

TICRR/TRESLIN is an essential protein required for the initiation of DNA replication. Here, Sansam et al. report that TICRR physically interacts with the acetyl-histone binding bromodomain and extraterminal (BET) proteins BRD2 and BRD4. Abrogation of this interaction impairs TICRR binding to acetylated chromatin and disrupts the spatiotemporal order of DNA replication.
Keywords: DNA replication, histone acetylation, chromatin, bromodomain, DNA replication initiation, epigenetic
Abstract
DNA replication origins in hyperacetylated euchromatin fire preferentially during early S phase. However, how acetylation controls DNA replication timing is unknown. TICRR/TRESLIN is an essential protein required for the initiation of DNA replication. Here, we report that TICRR physically interacts with the acetyl-histone binding bromodomain (BRD) and extraterminal (BET) proteins BRD2 and BRD4. Abrogation of this interaction impairs TICRR binding to acetylated chromatin and disrupts normal S-phase progression. Our data reveal a novel function for BET proteins and establish the TICRR–BET interaction as a potential mechanism for epigenetic control of DNA replication.
Tens of thousands of replication forks coordinately operate to ensure that the human genome is replicated accurately and entirely during every round of cell division. Replication forks do not initiate simultaneously at the start of S phase. Instead, they initiate throughout S phase according to a stereotypical spatiotemporal program. Cells that simultaneously activate too many forks rapidly accumulate DNA damage, while cells with extreme delays in replication fork initiation display chromosomal instability (Smith et al. 2001; Di Micco et al. 2006). Deregulation of replication fork initiation causes genome instability in cancer, yet we know little about the mechanisms that determine where and how many replication forks initiate at different times during S phase.
DNA replication forks launch upon the coordinated assembly of initiation factors at origins within the context of chromatin. Origins compete for limiting amounts of replication initiation factors, and chromatin structure affects when, where, and how many forks initiate (Simon et al. 2001; Karnani et al. 2010; Mantiero et al. 2011; Tanaka et al. 2011; Rhind and Gilbert 2013; Lubelsky et al. 2014). In particular, histone acetylation causes earlier and more efficient replication fork initiation (Vogelauer et al. 2002; Aggarwal and Calvi 2004; Aparicio et al. 2004; Kemp et al. 2005; Casas-Delucchi et al. 2011). Although the mechanism by which chromatin acetylation controls DNA replication initiation is unknown, the prevailing hypothesis is that it stimulates DNA replication by opening chromatin structure (Gindin et al. 2014).
In addition to relaxing chromatin structure, acetylated histones directly bind transcriptional regulatory proteins with acetyl-lysine-selective recognition modules called bromodomains (BRDs) (Garcia-Ramirez et al. 1995; Tse et al. 1998; Dhalluin et al. 1999; Fujisawa and Filippakopoulos 2017). BRD proteins stimulate transcription by recruiting multiprotein complexes to acetylated chromatin (Rahman et al. 2011; Fujisawa and Filippakopoulos 2017). We found that the BRD and extraterminal (ET) motif (BET) proteins BRD2 and BRD4 physically interact with the limiting replication initiation protein TICRR (also known as TRESLIN in vertebrates and Sld3 in yeast) (Kumagai et al. 2010; Sansam et al. 2010; Tanaka et al. 2011; Collart et al. 2013). TICRR–BET interaction abrogation reduces TICRR binding to acetylated chromatin and disrupts the normal spatiotemporal order of DNA replication within the nucleus of one cell type and inhibits overall DNA synthesis rates in a second. These results indicate that chromatin acetylation controls the efficiency and timing of DNA replication initiation by recruiting a key DNA replication factor via BRD proteins.
Results and Discussion
TICRR is a critical DNA replication initiation regulator (Kumagai et al. 2010; Sansam et al. 2010). Cyclin-dependent kinases (CDKs) control S-phase progression via TICRR phosphorylation, and TICRR expression levels determine S-phase length (Collart et al. 2013; Sansam et al. 2015; Charrasse et al. 2017). The TICRR N terminus contains essential CDK phosphorylated residues and critical binding sites for the DNA topoisomerase 2-binding protein (TopBP1) and the MDM2-binding protein (MTBP) (Fig. 1A; Sansam et al. 2010; Kumagai et al. 2011; Boos et al. 2013). The biological roles of the TICRR C terminus are largely unknown, although its evolutionary conservation suggests that it is functionally significant (Supplemental Fig. S1A; Sansam et al. 2010). Indeed, the Chk1 kinase interacts with part of the C-terminal conserved sequence to regulate replication fork initiation (Fig. 1A; Guo et al. 2015).
Figure 1.
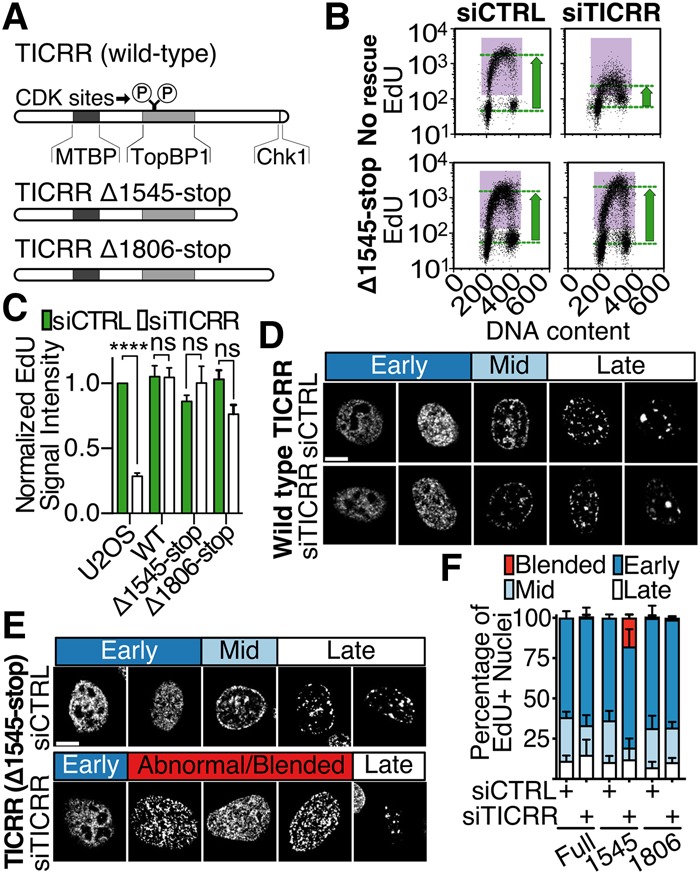
The C terminus of TICRR is necessary for normal spatiotemporal patterns of DNA replication. (A) Diagram of TICRR proteins used to rescue endogenous TICRR knockdown. CDK phosphorylation sites (Ser969 and Thr1001) and regions interacting with MTBP, TOPBP1, or Chk1 are marked. (B) EdU incorporation and DNA content were measured by flow cytometry following siRNA knockdown of TICRR. (C) Quantitation of data in B. EdU signal intensity was normalized to that of U2OS cells. Mean ± SD. (****) P = 0.0001. (D) Patterns of EdU-labeled replication sites in control (siCTRL) or TICRR (siTICRR) siRNA transfected cells expressing siRNA-resistant TICRR transgenes. Patterns characteristic of early, mid, and late S phase are shown. (E) Representative EdU-labeled nuclei from cells expressing the Δ1545-stop transgene. Abnormal patterns observed in siTICRR transfected cells are shown. (F) U2OS cells expressing full-length (full) or Δ1545-stop or Δ1806-stop truncations were transfected with siCTRL or siTICRR, and then patterns were counted.
To determine whether additional conserved TICRR C-terminal sequences regulate DNA replication initiation, we performed genetic rescue experiments in human cell culture. Specifically, we induced expression of siRNA-resistant wild-type or mutant TICRR transgenes in U2OS cells before knockdown of the endogenous TICRR mRNA and analyzed DNA replication phenotypes (Supplemental Fig. S1B–D). As expected, TICRR knockdown cells displayed reduced thymidine analog (EdU) incorporation (Fig. 1B,C; Supplemental Fig. S1E; Sansam et al. 2015). This defect was rescued in TICRR knockdown rescue cells expressing a wild-type (TICRR-kd-WT) or a C-terminally truncated (TICRR-kd-Δ1545-stop) transgene (Fig. 1A–C; Supplemental Fig. S1E), consistent with previous work showing that the human TICRR C terminus is nonessential for DNA synthesis in U2OS cells (Kumagai et al. 2011).
Next, we investigated whether the TICRR C terminus regulates the spatiotemporal replication initiation patterns. The higher eukaryotic DNA replication program is divided into three stages, which can be visualized by pulse-labeling cells with EdU or by immunofluorescent-labeling the replication factor PCNA (O'Keefe et al. 1992). At the start of S phase, euchromatin replicates in small foci within the nuclear interior. During mid-S phase, heterochromatin replicates in spots around the nuclear periphery and the nucleoli. At the end of S phase, heterochromatin located in larger interior nuclear patches replicates (Fig. 1D). We examined the spatial replication patterns in asynchronously growing TICRR knockdown and rescue cells. TICRR-kd-WT rescue cells displayed spatial replication patterns similar to those of control cells (Fig. 1D,F). In contrast, TICRR-kd-Δ1545-stop rescue cells displayed an almost complete absence of nuclei with the normal mid-S-phase replication pattern (P < 0.0001, two-way analysis of variance [ANOVA]). Furthermore, they displayed abnormal patterns consisting of the mid-S-phase pattern with numerous foci in the nuclear interior that resembled those of early S phase (Fig. 1E,F; Supplemental Fig. S2).
To determine whether the abnormal replication patterns in TICRR-kd-Δ1545-stop rescue cells were caused by loss of the Chk1 interaction, we examined TICRR knockdown cells rescued with a TICRR mutant that retains most of the C terminus but not the Chk1-binding domain (Δ1806-stop) (Fig. 1A; Guo et al. 2015). Like TICRR-kd-Δ1545-stop rescue cells, TICRR-kd-Δ1806-stop rescue cells displayed normal levels of EdU incorporation (Fig. 1C; Supplemental Fig. S1E). However, unlike TICRR-kd-Δ1545-stop rescue cells, TICRR-kd-Δ1806-stop rescue cells displayed normal spatiotemporal patterns of DNA replication (Fig. 1F). Thus, the TICRR C-terminal half is required for normal spatiotemporal regulation of DNA replication, and novel protein interactions might be critical for this function.
To understand how TICRR contributes to the spatiotemporal organization of DNA replication, we searched for proteins that physically interact with its C terminus. Through a yeast two-hybrid screen using a human TICRR fragment (amino acids 1059–1910) as bait against a human breast epithelial cDNA library, we identified BRD2 and BRD4 as interactors (Supplemental Fig. S3A). BRD proteins bind histone acetyl-lysine residues and mediate protein interactions with acetylated chromatin (Dhalluin et al. 1999; Fujisawa and Filippakopoulos 2017). BRD2 and BRD4 contain two BRDs and an ET domain (Fig. 2A; Taniguchi 2016). Our yeast two-hybrid specifically identified the BRD2 and four ET domains as interacting with the TICRR C terminus (Fig. 2A; Supplemental Fig. S3A). The ET domain mediates interactions between BET protein family members and other proteins (Rahman et al. 2011).
Figure 2.
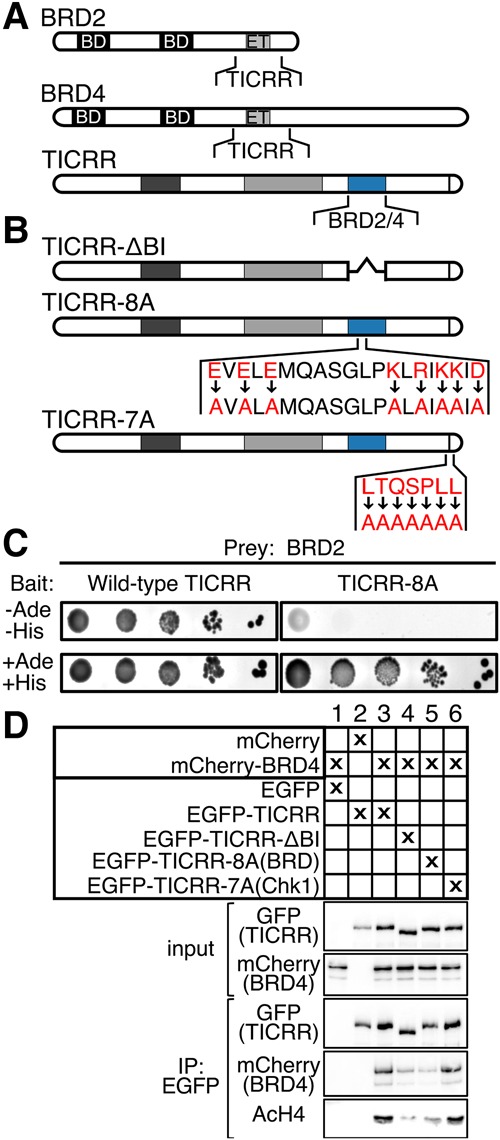
TICRR interacts with BRD2 and BRD4 through a conserved cluster of amino acids. (A) Diagrams showing BRD2, BRD4, and TICRR regions that interacted in yeast two-hybrid assays (see Supplemental Fig. S5). (B) TICRR mutations used in C and D. (C, left to right) Yeast two-hybrid serial dilutions. Growth without adenine (Ade) and histidine (His) indicated a bait–prey interaction. TICRR fragments (Val1484–Cys1646) and BRD2 (Ser561–Pro706) were used as bait and prey, respectively. (D) Immunoblots of anti-GFP immunoprecipitations from cells expressing the indicated mCherry and EGFP fusion proteins. Input lysate (2.5% of total) or anti-EGFP immunoprecipitate (30% of total) blots were probed with EGFP (for TICRR), mCherry (for BRD4), or endogenous acetylated histone H4 (AcH4) antibodies.
To define the TICRR C-terminal sequences that interact with BRD2/4, we performed additional yeast two-hybrid tests. The BRD2 ET domain interacted with TICRR fragments containing amino acids 1485–1660 (Supplemental Fig. S3B,C). By comparing vertebrate sequences, we found that this TICRR–BRD2/4 interaction domain (TICRR-BI) contains eight charged residues in a stretch of 19 conserved amino acids (Supplemental Fig. S3D). To test whether these residues were necessary for the interaction, we mutated them to alanine within the TICRR 1059–1910 bait (TICRR-8A). Indeed, TICRR-8A did not interact with the BRD2 ET domain in the yeast two-hybrid (Fig. 2B,C), suggesting that these eight residues are required for the TICRR–BRD2 interaction (Fig. 2B,C).
Next, we examined whether TICRR and BRD4 physically interact in human cells. We treated U2OS nuclear extracts with benzonase nuclease to eliminate the detection of an indirect interaction through DNA. We found that endogenous TICRR coimmunoprecipitated with BRD4 (Supplemental Fig. S3E). To verify that the TICRR-BI was necessary for the TICRR–BRD4 interaction, we performed coimmunoprecipitation experiments with epitope-tagged proteins. We found that mCherry-BRD4 efficiently coimmunoprecipitated with EGFP-TICRR but not with EGFP-TICRR-ΔBI, which lacks amino acids 1485–1660, or EGFP-TICRR-8A, which is the full-length TICRR protein with eight amino acid substitutions (Fig. 2B,D). In contrast, mCherry-BRD4 efficiently coimmunoprecipitated with EGFP-TICRR-7A, a mutant with substitutions in the Chk1 interaction site (Fig. 2B,D).
Since BRD proteins bind to acetylated histone H4, we tested whether EGFP-TICRR might coimmunoprecipitate with acetylated histones via BRD2/4 (Dhalluin et al. 1999). Indeed, endogenous acetylated histone H4 coimmunoprecipitated with EGFP-TICRR, and the interaction was reduced with EGFP-TICRR-ΔBI or EGFP-TICRR-8A (Fig. 2D). To examine whether the BET interaction affects TICRR chromatin binding, we took two approaches. First, we washed nuclei of U2OS cells expressing EGFP-TICRR or EGFP-TICRR-8A with increasing salt and quantified TICRR on immunoblots. Consistent with its binding to chromatin, endogenous TICRR and EGFP-TICRR resisted extraction by NaCl below 300 mM (Supplemental Fig. S4). In contrast, EGFP-TICRR-8A was extracted at 150 mM NaCl, indicating a weaker interaction (Supplemental Fig. S4). We also tested whether the BET interaction influences TICRR chromatin binding in live cells by evaluating EGFP-TICRR mobility using fluorescence recovery after photobleaching (FRAP) (French et al. 2008; Filippakopoulos et al. 2010). FRAP in EGFP-TICRR cells took more than twice as long as in EGFP-TICRR-8A cells (230 sec vs. 100 sec for 50% recovery), indicating increased mobility of the TICRR-8A mutant (Fig. 3A–C). Notably, photobleached EGFP-TICRR fluorescence recovers slower in early S phase than in late (P < 0.0001), but its late S-phase recovery is still slower than that of EGFP-TICRR-8A (Fig. 3D). These data show that chromatin association of the TICRR-8A mutant was reduced compared with wild-type TICRR, and together suggest that BET proteins recruit TICRR to acetylated chromatin.
Figure 3.
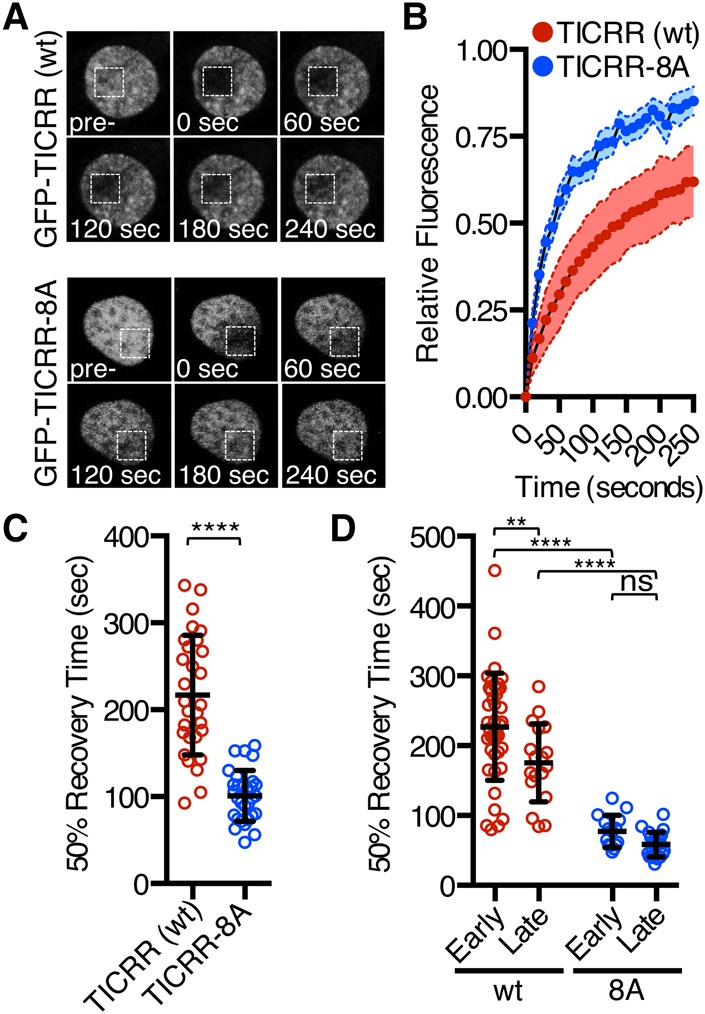
BET proteins recruit TICRR to chromatin. (A) Representative nuclei of cells expressing EGFP-tagged TICRR or TICRR-8A. The square marks the area photobleached. Images were taken at the indicated times after photobleaching. (B) Relative fluorescence in the bleached region for replicate FRAP experiments in which images were taken every 10 sec. (C) Times to 50% fluorescence recovery for experiments shown in B. Fifty percent recovery times were calculated from fitted curves. Mean ± SD. (****) P < 0.0001. (D) Times to 50% recovery for FRAP of synchronized cells in early or late S phase. Mean ± SD. (**) P = 0.0042; (****) P < 0.0001.
To evaluate the function of the TICRR-8A mutant, we generated U2OS cells expressing an siRNA-resistant TICRR-8A transgene. Consistent with the TICRR-Δ1545-stop mutant, the TICRR-8A mutant largely rescued the DNA synthesis defect in TICRR knockdown cells (TICRR-kd-8A) (Fig. 4A,B). To determine whether the TICRR-kd-8A rescue cells had slower S-phase progression over a longer time course, we followed the cell cycle progression of EdU-labeled cells over 5 h. We found that the EdU-labeled TICRR-kd-8A rescue cells entered and exited S phase at the same rate as control cells (Supplemental Fig. S5A–C). Thus, despite affecting the mobility of TICRR in the nucleus, impairing the TICRR–BET protein interaction did not alter the overall rate of DNA synthesis.
Figure 4.
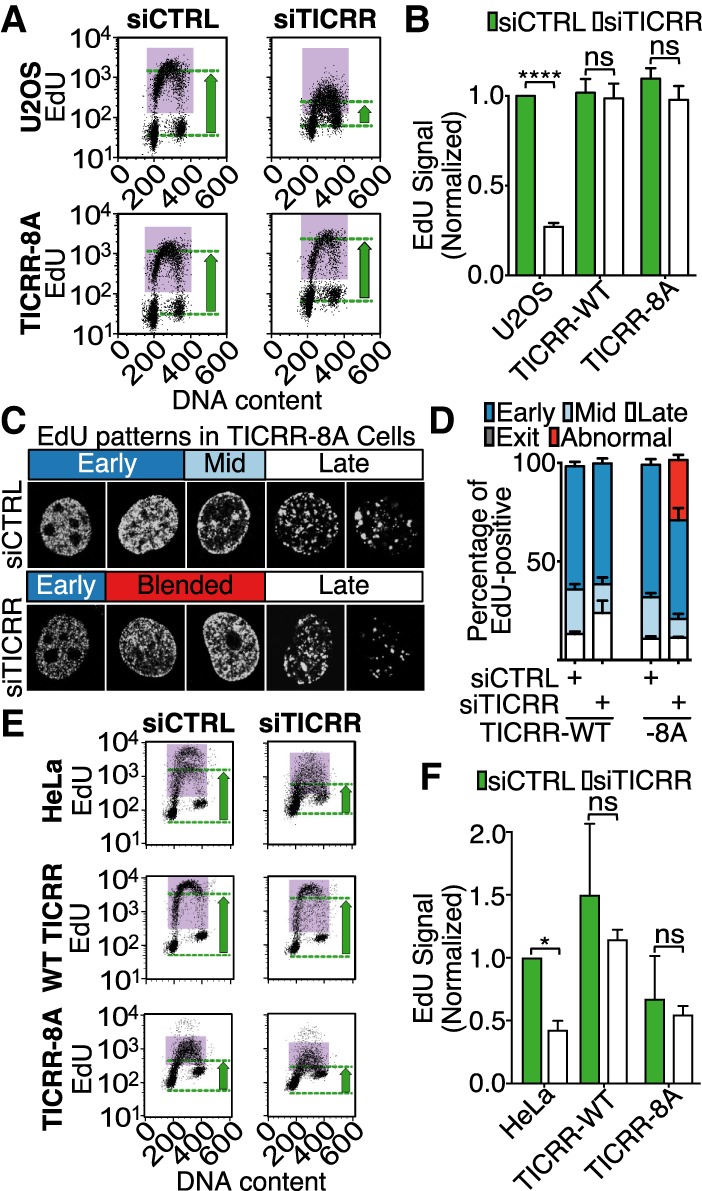
The TICRR–BET interaction is required for normal progression through the replication timing program. (A) EdU incorporation and DNA content in U2OS cells or cells expressing siRNA-resistant TICRR-8A were measured by flow cytometry following TICRR knockdown. The purple boxes mark EdU-positive cells, and the green arrows indicate EdU levels. (B) Quantitation of data in A. EdU signal was normalized to that of U2OS. Mean ± SD. (****) P = 0.0001. (C) Patterns of EdU-labeled replication sites in control (siCTRL) or TICRR (siTICRR) siRNA transfected cells expressing the siRNA-resistant TICRR-8A transgene. Patterns characteristic of early, mid, and late S phase and abnormal patterns are shown. (D) Proportions of cells in C with the indicated replication patterns. (E,F) Representative profiles of EdU incorporation and DNA content in HeLa cells (E) and quantitation for three experiments (F) as in B. Mean ± SD. P < 0.05.
Given that chromatin acetylation has been linked to spatiotemporal control of replication, we hypothesized that the abnormal replication patterns in the TICRR-kd-Δ1545-stop rescue cells were caused by disruption of the TICRR–BET interactions (Casas-Delucchi et al. 2011). We scored TICRR-kd-8A rescue cells for early, mid, or late S-phase replication patterns and indeed found a reduction of nuclei with the mid-S-phase pattern (P < 0.0001, two-way ANOVA) and the appearance of nuclei with abnormal patterns, which appeared to be a blending of early and mid-S phase patterns in which there was a ring of replication foci around the periphery of the nucleus, characteristic of mid-S phase, with a high number of small foci in the nuclear interior, characteristic of early S phase (Fig. 4C,D; Supplemental Fig. S6A). To quantify the pattern blending, we identified nuclei with peripheral EdU staining and measured EdU fluorescence intensity across the widths of those nuclei. Consistent with our qualitative scoring, we found that TICRR-kd-8A rescue nuclei with peripheral EdU also had a higher level of EdU signal in the nuclear interior than control cells (Supplemental Fig. S6B). This is consistent with a higher level of simultaneous early and mid-S-phase DNA replication in the TICRR-kd-8A rescue cells.
The early/mid blended patterns could be a result of a delay in the completion of euchromatin replication and/or precocious replication of peripheral heterochromatin. To uncover when during S phase the abnormal patterns in TICRR-kd-8A rescue cells arise, we visualized progression through the timing program using a double-labeling protocol (Supplemental Fig. 7; Sansam et al. 2015). Replication focus patterns at the start of each experiment were stably marked with EdU. After washing out unincorporated EdU, we incubated the cells for up to 8 h and fixed them so that sites of ongoing replication at the later time point could be visualized by immunofluorescent labeling of PCNA. We then scored both the EdU and PCNA focus patterns as early, mid, late, or abnormal in individual cells.
We focused on cells displaying the early S-phase pattern with the first label (EdU) and determined how replication patterns in those cells changed over time by evaluating the PCNA localization. In control cells (TICRR-kd-WT rescue cells), we observed the expected early-to-mid-to-late progression of replication focus patterns (Supplemental Fig. S7A,B). The EdU and PCNA labeling were identical and reflected early patterns in all cells at time 0. Control cells with mid patterns appeared at 2 h and increased between 4 and 8 h. By 8 h, many of the cells with early EdU patterns displayed either a late pattern of PCNA staining or no PCNA staining, indicating that they had exited S phase. In contrast, the TICRR-kd-8A rescue cells progressed through the spatial patterns abnormally. Instead of progressing to mid-S phase, the TICRR-kd-8A rescue cells progressed from the early pattern to the abnormal blended pattern, which increased in proportion over time (Supplemental Fig. S7A,C). Compared with control cells, a smaller proportion of TICRR-kd-8A rescue cells progressed into mid-S phase by 8 h (X2 [4, n = 277] = 30.09, P < 0.0001). These results indicate that in the TICRR-kd-8A cells, the early S-phase replication pattern is not resolved before replication at the nuclear periphery begins.
The slower resolution of the early S-phase replication pattern in the TICRR-kd-8A cells indicated that the rate of euchromatin replication was impaired, yet this seems to contradict our observation that overall EdU incorporation rates were not reduced. One possibility is that FACS analysis of EdU incorporation is not sensitive enough to detect the level of DNA replication change in TICRR-kd-8A U2OS cells. We wondered whether another cell type might be more dependent on the TICRR–BET interaction and would yield a stronger DNA synthesis phenotype. U2OS cells have a relatively long S phase in which there is a high level of temporal separation between early, mid, and late S phase. Furthermore, we showed that overexpression of wild-type TICRR by itself is insufficient to stimulate DNA synthesis in U2OS cells, indicating that its expression level is not limiting in that cell type. We found that HeLa cells have a shorter S phase, and the expression level of TICRR limits the rate of DNA synthesis. Thus, we generated HeLa cell lines stably expressing wild-type or TICRR-8A transgenes from a single locus. Overexpression of wild-type TICRR in HeLa cells stimulated DNA synthesis by itself and rescued the DNA synthesis defect caused by endogenous TICRR knockdown (Fig. 4E,F). In contrast, overexpression of TICRR-8A failed to rescue and even suppressed EdU incorporation by itself, indicating that it is a dominant-negative mutant (Fig. 4E,F; Supplemental Fig. S8). Together, our analyses show that the TICRR–BET interaction is important for DNA replication, and, in U2OS cells, the effect preferentially manifests as a delay in the completion of early replication.
We next asked whether inhibiting BET proteins would phenocopy the TICRR-8A mutant. The BET protein family is comprised of four highly similar paralogs, two of which were identified in our yeast two-hybrid screen as TICRR interactors. Therefore, we used MZ1, a small molecule that induces degradation of all BET proteins (Zengerle et al. 2015). MZ1 treatment of U2OS cells for 3 h caused nearly complete loss of BRD4 (Fig. 5A). To minimize effects before S-phase onset, we added MZ1 to U2OS cells released from a thymidine block and monitored S-phase progression at 3, 6, 9, and 12 h (Fig. 5B–D). The MZ1-treated cells synthesized DNA and exited S phase slower than cells treated with the cis-MZ1 control compound (Fig. 5B,D). MZ1-treated cells also progressed slower through the timing patterns (Fig. 5C). Like the TICRR-kd-8A rescue U2OS cells, the MZ1-treated cells delayed progression into mid-S phase, and a higher fraction of MZ1-treated cells had early/mid blended spatiotemporal patterns (Fig. 5C).
Figure 5.
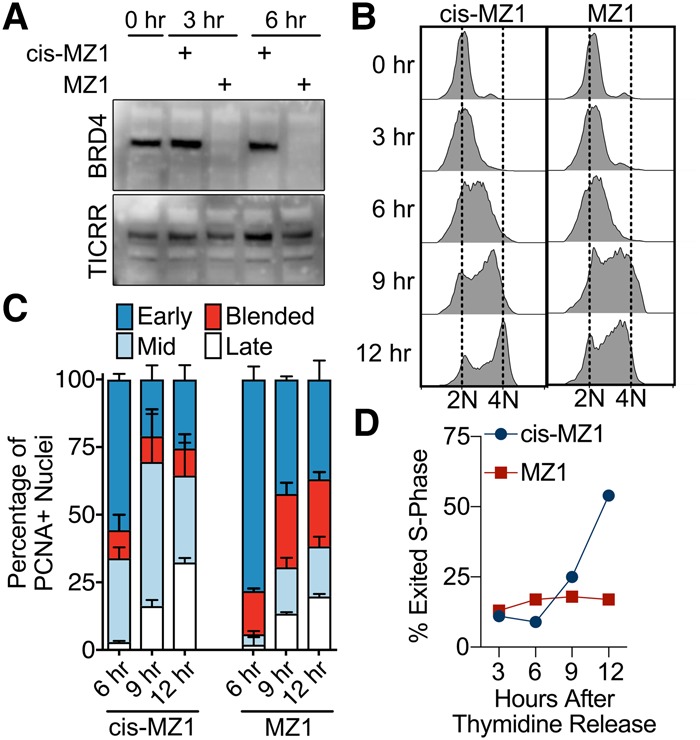
The BET inhibitor affects S-phase progression. (A) BRD4 and TICRR immunoblots after MZ1 or cis-MZ1 treatment of U2OS cells for the indicated times. (B–D) Synchronized U2OS cells were released into S phase with 1 µM MZ1 or cis-MZ1 and harvested at the indicated times for FACS DNA content analysis (B), spatiotemporal replication pattern scoring (C), or S-phase exit (D). DNA replication foci were detected using PCNA immunofluorescence in C and D.
Prior to this study, Rif1 was the only known site-specific regulator of DNA replication initiation and timing in mammals (Cornacchia et al. 2012; Yamazaki et al. 2012). The TICRR–BET interactions that we describe here provide the first evidence that chromatin acetylation directly regulates DNA replication by physically recruiting replication factors. Where TICRR binds in the genome is unknown, but our model predicts that it preferentially binds to acetylated chromatin through the BET–protein interaction. Breaking the TICRR–BET interaction in U2OS cells appeared to preferentially affect the synthesis of euchromatin, while it had a stronger effect on overall DNA synthesis in HeLa cells. Additional work is needed to understand why some cell types would be more dependent on the TICRR–BET interaction than others, but our data suggest that TICRR can be recruited to origins through multiple mechanisms. Epigenetic control of DNA replication timing may influence transcriptional states and minimize collisions between transcription and DNA replication machinery (Lande-Diner et al. 2009; Petryk et al. 2016). Therefore, the discovery of the TICRR–BET interaction opens experimental avenues for examining how epigenetic control of replication influences transcription and genome stability.
Materials and methods
Cell synchronization
U2OS cells were synchronized by sequential treatment with 100 ng/mL nocadazole for 12 h and 2 mM thymidine for 18 h before releasing into fresh medium with 1 µM MZ1 or cis-MZ1 (R&D Systems).
Immunofluorescence
EdU labeling and the EdU/PCNA double-labeling experiments were done as described in Sansam et al. (2015). Image names were coded, and the images were mixed before scoring based on replication patterns defined in O'Keefe et al. (1992). For quantitative analysis, four independent measurements of the fluorescence intensity at each pixel across the nucleus of the cell were calculated (ImageJ).
Statistical analysis
Mean EdU intensity in Figures 1C and 4, B and F, was calculated from multiple independent experiments, and differences between means were assessed with two-way ANOVA. Error bars represent standard deviations of the mean. P-values were corrected using the Bonferroni method. For Figure 1C, U2OS n = 8, TICRR wild-type n = 8, TICRR-Δ1545-stop n = 6, and TICRR-Δ1806-stop n = 5. For Figure 4B, U2OS n = 7, TICRR wild-type n = 9, and TICRR-8A n = 5.
In Figures 1F, 4D, and 5C, means of percentages from three independent experiments are plotted as stacked bars. The error bars represent standard deviations of the means. For each experiment, at least 100 EdU-positive cells were scored for every cell line and condition (siCTRL, siTICRR, and MZ1). Differences between means were evaluated using two-way ANOVA. The P-values were corrected using the Bonferroni post-test.
Supplementary Material
Acknowledgments
We thank L. Thompson, G. Gorbsky, and D. Dawson for discussion. This work was supported by National Institutes of Health National Institute of General Medical Sciences grants 5P20GM103636-02 (including Oklahoma Medical Research Foundation Imaging Core support) and 1R01GM121703, and a Health Research Program Award from the Oklahoma Center for the Advancement of Science and Technology.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.306464.117.
References
- Aggarwal BD, Calvi BR. 2004. Chromatin regulates origin activity in Drosophila follicle cells. Nature 430: 372–376. [DOI] [PubMed] [Google Scholar]
- Aparicio JG, Viggiani CJ, Gibson DG, Aparicio OM. 2004. The Rpd3–Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol 24: 4769–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos D, Yekezare M, Diffley JF. 2013. Identification of a heteromeric complex that promotes DNA replication origin firing in human cells. Science 340: 981–984. [DOI] [PubMed] [Google Scholar]
- Casas-Delucchi CS, van Bemmel JG, Haase S, Herce HD, Nowak D, Meilinger D, Stear JH, Leonhardt H, Cardoso MC. 2011. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res 40: 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrasse S, Gharbi-Ayachi A, Burgess A, Vera J, Hached K, Raynaud P, Schwob E, Lorca T, Castro A. 2017. Ensa controls S-phase length by modulating Treslin levels. Nat Commun 8: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart C, Allen GE, Bradshaw CR, Smith JC, Zegerman P. 2013. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 341: 893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB. 2012. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 31: 3678–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. 1999. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399: 491–496. [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638–642. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. 2010. Selective inhibition of BET bromodomains. Nature 468: 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, et al. 2008. BRD–NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 27: 2237–2242. [DOI] [PubMed] [Google Scholar]
- Fujisawa T, Filippakopoulos P. 2017. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18: 246–262. [DOI] [PubMed] [Google Scholar]
- Garcia-Ramirez M, Rocchini C, Ausio J. 1995. Modulation of chromatin folding by histone acetylation. J Biol Chem 270: 17923–17928. [DOI] [PubMed] [Google Scholar]
- Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS, Bilke S. 2014. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol Syst Biol 10: 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Kumagai A, Schlacher K, Shevchenko A, Dunphy WG. 2015. Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol Cell 57: 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani N, Taylor CM, Malhotra A, Dutta A. 2010. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell 21: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Ghosh M, Liu G, Leffak M. 2005. The histone deacetylase inhibitor trichostatin A alters the pattern of DNA replication origin activity in human cells. Nucleic Acids Res 33: 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Shevchenko A, Dunphy WG. 2010. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 140: 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. 2011. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J Cell Biol 193: 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande-Diner L, Zhang J, Cedar H. 2009. Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol Cell 34: 767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelsky Y, Prinz JA, DeNapoli L, Li Y, Belsky JA, MacAlpine DM. 2014. DNA replication and transcription programs respond to the same chromatin cues. Genome Res 24: 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 30: 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Henderson SC, Spector DL. 1992. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific α-satellite DNA sequences. J Cell Biol 116: 1095–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d'Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen CL, Hyrien O. 2016. Replication landscape of the human genome. Nat Commun 7: 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM. 2011. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol 31: 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Biol 5: a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CL, Cruz NM, Danielian PS, Amsterdam A, Lau ML, Hopkins N, Lees JA. 2010. A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev 24: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CG, Goins D, Siefert JC, Clowdus EA, Sansam CL. 2015. Cyclin-dependent kinase regulates the length of S phase through TICRR/TRESLIN phosphorylation. Genes Dev 29: 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon I, Tenzen T, Mostoslavsky R, Fibach E, Lande L, Milot E, Gribnau J, Grosveld F, Fraser P, Cedar H. 2001. Developmental regulation of DNA replication timing at the human β globin locus. EMBO J 20: 6150–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L, Plug A, Thayer M. 2001. Delayed replication timing leads to delayed mitotic chromosome condensation and chromosomal instability of chromosome translocations. Proc Natl Acad Sci 98: 13300–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Nakato R, Katou Y, Shirahige K, Araki H. 2011. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr Biol 21: 2055–2063. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y. 2016. The bromodomain and extra-terminal domain (BET) family: functional anatomy of BET paralogous proteins. Int J Mol Sci 17: E1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Sera T, Wolffe AP, Hansen JC. 1998. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol 18: 4629–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. 2002. Histone acetylation regulates the time of replication origin firing. Mol Cell 10: 1223–1233. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. 2012. Rif1 regulates the replication timing domains on the human genome. EMBO J 31: 3667–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M, Chan KH, Ciulli A. 2015. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol 10: 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.