect, could help to account for this missing heritability (BOX 2). In support of this possibility, NPHPor BBS-affected individuals can have lesions in multiple genes108,109.
Is the transition zone a lipid gate for ciliary trafficking?
How the transition zone is organized and functions to control ciliary composition is mostly unknown, but will be key to understanding various ciliopathies. A plausible hypothesis is that structural proteins that form the Y-links organize protein complexes at the transition zone membrane (the so-called ciliary necklace) establish a lipid microdomain that is involved in partitioning the ciliary domains from the extraciliary domains4,99.
The presence of a barrier at the base of cilia implies that there are trafficking systems involved in transiting this partition. One such trafficking system, which we refer to here as lipidated protein intraflagellar targeting (LIFT), is specific for proteins that are modified with lipids (for example, farnesylation and myristoylation). LIFT involves several components, including UNC119, PDE6D, RP2, ARL3 and ARL13B30,110, and is disrupted in ciliopathies such as RP, rod–cone dystrophy and JBTS (FIG. 5; see Supplementary information S1 (table)). Interestingly, JBTS is associated with multiple transition zone proteins100,102,111–113, suggesting a functional association between the transition zone and this lipidated protein trafficking system. Another key trafficking system that transits the transition zone is IFT.
IFT–BBSome trafficking defects in ciliopathies
In 1993, the Rosenbaum laboratory observed that in C. reinhardtii flagella, particles moved bidirectionally between the basal body and the tip of the axoneme114. The machinery involved in this process, IFT, was subsequently found to be powered by kinesin and dynein molecular motors, and to comprise two ‘core’ multiprotein subcomplexes (IFT-A and IFT-B), as well as an associated BBSome complex that mediates ciliary cargo transport16,18,115 (FIG. 5).
Most IFT subcomplex A/B subunits are linked to ciliopathies
In vertebrates, IFT is essential for cilium biogenesis and, consequently, embryonic development16,18,116. Many mouse mutations that affect IFT components cause embryonic midor late-gestation arrest with mispatterning of Hedgehog-dependent tissues, such as the neural tube and limb buds116. In humans, mutations in IFT genes that cause ciliopathies often affect the skeletal system (FIG. 5; see mation S1 (table)). Mutations in genes encoding several components of the IFT retrograde motor dynein-2 (DYNC2H1, DYNC2LI1, TCTEX1D2, WDR34 and WDR60) and the IFT-A subcomplex (IFT43, IFT121 (also known as WDR35), IFT122, IFT139 (also known as TTC21B), IFT140 and IFT144) are associated with several skeletal ciliopathies, including Jeune asphyxiating thoracic dystrophy (JATD), cranioectodermal dysplasia (CED; also known as Sensenbrenner syndrome) and short-rib polydactyly syndrome117–125. Mutations in IFT subunits are also linked to other diseases, including RP, NPHP and SLSN126,127 (Supplementary information S1 (table)).
The disruption of several IFT-B subunits is similarly associated with an overlapping subset of ciliopathies (FIG. 5). For example, hypomorphic mutations in IFT172 result in the skeletal ciliopathies JATD and Mainzer–Saldino syndrome128, or VACTERL (vertebral anomalies, anal atresia, cardiac defects, tracheoesophageal fistula and/or esophageal atresia, renal and radial anomalies and limb defects) associated with hydrocephalus129. Mutations in IFT52 and IFT80 also cause skeletal ciliopathies130,131. The disruption of IFT57 is associated with OFD, as well as short stature and brachymesophalangia132.
To understand the molecular basis of different IFTassociated ciliopathies, researchers are studying how mutations in different IFT subunits cause defects in the transport of specific cargo16,115. For example, the IFT-Aassociated protein TULP3 facilitates the transport of specific GPCRs to cilia133. LZTFL1 and IFT27, which are both associated with the IFT-B subcomplex, are implicated in the transport of Hedgehog signalling proteins134,135. Of note, both LZTFL1 and IFT27 are linked to BBS in humans136,137.
Possible additional links between IFT proteins and ciliopathies
Mouse models suggest that genes encoding other IFT components are good candidates for orphan ciliopathies (Supplementary information S1 (table)). For example, mouse IFT46 is essential for brain, neural tube and heart development138. A hypomorphic mutation in mouse Ift88 results in kidney cyst formation, suggesting that cilia modulate kidney epithelial growth and organization139. IFT components (DYNC2H1, IFT74 and IFT140) were also uncovered in a mouse mutagenesis screen for congenital heart defects140. Of note, these were among a high proportion of ciliary genes to be identified, which confirms the importance of motile and non-motile cilia, and the specification of left–right asymmetry, in the origin of congenital heart defects141,142.
BBS proteins: connecting signalling defects to ciliopathies
BBS arises from the disruption of BBSome components (BBS1, BBS2, BBS4, BBS5, BBS7 and BBS8), or disruption of BBSome trafficking (ARL6; also known as BBS3)143 or assembly (BBS6, BBS10 and BBS12)42 (FIG. 5; see Supplementary information S1 (table)). The broad phenotypic range of BBS — which includes retinal degeneration, cystic kidneys, obesity, polydactyly and cognitive impairment — may be explained by its crucial role in the transport of diverse ciliary cargoes.
Three BBS-dependent cargoes are dopamine receptor 1 (DR1)144, somatostatin receptor 3 (SSTR3)145 and melanin-concentrating hormone receptor 1 (MCHR1)145. Aberrant ciliary protein localization is the probable aetiology of BBS-associated phenotypes; for example, polydactyly may arise from impaired Hedgehog signalling146. However, many clinical presentations still have unclear molecular aetiologies and could be multifactorial. For example, obesity in BBS may result from hypothalamic dysfunction and satiety defects owing to the mislocalization of the NPY receptor MCHR1 and, potentially, the mislocalization of the leptin receptor145,147,148. Similarly, retinal degeneration may be caused by inefficient opsin trafficking31.
Regulation of IFT-BBSome trafficking and links to ciliopathies
Understanding the molecular basis of ciliopathies will require a deeper understanding of how IFT particles and the BBSome assemble and function to regulate the trafficking of ciliary cargoes, including GPCRs. Evidence that several IFT and BBSome proteins are evolutionarily related to vesicle coat proteins may be instructive, as protein functions may have parallels to vesicle trafficking18,149,150. The study of ciliopathies is likely to identify new core or regulatory players in these processes and to lead to important insights. For example, kinases that influence cilium length by regulating IFT include ICK, MAK and MOK, with ICK associated with lethal endocrine-cerebro-osteodysplasia and shortrib polydactyly syndrome, and MAK associated with RP151,152 (FIGS 3,5). Mutations in NEK1 cause short-rib polydactyly syndrome with brain malformations and kidney cysts153, pointing to an additional possible association between this NIMA-related kinase and IFT. The ciliopathy-associated ciliogenesis and planar polarity effector (CPLANE) complex, which participates in basal body recruitment of the IFT machinery, was recently associated with JBTS, OFD and SRPS154; this respresents another example of how the genetics of ciliopathies and cell biological insights into ciliogenesis inform each other.
Second-order ciliopathies
Most ciliogenic and ciliopathy proteins are components of the cilium, basal body or centriolar satellites. We use the term first-order ciliopathies for those associated with these proteins to reflect their local requirement at the basal body or cilium (BOX 1). However, non-ciliary proteins can also participate in ciliary functions and can be associated with ciliopathies. For example, transcription factors (such as RFX2, RFX3 and RFX4) that regulate the expression of ciliary genes are not cilium-localized but are crucial for cilium formation and function48. As another example, some ciliary complexes must be pre-assembled in the cytosol before being incorporated into the cilium. The PCD-associated proteins DNAAF2, DNAAF3 and DYX1C1 mediate the cytosolic assembly of axonemal dynein complexes that are crucial for ciliary motility22–24. These ciliopathies can be regarded as being secondary (second-order) to ciliary processes (BOX 1).
Second-order ciliopathies will continue to be uncovered. For example, mutations in the gene encoding the Golgi-localized glycosyltransferase GALNT11 perturb Notch signalling and alter motile and non-motile cilia ratios in Xenopus laevis, leading to laterality and heart defects155.
Ciliary proteins with extra-ciliary functions
Given the wide distribution of cilia in extant phyla, the last eukaryotic common ancestor (LECA) probably had cilia with essentially complete IFT–BBSome and transition zone systems1,156. Interestingly, there is some evidence that, as metazoans evolved specialized cell types, ancient ciliary proteins acquired novel functions.
For example, EFHC1 is widely conserved in ciliated eukaryotes that have motile cilia and is required for ciliary motility in mammalian cells157. However, C. elegans, which lacks motile cilia, has an orthologue of EFHC1 (REF. 158), and the D. melanogaster EFHC1 orthologue regulates the morphogenesis of neurons that lack cilia159. As in C. elegans, mammalian EFHC1 is expressed by cells with non-motile cilia, including those in the brain160. The function of EFHC1 in neurons is unclear but important, as mutations in this protein predispose humans to juvenile epilepsy157. Thus, the ancient roles of EFHC1 within motile cilia may have been more recently adapted in several non-ciliary functions that are relevant to neuronal or brain function.
Similarly, other ciliary proteins may have acquired extraciliary functions; for example, IFT20 may transport PKD2 from the Golgi to the cilium, and may enable trafficking to the immunological synapse161,162. When a cytotoxic T cell, which is unciliated, engages a target cell, its centrosome docks at the cell periphery using distal appendages, similar to those of the basal body. At this subcellular position, the centrosome directs polarized vesicle trafficking to create a functional immunological synapse163. Therefore, structures and proteins that have been implicated in cilium function — distal appendages, IFT20, and the small GTPase RAB29 that colocalizes with RAB8, RAB11 and IFT20 — are also associated with immunological synapse assembly and function164. At least in the mouse, the disruption of an established ciliary protein — surprisingly, one associated with cilium motility (SPAG6) — impairs immune synapse function165.
However, the cytotoxic T cell centrosome does not build a transition zone or extend an axoneme, so although there are similarities in the organization of the cilium and the immunological synapse and they use some of the same machinery, there are also pronounced structural and functional differences. It will be interesting to determine whether other cell type-specific centrosome-associated functions represent divergent functions for the ciliary machinery. IFT20 may be particularly versatile in its functions as, in addition to its roles in trafficking cargo within the cilium and to the immunological synapse, it contributes to intracellular transport of collagen166.
Thus, the analysis of proteins with established roles in cilia may need to take into consideration the possibility that such proteins participate in other cellular processes in both ciliated and non-ciliated cells. Shedding light on the combination of ciliary and cilium-independent functions of proteins may be helpful in explaining the complete molecular aetiology of the associated diseases.
Discovery of ciliopathy-associated proteins
Using the list of manually curated cilium-associated components (the current gold standard) published by the SysCilia consortium as a starting point, we compiled a list of 428 human proteins that are associated with cilia (by localization and/or function), and found that 187 of them are linked to ciliopathies (Supplementary information S1 (table)). Of note, since the publication of this gold standard list in 2013, at least 50 additional cilium-associated proteins have been identified; approximately 50% are linked to ciliopathies, highlighting the crucial importance of cilia in human disease.
Identification of ciliary proteins and ciliopathy candidates
A wide range of complementary studies aimed at uncovering the ‘ciliome’ suggest that additional basal body and ciliary proteins will be discovered, and some of these proteins might be associated with one or more ciliopathies. Such studies, compiled in the CilDB database167, include proteomics studies of isolated motile and non-motile cilia97,168,169, comparative genomics studies of ciliated versus non-ciliated organisms96,150, gene expression studies showing the upregulation of genes during cilium formation or changes in expression in mutants170,171, and the identification of RFX transcription factor target genes (through bioinformatic searches for X-box regulatory motifs and uncovering genes regulated by the nematode RFX transcription factor orthologue DAF-19)56,170. The CilDB database can be searched using Boolean logic for the presence or absence of a given protein in different studies and organisms, and can help to identify candidate ciliary proteins and ciliopathy proteins.
The refinement of the ciliome, which now comprises more than 420 proteins (mostly, but not exclusively human) (Supplementary information S1 (table)), is on-going. Furthermore, studies in model organisms continue to identify ciliopathy candidates and to provide insights into ciliary function. For example, the discoveries that C. elegans TMEM-218 functions at the transition zone172, and that the mouse Tmem218 mutant exhibits kidney cysts and retinal degeneration173, suggest that this gene is an excellent candidate gene to underlie SLSN or a related ciliopathy. Interactomes of established ciliary proteins can also identify new ciliary proteins and ciliopathy candidates100,174.
The various approaches for identifying ciliary proteins all have limitations. TMEM80, for example, was not implicated as a ciliary protein in any study included in the CilDB database before being revealed as a transition zone component, based on its homology to known ciliary proteins (TMEM17 and TMEM216)172. Thus, complementary and novel approaches are useful for identifying new ciliary proteins. A promising technique is proximity-dependent protein identification, in which a given protein is fused to an enzyme that can tag (for example, biotinylate) nearby interaction partners for subsequent identification by mass spectrometry98. Similarly, model organism genetic or genome-wide RNA interference (RNAi) screens can uncover, in an unbiased manner, new genes that are required for cilia function140,175,176.
Confirmation of novel ciliopathy genes and ciliopathies
As whole-genome sequencing continues to become more tractable, we expect that novel mutations that are associated with ciliopathies will be readily identified. Baker and Beales177 predicted in 2009 that more than 72 syndromes were possible ciliopathies. Some of their candidates have since been confirmed to be linked to ciliary dysfunction, including hydrolethalus syndrome, which is caused by mutations in TALPID3, KIF7 and HYLS1 (REFS 78,80,85,178,179). The endocrine-cerebroosteodysplasia syndrome was shown to result from mutations in ICK178, which encodes a kinase that is involved in the control of IFT180. Walker–Warburg (WWS) syndrome was a suspected but unproven ciliopathy; the B3GNT1 (also known as B4GNT1) glycosyltransferase implicated in this disorder is now known to influence ciliated cell function in C. elegans175. The 241 candidates listed in Supplementary information S1 (table) may reveal additional connections to known or novel ciliary disorders. Functional analysis of novel ciliopathy proteins will further increase our knowledge of the signalling, physiological and developmental functions of cilia.
Conclusions and perspectives
The known connections between cilia and human disease will continue to increase, and are likely to include additional diseases that are not specifically — or traditionally — thought of as ciliopathies, such as cancer and congenital heart defects. Ciliopathy research will provide new, valuable insights into the fundamental biology of cilia. Furthermore, the discovery of rare disease variants of essential genes may help to unveil unanticipated roles in ciliogenesis. For example, mutations in the BUBR1 mitotic spindle checkpoint regulator are associated with aneuploidy, cancer predisposition and impaired ciliogenesis181. New tools (beyond loss-of-function approaches) may be required to understand whether non-ciliary proteins underlying common diseases, such as cancer, have ciliary functions.
Notwithstanding such important advances, understanding the molecular functions of human ciliopathyassociated proteins, and deciphering their mechanistic roles within a complex, network and pathway, remains challenging, and the use of model organisms to dissect the roles of ciliary proteins and to model the effects of mutations remains essential. Studies in mammalian (mouse) and vertebrate (zebrafish) model systems must continue to be complemented by research in C. elegans and D. melanogaster, and in ciliated protists such as C. reinhardtii, Trypanosoma brucei and Tetrahymena thermophila. Connections between clinician scientists and model organism researchers through organizations such as the Rare Diseases: Models & Mechanisms (RDMM) network182 will help to use human clinical and genomic data to uncover how cilia function in physiology and development.
Supplementary Material
Acknowledgments
The authors apologise for not citing numerous important studies relevant to this vast and growing area of biology, due to space restrictions. Funding for this work was provided by the Canadian Institutes of Health Research (CIHR; grants MOP142243 and MOP82870 to MRL) and grants AR054396 and GM095941 from the NIH to JFR. MRL acknowledges a senior scholar award from the Michael Smith Foundation for Health Research (MSFHR).
GLOSSARY
- Dynein-2
Molecular motor involved in the retrograde (tip-to-base) transport of the IFT machinery.
- Kinesin-2
Heterotrimeric molecular motor required for the anterograde (base-to-tip) transport of the IFT machinery.
- Centriolar satellites
Electron-dense puncta found at the periphery of centrosomes or basal bodies. May function as a temporary hub for several proteins that are required for the proper formation and function of cilia.
- Exocyst complex
Protein complex involved in targeting Golgi-derived vesicles to the plasma membrane.
- Mosaicism
Two or more cell populations with different genotypes in one single individual.
- Mother centriole
Centriolar structure that is remodeled into a basal body prior to the onset of cilium formation.
- Septins
Proteins which create barriers between different membrane compartments in several contexts, including possibly at the base of cilia.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
References
- 1.Carvalho-Santos Z, Azimzadeh J, Pereira-Leal JB, Bettencourt-Dias M. Evolution: Tracing the origins of centrioles, cilia, and flagella. J Cell Biol. 2011;194:165–175. doi: 10.1083/jcb.201011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fisch C, Dupuis-Williams P. Ultrastructure of cilia and flagella back to the future! Biol Cell. 2011;103:249–270. doi: 10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- 3.Nachury MV. How do cilia organize signalling cascades. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13:608–618. doi: 10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Gonzalo FR, Reiter JF. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol. 2012;197:697–709. doi: 10.1083/jcb.201111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awata J, et al. NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J Cell Sci. 2014;127:4714–4727. doi: 10.1242/jcs.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basiri ML, et al. A migrating ciliary gate compartmentalizes the site of axoneme assembly in Drosophila spermatids. Curr Biol. 2014;24:2622–2631. doi: 10.1016/j.cub.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chih B, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Gonzalo FR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams CL, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Q, et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–439. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breslow DK, Koslover EF, Seydel F, Spakowitz AJ, Nachury MV. An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. J Cell Biol. 2013;203:129–147. doi: 10.1083/jcb.201212024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin YC, et al. Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat Chem Biol. 2013;9:437–443. doi: 10.1038/nchembio.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Najafi M, Maza NA, Calvert PD. Steric volume exclusion sets soluble protein concentrations in photoreceptor sensory cilia. Proc Natl Acad Sci U S A. 2012;109:203–208. doi: 10.1073/pnas.1115109109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.TOKUYASU K, YAMADA E. The fine structure of the retina studied with the electron microscope. IV. Morphogenesis of outer segments of retinal rods. J Biophys Biochem Cytol. 1959;6:225–230. doi: 10.1083/jcb.6.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mourão A, Christensen ST, Lorentzen E. The intraflagellar transport machinery in ciliary signaling. Curr Opin Struct Biol. 2016;41:98–108. doi: 10.1016/j.sbi.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Bhogaraju S, et al. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science. 2013;341:1009–1012. doi: 10.1126/science.1240985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat Cell Biol. 2013;15:1387–1397. doi: 10.1038/ncb2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao C, Malicki J. Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 2011;30:2532–2544. doi: 10.1038/emboj.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horani A, Ferkol TW, Dutcher SK, Brody SL. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev. 2016;18:18–24. doi: 10.1016/j.prrv.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dasgupta A, Amack JD. Cilia in vertebrate left-right patterning. Philos Trans R Soc Lond B Biol Sci. 2016;371 doi: 10.1098/rstb.2015.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchison HM, et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat Genet. 2012;44:381–9. S1. doi: 10.1038/ng.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarkar A, et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat Genet. 2013;45:995–1003. doi: 10.1038/ng.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omran H, et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bloodgood RA. Sensory reception is an attribute of both primary cilia and motile cilia. J Cell Sci. 2010;123:505–509. doi: 10.1242/jcs.066308. [DOI] [PubMed] [Google Scholar]
- 26.Wheway G, Parry DA, Johnson CA. The role of primary cilia in the development and disease of the retina. Organogenesis. 2014;10:69–85. doi: 10.4161/org.26710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McEwen DP, Jenkins PM, Martens JR. Olfactory cilia: our direct neuronal connection to the external world. Curr Top Dev Biol. 2008;85:333–370. doi: 10.1016/S0070-2153(08)00812-0. [DOI] [PubMed] [Google Scholar]
- 28.Schou KB, Pedersen LB, Christensen ST. Ins and outs of GPCR signaling in primary cilia. EMBO Rep. 2015;16:1099–1113. doi: 10.15252/embr.201540530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ozgül RK, et al. Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am J Hum Genet. 2011;89:253–264. doi: 10.1016/j.ajhg.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas S, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014;35:137–146. doi: 10.1002/humu.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimura DY, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, et al. Mistrafficking of prenylated proteins causes retinitis pigmentosa 2. FASEB J. 2015;29:932–942. doi: 10.1096/fj.14-257915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunte MM, et al. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53:3792–3800. doi: 10.1167/iovs.11-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenkins PM, McEwen DP, Martens JR. Olfactory cilia: linking sensory cilia function and human disease. Chem Senses. 2009;34:451–464. doi: 10.1093/chemse/bjp020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulaga HM, et al. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet. 2004;36:994–998. doi: 10.1038/ng1418. [DOI] [PubMed] [Google Scholar]
- 36.McEwen DP, et al. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc. Natl. Acad. Sci. USA. 2007;104:15917–15922. doi: 10.1073/pnas.0704140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 38.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 39.Corbit KC, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 40.Haycraft CJ, et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos N, Reiter JF. A central region of Gli2 regulates its localization to the primary cilium and transcriptional activity. J Cell Sci. 2014;127:1500–1510. doi: 10.1242/jcs.139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q, Yu D, Seo S, Stone EM, Sheffield VC. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. J Biol Chem. 2012;287:20625–20635. doi: 10.1074/jbc.M112.341487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caparrós-Martín JA, et al. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum Mol Genet. 2013;22:124–139. doi: 10.1093/hmg/dds409. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay S, et al. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152:210–223. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz-Perez VL, Goodship JA. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands. Am J Med Genet C Semin Med Genet. 2009;151C:341–351. doi: 10.1002/ajmg.c.30226. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez GM, et al. Hedgehog Signaling Regulates the Ciliary Transport of Odorant Receptors in Drosophila. Cell Rep. 2016;14:464–470. doi: 10.1016/j.celrep.2015.12.059. [DOI] [PubMed] [Google Scholar]
- 47.Bijlsma MF, Damhofer H, Roelink H. Hedgehog-stimulated chemotaxis is mediated by smoothened located outside the primary cilium. Sci Signal. 2012;5:ra60. doi: 10.1126/scisignal.2002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choksi SP, Lauter G, Swoboda P, Roy S. Switching on cilia: transcriptional networks regulating ciliogenesis. Development. 2014;141:1427–1441. doi: 10.1242/dev.074666. [DOI] [PubMed] [Google Scholar]
- 49.Swoboda P, Adler HT, Thomas JH. The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. elegans. Mol Cell. 2000;5:411–421. doi: 10.1016/s1097-2765(00)80436-0. [DOI] [PubMed] [Google Scholar]
- 50.Chung MI, et al. RFX2 is broadly required for ciliogenesis during vertebrate development. Dev Biol. 2012;363:155–165. doi: 10.1016/j.ydbio.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonnafe E, et al. The transcription factor RFX3 directs nodal cilium development and left-right asymmetry specification. Mol Cell Biol. 2004;24:4417–4427. doi: 10.1128/MCB.24.10.4417-4427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El Zein L, et al. RFX3 governs growth and beating efficiency of motile cilia in mouse and controls the expression of genes involved in human ciliopathies. J Cell Sci. 2009;122:3180–3189. doi: 10.1242/jcs.048348. [DOI] [PubMed] [Google Scholar]
- 53.Ashique AM, et al. The Rfx4 transcription factor modulates Shh signaling by regional control of ciliogenesis. Sci Signal. 2009;2:ra70. doi: 10.1126/scisignal.2000602. [DOI] [PubMed] [Google Scholar]
- 54.Manojlovic Z, Earwood R, Kato A, Stefanovic B, Kato Y. RFX7 is required for the formation of cilia in the neural tube. Mech Dev. 2014;132:28–37. doi: 10.1016/j.mod.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dubruille R, et al. Drosophila regulatory factor X is necessary for ciliated sensory neuron differentiation. Development. 2002;129:5487–5498. doi: 10.1242/dev.00148. [DOI] [PubMed] [Google Scholar]
- 56.Blacque OE, et al. Functional genomics of the cilium, a sensory organelle. Curr. Biol. 2005;15:935–941. doi: 10.1016/j.cub.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 57.You Y, et al. Role of f-box factor foxj1 in differentiation of ciliated airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L650–7. doi: 10.1152/ajplung.00170.2003. [DOI] [PubMed] [Google Scholar]
- 58.Chen S, et al. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19:1017–1030. doi: 10.1016/s0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- 59.Kyrousi C, et al. Mcidas and GemC1 are key regulators for the generation of multiciliated ependymal cells in the adult neurogenic niche. Development. 2015;142:3661–3674. doi: 10.1242/dev.126342. [DOI] [PubMed] [Google Scholar]
- 60.Beckers A, Alten L, Viebahn C, Andre P, Gossler A. The mouse homeobox gene Noto regulates node morphogenesis, notochordal ciliogenesis, and left right patterning. Proc Natl Acad Sci U S A. 2007;104:15765–15770. doi: 10.1073/pnas.0704344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swaroop A, et al. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8:299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- 62.Nemajerova A, et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 2016;30:1300–1312. doi: 10.1101/gad.279836.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boon M, et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun. 2014;5:4418. doi: 10.1038/ncomms5418. [DOI] [PubMed] [Google Scholar]
- 64.Wallmeier J, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. 2014;46:646–651. doi: 10.1038/ng.2961. [DOI] [PubMed] [Google Scholar]
- 65.SOROKIN S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nachury MV, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 67.Mazelova J, et al. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28:183–192. doi: 10.1038/emboj.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knödler A, et al. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A. 2010;107:6346–6351. doi: 10.1073/pnas.1002401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu Q, et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol. 2015;17:228–240. doi: 10.1038/ncb3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogers KK, et al. The exocyst localizes to the primary cilium in MDCK cells. Biochem Biophys Res Commun. 2004;319:138–143. doi: 10.1016/j.bbrc.2004.04.165. [DOI] [PubMed] [Google Scholar]
- 71.Zuo X, Guo W, Lipschutz JH. The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell. 2009;20:2522–2529. doi: 10.1091/mbc.E08-07-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joo K, et al. CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc Natl Acad Sci U S A. 2013;110:5987–5992. doi: 10.1073/pnas.1220927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmidt KN, et al. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J Cell Biol. 2012;199:1083–1101. doi: 10.1083/jcb.201202126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanos BE, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27:163–168. doi: 10.1101/gad.207043.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Failler M, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet. 2014;94:905–914. doi: 10.1016/j.ajhg.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chaki M, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adly N, Alhashem A, Ammari A, Alkuraya FS. Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Hum Mutat. 2014;35:36–40. doi: 10.1002/humu.22477. [DOI] [PubMed] [Google Scholar]
- 78.Mee L, et al. Hydrolethalus syndrome is caused by a missense mutation in a novel gene HYLS1. Hum Mol Genet. 2005;14:1475–1488. doi: 10.1093/hmg/ddi157. [DOI] [PubMed] [Google Scholar]
- 79.Oka M, et al. A novel HYLS1 homozygous mutation in living siblings with Joubert syndrome. Clin Genet. 2016;89:739–743. doi: 10.1111/cge.12752. [DOI] [PubMed] [Google Scholar]
- 80.Dammermann A, et al. The hydrolethalus syndrome protein HYLS-1 links core centriole structure to cilia formation. Genes Dev. 2009;23:2046–2059. doi: 10.1101/gad.1810409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ferrante MI, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68:569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Webb TR, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum Mol Genet. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Coene KL, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thauvin-Robinet C, et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet. 2014;46:905–911. doi: 10.1038/ng.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alby C, et al. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. Am J Hum Genet. 2015;97:311–318. doi: 10.1016/j.ajhg.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lopes CA, et al. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J Cell Sci. 2011;124:600–612. doi: 10.1242/jcs.077156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye X, Zeng H, Ning G, Reiter JF, Liu A. C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci U S A. 2014;111:2164–2169. doi: 10.1073/pnas.1318737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kobayashi T, Kim S, Lin YC, Inoue T, Dynlacht BD. The CP110-interacting proteins Talpid3 and Cep290 play overlapping and distinct roles in cilia assembly. J Cell Biol. 2014;204:215–229. doi: 10.1083/jcb.201304153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hori A, Toda T. Regulation of centriolar satellite integrity and its physiology. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2315-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee JY, Stearns T. FOP is a centriolar satellite protein involved in ciliogenesis. PLoS One. 2013;8:e58589. doi: 10.1371/journal.pone.0058589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Silva E, et al. Ccdc11 is a novel centriolar satellite protein essential for ciliogenesis and establishment of left-right asymmetry. Mol Biol Cell. 2016;27:48–63. doi: 10.1091/mbc.E15-07-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH. Cortical neurogenesis in the absence of centrioles. Nat Neurosci. 2014;17:1528–1535. doi: 10.1038/nn.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang L, Hou S, Han YG. Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat Neurosci. 2016;19:888–896. doi: 10.1038/nn.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kodani A, et al. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife. 2015;4 doi: 10.7554/eLife.07519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kilburn CL, et al. New Tetrahymena basal body protein components identify basal body domain structure. J Cell Biol. 2007;178:905–912. doi: 10.1083/jcb.200703109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li JB, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 97.Liu Q, et al. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics. 2007;6:1299–1317. doi: 10.1074/mcp.M700054-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gupta GD, et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell. 2015;163:1484–1499. doi: 10.1016/j.cell.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jensen VL, et al. Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J. 2015;34:2537–2556. doi: 10.15252/embj.201488044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sang L, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roberson EC, et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. 2015;209:129–142. doi: 10.1083/jcb.201411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang L, et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang L, Lipschutz JH. Cilia and polycystic kidney disease, kith and kin. Birth Defects Res C Embryo Today. 2014;102:174–185. doi: 10.1002/bdrc.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Valente EM, et al. Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann Neurol. 2005;57:513–519. doi: 10.1002/ana.20422. [DOI] [PubMed] [Google Scholar]
- 105.Leitch CC, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 106.Yee LE, et al. Conserved Genetic Interactions between Ciliopathy Complexes Cooperatively Support Ciliogenesis and Ciliary Signaling. PLoS Genet. 2015;11:e1005627. doi: 10.1371/journal.pgen.1005627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Katsanis N, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 109.Hoefele J, et al. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 110.Wright KJ, et al. An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 2011;25:2347–2360. doi: 10.1101/gad.173443.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Srour M, et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet. 2012;49:636–641. doi: 10.1136/jmedgenet-2012-101132. [DOI] [PubMed] [Google Scholar]
- 112.Damerla RR, et al. Novel Jbts17 mutant mouse model of Joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum Mol Genet. 2015;24:3994–4005. doi: 10.1093/hmg/ddv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lambacher NJ, et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol. 2016;18:122–131. doi: 10.1038/ncb3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA. 1993;90:5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bhogaraju S, Engel BD, Lorentzen E. Intraflagellar transport complex structure and cargo interactions. Cilia. 2013;2:10. doi: 10.1186/2046-2530-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bangs F, Anderson KV. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb Perspect Biol. 2016 doi: 10.1101/cshperspect.a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gilissen C, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arts HH, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 119.Ashe A, et al. Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum Mol Genet. 2012;21:1808–1823. doi: 10.1093/hmg/ddr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schmidts M, et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum Mutat. 2013;34:714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schmidts M, et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J Med Genet. 2013;50:309–323. doi: 10.1136/jmedgenet-2012-101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmidts M, et al. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2013;93:932–944. doi: 10.1016/j.ajhg.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Taylor SP, et al. Mutations in DYNC2LI1 disrupt cilia function and cause short rib polydactyly syndrome. Nat Commun. 2015;6:7092. doi: 10.1038/ncomms8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gholkar AA, et al. Tctex1d2 associates with short-rib polydactyly syndrome proteins and is required for ciliogenesis. Cell Cycle. 2015;14:1116–1125. doi: 10.4161/15384101.2014.985066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moosa S, et al. Novel IFT122 mutations in three Argentinian patients with cranioectodermal dysplasia: Expanding the mutational spectrum. Am J Med Genet A. 2016;170A:1295–1301. doi: 10.1002/ajmg.a.37570. [DOI] [PubMed] [Google Scholar]
- 126.Davis EE, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coussa RG, et al. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet. 2013;84:150–159. doi: 10.1111/cge.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Halbritter J, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93:915–925. doi: 10.1016/j.ajhg.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Friedland-Little JM, et al. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum Mol Genet. 2011;20:3725–3737. doi: 10.1093/hmg/ddr241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Beales PL, et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 131.Zhang W, et al. IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thevenon J, et al. Autosomal recessive IFT57 hypomorphic mutation cause ciliary transport defect in unclassified oral-facial-digital syndrome with short stature and brachymesophalangia. Clin Genet. 2016 doi: 10.1111/cge.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mukhopadhyay S, et al. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24:2180–2193. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Seo S, et al. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011;7:e1002358. doi: 10.1371/journal.pgen.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eguether T, et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell. 2014;31:279–290. doi: 10.1016/j.devcel.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marion V, et al. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet--Biedl syndrome with situs inversus and insertional polydactyly. J Med Genet. 2012;49:317–321. doi: 10.1136/jmedgenet-2012-100737. [DOI] [PubMed] [Google Scholar]
- 137.Aldahmesh MA, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014;23:3307–3315. doi: 10.1093/hmg/ddu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee MS, et al. IFT46 plays an essential role in cilia development. Dev Biol. 2015;400:248–257. doi: 10.1016/j.ydbio.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pazour GJ, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li Y, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520–524. doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Koefoed K, Veland IR, Pedersen LB, Larsen LA, Christensen ST. Cilia and coordination of signaling networks during heart development. Organogenesis. 2014;10:108–125. doi: 10.4161/org.27483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Harrison MJ, Shapiro AJ, Kennedy MP. Congenital Heart Disease and Primary Ciliary Dyskinesia. Paediatr Respir Rev. 2016;18:25–32. doi: 10.1016/j.prrv.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 143.Liew GM, et al. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev Cell. 2014;31:265–278. doi: 10.1016/j.devcel.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Domire JS, et al. Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci. 2011;68:2951–2960. doi: 10.1007/s00018-010-0603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang Q, Seo S, Bugge K, Stone EM, Sheffield VC. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum Mol Genet. 2012;21:1945–1953. doi: 10.1093/hmg/dds004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Seo S, et al. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet. 2009;18:1323–1331. doi: 10.1093/hmg/ddp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Loktev AV, Jackson PK. Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep. 2013;5:1316–1329. doi: 10.1016/j.celrep.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 149.van Dam TJ, et al. Evolution of modular intraflagellar transport from a coatomer-like progenitor. Proc Natl Acad Sci USA. 2013;110:6943–6948. doi: 10.1073/pnas.1221011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Avidor-Reiss T, et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 151.Stone EM, et al. Autosomal recessive retinitis pigmentosa caused by mutations in the MAK gene. Invest Ophthalmol Vis Sci. 2011;52:9665–9673. doi: 10.1167/iovs.11-8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Paige Taylor S, et al. An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thiel C, et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Toriyama M, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet. 2016;48:648–656. doi: 10.1038/ng.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Boskovski MT, et al. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature. 2013;504:456–459. doi: 10.1038/nature12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mitchell DR. The evolution of eukaryotic cilia and flagella as motile and sensory organelles. Adv Exp Med Biol. 2007;607:130–140. doi: 10.1007/978-0-387-74021-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Suzuki T, et al. Efhc1 deficiency causes spontaneous myoclonus and increased seizure susceptibility. Hum Mol Genet. 2009;18:1099–1109. doi: 10.1093/hmg/ddp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Loucks CM, et al. PACRG, a protein linked to ciliary motility, mediates cellular signaling. Mol Biol Cell. 2016;27:2133–2144. doi: 10.1091/mbc.E15-07-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rossetto MG, et al. Defhc1.1, a homologue of the juvenile myoclonic gene EFHC1, modulates architecture and basal activity of the neuromuscular junction in Drosophila. Hum Mol Genet. 2011;20:4248–4257. doi: 10.1093/hmg/ddr352. [DOI] [PubMed] [Google Scholar]
- 160.Léon C, et al. Distribution of EFHC1 or Myoclonin 1 in mouse neural structures. Epilepsy Res. 2010;88:196–207. doi: 10.1016/j.eplepsyres.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 161.Follit JA, Tuft RA, Fogarty KE, Pazour GJ. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Finetti F, et al. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol. 2009;11:1332–1339. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stinchcombe JC, et al. Mother Centriole Distal Appendages Mediate Centrosome Docking at the Immunological Synapse and Reveal Mechanistic Parallels with Ciliogenesis. Curr Biol. 2015;25:3239–3244. doi: 10.1016/j.cub.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Onnis A, et al. The small GTPase Rab29 is a common regulator of immune synapse assembly and ciliogenesis. Cell Death Differ. 2015;22:1687–1699. doi: 10.1038/cdd.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cooley LF, et al. Impaired immunological synapse in sperm associated antigen 6 (SPAG6) deficient mice. Sci Rep. 2016;6:25840. doi: 10.1038/srep25840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Noda K, Kitami M, Kitami K, Kaku M, Komatsu Y. Canonical and noncanonical intraflagellar transport regulates craniofacial skeletal development. Proc Natl Acad Sci U S A. 2016;113:E2589–97. doi: 10.1073/pnas.1519458113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Arnaiz O, Cohen J, Tassin AM, Koll F. Remodeling Cildb, a popular database for cilia and links for ciliopathies. Cilia. 2014;3:9. doi: 10.1186/2046-2530-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ishikawa H, Thompson J, Yates JR, Marshall WF. Proteomic analysis of mammalian primary cilia. Curr Biol. 2012;22:414–419. doi: 10.1016/j.cub.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Phirke P, et al. Transcriptional profiling of C. elegans DAF-19 uncovers a ciliary base-associated protein and a CDK/CCRK/LF2p-related kinase required for intraflagellar transport. Dev Biol. 2011;357:235–247. doi: 10.1016/j.ydbio.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jensen VL, et al. Whole-Organism Developmental Expression Profiling Identifies RAB-28 as a Novel Ciliary GTPase Associated with the BBSome and Intraflagellar Transport. PLoS Genet. 2016;12:e1006469. doi: 10.1371/journal.pgen.1006469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li C, et al. MKS5 and CEP290 Dependent Assembly Pathway of the Ciliary Transition Zone. PLoS Biol. 2016;14:e1002416. doi: 10.1371/journal.pbio.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vogel P, et al. Nephronophthisis and retinal degeneration in tmem218-/mice: a novel mouse model for Senior-Løken syndrome. Vet Pathol. 2015;52:580–595. doi: 10.1177/0300985814547392. [DOI] [PubMed] [Google Scholar]
- 174.Boldt K, et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun. 2016;7:11491. doi: 10.1038/ncomms11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Timbers TA, et al. Accelerating Gene Discovery by Phenotyping Whole-Genome Sequenced Multi-mutation Strains and Using the Sequence Kernel Association Test (SKAT) PLoS Genet. 2016;12:e1006235. doi: 10.1371/journal.pgen.1006235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wheway G, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol. 2015;17:1074–1087. doi: 10.1038/ncb3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 178.Oud MM, et al. A novel ICK mutation causes ciliary disruption and lethal endocrine-cerebro-osteodysplasia syndrome. Cilia. 2016;5:8. doi: 10.1186/s13630-016-0029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Putoux A, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011;43:601–606. doi: 10.1038/ng.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Broekhuis JR, Verhey KJ, Jansen G. Regulation of cilium length and intraflagellar transport by the RCK-kinases ICK and MOK in renal epithelial cells. PLoS One. 2014;9:e108470. doi: 10.1371/journal.pone.0108470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Miyamoto T, et al. Insufficiency of BUBR1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Hum Mol Genet. 2011;20:2058–2070. doi: 10.1093/hmg/ddr090. [DOI] [PubMed] [Google Scholar]
- 182.Foley KE. Model network: Canadian program aims to generate models for rare disease. Nat Med. 2015;21:1242–1243. doi: 10.1038/nm1115-1242. [DOI] [PubMed] [Google Scholar]
- 183.Cornillie FJ, Lauweryns JM, Corbeel L. Atypical bronchial cilia in children with recurrent respiratory tract infections. A comparative ultrastructural study. Pathol Res Pract. 1984;178:595–604. doi: 10.1016/S0344-0338(84)80093-X. [DOI] [PubMed] [Google Scholar]
- 184.Inglis PN, Boroevich KA, Leroux MR. Piecing together a ciliome. Trends Genet. 2006;22:491–500. doi: 10.1016/j.tig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 185.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 186.Valente EM, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 187.Song R, et al. miR-34/449 miRNAs are required for motile ciliogenesis by repressing cp110. Nature. 2014;510:115–120. doi: 10.1038/nature13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mougou-Zerelli S, et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum Mutat. 2009;30:1574–1582. doi: 10.1002/humu.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Maglic D, et al. TMEM231 Gene Conversion Associated with Joubert and Meckel-Gruber Syndromes in the Same Family. Hum Mutat. 2016 doi: 10.1002/humu.23054. [DOI] [PubMed] [Google Scholar]
- 190.Bujakowska KM, et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum Mol Genet. 2015;24:230–242. doi: 10.1093/hmg/ddu441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sayer JA, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 192.Baala L, et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Perrault I, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- 194.Craige B, et al. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Klinger M, et al. The novel centriolar satellite protein SSX2IP targets Cep290 to the ciliary transition zone. Mol Biol Cell. 2014;25:495–507. doi: 10.1091/mbc.E13-09-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pretorius PR, et al. Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet. 2010;6:e1000884. doi: 10.1371/journal.pgen.1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Doherty D, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis) J Med Genet. 2010;47:8–21. doi: 10.1136/jmg.2009.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Delous M, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 199.Dryja TP, et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hameed A, et al. Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet. 2003;40:616–619. doi: 10.1136/jmg.40.8.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.