

Abstract
The human 20S proteasome inhibitor scytonemide A (1), a macrocyclic imine originally isolated from the cyanobacterium Scytonema hofmanni, was synthesized via a biomimetic solid-phase peptide synthesis (SPPS) approach employing the Weinreb AM resin. Utilizing this approach, cyclization of the protected heptapeptide via formation of the imine bond occurred spontaneously upon cleavage from the resin in the presence of a reducing agent and subsequent aqueous work-up. The final deprotection step necessary to produce the natural product was accomplished under slightly basic conditions, facilitating cleavage of the silyl ether group while leaving the macrocycle intact. Purification of the synthetic scytonemide A was accomplished via normal phase flash column chromatography, potentially facilitating larger scale preparation of the compound necessary for future mechanistic and SAR studies. The structure of the target compound was confirmed by NMR spectroscopy, which also shed light on differences in the spectroscopic data obtained for the synthetic and natural scytonemide A samples for some of the amide and alcohol signals in the 1H NMR spectrum.
Graphical Abstract
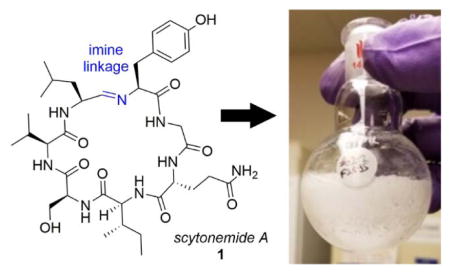
In 2010, Krunic et al.1 reported the isolation of scytonemides A (1) and B from the cultured freshwater cyanobacterium Scytonema hofmanni (UTEX 1834). Scytonemide A (1), a novel cyclic peptide containing an unusual imine linkage, was found to potently inhibit human 20S proteasome chymotrypsin catalytic activity in vitro with an IC50 of 96 nM.1 Although it has not yet been established whether the imine moiety is responsible for this activity, this structural motif does make scytonemide A an interesting and potentially challenging target for chemical synthesis. In addition to scytonemide A, imine linkages have only been observed in a small number of naturally occurring macrocycles, namely in nostocyclopeptides A1 (2), A2 (3),2 M1,3 and koranimine (4).4 Although scytonemide A, 2, and 3 are fairly homologous with respect to their amino acid sequences, nostocyclopetptide M1 and 4, despite also being heptapeptides, vary more dramatically.
In addition to the imine functionality, the cyclic nature of this natural product also makes it a potentially attractive target from a drug discovery and development perspective. In general, macrocyclic peptides have received significant attention recently due to their large surface areas, limited conformational flexibility, and promising drug properties, suggesting the ability to better inhibit protein-protein interactions.5–8 As further evidence of this trend, Malins et al.9 recently published the synthesis of a series of peptide macrocycles, utilizing the cyclic imines 1 and 4 as intermediates in the production of more stable amine adducts with non-native motifs. This synthesis required the preparation of the linear amino aldehyde precursor of 1 which was then cyclized via titration with triethylamine (TEA) in deuterated methanol. Ultimately, the key cyclization event effectively exploited the pH dependent equilibrium between the cyclic and acyclic forms of these peptides previously demonstrated with the nostocyclopeptides.10
Unfortunately, in the case of scytonemide A, further development as a potential proteasome inhibitor has been hampered by the relatively small quantities that can be successfully isolated from the cyanobacterium (i.e., 1 mg from 9 L of culture).1 In addition, despite the efficient syntheses and subsequent transformations of the imines noted above,8 the purification of scytonemide A itself was not reported as a part of this effort. With this in mind, we were interested in developing an alternative method for the preparation of scytonemide A that could produce the quantities of the natural product necessary for biological testing. In considering the total synthesis of 1, two primary goals for this effort were established, namely the efficient and rapid synthesis of the natural product and the isolation of the desired compound without the need for high pressure liquid chromatography (HPLC) purification.
RESULTS AND DISCUSSION
Similar to the approaches of Malins9 and Kopp,10 solid-phase peptide synthesis (SPPS) methodology was envisioned to facilitate rapid access to the heptapeptide precursor of 1. In this case, however, inspiration for the choice of resin/linker was drawn from the biosynthesis of nostocyclopeptide A1 (Figure 1A), proceeding via reductive cleavage from the non-ribosomal peptide synthetase (NRPS).11
Figure 1.

A) Biosynthesis of 2 via reductive cleavage of peptide from Ncp non-ribosomal peptide synthetase (NRPS). B) “Biomimetic” formation of 1 via cleavage of peptide from Weinreb resin.
In studies of the mechanism through which this process takes place, reductive cleavage of thioester-containing peptidyl-CoA substrates designed to mimic the natural system were shown to result in direct cyclization (self-assembly) to form the imines as detected by HPLC-MS in both the presence and absence of the recombinant NRPS reductase domain and the adjacent peptidyl-carrier protein.10 This study demonstrated that the cyclization reaction was not dependent on the enzyme itself. By analogy, therefore, a reductive cleavage of the heptapeptide directly from a solid-phase resin (Figure 1B) would also be expected to result in formation of the desired imine.
With this in mind, the Weinreb aminomethyl (AM) resin was chosen for the synthesis since reduction of the N-methoxy-N-alkylamide linkage in the presence of a reducing agent has been shown to provide the corresponding aldehyde through the unique reactivity of the Weinreb amide motif.12,13 Since the introduction of the Weinreb amide resin,14 it has been recognized as a useful tool for the preparation of amino and peptidyl aldehydes.15–18 Since the introduction of the Weinreb amide resin,14 it has been recognized as a useful tool for the preparation of amino and peptidyl aldehydes.15–18 For instance, fellutamide B, another peptidyl aldehyde with proteasome inhibitory activity, was synthesized using a Weinreb amide resin approach.19 As opposed to these applications, cleavage of the scytonemide A heptapeptide from this resin was expected to generate the desired peptidyl aldehyde precursor that would subsequently undergo intramolecular condensation with the free N-terminal amine to provide the desired cyclic imine.
In practice, the initial route taken to synthesize 1 employing the Weinreb resin is illustrated in Scheme 1. The on-resin heptapeptide 5a was assembled following standard SPPS protocols.19 During this process, the Ser, D-Gln, and Tyr residues were introduced with the relatively stable t-butyl and trityl protecting groups due to the potentially harsh basic and reductive conditions of the subsequent cleavage step and to provide increased lipophilicity for the extraction and purification of the cyclized product upon reduction.
Scheme 1.

Total synthesis of 1 using Weinreb AM resin and acid-labile protecting groups.
Once assembled, the terminal Fmoc group of heptapeptide 5a was removed with piperidine. The N-deprotected resin-bound amino peptide was then treated with excess lithium aluminum hydride (LAH) to liberate the desired peptidyl aldehyde from the resin. This type of reduction has also been reported by Payne and coworkers in their total synthesis of fellutamide B.19 The selective reduction of the Weinreb amide in these transformations is particularly remarkable due to both the heterogeneous nature of the reducing agent and the presence of other potentially reactive carbonyl groups. Analysis of the product mixture using mass spectrometry indicated the direct formation of the cyclic imine 6a, which could be purified using normal phase silica-gel column chromatography. The formation of the imine was further supported by the presence of a doublet (J = 6.9 Hz) at δH 6.94 in the 1H NMR spectrum. This doublet showed a correlation with the carbon at δC 165.7 in an HSQC experiment (Supporting Information, Page S4), correlating with the data reported for the imine signal of scytonemide A itself.1 Surprisingly, analysis of the crude reaction mixture by 1H NMR showed no diagnostic signal for the presence of the aldehyde, suggesting that the condensation reaction appears to be favored under conditions in which the aldehyde is generated in the presence of the basic N-terminal amine. The facile nature of this ring closure may be explained by drawing comparisons between compound 6 and the closely related structures of the nostocyclopeptides. The cyclization of the nostocyclopeptides has been suggested to be aided by the “elaborated prefolding”20 of the acyclic aldehyde precursor of 2, allowing it to adopt a “product-like” conformation in solution, bringing the aldehyde and amine functionality into close proximity and facilitating the imine self-assembly.10
Interestingly, the yield of the product obtained could be improved by repeating the reduction step two additional times using the same resin, cleanly providing quantities of the desired imine upon each treatment. Purification of these combined fractions via column chromatography provided an overall yield of 31% based on the theoretical loading of the resin (0.441 mmol/g). The successful re-subjection of the resin to obtain additional macrocyclic product suggests that the steric environment of the resin hinders nucleophilic attack of the Weinreb amide motif to some degree in the presence of the attached heptapeptide.
With compound 6a in hand, at this stage all that remained in order to complete the synthesis of 1 was the global deprotection of the acid-labile side chain protecting groups. Unfortunately, but not unexpectedly, treatment of 6a with trifluoroacetic acid (TFA) resulted in hydrolysis of the imine and the formation of a complex mixture of cyclic and acyclic products as observed by MS analysis. Subjection of this mixture to solid Na2CO3 in MeOH successfully regenerated the imine, although small amounts of byproducts (possibly epimers) with similar retention times and identical molecular weights were also observed in the HPLC trace for the product mixture. HPLC purification was able to successfully provide 1.1 mg of 1 from a 7 mg sample, but the low yield led us reconsider the synthetic route, specifically with regard to the protecting groups employed on the Tyr1, D-Gln3, and Ser5 residues.
Initial attempts to address the requirements for the protecting groups for the LAH reduction step focused on the sequential removal of the trityl and then t-butyl groups. During these studies, it was shown that the trityl protecting group on the D-Gln3 residue was not necessary for peptide synthesis21 or its cleavage from the resin, as the efficiencies of the subsequent SPPS coupling steps were not affected and the cyclic imine 6b could still obtained through the reduction of 5b without undesired reduction of the unprotected amide. Removal of the remaining t-butyl protecting groups at this stage, however, remained problematic. Under mild acidic conditions using trifluoroethanol (TFE), the t-butyl groups were unaffected. Employing stronger conditions (TFA) once again resulted in undesired hydrolysis of the imine. Based on the difficulty in removal of these t-butyl groups after the imine was formed, a global deprotection step was carried out on the resin to remove these groups to provide 5c. Treatment of this material with LAH resulted in a complex mixture of highly polar products. Although 1 could be detected by TLC and MS analyses, purification of the desired compound from the mixture proved to be difficult without the use of HPLC.
Therefore, a new strategy was required in order to effectively produce scytonemide A. It was obvious from the results obtained thus far that the cyclization event could be successfully accomplished with the aid of protecting groups, but the groups employed would need to be revised in order to maintain the desired levels of lipophilicity to aid in chromatographic separation of this product and would also need to be cleaved under non-acidic conditions in order to keep the imine moiety intact. Silyl protecting groups ideally fit these criteria due to their strong lipophilic properties, relative stability to the coupling steps, and cleavage under mild, non-acidic conditions with tetrabutylammonium fluoride (TBAF). For this reason, the t-butyldimethylsilyl (TBS) protected ethers were targeted for both the Ser and Tyr residues.
The TBS-protected Fmoc-Tyr-OH required for the synthesis of the heptapeptide was prepared according to the procedure of Taleski et al.22 This same procedure was also modified to produce the corresponding TBS-protected Fmoc-Ser-OH (Scheme 2). For the preparation of this compound, commercially available Fmoc-Ser(tBu)-OH (7) was first converted into the allyl ester 8. The t-butyl ether was then deprotected with TFA and the primary alcohol was re-protected as the TBS ether 9. Removal of the allyl group from 9 was affected with Pd(PPh3)4 to form the desired amino acid 10, which was then used for preparation of the heptapeptide.
Scheme 2.
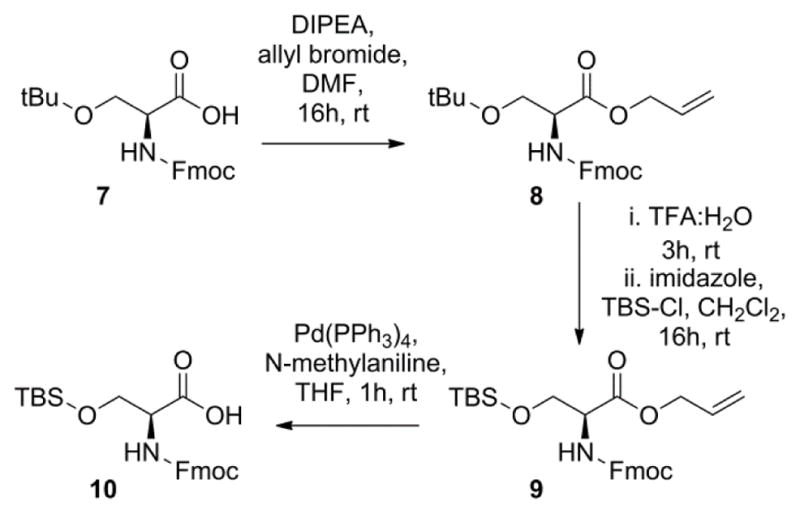
Synthesis of Fmoc-Ser(TBS)-OH via application of the protocol of Taleski et al.22
With the TBS-protected tyrosine and serine residues in hand, the heptapeptide was once again assembled utilizing these amino acids to produce 11a (Scheme 3), applying the same methodology used to produce 5. Subjection of 11a to LAH was expected to yield the bis-silylated macrocycle. Once again, the reduction proceeded smoothly and an imine macrocycle was obtained as the major product. Analysis by mass spectrometry, however, indicated formation of the mono-silylated imine product 12a as opposed to the bis-protected product. In an effort to determine which silyl group had been lost during the course of this reaction, further spectroscopic data was needed.
Scheme 3.

Revised protecting group strategy using silyl-protected Ser and Tyr for completion of the total synthesis of 1.
A comparison of the 1H NMR and 13C NMR spectroscopic data of silylated peptide 12a with the isolated 1 indicated little to no difference in the Ser5 3a (ΔδH = 0.00 ppm) and 3b (ΔδH = 0.01 ppm) proton shifts and 3a/b carbon (ΔδC =0.01 ppm) shifts. In contrast, clear differences in the chemical shifts between the tyrosine 5/9 (ΔδH = 0.11 ppm) and 6/8 (ΔδH = 0.09 ppm) protons and 5/9 (ΔδC = 0.26 ppm) and 6/8 (ΔδC = 4.54 ppm) carbons were observed, suggesting that the silyl protecting group had been removed from the Ser5 residue. A selective 1D NOESY experiment also revealed a through space correlation between the methyl protons of the TBS group and the protons on Tyr1 positions 6/8 (Figure S2). In addition, an HMBC experiment revealed that an exchangeable proton, observed as a triplet around 5.55 ppm, was found to have 2J and 3J correlations with Ser5-C2 and Ser5-C3, indicating that this was the Ser5 alcohol proton (Figure S8). Overall, these spectroscopic data confirmed the structure of the mono-silylated imine 12a, verifying the presence of the TBS group on the Tyr1 residue. The loss of the protecting group during the course of this reaction is interesting because TBS ethers are typically compatible with reductive reaction conditions. There have been reports, however, of the loss of TBS groups in the presence of LAH when other heteroatoms are proximal to the silyl ether.23–26 Mechanistically, de Vries and coworkers suggested that this type of deprotection proceeds through an intramolecular hydride delivery from an aluminum species coordinated to the nearby heteroatom group through either a deprotonation or reduction event.23 In the present case, it is conceivable that the silyl group removal is affected by coordination of the aluminum species to the N-atom of the Ser residue, resulting in a seven-membered transition state for delivery of the hydride.
In an effort to improve the stability of the silyl protecting group on the Ser residue, the more stable and sterically encumbered triisopropylsilyl (TIPS) ether (13) was also prepared and introduced into the heptapeptide. In this case, treatment with LAH resulted in the production of two products in an approximate ratio of 2.4:1. The major product in this case was still found to be imine 12a, corresponding to removal of the TIPS group. In addition, however, the bis-silylated product 12b could be isolated in this case, reflecting the attenuated reactivity of the TIPS group towards reductive deprotection as compared to the TBS moiety. Because 12b was obtained in lower yields, however, imine 12a obtained from reduction of 11a was taken on to compound 1.
To complete the synthesis of scytonemide A, therefore, silyl ether 12a was subjected to standard silyl deprotection conditions using TBAF. These slightly basic reaction conditions cleanly provided the deprotected material in 51% yield without affecting ring opening or epimerization, based on the presence of a single imine signal in the 1H NMR. Importantly, this product could be cleanly isolated as a white solid using normal phase silica gel column chromatography and did not require HPLC purification. The 1H and 13C NMR data1 for the natural scytonemide A and the synthetic sample are shown in Table 2. Although the resonances observed in the 13C spectrum agree with the reported values, the 1H NMR spectra of the product obtained did not completely match the reported isolation spectra. This was quite evident when the spectra were overlaid (Figure S10) as a few of the previously reported signals in the aromatic/amide region were seen to shift differently and two “new” resonances were observed at 5.55 and 9.15 ppm. Although differences were noted in comparison to the isolated material, the obtained spectrum of the synthetic material was identical to the spectrum reported by Malins et al.9 Based on the similarities in the 13C spectra and the alkyl region of the 1H spectra reported for all three samples, we were relatively confident that the synthetic material was in fact scytonemide A. In order to explain the observed differences and to unambiguously confirm the identity of the synthesized product, however, more extensive NMR experiments were undertaken.
In an effort to examine and assign the protons in the aromatic/amide region, an HSQC experiment was used to confirm the presence of the macrocyclic imine and identify which protons lacked a carbon correlation, suggesting that these were “exchangeable” heteroatom protons (Figure 2 and Page S27). Based on this experiment, the imine and tyrosine protons could be easily identified and the fact that only one macrocyclic imine signal existed in this sample was confirmed. The remaining protons were likely amide -NH or possibly alcohol –OH protons. This was further supported by the disappearance of these signals, as well as the signal at 5.55 ppm, in deuterated methanol (Page S28). Further comparison of the silylated precursor 12a and the final product, along with HMBC (Figures S3-S8) and selective TOCSY spectra (Figure S9) for these compounds facilitated identification of individual amide and alcohol protons (Figure S1), and confirmed the carbon framework of scytonemide A.
Figure 2.

Expanded view of HSQC spectrum of 1 showing the cross-peak corresponding to the diagnostic imine moiety.
At this stage, the “new” signals at 5.55 and 9.15 ppm remained to be assigned. As all of the amides were accounted for, it seemed likely that these two protons corresponded to the Ser and Tyr –OH signals. Neither of these resonances had been reported in the spectrum of the isolated material, although very broad signals are visible in these regions in the 1H NMR spectrum provided in the Supporting Information of the isolation paper. A triplet (5.58 (t, J = 5.4 Hz, 1H) similar to the splitting of the signal at 5.55 ppm of scytonemide A also appeared in the 1H spectrum of compound 12a, but the singlet at 9.15 was not present in that spectrum. Based on the assignment of the silyl group on the Tyr residue of 12a and the observed splitting pattern, this suggested that the triplet was the Ser –OH proton. This was confirmed by a selective TOCSY experiment in which the triplet was irradiated, providing correlations with the Ser protons at 3.62, 3.89, and 4.90 ppm (Figure 3, Table 1). The remaining singlet at 9.15 ppm is likely the phenol signal of the Tyr residue. Despite the fact that no correlations were observed for this proton in the HMBC and TOCSY experiments, analogy for this assignment can be drawn from the reported spectrum of scytonemide B,1 which possesses a similar tyrosine -OH signal reported as a singlet at 9.24 ppm.
Figure 3.

1D TOCSY experiment, with irradiation of Ser OH (blue), overlaid with spectrum of 1 (red).
Table 1.
Comparison of 1H and 13C NMR Data of Naturally Occurring Scytonemide A with Synthesized Scytonemide A in DMSO-d6 a
| Natural scytonemide Ab | Synthetic 1 | ||||
|---|---|---|---|---|---|
| position | δC, type | δH, mult., (J in Hz) | δC, type | δH, mult., (J in Hz) | |
| Tyr1 | 1 | 172.93, C | 172.39, C | ||
| 2 | 74.0, CH | 3.53, dd (11.3, 2.8) | 73.5, CH | 3.54, dd (11.2, 2.4) | |
| 3a | 40.1, CH2 | 3.04, dd (13.5, 2.4) | 39.45, CH2 | 3.05, dd (13.4, 2.4) | |
| 3b | 2.48, ddd (13.5, 13.5, 2.4) | 2.48, dd (13.4, 11.2) | |||
| 4 | 127.6, C | 127.32, C | |||
| 5/9 | 131.4, CH | 6.86, d (8.5) | 130.91, CH | 6.86, d (8.5) | |
| 6/8 | 115.3, CH | 6.61, d (8.5) | 114.78, CH | 6.61, d (8.5) | |
| 7 | 156.4, C | 155.8, C | |||
| OH | 9.15, s | ||||
| Gly2 | 1 | 171.28, C | 170.84, C | ||
| 2a | 40.6, CH2 | 4.32, m | 39.88, CH2 | 4.32, dd (16.8,10.3) | |
| 2b | 3.42, m | 3.42, dd (16.8, 2.8) | |||
| NH | 7.78, dd (10.0, 2.6) | 7.75, dd (10.3, 2.8) | |||
| D-Gln3 | 1 | 172.58, C | 172.14, C | ||
| 2 | 53.8, CH | 4.33 | 53.52, CH | 4.31, dt (6.8, 6.5) | |
| 3a | 25.7, CH2 | 1.85, m | 25.23, CH2 | 1.85, m | |
| 3b | 1.76, m | 1.76, m | |||
| 4a | 31.8, CH2 | 2.11 | 31.26, CH2 | 2.11, ddd (15.5, 10.3, 5.6) | |
| 4b | 1.99 | 1.99, ddd (15.5, 10.3, 5.6) | |||
| 5 | 173.68, C | 173.20, C | |||
| NH | 8.55, d (6.3) | 8.54, d (6.5) | |||
| NH2 | 7.33, br s | 7.31, br s | |||
| 6.78, br s | 6.77, br s | ||||
| Ile4 | 1 | 170.15, C | 169.75, C | ||
| 2 | 58.0, CH | 4.17, dd (9.2, 3.9) | 57.51, CH | 4.18, dd (9.1, 3.9) | |
| 3 | 35.7, CH | 2.06, m | 35.22, CH | 2.06, m | |
| 3-Me | 16.4, CH3 | 0.86, d (7.0) | 15.95, CH3 | 0.86, d (7.0) | |
| 4 | 24.1, CH2 | 1.28, m | 23.61, CH2 | 1.28, m | |
| 5 | 12.5, CH3 | 0.83, m | 11.96, CH3 | 0.84, t (7.4) | |
| NH | 8.17, d (9.3) | 8.17, d (9.1) | |||
| Ser5 | 1 | 172.56, C | 172.06, C | ||
| 2 | 53.0, CH | 4.93, m | 52.58, CH | 4.90, td (9.5, 6.5) | |
| 3a | 62.8, CH2 | 3.87, dd (10.0, 10.0) | 62.22, CH2 | 3.89, ddd (9.9, 9.5, 5.8) | |
| 3b | 3.60, dd (9.8, 6.0) | 3.62, ddd (9.9, 6.5, 5.8) | |||
| OH | 5.55, t (5.8) | ||||
| NH | 7.36, d (9.0) | 7.37, d (9.5) | |||
| Val6 | 1 | 170.31, C | 169.77, C | ||
| 2 | 59.7, CH | 4.01, dd (7.7, 4.2) | 59.18, CH | 4.01, dd (7.6, 4.2) | |
| 3 | 29.0, CH | 2.22, m | 28.44, CH | 2.24, sept d (7.0, 4.2) | |
| 3-Me | 17.4, CH3 | 0.90 d (7.0) | 16.97, CH3 | 0.89, d (7.0) | |
| 4 | 19.7, CH3 | 0.93, d (7.0) | 19.18, CH3 | 0.93, d (7.0) | |
| NH | 8.40, br s | 8.16, d (7.6) | |||
| Leu7 | 1 | 166.0, CH | 6.80, d (1.8) | 165.60, CH | 6.81, d (1.8) |
| 2 | 49.6, CH | 4.37, m | 49.11, CH | 4.37, dddd (10.6, 9.3, 4.7, 1.8) | |
| 3a | 40.6, CH2 | 1.05 | 39.80, CH2 | 1.05, ddd, (13.6, 10.6, 5.3) | |
| 3b | 0.81 | 0.81, m | |||
| 4 | 24.8, CH | 1.29, m | 24.26, CH | 1.29, m | |
| 4-Me | 23.4, CH3 | 0.80, d (6.7) | 22.82, CH3 | 0.80, d (6.7) | |
| 5 | 21.7, CH3 | 0.72, d (6.5) | 21.16, CH3 | 0.72, d (6.5) | |
| NH | 7.90, d (9.4) | 7.78, d (9.3) | |||
Frequency: Natural 900 MHz for 1H, 226 MHz for 13C, synthetic 700 MHz for 1H, 176 MHz for 13C.
J. Nat. Prod. 2010, 73, 1927–1932.
Altogether, the spectroscopic data reported herein confirm the structural assignment of the synthetic material as scytonemide A. A complete NMR assignment of scytonemide A (1) is summarized in Table 1. In accordance with our plans from the outset of the project, purification of the desired product was successfully achieved using normal phase silica gel flash column chromatography. The overall synthetic strategy is expected to facilitate the preparation of the quantities of the natural product necessary for further biological evaluation of its proteasome inhibitory activity and subsequent preparation of structural analogues.
EXPERIMENTAL SECTION
General Experimental Procedures
Melting points were recorded using a Thomas Hoover Melting Point Capillary Apparatus. Optical rotation was measured on an Anton Paar MCP 150 Polarimeter. UV absorption spectra were measured on a Hitachi U2910 spectrophotometer. Infrared (IR) absorption spectra were recorded on a Thermo Nicolet 6700 FT-IR spectrometer. NMR spectra were recorded at 300 K using Bruker AV 300 MHz, AVIII 400 MHz, or Ascend 700 MHz NMR. 1H NMR chemical shifts are reported in parts per million (ppm) and are referenced to the solvent residual signals: CDCl3 (δ = 7.26 ppm) and DMSO-d6 (δ = 2.50). 13C-NMR chemical shifts are reported in ppm and are referenced to the solvent residual signal signals: CDCl3 (δ = 77.16 ppm) and DMSO-d6 (δ = 39.52). Electrospray ionization mass spectra (ESI-MS) were recorded on a Thermo LTQ Orbitrap mass spectrometer. Thin layer chromatography was performed on Sorbtech UV254 aluminum backed sheets. The compounds were visualized by UV light at 254 nm or by staining with KMnO4. Flash column chromatography was carried out using Sorbtech 40–63 μm silica gel with solvents as described. Ratios of solvent systems used for flash chromatography are expressed in v/v as specified. LC-MS was performed using Waters 2795 HPLC with Waters 2996 Photodiode Array Detector operating at 200–800 nm, and compounds were loaded using MeCN or MeOH. An Ace Excel 3 C18-PFP column (150 x 4.6 mm, 1.7 um particle size) was used with a flow rate of 1 mL·min−1. Compounds were loaded in MeCN or MeOH. Separations involved a mobile phase of H2O (solvent A) and MeOH (solvent B) using a linear gradient of 5%–95% solvent B over 25 min. All reactions were performed at room temperature unless otherwise stated. Commercially available chemicals were used as purchased. Ice/water was used as the temperature bath to achieve 0 °C. Dry THF and CH2Cl2 were obtained from Innovative Technology PureSolv system.
General Procedure for Solid Phase Peptide Synthesis (SPPS) using the Weinreb Aminomethyl (AM) Resin
Loading
Nα-Fmoc-N-methoxy-β-alanine AM resin (Weinreb AM resin) (1.0 equiv., substitution = 0.4–0.8 mmol/g) was swollen in CH2Cl2 (5 mL) for 20 min. The resin was then soaked in a solution of piperidine/DMF (4.86 mL, 1:9 v/v) and agitated for five minutes. The solvent was then filtered off and the procedure was then repeated for 10 min. The resin was then filtered with DMF (2 x 5 mL), CH2Cl2 (2 x 5 mL), and DMF (2 x 5 mL). A Chloranil test was then applied. After a positive Chloranil test (colored beads), the resin was soaked in a solution of Fmoc-Leu-OH (3.0 equiv.), 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate (HATU) (2.9 equiv.), and N,N-Diisopropylethylamine (DIPEA) (3.5 equiv.) in DMF (0.2 M relative to Fmoc-Leu-OH). The beads were then agitated for 1 h. The solvent was then filtered off and the procedure was then repeated for 1 h. The resin was then filtered with DMF (2 x 5 mL), CH2Cl2 (2 x 5 mL), and DMF (2 x 5 mL). Chloranil test was then applied to ensure proper coupling.
Loading Efficiency
Loading efficiency was determined in accordance with the procedure reported by Malins et al.,9 employing the extinction coefficient reported by Eissler et al.27 The Fmoc-Leu loaded Weinreb resin was shaken in a solution of piperidine/DMF (3 mL, 1:9 v/v) for 5 min. The solvent was filtered off and the procedure was then repeated for 10 min. The filtrates were then combined and an aliquot (150 μL) was diluted 20-fold to 3 mL. 300 μL of this solution was diluted 10-fold to 3 mL and placed in a quartz cuvette to measure UV absorbance of the piperidine-fulvene adduct (λ = 289.8 nm, εc = 6089 M−1 cm−1) for quantification of the Leu residue loaded onto the resin. The theoretical maximum yields of the isolated peptides are based on the numerical value obtained from this resin loading, as opposed to the theoretical loading reported by the manufacturer.
Fmoc Deprotection
The resin was then shaken in a solution of piperidine/DMF (3 mL, 1:9 v/v) for 5 min and then repeated for 10 min. The resin was filtered and washed with DMF (2 x 5 mL), CH2Cl2 (2 x 5 mL), and DMF (2 x 5 mL). A Chloranil test was applied to ensure deprotection had occurred. The Fmoc deprotection process was repeated after each amino acid coupling.
Amino Acid Coupling
To the resin (1.0 equiv.) was added a solution of amino acid (3.0 equiv.), HATU (2.9 equiv.), and DIPEA (3.5 equiv.) in DMF (0.2 M relative to amino acid) was added to the resin and agitated for 15 min. The solvent was then filtered off and the procedure was then repeated for 15 min. The resin was filtered and washed with DMF (2 x 5 mL), CH2Cl2 (2 x 5 mL) and DMF (2 x 5 mL). The Chloranil test was applied to ensure coupling had occurred.
Chloranil Test:28
After each Fmoc deprotection and amino acid coupling step, the Chloranil test for free amines was used to ensure that the Fmoc group had been removed or the proper amino acid had been coupled. Solutions of acetaldehyde/DMF (2% v/v) and chloranil/DMF (2% m/v) were prepared. After filtration of the resin, a glass capillary tube was used to remove a small amount of the beads. The capillary tube was then placed into an Eppendorf tube. The acetaldehyde/DMF was then added dropwise (100 μL), followed by the chloranil/DMF (100 μL). After chloranil/DMF addition, the Eppendorf tubes were slightly shaken to ensure mixing with the resin, then allowed to stand for 1 min. The resin was visually checked for color development: deep blue colored beads indicated free amine (complete Fmoc deprotection), while colorless/clear beads indicated no free amine (complete coupling of the amino acid).
General Procedure for Cleavage from Weinreb AM Resin
The on-resin heptapeptide (1.0 equiv.) was swollen in dry THF (0.05 M resin) and cooled to 0 °C. LiAlH4 (11.0 equiv.) was added portionwise and the mixture was allowed to stir for 2.25 h. The mixture was again cooled to 0 °C and diluted with EtOAc (5 mL). The mixture was then quenched with saturated Rochelle’s salt solution (5 mL) and allowed to stir for 15 min to ensure quenching. The mixture was then filtered using a fritted filter to remove any solid particulates. The resulting filtrate was extracted three times using EtOAc. The combined EtOAc fractions were concentrated in vacuo to yield the desired cyclic imine.
Tyr(tBu)1-Gly2-D-Gln(trt)3-Ile4-Ser-(tBu)5-Val6-Leu7 cyclic imine (6a)
Fmoc-Leu loaded Weinreb AM resin (1000 mg, substitution = 0.331 mmol) was sequentially coupled with Fmoc-Val-OH, Fmoc-Ser(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Gln(trt)-OH, Fmoc-Gly-OH, and Fmoc-Tyr(tBu)-OH according to the SPPS protocol. Fmoc deprotection was performed as stated to produce the free amine on the Tyr1 residue. Cleavage from the resin as detailed yielded the crude product. The crude residue was then purified via flash chromatography (eluent: 44:40:10:5:1 EtOAc:hexanes:CH2Cl2:MeOH:TEA) to yield the desired cyclic imine 6a as an off-white powder (146.9 mg, 40% yield). mp. 138–140 °C; IR (neat): 3305, 2967, 1655, 1529 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 10.1 Hz, 1H), 7.93 (d, J = 5.1 Hz, 1H), 7.44 (dd, J = 9.6, 3.6 Hz, 1H), 7.36 – 7.15 (m, 15H), 7.06 (d, J = 6.3 Hz, 1H), 6.94 (d, J = 1.9 Hz, 1H), 6.91 (d, J = 8.5 Hz, 2H), 6.86 – 6.80 (m, 3H), 6.69 (d, J = 9.5 Hz, 1H), 6.44 (d, J = 9.2 Hz, 1H), 4.64 (tdd, J = 10.6, 4.23, 1.8 Hz, 1H), 4.50 (dd, J = 10.1, 3.2 Hz, 1H), 4.47 – 4.41 (m, 1H), 4.33 (dd, J = 9.2, 3.3 Hz, 1H), 4.26 (dd, J = 16.4, 9.6 Hz, 1H), 4.15 (p, J = 4.99 Hz, 1H), 3.88 (dd, J = 10.2, 4.8 Hz, 1H), 3.77 (dd, J = 11.2, 2.6 Hz, 1H), 3.48 – 3.38 (m, 2H), 3.27 (dd, J = 16.4, 3.6 Hz, 1H), 2.79 – 2.61 (m, 2H), 2.56 – 2.46 (m, 1H), 2.41 – 2.30 (m, 1H), 2.30 – 2.18 (m, 2H), 2.05 – 1.92 (m, 1H), 1.51 – 1.39 (m, 1H), 1.34 (s, 9H), 1.30 (s, 9H), 1.33 – 1.28 (m, 1H), 1.22 – 1.11 (m, 1H), 0.97 (d, J = 6.9 Hz, 3H), 1.02 – 0.93 (m, 2H), 0.93 – 0.85 (m, 9H), 0.83 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 173.31, 172.68, 172.66, 171.30, 171.01, 170.94, 170.88, 165.7, 154.0, 144.3, 132.5, 130.1, 128.7, 128.3, 127.4, 124.1, 78.4, 76.2, 74.5, 71.0, 61.8, 59.3, 57.7, 54.4, 53.6, 50.2, 41.4, 40.7, 40.4, 36.6, 33.1, 29.0, 28.8, 28.0, 25.0, 24.2, 23.2, 21.2, 19.8, 16.7, 16.5, 12.0. HRESIMS m/z 1121.6438 [M+Na]+ (calcd C63H86N8O9Na, 1121.6410).
(S)-allyl 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butoxy)propanoate (8)
To a cooled solution of Fmoc-L-Ser(tBu) (4.00 g, 10.43 mmol) in DMF (9.5 mL) at 0 °C was added dropwise N,N-diisopropylethylamine (DIPEA) (3.64 mL, 20.9 mmol) followed by allyl bromide (1.81 mL, 209 mmol). The mixture was allowed to warm to room temperature and stirred for 16 h. The reaction was then diluted with EtOAc (25 mL). The solution was washed with H2O (3 x 25 mL), dried (Na2SO4) and concentrated in vacuo. The resulting solid was purified by column chromatography (eluent: 90:10 hexanes:EtOAc) to afford the desired allyl ester 8 (3.75 g, 85%) as a white powder. IR (neat): 3443, 3338, 2973, 1726, 1508, 1198, 1103, 1085, 759, 740 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.5 Hz, 2H), 7.62 (t, J = 7.0 Hz, 2H), 7.43 (t, J = 7.4 Hz, 2H), 7.35 (t, J = 7.4 Hz, 2H), 5.95 (ddd, J = 22.7, 10.8, 5.6 Hz, 1H), 5.71 (d, J = 9.0 Hz, 1H), 5.38 (dd, J = 17.2, 1.2 Hz, 1H), 5.27 (dd, J = 10.5, 0.96 Hz, 1H), 4.70 (dd, J = 5.6, 0.98 Hz, 2H), 4.56 (dt, J = 8.9, 2.9 Hz, 1H), 4.52-4.34 (m, 2H), 4.29 (t, J = 7.2 Hz, 1H), 3.90 (dd, J = 9.0, 2.8 Hz, 1H), 3.65 (dd, J = 9.0, 3.1 Hz, 1H), 1.24 – 1.13 (br s, 9H); 13C NMR (101 MHz, CDCl3) δ 170.36, 156.15, 144.02, 143.84, 141.31, 141.30, 131.72, 127.71, 127.09, 127.07, 125.22, 125.17, 119.98, 118.48, 73.50, 67.21, 65.97, 62.15, 54.73, 47.18, 27.33; HRESIMS m/z 424.2108 [M+H]+ (calcd C25H30NO5, 424.2119), 446.1922 [M+Na]+ (cacld C25H29NO5Na, 446.1938).
(S)-allyl 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-((tertbutyldimethylsilyl)oxy) propanoate (9)
To a solution of TFA:H2O (4:1 v/v, 34 mL) was added 8 (3.27 g, 7.72 mmol). After 2 h the mixture was concentrated in vacuo to afford the desired alcohol intermediate as a yellow solid. This solid was immediately redissolved into CH2Cl2 (38.6 mL), followed by the addition of imidazole (1.68 g, 24.7 mmol). The mixture was then cooled to 0 °C prior to the addition of TBS-Cl (2.91 g, 19.3 mmol). The reaction was then allowed to stir for 16 h before quenching with H2O. Extraction was performed using CH2Cl2 (3 x 75 mL). The organic fractions were then combined, dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by column chromatography (eluent 90:10 v/v hexanes:EtOAc) to afford the TBS protected ether 9 (3.56 g, 95.8%) as a white powder. IR (neat): 3443, 3346, 2952, 2930, 1727, 1507, 1253, 1198, 837, 740 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.5 Hz, 2H), 7.65 (t, J = 8.0 Hz, 2H, ), 7.43 (t, J = 7.5 Hz, 2H), 7.34 (t, J = 7.4 Hz, 2H), 5.95 (ddd, J = 22.7, 10.9, 5.8 Hz, 1H), 5.69 (d, J = 8.6 Hz, 1H), 5.33 (dd, J = 37.0, 13.8 Hz, 2H), 4.70 (d, J = 5.70 Hz, 2H), 4.50 (dt, J = 8.70, 2.60 Hz, 1H), 4.47-4.35 (m, 2H), 4.29 (t, J = 7.3 Hz, 1H), 4.14 (dd, J = 10.1, 2.5 Hz, 1H), 3.97 – 3.86 (m, 1H), 0.96 – 0.85 (br s, 9H), 0.13 – 0.00 (d, 6H); 13C NMR (101 MHz, CDCl3) δ 170.17, 155.96, 144.00, 143.79, 141.32, 141.31, 131.58, 127.72, 127.09, 127.07, 125.22, 125.15, 119.99, 118.81, 67.24, 66.13, 63.69, 56.04, 47.15, 25.75, 18.23, −5.50, −5.56; HRESIMS m/z 482.2348 [M+H]+ (calcd C27H36NO5Si, 482.2357), 504.2163 [M+Na]+ (cacld C27H35NO5SiNa, 504.2177).
(S)-2-((((9H-fluoren-9-yl)methoxy)-carbonyl)amino)-3-((tertbutyldimethylsilyl)oxy) propanoic acid (10)
Compound 10 was prepared according to the procedure reported by Gagnon et al.29 1H NMR and 13C NMR spectra were in agreement with the previously reported data.
Tyr(TBS)1-Gly2-D-Gln3-Ile4-Ser5-Val6 -Leu7 cyclic imine (12a)
Fmoc-Leu loaded Weinreb AM resin (1000 mg, substitution = 0.295 mmol) was sequentially coupled with Fmoc-Val-OH, Fmoc-Ser(TBS)-OH, Fmoc-Ile-OH, Fmoc-D-Gln-OH, Fmoc-Gly-OH, and Fmoc-Tyr(TBS)-OH according to the SPPS protocol. Fmoc deprotection was performed as stated to produce the free amine on the Tyr1 residue. Cleavage from the resin as detailed yielded the crude product. The product was then purified via flash chromatography (eluent: 60:10:5:2.5 v/v EtOAc:MeCN:MeOH:H2O) to yield the desired cyclic imine 12a as an off-white powder (67.7 mg, 24% yield). IR (neat): 3296, 2958, 2929, 2857, 2539, 1647, 1541, 1510, 1258, 838 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.54 (d, J = 6.4 Hz, 1H), 8.24 – 8.11 (m, 2H), 7.86 – 7.71 (m, 2H), 7.38 (d, J = 9.7 Hz, 1H), 7.31 (s, 1H), 6.97 (d, J = 8.5 Hz, 2H), 6.79 (d, J = 1.7 Hz, 1H), 6.77 (s, 1H), 6.70 (d, J = 8.4 Hz, 2H), 5.58 (t, J = 5.4 Hz, 1H), 4.92 (td, J = 9.9, 6.1 Hz, 1H), 4.42 – 4.27 (m, 3H), 4.19 (dd, J = 9.2, 3.9 Hz, 1H), 4.02 (dd, J = 7.6, 4.2 Hz, 1H), 3.89 (td, J = 9.7, 6.7 Hz, 1H), 3.79 – 3.60 (m, 1H), 3.58 (dd, J = 11.2, 2.5 Hz, 1H), 3.43 (dd, J = 16.7, 2.5 Hz, 1H), 3.11 (dd, J = 13.2, 2.3 Hz, 1H), 2.60-2.55 (m, 1H), 2.31 – 2.19 (m, 1H), 2.18 – 1.95 (m, 3H), 1.81 – 1.71 (m, 2H), 1.45 – 1.26 (m, 3H), 1.11 – 0.98 (m, 1H), 0.94 (s, 9H), 0.92 (d, J = 6.3 Hz, 3H), 0.90-0.84 (m, 7H), 0.83 (s, 3H), 0.81 (d, J = 6.7 Hz, 3H), 0.73 (d, J = 6.5 Hz, 3H), 0.16 (s, J = 3.0 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 173.23, 172.32, 172.13, 172.06, 170.82, 169.82, 169.76, 165.69, 153.52, 131.17, 130.13, 119.32, 73.29, 62.23, 59.21, 57.52, 53.33, 52.52, 49.21, (39.90, 39.72, 39.41 HSQC), 35.23, 31.26, 28.45, 25.75, 25.52, 25.25, 24.29, 23.62, 23.03, 20.92, 19.18, 16.99, 15.95, 11.95, −4.47, −4.48; HRESIMS m/z 881.4924 [M+Na]+ (calcd C42H70N8O9SiNa, 881.4927).
Tyr(TBS)1-Gly2-D-Gln3-Ile4-Ser(TIPS)5-Val6-Leu7 cyclic imine (12b). )
Fmoc-Leu loaded Weinreb AM resin (1000 mg, substitution = 0.295 mmol) was sequentially coupled with Fmoc-Val-OH, Fmoc-Ser(TBS)-OH, Fmoc-Ile-OH, Fmoc-D-Gln-OH, Fmoc-Gly-OH, and Fmoc-Tyr(TBS)-OH according to the SPPS protocol. Fmoc deprotection was performed as stated to produce the free amine on the Tyr1 residue. Cleavage from the resin as detailed yielded the crude product. The product was then purified via flash chromatography (eluent: 70:10:5:2.5 v/v EtOAc:MeCN:MeOH:H2O) to yield the desired cyclic imine 12b as an off-white powder (19.8 mg, 7% yield). IR (neat): 3305, 2959, 2857, 2478, 1651, 1510, 1462, 1261, 918 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 6.0 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.91 – 7.84 (m, 1H), 7.68 (s, 1H), 7.52 (d, J = 8.7 Hz, 1H), 7.33 (s, 1H), 7.01 (d, J = 1.7 Hz, 1H), 6.91 (d, J = 8.4 Hz, 2H), 6.78 (s, J = 14.8 Hz, 1H), 6.73 (s, 1H), 6.70 (d, J = 8.4 Hz, 2H), 4.90 – 4.81 (m, 1H), 4.38 – 4.29 (m, 3H), 4.12 (dd, J = 8.5, 4.0 Hz, 1H), 4.05 (d, J = 6.5 Hz, 1H), 3.83 (dd, J = 13.2, 7.9 Hz, 2H), 3.63 (dd, J = 10.7, 2.6 Hz, 1H), 3.43 (dd, J = 16.5, 5.5 Hz, 1H), 3.15 – 3.11 (m, 1H), 2.53 (d, J = 2.4 Hz, 1H), 2.22 (ddd, J = 15.5, 11.7. 5.3 Hz, 2H), 2.14 (dd, J = 9.5, 6.3 Hz, 1H) 2.02 (ddd, J = 9.9, 7.9, 4.3 Hz, 3H), 1.84 – 1.77 (m, 2H), 1.45 – 1.36 (m, 3H), 1.05 (d, J = 3.1 Hz, 9H), 0.94 (s, 18H), 0.85 (d, J = 3.7 Hz, 3H), 0.83 (d, J = 4.4 Hz, 6H), 0.78 (d, J = 6.5 Hz, 5H), 0.16 (s, 6H). 13C NMR (176 MHz, DMSO-d6) δ 173.42, 172.51, 171.58, 171.51, 170.33, 170.24, 170.07, 166.27, 166.26, 153.61, 130.78, 130.71, 130.22, 119.79, 119.35, 119.30, 73.76, 64.31, 58.27, 53.16, 49.74, 45.61, 40.49, 40.02, 35.34, 35.32, 31.27, 28.75, 25.58 25.55, 24.29, 23.44, 22.85, 21.17, 19.11, 18.07, 17.95, 17.94, 17.88, 17.80, 17.79, 15.63, 11.80, 11.66, 11.40, 11.35, 8.60, −4.49, −4.52., HRESIMS m/z 1037.6237 [M+Na]+ (calcd C51H90N8O9Si2Na, 1037.6262).
(S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-((triisopropylsilyl)oxy)propanoic acid (13)
Compound 13 was prepared according to the procedure reported by Aguda et al.30 1H NMR and 13C NMR spectra were in agreement with the previously reported data.
Scytonemide A (1)
Cyclic imine 12a (25 mg, 0.03 mmol) was dissolved in MeOH (3 mL). TBAF (33 μL, 0.033 mmol) was then added dropwise. Reaction mixture was allowed to stir for 16 h. Solvent was then removed in vacuo, yielding an off-white powder. Trituration with hexanes (3 x 5 mL) was then performed to remove any nonpolar impurities. Crude material was then purified by column chromatography (eluent 60:10:5:2.5 v/v EtOAc:MeCN:MeOH:H2O) to yield desired product 1 (11.5 mg, 51%) as a white solid. – 14 (c 0.05, MeOH), isolated scytonemide A – 15 (c 0.0013, MeOH)1; IR (KBr): 3613, 3346, 2962, 2365, 1647, 1543, 1519, 1416, 1236, 1068 cm−1; Table 1 for 1H NMR and 13C NMR data; HRESIMS m/z 767.4024 [M+Na]+ (calcd C36H56N8O9Na, 767.4063).
Supplementary Material
Acknowledgments
The authors would like to thank the NIH for funding provided through the 2P01 CA125066 program project [Project 1 (LHR), Project 2 (JO), and Core B (JRF)], a CBIP Fellowship (GM08512) for TAW, and a Fellowship (5T32 AT007533) for PS. We also would like to thank Dr. Craig McElroy, Dr. Chunhua Yuan, and the OSU CCIC for assistance with acquisition of NMR data.
Footnotes
The authors declare no competing financial interest.
DEDICATION
Dedicated to Dr. Susan Band Horwitz, of Albert Einstein College of Medicine, Bronx, NY, for her pioneering work on bioactive natural products.
The Supporting Information is available free of charge on the ACS Publications website at DOI: ##.####/acs.jnatprod.#######.
1H and 13C NMR spectra for compounds 8, 9, and 12b; 1H, 13C, HSQC, and HMBC NMR spectra for imines 6a and 12a; and 1H, 13C, HSQC, and selective TOCSY NMR spectra for scytonemide A (1) (PDF)
References
- 1.Krunic A, Vallat A, Mo S, Lantvit DD, Swanson SM, Orjala J. J Nat Prod. 2010;73:1927–1932. doi: 10.1021/np100600z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golakoti T, Yoshida WY, Chaganty S, Moore RE. J Nat Prod. 2001;64:54–59. doi: 10.1021/np000316k. [DOI] [PubMed] [Google Scholar]
- 3.Jokela J, Herfindal L, Wahlsten M, Permi P, Selheim F, Vasconçelos V, Døskeland SO, Sivonen K. ChemBioChem. 2010;11:1594–1599. doi: 10.1002/cbic.201000179. [DOI] [PubMed] [Google Scholar]
- 4.Evans BS, Ntai I, Chen Y, Robinson SJ, Kelleher NL. J Am Chem Soc. 2011;133:7316–7319. doi: 10.1021/ja2015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott JD, Li SW, Brunskill APJ, Chen X, Cox K, Cumming JN, Forman M, Gilbert EJ, Hodgson RA, Hyde LA, Jiang Q, Iserloh U, Kazakevich I, Kuvelkar R, Mei H, Meredith J, Misiaszek J, Orth P, Rossiter LM, Slater M, Stone J, Strickland CO, Voigt JH, Wang G, Wang H, Wu Y, Greenlee WJ, Parker EM, Kennedy ME, Stamford AW. J Med Chem. 2016;59:10435–10450. doi: 10.1021/acs.jmedchem.6b00307. [DOI] [PubMed] [Google Scholar]
- 6.Wójcik P, Berlicki Ł. Bioorg Med Chem Lett. 2016;26:707–713. doi: 10.1016/j.bmcl.2015.12.084. [DOI] [PubMed] [Google Scholar]
- 7.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Angew Chem Int Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di L. AAPS J. 2014;17:134–143. doi: 10.1208/s12248-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malins LR, deGruyter JN, Robbins KJ, Scola PM, Eastgate MD, Ghadiri MR, Baran PS. J Am Chem Soc. 2017;139:5233–5241. doi: 10.1021/jacs.7b01624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopp F, Mahlert C, Grünewald J, Marahiel MA. J Am Chem Soc. 2006;128:16478–16479. doi: 10.1021/ja0667458. [DOI] [PubMed] [Google Scholar]
- 11.Becker JE, Moore RE, Moore BS. Gene. 2004;325:35–42. doi: 10.1016/j.gene.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 12.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 13.Mentzel M, Hoffmann HMR. Adv Synth Catal. 1997;339:517–524. [Google Scholar]
- 14.Fehrentz JA, Paris M, Heitz A, Velek J, Liu CF, Winternitz F, Martinez J. Tetrahedron Lett. 1995;36:7871–7874. [Google Scholar]
- 15.Boas U, Brask J, Jensen K. J Chem Rev. 2009;109:2092–2118. doi: 10.1021/cr068206r. [DOI] [PubMed] [Google Scholar]
- 16.Dinh TQ, Armstrong RW. Tetrahedron Lett. 1996;37:1161–1164. [Google Scholar]
- 17.Salvino JM, Mervic M, Mason HJ, Kiesow T, Teager D, Airey J, Labaudiniere R. J Org Chem. 1999;64:1823–1830. doi: 10.1021/jo981431r. [DOI] [PubMed] [Google Scholar]
- 18.O’Donnell MJ, Drew MD, Pottorf RS, Scott WL. J Comb Chem. 2000;2:172–181. doi: 10.1021/cc990071y. [DOI] [PubMed] [Google Scholar]
- 19.Giltrap A, Cergol K, Pang A, Britton W, Payne R. Mar Drugs. 2013;11:2382–2697. doi: 10.3390/md11072382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopp F. Macrocyclization and Fatty Acid Modification during the Synthesis of Nonribosomal Peptides. Universitätsbibliothek Marburg; 2008. [Google Scholar]
- 21.Isidro-Llobet A, Álvarez M, Albericio F. Chem Rev. 2009;109:2455–2504. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- 22.Taleski D, Butler SJ, Stone MJ, Payne R. J Chem – Asian J. 2011;6:1316–1320. doi: 10.1002/asia.201100232. [DOI] [PubMed] [Google Scholar]
- 23.de Vries EFJ, Brussee J, van der Gen A. J Org Chem. 1994;59:7133–7137. [Google Scholar]
- 24.Brussee J, Dofferhoff F, Kruse CG, Van Der Gen A. Tetrahedron. 1990;46:1653–1658. [Google Scholar]
- 25.Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K, Sorensen E. J Nature. 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- 26.Colvin EW, Beck AK, Seebach D. Helv Chim Acta. 1981;64:2264–2271. [Google Scholar]
- 27.Eissler S, Kley M, Bächle D, Loidl G, Meier T, Samson D. J Pept Sci. 2017;23:757–762. doi: 10.1002/psc.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vojkovsky T. Pept Res. 1995;8:236–237. [PubMed] [Google Scholar]
- 29.Gagnon D, Lauzon S, Godbout C, Spino C. Org Lett. 2005;7:4769–4771. doi: 10.1021/ol052034n. [DOI] [PubMed] [Google Scholar]
- 30.Aguda AH, Lavallee V, Cheng P, Bott TM, Meimetis LG, Law S, Nguyen NT, Williams DE, Kaleta J, Villanueva I, Davies J, Andersen RJ, Brayer GD, Brömme D. J Nat Prod. 2016;79:1962–1970. doi: 10.1021/acs.jnatprod.6b00215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.