

Abstract
Motivation: Antimicrobial peptides (AMPs) are innate immune molecules that exhibit activities against a range of microbes, including bacteria, fungi, viruses and protozoa. Recent increases in microbial resistance against current drugs has led to a concomitant increase in the need for novel antimicrobial agents. Over the last decade, a number of AMP prediction tools have been designed and made freely available online. These AMP prediction tools show potential to discriminate AMPs from non-AMPs, but the relative quality of the predictions produced by the various tools is difficult to quantify.
Results: We compiled two sets of AMP and non-AMP peptides, separated into three categories—antimicrobial, antibacterial and bacteriocins. Using these benchmark data sets, we carried out a systematic evaluation of ten publicly available AMP prediction methods. Among the six general AMP prediction tools—ADAM, CAMPR3(RF), CAMPR3(SVM), MLAMP, DBAASP and MLAMP—we find that CAMPR3(RF) provides a statistically significant improvement in performance, as measured by the area under the receiver operating characteristic (ROC) curve, relative to the other five methods. Surprisingly, for antibacterial prediction, the original AntiBP method significantly outperforms its successor, AntiBP2 based on one benchmark dataset. The two bacteriocin prediction tools, BAGEL3 and BACTIBASE, both provide very good performance and BAGEL3 outperforms its predecessor, BACTIBASE, on the larger of the two benchmarks.
Contact: gaberemu@ngha.med.sa or william-noble@uw.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The recently increasing incidence of antimicrobial resistant strains of many pathogens suggests that the utility of conventional drugs against these diseases is decreasing. Antimicrobial peptides (AMPs) offer a promising alternative to conventional drugs. AMPs are short peptides, typically less than 50 amino acid residues in length, that are produced by a wide variety of organisms, including bacteria, insects, amphibians, mammals and plants, to protect the organism against various pathogens. AMPs have been shown to have activities against microbes responsible for malaria (Kückelhaus et al., 2009; Vale et al., 2014), trypanosomiasis (Pinto et al., 2014), leishmaniasis (Kückelhaus et al., 2009; Pinto et al., 2014), tuberculosis (Kapoor et al., 2011; Ziqing et al., 2011; Ramón-García et al., 2013) and HIV AIDS (Chen et al., 2012; Tincho et al., 2016). Furthermore, recent evidence suggests that such peptides can also be useful in combating cancer (Hoskin and Ramamoorthy, 2008; Gaspar et al., 2013) and diabetes (Conlon et al., 2014).
Given the therapeutic potential of AMPs, there is an urgent need to identify novel AMPs. On the one hand, as increasingly diverse species are sequenced, the pool of potential AMP sequences grows. On the other hand, experimental methods for characterizing AMPs are costly, both in terms of time and resources. Thus, there is a need for computational tools capable of predicting, from a given set of peptide sequences, which ones are more likely to function as AMPs.
Over the past decade, a large number of AMP prediction methods have been proposed (reviewed in Porto et al., 2012). Some methods, such as ADAM (Li and Godzik, 2015) and CAMPR3 (Waghu et al., 2016), are designed to identify any variety of AMPs, whereas other tools focus on specific subclasses of AMPs. For example, AntiBP (Lata et al., 2007) and its successor AntiBP2 (Lata et al., 2010) concentrate on predicting antibacterial peptides, i.e. peptides that specifically target bacteria. Such peptides can be used to enhance the therapeutic effects of infection treatments and decrease the prevalence of resistant strains. BAGEL (de Jong et al., 2006) and BACTIBASE (Hammami et al., 2007) focus on bacteriocins, a subclass of antibacterials that are themselves derived from bacteria (Arthur et al., 2014).
Predictions from these methods have already proved to be valuable as hypothesis generators for use in informing lower throughput experimental approaches. For example, BAGEL3 and BACTIBASE were used to classify unknown bacteriocins encrypted by 317 microbial genomes in the human intestine (Drissi et al., 2015). Singh et al. characterized a new pediocin-like bacteriocin that is active in reduced conditions, using both BACTIBASE and CAMPR3 (Singh et al., 2014). The authors found out that the novel sequence did not exhibit significant similarity with known pediocin-like bacteriocins or other bacterial AMPs available in the BACTIBASE and CAMPR3 databases. In another study, a novel bacteriocin produced by Streptococcus mutans was characterized with the help of the APD2 (Wang et al., 2009) and AntiBP2 servers (Nicolas, 2011). The AMP activity of a mature peptide from Drosophila virilis was predicted using CAMPR3 (Waghu et al., 2016), AntiBP2 (Lata et al., 2010) and AMPA (Torrent et al., 2012; Seto and Tamura, 2013), and it was further shown that this peptide is divergent from the peptide produced by Drosophila melanogaster. Finally, a new peptide found in milk protein using the CAMPR3 tool was subsequently validated to possess antimicrobial activity (Dziuba and Dziuba, 2014).
As is common in many bioinformatics enterprises, most publications describing a new AMP predictor claim that the new tool is superior to all existing methods. Yet these claims may be somewhat biased because the comparison is carried out by the developers of the tool itself. In addition, performance results from one study to the next are not comparable to one another because the benchmark data sets vary. The purpose of the present study is to carry out an unbiased evaluation of existing tools using two high quality benchmark datasets.
For this study, we focus on predictors that have been made available as open access web portals. We compare the predictive accuracy of ten AMP predictors using data from the DAMPD (Seshadri et al., 2012) and APD3 (Wang et al., 2016) databases and a randomly generated set of non-AMP examples. The tools include six AMP predictors [CAMPR3 using either a random forest or support vector machine classifier (Waghu et al., 2016), ADAM (Li and Godzik, 2015), AMPA (Torrent et al., 2012), DBAASP (Vishnepolesky and Pirtskhalava, 2014) and MLAMP (Lin and Xu, 2016)], two antibacterial prediction tools [AntiBP (Lata et al., 2007) and [AntiBP2 (Lata et al., 2010)], and two bacteriocin prediction tools [BAGEL3 (van Heel et al., 2013) and BACTIBASE (Hammami et al., 2007)]. Our results indicate that, among AMP predictors, the CAMPR3(RF) tool provides the best performance. For prediction of antibacterial peptides, AntiBP performs much better than its successor, AntiBP2 and for bacteriocins, BAGEL3 provides higher quality predictions than BACTIBASE.
2 Materials and methods
The methodology adopted in the study is described in the following sections and is shown in Figure 1.
Fig. 1.

Methodology employed in comparing and ranking various AMP tools
2.1 Validation dataset
Two benchmark data sets were used in the analysis, downloaded from the DAMPD (Seshadri et al., 2012) and APD3 (Wang et al., 2016) databases. These sets consist of, respectively, 1232 and 2338 manually curated, experimentally verified AMP peptides. The AMPs in DAMPD consist of mature and propeptide regions, while APD3 consists only of mature peptides less than 100 residues as well as important human antimicrobial proteins greater than 100 residues, such as lysozyme, eosinophil-derived neurotoxin (RNase 2), eosinophil cationic protein (RNase 3), regenerating gene family protein III (Reg III) and psoriasin. These proteins are important since they show activities against parasites, viruses and uropathogens (Wang, 2014). To reduce potential bias in the data set, we removed highly similar sequences using the CD-HIT software version 4.6.1 (Li and Godzik, 2006). Applying a 90% maximum sequence identity threshold reduced the data sets to 547 and 1713 sequences for the DAMPD and APD3, respectively. The DAMPD set consists of 313 antibacterial peptides, 31 bacteriocins and 234 other AMPs, and the APD3 set consists of 1446 antibacterial peptides, 154 bacteriocins and 113 other AMPs. These sequences comprise the positive sets for our experiments.
To test the discriminative power of the predictors, we also constructed a matched set of non-AMP sequences. To do so, for each positive sequence, we randomly extracted from a real protein database a peptide sequence of the same length. For the DAMPD benchmark, this was done by downloading non-AMP sequences from the UniProt database (UniProt Consortium, 2015) version 2016_04, which contains 4 265 218 proteins, using the query, (golgi OR cytoplasm OR ‘endoplasmic reticulum’ OR mitochondria) AND NOT antimicrobial. For the APD3 benchmark, we used UniProt version 2016_12, which contains 548 843 proteins, and the query NOT antimicrobial AND reviewed:yes. In each case, we concatenated all of the resulting proteins, and then selected starting positions uniformly at random along the resulting sequence. To improve the resolution of our discriminative scores, we extracted six non-AMP examples for each AMP example. Hence, the final collection consisted of 2735 (8565), 1565 (7230) and 155 (770) non-AMP sequences for the AMP, antibacterial and bacteriocin sets extracted from DAMPD (APD3), respectively. These dataset are available in Supplementary Tables S1 and S2.
2.2 Prediction tools
Many methods for predicting AMPs have been described in the scientific literature, including methods focused on prediction of bacteriocins (de Jong et al., 2006; Hammami et al., 2007), antibacterial peptides (Lata et al., 2007, 2010) and general prediction of antimcrobial peptides (Fjell et al. 2007; Wang et al. 2011; Joseph et al. 2012; Fernandes et al. 2012; Vishnepolsky and Pirtskhalava 2014; Ng et al. 2015; Waghu et al. 2016). We analyzed the performance of the ten web-accessible AMP predictors (Table 1).
Table 1.
Web-accessible AMP predictors
| Name | Category | Year | URL | Batch? | References |
|---|---|---|---|---|---|
| BAGEL3 | Bacteriocin | 2013 | bagel2.molgenrug.nl/index.php/bagel3 | ✓ | van Heel et al. (2013) |
| BACTIBASE | Bacteriocin | 2014 | http://bactibase.pfba-lab-tun.org/main.php | ✓ | Hammami et al. (2007) |
| AntiBP | Antibacterial | 2007 | http://www.imtech.res.in/raghava/antibp/ | Lata et al. (2007) | |
| AntiBP2 | Antibacterial | 2010 | http://www.imtech.res.in/raghava/antibp2/ | Lata et al. (2010) | |
| AMPA | Antimicrobial | 2012 | http://tcoffee.crg.cat/apps/ampa/do | ✓ | Torrent et al. (2012) |
| DBAASP | Antimicrobial | 2014 | http://dbaasp.org/prediction | ✓ | Vishnepolesky and Pirtskhalava (2014) |
| ADAM | Antimicrobial | 2015 | http://bioinformatics.cs.ntou.edu.tw/ADAM/ | ✓ | Li and Godzik (2015) |
| MLAMP | Antimicrobial | 2016 | http://www.jci-bioinfo.cn/MLAMP | ✓ | Lin and Xu (2016) |
| CAMPR3(RF) | Antimicrobial | 2016 | http://www.camp.bicnirrh.res.in/prediction.php | ✓ | Waghu et al. (2016) |
| CAMPR3(SVM) | Antimicrobial | 2016 | http://www.camp.bicnirrh.res.in/prediction.php | ✓ | Waghu et al. (2016) |
Note: The ‘Batch?’ column indicates whether multiple sequences can be submitted at once to the server.
BAGEL3 was created to store experimentally validated bacteriocins as well as to predict three types of bacteriocin: small modified bacteriocins, small unmodified bacteriocins and bacteriocins larger than 10 kD. It employs BLAST (Altschul et al., 1990) to search for motifs in a database of bacteriocins. The server allows the user to submit multiple entries at once and has been updated recently as BAGEL3 (van Heel et al., 2013). To assess this webserver, we queried the database by selecting the option ‘BLAST Bacteriocins’ in the main menu and setting ‘BLASTP maximum number of hits’ to 1. Because the server requires that the user specify a bacteriocin subtype at query time, we ran the queries twice, once using the ‘Bacteriocin Type IA’ and once using ‘Bacteriocin Type II.’ We took the minimum of the resulting two E-values as the prediction for each sequence.
BACTIBASE is a database that stores bacteriocins and allows users to predict bacteriocins using profile hidden Markov models (Eddy, 1998) of each bacteriocin family (de Jong et al. 2006; van Heel et al. 2013). In order to predict the Bacteriocins families, one is required to choose ‘Hidden Markov Models’ from the drop-down menu in the tool section. BACTIBASE allows searching using batch, and we submitted the query sequences by selecting ‘Classic’ as the classification scheme.
AntiBP is an antibacterial prediction tool that makes predictions using support vector machines (SVMs) (Vapnik, 1995), artificial neural networks (Bishop, 1995) or quantitative matrices (Lata et al., 2007). In these methods, the training of the model was based on three feature extraction approaches, focusing on the 15 N-terminal residues, 15 C-terminal residues and a combination of 15 N- and 15 C-terminal residues (Lata et al., 2007). The dataset was extracted from the APD2 database (Wang et al., 2009). The parameter settings chosen are as follows: Terminus = ‘NC Termini,’ SVM threshold = 0 and Sequence Format = ‘Amino Acid Sequence in Single letter code.’ We selected the SVM model for AntiBP, since the SVM coupled with NC termini features was declared by the authors to be the best-performing model.
AntiBP2 is an extension of AntiBP in which the SVM model was created using a larger set of antibacterial data than what was used in AntiBP. The parameters chosen are as follows: Sequence format is ‘Amino acid sequence in sequence letter code’, Terminus = ‘NC termini’ and Method = ‘SVM’ with threshold set to zero (Lata et al., 2010).
ADAM is a database of AMPs that contains 7007 sequences. The data is extracted from twelve databases (APD2 (Wang et al., 2009), AVpred (Thakur et al., 2012), BACTIBASE (Hammami et al., 2007), BAGEL3 (van Heel et al., 2013), CAMP (Thomas et al., 2010), PenBase (Gueguen et al., 2006), PhytAMP (Hammami et al., 2009), etc.). The server allow users to predict sequences using an SVM and hidden Markov model. The SVM model is trained on the AMP sequences in the database, using amino acid composition as features.
CAMPR3 is a database of experimentally validated AMPs. It was created because most existing database do not consist of experimentally verified AMPs. CAMPR3 includes AMP prediction tools based on random forests (Breiman, 2001), SVMs and discriminant analysis (Karatzoglou et al., 2004). The predictors take as input a sequence matrix converted into physicochemical properties and structural characteristics of amino acids, as well as dipeptide and tripeptide frequencies of the reduced alphabets. In this project, we choose CAMPR3(SVM) and CAMPR3(RF), since they were previously determined to be the best-performing AMP prediction methods (Waghu et al., 2016).
AMPA is an AMP prediction tool that employs an ‘antimicrobial propensity scale’ derived from high-throughput screening results from the AMP bactenicin 2A (Torrent et al., 2012). The parameter settings we used are as follows: Window size = ‘7’ and Threshold value = 0.225.
DBAASP is an AMP prediction tool that is based on a simple algorithm that evaluates the efficacies of the characteristics as descriptors. The tool offers no parameter settings on the submission page (Vishnepolesky and Pirtskhalava, 2014).
MLAMP is a two-level AMP prediction tool that first identifies whether a peptide is an AMP and then proceeds to classify it into a subcategory. The tool employs a synthetic minority over-sampling technique for use on imbalanced and multi-label datasets, referred to as the ML-SMOTE algorithm (Lin and Xu, 2016). Training data was extracted from the APD2 database (Wang et al., 2009). Like DBAASP, this tool offers no user-settable parameters.
Some of the AMP tools require that queries be submitted individually (Table 1). In these cases, we use the Python module ‘mechanize’ (https://pypi.python.org/pypi/mechanize) to submit multiple queries in series.
2.3 Performance measures
To compare the AMP prediction tools, we used two different types of performance measures: threshold-based and rank-based.
For the threshold-based measures, we used the thresholds provided by each tool. By comparing each predicted label with the true label we assigned each prediction to one of four classes: true positive (TP), false positive (FP), true negative (TN), or false negative (FN). We then considered four complementary performance measures: sensitivity (TP/(TP + FN)), specificity (TN/(TN + FP)), precision (TP/(TP + FP)) and balanced accuracy:
| (1) |
For the rank-based comparison, we used receiver operating characteristic (ROC) curves, which plot sensitivity as a function of (1 - specificity) for varying decision thresholds. For quantitative comparison of two ROC curves, we compute the area under the curve. The statistical significance of differences between two ROC areas was assessed using the DeLong test (DeLong et al., 1988), calculated using the pROC package in R (Robin et al., 2011).
3 Results
3.1 Threshold-based comparison
When we compare the thresholded predictions made by each of the ten methods on the two benchmarks (Tables 2 and 3), some clear trends are apparent. For the general AMP prediction task, the two CAMPR3 methods, based on random forests and SVM classifiers, outperform the competing ADAM, MLAMP, AMPA and DBAASP methods by all four metrics—sensitivity, specificity, precision and balanced accuracy. For the prediction of antibacterial peptides, we find that the ‘improved’ AntiBP2 does not actually outperform its predecessor, AntiBP, on the DAMPD benchmark. Indeed, the accuracy of AntiBP2 is much lower, barely above the 50% that would be expected from a random classifier. However, on the APD3 benchmark, which we analyzed several months after the DAMPD benchmark, AntiBP failed to return any results. Finally, for the prediction of bacteriocins, both BAGEL3 and BACTIBASE do very well, achieving specificities of 100% and balanced accuracies of 96.77% and 91.93%, respectively, on the DAMPD benchmark dataset. However, BAGEL3 consistently outperforms BACTIBASE on the APD3 benchmark dataset. For this task, the consistently high performance measures suggests that identification of this specific subclass of AMPs is a relatively easy task.
Table 2.
Summary of threshold-based results for the DAMPD benchmark dataset
| Tool | TP | FP | FN | TN | Total | Sens (%) | Spec (%) | Prec (%) | Bal Acc (%) |
|---|---|---|---|---|---|---|---|---|---|
| CAMPR3(RF) | 505 | 748 | 42 | 1987 | 3282 | 92.32 | 72.65 | 40.30 | 82.49 |
| CAMPR3(SVM) | 493 | 763 | 54 | 1972 | 3282 | 90.13 | 72.10 | 39.25 | 81.11 |
| ADAM | 460 | 851 | 87 | 1884 | 3282 | 84.09 | 68.88 | 35.09 | 76.49 |
| MLAMP | 348 | 485 | 199 | 2250 | 3282 | 63.62 | 82.27 | 41.78 | 72.94 |
| DBAASP | 121 | 195 | 426 | 2540 | 3282 | 22.12 | 92.87 | 38.28 | 57.49 |
| AMPA | 267 | 416 | 280 | 2319 | 3282 | 48.81 | 84.79 | 39.09 | 66.80 |
| AntiBP | 281 | 860 | 32 | 705 | 1878 | 89.78 | 45.05 | 24.63 | 67.41 |
| AntiBP2 | 272 | 1315 | 41 | 250 | 1878 | 86.90 | 15.97 | 17.14 | 51.44 |
| BAGEL3 | 29 | 0 | 2 | 155 | 186 | 93.55 | 100.0 | 100.0 | 96.77 |
| BACTIBASE | 26 | 0 | 5 | 155 | 186 | 83.87 | 100.0 | 100.0 | 91.93 |
Table 3.
Summary of threshold-based results (APD3 dataset)
| Tool | TP | FP | FN | TN | Total | Sens (%) | Spec (%) | Prec (%) | Bal Acc (%) |
|---|---|---|---|---|---|---|---|---|---|
| CAMPR3(RF) | 1624 | 1419 | 89 | 7146 | 10278 | 94.80 | 83.43 | 53.37 | 89.11 |
| CAMPR3(SVM) | 1552 | 1578 | 161 | 6987 | 10 278 | 90.60 | 81.58 | 49.58 | 86.09 |
| ADAM | 1560 | 3292 | 153 | 5273 | 10 278 | 91.07 | 61.56 | 32.15 | 76.32 |
| MLAMP | 1295 | 1901 | 418 | 6664 | 10 278 | 75.59 | 77.81 | 40.52 | 76.70 |
| DBAASP | 1076 | 715 | 637 | 7850 | 10 278 | 62.81 | 91.65 | 60.08 | 77.23 |
| AMPA | 671 | 862 | 1042 | 7703 | 10 278 | 39.17 | 89.94 | 43.77 | 64.55 |
| AntiBP2 | 963 | 5350 | 483 | 1880 | 8676 | 66.59 | 26.00 | 15.25 | 46.30 |
| BAGEL3 | 133 | 0 | 21 | 770 | 924 | 86.36 | 100.0 | 100.0 | 93.18 |
| BACTIBASE | 60 | 0 | 94 | 770 | 924 | 38.96 | 100.0 | 100.0 | 69.48 |
It is instructive to compare the results of this analysis to similar values reported in previous studies (Table 4). Although the values are not directly comparable because different benchmark data sets were employed in each case, the general trend is clear: the values reported by the authors are almost always higher than the values reported here, often by a fairly large amount. The only case where the two values are similar is for BAGEL3 (91.5 versus 91.93%), where the task is apparently quite easy.
Table 4.
Comparison of performance in the original publication with this study
| Original benchmark |
Current study |
||||||
|---|---|---|---|---|---|---|---|
| Tool | Accuracy (%) | Sensitivity (%) | Specificity (%) | Bal. accuracy (%) | Sensitivity (%) | Specificity (%) | |
| CAMPR3(RF) | 99.00 | — | — | 82.49 (89.11) | 92.32 (94.80) | 72.65 (83.43) | |
| CAMPR3(SVM) | 91.50 | — | — | 81.11 (86.09) | 90.13 (90.60) | 72.10 (81.58) | |
| ADAM | — | — | — | 76.49 (76.32) | 84.09 (91.07) | 68.88 (61.56) | |
| MLAMP | 94.70 | 97.30 | 92.1 | 72.94 (76.70) | 63.62 (75.59) | 82.27 (77.81) | |
| DBAASP | 90.20 | 84.03 | 93.00 | 57.49 (77.23) | 22.12 (62.81) | 92.87 (91.65) | |
| AMPA | 80.00 | — | — | 66.80 (64.55) | 48.81 (39.17) | 84.79 (89.94) | |
| AntiBP | — | 92.11 | — | 67.41 (N/A) | 89.78 (N/A) | 45.05 (N/A) | |
| AntiBP2 | 91.64 | 92.22 | — | 51.44 (46.30) | 86.90 (66.59) | 15.97 (26.00) | |
| BAGEL3 | — | — | — | 96.77 (93.18) | 93.55 (86.36) | 100.00 (100.00) | |
| BACTIBASE | 91.50 | — | — | 91.93 (69.48) | 83.87 (38.96) | 100.00 (100.00) | |
Note: Results from the current study are reported as value 1 (value 2), where value 1 is relative to the DAMPD benchmark, and value 2 is relative to the APD3 benchmark. AntiBP did not return results for the APD3 benchmark.
3.2 Rank-based comparison
A significant drawback to a threshold-based comparison is that there is no way to assign a statistical confidence estimate to the observed difference between two methods based on a single measurement. To address this problem, we turn instead to a rank-based assessment, using ROC curves (Fig. 2). Qualitatively, the results of the ROC analysis mirror those of the threshold-based analysis. However, with ROC analysis we can use DeLong’s test to assign a P value to the observed differences (Tables 5 and 6). This analysis allows us to confirm that the best-performing AMP predictor is CAMPR3(RF), followed by CAMPR3(SVM) and then ADAM. We also confirm that AntiBP performs significantly better than AntiBP2 on the DAMPD benchmark, and BAGEL3 significantly outperforms BACTIBASE only on the APD3 benchmark.
Fig. 2.

Receiver operating characteristic curves for the 10 methods, separated by classification task. Panels on the left are for the DAMPD3 benchmark, and on the right are for APD3. For reference, each plot includes the line y = x, which corresponds to the performance of a random classifier. Each series is marked with a single point, indicating the location of the decision threshold selected by the method. In the key, the numeric values next to the name of each method are the corresponding ROC areas (Color version of this figure is available at Bioinformatics online.)
Table 5.
Comparison of ROC AUC using DeLong’s test (DAMPD dataset)
| Task | Method 1 | Method 2 | AUC 1 | AUC 2 | Difference | P value |
|---|---|---|---|---|---|---|
| Antimicrobial | CAMPR3(RF) | ADAM | 0.883 | 0.839 | 0.044 | 7.46e−5 |
| Antimicrobial | CAMPR3(RF) | CAMPR3(SVM) | 0.883 | 0.813 | 0.070 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | MLAMP | 0.883 | 0.780 | 0.103 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | DBAASP | 0.883 | 0.575 | 0.308 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | AMPA | 0.883 | 0.654 | 0.229 | 2.2e−16 |
| Antibacterial | AntiBP | AntiBP2 | 0.797 | 0.544 | 0.253 | 2.2e−16 |
| Bacteriocin | BACTIBASE | BAGEL3 | 0.984 | 0.952 | 0.032 | 0.375 |
Table 6.
Comparison of ROC AUC using DeLong’s test (APD3 dataset)
| Task | Method 1 | Method 2 | AUC 1 | AUC 2 | Difference | P value |
|---|---|---|---|---|---|---|
| Antimicrobial | CAMPR3(RF) | ADAM | 0.953 | 0.877 | 0.076 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | CAMPR3(SVM) | 0.953 | 0.919 | 0.034 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | MLAMP | 0.953 | 0.839 | 0.114 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | DBAASP | 0.953 | 0.766 | 0.187 | 2.2e−16 |
| Antimicrobial | CAMPR3(RF) | AMPA | 0.953 | 0.639 | 0.314 | 2.2e−16 |
| Antibacterial | AntiBP | AntiBP2 | — | 0.465 | — | NA |
| Bacteriocin | BAGEL3 | BACTIBASE | 0.929 | 0.827 | 0.102 | 2.2e−16 |
Qualitative assessment of the shape of an ROC curve can also provide insight into how a classifier is behaving. For example, the DBAASP curve consists of two straight line segments, because this classifer provides only a binary score. Also, the curve for CAMPR3(SVM) has a strange shape in the lower left portion of the plot. Investigation of the sequences in this area shows that they correspond to the longer sequences in the benchmark. All such sequences are assigned very high scores (close to 1.0) by CAMPR3(SVM), regardless of whether they are AMPs or not. To better understand this phenomenon, we plotted, for each classifier, the predicted score of a given AMP as a function of the sequence length. The resulting plots (Fig. 3 and Supplementary Fig. S1) are quite striking. We see, for example, that CAMPR3(SVM) is largely indifferent to sequence length until the query is 100 amino acids long, at which point the sequences are all assigned scores very close to 1.0. CAMPR3(RF) shows a similar, though not quite as pronounced, trend. Some of the other predictors, including ADAM, MLAMP and BAGEL3, show length dependencies.
Fig. 3.

Dependence of scores on sequence length (DAMPD dataset). In each panel, a point corresponds to an AMP or non-AMP peptide from the AMP (top row), antibacterial (middle row) or bacteriocin (bottom row) data set. The figures plot the score assigned to a peptide by a given prediction method as a function of the peptide length. Note that the BACTIBASE and BAGEL3 scores have been transformed (Color version of this figure is available at Bioinformatics online.)
This length dependency is almost certainly a result of the choice of data sets used to train these classifiers. For example, the CAMPR3 methods were trained using positive sequences with lengths 10–80 amino acids, and the non-AMP sequences were truncated to be in the same range (Thomas et al., 2010).
This observation naturally leads to the question of whether longer query peptides should be included in the DAMPD benchmark data set. In practice, some longer peptides do exhibit antimicrobial function. For example, the bombinin peptide is 137 amino acids long, consisting of a signal peptide, a propeptide that is cleaved during maturation or activation and the primary amino acid chain (http://www.uniprot.org/uniprot/P29006). Another example is the apidaecins, which are ∼280 amino acids long and consist of multiple AMP domains (http://www.uniprot.org/uniprot/Q06602). In the context of searching for AMPs, antibacterial peptides or bacteriocins, the ability to consider longer peptides will clearly be beneficial. In contrast to APD3, we have employed mostly mature peptides in order to compare the results to DAMPD benchmark dataset.
Nonetheless, to understand how the inclusion of longer peptides affected our results, we re-did the ROC analysis for the DAMPD benchmark, eliminating from our benchmark all peptides longer than 100 amino acids. The results (Fig. 4) confirm that in this setting CAMPR3(SVM) performs much more similarly to CAMPR3(RF).
Fig. 4.

Receiver operating characteristic for the AMP prediction task, with queries limited to 100 amino acids in length (DAMPD and APD3 datasets) (Color version of this figure is available at Bioinformatics online.)
The remaining mystery is why the second generation AntiBP2 performs so much worse than its predecessor, AntiBP. A comparison of the two scores on AMP and non-AMP examples from our benchmark shows a trend consistent with the previous analyses: AntiBP generally succeeds in separating AMPs from non-AMPs, whereas AntiBP2 does not (Fig. 5). Most striking is the very low correlation (Pearson r = 0.293) between the two sets of predictions. Based on this analysis, we can only conclude that AntiBP2 is not working properly at this time. On the other hand, our inability to obtain results from AntiBP on the APD3 benchmark implies that neither method is currently useful.
Fig. 5.
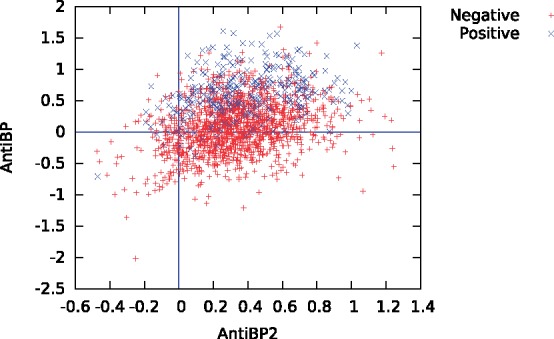
Scatter plot for prediction scores from AntiBP versus AntiBP2 (DAMPD dataset)
4 Discussion
This study aims to compare empirically ten AMP prediction tools that are freely accessible as web portals. We find that, among general predictors, the CAMPR3(RF) tool provides the best performance. This may be due to the large dataset used in training the CAMPR3(RF) model. For prediction of antibacterial proteins, AntiBP performs much better than its successor, AntiBP2, but the AntiBP web server seems recently to have stopped functioning correctly. For bacteriocins, both BAGEL3 and BACTIBASE work very well. BAGEL3 has recently updated its database with new bacteriocin data. Accordingly, BAGEL3 significantly outperforms BACTIBASE on the APD3 dataset, though this difference is not statistically significant for the smaller DAMPD dataset. Generally, our rankings are consistent with the findings by Porto et al. (2012), who used author-reported results to rank CAMPR3(RF) and AntiBP as the best AMP and antibacterial predictors, respectively.
In addition to these overall trends, we noted that the predictions produced by many tools exhibit a strong length dependence, which has the potential to confound some types of analysis. We also note that, among the ten prediction tools that we have compared in this study, only four—BAGEL3, CAMPR3(RF), CAMPR3(SVM) and ADAM—can be used to make predictions across complete proteomes, since these tools allow batch queries. However, AntiBP and AntiBP2 are not meant for genomic predictions.
Many possible improvements to these tools may be explored in the future. For example, all of the tools, except BAGEL3 and BACTIBASE, generate features for the classifier using either amino acid composition, dipeptide composition, or physicochemical properties of the amino acids. Such features necessarily fail to represent motif-like features that may be important for AMP function. Furthermore, physicochemical features are based on averages and hence suffer from degeneracy (Andreu and Torrent, 2015). This effect may be ameliorated by using complex prime numerical representation of amino acids (Chen et al., 2016). Another direction for improvement lies in feature selection procedures. The CAMPR3 models employ 257 hand-picked features. A more rigorous feature selection procedure could potentially yield improved performance. Conversely, ADAM is trained on 7007 AMP sequences using only amino acid composition as features. Expanding on this feature space may be beneficial.
In this study, our rank-based analysis relies upon ROC analysis. A key characteristic of the ROC curve is that it normalizes away any imbalance between the sizes of the two classes. A complementary, rank-based procedure employs precision-recall curves, which retain information about this class imbalance (Davis and Goadrich, 2006). While a precision-recall analysis would clearly be of interest in the current setting, carrying out such an analysis would require an estimate of the ‘true’ ratio of class sizes (i.e. the number of AMP versus non-AMP sequences). This ratio varies depending upon the application setting. For this reason, and because of the availability of well-developed statistical methods for comparing ROC curves, we have chosen to employ ROC analysis in the current study.
One caveat to our current study is the potential for overlap between our benchmark data set and the sequences used to train the various methods in the study. For a truly fair comparison, we would need to ensure that no such overlap occurs. Unfortunately, in practice, such a constraint is impossible to enforce. As more AMP sequences are identified and validated, a more unbiased benchmark will be possible, though of course those newly identified AMPs will also likely be incorporated into the training sets of future releases of these tools.
A second issue that we have not addressed is the calibration of the prediction scores. An ROC curve measures the quality of a ranking but does not address whether the score itself is interpretable. An ideal score will have a probabilistic interpretation. For example, MLAMP, AMPA and the two CAMPR3 methods provide predictions as the posterior probability of a given class label, and BACTIBASE and BAGEL3 report results as E values. AntiBP and AntiBP2 in contrast, output unitless discriminant scores and DBAASP only provides a binary prediction. Clearly, an important question for future work is the extent to which the scores produced by AMP prediction methods are interpretable; i.e. if we consider all the predictions to which CAMPR3(RF) assigns a posterior probability of 80%, are 80% of these predictions actually correct?
Porto et al. emphasized the need for non-AMP sets with which to test the specificity of AMP prediction models (Porto et al., 2012). Our benchmark dataset, which includes both AMP and non-AMP sequences, addresses this need and is available as Supplementary Tables S1 and S2.
Funding
This work has been supported by King Abdullah International Medical Research Center grant #RC16/089 and by National Institutes of Health award P41 GM103533.
Conflict of Interest: none declared.
Supplementary Material
References
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andreu D., Torrent M. (2015) Prediction of Bioactive Peptides Using Artificial Neural Networks, pp. 101–118. Springer, New York. [DOI] [PubMed] [Google Scholar]
- Arthur T.D. et al. (2014) On bacteriocin delivery systems and potential applications. Future Microbiology, 9, 235–248. [DOI] [PubMed] [Google Scholar]
- Bishop C. (1995) Neural Networks for Pattern Recognition. Oxford UP, Oxford, UK. [Google Scholar]
- Breiman L. (2001) Random forest. Machine Learning, 45, 5–32. [Google Scholar]
- Chen D. et al. (2016) A complex prime numerical representation of amino acids for protein function comparison. Comput. Biol., 23, 669–677. [DOI] [PubMed] [Google Scholar]
- Chen Y. et al. (2012) Anti-HIV-1 activity of a new scorpion venom peptide derivative Kn2-7. PLoS One, 7, e34947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon J.M. et al. (2014) Potential therapeutic applications of multifunctional host-defense peptides from frog skin as anti-cancer, anti-viral, immunomodulatory, and anti-diabetic agents. Peptides, 57, 67–77. [DOI] [PubMed] [Google Scholar]
- Davis J., Goadrich M. (2006). The relationship between precision-recall and ROC curves. In Proceedings of the International Conference on Machine Learning.
- de Jong A. et al. (2006) BAGEL: a web-based bacteriocin genome mining tool. Nucleic Acids Res., 34 (Web Server issue), W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong E.R. et al. (1988) Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics, 44, 837–845. [PubMed] [Google Scholar]
- Drissi F. et al. (2015) Common occurrence of antibacterial agents in human intestinal microbiota. Front. Microbiol., 6, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziuba B., Dziuba M. (2014) New milk protein-derived peptides with potential antimicrobial activity: An approach based on bioinformatic studies. Int. J. Mol. Sci., 15, 14531.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. (1998) Profile hidden markov models. Bioinformatics, 14, 755–763. [DOI] [PubMed] [Google Scholar]
- Fernandes F.C. et al. (2012) Prediction of antimicrobial peptides based on the adaptive neuro-fuzzy inference system application. Biopolymers, 98, 280–287. [DOI] [PubMed] [Google Scholar]
- Fjell C.D. et al. (2007) AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics, 23, 1148–1155. [DOI] [PubMed] [Google Scholar]
- Gaspar D. et al. (2013) From antimicrobial to anticancer peptides. A review. Front. Microbiol., 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen Y. et al. (2006) PenBase, the shrimp antimicrobial peptide penaeidin database: sequence-based classification and recommended nomenclature. Dev. Comput. Immunol., 30, 283–288. [DOI] [PubMed] [Google Scholar]
- Hammami R. et al. (2007) BACTIBASE: a new web-accessible database for bacteriocin characterization. BMC Microbiol., 7, 89.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammami R. et al. (2009) PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res., 2009, (Database issue), D963–D968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskin D.W., Ramamoorthy A. (2008) Studies on anticancer activities of antimicrobial peptides. Biochimica Et Biophysica Acta (BBA) Biomembranes, 1778, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S. et al. (2012) ClassAMP: a prediction tool for classification of antimicrobial peptides. IEEE/ACM Trans. Comput. Biol. Bioinform., 9, 1535–1538. [DOI] [PubMed] [Google Scholar]
- Kapoor R. et al. (2011) Efficacy of antimicrobial peptoids against Mycobacterium tuberculosis. Antimicrobial Agents Chemother., 55, 3058–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatzoglou A. et al. (2004). kernlab—an S4 package for kernel methods in R.
- Kückelhaus S.A.S. et al. (2009) Antiplasmodial and antileishmanial activities of phylloseptin-1, an antimicrobial peptide from the skin secretion of Phyllomedusa azurea (Amphibia). Exp. Parasitol., 123, 11–16. [DOI] [PubMed] [Google Scholar]
- Lata S. et al. (2007) Analysis and prediction of antibacterial peptides. BMC Bioinformatics, 8, 263.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S. et al. (2010) AntiBP2: improved version of antibacterial peptide prediction. BMC Bioinformatics, 11, S19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A. (2006) CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li W., Godzik A. (2015) A large-scale structural classification of antimicrobial peptides. BioMed Res. Int. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Xu D. (2016) Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics, 32, 130–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng X.Y. et al. (2015) Prediction of antimicrobial peptides based on sequence alignment and support vector machine-pairwise algorithm utilizing LZ-complexity. Biomed Res. Int., 2015, 212715.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas G.G. (2011) Detection of putative new mutacins by bioinformatic analysis using available web tools. BioData Mining, 4, 22–22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto E.G. et al. (2014) Antileishmanial and antitrypanosomal activity of the cutaneous secretion of Siphonops annulatus. J. Venomous Animals Toxins Tropical Dis., 20, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto W. et al. (2012) Protein Structure, volume 1, chapter Prediction and rational design of antimicrobial peptides, pp. 101–118. InTech.
- Ramón-García S. et al. (2013) Targeting Mycobacterium tuberculosis and other microbial pathogens using improved synthetic antibacterial peptides. Antimicrobial Agents Chemother., 57, 2295–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X. et al. (2011) pROC: an open-source package for R and S+ to analyze and compare roc curves. BMC Bioinformatics, 12, 8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S.V. et al. (2012) DAMPD: a manually curated antimicrobial peptide database. Nucleic Acids Res., 40, D1108–D1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto Y., Tamura K. (2013) Extensive differences in antifungal immune response in two Drosophila species revealed by comparative transcriptome analysis. Int. J. Genomics, 2013, 15.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P.K. et al. (2014) A non-pediocin low molecular weight antimicrobial peptide produced by Pediococcus pentosaceus strain IE-3 shows increased activity under reducing environment. BMC Microbiol., 14, 226.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur N. et al. (2012) AVPpred: collection and prediction of highly effective antiviral peptides. Nucleic Acids Res., 40, (Web Server issue), W199–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. et al. (2010) CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res., 38, (Database issue), D774–D780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tincho M.B. et al. (2016) In silico identification and molecular validation of putative antimicrobial peptides for hiv therapy. J. AIDS Clin. Res., 7, 1–11. [Google Scholar]
- Torrent M. et al. (2012) AMPA: an automated web server for prediction of protein antimicrobial regions. Bioinformatics, 28, 130–131. [DOI] [PubMed] [Google Scholar]
- The Uniprot Consortium. (2015) UniProt: a hub for protein information. Nucleic Acids Res., 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale N. et al. (2014) Antimicrobial peptides: a new class of antimalarial drugs?. Front. Pharmacol., 5, 275.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel A.J. et al. (2013) BAGEL3: automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res., 41, W448–W453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vapnik V. (1995) The Nature of Statistical Learning Theory. Springer Science & Business Media. [Google Scholar]
- Vishnepolesky B., Pirtskhalava M. (2014) Prediction of linear cationic antimicrobial peptides based on characteristics responsible for their interaction with the membranes. J. Chem. Inform. Model., 54, 1512–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghu F.H. et al. (2016) CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res., 44, D1094–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. (2014) Human antimicrobial peptides and proteins. Pharmaceuticals, 7, 545–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. et al. (2009) APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res., 37(Suppl. 1), D933–D937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. et al. (2016) APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res., 44, D1087–D1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. (2011) Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS One, 6, e18476.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziqing J. et al. (2011) Anti-tuberculosis activity of alpha-helical antimicrobial peptides: De novo designed L- and D-enantiomers versus L- and D-LL37. Protein and Peptide. Letters, 18, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.