

Zhang et al. identified two distinct neuroendocrine signaling circuits by which the worm nervous system senses cool and warm environmental temperatures through cool- and warm-sensitive neurons and then signals the gut to extend and shorten life span, respectively.
Keywords: aging, C. elegans, longevity
Abstract
Tissue–tissue communications are integral to organismal aging, orchestrating a body-wide aging process. The brain plays a key role in this process by detecting and processing signals from the environment and then communicating them to distal tissues such as the gut to regulate longevity. How this is achieved, however, is poorly understood. Here, using Caenorhabditis elegans as a model, we identified two distinct neuroendocrine signaling circuits by which the worm nervous system senses cool and warm environmental temperatures through cool- and warm-sensitive neurons and then signals the gut to extend and shorten life span, respectively. The prolongevity “cool” circuit uses the small neurotransmitters glutamate and serotonin, whereas the anti-longevity “warm” circuit is mediated by insulin-like neuropeptides. Both types of neuroendocrine signals converge on the gut through their cognate receptors to differentially regulate the transcription factor DAF-16/FOXO, leading to opposing outcomes in longevity. Our study illustrates how the brain detects and processes environmental signals to bidirectionally regulate longevity by signaling the gut.
Aging is a body-wide process. As animals and humans get older, all of the tissues/organs age and lose functions, albeit at different rates, resulting in an increased probability of death (Sawhney et al. 2005; Sehl et al. 2005). Tissue–tissue communications play a key role in choreographing such a body-wide process (Apfeld and Kenyon 1998; Libina et al. 2003; Alcedo and Kenyon 2004; Murphy et al. 2007; Medkour et al. 2016). Work in the past two decades has greatly advanced our understanding of how various genes modulate longevity by regulating the physiology of individual organelles, cells, and tissues (Kenyon 2010; Fontana and Partridge 2015). In contrast, relatively little is known about how the cross-talk between different cell types/tissues affects longevity and how genes regulate this process.
The brain is uniquely positioned to coordinate tissue–tissue communications in longevity signaling for a number of reasons. First, the brain is specialized in mediating intercellular communications by forming direct synaptic connections between neurons and with other tissues (e.g., neuromuscular junctions) as well as by secreting chemicals to signal distal tissues through long-range neuroendocrine signaling. Second, as the brain mediates the interaction between the organism and its surrounding environment by sensing and processing environmental cues, it sits at a key position to integrate environmental signals and internal physiological states to modulate longevity (Allen et al. 2015). Indeed, recent work in model organisms such as Caenorhabditis elegans has pointed to an increasingly important role for the brain in regulating longevity by signaling distal tissues, particularly the gut (Alcedo and Kenyon 2004; Durieux et al. 2011; Taylor and Dillin 2013; Burkewitz et al. 2015; Leiser et al. 2015; Medkour et al. 2016). However, how such brain–gut communications shape longevity is not well understood.
Temperature and diet are the two primary environmental factors that affect aging (Kenyon 2010; Xiao et al. 2015). Both environmental and core body temperatures modulate the life span of poikilotherms such as worms, flies, and fish as well as homeotherms such as rodents (Holloszy and Smith 1986; Conti et al. 2006; Xiao et al. 2015). Lower temperatures promote life span, while higher temperatures reduce it. Interestingly, temperature-dependent life span regulation is not simply a passive thermodynamic phenomenon but rather a process that is actively regulated by genes (Lee and Kenyon 2009; Xiao et al. 2013; Zhang et al. 2015). However, it remains elusive as to how the nervous system detects and processes temperature information from the environment and then signals distal tissues in the body to regulate longevity.
Here, we attempted to address these questions in C. elegans, a widely used genetic model for the study of the biology of aging. We identified two distinct neuroendocrine signaling circuits through which the worm nervous system detects and processes both cool and warm temperature signals to bidirectionally regulate longevity by cell-nonautonomously signaling the distal tissue gut. Furthermore, we elucidated the underlying molecular mechanisms by identifying the signaling molecules (i.e., neurotransmitters and neuropeptides) and their cognate receptors used by the two circuits for information processing. Our results define the molecular, cellular, and circuit mechanisms by which brain–gut communications modulate longevity in a genetic model organism.
Results
As a first step to investigate how the C. elegans nervous system detects and processes environmental signals to regulate longevity through tissue–tissue communications, we searched for an environmental factor known to affect aging. Temperature and diet are the two primary environmental factors that modulate longevity (Kenyon 2010; Xiao et al. 2015). We decided to focus on temperature for a number of considerations. First, compared with a diet that contains multiple longevity-affecting factors such as nutrients, tastants, and odorants (Alcedo and Kenyon 2004; Allen et al. 2015), temperature is much simpler in nature. Second, it is relatively easier to deliver and control temperature. Third, it is well known that the nervous system can sense temperature cues through specific thermosensory neurons.
IL1 neurons are cool-sensitive and can promote life span at lower temperatures
We first explored how the nervous system detects and processes low-temperature signals from the environment to promote longevity. We hypothesized that some cool-sensitive neurons may sense a temperature drop in the environment to extend life span by signaling distal tissues. However, the identities of cool-sensitive neurons in C. elegans remain largely unknown. To identify such cool-sensitive neurons, we turned our attention to TRPA-1, a cooling-activated channel known to promote life span at lower temperatures (Xiao et al. 2013). Although TRPA-1 can act in neurons as well as other tissues such as the intestine to regulate life span (Xiao et al. 2013), we took advantage of its role in the nervous system because it would offer us an opportunity to identify cool-sensitive neurons. We reasoned that the neurons in which TRPA-1 acts to extend life span would be candidate cool-sensitive neurons.
Three temperatures are commonly used to culture worms in the laboratory: 25°C, 20°C, and 15°C. We first verified the temperature-dependent life span phenotype of trpa-1 mutant worms. As reported previously (Xiao et al. 2013), trpa-1 mutant worms were short-lived at lower temperatures such as 20°C and 15°C but lived a normal life span at higher temperatures such as 25°C (Fig. 1A,B; Supplemental Fig. S1A; Supplemental Table S1). Conversely, overexpression of TRPA-1 as a transgene in neurons using a pan-neuron promoter extended life span at 20°C and 15°C, but the same transgene had no notable effect on life span at 25°C (Fig. 1A,B; Supplemental Fig. S1A; Supplemental Table S1). This is consistent with the fact that TRPA-1 is a cool-sensitive channel that is activated when the temperature decreases to ∼20°C or lower but remains inactive at higher temperatures such as 25°C (Chatzigeorgiou et al. 2010). Since TRPA-1 can promote life span at both 20°C and 15°C, for simplicity, we focused on 20°C for further characterizations.
Figure 1.

The cool-sensitive IL1 neurons signal the intestine to extend life span at lower temperatures. (A,B) trpa-1 mutant worms are short-lived at lower but not higher temperatures, and transgenic expression of TRPA-1 in neurons extends life span at lower but not higher temperatures. The pan-neuronal promoter Prgef-1 was used to drive expression of TRPA-1 as a transgene in neurons. (C) Transgenic expression of TRPA-1 in sensory neurons extends life span at lower temperatures. The pan-sensory neuron promoter Posm-6 was used to drive expression of TRPA-1 as a transgene in sensory neurons. (D) Transgenic expression of TRPA-1 in motor neurons and a group of interneurons does not extend life span. The acr-2 and unc-25 promoter drives expression in cholinergic and GABAergic motor neurons, respectively, while the glr-1 promoter directs expression in interneurons. (E,F) Transgenic expression of TRPA-1 in IL1 neurons extends life span at lower (E) but not higher (F) temperatures. A segment of aqp-6 promoter was used to drive expression specifically in IL1 neurons. (G) Transgenic expression of TRPA-1 in IL1 neurons is sufficient to rescue the short-lived phenotype of trpa-1 mutant worms at lower temperatures. (H–J) IL1 neurons are cool-sensitive and depend on TRPA-1 for cool sensation. The genetically encoded calcium sensor GCaMP6(f) was expressed as a transgene in IL1 neurons. DsRed was coexpressed as an internal reference marker to enable ratiometric imaging. While IL1 neurons did not respond to warming (H), cooling evoked robust calcium transients in these neurons (I). Shades along the traces in H and I represent error bars (SEM). Bar graphs in J summarize the data in I. n ≥ 7. (**) P < 0.005, ANOVA with Bonferroni test. (K) Blocking the output of IL1 neurons shortens life span at lower temperatures. Tetanus toxin (TeTx) was expressed as a transgene in IL1 neurons to block the exocytosis from IL1 neurons. (L) Enhancing the output of IL1 neurons extends life span. The gain-of-function form of Drosophila syntaxin, known to promote exocytosis, was expressed in IL1 neurons as a transgene to enhance the output from IL1 neurons. (M) Extension of life span by IL1 neurons requires DAF-16. Loss of DAF-16 blocked the life span-extending effect of IL1 neurons. (N,O) DAF-16 acts in the intestine instead of neurons to mediate the life span-extending effect of IL1 neurons. daf-16 cDNA expressed as a transgene in the intestine using the ges-1 promoter rescued the phenotype of daf-16 mutant worms (N), while its expression in neurons using the rgef-1 promoter did not (O).
To identify the putative cool-sensitive neurons in which TRPA-1 acts to promote life span at lower temperatures, we tested a number of neuron-type-specific promoters. Transgenic expression of TRPA-1 in sensory neurons using the pan-sensory neuron promoter osm-6 extended life span (Fig. 1C), consistent with the notion that TRPA-1 acts in sensory neurons. As a control, transgenic expression of TRPA-1 in motor neurons and a group of interneurons did not have an effect (Fig. 1D). Expression of TRPA-1 as a transgene in PVD, a putative cool-sensitive neuron (Chatzigeorgiou et al. 2010), also had no effect (Supplemental Fig. S1J,K). As the reported expression pattern of TRPA-1 could be incomplete, to pinpoint in which sensory neurons TRPA-1 functions, we took an unbiased approach by examining each of the 23 types of sensory neurons (56 neurons in total) marked by the osm-6 promoter for their ability to extend life span upon expression of TRPA-1. To this end, we tested 10 different sensory neuron-specific promoters (Supplemental Fig. S1K) and found that expression of TRPA-1 in a single type of sensory neuron, IL1, was sufficient to extend life span in wild-type worms at low but not high temperatures (Fig. 1E,F). None of the other tested sensory neurons had such an effect (Supplemental Fig. S1B–K). Notably, TRPA-1 is found to be expressed in IL1 neurons (Kindt et al. 2007). We also found that transgenic expression of TRPA-1 in IL1 neurons alone in a trpa-1 mutant background was sufficient to rescue the short-lived phenotype of trpa-1 mutant worms at lower temperatures (Fig. 1G). These data demonstrate that TRPA-1 can act in IL1 neurons to promote life span at lower temperatures, suggesting that IL1 neurons are cool-sensitive.
To provide further evidence that IL1 neurons are cool-sensitive, we recorded the activity of these neurons by calcium imaging. Remarkably, cooling evoked robust calcium transients in IL1 neurons, while heating did not have a notable effect, suggesting that these neurons are indeed cool-sensitive (Fig. 1H–J). The cooling-triggered calcium transients in IL1 neurons were nearly absent in trpa-1 mutant worms, a defect that can be fully rescued by transgenic expression of wild-type trpa-1 cDNA specifically in IL1 neurons (Fig. 1I,J). These data identify IL1 neurons as cool-sensitive neurons and also show that TRPA-1 is the primary sensory channel mediating cool sensation in IL1 neurons.
IL1 neurons signal the intestine to promote life span at lower temperatures
How do the cool-sensitive IL1 neurons promote life span at lower temperatures? Based on our model, IL1 neurons may transmit temperature information by signaling other tissues in a cell-nonautonomous mode. If so, blocking such signaling should render worms short-lived at lower temperatures but would have no effect on life span at higher temperatures. To test this, we ablated the output of IL1 neurons by expressing tetanus toxin (TeTx) as a transgene specifically in IL1 neurons. TeTx cleaves the SNARE protein synaptobrevin, thereby blocking the exocytosis (neurotransmission) from the neuron of interest (Link et al. 1992). We found that such functional ablation of IL1 neurons shortened life span only at 20°C but had no effect at 25°C (Fig. 1K; Supplemental Fig. S1M), supporting our model. We also performed the converse experiment by enhancing the output of IL1 neurons. To do so, we expressed in IL1 neurons a transgene expressing a gain-of-function form of the SNARE protein syntaxin, syntaxin(T254I), which enhances exocytosis (neurotransmission) without causing vesicle depletion (Lagow et al. 2007). This transgene extended life span (Fig. 1L), indicating that enhancing the output from IL1 neurons promoted longevity. These results demonstrate that the cool-sensitive IL1 neurons promote longevity at lower temperatures through neurotransmission, presumably by signaling other neurons and/or distal tissues.
We next went on to identify the downstream target tissues to which IL1 neurons transmit longevity signals through neurotransmission. Considering that longevity pathways tend to ultimately converge on a handful of transcription factors (Kenyon 2010), we first sought to identify a transcription factor, if present, that functions downstream from IL1 neurons to promote life span. We reasoned that identifying the site of action of such a transcription factor would then provide us an opportunity to uncover the target tissues to which IL1 neurons transmit longevity signals. Loss of DAF-16/FOXO, a key life span regulator, abolished the ability of IL1 neurons to promote life span (Fig. 1M), demonstrating that DAF-16 is required for IL1 neurons to promote life span at lower temperatures. This is consistent with the view that TRPA-1 acts upstream of DAF-16 to promote longevity (Xiao et al. 2013). In contrast, deficiency in other longevity-regulating transcription factors, such as HSF-1, SKN-1, PHA-4, and DAF-12, had no notable effect (Supplemental Fig. S2A–F). In addition, IL1 neuron activation did not seem to promote the overall level of unfolded protein responses or heat-shock responses, as suggested by reporter assays (Supplemental Fig. S2G). DAF-16 is known to regulate life span primarily in the intestine and neurons (Libina et al. 2003). To ascertain in which tissue DAF-16 functions, we performed rescuing experiments. The daf-16 mutant phenotype can be fully rescued by transgenic expression of daf-16 cDNA specifically in the intestine using an intestine-specific promoter (Fig. 1N), while neuronal expression of daf-16 cDNA did not have any effect (Fig. 1O). Thus, DAF-16 appears to function in the intestine to mediate the life span-extending effect of IL1 neurons, suggesting that the intestine is a target tissue to which IL1 neurons send longevity signals. This also suggests that IL1 neurons extend life span at lower temperatures by stimulating DAF-16 in the intestine. It is worth noting that, similar to the nervous system, the intestine is also a signaling center that integrates and disseminates longevity signals (Medkour et al. 2016).
IL1 neurons promote life span at lower temperatures through glutamate-mediated neurotransmission
We next wondered how IL1 neurons signal the intestine through neurotransmission. Presumably, at lower temperatures, the cool-sensitive IL1 neurons may release some signaling molecules that target their cognate receptors in the intestine. Two major types of signaling molecules are usually released by neurons: small neurotransmitters and neuropeptides. IL1 neurons store both small neurotransmitters and neuropeptides (White et al. 1986; Lee et al. 1999). To determine which types of signaling molecules might be released by IL1 neurons to regulate life span, we tested unc-13 and unc-31 mutant worms, which are defective in secreting small neurotransmitters and neuropeptides, respectively (Richmond et al. 1999; Speese et al. 2007). Loss of unc-13 blocked the ability of IL1 neurons to promote life span at lower temperatures (Fig. 2A), whereas no defect was observed in unc-31 mutant worms (Fig. 2B). RNAi of unc-13 and unc-31 specifically in IL1 neurons using a transgene yielded a similar result (Fig. 2C,D). Thus, small neurotransmitter rather than neuropeptide signaling is required for IL1 neurons to promote life span at lower temperatures, suggesting that small neurotransmitters may serve as signaling molecules to convey temperature information from IL1 neurons to the intestine.
Figure 2.

Glutamate and the metabotropic glutamate receptor MGL-1 mediate the life span-extending effect of IL1 neurons. (A,B) Mutant analyses show that small neurotransmitters but not neuropeptides are required for IL1 neurons to promote life span at lower temperatures. UNC-13 was required for IL1 neurons to extend life span (A), while UNC-31 was not (B). (C,D) Neuron-specific RNAi analyses show that small neurotransmitters but not neuropeptides are required in IL1 neurons to promote life span at lower temperatures. Knockdown of UNC-13 specifically in IL1 neurons by RNAi, which is expressed as a transgene in these neurons, prevented IL1 neurons from extending life span (C), while RNAi knockdown of UNC-31 did not (D). (E,F) Identification of glutamate as a neurotransmitter required for IL1 neurons to promote longevity at lower temperatures. (E) Mutations in eat-4 blocked the ability of IL1 neurons to promote life span. (F) RNAi of eat-4 in IL1 neurons using a transgene blocked the ability of IL1 neurons to promote life span. (G) A table listing all of the 19 glutamate receptor genes encoded by the C. elegans genome. (H) The metabotropic glutamate receptor MGL-1 is required for IL1 neurons to promote life span at lower temperatures.
IL1 neurons are known to be glutamatergic (Lee et al. 1999), suggesting that glutamate may be the neurotransmitter that conveys temperature information from IL1 neurons to the intestine. We thus examined eat-4 mutant worms that are deficient in glutamate release (Lee et al. 1999). eat-4 encodes the sole C. elegans ortholog of vesicular glutamate transporter (Lee et al. 1999). Mutations in eat-4 abolished the ability of IL1 neurons to promote life span at lower temperatures (Fig. 2E). A similar result was obtained with RNAi knockdown of the eat-4 gene specifically in IL1 neurons using a transgene (Fig. 2F). These experiments identify a key role for glutamate, suggesting that glutamate is the signaling molecule that conveys temperature information from IL1 neurons to the intestine.
The glutamate receptor MGL-1 is required for transmitting longevity signals from IL1 neurons to the intestine
To identify the glutamate receptor that might function in the intestine to mediate the life span-extending effect of IL1 neurons, we examined all of the glutamate receptor genes. The C. elegans genome encodes a total of 19 glutamate receptors, falling into four classes: NMDA-type and AMPA/Kainate-type ionotropic glutamate receptors, glutamate-gated chloride channels, and metabotropic glutamate receptors (Fig. 2G). By screening mutant strains lacking each of these 19 glutamate receptors, we found that the metabotropic glutamate receptor gene mgl-1 was required for IL1 neurons to promote life span at lower temperatures (Fig. 2H). None of the other 18 glutamate receptors had such an effect (Supplemental Fig. S3). This suggests MGL-1 as the glutamate receptor that may act in the intestine to transmit longevity signals from IL1 neurons.
Identification of serotonin as another key neurotransmitter required for transmitting longevity signals from IL1 neurons to the intestine
Surprisingly, MGL-1 is not expressed in the intestine, although it is found in many neurons (Greer et al. 2008; Dillon et al. 2015). Apparently, longevity signals are probably mediated by some other neurons that express MGL-1. This elusive neuron may then signal the intestine to promote life span at lower temperatures. To identify such a neuron, we took a rather unconventional approach by first identifying the small neurotransmitter used by this neuron, as neuropeptide signaling is not required for transmitting longevity signals from IL1 neurons (Fig. 2B). To do so, we examined mutant worms lacking each of the major neurotransmitters, including GABA (unc-25 mutant), dopamine (cat-2 mutant), octopamine (tbh-1 mutant), tyramine (tdc-1 mutant), ACh (unc-17 mutant), and serotonin (tph-1 mutant) (Fig. 3A–F). Loss of serotonin prevented IL1 neurons from promoting life span at lower temperatures (Fig. 3F), while deficiency in other neurotransmitters did not (Fig. 3A–E). This identifies serotonin as another essential neurotransmitter that conveys longevity signals from IL1 neurons to the intestine.
Figure 3.

MGL-1 acts in the serotoninergic neuron NSM to promote life span. (A–E) GABA, dopamine, octopamine, tyramine, and ACh are not required for IL1 neurons to promote life span at lower temperatures. Mutations in unc-25 (A), cat-2 (B), tbh-1 (C), tdc-1 (D), and unc-17 (E) blocked GABA, dopamine, octopamine, tyramine, and ACh neurotransmission, respectively. (F) Serotonin is required for IL1 neurons to promote life span at lower temperatures. tph-1 encodes the C. elegans tryptophan hydroxylase required for serotonin synthesis. (G) MGL-1 acts in serotonin neurons to mediate the life span-extending effect of IL1 neurons. The mgl-1 mutant phenotype can be rescued by transgenic expression of mgl-1 cDNA in serotonin neurons using the tph-1 promoter. (H) MGL-1 acts in the serotonin neuron NSM but not ADF to mediate the life span-extending effect of IL1 neurons. The mgl-1 mutant phenotype can be rescued by transgenic expression of mgl-1 cDNA in NSM but not ADF neurons using neuron-specific promoters. For comparison, the life span curves for xuEx1670 and mgl-1; xuEx1670 from G are duplicated in H. The life span data in G and H were acquired at the same time. (I) Functional ablation of NSM neurons blocked the ability of IL1 neurons to extend life span at lower temperatures. TeTx was expressed as a transgene in NSM neurons to functionally silence their output. (J) Transgenic expression of MGL-1 in NSM neurons is sufficient to promote life span in wild-type worms at lower temperatures, and this effect requires DAF-16. (K) Enhancing the output of NSM neurons extends life span, and this life span-extending effect requires DAF-16. The gain-of-function form of Drosophila syntaxin, known to promote exocytosis, was expressed in NSM neurons as a transgene to enhance the output from NSM neurons.
MGL-1 acts in the serotoninergic neuron NSM to transmit longevity signals from IL1 neurons to the intestine
The identification of serotonin as a key neurotransmitter raises the possibility that the metabotropic glutamate receptor MGL-1 may function in serotoninergic neurons. If so, serotoninergic neurons would serve as the “interneurons” that relay temperature information from IL1 neurons to the intestine. Indeed, MGL-1 is known to be expressed in the serotoninergic neuron NSM (Greer et al. 2008; Dillon et al. 2015). Importantly, transgenic expression of mgl-1 cDNA in serotonin neurons using the tph-1 promoter rescued the mgl-1 mutant phenotype (Fig. 3G). In addition, the mgl-1 mutant phenotype can be fully rescued by transgenic expression of mgl-1 cDNA specifically in NSM (Fig. 3H), while such expression in the other major serotoninergic neuron, ADF, cannot (Fig. 3H). Thus, MGL-1 acts in the serotoninergic neuron NSM to transmit longevity signals from IL1 neurons. Furthermore, functional ablation of NSM neurons by transgenic expression of TeTx in these neurons not only shortened life span but also fully suppressed the life span-extending effect of IL1 neurons, suggesting that NSM neurons act downstream from IL1 neurons to promote life span (Fig. 3I). This set of data identifies NSM neurons as the “interneurons” that relay temperature information from the cool-sensitive IL1 neurons to the intestine.
To provide further evidence, we performed the converse experiment by overexpressing MGL-1 in NSM neurons and found that MGL-1 can promote life span (Fig. 3J). In addition, enhancing the output of NSM neurons by transgenic expression of syntaxin(T254I) in these neurons prolonged life span (Fig. 3K). Thus, MGL-1 not only is required for IL1 neurons to promote life span at lower temperatures but also can promote life span on its own by acting in NSM neurons. Importantly, this life span-extending effect was fully suppressed by mutations in tph-1 (Supplemental Fig. S4A), a gene necessary for serotonin synthesis (Sze et al. 2000), providing further evidence supporting a role for serotonin. Moreover, loss of the transcription factor DAF-16 abolished the ability of the MGL-1 glutamate receptor as well as NSM neurons to promote life span (Fig. 3J,K). As DAF-16 acts in the intestine, these results are consistent with the model that the glutamate receptor MGL-1 acts in the serotoninergic NSM neurons to relay longevity signals from IL1 neurons to the intestine, presumably by releasing serotonin.
The serotonin receptor SER-7 acts in the intestine to mediate the life span-extending effect of IL1 and NSM neurons
It is conceivable that serotonin released from NSM neurons may exert its effect by targeting a serotonin receptor expressed in the intestine. We therefore set out to identify such a serotonin receptor. A total of five serotonin receptors are found in C. elegans, falling into two categories (Chase and Koelle 2007): metabotropic serotonin receptors (SER-1, SER-4, SER-5, and SER-7) and a serotonin-gated chloride channel (MOD-1) (Fig. 4A). In ser-7 mutant worms, IL1 and NSM neurons can no longer promote life span at lower temperatures (Fig. 4B), indicating a requirement for SER-7. Mutations in the other four serotonin receptors had no effect (Supplemental Fig. S4B–E). This suggests SER-7 as the serotonin receptor in the intestine.
Figure 4.
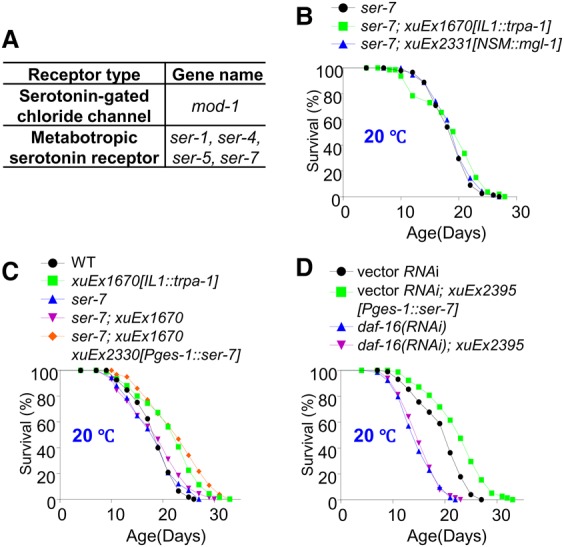
The serotonin receptor SER-7 acts in the intestine to mediate the life span-extending effect of IL1 and NSM neurons. (A) A table listing all of the five serotonin receptor genes encoded by the C. elegans genome. (B) IL1 and NSM neuron-dependent life span extension requires the serotonin receptor SER-7. IL1 and NSM neurons can no longer extend life span in ser-7 mutant worms. (C) SER-7 acts in the intestine to mediate the life span-extending effect of IL1 neurons. The phenotype of ser-7 mutant worms was rescued by transgenic expression of ser-7 cDNA in the intestine using the intestine-specific promoter Pges-1. (D) Transgenic expression of SER-7 in the intestine is sufficient to promote life span in wild-type worms, and this effect requires DAF-16. SER-7 expressed as a transgene in the intestine using the ges-1 promoter extended life span, which can be blocked by daf-16 RNAi.
Notably, SER-7 is known to be expressed in the intestine (Supplemental Fig. S4G; Hunt-Newbury et al. 2007). Indeed, the ser-7 mutant phenotype can be fully rescued by transgenic expression of ser-7 cDNA specifically in the intestine (Fig. 4C), providing additional evidence supporting SER-7 as the serotonin receptor in the intestine. Further evidence came from the experiment in which we overexpressed SER-7 as a transgene specifically in the intestine (Fig. 4D). Such a transgene extended life span (Fig. 4D), an effect that can be suppressed by daf-16 RNAi (Fig. 4D), consistent with the notion that DAF-16 acts at the very downstream to promote life span. As a control, RNAi of fmo-2, a gene implicated in SER-7 signaling (Leiser et al. 2015), had no effect (Supplemental Fig. S4H,I). Thus, SER-7 not only is required for both IL1 and NSM neurons to promote longevity at lower temperatures but also can prolong life span on its own by acting in the intestine. These observations together identify SER-7 as the serotonin receptor that acts in the intestine to mediate the life span-extending effect of IL1 and NSM neurons.
Taken together, the above results suggest a model in which the nervous system senses and processes low-temperature signals and then signal the distal intestine tissue to extend life span through a signaling circuit (Fig. 7K, below). This “cool” circuit is formed by the glutamatergic IL1 neurons, serotoninergic NSM neurons, and the intestine and uses the neurotransmitters glutamate and serotonin to transmit longevity signals (Fig. 7K). As IL1, NSM, and the intestine do not form direct synaptic connections with each other, this circuit shall be neuroendocrine in nature. We suggest that in this neuroendocrine signaling circuit, the IL1 sensory neurons detect temperature signals through the cooling-activated TRPA-1 channel and then turn on the glutamate receptor MGL-1 in the NSM “interneurons,” which in turn signal the serotonin receptor SER-7 in the intestine, leading to the activation of the transcription factor DAF-16 and thereby the extension of life span (Fig. 7K).
Figure 7.
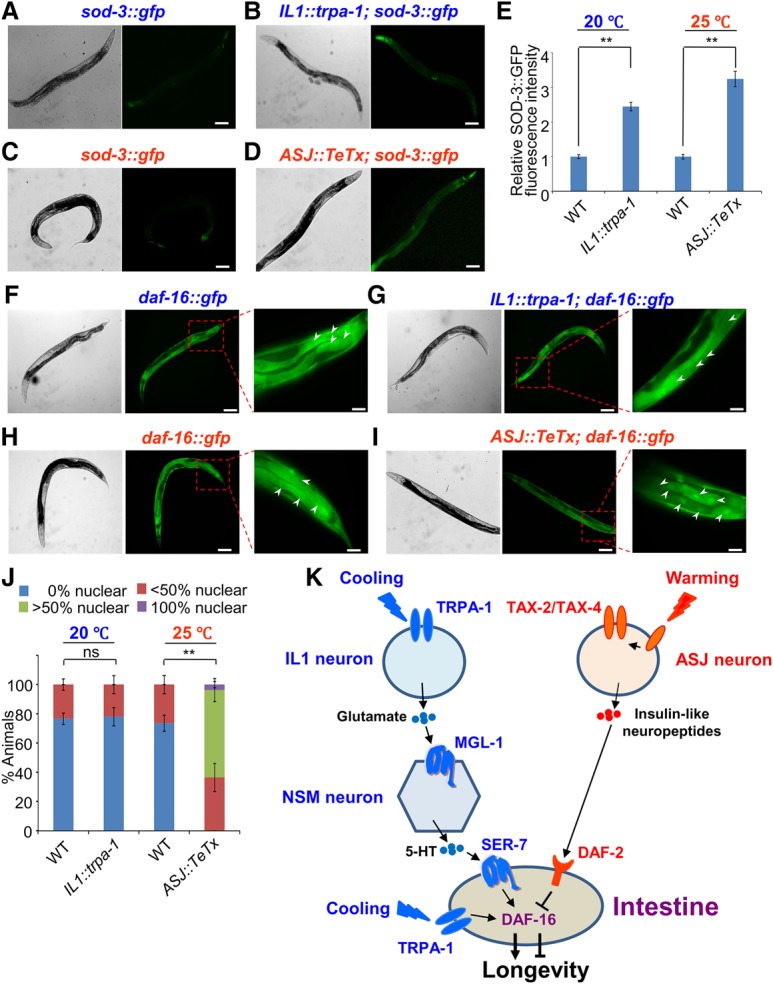
The cool-sensitive IL1 neurons and warm-sensitive ASJ neurons differentially regulate DAF-16 in the intestine. (A,B) Stimulation of IL1 neurons promotes the expression of the DAF-16 target gene sod-3 at lower temperatures. Sample images. (Left) DIC. (Right) Fluorescence. (A) The sod-3::gfp transgene was expressed at a very low level in control worms. (B) Simulation of IL1 neurons by the IL1::trpa-1 transgene promoted the expression of sod-3::gfp at lower temperatures. sod-3::gfp expression can be seen in multiple tissues, including body wall muscles, pharyngeal muscles, the intestine, neurons, and the cuticle. Bars, 100 µm. No such effect was observed at higher temperature (25°C) (see Supplemental Fig. S6A–C). (C,D) Inhibition of ASJ neuron output promotes the expression of the DAF-16 target gene sod-3 at higher temperatures. Sample images. (Left) DIC. (Right) Fluorescence. (C) The sod-3::gfp transgene was expressed at a very low level in control worms. (D) Inhibition of ASJ neuron output by the ASJ::TeTx transgene promoted the expression of sod-3::gfp at higher temperatures. sod-3::gfp expression can be seen in multiple tissues, including body wall muscles, pharyngeal muscles, the intestine, neurons, and the cuticle. Bars, 100 µm. No such effect was observed at 20°C (see Supplemental Fig. S6D–F). (E) Bar graph summarizing the data in A–D. Error bars indicate SEM. n ≥ 33. (**) P < 0.001, t-test. (F,G) Stimulation of IL1 neurons does not notably affect the subcellular localization pattern of DAF-16 in intestinal cells at lower temperatures. (F) DAF-16::GFP was localized to the cytoplasm as well as the nucleus. (G) Stimulation of IL1 neurons by the IL1::trpa-1 transgene did not alter the subcellular localization pattern of DAF-16::GFP. Sample images. (Left) DIC. (Middle) Fluorescence. (Right) Fluorescence under a higher magnification. Arrowheads point to the nuclei of intestinal cells. Bars, 25 µm. (H,I) Inhibition of ASJ neuron output promotes nuclear translocation of DAF-16 in intestinal cells at higher temperatures. (H) DAF-16::GFP was localized to the cytoplasm as well as the nucleus. (I) Inhibition of ASJ neuron output by the ASJ::TeTx transgene promoted nuclear translocation of DAF-16::GFP. Sample images. (Left) DIC. (Middle) Fluorescence. (Right) Fluorescence under a higher magnification. Arrowheads point to the nuclei of intestinal cells. Bars, 25 µm. No such nuclear translocation effect was observed at 20°C (see Supplemental Fig. S7C–F). (J) Bar graph summarizing the data in F–I. We grouped animals into four categories based on the subcellular localization pattern of DAF-16::GFP in intestinal cells (0% nuclear, <50% nuclear, >50% nuclear, and 100% nuclear), with each representing the percentage of intestinal cells showing enrichment of DAF-16::GFP fluorescence in the nucleus. There is no significant difference between wild-type (daf-16::gfp) and IL1::trpa-1 transgenic worms. P = 0.999, Pearson's χ2 test. In contrast, there is a significant difference between wild-type (daf-16::gfp) and ASJ::TeTx transgenic worms. (**) P < 0.001, Pearson's χ2 test. Error bars indicate SEM. n ≥ 76. (K) A schematic model illustrating the two neuron–intestine signaling circuits that transform environmental temperature inputs into opposing longevity outputs by differentially regulating DAF-16 in the intestine. Note that cooling can also activate intestinal TRPA-1 to promote DAF-16 activity via a distinct signaling pathway (Xiao et al. 2013).
The warm-sensitive ASJ neurons shorten life span at higher temperatures
Having characterized the mechanisms by which the nervous system communicates with the intestine to promote life span at lower temperatures, we then set out to explore how higher temperatures shorten life span. We hypothesized that some warm-sensitive neurons may detect temperature increases in the environment to shorten life span by signaling distal tissues. Unlike cool-sensitive neurons whose identities are largely unknown, some warm-sensitive neurons have been identified in C. elegans. Among them, AFD neurons are best characterized (Mori and Ohshima 1995). However, as AFD neurons maintain rather than shorten life span (Lee and Kenyon 2009; Chen et al. 2016), we turned our attention to another major type of warm-sensitive neuron: ASJ (Ohta et al. 2014). Consistent with previous results (Ohta et al. 2014), we found that ASJ neurons are indeed warm-sensitive (Fig. 5A,B). The transduction channel in ASJ neurons is the TAX-2/TAX-4 heteromeric channel (Liu et al. 2010; Ohta et al. 2014), although the warm sensor is currently unknown. Indeed, the warming-induced calcium transients in ASJ neurons were nearly eliminated in mutant worms lacking TAX-2 and TAX-4 (Fig. 5A,B). ASJ neurons are known to regulate food-dependent longevity and cold tolerance responses (Ohta et al. 2014; Artan et al. 2016), but it is unclear whether these neurons play a role in temperature-mediated longevity control. We therefore functionally ablated ASJ neurons by expressing TeTx as a transgene in these neurons and found that blocking the output of ASJ neurons extended life span in a temperature-dependent manner (Fig. 5C,D); namely, these animals were long-lived at higher temperatures (25°C) (Fig. 5C) but exhibited a normal life span at lower temperatures (20°C) (Fig. 5D), a phenomenon opposite to that observed with the cool-sensitive IL1 neurons. In addition, blunting the output of ASJ neurons by RNAi of TAX-2 and TAX-4 specifically in these neurons produced a similar result (Fig. 5E; Supplemental Fig. S5A). We also conducted the converse experiment by enhancing the output of ASJ neurons. To do so, we expressed syntaxin(T254I) as a transgene in ASJ neurons and found that this transgene shortened life span (Fig. 5F). We thus conclude that the warm-sensitive ASJ neurons shorten life span at higher temperatures and do so through neurotransmission, presumably by signaling other tissues and/or neurons.
Figure 5.

The warm-sensitive ASJ neurons signal the intestine to shorten life span at higher temperatures. (A,B) ASJ neurons are warm-sensitive and depend on the TAX-2/TAX-4 channel for temperature transduction. The genetically encoded calcium sensor GCaMP6(f) was expressed as a transgene in ASJ neurons. DsRed was coexpressed as an internal reference marker to enable ratiometric imaging. (A) Warming evoked robust calcium transients in ASJ neurons. Shades along the traces in A represent error bars (SEM). Bar graphs in B summarize the data in A. n ≥ 6. (**) P < 0.005, ANOVA with Bonferroni test. (C,D) ASJ neurons shorten life span at higher but not lower temperatures. Blocking the output of ASJ neurons by TeTx expressed as a transgene specifically in ASJ neurons extended life span only at higher temperatures (C), but no such effect was observed at lower temperatures (D). (E) The TAX-2/TAX-4 channel is important for mediating the life span-shortening effect of ASJ neurons. RNAi of tax-2 and tax-4 genes specifically in ASJ neurons using a transgene extended life span at higher temperatures. (F) Enhancing the output of ASJ neurons shortened life span. A gain-of-function form of Drosophila syntaxin was expressed as a transgene specifically in ASJ neurons. (G) DAF-16 acts in the intestine, but not neurons, to mediate the life span-shortening effect of ASJ neurons. Mutations in daf-16 suppressed the long-lived phenotype of ASJ::TeTx worms, in which the output of ASJ neurons was blocked by a TeTx transgene expressed in ASJ neurons. This daf-16 mutant phenotype was rescued by transgenic expression of daf-16 cDNA specifically in the intestine, but not in neurons, using the intestine promoter Pges-1 and the pan-neuron promoter Prgef-1, respectively.
ASJ neurons signal the intestine to shorten life span at higher temperatures
To identify the target tissues to which ASJ neurons transmit longevity signals, we took an approach similar to that used to map the IL1 neuron signaling circuit by first identifying the transcription factor that acts downstream from ASJ neurons to regulate life span. Similarly, loss of DAF-16 abolished the ability of the ASJ::TeTx transgene to promote life span (Fig. 5G), indicating that DAF-16 is required for ASJ neurons to suppress life span at higher temperatures. As a control, no such effect was observed with other longevity-regulating transcription factors such as HSF-1, SKN-1, PHA-4, and DAF-12 (Supplemental Fig. S5D–I). The ASJ::TeTx transgene did not promote the overall level of unfolded protein responses or heat-shock responses, as shown by reporter assays (Supplemental Fig. S5J). The daf-16 mutant phenotype can be rescued by transgenic expression of daf-16 cDNA specifically in the intestine, while neuronal expression of daf-16 cDNA did not rescue the phenotype (Fig. 5G). Thus, DAF-16 acts in the intestine to mediate the life span-shortening effect of ASJ neurons, suggesting that the intestine is a target tissue to which ASJ neurons transmit longevity signals. This also suggests that ASJ neurons shorten life span at higher temperatures, probably by inhibiting DAF-16 in the intestine.
ASJ neurons shorten life span at higher temperatures through insulin-like neuropeptides and their receptor, DAF-2
Similar to IL1 neurons, ASJ neurons store both small neurotransmitters and neuropeptides (White et al. 1986; Pereira et al. 2015). To determine which type of signaling molecule is used by ASJ neurons to communicate with the intestine, we examined worms deficient in UNC-13 and UNC-31, which are required for the secretion of small neurotransmitters and neuropeptides, respectively (Richmond et al. 1999; Speese et al. 2007). RNAi of unc-31 in ASJ neurons using a transgene extended life span at higher but not lower temperatures (Fig. 6A; Supplemental Fig. S5B), while unc-13 RNAi had no effect (Fig. 6A; Supplemental Fig. S5B). Apparently, ASJ neurons rely on neuropeptides rather than small neurotransmitters to signal the intestine, a feature opposite to that pertaining to IL1 neurons.
Figure 6.

The warm-sensitive ASJ neurons shorten life span through insulin-like neuropeptides and their cognate receptor, DAF-2. (A) RNAi of unc-31, but not unc-13, in ASJ neurons extends life span at higher temperatures. unc-31 and unc-13 RNAi was introduced as a transgene in ASJ neurons. (B) RNAi of the insulin genes ins-6 and daf-28 in ASJ neurons extends life span at higher temperatures, and this effect requires daf-16. ins-6 and daf-28 RNAi was introduced as a transgene in ASJ neurons. (C) The life span-shortening effect of ASJ neurons requires INS-6 and DAF-28. Loss of ins-6 and daf-28 can block the life span-shortening effect of a syntaxin(T254I) transgene expressed in ASJ neurons. (D) The long-lived life span phenotype of daf-2 mutant and ASJ::TeTx transgenic worms is non-additive. (E) The short-lived life span phenotype of ASJ::syntaxin(T254I) transgenic worms can be suppressed by daf-2 mutation. (F) DAF-2 acts in the intestine to mediate the life span-shortening effect of ASJ neurons. Transgenic expression of daf-2 cDNA in the intestine (Pges-1::daf-2), but not in neurons (Prgef-1::daf-2), restored the ability of the ASJ::syntaxin(T254I) transgene to shorten life span in daf-2 mutant worms.
The insulin-like peptides INS-6 and DAF-28 are the two major neuropeptides expressed in ASJ neurons (Ohta et al. 2014; Artan et al. 2016). RNAi of ins-6 and daf-28 specifically in ASJ neurons extended life span at 25°C but had no effect at 20°C, and this temperature-dependent life span-extending effect can also be suppressed by daf-16 RNAi (Fig. 6B; Supplemental Fig. S5C). This result identifies an important role for these two insulins in mediating the life span-shortening effect of ASJ neurons at higher temperatures. As indicated above, enhancing the output of ASJ neurons by a syntaxin(T254I) transgene shortened life span (Fig. 5F). Interestingly, loss of ins-6 and daf-28 can suppress this short-lived phenotype (Fig. 6C), consistent with the notion that these two insulins are the signaling molecules released by ASJ neurons to shorten life span at higher temperatures. As the C. elegans genome encodes only one insulin/IGF-1 receptor, DAF-2 (Kimura et al. 1997), these data also suggest that ASJ neurons may act upstream of DAF-2 to shorten life span. In support of this model, the long-lived phenotype of daf-2 mutant and ASJ::TeTx transgenic worms was nonadditive (Fig. 6D). More importantly, the short-lived phenotype of ASJ::syntaxin(T254I) transgenic worms was fully suppressed by mutations in daf-2 (Fig. 6E), further suggesting that ASJ neurons act upstream of DAF-2 in the pathway. Together, these observations point to a model in which ASJ neurons transmit temperature information to the intestine through insulin-like neuropeptides and their cognate receptor, DAF-2.
DAF-2 acts in the intestine to mediate the life span-shortening effect of ASJ neurons
Given that ASJ neurons signal the intestine to regulate DAF-16, we wondered whether DAF-2 also acts in the intestine. Alternatively, DAF-2 may function in some “interneurons,” which would then relay longevity signals to the intestine. Notably, DAF-2 has been shown to function in both neurons and the intestine (Wolkow et al. 2000). To distinguish between these two possibilities, we sought to determine the site of action of DAF-2 in mediating the life span-shortening effect of ASJ neurons. While transgenic expression of daf-2 cDNA in either neurons or the intestine can partially rescue the long-lived life span phenotype of daf-2 mutant worms (Fig. 6F), only intestinal expression of daf-2 cDNA was able to restore the ability of ASJ::syntaxin(T254I) transgene to shorten life span at higher temperatures (Fig. 6F). Neuronal expression of daf-2 cDNA did not have such an effect (Fig. 6F). This demonstrates that DAF-2 acts in the intestine to mediate the life span-shortening effect of ASJ neurons.
Collectively, this set of data suggests a model in which the nervous system senses and processes temperature signals and then communicates with the distal intestine tissue to shorten life span at higher temperatures by using another neuroendocrine signaling circuit (Fig. 7K). This “warm” circuit is formed by the warm-sensitive ASJ neurons and the intestine and relies on insulin-like neuropeptides rather than small neurotransmitters to transmit longevity signals from ASJ neurons to the intestine (Fig. 7K). We suggest that ASJ neurons detect and process temperature information through the TAX-2/TAX-4 transduction channel, which in turn signals the insulin/IGF-1 receptor DAF-2 in the intestine, leading to the inhibition of the transcription factor DAF-16 and thereby the suppression of life span (Fig. 7K).
The ‘cool’ and ‘warm’ neuroendocrine signaling circuits converge on the intestine to differentially regulate DAF-16
Our data suggest that the “cool” and “warm” neuroendocrine signaling circuits converge on the intestine to regulate DAF-16, with the former stimulating and the latter inhibiting it (Fig. 7K). To test whether DAF-16 is indeed differentially regulated by the two neuroendocrine signaling circuits, we examined sod-3, a well-characterized DAF-16 target gene (Honda and Honda 1999; Lee et al. 2003; Murphy et al. 2003). Stimulation of IL1 neurons by TRPA-1 at lower temperatures promoted the expression level of sod-3 (Fig. 7A,B,E; Supplemental Fig. S7A), while no such effect was detected at higher temperatures (Supplemental Figs. S6A–C, S7A), indicating that the “cool” circuit promoted DAF-16 activity. In contrast, inhibition of ASJ neuron output by TeTx promoted the expression level of sod-3 only at higher but not lower temperatures (Fig. 7C–E; Supplemental Figs. S6D–F, S7B), indicating that the “warm” circuit suppressed DAF-16 activity. Notably, the increase in SOD-3::GFP expression level was observed globally in nearly every tissue throughout the body (Fig. 7B,D), suggesting that the activity of DAF-16 in the intestine can be disseminated to other tissues, a phenomenon that has also been observed under other conditions (Libina et al. 2003; Murphy et al. 2007; Zhang et al. 2013). In support of this model, RNAi knockdown of DAF-16 locally in the intestine using a transgene blocked the ability of IL1 and ASJ neurons to globally up-regulate SOD-3::GFP expression in other tissues (Supplemental Fig. S6G–P). This observation is consistent with the notion that the intestine is also a longevity signaling center.
The question arises of how the two neuroendocrine signaling circuits might differentially regulate the same protein—DAF-16—in the intestine. DAF-16 is best known to be regulated mainly by two mechanisms. Specifically, pathways such as insulin signaling regulate DAF-16 primarily by inhibiting its translocation to the nucleus (Henderson and Johnson 2001; Lin et al. 2001), while some other longevity pathways regulate DAF-16 function within the nucleus without affecting its nuclear translocation (Wolff et al. 2006; Li et al. 2008; Alam et al. 2010). Indeed, we found that inhibition of ASJ neuron output by the ASJ::TeTx transgene promoted the translocation of DAF-16::GFP to the nucleus in the intestine only at 25°C but not at 20°C (Fig. 7H–J; Supplemental Fig. S7C–F), indicating that ASJ neurons inhibit DAF-16's nuclear translocation at higher temperatures. In contrast, stimulation of IL1 neurons by the trpa-1 transgene did not have a notable effect on DAF-16 nuclear translocation (Fig. 7F,G,J), and neither did mutations in trpa-1 (Supplemental Fig. S7G–K). This suggests that IL1 neurons may primarily regulate DAF-16 function within the nucleus. No notable change in the expression level of daf-16 was detected under either condition (Supplemental Fig. S7A,B). Thus, IL1 neurons and ASJ neurons appear to use distinct mechanisms to regulate DAF-16 function. We conclude that the “cool” and “warm” neuroendocrine signaling circuits converge on the intestine to differentially regulate DAF-16, with the former promoting its function and the latter inhibiting it, thereby leading to opposing outcomes in longevity.
Discussion
Research in the past two decades has led to rapid progress in understanding how longevity genes and pathways regulate the function of individual organelles, cells, and tissues. Nevertheless, aging in multicellular organisms is a body-wide process involving the coordination of multiple cell types and tissues (Apfeld and Kenyon 1998; Libina et al. 2003; Alcedo and Kenyon 2004; Murphy et al. 2007; Medkour et al. 2016). Such coordination requires cross-talk between different cell types and tissues throughout the body. Despite its importance, little is known about how cell–cell and tissue–tissue communications occur and how they contribute to longevity modulation. In the present study, using C. elegans as a model, we characterized how the brain communicates with the distal gut tissue (intestine) through neuroendocrine signaling to bidirectionally regulate life span. We identified two brain–gut neuroendocrine signaling circuits through which worms detect and transform environmental temperature inputs into opposing longevity outputs. We also identified the signaling molecules and their cognate receptors used by the two circuits for information processing. To the best of our knowledge, this represents the first comprehensive study illustrating how tissue–tissue communications bidirectionally regulate longevity at the molecular, cellular, and circuit levels in any organisms.
The two circuits share similarities but also exhibit interesting distinctions (Fig. 7K). They both use neuroendocrine signaling to mediate tissue–tissue communications but use distinct types of signaling molecules, with the “cool” circuit depending on small neurotransmitters (i.e., glutamate and serotonin) and the “warm” circuit relying on neuropeptides (i.e., insulins) (Fig. 7K). In addition, while both circuits converge on the intestine to regulate DAF-16, they do so through distinct mechanisms. The exact mechanisms by which the two circuits converge on the intestine to differentially regulate DAF-16 are unclear at present. Considering that the insulin/IGF1 receptor DAF-2 acts in the intestine to mediate the effect of the “warm” circuit, it is conceivable that the PI3K–AKT signaling pathway, which is known to function downstream from DAF-2 (Kenyon 2010), may regulate the nuclear translocation of DAF-16 in the intestine. In the case of the “cool” circuit, it is presently unclear how it regulates the nuclear activity of DAF-16 in the intestine. Interestingly, some of the genes such as pkc-2 that are important for intestinal TRPA-1 to regulate DAF-16 in the intestine (Xiao et al. 2013) are not required for neuronal TRPA-1 to extend life span (B Zhang and XZS Xu, unpubl.). This suggests that a distinct signaling mechanism may act in the intestine to regulate DAF-16 to mediate the life span-extending effect of the “cool” circuit. Last, additional neurons/cells may function in the two signaling circuits, but their identities remain to be uncovered. Future work is needed to address these questions.
Recent studies have revealed an increasingly important role for the brain in longevity as well as stress signaling. For example, studies in C. elegans show that ER and mitochondria stress signals in neurons can be propagated to the intestine (Durieux et al. 2011; Taylor and Dillin 2013; Berendzen et al. 2016), and neuronal AMPK and HIF-1 signals can be spread to other tissues through neuroendocrine signaling (Burkewitz et al. 2015; Leiser et al. 2015); nevertheless, the identities of the neurons involved, the physiological signals triggering the signaling, and the underlying molecular, neuronal, and circuit mechanisms are not well understood. Our results present an example illustrating how specific types of sensory neurons detect and process environmental signals and then transmit them to downstream interneurons and the distal intestine tissue through neuroendocrine signaling by releasing specific signaling molecules such as small neurotransmitters and neuropeptides, which then exert their effects by targeting their cognate receptors expressed in interneurons and the intestine. It is noteworthy that this type of mechanism adopted by animals to regulate longevity bears remarkable similarities to those used to detect and process sensory information to regulate animal behavior, with the exception that the output of the former is longevity, while the output of the latter is behavior. This highlights the versatility of the nervous system.
Notably, the intestine is another key longevity signaling center in C. elegans (Kenyon 2010; Medkour et al. 2016). Specifically, longevity signals can be further propagated from the intestine to other tissues in the body (Libina et al. 2003). It is therefore conceivable that temperature-dependent longevity signals ultimately would be spread to other tissues from the intestine. Consistent with this model, we found that the DAF-16 target gene sod-3 is indeed up-regulated in multiple tissues in response to temperature signals, and such global up-regulation requires DAF-16 in the intestine. Strikingly, both the brain and the intestine also play a critical role in aging in mice and flies (Mattson et al. 2002; Taguchi et al. 2007; Rera et al. 2013; Nicoletti 2015), highlighting the importance of these two tissues in coordinating longevity signaling. We propose that the brain and the gut may form a signaling axis that detects, processes, integrates, and disseminates longevity signals in not only C. elegans but probably many other organisms.
Materials and methods
Strains
The strains used in this study are listed in Supplemental Table S2.
Life span assay
All mutant alleles were outcrossed into the wild-type (N2) background at least six times prior to life span analysis. At least three independent transgenic lines were assayed to verify the effect of each transgene, and data from one representative line is shown in the report. Life span experiments were performed as described previously (Xiao et al. 2013; Zhang et al. 2015). We scored the first day of adulthood as day 1. For each life span experiment, 80–160 worms were assayed, and worms were transferred every 2–3 d to fresh 60 mm NGM (nematode growth medium) plates. Worm strains were first maintained at 20°C, and L4 larvae were then moved to designated temperatures to assay adult life span. Those worms (10%–35% of total population) that crawled off the plate, exploded, or bagged were censored for the accuracy of data interpretation. For RNAi experiments, we included 25 µg/mL carbenicillin and 1 mM IPTG in NGM plates. HT115 bacteria containing either empty vector L4440 or RNAi plasmids were seeded on RNAi plates 2 d prior to the experiment. All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, Inc.). P-values were calculated using the log rank (Kaplan-Meier) method. All life span experiments were repeated at least twice. Detailed data analysis is in Supplemental Table S1.
Calcium imaging and microscopy
Calcium imaging experiments were conducted on an Olympus upright microscope (BX51WI) under a 60× objective as described previously (Feng et al. 2006; Li et al. 2006). Worms were immobilized on an agarose pad and incubated in bath solution (10 mM HEPES at pH 7.4, 5 mM KCl, 145 mM NaCl, 1.2 mM MgCl2, 2.5 mM CaC12, 10 mM glucose). Bath solution was perfused toward the nose of the worm. The temperature of the bath solution was controlled by a bipolar in-line temperature controller (Warner Instruments) triggered by an Axon 700B amplifier. A Roper CoolSnap CCD camera was used to acquire images, which were processed by MetaFluor (Molecular Devices). To enable ratiometric imaging, GCaMP6(f) and DsRed plasmids were coinjected to generate transgenic lines, and the peak percentage change in the ratio of GCaMP/DsRed fluorescence in the neuron of interest was quantified.
To quantify SOD-3::GFP and DAF-16::GFP fluorescence, worms were immobilized on an agarose pad with 5 mM sodium azide, and images were acquired on an Olympus upright microscope (BX51) under 10× and 40× objectives by MetaMorph (Molecular Devices) and processed by ImageJ.
Quantitative RT–PCR
Total RNA (100–300 worms) was prepared with TRI reagent (Life Technologies). We conducted quantitative PCR with SYBR Green (Life Technologies) according to the manufacturer's protocol. act-1 (actin) was used as an internal reference for normalization, and the ΔΔCt method was adopted to analyze quantitative PCR data.
Supplementary Material
Acknowledgments
We thank Ao-Lin Hsu and Rene Garcia for providing reagents, and Teng Yu for technical assistance. Some strains were obtained from the Caenorhabditis Genetics Center and Knockout Consortiums in the USA and Japan. This work was supported by the National Natural Science Foundation of China (31420103909, 81720108031, and 31721002 to J.L., and 31600974 to B.Z.), the Program of Introducing Talents of Discipline to the Universities from the Ministry of Education (B08029 to J.L.), the Program for Changjiang Scholars and Innovative Research Team in University (IRT13016 to J.L.), the Natural Science Foundation of Hubei province (2014CFA010 to J.L.), and grants from the National Institute on Aging and National Institute of General Medical Sciences (to X.Z.S.X.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.309625.117.
References
- Alam H, Williams TW, Dumas KJ, Guo C, Yoshina S, Mitani S, Hu PJ. 2010. EAK-7 controls development and life span by regulating nuclear DAF-16/FoxO activity. Cell Metab 12: 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcedo J, Kenyon C. 2004. Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron 41: 45–55. [DOI] [PubMed] [Google Scholar]
- Allen EN, Ren J, Zhang Y, Alcedo J. 2015. Sensory systems: their impact on C. elegans survival. Neuroscience 296: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. 1998. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell 95: 199–210. [DOI] [PubMed] [Google Scholar]
- Artan M, Jeong DE, Lee D, Kim YI, Son HG, Husain Z, Kim J, Altintas O, Kim K, Alcedo J, et al. 2016. Food-derived sensory cues modulate longevity via distinct neuroendocrine insulin-like peptides. Genes Dev 30: 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, Liu Y, Dillin A. 2016. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166: 1553–1563.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Morantte I, Weir HJ, Yeo R, Zhang Y, Huynh FK, Ilkayeva OR, Hirschey MD, Grant AR, Mair WB. 2015. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell 160: 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Koelle MR. 2007. Biogenic amine neurotransmitters in C. elegans. WormBook 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzigeorgiou M, Yoo S, Watson JD, Lee WH, Spencer WC, Kindt KS, Hwang SW, Miller DM III, Treinin M, Driscoll M, et al. 2010. Specific roles for DEG/ENaC and TRP channels in touch and thermosensation in C. elegans nociceptors. Nat Neurosci 13: 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Chen HJ, Tseng WC, Hsu JM, Huang TT, Chen CH, Pan CL. 2016. A C. elegans thermosensory circuit regulates longevity through crh-1/CREB-dependent flp-6 neuropeptide signaling. Dev Cell 39: 209–223. [DOI] [PubMed] [Google Scholar]
- Conti B, Sanchez-Alavez M, Winsky-Sommerer R, Morale MC, Lucero J, Brownell S, Fabre V, Huitron-Resendiz S, Henriksen S, Zorrilla EP, et al. 2006. Transgenic mice with a reduced core body temperature have an increased life span. Science 314: 825–828. [DOI] [PubMed] [Google Scholar]
- Dillon J, Franks CJ, Murray C, Edwards RJ, Calahorro F, Ishihara T, Katsura I, Holden-Dye L, O'Connor V. 2015. Metabotropic glutamate receptors: modulators of context-dependent feeding behaviour in C. elegans. J Biol Chem 290: 15052–15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. 2011. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Li W, Ward A, Piggott BJ, Larkspur E, Sternberg PW, Xu XZS. 2006. A C. elegans model of nicotine-dependent behavior: regulation by TRP family channels. Cell 127: 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Partridge L. 2015. Promoting health and longevity through diet: from model organisms to humans. Cell 161: 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer ER, Perez CL, Van Gilst MR, Lee BH, Ashrafi K. 2008. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab 8: 118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. 2001. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol 11: 1975–1980. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Smith EK. 1986. Longevity of cold-exposed rats: a reevaluation of the ‘rate-of-living theory’. J Appl Physiol 61: 1656–1660. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. 1999. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13: 1385–1393. [PubMed] [Google Scholar]
- Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, Halfnight E, Lee D, Lin J, Lorch A, et al. 2007. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol 5: e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. 2010. The genetics of ageing. Nature 464: 504–512. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. 1997. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277: 942–946. [DOI] [PubMed] [Google Scholar]
- Kindt KS, Viswanath V, Macpherson L, Quast K, Hu H, Patapoutian A, Schafer WR. 2007. Caenorhabditis elegans TRPA-1 functions in mechanosensation. Nat Neurosci 10: 568–577. [DOI] [PubMed] [Google Scholar]
- Lagow RD, Bao H, Cohen EN, Daniels RW, Zuzek A, Williams WH, Macleod GT, Sutton RB, Zhang B. 2007. Modification of a hydrophobic layer by a point mutation in syntaxin 1A regulates the rate of synaptic vesicle fusion. PLoS Biol 5: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Kenyon C. 2009. Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans. Curr Biol 19: 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L. 1999. EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci 19: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G. 2003. DAF-16 target genes that control C. elegans life-span and metabolism. Science 300: 644–647. [DOI] [PubMed] [Google Scholar]
- Leiser SF, Miller H, Rossner R, Fletcher M, Leonard A, Primitivo M, Rintala N, Ramos FJ, Miller DL, Kaeberlein M. 2015. Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science 350: 1375–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Feng Z, Sternberg PW, Xu XZS. 2006. A C. elegans stretch receptor neuron revealed by a mechanosensitive TRP channel homologue. Nature 440: 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ebata A, Dong Y, Rizki G, Iwata T, Lee SS. 2008. Caenorhabditis elegans HCF-1 functions in longevity maintenance as a DAF-16 regulator. PLoS Biol 6: e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. 2003. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115: 489–502. [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. 2001. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28: 139–145. [DOI] [PubMed] [Google Scholar]
- Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Sudhof TC, Niemann H, Jahn R. 1992. Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem Biophys Res Commun 189: 1017–1023. [DOI] [PubMed] [Google Scholar]
- Liu J, Ward A, Gao J, Dong Y, Nishio N, Inada H, Kang L, Yu Y, Ma D, Xu T, et al. 2010. C. elegans phototransduction requires a G protein–dependent cGMP pathway and a taste receptor homolog. Nat Neurosci 13: 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Maswood N. 2002. How does the brain control lifespan? Ageing Res Rev 1: 155–165. [DOI] [PubMed] [Google Scholar]
- Medkour Y, Svistkova V, Titorenko VI. 2016. Cell-nonautonomous mechanisms underlying cellular and organismal aging. Int Rev Cell Mol Biol 321: 259–297. [DOI] [PubMed] [Google Scholar]
- Mori I, Ohshima Y. 1995. Neural regulation of thermotaxis in Caenorhabditis elegans. Nature 376: 344–348. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. 2003. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283. [DOI] [PubMed] [Google Scholar]
- Murphy CT, Lee SJ, Kenyon C. 2007. Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc Natl Acad Sci 104: 19046–19050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti C. 2015. Age-associated changes of the intestinal epithelial barrier: local and systemic implications. Expert Rev Gastroenterol Hepatol 9: 1467–1469. [DOI] [PubMed] [Google Scholar]
- Ohta A, Ujisawa T, Sonoda S, Kuhara A. 2014. Light and pheromone-sensing neurons regulates cold habituation through insulin signalling in Caenorhabditis elegans. Nat Commun 5: 4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Kratsios P, Serrano-Saiz E, Sheftel H, Mayo AE, Hall DH, White JG, LeBoeuf B, Garcia LR, Alon U, et al. 2015. A cellular and regulatory map of the cholinergic nervous system of C. elegans. Elife 4: e12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rera M, Azizi MJ, Walker DW. 2013. Organ-specific mediation of lifespan extension: more than a gut feeling? Ageing Res Rev 12: 436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. 1999. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci 2: 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawhney R, Sehl M, Naeim A. 2005. Physiologic aspects of aging: impact on cancer management and decision making, part I. Cancer J 11: 449–460. [DOI] [PubMed] [Google Scholar]
- Sehl M, Sawhney R, Naeim A. 2005. Physiologic aspects of aging: impact on cancer management and decision making, part II. Cancer J 11: 461–473. [DOI] [PubMed] [Google Scholar]
- Speese S, Petrie M, Schuske K, Ailion M, Ann K, Iwasaki K, Jorgensen EM, Martin TF. 2007. UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J Neurosci 27: 6150–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. 2000. Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature 403: 560–564. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. 2007. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317: 369–372. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. 2013. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153: 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. 1986. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B 314: 1–340. [DOI] [PubMed] [Google Scholar]
- Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A. 2006. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell 124: 1039–1053. [DOI] [PubMed] [Google Scholar]
- Wolkow CA, Kimura KD, Lee MS, Ruvkun G. 2000. Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science 290: 147–150. [DOI] [PubMed] [Google Scholar]
- Xiao R, Zhang B, Dong Y, Gong J, Xu T, Liu J, Xu XZS. 2013. A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell 152: 806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Liu J, Xu XZ. 2015. Thermosensation and longevity. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 201: 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Judy M, Lee SJ, Kenyon C. 2013. Direct and indirect gene regulation by a life-extending FOXO protein in C. elegans: roles for GATA factors and lipid gene regulators. Cell Metab 17: 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Xiao R, Ronan EA, He Y, Hsu AL, Liu J, Xu XZ. 2015. Environmental temperature differentially modulates C. elegans longevity through a thermosensitive TRP channel. Cell Rep 11: 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.