

ABSTRACT
Our previous study demonstrated that neutrophils play an important role in host defense against Coxiella burnetii infection in mice. In this study, avirulent strain C. burnetii Nine Mile phase II (NMII) was used to examine if C. burnetii can modulate mouse bone marrow-derived neutrophil apoptosis. The results indicated that NMII can inhibit neutrophil apoptosis. Western blotting demonstrated that caspase-3 cleavage was decreased in NMII-infected neutrophils, while phosphorylated mitogen-activated protein kinase (MAPK) p38 and extracellular signal-regulated kinase 1 (Erk1) were increased. Additionally, p38, Erk1/2, phosphoinositide 3-kinase (PI3K), or NF-κB inhibitors reduced the ability of NMII to inhibit neutrophil apoptosis. These results suggest that NMII-mediated inhibition of neutrophil apoptosis depends on its ability to activate neutrophil MAPK pathways. Antiapoptotic protein myeloid cell leukemia-1 (Mcl-1) was significantly increased in NMII-infected neutrophils, and an Mcl-1 inhibitor significantly reduced the ability of NMII to inhibit neutrophil apoptosis. Mcl-1 protein stability was enhanced by phosphorylation at Thr-163 by Erk, and the protein levels were regulated by p38, Erk, PI3K, and NF-κB. Furthermore, the observation that a type IV secretion system (T4SS)-deficient dotA mutant showed a significantly reduced ability to inhibit neutrophil apoptosis compared to wild-type (WT) NMII suggests that T4SS-secreted factors may be involved in NMII-induced inhibition of neutrophil apoptosis. Collectively, these results demonstrate that NMII inhibits neutrophil apoptosis through inhibition of caspase-3 cleavage and activation of MAPK survival pathways with subsequent expression and stabilization of antiapoptotic protein Mcl-1, a process that may partially require the T4SS.
KEYWORDS: Coxiella burnetii, Mcl-1, NF-κB, PI3K, apoptosis, caspase-3, neutrophils
INTRODUCTION
Coxiella burnetii is an obligate intracellular Gram-negative bacterium that causes the zoonotic disease Q fever in humans. Q fever commonly manifests as an acute flu-like illness characterized by high fever with chills and atypical pneumonia, although infection may persist to a chronic disease that can be fatal if left untreated (1). A recent outbreak in the Netherlands from 2007 to 2011 resulted in more than 3,500 reported clinical Q fever cases (2), highlighting the significant threat that this globally distributed pathogen poses to public health. Antibiotic treatment for acute Q fever is most effective when initiated within the first 3 days of illness; however, the accurate early diagnosis of Q fever is difficult and often overlooked due to nondescript flu-like symptoms. Chronic Q fever is much more difficult to treat effectively, often requiring treatment with multiple antibiotics for several years (3). Therefore, it is necessary to discover novel drugs and alternative strategies to control C. burnetii infections.
C. burnetii undergoes lipopolysaccharide (LPS) phase variation, whereby virulent phase I organisms, such as the Nine Mile phase I (NMI) strain (with smooth LPS), convert to phase II organisms, such as the avirulent Nine Mile phase II (NMII) strain (with rough LPS), upon serial passage in eggs, tissue culture, or synthetic media (4, 5). NMI is able to replicate in immunocompetent animals and cause disease, while NMII is rapidly cleared in immunocompetent animals and does not cause disease (6). Understanding the mechanisms of host cell-pathogen interactions may reveal important insights that lead to the development of novel therapeutic strategies for treatment of C. burnetii infections. Neutrophils are major effector cells of the innate immune system with a primary role in resistance against extracellular pathogens. However, accumulating evidence suggests that neutrophils also play a protective role in host defense against intracellular microbial pathogens (7). The observation that the selective depletion of neutrophils results in the delayed clearance of bacterial infections caused by Legionella pneumophila and Klebsiella pneumoniae (8–10) indicates that neutrophils play an important role in host defense against intracellular bacterial pathogens. Interestingly, our previous study demonstrated that C. burnetii intranasal infection induced more severe disease in neutrophil-depleted mice, suggesting that neutrophils play an important role in host defense against C. burnetii pulmonary infection (11). However, the mechanisms of interaction between neutrophils and C. burnetii are not well understood.
Neutrophils have a very short life span because they are preprogrammed to undergo apoptosis 18 to 24 h after being released into the circulation (12). However, it has been shown that neutrophil apoptosis can be modulated by many microbial pathogens (13). Induction of neutrophil apoptosis following phagocytosis of bacteria is a well-known phenomenon first described for Escherichia coli (14) but subsequently reported for many other bacteria (15, 16). In contrast, some intracellular pathogens, such as Anaplasma phagocytophilum (17), Leishmania major (18), and Chlamydia pneumoniae (19), have been reported to inhibit neutrophil apoptosis. C. burnetii is capable of differentially modulating host cell apoptosis, depending on the conditions. One study indicated that both the virulent strain C. burnetii NMI and the avirulent strain C. burnetii NMII can partially prevent exogenously induced apoptosis in differentiated THP-1 cells and primary monkey alveolar macrophages (20). Similarly, a recent report also demonstrated the ability of NMII to inhibit exogenously induced apoptosis in Chinese hamster ovary and HeLa cells in late stages of infection (21). These observations suggest that C. burnetii-infected cells are able to resist apoptosis in the presence of exogenously applied apoptotic stimuli, which may be important for C. burnetii to establish a persistent infection in vitro. In contrast, our recent studies found that NMII can induce apoptosis in undifferentiated THP-1 cells through a caspase-3-independent pathway (22). We also found that NMII-infected murine peritoneal B1a cells undergo caspase-1-dependent pyroptosis through activation of Toll-like receptor 2 and NLRP3 signaling pathways (23). However, it remains unclear whether C. burnetii can modulate neutrophil apoptosis.
In this study, we examined if C. burnetii NMII can modulate mouse bone marrow-derived neutrophil apoptotic signaling pathways. Our results demonstrated that NMII infection inhibits caspase-3 cleavage, activates mitogen-activated protein kinase (MAPK) signaling pathways, and induces expression of antiapoptotic protein myeloid cell leukemia-1 (Mcl-1), thus delaying neutrophil apoptosis.
RESULTS
C. burnetii NMII infects primary mouse BMNs.
Our previous study demonstrated that C. burnetii NMII is able to infect mouse bone marrow neutrophils (BMNs) (24). In this study, an immunofluorescence assay (IFA) was used to determine the percentage of NMII-infected BMNs at different time points. As shown in Fig. 1A, C. burnetii (green) was observed only in NMII-infected BMNs. Colocalization of C. burnetii with lysosome-associated membrane protein 1 (LAMP-1) was observed in Ly-6G-positive BMNs (Fig. 1B), confirming that the bacteria were inside neutrophils. Figure 1C shows the percentage of NMII-infected neutrophils. Approximately 13% of BMNs were infected with NMII after 4 h, while the percentage of infected BMNs increased to 44% after 18 h. The percentage of NMII-infected neutrophils was not significantly different at between 18 and 24 h postinfection (hpi) (data not shown). Thus, the percentage of NMII-infected neutrophils is comparable to that in our previous study (24).
FIG 1.

NMII infects primary mouse BMNs. BMNs were infected with NMII at an MOI of 100 and stained by IFA using antibodies against C. burnetii (C.b; green), Ly-6G PE (red), and LAMP-1–Alexa Fluor 647 (AF; purple) at 18 h postinfection (hpi). DAPI was used to stain host cell DNA (blue). (A) NMII colocalized with host cell nuclear DNA. The circled portion is expanded. (B) NMII colocalized with a marker of host cell lysosomal membranes, LAMP-1, inside Ly-6G-positive neutrophils. (C) The infection rate was determined at 4 and 18 hpi by counting the number of cells positively labeled with C. burnetii antibodies. A total of 200 cells were counted per coverslip in three separate experiments for each time point. Results are expressed as the percentage of infected cells compared to the percentage of uninfected cells. Error bars represent the standard deviation from the mean. **, P < 0.01.
NMII infection inhibits DNA fragmentation in BMNs.
DNA fragmentation is a major biochemical feature of apoptosis. To determine if NMII can modulate neutrophil apoptosis, terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was used to examine whether NMII infection altered DNA fragmentation in neutrophils. As shown in Fig. 2A, fewer TUNEL-positive cells were observed in NMII-infected neutrophils than in uninfected neutrophils at 18 hpi. NMII-infected neutrophils possessed significantly fewer TUNEL-positive cells at both 4 and 18 hpi (Fig. 2B). These results suggest that NMII can inhibit neutrophil apoptosis.
FIG 2.

NMII inhibits DNA fragmentation in primary mouse BMNs. BMNs were infected with NMII at an MOI of 100 for 18 h. (A) Uninfected and infected cells were stained with TUNEL (red) to detect DNA fragmentation. Cells were also labeled by IFA using antibodies directed against C. burnetii (green). DAPI was used to stain DNA (blue). Uninfected neutrophils exhibited significant DNA fragmentation, while there were no detectable TUNEL-positive cells during NMII infection. (B) The percentage of apoptotic cells was determined at 4 and 18 hpi by counting the number of TUNEL-positive cells. A total of 200 cells per coverslip were counted in three separate experiments for each time point. Results are expressed as percent apoptosis, and error bars represent the standard deviation from the mean. *, P < 0.05; **, P < 0.01.
NMII infection inhibits neutrophil apoptosis in a dose- and time-dependent manner via inhibition of caspase-3 activity.
To further characterize the ability of NMII to inhibit neutrophil apoptosis, we first examined if the infection dose affected the ability of NMII to inhibit neutrophil apoptosis. IFA was used to determine the percentage of C. burnetii-infected BMNs at 24 hpi at a multiplicity of infection (MOI) of 25, 50, 100, 200, or 500. As shown in Fig. 3A, the infection rate was increased in a dose-dependent manner up to an MOI of 100, at which point the dose-response was saturated. Similarly, NMII bacteria could inhibit neutrophil apoptosis in a dose-dependent manner up to an MOI of 100, at which point maximum inhibition was reached (Fig. 3B). These results indicate that the ability of NMII to inhibit neutrophil apoptosis correlates with the infection rate. Next, a quantitative 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was used to compare the viability of uninfected and NMII-infected neutrophils. To determine the time frame for NMII-induced inhibition of neutrophil apoptosis, neutrophils were infected with NMII at an MOI of 100 and the infection rate and apoptosis were examined by IFA and MTS assay at 24, 48, 72, and 96 hpi. As shown in Fig. 3C, there were no significant changes in the infection rate at 24, 48, 72, and 96 hpi. Compared to the level of apoptosis seen in uninfected neutrophils, NMII infection significantly inhibited neutrophil apoptosis beginning at 24 h and continuing through 96 hpi (Fig. 3D). These results further demonstrate the ability of NMII to inhibit neutrophil apoptosis in a dose-dependent manner for a prolonged period. Heat-killed NMII was unable to inhibit neutrophil apoptosis (data now shown), suggesting that viable bacteria are required for apoptosis inhibition.
FIG 3.

NMII inhibits BMN apoptosis in a dose- and time-dependent manner via inhibition of caspase-3 activity. BMNs were infected with NMII at different MOIs and incubated for different time points after infection. (A) At 24 hpi, the percentage of BMNs infected with C. burnetii NMII at an MOI of 25, 50, 100, 200, or 500 was determined by IFA in the same manner described in the legend to Fig. 1B. The results are expressed as the percentage of infected cells compared to the percentage of uninfected cells. A maximum infection rate of ∼40 to 50% was reached at an MOI of 100 and was not increased at doses beyond this dose. (B) The percent cell death of uninfected BMNs and BMNs infected with NMII at an MOI of 25, 50, 100, 200, or 500 was measured by the MTS assay at 24 hpi. NMII could inhibit BMN apoptosis in a dose-dependent manner up to an MOI of 100, where maximum inhibition was reached. (C) The percentage of BMNs infected with C. burnetii NMII at an MOI of 100 was determined by IFA at 24, 48, 72, and 96 hpi. A maximum infection rate of ∼50% occurred as early as 24 h. (D) The percent cell death of uninfected BMNs or BMNs infected with NMII at an MOI of 100 was measured by the MTS assay at 24, 48, 72, and 96 hpi. NMII could significantly inhibit neutrophil apoptosis beginning at 24 hpi and continuing through 96 hpi. (E) The levels of cleaved caspase-3 (Casp-3; 17- and 19-kDa bands) between uninfected BMNs and BMNs infected with NMII at an MOI of 100 were compared by Western blotting at 8, 18, 38, and 48 hpi. (F) The fold reduction of the intensity units of cleaved caspase-3 protein bands at different times postinfection was calculated using densitometry software and normalized to the intensity for the loading controls. Error bars represent the standard deviation from the mean. *, P < 0.05; ***, P < 0.001. 0hr, 0-h time point, fresh uninfected BMNs; CU, uninfected control BMNs incubated for the indicated time period; +, NMII-infected BMNs; Actin, protein loading control.
It has been shown that activation of caspase-3 is critical for apoptosis in neutrophils (25). To determine if NMII infection affected the activity of caspase-3 in neutrophils, the levels of cleaved caspase-3 in uninfected and NMII-infected neutrophils were compared by Western blotting. As shown in Fig. 3E, the strengths of the cleaved caspase-3 protein bands were similar between uninfected and NMII-infected neutrophils at 8 hpi, but the cleaved caspase-3 protein bands in NMII-infected neutrophils were weaker than those in uninfected neutrophils at 18, 38, and 48 hpi. In addition, the intensity units of the cleaved caspase-3 protein bands were significantly decreased in NMII-infected neutrophils compared to those in uninfected neutrophils at 38 hpi (49% for the 17-kDa band, 74% for the 19-kDa band) and 48 hpi (51% for the 17-kDa band, 82% for the 19-kDa band) (Fig. 3F). These results indicate that NMII infection can significantly reduce the levels of cleaved caspase-3, suggesting that NMII-induced inhibition of neutrophil apoptosis may depend on its ability to reduce the activity of caspase-3.
NMII infection in neutrophils leads to activation of MAPK p38 and Erk1.
MAPK signaling pathways promote cell survival by one of two mechanisms: posttranslational modification and inactivation of proapoptotic proteins, such as Bad/Bim, or increased transcription/translation of prosurvival proteins, such as Bcl-XL/Mcl-1. Neutrophil apoptosis has been demonstrated to be tightly regulated by a complex network of signaling pathways through controlled expression and degradation of key signaling molecules, including Bcl-2 family proteins, MAPKs, NF-κB, and caspases (26–29). To determine whether NMII inhibition of neutrophil apoptosis was dependent on its ability to promote cell survival pathways, we next evaluated the phosphorylation of p38 and extracellular signal-regulated kinase 1/2 (Erk1/2) MAPKs by Western blotting. As shown in Fig. 4A and B, larger amounts of phosphorylated p38 and Erk1 MAPKs were detected in NMII-infected neutrophils than in the uninfected controls. The intensity units of the phosphorylated p38 (Fig. 4C) and Erk1 (Fig. 4D) protein bands in NMII-infected neutrophils were significantly higher than those in uninfected neutrophils. These results indicate that NMII bacteria activate p38 and Erk1 MAPKs during infection of neutrophils, suggesting that NMII inhibition of neutrophil apoptosis may depend on the promotion of cell survival pathways. To confirm that activation of the p38 and Erk1 MAPKs promotes neutrophil survival, we examined by the MTS assay if treatment of NMII-infected neutrophils with p38 or Erk1/2 inhibitors would affect the ability of NMII to inhibit neutrophil apoptosis. The results indicated that both p38 (Fig. 4E) and Erk1/2 (Fig. 4F) inhibitors significantly reduced the ability of NMII to inhibit neutrophil apoptosis in a dose-dependent manner. Additionally, the Erk1/2 inhibitor (PD98059) appeared to be more effective than the p38 inhibitor (SB203580). Collectively, these results support the hypothesis that NMII inhibition of neutrophil apoptosis depends on its ability to promote cell survival pathways via activation of p38 and Erk1 MAPKs.
FIG 4.

NMII activates MAPK p38 and Erk1 for inhibition of neutrophil apoptosis. (A) Analysis of phosphorylated p38 in BMNs infected with NMII at an MOI of 100 by Western blotting at 8, 18, and 38 hpi. (B) Analysis of phosphorylated Erk1/2 in BMNs infected with NMII at an MOI of 100 by Western blotting at 8, 18, 38, and 48 hpi. (C) Fold increase in the number of intensity units of the phosphorylated p38 protein band. (D) Fold increase in the number of intensity units of the phosphorylated Erk1 protein band. (E) BMNs were untreated or treated with 5 or 10 μM the p38 inhibitor SB203580 for 1 h prior to infection with NMII at an MOI of 100. The percent cell death of BMNs with different treatments was measured by the MTS assay at 18 hpi. (F) BMNs were untreated or treated with 25 or 50 μM the Erk1/2 inhibitor PD98059 for 1 h prior to infection with NMII at an MOI of 100. The percent cell death of BMNs receiving different treatments was measured by the MTS assay at 18 hpi. Error bars represent the standard deviation from the mean. *, P < 0.05; **, P < 0.01; N.S, not significant. P-p38, phosphorylated p38; P-Erk1 and P-Erk2, phosphorylated Erk1 and Erk2, respectively; SB, SB203580; PD, PD98059; C, 0-h time point, fresh uninfected BMNs; CU, uninfected control BMNs incubated for the indicated times; CU+DMSO, inhibitor buffer (dimethyl sulfoxide [DMSO])-treated uninfected BMNs.
Both PI3K and NF-κB are involved in NMII-induced inhibition of neutrophil apoptosis.
The phosphoinositide 3-kinase (PI3K) pathway is an important survival pathway activated by growth factors, cytokines, toxins, and pathogens. PI3K activates the downstream kinase Akt, an important player in survival signaling. Akt increases the transcription/translation of survival proteins, such as Bcl-XL and Mcl-1, through activation of NF-κB. Akt also inactivates proapoptotic proteins, such as Bad, by phosphorylation (27, 28). To determine if NMII-induced neutrophil survival was dependent on activation of PI3K, we tested whether treatment of NMII-infected neutrophils with wortmannin, a PI3K inhibitor, affected the ability of NMII to inhibit neutrophil apoptosis by the MTS assay. As shown in Fig. 5A, wortmannin treatment significantly reduced the ability of NMII to inhibit neutrophil apoptosis in NMII-infected neutrophils compared to no treatment. This effect was dose dependent, as increasing concentrations of wortmannin significantly increased the percentage of apoptotic neutrophils. This suggests that NMII infection triggers activation of PI3K, which is critical for promotion of neutrophil survival. PI3K activation may be the upstream signal for activation of neutrophil survival pathways. Next, to determine whether NMII induced neutrophil survival through activation of transcription factor NF-κB, we examined whether treatment of NMII-infected neutrophils with the NF-κB inhibitor SN50 affected the ability of NMII to inhibit neutrophil apoptosis by the MTS assay. The results indicated that the NF-κB inhibitor was also able to partially reduce the ability of NMII to inhibit neutrophil apoptosis in a dose-dependent manner (Fig. 5B). Activation of NF-κB may be involved in the NMII-induced promotion of neutrophil survival, and NF-κB signaling may be the downstream signal for activation of neutrophil survival pathways.
FIG 5.
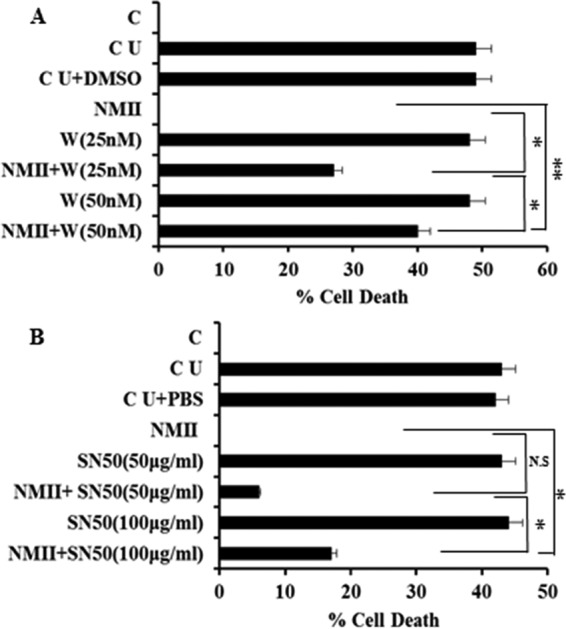
Both PI3K and NF-κB are involved in NMII-induced inhibition of neutrophil apoptosis. (A) BMNs were untreated or treated with 25 or 50 nM the PI3K inhibitor wortmannin for 1 h prior to infection with NMII at an MOI of 100. The percent cell death of BMNs receiving different treatments was measured by the MTS assay at 18 hpi. (B) BMNs were untreated or treated with 50 or 100 μg/ml of NF-κB inhibitor SN50 for 1 h prior to infection with NMII at an MOI of 100. The percent cell death of BMNs receiving different treatments was measured by the MTS assay at 18 hpi. Error bars represent the standard deviation from the mean. *, P < 0.05; **, P < 0.01; N.S, not significant. C, 0-h time point, fresh uninfected BMNs; CU, uninfected BMNs; CU+DMSO, wortmannin inhibitor buffer (dimethyl sulfoxide [DMSO])-treated uninfected BMNs; W, wortmannin; CU+PBS, SN50 inhibitor buffer-treated uninfected BMNs.
NMII-induced inhibition of neutrophil apoptosis depends on expression of the antiapoptotic protein Mcl-1.
It has been shown that the antiapoptotic protein Mcl-1 is required for neutrophil survival (30). To determine if Mcl-1 is involved in promoting neutrophil survival during NMII infection, we examined by Western blotting whether the Mcl-1 protein is expressed in NMII-infected neutrophils. As shown in Fig. 6A, elevated Mcl-1 protein levels were detected in NMII-infected neutrophils at 18, 38, and 48 hpi. In addition, we also compared the Mcl-1 protein intensity units between uninfected and NMII-infected neutrophils. As shown in Fig. 6B, the expression of Mcl-1 was 6.4-, 6.6-, and 6.2-fold higher in NMII-infected neutrophils than in uninfected neutrophils at 18, 38, and 48 hpi, respectively. These results indicate that the expression of Mcl-1 in neutrophils was significantly increased in response to NMII infection, suggesting that Mcl-1 may also contribute to NMII-induced inhibition of neutrophil apoptosis. To further demonstrate the role of Mcl-1 in NMII-induced inhibition of neutrophil apoptosis, we examined if treatment of NMII-infected neutrophils with a specific irreversible Mcl-1 inhibitor would affect the ability of NMII to inhibit neutrophil apoptosis by MTS assay. As shown in Fig. 6C, 45% cell death was detected in untreated and uninfected neutrophils, whereas the changes in NMII-infected neutrophils were undetectable. Levels of apoptosis of 19% and 49% were detected with 10 and 25 μM Mcl-1 inhibitor, respectively. These results indicate that the Mcl-1 inhibitor blocks NMII-induced neutrophil survival in a concentration-dependent manner, with the higher concentration of the Mcl-1 inhibitor almost completely blocking NMII-mediated neutrophil survival. Collectively, these data support a critical role for Mcl-1 in NMII-induced inhibition of neutrophil apoptosis.
FIG 6.
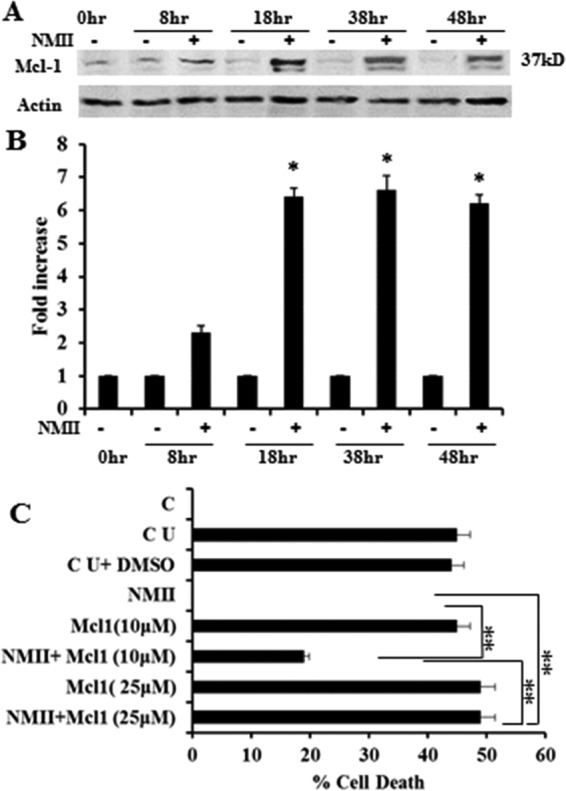
Antiapoptosis protein Mcl-1 is critical for NMII-induced inhibition of neutrophil apoptosis. (A) Detection of Mcl-1 in BMNs infected with NMII at an MOI of 100 by Western blotting at 8, 18, 38, and 48 hpi. (B) Fold induction of the intensity units of the Mcl-1 protein band. (C) BMNs were untreated or treated with 10 or 25 μM Mcl-1 inhibitor for 1 h prior to infection with NMII at an MOI of 100. The percent cell death of BMNs receiving different treatments was measured by the MTS assay at 18 hpi. Error bars represent the standard deviation from the mean. *, P < 0.05; **, P < 0.01. C, 0-h time point, fresh uninfected BMNs; CU, uninfected control BMNs; CU+DMSO, inhibitor buffer (dimethyl sulfoxide [DMSO])-treated uninfected BMNs; +, NMII-infected BMNs; Actin, protein loading control.
NMII-induced Mcl-1 expression is decreased in the presence of inhibitors, and the Erk pathway stabilizes the Mcl-1 protein from degradation by phosphorylating Thr-163.
To verify that the delay of apoptosis in neutrophils was only due to inhibition of signaling pathways, rather than a reduction in the NMII infection rate, we examined if the inhibitors affected the infection rate. As shown in Fig. 7A, there was no significant difference in the NMII infection rate between inhibitor-treated and untreated neutrophils. This result suggests that the ablation of NMII-mediated inhibition of apoptosis after treatment with inhibitors was not due to a reduction of the infection rate.
FIG 7.

Analysis of Mcl-1 protein levels in the presence of different inhibitors and Mcl-1 protein stabilization. (A) At 18 h hpi, the percentage of NMII-infected BMNs in the presence of different inhibitors was determined by IFA in the same manner described in the legend to Fig. 1B. (B) Analysis of phosphorylated Mcl-1 (P-Mcl-1; Ser-159/Thr-163) in BMNs infected with NMII at an MOI of 100 by Western blotting at 8, 18, 38, and 48 hpi. (C) Fold increase in the number of intensity units of the phosphorylated Mcl-1 protein band. (D) Detection of Mcl-1 in BMNs infected with NMII at an MOI of 100 by Western blotting at 18 hpi after treatment with SB203580 (SB; 10 μM), PD98059 (PD; 50 μM), wortmannin (W; 50 nM), SN50 (100 μg/ml), MIM1 (25 μM), or the inhibitor control or in untreated cells. (E) Percentage of the number of intensity units of the Mcl-1 protein band after treatment with inhibitors. *, P < 0.05; N.S, not significant. 0hr, 0-h time point, fresh uninfected BMNs; −, uninfected control inhibitor buffer (dimethyl sulfoxide [DMSO])-treated uninfected BMNs; +, NMII-infected BMNs; Actin, protein loading control.
It is known that activation of the Erk survival pathway can stabilize short-lived antiapoptotic proteins, such as Mcl-1, by phosphorylating Thr-163. To determine whether Mcl-1 was stabilized by Erk during NMII infection, we analyzed the levels of phosphorylated Mcl-1 by Western blotting using a phosphorylation-specific antibody (Ser-159/Thr-163) against Mcl-1. As shown in Fig. 7B, compared to the levels observed in NMII-infected neutrophils at 0 hpi, elevated levels of phosphorylated Mcl-1 were observed in NMII-infected neutrophils at 8, 18, 38, and 48 hpi. In addition, the intensity units of the phosphorylated Mcl-1 significantly increased in NMII-infected neutrophils at 18, 38, and 48 hpi compared to those in NMII-infected neutrophils at 0 hpi (Fig. 7C). These results indicate that the stabilization of Mcl-1 occurred during NMII infection in neutrophils.
As shown above, our results demonstrate that NMII infection can activate p38, Erk, PI3K, and NF-κB in neutrophils and that inhibition of p38, Erk, PI3K, or NF-κB by specific inhibitors decreases the ability of NMII to inhibit neutrophil apoptosis. Since activation of p38, Erk, PI3K, and NF-κB can regulate the expression of Mcl-1, we analyzed the levels of Mcl-1 in NMII-infected neutrophils untreated or treated with an p38 inhibitor (SB203580), an Erk inhibitor (PD98059), a PI3K inhibitor (wortmannin), an NF-κB inhibitor (SN50), or an Mcl-1 inhibitor (MIM1) by Western blotting. As shown in Fig. 7D, the levels of Mcl-1 decreased in NMII-infected neutrophils treated with each inhibitor compared to the levels in the untreated controls. Additionally, the intensity units of Mcl-1 significantly decreased in NMII-infected neutrophils treated with each inhibitor (Fig. 7E). These results suggest that the p38, Erk, PI3K, and NF-κB signaling pathways can regulate the expression of Mcl-1 in NMII-infected neutrophils. Activation of p38, Erk, PI3K, and NF-κB and subsequent expression and/or stabilization of Mcl-1 may be an important mechanism for NMII to inhibit neutrophil apoptosis.
NMII-induced inhibition of neutrophil apoptosis is partially dependent on its functional T4SS.
C. burnetii possesses a type IV secretion system (T4SS), and it has been shown that T4SS-secreted effectors can modulate host cell apoptotic signaling (31–35). To determine whether T4SS effectors might be involved in NMII-induced inhibition of neutrophil apoptosis, we investigated if an NMII dotA mutant, which lacks a functional T4SS, could also inhibit neutrophil apoptosis. First, we used IFA to determine if dotA mutant bacteria could infect BMNs. As shown in Fig. 8A, C. burnetii (green) was observed in dotA mutant-infected BMNs, suggesting that dotA mutant bacteria could infect neutrophils. Figure 8B shows the percentage of dotA mutant-infected neutrophils. Approximately 9% of BMNs were infected with NMII dotA after 4 h, while the percentage of infected cells increased to 42% after 18 h. Thus, the percentage of NMII dotA mutant-infected BMNs was similar to the percentage of wild-type (WT) NMII-infected BMNs (24). To determine if the T4SS is required for NMII-induced inhibition of neutrophil apoptosis, we examined whether the dotA mutant could alter DNA fragmentation in BMNs by TUNEL staining. As shown in Fig. 9A and B, fewer TUNEL-positive cells were observed in dotA mutant-infected neutrophils than uninfected neutrophils at 18 hpi. These results indicate that the NMII dotA mutant can also inhibit neutrophil apoptosis. Furthermore, to determine whether there were differences between dotA mutant and WT NMII bacteria in their ability to inhibit neutrophil apoptosis, the quantitative MTS assay was used to compare cell viability between dotA mutant-infected and NMII-infected neutrophils. As shown in Fig. 9C, the level of apoptosis in dotA mutant-infected neutrophils was significantly lower than that in uninfected neutrophils; however, it was significantly higher than that in NMII-infected neutrophils. These results suggest that T4SS effectors may be partially involved in NMII-induced neutrophil survival.
FIG 8.

The C. burnetii NMII dotA mutant infects primary mouse BMNs. BMNs were infected with the NMII dotA mutant at an MOI of 100 and stained by IFA using antibodies against C. burnetii (green) and LAMP-1 (red) at 18 h hpi. DAPI was used to stain host cell DNA (blue). (A) C. burnetii (green) was observed in dotA mutant-infected BMNs. (B) The NMII dotA mutant infection rate was determined at 4 and 18 hpi by counting the number of BMNs positively labeled with C. burnetii antibodies. Results are expressed as the percentage of infected BMNs compared to the percentage of uninfected BMNs. Error bars represent the standard deviation from the mean. *, P < 0.05; **, P < 0.01.
FIG 9.

The NMII dotA mutant partially inhibits neutrophil apoptosis. BMNs were infected with the NMII dotA mutant at an MOI of 100 for 18 h. (A) Uninfected and infected BMNs were stained with TUNEL (red) to detect DNA fragmentation. BMNs were also labeled by IFA using antibodies directed against C. burnetii (green). DAPI was used to stain DNA (blue). Uninfected BMNs exhibited significant DNA fragmentation, while there were fewer TUNEL-positive cells in WT strain NMII-infected BMNs than in NMII dotA mutant-infected BMNs. (B) The percentage of apoptotic cells was determined at 18 hpi by counting the number of TUNEL-positive cells. (C) The percent cell death of uninfected BMNs and BMNs infected with WT strain NMII or the NMII dotA mutant at an MOI of 100 was measured by the MTS assay at 18 hpi. Results are expressed as percent cell death, and error bars represent the standard deviation from the mean. *, P < 0.05; **, P < 0.01. CU, uninfected control BMNs.
DISCUSSION
In this study, we found that avirulent NMII bacteria are capable of inhibiting BMN apoptosis. To understand the molecular mechanisms underlying NMII-induced inhibition of neutrophil apoptosis, we examined whether NMII could modulate cell survival and/or antiapoptotic signaling pathways during infection of BMNs. Our results indicate that NMII bacteria can inhibit the activity of caspase-3; activate PI3K, p38 MAPK, Erk1, and NF-κB; and increase the expression of the antiapoptotic protein Mcl-1. In addition, the observation that the T4SS-deficient NMII dotA mutant showed significantly lower levels of inhibition of apoptosis than WT strain NMII suggests that T4SS effectors may also be involved in NMII-induced inhibition of neutrophil apoptosis.
Generally, neutrophils constitutively undergo apoptosis, and apoptotic neutrophils are subsequently removed by scavenger macrophages, resulting in resolution of the inflammatory response without releasing cytotoxic molecules that damage host tissues (12). Although apoptosis is an intrinsic cell process, neutrophil apoptosis can be altered during the course of inflammatory responses by multiple extrinsic signals from the inflammatory microenvironment (27–29). For example, the recruitment of neutrophils into infected tissues is often associated with prolongation of their life span through a delay of apoptosis, which allows neutrophils to effectively perform their microbicidal functions (36, 37). In addition, the neutrophil life span can be extended in vitro by incubation with either proinflammatory cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) (38), interleukin-8 (IL-8) (39), IL-1β (40), and glucocorticoids (41), or bacterial products, such as LPS and N-formyl-methionyl-leucyl-phenylalanine (40, 42). Interestingly, we found that NMII can delay neutrophil apoptosis in vitro in a dose-dependent manner for as long as 96 hpi. The ability to inhibit neutrophil apoptosis has been described for several other intracellular pathogens. Yoshiie et al. reported that A. phagocytophilum-infected neutrophils delayed apoptosis to 2 days postinfection (43). Aga et al. also observed that L. major-infected neutrophils can survive up to 2 days after infection (18). Like C. burnetii, van Zandbergen et al. showed that C. pneumoniae-infected neutrophils survived as long as 90 hpi (19). Thus, the longevity of neutrophil survival appears to vary between pathogens. The reasons why different pathogens induce distinct longevities of neutrophil survival remain unknown. However, extension of the neutrophil life span may be a crucial mechanism of escape from host defense for these intracellular pathogens to establish infection in their hosts. In addition, since neutrophils are short-lived cells, the ability to inhibit neutrophil apoptosis may be crucial for invading intracellular pathogens to maintain the intracellular niche for their survival and replication. This hypothesis is supported by the observation that A. phagocytophilum, L. major, and C. pneumoniae not only survive but also multiply in the hostile environment of neutrophils (18, 19, 43). In contrast, we did not detect C. burnetii replication in NMII-infected BMNs (data not shown). Our previous study (24) also demonstrated that both the virulent strain C. burnetii NMI and the avirulent strain C. burnetii NMII were able to infect and survive in mouse BMNs; however, bacterial replication was not detected. Although the reason why C. burnetii was unable to replicate in neutrophils remains unclear, the observation that the C. burnetii bacteria inside neutrophils can infect and replicate within macrophages suggests that neutrophils cannot kill C. burnetii. Thus, C. burnetii may be using infection of neutrophils as an immune-evasive strategy to infect macrophages. This phenomenon may be similar to L. major using neutrophils as Trojan horses to enter its favorable host cells for replication (18).
It has been demonstrated that activation of caspase-3 is critical for neutrophil apoptosis (25). Freshly isolated neutrophils contain high levels of procaspase-3, which is cleaved during apoptosis, giving rise to the enzymatically active caspase-3 (44). In addition, inhibition of the activation of caspase-3 was found in L. major-infected human neutrophils (18), and C. pneumoniae-mediated delay of neutrophil apoptosis was demonstrated to be associated with a decrease in caspase-3 activity in infected neutrophils (19). In this study, we found that the levels of cleaved caspase-3 were significantly reduced in NMII-infected neutrophils compared to the levels in uninfected cells, suggesting that NMII-induced inhibition of neutrophil apoptosis depends on its ability to reduce the activity of caspase-3, resulting in prolongation of neutrophil survival. Thus, the intracellular pathogen-induced delay of neutrophil apoptosis may be through a common mechanism that involves inhibiting the activity of caspase-3.
The molecular mechanisms utilized by pathogenic bacteria to prolong the life span of neutrophils remains an important research topic. Neutrophil apoptosis has been demonstrated to be tightly regulated by a complex network of signaling pathways through controlled expression and degradation of key signaling molecules, including Bcl-2 family proteins, MAPKs, NF-κB, and caspases (26–29). To determine whether NMII modulated neutrophil apoptosis by promoting cell survival pathways, the activities of the kinases MAPK p38 and Erk1/2 in NMII-infected neutrophils were analyzed by Western blotting. Compared to the levels in uninfected controls, elevated levels of activated p38 and Erk1 kinases were detected in NMII-infected neutrophils, suggesting that the promotion of survival pathways may be involved in NMII-induced inhibition of neutrophil apoptosis. This hypothesis is supported by the observation that both p38 and Erk1/2 inhibitors were able to significantly reduce the ability of NMII to inhibit neutrophil apoptosis in a dose-dependent manner. Additionally, the Erk1/2 inhibitor PD98059 resulted in higher levels of cell death than the p38 inhibitor SB203580, suggesting that the kinase Erk1 may play a more important role than p38 in promoting neutrophil survival during NMII infection. Klein et al. (45) also reported that the Erk inhibitor PD98059, but not the p38 inhibitor SB203580, reduced the ability of GM-CSF to inhibit neutrophil apoptosis. Our results corroborate these published data, indicating that Erk1 may play a more critical role than p38 in promoting neutrophil survival.
A. phagocytophilum-mediated prevention of neutrophil apoptosis has been demonstrated to be regulated by cell survival pathways via activation of PI3K/Akt, p38 MAPK, Erk, and NF-κB (46–48). Similarly, the observation that inhibition of PI3K upstream of Akt increases the apoptosis of HeLa cells during C. trachomatis infection suggests that the PI3K/Akt signaling pathway is critical for the C. trachomatis-mediated prevention of apoptosis (49, 50). In this study, we used the PI3K inhibitor wortmannin to determine by the MTS assay whether PI3K is involved in NMII-induced neutrophil survival. The observation that treatment of NMII-infected BMNs with wortmannin significantly blocked the ability of NMII bacteria to promote neutrophil survival in a dose-dependent manner suggests that NMII infection triggers activation of PI3K. This activation is required for promotion of neutrophil survival, and PI3K signaling may be the upstream signal for activation of neutrophil survival pathways. Previous studies (21, 51) also demonstrated that C. burnetii prevents intrinsic apoptosis by activating prosurvival Erk1/2 and Akt signaling pathways in THP-1 cells. Although the current study did not rule out the possibility that Akt is activated in NMII-infected neutrophils, our results suggest that NMII delays neutrophil apoptosis by promoting cell survival via activation of the PI3K, p38, and Erk1 signaling pathways.
Activation of antiapoptotic transcription factor NF-κB for the prevention of host cell apoptosis has been reported for several intracellular pathogens, including Mycobacterium tuberculosis (52), A. phagocytophilum (47), C. pneumoniae (53), Rickettsia rickettsii (54), and L. pneumophila (55, 56). In contrast, Bartonella henselae (57, 58) failed to induce NF-κB to inhibit apoptosis, suggesting that B. henselae-mediated inhibition of apoptosis is independent of NF-κB. In this study, we examined by the MTS assay whether treatment of NMII-infected neutrophils with the NF-κB inhibitor SN50 would affect the ability of NMII to promote neutrophil survival. The results indicated that SN50 is able to partially reduce the ability of NMII to promote neutrophil survival in a dose-dependent manner, suggesting that activation of NF-κB is partially involved in NMII-induced neutrophil survival and that NF-κB signaling may be the downstream signal for activating neutrophil survival pathways.
Bcl-2 family proteins have been shown to play critical roles in regulating neutrophil apoptosis (59). Mcl-1 is a key Bcl2 family member protein located in the nucleus and cytoplasm. When senescent neutrophils undergo apoptosis, the levels of Mcl-1 protein decrease rapidly due to proteasomal degradation (60). Mcl-1 as an antiapoptotic protein has been shown to be absolutely necessary for the survival of neutrophils, as Mcl-1 conditional knockout mice have severe defects in neutrophil survival (30). To determine if Mcl-1 is involved in promoting neutrophil survival during NMII infection, we examined by Western blotting whether the Mcl-1 protein is expressed in NMII-infected neutrophils. The results indicated that the expression of Mcl-1 in neutrophils is significantly increased in response to NMII infection, suggesting that Mcl-1 maybe also be involved in NMII-mediated neutrophil survival. In addition, treatment of NMII-infected neutrophils with a specific irreversible Mcl-1 inhibitor completely blocked NMII-mediated neutrophil survival. Collectively, these results suggest that Mcl-1 plays a critical role in NMII-induced inhibition of neutrophil apoptosis.
Mcl-1 is a short-lived protein that is regulated at the transcriptional and posttranslational level. It has an extended amino-terminal PEST sequence (enriched in proline, glutamic acid, serine, and threonine), which is responsible for its relatively short half-life (61, 62). Posttranslationally, Mcl-1 is phosphorylated at Thr-163 by Erk, which enhances its stability via prevention of degradation (63). However, phosphorylation at Thr-163 may prime phosphorylation at Ser-159 by glycogen synthase kinase-3 (GSK-3), which leads to Mcl-1 destabilization and degradation. It has been shown that activation of the PI3K pathway leads to inactivation of GSK-3, leading to inhibition of phosphorylation at Ser-159, resulting in the enhanced stability of Mcl-1 (64). Thus, the Erk and PI3K pathways play critical roles in stabilizing Mcl-1 and prolonging cell survival. The observation that NMII induced the phosphorylation of Mcl-1 at Thr-163 suggests that the Mcl-1 protein is stabilized by Erk. Furthermore, the observation that a PI3K inhibitor abolished NMII-mediated inhibition of apoptosis suggests an important role for PI3K activation. As mentioned above, PI3K activation can lead to GSK-3 inactivation, protecting Mcl-1 from Ser-159 destabilization. These observations further imply that both the Erk and PI3K pathways are critical for enhancing the viability of neutrophils. The role of p38 in apoptosis and cell survival depends on the cell type and the stimulus. It may function both upstream and downstream of caspases and is activated in vivo by environmental stress and inflammatory cytokines. It has been shown that the oxidative stress induced by paraquat (an herbicide) can inhibit human neutrophil apoptosis, which involves p38, NF-κB, tumor necrosis factor, and IL-6 (65). Our results demonstrate that (i) p38 is activated in NMII-infected neutrophils, (ii) a p38 inhibitor significantly reduces the ability of NMII to inhibit neutrophil apoptosis in a dose-dependent manner, and (iii) a p38 inhibitor is able to reduce the levels of the Mcl-1 protein in NMII-infected neutrophils. These findings suggest that p38 plays an important role in NMII-induced inhibition of neutrophil apoptosis via regulation of the levels of the Mcl-1 protein. Overall, the MAPK and PI3K pathways act in parallel to stabilize the antiapoptotic protein Mcl-1 via phosphorylation.
Activation of the MAPK (p38, Erk) and PI3K pathways further leads to activation of the downstream transcription factor NF-κB, which is an important player in enhancing the antiapoptotic proteins (27). The observation that the NF-κB inhibitor SN50 reduces the ability of NMII to inhibit neutrophil apoptosis and decreases the levels of the Mcl-1 protein in NMII-infected neutrophils suggests that activation of NF-κB and subsequent expression of the Mcl-1 protein may also be an important mechanism for NMII-induced inhibition of neutrophil apoptosis. Collectively, these data suggest that activation of p38, Erk, PI3K, and NF-κB to express and/or stabilize the Mcl-1 protein may be critical for NMII-induced inhibition of neutrophil apoptosis.
T4SS effector proteins have been demonstrated to modulate host cell apoptotic signaling (32). It has been shown that expression of the C. burnetii T4SS protein CBU0388 enhances the MAPK pathway in the yeast Saccharomyces cerevisiae (66). To determine whether T4SS effectors are required for NMII-induced inhibition of neutrophil apoptosis, we examined if there were differences between a T4SS-deficient dotA mutant and WT strain NMII in their ability to inhibit neutrophil apoptosis by TUNEL staining and the MTS assay. Interestingly, even though the dotA mutant and WT NMII bacteria have similar infection rates in neutrophils, the number of dotA mutant-infected apoptotic cells was significantly higher than the number of NMII-infected neutrophils. However, the number of apoptotic cells in dotA mutant-infected neutrophils was significantly lower than the number of uninfected neutrophils, which suggests that T4SS effectors partially contribute to NMII-induced inhibition of neutrophil apoptosis. This partial prevention of apoptosis by the NMII dotA mutant, which does not secrete bacterial effector proteins inside the host cell, could be attributed to the activation of PI3K, which is a membrane-derived process that occurs when the NMII dotA mutant binds to the integrin receptor at the start of phagocytosis. In addition, activation of downstream transcription factor NF-κB and subsequent expression of antiapoptotic protein Mcl-1 may be also involved in NMII dotA mutant-induced inhibition of neutrophil apoptosis. Further studies are necessary to decipher this mechanism.
In summary, this study demonstrated that NMII bacteria can inhibit neutrophil apoptosis via their ability to decrease the activity of caspase-3, activate the PI3K survival pathway, and induce the expression and/or stabilization of antiapoptotic protein Mcl-1. Inhibitors of the MAPK and PI3K pathways reduce the expression of the Mcl-1 protein, suggesting their critical role in survival. Future studies are necessary to investigate whether C. burnetii-mediated inhibition of neutrophil apoptosis plays an important role during C. burnetii infection in vivo and to identify the bacterial factors that are responsible for NMII-induced inhibition of neutrophil apoptosis.
MATERIALS AND METHODS
Reagents.
Phospho-p38 (Thr-180/Tyr-182) antibody and p38 antibody (Cell Signaling Technologies, Danvers, MA), phospho-Erk1/2 (pT202/pY204) antibody (BD Transduction Laboratories, San Jose, CA), Mcl-1 antibody (BioLegend, San Diego, CA), anti-β-actin antibody (Santa Cruz Biotechnology, Dallas, TX), and phospho-Mcl-1 (Ser-159/Thr-163) (Cell Signaling Technologies) were used for Western blotting. The MAPK p38 inhibitor (SB203580) and Mcl-1 inhibitor (MIM1) were purchased from EMD Millipore Corp (Billerica, MA). The Erk1/2 inhibitor (PD98059) and NF-κB inhibitor (SN50) were obtained from Cell Signaling Technologies and BioVision (Milpitas, CA), respectively. RPMI and other cell culture reagents were obtained from Thermo Fisher Scientific (Waltham, MA). All remaining chemicals were obtained from Sigma (St. Louis, MO).
Bacteria.
C. burnetii NMII clone 4 (strain RSA 439) was propagated in L929 cells and purified by density gradient centrifugation as described previously (67). The T4SS-deficient strain NMII (a dot icmA mutant with a defect in organelle trafficking/intracellular multiplication), a kind gift from Paul Beare, was propagated in acidified citrate cysteine medium-2 (ACCM-2) (68). Heat-killed NMII was generated by boiling in a water bath for 10 min.
Animals.
Six- to 8-week-old female BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in microisolator cages at a conventional animal facility at the University of Missouri. All experiments involving the use of animals were conducted in accordance with the animal care and use guidelines, and all animal use protocols were approved by the Animal Care and Use Committee at the University of Missouri.
Isolation of BMNs.
Bone marrow neutrophils (BMNs) were isolated as described previously (69). Briefly, mice were euthanized, their femurs and tibias were removed surgically, outer connective tissue was cleaned off, and the ends of the femurs and tibias were cut off to expose the bone marrow. Three milliliters of Hanks' balanced salt solution (HBSS; HyClone Labs, Logan, UT) was flushed through the bone by use of a syringe, and the bone marrow was collected in a tube. Cells were pelleted by centrifugation at 500 × g for 10 min. Red blood cells were lysed by incubating the pelleted cells with ammonium chloride-potassium (ACK) lysis buffer (Lonza, Walkersville, MD) at room temperature for 5 min. The cell suspension was then layered onto a discontinuous density gradient (55%, 65%, 75%) prepared with Percoll solution (GE Healthcare, Pittsburg, PA) and separated by centrifugation at 500 × g for 30 min. BMNs were harvested between the 65 and 75% layers and counted, and their purity was confirmed by immunofluorescence assay (IFA) with the neutrophil marker Ly-6G and Giemsa staining. As shown in Fig. S1 in the supplemental material, the purity of freshly isolated BMNs was >96%.
C. burnetii infection and cell lysate preparation.
A total of 2 × 106 freshly isolated BMNs were placed in each well of a 24-well plate containing RPMI and 2.5% fetal bovine serum, infected with NMII at a multiplicity of infection (MOI) of 100 or left uninfected, and incubated at 37°C in 5% CO2. At 8, 18, 38, and 48 h postinfection (hpi), the cells were pelleted by centrifugation at 500 × g for 10 min. Proteins were extracted from the pelleted cells by adding lysis buffer, prepared using the M-PER mammalian protein extraction reagent (Thermo Fisher Scientific) mixed with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific) according to the manufacturer's specifications. The lysates were mixed thoroughly and clarified by centrifugation at 10,000 rpm for 10 min. The soluble fraction was collected and used for protein quantification and Western blotting.
Western blotting.
The protein concentration was measured using a Pierce bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL). Equal amounts of protein (30 to 75 μg/sample) were separated by SDS-PAGE with 12% or 15% polyacrylamide gels and then transferred onto nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in Tris buffer (pH 7.5) containing 200 mM Tris, 1.38 M NaCl, and 0.1% Tween 20 for 2 h and then incubated with a primary antibody specific for p38, phospho-p38, phospho-Erk1/2, phospho-Mcl-1, or anti-β-actin at 4°C overnight in 5% bovine serum albumin. The bands were visualized using an enhanced chemiluminescence Western blot detection kit (Thermo Fisher Scientific), and chemiluminescent protein bands were captured on autoradiography film. Band intensity units were quantified using Image Studio software (Li-Cor), and the fold change in intensity was calculated by normalization to that for the loading controls and subtracting the background levels. Each experiment was repeated at least three times.
MTS assay.
Neutrophil death was measured indirectly using a CellTiter 96 AQueous one-solution cell proliferation assay (Promega, Madison, WI). The absorbance is directly proportional to the number of living cells in the culture. Neutrophils freshly isolated from BALB/c mice were added to a 96-well plate at 5 × 104 cells/well, infected with NMII at an MOI of 25, 50, 100, 200, or 500, and incubated at 37°C in 5% CO2 for 24, 48, 72, or 96 h. At each time point, 20 μl of MTS solution was added to each well and the plate was incubated at 37°C for 1 to 4 h until color developed. The absorbance at 490 nm was measured using a Molecular Devices SpectraMax Plus plate reader and SoftMax software, and the percent cell death was calculated using the following formula: [(average OD490 for uninfected cells − average OD490 for infected cells)/average OD490 for uninfected cells] · 100, where OD490 is the optical density at 490 nm. The background absorbance at 630 nm was measured, and the value was subtracted from the sample values. All experiments were repeated three times.
Inhibitor treatment.
A total of 5 × 104 freshly isolated BMNs were added into each well of a 96-well plate, treated with 5 or 10 μM the p38 inhibitor (SB203580), 10 or 25 μM the Mcl-1 inhibitor (MIM1), 25 or 50 μM the Erk1/2 inhibitor (PD98059), 50 or 100 μg/ml the NF-κB inhibitor (SN50), 25 or 50 nM wortmannin, or each inhibitor control at 37°C in 5% CO2 for 1 h prior to infection with NMII. The inhibitory activity was measured by the MTS assay described above. For Western blotting, 4 × 106 cells were left untreated or treated with SB203580 (10 μM), PD98059 (50 μM), wortmannin (50 nM), SN50 (100 μg/ml), MIM1 (25 μM), or the inhibitor control for 1 h, infected with NMII (MOI, 100), and then incubated for 18 h at 37°C in 5% CO2. Cell lysates were prepared and clarified, and 50 to 100 μg of protein was separated by 12% SDS-PAGE, as described above.
IFA.
A total of 1 × 106 freshly isolated BMNs were placed into each well of a 24-well plate containing poly-d-lysine-coated coverslips, infected with NMII at an MOI of 100, and incubated at 37°C in 5% CO2. At 4 and 18 hpi, the cells were washed with sterile 1× phosphate-buffered saline (PBS), fixed with 2% paraformaldehyde (PFA), and permeabilized with ice-cold (−20°C) methanol. The cells were stained intracellularly with rabbit anti-Coxiella polyclonal antibodies for 1 h at room temperature and then stained with CD107a-phycoerythrin (PE) (lysosome-associated membrane protein 1 [LAMP-1]), Ly-6G, and goat anti-rabbit IgG-fluorescein isothiocyanate (FITC) for 1 h at room temperature. The nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) for 5 min at 4°C. Coverslips were mounted in ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA) and allowed to cure overnight at room temperature. Microscopic analysis was performed using an Olympus IX70 inverted microscope.
TUNEL staining.
TUNEL staining was performed using an in situ apoptosis detection kit (TMR red; Roche, Indianapolis, IN) per the manufacturer's specifications. Briefly, following fixation and permeabilization as described above, the cells were washed and the TUNEL reaction mix was added for 1 h at 37°C in the dark. The cells were washed twice with 1× PBS and visualized using an Olympus IX70 inverted microscope.
Statistical analysis.
Statistical analysis was performed using Prism (version 5.0) software (GraphPad Software Inc., San Diego, CA). For p38, Erk1, and Mcl-1 activation, the significance of the fold induction was analyzed using the t test. For inhibitor treatment studies and infection rate, statistical significance was calculated using a two-tailed t test. The differences were considered significant at P values of ≤0.05.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIAID/NIH grant RO1AI083364 to G.Z. and a DTRA funding grant HDTRA1-13-0003 subcontract to G.Z.
We thank Alexander Jurkevich and Frank Baker of the MU Molecular Cytology Core for their assistance with microscopy. We also thank Nicholas Olivarez for critical reading and editing of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00504-17.
REFERENCES
- 1.Raoult D, Marrie T, Mege J. 2005. Natural history and pathophysiology of Q fever. Lancet Infect Dis 5:219–226. doi: 10.1016/S1473-3099(05)70052-9. [DOI] [PubMed] [Google Scholar]
- 2.Dijkstra F, van der Hoek W, Wijers N, Schimmer B, Rietveld A, Wijkmans CJ, Vellema P, Schneeberger PM. 2012. The 2007-2010 Q fever epidemic in The Netherlands: characteristics of notified acute Q fever patients and the association with dairy goat farming. FEMS Immunol Med Microbiol 64:3–12. doi: 10.1111/j.1574-695X.2011.00876.x. [DOI] [PubMed] [Google Scholar]
- 3.Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. 1999. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med 159:167–173. doi: 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- 4.Hackstadt T, Peacock MG, Hitchcock PJ, Cole RL. 1985. Lipopolysaccharide variation in Coxiella burnetii: intrastrain heterogeneity in structure and antigenicity. Infect Immun 48:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kersh GJ, Oliver LD, Self JS, Fitzpatrick KA, Massung RF. 2011. Virulence of pathogenic Coxiella burnetii strains after growth in the absence of host cells. Vector Borne Zoonotic Dis 11:1433–1438. doi: 10.1089/vbz.2011.0670. [DOI] [PubMed] [Google Scholar]
- 6.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A, Cassatella MA, Costantini C, Jaillon S. 2011. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 8.Garvy BA, Harmsen AG. 1996. The importance of neutrophils in resistance to pneumococcal pneumonia in adult and neonatal mice. Inflammation 20:499–512. doi: 10.1007/BF01487042. [DOI] [PubMed] [Google Scholar]
- 9.Jeyaseelan S, Young SK, Yamamoto M, Arndt PG, Akira S, Kolls JK, Worthen GS. 2006. Toll/IL-1R domain-containing adaptor protein (TIRAP) is a critical mediator of antibacterial defense in the lung against Klebsiella pneumoniae but not Pseudomonas aeruginosa. J Immunol 177:538–547. doi: 10.4049/jimmunol.177.1.538. [DOI] [PubMed] [Google Scholar]
- 10.Tateda K, Moore TA, Deng JC, Newstead MW, Zeng X, Matsukawa A, Swanson MS, Yamaguchi K, Standiford TJ. 2001. Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. J Immunol 166:3355–3361. doi: 10.4049/jimmunol.166.5.3355. [DOI] [PubMed] [Google Scholar]
- 11.Elliott A, Schoenlaub L, Freches D, Mitchell W, Zhang G. 2015. Neutrophils play an important role in protective immunity against Coxiella burnetii infection. Infect Immun 83:3104–3113. doi: 10.1128/IAI.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. 1989. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest 83:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskay T, van Zandbergen G, Solbach W. 2008. Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology 213:183–191. doi: 10.1016/j.imbio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. 1996. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol 156:3986–3992. [PubMed] [Google Scholar]
- 15.Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, DeLeo FR. 2003. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci U S A 100:10948–10953. doi: 10.1073/pnas.1833375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLeo FR. 2004. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9:399–413. doi: 10.1023/B:APPT.0000031448.64969.fa. [DOI] [PubMed] [Google Scholar]
- 17.Scaife H, Woldehiwet Z, Hart CA, Edwards SW. 2003. Anaplasma phagocytophilum reduces neutrophil apoptosis in vivo. Infect Immun 71:1995–2001. doi: 10.1128/IAI.71.4.1995-2001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aga E, Katschinski DM, van Zandbergen G, Laufs H, Hansen B, Muller K, Solbach W, Laskay T. 2002. Inhibition of the spontaneous apoptosis of neutrophil granulocytes by the intracellular parasite Leishmania major. J Immunol 169:898–905. doi: 10.4049/jimmunol.169.2.898. [DOI] [PubMed] [Google Scholar]
- 19.van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, Klinger M, Brade H, Dalhoff K, Maass M, Solbach W, Laskay T. 2004. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol 172:1768–1776. doi: 10.4049/jimmunol.172.3.1768. [DOI] [PubMed] [Google Scholar]
- 20.Voth DE, Howe D, Heinzen RA. 2007. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun 75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luhrmann A, Roy CR. 2007. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect Immun 75:5282–5289. doi: 10.1128/IAI.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Zhang G, Hendrix LR, Tesh VL, Samuel JE. 2012. Coxiella burnetii induces apoptosis during early stage infection via a caspase-independent pathway in human monocytic THP-1 cells. PLoS One 7:e30841. doi: 10.1371/journal.pone.0030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenlaub L, Cherla R, Zhang Y, Zhang G. 2016. Coxiella burnetii avirulent Nine Mile phase II induces caspase-1-dependent pyroptosis in murine peritoneal B1a B Cells. Infect Immun 84:3638–3654. doi: 10.1128/IAI.00694-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elliott A, Peng Y, Zhang G. 2013. Coxiella burnetii interaction with neutrophils and macrophages in vitro and in SCID mice following aerosol infection. Infect Immun 81:4604–4614. doi: 10.1128/IAI.00973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daigle I, Simon HU. 2001. Critical role for caspases 3 and 8 in neutrophil but not eosinophil apoptosis. Int Arch Allergy Immunol 126:147–156. doi: 10.1159/000049506. [DOI] [PubMed] [Google Scholar]
- 26.Savill J, Dransfield I, Gregory C, Haslett C. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 27.Luo HR, Loison F. 2008. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 83:288–295. doi: 10.1002/ajh.21078. [DOI] [PubMed] [Google Scholar]
- 28.Simon HU. 2003. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev 193:101–110. doi: 10.1034/j.1600-065X.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 29.El Kebir D, Filep JG. 2010. Role of neutrophil apoptosis in the resolution of inflammation. ScientificWorldJournal 10:1731–1748. doi: 10.1100/tsw.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dzhagalov I, St John A, He YW. 2007. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 109:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moffatt JH, Newton P, Newton HJ. 2015. Coxiella burnetii: turning hostility into a home. Cell Microbiol 17:621–631. doi: 10.1111/cmi.12432. [DOI] [PubMed] [Google Scholar]
- 32.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. 2013. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol 11:561–573. doi: 10.1038/nrmicro3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bisle S, Klingenbeck L, Borges V, Sobotta K, Schulze-Luehrmann J, Menge C, Heydel C, Gomes JP, Luhrmann A. 2016. The inhibition of the apoptosis pathway by the Coxiella burnetii effector protein CaeA requires the EK repetition motif, but is independent of survivin. Virulence 7:400–412. doi: 10.1080/21505594.2016.1139280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klingenbeck L, Eckart RA, Berens C, Luhrmann A. 2013. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol 15:675–687. doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- 35.Luhrmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A 107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan C. 2006. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 37.Nauseef WM. 2007. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 38.Cox G, Gauldie J, Jordana M. 1992. Bronchial epithelial cell-derived cytokines (G-CSF and GM-CSF) promote the survival of peripheral blood neutrophils in vitro. Am J Respir Cell Mol Biol 7:507–513. doi: 10.1165/ajrcmb/7.5.507. [DOI] [PubMed] [Google Scholar]
- 39.Kettritz R, Gaido ML, Haller H, Luft FC, Jennette CJ, Falk RJ. 1998. Interleukin-8 delays spontaneous and tumor necrosis factor-alpha-mediated apoptosis of human neutrophils. Kidney Int 53:84–91. doi: 10.1046/j.1523-1755.1998.00741.x. [DOI] [PubMed] [Google Scholar]
- 40.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. 1992. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80:2012–2020. [PubMed] [Google Scholar]
- 41.Liles WC, Dale DC, Klebanoff SJ. 1995. Glucocorticoids inhibit apoptosis of human neutrophils. Blood 86:3181–3188. [PubMed] [Google Scholar]
- 42.Lee A, Whyte MK, Haslett C. 1993. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol 54:283–288. [PubMed] [Google Scholar]
- 43.Yoshiie K, Kim HY, Mott J, Rikihisa Y. 2000. Intracellular infection by the human granulocytic ehrlichiosis agent inhibits human neutrophil apoptosis. Infect Immun 68:1125–1133. doi: 10.1128/IAI.68.3.1125-1133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanghavi DM, Thelen M, Thornberry NA, Casciola-Rosen L, Rosen A. 1998. Caspase-mediated proteolysis during apoptosis: insights from apoptotic neutrophils. FEBS Lett 422:179–184. doi: 10.1016/S0014-5793(98)00004-0. [DOI] [PubMed] [Google Scholar]
- 45.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR. 2000. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol 164:4286–4291. doi: 10.4049/jimmunol.164.8.4286. [DOI] [PubMed] [Google Scholar]
- 46.Choi KS, Park JT, Dumler JS. 2005. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect Immun 73:8209–8218. doi: 10.1128/IAI.73.12.8209-8218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HC, Goodman JL. 2006. Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics 88:496–503. doi: 10.1016/j.ygeno.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Ge Y, Rikihisa Y. 2006. Anaplasma phagocytophilum delays spontaneous human neutrophil apoptosis by modulation of multiple apoptotic pathways. Cell Microbiol 8:1406–1416. doi: 10.1111/j.1462-5822.2006.00720.x. [DOI] [PubMed] [Google Scholar]
- 49.Scidmore MA, Hackstadt T. 2001. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol 39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- 50.Verbeke P, Welter-Stahl L, Ying S, Hansen J, Hacker G, Darville T, Ojcius DM. 2006. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog 2:e45. doi: 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voth DE, Heinzen RA. 2009. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun 77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhiman R, Raje M, Majumdar S. 2007. Differential expression of NF-kappaB in mycobacteria infected THP-1 affects apoptosis. Biochim Biophys Acta 1770:649–658. doi: 10.1016/j.bbagen.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 53.Wahl C, Oswald F, Simnacher U, Weiss S, Marre R, Essig A. 2001. Survival of Chlamydia pneumoniae-infected Mono Mac 6 cells is dependent on NF-kappaB binding activity. Infect Immun 69:7039–7045. doi: 10.1128/IAI.69.11.7039-7045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clifton DR, Goss RA, Sahni SK, van Antwerp D, Baggs RB, Marder VJ, Silverman DJ, Sporn LA. 1998. NF-kappa B-dependent inhibition of apoptosis is essential for host cell survival during Rickettsia rickettsii infection. Proc Natl Acad Sci U S A 95:4646–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, Kwaik YA. 2007. Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol 9:246–264. doi: 10.1111/j.1462-5822.2006.00785.x. [DOI] [PubMed] [Google Scholar]
- 56.Losick VP, Isberg RR. 2006. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J Exp Med 203:2177–2189. doi: 10.1084/jem.20060766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kempf VA, Schairer A, Neumann D, Grassl GA, Lauber K, Lebiedziejewski M, Schaller M, Kyme P, Wesselborg S, Autenrieth IB. 2005. Bartonella henselae inhibits apoptosis in Mono Mac 6 cells. Cell Microbiol 7:91–104. doi: 10.1111/j.1462-5822.2004.00440.x. [DOI] [PubMed] [Google Scholar]
- 58.Schmid MC, Scheidegger F, Dehio M, Balmelle-Devaux N, Schulein R, Guye P, Chennakesava CS, Biedermann B, Dehio C. 2006. A translocated bacterial protein protects vascular endothelial cells from apoptosis. PLoS Pathog 2:e115. doi: 10.1371/journal.ppat.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kennedy AD, DeLeo FR. 2009. Neutrophil apoptosis and the resolution of infection. Immunol Res 43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 60.Moulding DA, Akgul C, Derouet M, White MR, Edwards SW. 2001. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol 70:783–792. [PubMed] [Google Scholar]
- 61.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. 1993. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A 90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rogers S, Wells R, Rechsteiner M. 1986. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 63.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. 2004. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 23:5301–5315. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- 64.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. 2006. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Luo F, Zhao H. 2014. Paraquat-induced reactive oxygen species inhibit neutrophil apoptosis via a p38 MAPK/NF-kappaB-IL-6/TNF-alpha positive-feedback circuit. PLoS One 9:e93837. doi: 10.1371/journal.pone.0093837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lifshitz Z, Burstein D, Schwartz K, Shuman HA, Pupko T, Segal G. 2014. Identification of novel Coxiella burnetii Icm/Dot effectors and genetic analysis of their involvement in modulating a mitogen-activated protein kinase pathway. Infect Immun 82:3740–3752. doi: 10.1128/IAI.01729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ho T, Htwe KK, Yamasaki N, Zhang GQ, Ogawa M, Yamaguchi T, Fukushi H, Hirai K. 1995. Isolation of Coxiella burnetii from dairy cattle and ticks, and some characteristics of the isolates in Japan. Microbiol Immunol 39:663–671. doi: 10.1111/j.1348-0421.1995.tb03254.x. [DOI] [PubMed] [Google Scholar]
- 68.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang X, Goncalves R, Mosser DM. 2008. The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14:Unit 14.1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.