

Abstract
Natural products have served as powerful therapeutics against pathogenic bacteria since the golden age of antibiotics of the mid-20th century. However, the increasing frequency of antibiotic-resistant infections clearly demonstrates that new antibiotics are critical for modern medicine. Because combinatorial approaches have not yielded effective drugs, we propose that the development of new antibiotics around proven natural scaffolds is the best short-term solution to the rising crisis of antibiotic resistance. We analyze herein synthetic approaches aiming to reengineer natural products into potent antibiotics. Furthermore, we discuss approaches in modulating quorum sensing and biofilm formation as a nonlethal method, as well as narrowspectrum pathogen-specific antibiotics, which are of interest given new insights into the implications of disrupting the microbiome.
Graphical abstract

1. INTRODUCTION
Among the greatest achievements of humankind in recent history stands the discovery and production of penicillin as a life-saving antibiotic. However, nearly a century of unchecked usage has rendered the world’s supply of antibiotics severely weakened; Sir Alexander Fleming noted in his Nobel lecture that underdosage can apply the selective pressure that induces bacteria to evolve resistance to these drugs. In this review, we contrast the traditional method of semisynthetic modifications to natural products with modern synthetic approaches to develop new antibiotics around the privileged scaffolds that informed drug discovery for decades in order to overcome contemporary antibiotic resistance.
In the 90 years since the discovery of penicillin (1), natural products have provided a major foundation for the development of antibiotic drugs. The reliance on natural products to provide new molecular entities for virtually every disease is also well established.1 Of the nine antibiotic classes in Figure 1, six represent naturally occurring compounds, with only three (the sulfonamides, fluoroquinolones, and oxazolidinones) conceived entirely through synthetic chemistry. We note the impressive structural diversity and complexity within the natural product antibiotics especially when compared to the synthetic classes.
Figure 1.
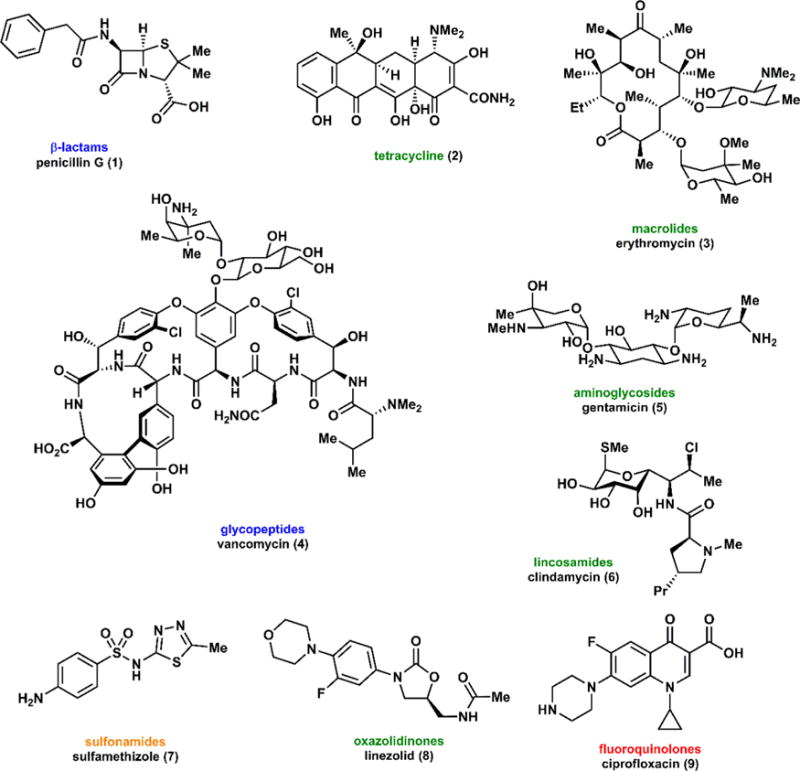
Representative classes of antibiotics of the modern era, excluding the arsenic-containing antibiotics of the early twentieth century. Color coding corresponds to the mechanisms of action in Figure 3.
Scientists have warned for decades that bacteria are rapidly evolving resistance to antibiotics.2–4 Resistance has proliferated due to a confluence of two key factors: the frequent prescription against infections of a nonbacterial nature, such as viral infections, and unregulated usage, which can lead to sublethal doses, permitting resistance to spread rapidly.5 We also observe that prescribing habits vary drastically from country to country; the United States is particularly likely to use recently developed antibiotics rapidly, possibly shortening their lifetime of efficacy.6 Analysis of the IMS Health Midas database indicated that between 2010 and 2014 consumption of antibiotics worldwide increased by 36%;7 the carbapenems and polymyxins, two “last-resort” drugs, have increased in usage by 45% and 13%, respectively. This resistance is extensively observed in hospitals where immunocompromised patients are particularly vulnerable.8 Hospital-acquired resistant infections have spread rapidly since the initial discovery of sulfonamide- and penicillin-resistant strains shortly after the introduction of these drugs in the 1930s and 1940s.9,10 In the U.S. and U.K. this problem has not abated, as nearly 40–60% of hospital-acquired S. aureus strains are methicillin-resistant.11 These public health threats will continue to rise without new antibiotics and meaningful changes in treating infections. Beyond prescription in humans, antibiotics find extensive use as prophylactic agricultural supplements to promote livestock growth and prevent diseases. It is estimated that the US livestock industry consumes a staggering 80% of antibiotics produced.5 Antibiotic-resistant strains of Salmonella have been identified in ground meat,12 and antibiotic use in livestock has been strongly linked to fluoroquinolone-resistant Salmonella.13 The need for new antibiotics is increasingly widely appreciated as a pressing concern by governments, scientists, and the general public.14,15 These factors, in tandem with the reduced research and development toward discovering new antibiotics, have worsened the recent eruption of antibiotic resistant bacterial populations across the globe. As the golden age of antibiotics has clearly ended, the most pessimistic view of the current state of affairs is that a postantibiotic era may be approaching.
As of 2013, the CDC identified that antibiotic resistance had contributed to at least two million illnesses and 23,000 deaths in the United States.11 Antibiotic resistance is also a global problem, with nearly 450,000 cases of drug-resistant tuberculosis and an estimated 170,000 deaths occurring in 2012 alone.16 Previously treatable infections are now serious concerns due to a lack of effective antibiotics, which is evident given the figures above. Furthermore, increased rates of world travel threaten to allow antibiotic resistance to spread rapidly.17 An overview of the estimated cases of antibiotic resistant infection and mortality in recent years demonstrates this frightening reality (Figure 2). Notably, antibiotic resistance correlates with high mortality; methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA) have estimated rates of 14% and 6.5%, respectively.11
Figure 2.

Total infections (gray) and deaths (black) in the US associated with various pathogenic bacteria.11 CRE = carbapenem-resistant enterococci; VRE = vancomycin-resistant enterococci; MDR = multidrug resistant.
1.1. Mechanisms of Action of Established Classes of Antibiotics
Antibiotics act on three primary targets within bacteria cells, with each class of drugs favoring one specific mode of action. These targets include the inhibition of (1) cell wall (peptidoglycan) synthesis; (2) protein synthesis (ribosome); or (3) DNA or RNA synthesis (DNA topoisomerase or RNA polymerase) (Figure 3). An excellent comprehensive summary of mechanisms of both action and resistance has been provided by Walsh and Wencewicz; however, we will also provide a brief overview.18,19
Figure 3.

Schematic representation of the three major mechanisms of action of widely used antibiotics, also noting the sulfa drugs.
1.1.1. Inhibition of Cell Wall Biosynthesis
The β-lactams, including penicillin (1) and the cephalosporins, inhibit the transpeptidation cross-linking step of peptidoglycan, the cell wall precursor. The strained four-membered β-lactam rings in these molecules are attacked by a serine hydroxyl group, acting as a suicide inhibitor against these critical enzymes. This acyl intermediate is slow to hydrolyze, which halts cell wall synthesis and weakens the reproducing cells.18 Vancomycin (4) also targets peptidoglycan synthesis by blocking transpeptidase from cross-linking the terminal D-Ala-D-Ala peptidyl tail. Instead of targeting an enzyme, vancomycin and other glycopeptides (refer to section 5) bind to this tail through a strong hydrogen-bonding network, preventing cell wall construction from proceeding. Uniquely, tunicamycin and other uridyl-peptide inhibitors act internally to block N-glycosylation in the lipid carrier cycle.
1.1.2. Inhibition of Protein Synthesis
Several classes of antibiotics target protein synthesis in the bacterial ribosome. The natural product-derived family of macrolides including erythromycin (3) targets the larger 50S subunit, binding in the polypeptide exit tunnel. Synthetically derived antibiotics such as the oxazolidinones, including linezolid (8), also bind to the 50S subunit. The tetracyclines, another class of natural product-derived antibiotics, bind to the smaller 30S subunit in its A-site (acceptor) for aminoacyl-tRNA, which contains the building blocks for translation.
1.1.3. Inhibition of DNA Replication
A third major target of antibiotics is nucleic acid replication and repair mechanisms. The synthetic antibiotic class of quinolones, including ciprofloxacin (9), targets DNA gyrase (DNA topoisomerase II), which assists in unwinding replicated DNA. These gyrases make cuts in both strands of DNA to relieve torsional stress in the replicating supercoil. Quinolones bind at these cleavage sites, stabilizing the DNA, and thus preventing further repair to the system.
1.1.4. Inhibition of Folate Biosynthesis
We also note the sulfonamide (sulfa) drugs, including sulfamethizole (9), which target folic acid pathways in bacteria. These drugs are structural mimics of p-aminobenzoic acid, a key precursor in the biosynthesis of folic acid.20 These drugs were among the first useful antibiotics, finding widespread use until the late 1940s when alternatives (such as penicillin) became available.21 Notably, a batch of elixir sulfanilamide-Massengill tainted with ethylene glycol led to the deaths of more than 100 people across the American South in 1937.22 This incident was a major impetus for the passage of the Federal Food, Drug, and Cosmetics Act in 1938, which established greater regulatory power of the US Food and Drug Administration (FDA).23 We note throughout this review that issues regarding safety are a major obstacle in antibiotic drug discovery and lead to the demise of many promising candidates.
1.2. Common Mechanisms of Antibiotic Resistance
Despite targeting many critical pathways, antibiotics have lost efficacy due to the evolution of resistance mechanisms in bacteria.18,24,25 It is hypothesized that antibiotics evolved as weapons for biological warfare between bacteria, which means that resistance has been developing for millennia. Because the “fittest” survive, these methods to resist antibiotic-induced death are passed on to the next generation through cell division, ensuring proliferation of the evolutionary advantages. Additionally, mechanisms of resistance are also shared among bacteria through horizontal gene transfer.26 While these systems are quite diverse across bacterial species, resistance mechanisms can be grouped according to some general similarities.27,28 In particular, bacteria have evolved three distinct mechanisms to resist antibiotics, including the reduced penetration of the drug via limited permeability and efflux pumps, mutation or modification of the binding target, and degradation of the drug itself (Figure 4).29–31 As the targeted processes are especially critical to bacterial life, there is considerable selection pressure, which drives the spread of resistance. Specific mechanisms of resistance relevant to each class of antibiotics we discuss will be addressed within the following sections; however, we provide a brief overview here.
Figure 4.

Schematic representation of general antibiotic resistance mechanisms.
1.2.1. Drug Efflux
Efflux pumps actively transport small molecules out of the bacterial cell. Found in both Gram-positive and Gram-negative bacteria, these pumps exhibit varying substrate scope and specificity. A tetracycline efflux system was among the first to be identified, which remains a critical factor in tetracycline resistance.32 Some pumps are known to remove various substrates, making these factors key contributors to multidrug resistance.33 These are classified into five major families which differ in structure, energy source, substrate specificity, and species distribution.34–36
1.2.2. Modification of the Target
Mutations or modifications of the binding sites of antibiotic targets are another common defense mechanism employed in resistance.29 Well-studied examples are the methylation of ribosomes to inhibit macrolide binding (section 3.2) and the mutation of the rifampin target RNA polymerase. In the first case, the methylation provides steric congestion, which clashes with the highly substituted macrolide backbone.29 In the second case, single point mutations are able to decrease the binding affinity of rifampin while maintaining the activity of the polymerase, allowing the bacteria to function normally.37
1.2.3. Drug Metabolism
The final key mechanism of resistance is the degradation or inactivation of the antibiotics themselves. This method relies on chemical transformations to destroy the inherent bioactivity of the compounds.29,33 The β-lactam antibiotics are particularly sensitive to inactivation, as bacteria have evolved β-lactamase enzymes to cleave the critical lactam ring. In addition, the aminoglycosides are also easily rendered inactive, as the amino and hydroxyl groups can be acetylated or phosphorylated leading to a reduction in binding affinity to ribosomal targets.38
1.3. Antibiotic Terminology
In line with calls, old and new, for standardized terminology across the broad field of antibiotics,39,40 we shall abide herein by the nomenclature described below. Susceptible bacteria are those which lack observable resistance to an antibiotic, which are often well-known drugs of the golden age of antibiotics, such as tetracycline, erythromycin, or penicillin. Strains of susceptible bacteria are commonly known as quality control (QC) strains. To discuss the efficacy of antibiotics in vivo, we frequently refer to minimum inhibitory concentrations (MIC), which is the lowest concentration of drug at which no bacterial growth is observed. When discussing efficacy of antibiotics against a particular biological target assayed in vitro, the IC50 is typically reported, which refers to the concentration at which a reduction by half of some measurable is observed. Given the inevitable clinical and epidemiological concerns that accompany any discussion of the need for antibiotics, we also intend to abide by Mendelson’s recent proposal.40 Accordingly, stewardship shall generally refer to the pressing need to use antibiotics responsibly at all levels of care, from the physician at the clinical level to policy decisions made at the regulatory level. We will avoid the use of the nonspecific term “antimicrobial” and discuss antibiotic resistance. Finally, Mendelson encourages a departure from the overused theme of a militaristic campaign against bacteria, especially given recent discoveries concerning symbiotic relationships with commensal bacteria (which greatly outnumber pathogens!).
1.4. Scope
Considering the multiple resistance mechanisms against natural product-derived antibiotics that have been observed, humankind must organize a multifaceted response to maintain a catalog of effective treatments. Two possible approaches of addressing this issue of resistance are (1) developing entirely new scaffolds or (2) modifying existing natural product scaffolds to extend their life. The second approach will be the major focus of this review. We analyze synthetic approaches to rejuvenate three key scaffolds, the macrolides, the tetracyclines, and the glycopeptides, all of which hail from the golden age of antibiotics. Furthermore, we outline approaches in developing small molecules derived from species involved in quorum sensing as a nonlethal approach. Finally, we discuss early stage investigations of other natural product antibiotics, especially those which demonstrate narrow-spectrum activity. We avoid discussions of biosynthetic tailoring of natural products; we note that van der Donk has recently cataloged biosynthetic studies of the lantibiotic peptides.41
2. STRATEGIES TOWARD ANALOG DEVELOPMENT
Due to a general lack of new antibacterial compounds in the drug discovery pipeline, extensive efforts are underway to fill this gap. There are three chemical approaches to generating new lead molecules (aside from efforts in new natural product isolation): (1) combinatorial chemistry generally resulting in primarily sp2-hybridized molecules,42,43 (2) methods which develop diverse libraries of complex scaffolds, and (3) modifications to previously identified antibiotic compounds (Figure 5). The first approach has been extensively employed by industry to provide lead compounds; however, it has been largely unsuccessful in generating new antibiotic scaffolds. In a recent review, Wright and co-workers note that “despite new genomic tools, the ability to identify high-priority targets using, for example, essential gene screens, and innovation in high-throughput screening [(HTS)] technologies that enables millions of compounds to be probed in a short period of time, no new antibiotic drugs have emerged.”44 Furthermore, in Lipinski’s landmark paper which set the rules for combinatorial chemistry, which so frequently generates the large libraries tested by HTS, it is stated plainly that antibiotics do not fit the paradigm of the medicinal chemistry approaches which might be more successfully employed to identify anticancer drugs.45 The second approach has seen extensive application in developing compounds with structurally complex and highly functionalized scaffolds. Methods such as diversity-oriented synthesis (DOS) and complexity-to-diversity (CtD) have been shown to furnish large numbers of compounds through relatively few chemical transformations in a highly divergent manner.46–50 In contrast to the traditional means of natural product development for antibiotics, these approaches are used to generate compounds which expand beyond the known antibacterial motifs, while avoiding the planar nature of molecules typically yielded via combinatorial chemistry.
Figure 5.

Traditional means of generating diverse compound libraries, including combinatorial and semisynthetic approaches.
2.1. Diverted Total Synthesis
In the 1990s, an intriguing family of natural products, the epothilones, was identified to have potent antitumor activity. While the epothilones shared a tubulin interference mechanism with the familiar taxol, scientists observed a much-improved potency against drug resistant cell lines. Given the lucrative prospects surrounding the epothilones, a number of synthetic laboratories began to pursue this scaffold, including the Danishefsky, Nicolaou, and Schinzer groups. A detailed summary of the historical context and synthetic efforts has been compiled nicely in Harran’s recent review.51
Following their synthetic work toward the natural products, the Danishefksy laboratory began to apply a medicinal chemistry approach, seeking to establish a structure–activity relationship (SAR) with the hope of ultimately arriving at an optimized clinical candidate.52 Comparison of epithilones B (10) and D (11), which differ only by the presence of the epoxide in 10, led to the insight that the removal of this molecular feature might reduce toxicity. To develop a thorough SAR, Danishefsky established an approach he terms “molecular editing”. In short, late-stage intermediates in a synthetic route en route to the natural product of interest can be diverted to access analogs which possess molecular features not possible through either semisynthetic or biosynthetic methods (Figure 6).53,54 This approach, known as diverted total synthesis (DTS), established that further unsaturation in the macrocycle enhanced potency and stability (Figure 7). Given the sensitivity of the allylic methyl group, installation of a trifluoromethyl group in its place also widened the therapeutic window, and the Danishefsky group arrived at the preclinical candidates fludelone (13) and iso-fludelone (14), compounds which would not exist absent the ingenuity of synthetic chemists.
Figure 6.

Innovative strategies in generating diversity through synthesis.
Figure 7.
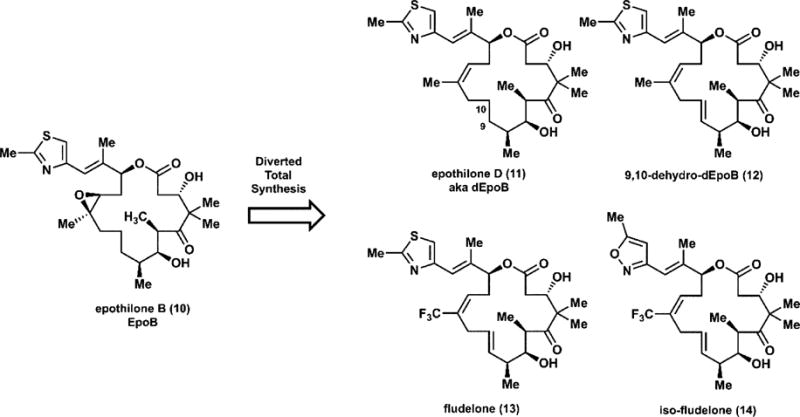
Diverted total synthesis leads to analogs of epothilone B with superior pharmacological characteristics.
We also note a highly analogous strategy, function-oriented synthesis (FOS), which aims to generate simplified analogs of natural products which retain or improve upon biological activity.55 Several highly successful drugs have been developed using this strategy, including Lipitor, which was designed from a naturally occurring statin.56
2.2. Diversity-Oriented Synthesis
The DOS strategy was originally introduced by Schreiber, who conceived the notion in the early 21st century that simple chiral building blocks could be taken through a few divergent transformations to generate a large degree of complexity (Figure 8).47 This could then be applied to similar starting materials to generate a library of substituted scaffolds. Additionally, because these intermediates would contain reactive functionalities, the libraries could be further diversified by following divergent pathways. Both Schreiber and others have acknowledged the utility of such a synthetic design, and DOS has been successfully employed to discover complex bioactive scaffolds.57
Figure 8.

Spring’s diversity-oriented synthesis seeking anti-MRSA compounds. MICs in μg/mL.
Utilizing DOS, Spring has discovered three compounds with inhibition activity against S. aureus, including two UK epidemic methicillin-resistant strains (EMRSA 15 and EMRSA 16).58 Starting from simple chiral building blocks, the authors performed up to four divergent transformations to generate a diverse series of scaffolds with varying substitution (Figure 8). After screening their library of 242 compounds, they identified three which demonstrated some antibacterial activity. The most potent compound, (±)-gemmacin (15), was weakly active against resistant strains including vancomycin-resistant enterococci (VRE) (Figure 8). It also showed low antifungal activity, indicating a degree of selectivity. Their brief study demonstrates the potential of accessing compounds with natural product-like complexity. While these compounds are not sufficiently potent for clinical applications, compounds identified through DOS may be useful starting points in drug discovery campaigns.
Unlike Spring’s DOS campaign, Sello developed a DOS strategy around previously identified antibiotic compounds.59 The enopeptin family (18, 19) was shown to have excellent antibacterial activity in vitro but limited activity in vivo due to its poor solubility (Figure 9). Extensive SAR studies by Bayer Pharmaceutical Research identified key hydrogen bonding interactions that constrained the peptidolactone core and identified a lead compound called acyldepsipeptide 4 (ADEP 4, 20). Sello’s group expanded on this work, developing a convergent strategy to illuminate the optimal tripeptide fragment for the macrocyclic core (Figure 9). By designing a retrosynthesis to provide each fragment separately, the authors synthesized eight compounds which varied in their bioactivity. Compounds 21 and 23 were the most potent against both MRSA and VRE strains. This study clearly demonstrates the potential for DOS studies to yield potent antibacterial compounds.
Figure 9.

DOS strategy to access enopeptin analogs. (a) Enopeptin antibiotics as inspiration. (b) Modifications to enopeptin scaffold with biological evaluation (MICs, μg/mL) against Gram-positive bacteria. ND = not determined.
2.3. Complexity to Diversity
While there are few examples of the CtD approach being directly applied to generate antibacterial compounds, this strategy is capable of creating diverse collections of compounds for screening libraries.48–50 Starting with an abundant and highly functionalized natural product, complexity is generated by ring distortion through cleavage, rearrangement, or ring fusion. Huigens recently used this approach to create a series of compounds from the indole alkaloid yohimbine (29).60 In four steps or fewer, yohimbine is transformed to a diverse array of scaffolds (Figure 10). While yohimbine has no antibacterial activity, the authors identified two compounds, 30 and 33, which offered slight inhibitory activity against S. aureus. This ring-distortion method demonstrates the ability to introduce antibacterial activity by introducing drastic structural modifications, and it certainly provides proof-of-concept for the method.
Figure 10.

Huigens’ ring distortion strategies to access yohimbine-based bioactive scaffolds.
2.4. Summary
These strategies offer an alternative to the classic means of antibiotic discovery, including new compound isolation and semisynthetic modifications of natural products. They allow access to unexplored chemical space which in turn provides the potential for new antibiotic targets to be discovered. One primary benefit of these strategies is that these diverse libraries can be accessed in rapid fashion as transformations can be performed on a diverse series of substrates introducing further chemical complexity. While these have not yet been successfully employed to develop an approved antibiotic, these strategies have certainly inspired chemists to move beyond the once popular combinatorial chemistry and provide more robust structures for probing biological activity.
3. MACROLIDES AND KETOLIDES–FROM FERMENTATION TO SYNTHESIS
The term “macrolide” was coined by Woodward to describe macrolactone glycosides. Erythromycin (3), the first clinically useful macrolide antibiotic, was isolated at the midpoint of the 20th century during the golden age of antibiotics from the actinomycete Saccharopolyspora erythrea.61 Early reports in medical journals were quick to indicate erythromycin’s excellent spectrum of activity against Gram-positive pathogens.62,63 The structure of erythromycin was first deduced by chemical testing,64 and X-ray studies defined the stereochemistry of this formidable skeleton.65 Despite its promising antibacterial potency, erythromycin was limited by its poor pharmacological profile, which spurred semisynthetic innovation beginning in the late 20th century and, more recently, totally synthetic approaches to both test hypotheses regarding resistance and binding as well as broadly explore unknown chemical space.
3.1. Mechanism of Action
The macrolide antibiotics target the 50S subunit of bacterial ribosome to inhibit protein synthesis. This portion of the ribosome contains the catalytic peptidyl transferase center, where translation is carried out. As the peptide is lengthened, the just-assembled end of the nascent peptide is extruded though an exit tunnel. Erythromycin and related compounds bind to the interior of this channel, which sterically blocks the growing peptide from continuing down the tunnel (Figure 11). The precise details of this inhibition were previously debated; it has been determined that erythromycin cannot bind to a ribosome in which protein synthesis is already underway.67,68 After binding to an empty ribosome, erythromycin then clogs the exit tunnel. When the bacterial machinery becomes jammed, it then ejects the incomplete peptidyl–tRNA complex. A crystal structure of erythromycin bound to the 50S ribosome clearly demonstrates the obstruction of the peptide exit tunnel (Figure 11).69
Figure 11.
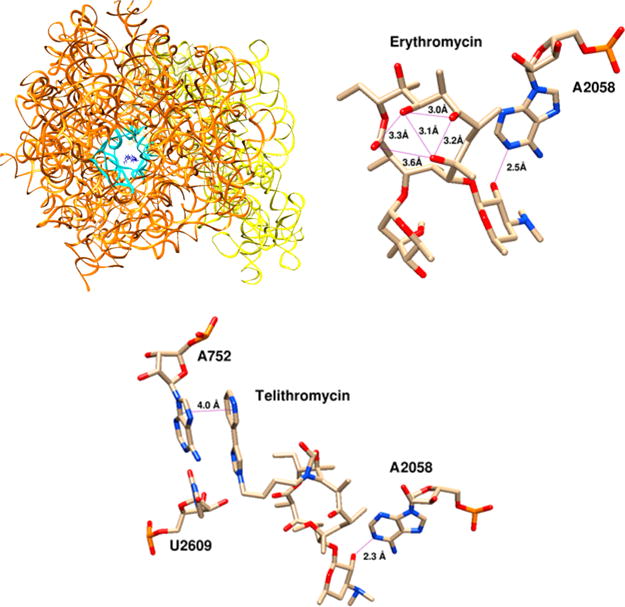
Crystal structure of erythromycin bound to E. coli ribosome with ribosomal proteins omitted. 30S subunit in yellow, 50S subunit in orange, erythromycin in blue. View down the axis of the nascent peptide exit tunnel, outlined in cyan. Key interactions of erythromycin and telithromycin with the ribosome. Erythromycin is conformationally rigid due to the avoidance of syn-pentane interactions between methyl groups;66 we also highlight hydrogen bonding across the macrocycle above. Both macrolides hydrogen-bond to A2058 through desosamine; telithromycin makes an additional π-stacking interaction with A752, explaining its enhanced binding affinity. Structures edited in Swiss PDB Viewer and rendered in UCSF Chimera from PDB IDs 4V7U (erythomycin) and 4V7S (telithromycin).
3.2. Resistance to Macrolides and Ketolides
Macrolide resistance is very well characterized and can be classified into three general approaches: active efflux, ribosomal modification, and drug modification. These are significant obstacles and are in large part the impetus for the semisynthetic innovations described briefly herein.
3.2.1. Efflux Pumps
A common method of avoiding the inhibitory effect of antibiotics is to actively remove them from the cell. An enzyme specific to the macrolides is encoded by mef (macrolide efflux) genes.70 These efflux pumps were first identified in Gram-positive species but have also been observed in various Gram-negative pathogens. These are capable of effluxing macrolides, as well as lincosamides and streptogramin B (this phenotype of resistance is generally known as MLSB or MLKSB, if ketolides are included). The Mef family of efflux pumps are generally not regarded as a major contributor to resistance (which can be measured by comparing the change in MIC between strains carrying this gene and those that do not); Mef is, however, a significant cause of the rise in macrolide-resistant streptococci, particularly S. pneumoniae.71,72 Also known are ATP-binding cassette (ABC) transporters, a superfamily of efflux pumps, which are known across the phylogenetic tree but have been identified to contribute to resistance against many antibiotics, including the macrolides, in bacteria.73 A macrolide-specific ABC-type pump, macAB, has been identified in E. coli,74 while the Msr family of macrolide pumps is known to have spread from Staphylococcus to other genera such as Streptococcus and Pseudomonas.75 Among Gram-negative bacteria, the RND family is a major class of multidrug efflux pumps, which partially account for the persistence of these pathogens in clinical settings.76,77 Recent studies have identified amino acid residues specifically responsible for macrolide resistance via the RND transporter complex AcrAB-TolC in E. coli.78
3.2.2. Ribosomal Mutation
Another observed method of resistance is to alter the erythromycin-binding site through mutation. The replacement of adenine 2058 (based on E. coli numbering) with guanine, which disrupts the key hydrogen-bonding interaction (Figure 11), is well-known to cause resistance to erythromycin and other macrolides with a >2,000-fold decrease in potency.79 Mutations at nearby nucleo-sides, like C2611G, have also been observed, though these make a relatively minor contribution to resistance; the increase in MIC is generally within 1 order of magnitude.79 A critical series of adenine residues (749–752) is also known; a deletion within this range reduces macrolide potency by a factor of 500. This effect is also observed in telithromycin (37), a ketolide. In addition to the nucleic acid backbone, the ribosome also incorporates proteins that assist in various functions; a mutation in the L22 ribosomal protein (G95D) also contributes to resistance.
3.2.3. Ribosomal Modification (Methylation)
Active efflux has a critical drawback—namely in the fitness cost. There is a considerable fitness cost to expressing these enzymes and maintaining active efflux, which is energy-dependent. Though these genes can be shared easily via horizontal gene transfer, constitutively expressed resistance can be a significant drain on various cellular processes. A murine model demonstrated that bacteria susceptible to antibiotics are more viable than a resistant variant.80 An ideal solution would be a system that can “turn on” in the presence of antibiotic to affect resistance without requiring considerable upkeep when not threatened.
Such an inducible mechanism of resistance has been observed which utilizes erythromycin’s binding to the ribosome to trigger expression of resistance, specifically dimethylation of the critical A2058.81–83 These Erm-type genes follow a short sequence, an open reading frame (ORF), which encodes a leader peptide.84 This leader peptide is constantly translated, which acts as a sensor. The genetic material that follows (which encodes Erm) is not accessible by the ribosome thanks to a conformation which shields the ribosomal binding site (RBS). When erythromycin is present, the blocked ribosome essentially becomes stuck on the ORF sequence. This induces a thermodynamically driven conformational change which frees the RBS and allows translation of Erm. Conveniently, this process is triggered at low concentrations of macrolide (0.1–10% of MIC), which ensures that there is sufficient unimpeded ribosome to translate the ribosomal methyltransferase from Erm.85 The S-adenosyl methionine-dependent methylase then acts upon A2058. This process yields a similar outcome compared to the A2058G mutation, though it is likely to spread via plasmid horizontal gene transfer.
3.2.4. Drug Degradation
A final mechanism of macrolide resistance features esterases which hydrolyze the C1–O14 ester in erythromycin. These erythromycin esterases (EreA,86 EreB,87 and the more recently identified EreC88) have been identified to provide high levels of resistance when encountered. Though not commonly observed, these enzymes have the potential to proliferate given that they have been found in integrons and transposons, which potentially allows these resistance traits to be shared rapidly via horizontal gene transfer. The Wright Laboratory at McMaster University recently performed a thorough characterization of EreA and EreB, finding that both enzymes act upon erythromycin (3) and clarithromycin (35), while EreB also acts on azithromycin.89 No activity toward telithromycin was observed, providing hope that the ketolides will retain activity should this mechanism of resistance become widespread.
3.3. History of Macrolide Development—Semisynthetic Innovations
For more than a half-century, semisynthetic innovation drove the macrolide family forward. Despite erythromycin’s antibacterial potency, unpleasant side effects were observed, including gastrointestinal disturbances and poor bioavailability. These were attributed to the compound’s instability in acid, as the C6 and C12 hydroxyl groups can form spiroketal 34 at the C9 ketone (Figure 12).91 To solve this problem, scientists at Taisho Pharmaceuticals in Tokyo developed a method to block this chemistry while retaining an active antibiotic. The solution was to methylate the C6 hydroxyl group, which is ordinarily straightforward chemistry. However, a brief inspection of erythromycin reveals numerous vulnerable alcohols. Ingeniously, Omura and co-workers converted the ketone to a substituted, bulky oxime. This alteration forced a conformational change rendering only the C6-alcohol accessible, permitting methylation to yield a more stable antibiotic, clarithromycin (35), which was approved for use in 1980.92 Contemporaneously, scientists at Pliva in Zagreb, Croatia, took erythromycin in a different direction, generating an oxime and forcing a Beckmann ring expansion, ultimately yielding a 15-membered azamacrolide from the 14-membered macrolide.93 This drug, azithromycin (36), found widespread clinical use and was the seventh-most prescribed drug in 2010. However, such frequent use can only accelerate the evolution of bacterial resistance; a recent report indicates that 76% of MRSA isolates are resistant to azithromycin.94 These reports of rising resistance drove scientists to two key innovations that would inform the next generation of macrolide antibiotics. First, the cladinose sugar bonded to the oxygen at C3 was found to be unnecessary, corroborated by the lack of contacts in the crystal structure (Figure 11). Removal of this group and oxidation to a C3-ketone was found to be optimal, giving rise to the ketolides. Furthermore, scientists at Abbott Laboratories introduced a cyclic carbamate at C11 and C12, with the included nitrogen serving as a functional handle.95 It was quickly discovered that incorporating a biaryl group tethered by a short alkyl chain could provide additional binding to the ribosome via π-stacking with C2644 (Figure 11), which explains the increased potency. These two key innovations allowed scientists at Aventis Pharmaceuticals to develop telithromycin (37), approved for use in 2004. Despite its antibiotic capabilities, the use of telithromycin has been recently discouraged because of severe side effects. The pyridyl-imidazole functionality, which bears considerable molecular resemblance to nicotine, was hypothesized to block nicotinic acetyl-choline receptors (nAChR) in the neuromuscular junction, optic nerve, and liver, which explains reports of myasthenia gravis (a neuromuscular disease), visual disturbances, and hepatotoxicity, respectively.96 In collaboration with the Sharpless group, scientists at Optimer Pharmaceuticals sought to replace the imidazole with the similar 1,2,3-triazole, easily introduced using azide–alkyne “Cu-click” chemistry. These triazoles were reasonably expected to withstand metabolism, given that these are often used as bioorthogonal linkages in chemical biology. Furthermore, the pyridine was replaced with an aniline ring, and with the installation of fluorine at C2, the next-generation candidate solithromycin (39) was born, which made it through the pipeline to Phase III trials. Despite their efforts, solithromycin, in a recent FDA report,97 was identified as inferior to the existing treatment of moxifloxacin against community-acquired bacterial pneumoniae (CABP). Furthermore, there are remaining concerns with hepatoxicity, indicating that the pyridyl-imidazole moiety may not be culpable as hypothesized previously. These issues led the FDA to reject solithromycin, and some industry scientists speculate that overcoming these concerns via further trials may be cost-prohibitive.98 Cempra has recently withdrawn the European equivalent of a new drug application; it is unclear whether or not this effort will continue.
Figure 12.

(a) Acid-driven decomposition of erythromycin to spiroketal necessitating drug modification. (b) Semisynthetic macrolides developed from erythromycin with year of FDA approval.90 (c) Semisynthetic ketolides at various stages in development. *To the best of our knowledge, the company sponsoring cethromycin, Advanced Life Sciences, ceased operations in 2011.
3.4. Macrolides and Ketolides via Total Synthesis
3.4.1. Woodward’s Posthumous Synthesis of Eryth-romycin A
While numerous routes to the erythronolides (the erythromycin aglycone) are reported, including work by Corey,99,100 we briefly discuss only total syntheses of complete, glycosylated macrolides to establish the state of the prior art. Naturally, the first total synthesis of erythromycin A was achieved by Woodward’s laboratory at Harvard. Their reports, published after the great chemist’s death, describe these efforts.101–103 Dithiohemiacetal 40 was elaborated in 9 steps to dithiodecalin 41, a point of divergence to access intermediates 42 and 43 (Scheme 1). The two were united by aldol chemistry with an additional 24 steps to complete seco-acid derivative 45 from 44. Macrocyclization was achieved using the Corey–Nicolaou protocol, a thermally driven addition of the alcohol to an activated thioester. Ten steps followed, which include two innovative glycosylation steps featuring modified Königs–Knorr glycosyl donors (47 and 48). This general method has informed nearly every synthetic effort toward macrolides and ketolides. The late Woodward and co-workers achieved the first and only total synthesis of erythromycin A in a longest linear sequence of 48 steps.
Scheme 1.

Summary of Key Steps in the First and Only Total Synthesis of Erythromycin A by Woodward
3.4.2. Martin’s Synthesis of Erythromycin B
In the following decade, the Martin Group at the University of Texas, Austin, reported a total synthesis of the closely related erythromycin B (53), which lacks oxidation at C12 (Scheme 2).104 Using chemistry developed previously, 2-ethylfuran (49) was converted to polyketide precursor 50, which undergoes relatively straightforward aldol chemistry to yield seco-acid 51. Lactonization using Yamaguchi’s method was particularly efficient with an impressive 93% yield. With the main scaffold in hand, only glycosylation and protecting group manipulations remained, ultimately furnishing erythromycin B in 30 steps from 49, or 23 from known material. Following this report, the Martin Laboratory published other work toward a more streamlined synthesis featuring changes in the order of operations in the endgame, notably the sequence in which macrolactonization and glycosylation are carried out.105,106
Scheme 2.

Summary of Key Steps in Martin’s 1997 Synthesis of Erythromycin B
3.4.3. Kang’s Synthesis of Azithromycin
Due to their pharmaceutical applications, semisynthetic macrolides have also been popular targets for synthetic campaigns. Kang in Daejong, South Korea, recently reported a convergent synthesis of azithromycin (Scheme 3).107 To construct the western half, vinylic triol 54 is desymmetrized and, using epoxide chemistry, was elaborated into amine precursor 55 in 11 total steps. The eastern half was prepared from chiral building block 56; further epoxide manipulation and crotylation chemistry followed to furnish 57. Notably, Kang and co-workers installed the desosamine relatively early in the synthesis in a distinctly abiotic fashion; the prior attempts constructed the macrocycle and then installed both the desosamine and cladinose functionalities in a biomimetic manner. Here, the authors found it advantageous to incorporate the desosamine sugar into the eastern fragment; this allowed for minimal use of protecting groups for a streamlined synthesis. The eastern fragment was elaborated into aldehyde 58, which was united with the amine precursor via reductive amination. Yamaguchi esterification, the second glycosylation, and a final deprotection completed azithromycin (36) in 8 steps in the longest linear sequence.
Scheme 3.
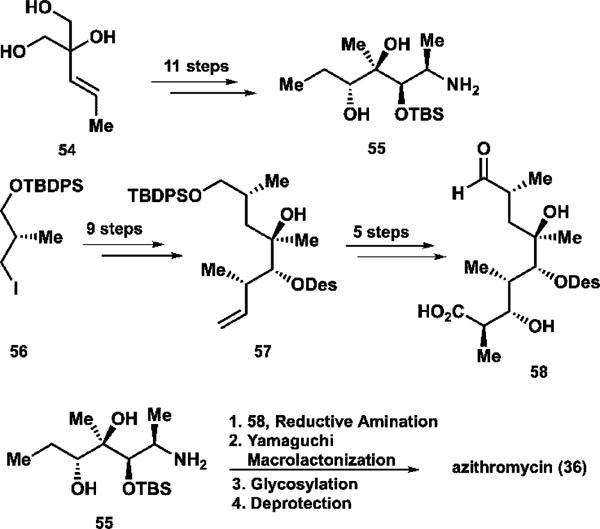
Summary of Key Steps in Kang’s 1997 Synthesis of Azithromycin
3.4.4. Summary
While these works have been great undertakings in the field of total synthesis, they have been carried out solely for the sake of making the molecule; these are not efficient routes to generate these macrolides. For example, there is no imaginable reason to synthesize erythromycin for pharmaceutical application when it is readily available from Nature via fermentation. Synthesis of the semisynthetic azithromycin is also not useful to medicine, as it is also readily prepared from natural material in just four steps. Furthermore, these works are not particularly amenable to analog development; these syntheses are target-oriented and are designed with the sole objective of reaching this target. Alteration of the target, like a desired analog to establish a structure–activity relationship, may require drastic changes to the synthetic strategy. This said, there has been immense evolution in synthetic methodology over the decades since Woodward’s work which enables elegant syntheses targeting novel macrolides and ketolides to overcome the issues of resistance and hepatotoxicity.
3.5. Efforts in Analog Development
Here we discuss three approaches to developing new macrolide and ketolide antibiotics, one very broadly focused at generating unprecedented structures and two aiming to engineer existing scaffolds to overcome the topological barrier introduced by ribosomal mutation as a mechanism of resistance and another which seeks to extend the range of the aryl side chains found in the ketolides to enhance ribosomal binding.
3.6. Myers’ Synthetic Campaign toward Unprecedented Ketolide Antibiotics
In 2016, the Myers laboratory at Harvard published an innovative route to designing and constructing more than 300 potential ketolide antibiotics, which, like their tetracycline cousins, possess molecular features which are not possible to introduce through semisynthesis.108 They utilize a highly modular approach in which each designed ketolide is constructed from eight simple and diversifiable starting materials. This approach incorporates strategies from diverted total synthesis (using late-stage intermediates as points of divergence), diversity-oriented synthesis (accessing a large region of chemical space in an efficient manner), and combinatorial chemistry (union of simple precursors to generate a library of drug candidates). Unlike in their tetracycline work (vide infra), there has been sufficient innovation in the field of macrolide total synthesis to develop this approach without radically reinventing a synthetic route. That said, the Myers group successfully draws inspiration from established work while using the latest in asymmetric methodology (some of which they developed) to streamline their route. Without delving into the syntheses of three-hundred-odd ketolides, we discuss here the syntheses of two novel azaketolides, which bear resemblance to azithromycin and diverge from a common Western precursor 65 (Scheme 4).
Scheme 4.

Syntheses of Key Intermediates en Route to 14- and 15-Membered Azaketolides by Myers
The Western precursor (Scheme 4a) began with the aldol reaction of ketone 59, the preparation of which has been optimized by the Myers group,109 with the acylated pseudoephenamine 60.110 Phosgene was then added to construct the cyclic carbamate early in the synthesis; this is notable as this carbamate is normally generated late in semisynthetic methods. Methyl Grignard generated in situ from methyllithium and isopropyl Grignard displaced the chiral auxiliary. The nitrogen in the carbamate was then alkylated to ultimately provide butyl azide 63 whose unique functionality would be accessed later. The ketone then underwent asymmetric reductive amination, and desilylation provided the key intermediate 65 in just 7 steps.
To construct 15-membered azaketolide 86, an analog of the well-known azithromycin (36), the eastern precursor (Scheme 4b) began with the addition of lithium reagent derived from 56 to ketone 66. The resultant alcohol 67 was then methylated in a straightforward manner—to point out another advantage of this totally synthetic route, recall that the methylation of erythromycin to clarithromycin required 6 steps to do so selectively. Oxidative cleavage of the acetonide yielded aldehyde 68. To install the functional handle for macrolactonization, dioxinone 69111 was added through a vinylogous Mukaiyama aldol reaction. The resulting alcohol was then glycosylated using a desosamine donor closely related to the one pioneered by Woodward. Finally, deprotection and oxidation with the Dess–Martin reagent completed eastern precursor 72.
Myers notes that a key advantage of this modular synthetic approach is that ketolides with unprecedented structural features can be constructed efficiently. When azithromycin is produced from erythromycin, the 14-membered macrolide undergoes ring expansion to the 15-membered semisynthetic drug. It would, therefore, be difficult to examine the pharmaceutical potential of a 14-membered azithromycin analog using semisynthetic techniques. Myers’ approach, unbeholden to Nature’s design, facilitates this exploration. The Eastern precursor en route to the 14-membered azaketolide (Scheme 4c) was constructed quite similarly to the 15-membered macrocycle, beginning with the addition of silyl enol ether 73 to ethyl pyruvate (74), guided by the chiral Pd-SegPhos. Following protection as the ketal, methylation of the alcohol and reduction of the ester furnished aldehyde 76. Similar steps followed, with the installation of the dioxolinone, glycosylation, and finally, deprotection of the ketal.
Thanks to the elegance of this synthesis, the final steps in the construction of these ketolides ran in parallel (Scheme 5), beginning with the union of the western and eastern fragments through reductive amination. The thermal macrolactonization followed using a method pioneered by Boeckman in the 1980s, which is an excellent general method to construct these formidable structures.112 Mechanistically, thermolysis of the dioxolinone yields the fragments acetone and an acyl ketene, which is a powerful acylating agent. Addition of the alcohol with proton-transfer steps then completes the β-keto lactone, and stereochemistry is controlled at C2 inductively to avoid a syn-pentane interaction with the methyl group at C4. With the macrocycle complete, all that remained was to “click” on the alkynyl aniline to furnish the 1,2,3-triazole, inspired by the work done on the semisynthetic solithromycin, yielding the 14- and 15-membered azaketolides both with a remarkable ten steps in the longest linear sequence.
Scheme 5.

Myers’ Completion of Azaketolides 83 and 86 via Identical Endgames
The modular nature of this synthesis provides the means to introduce nearly any imaginable structural feature onto a variety of macrocyclic scaffolds (Figure 13). Some of these function-alities are well-precedented in the prior art; others are rather unexpected. Broadly, the structures encompass 14-, 15-, and 16-membered azaketolides, 14-membered ketolides, and several novel structures that do not classify particularly well. Some structural features include heterocyclic modification, removal of skeletal methyl groups, desosamine alteration, inversion of various stereocenters, and unsaturation of the alkyl chain tethering the triazole. The triazole-aniline from solithromycin is reserved as the default functionality; introduction of other heterocycles or substituents is possible and straightforward. The Myers group also recently developed a method to construct a variety of desosamine variants, which provides an additional route of diversification.113
Figure 13.
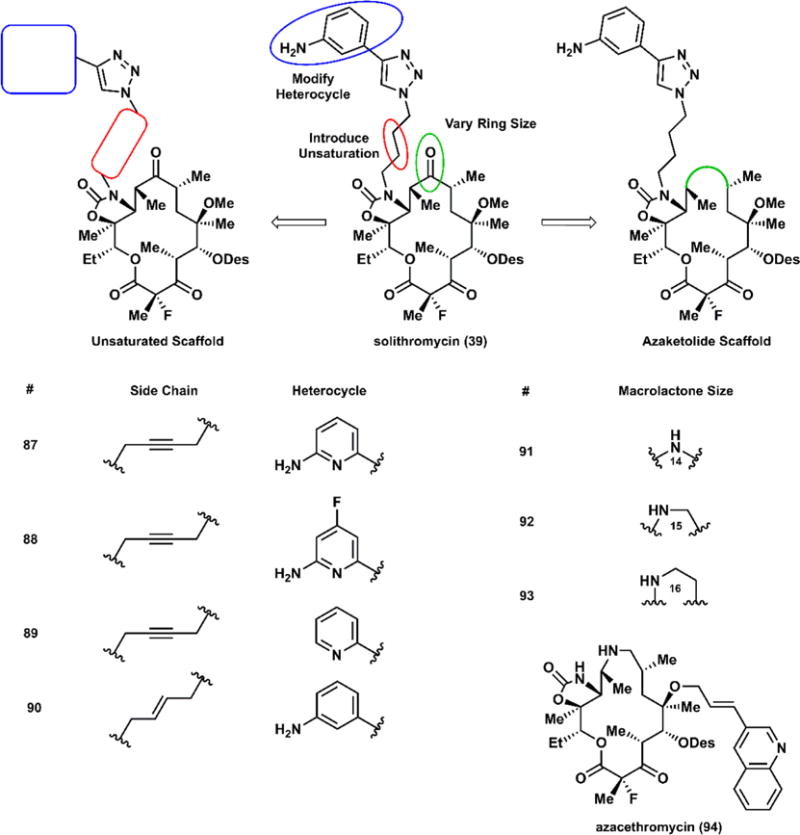
Rationale in designing new fully synthetic macrolides; selected analogs fall into two general categories: ketolides bearing an unsaturated side chain with varying heterocycles, and azaketolides, including an azacethromycin analog.
Each macrolide was screened against a panel of Gram-positive and Gram-negative bacteria, some of which are characterized by particular resistance phenotypes, including those discussed in Section 3.2. In the interest of brevity, we summarize in Table 1 biological assays of selected compounds to highlight a preliminary structure–activity relationship. Ketolides bearing an unsaturated side chain outperformed solithromycin in nearly all strains tested, showing promising activity against bacteria expressing cErmA, MsrA, ErmB, and MefA, common resistance genes expressing ribosomal methylases and efflux pumps. The novel azaketolides, including an azacethromycin analog (94), generally do not provide an improvement in potency over solithromycin against any of these resistance phenotypes. These compounds were also assayed against Gram-negative pathogens; modest improvements were observed, though no compound exhibited an MIC below 2 μg/mL.
Table 1.
Antimicrobial Assays of Selected Analogs against Gram-Positive Strainsa
| S. aureus | S. pneumoniae | E. faecalis | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| ATCC 29213 | BAA-977 | MP513 | NRS384 | ATCC 49619 | UNT-042 | ATCC 29212 | UNT-047 | |
| Compound | QC | iErmA | cErmA | MsrA | QC | ErmB, MefA | QC | ErmB |
| 87 | 0.06 | 0.06 | 16 | 0.06 | <0.03 | <0.03 | 0.03 | 1 |
| 88 | <0.03 | 0.06 | 16 | 0.125 | <0.03 | <0.03 | 0.03 | 2 |
| 89 | <0.03 | 0.03 | 64 | 0.06 | <0.03 | <0.03 | 0.03 | 2 |
| 90 | 0.06 | 0.06 | 64 | 0.125 | <0.03 | <0.03 | <0.03 | 4 |
| 91 | 0.5 | 0.5 | >64 | 1 | <0.03 | 2 | 0.125 | >64 |
| 92 | 0.25 | 0.5 | 64 | 1 | <0.03 | 0.125 | 0.06 | 32 |
| 93 | 4 | 4 | 64 | 8 | 0.06 | 8 | 0.5 | 64 |
| 94 | 0.25 | 0.5 | 64 | 0.5 | <0.03 | 0.5 | 0.25 | >64 |
| Solithromycin (39) | 0.125 | <0.03 | >64 | 0.25 | <0.03 | 0.25 | <0.03 | 32 |
![]()
MICs in μg/mL. Strains listed under the species name with resistance gene. Colors denote the x-fold change relative to solithromycin. For the sake of simplicity, the color scale ignores upper and lower limits on MICs.
Given the potential of this technology, Prof. Myers founded a startup, Macrolide Pharmaceuticals, to expand drug discovery efforts and explore the possibility of clinical applications of fully synthetic macrolides. Furthermore, Macrolide has reached an agreement with Cempra Pharmaceuticals to develop a totally synthetic manufacturing approach of solithromycin, hoping to reach a more efficient route than the current semisynthetic method, though this arrangement is likely inconsequential given the issues faced by solithromycin.
3.7. Andrade’s Synthesis of the 4-Desmethyl Telithromycins
The availability of high-quality crystal structures has been highly beneficial to medicinal chemists; the ability to inspect and examine the interactions between a drug and its target is invaluable. In 2005, the Steitz laboratory at Yale published compelling evidence that the critical A2058G (using E. coli numbering) mutation in the 50S ribosome disfavors macrolide binding due to a steric clash between the guanine N2 and the macrolide C4 methyl group.114 While Steitz noted that reversal of this mutation would restore normal binding, the Andrade group at Temple University then sought to leverage the power of total synthesis to effectively “mutate” telithromycin to remove the C4 methyl group,115 planning the first synthetic effort to radically alter the carbon skeleton of the macrolides. Furthermore, the Andrade group designed syntheses to explore the effect of removing the methyl groups at C8 and C10, both of which flank the ketone at C9.116–118 In the interest of brevity, we outline the culmination of their campaign with the synthesis of 4-desmethyl telithromycin (110) (Scheme 6).119
Scheme 6.
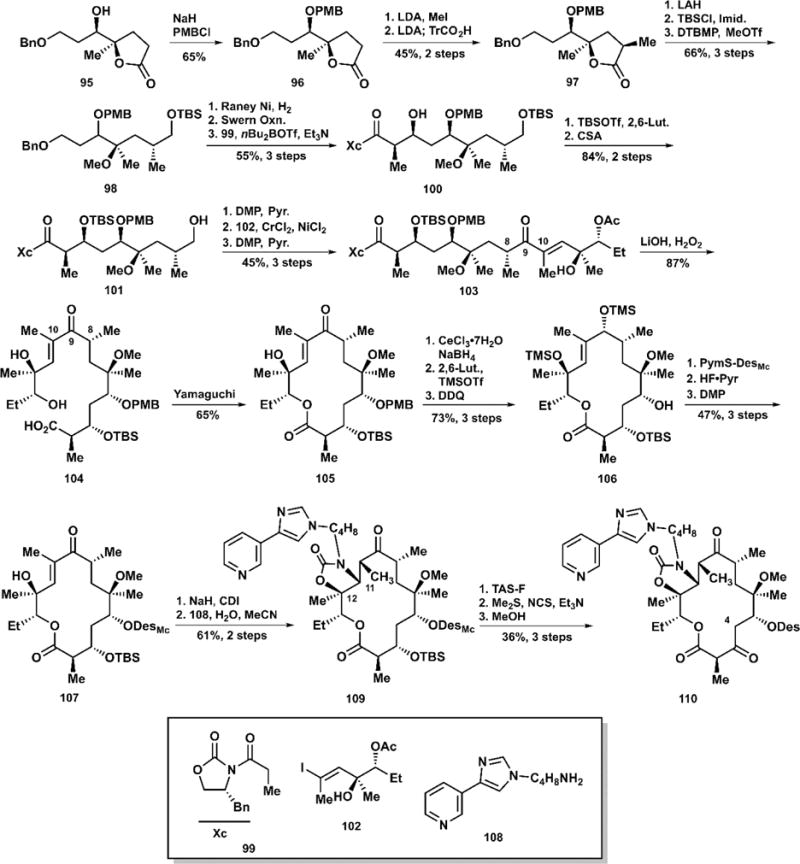
Andrade’s Total Synthesis of 4-Desmethyl Telithromycin (110)
The synthesis began with the PMB-protection of lactone 95, followed by the lithium diisopropylamide-mediated methylation of 96. Interestingly, the Andrade group reported further treatment with LDA to regenerate an enolate followed by quenching with triphenylacetic acid to yield methylated lactone 97 in 45% over 2 steps. Conceivably, this allowed for isomerization to the desired stereochemical configuration. At this point, the lactone was opened by lithium aluminum hydride; sequential treatment with tert-butyldimethylsilyl chloride followed by methyl triflate protected key functionalities. Hydrogenolysis with Raney nickel cleaved the benzyl ether. Swern oxidation set up the Evans aldol addition of propionamide to extend the carbon chain en route to the seco acid. Following another protection/deprotection sequence, oxidation with the Dess–Martin periodinane furnished an intermediate aldehyde. In their prior work on their di- and tri-desmethyl analogs, Andrade and co-workers established the use of the Nozaki–Hiyama–Kishi reaction to forge the connection between carbons 9 and 10, which is exemplified here with the addition of vinyl iodide 102. Oxidation with the Dess–Martin reagent and oxidative removal of the Evans auxiliary yielded seco acid precursor 104. As is typically the case in macrolide formation, Yamaguchi’s macrolactonization was preferred. To affect efficient glycosylation, the Andrade group found it necessary to reduce the ketone at C9 using sodium borohydride with cerium(III) chloride and protect the resultant alcohol as a silyl ether. Removal of the PMB group at C5 with DDQ exposed the alcohol to glycosylation via Woodward’s thiopyrimidyl desosamine donor. Upon successful glycosylation, desilylation and reoxidation restored the ketone functionality at C9. Treatment with carbonyl diimidazole acylated the alcohol at C12; addition of the pyridyl imidazole butylamine furnished cyclic carbamate following Michael addition to C11. Cleavage of the silyl ether at C3 with TAS-F followed by Corey–Kim oxidation installs the ketolide, and gentle removal of the methyl carbonate group on the desosamine yields 4-desmethyl telithromycin (110) in 27 steps from lactone 95.
To verify their hypothesis, the Andrade group tested their desmethyl telithromycin analogs against both Gram-positive and Gram-negative bacteria with varying phenotypes of resistance, including A2058 mutations.115 The results of these assays are summarized in Table 2. Against the Gram-positive S. aureus, all compounds, including telithromycin, were ineffective. In E. coli, however, the 4-desmethyl analog is just as potent as telithromycin, a promising result. When tested against the A2058G mutation these analogs were designed to overcome, all compounds lost potency. While the 4-desmethyl was the most active out of all analogs, it had just one-fourth the efficacy of telithromycin. While not an ideal result in terms of overcoming this resistance, Andrade observes that the addition of methyl groups around the macrocycle appears to enhance activity, perhaps by forcing the ketolide into the conformation preferred for binding.
Table 2.
Summary of Biological Assaysa
| Methyl deletions | S. aureus | E. coli | |||
|---|---|---|---|---|---|
| A2058T | ermA | wt | A2058G | ||
| Telithromycin (37) | >128 | >128 | 0.5 | 1 | |
| 111 | 4,8,10 | 32 | >128 | 32 | 64 |
| 112 | 4,10 | >256 | <128 | 8 | 16 |
| 113 | 4,8 | >256 | >64 | 4 | 32 |
| 110 | 4 | >256 | >128 | 0.5 | 4 |
![]()
MICs given in μg/mL, and relative activity measured against telithromycin.
3.8. Oyelere’s Engineered Azithromycin Analogs
In order to understand the process and mechanism of protein expression more clearly, biochemists have studied the ribosome, a critical piece of bacterial machinery, with great interest. These investigations uncovered a regulatory peptide encoded by SecM. During its expression, the growing peptide binds somewhat tightly to nucleotides in the exit tunnel, inhibiting protein synthesis, which in turn up-regulates the expression of SecA, a protein translocase. The mechanism of SecM binding has been examined via cryo-electron microscopy (cryo-EM), due to the difficulty in cocrystallizing the necessary elements.120 The Oyelere group at Georgia Tech were recently inspired by these findings and sought to leverage native bacterial machinery to develop direly needed antibiotics.121
The focal point of this effort is to mimic the binding of SecM to the ribosomal exit tunnel—in particular, a key π-stacking interaction between W155 of SecM and A751 of the ribosome. Designing antibiotics to make additional contacts to the ribosome is a proven strategy; such molecular engineering gave rise to telithromycin and candidates such as cethromycin and solithromycin. To test this hypothesis, the Oyelere group developed a series of rationally designed semisynthetic analogs of azithromycin, consisting of a variable linker, a triazole moiety to facilitate late-stage combination by Cu-click chemistry, and various functionalized indole rings to mimic tryptophan 155 of SecM (Scheme 7).
Scheme 7.

Oyelere’s Semisyntheses of Extended-Range Azithromycin Analogs
Beginning from the available azathramycin (114), which lacks the N-methyl group of azithromycin (36) at N9a, one of either of two alkynyl linkages (115 or 116) were introduced via reductive amination. The key variable here was the rigidity of the carbon chain, which might influence whether the indole functionality can adopt the correct conformation for π-stacking. The extended azithromycin analogs were completed upon addition of the indole bearing an alkyl azide tether, which can vary in placement around the indole. Substitution at C6 permitted modulation of the ring’s electronics, while methylation of nitrogen and saturation of the five-membered ring provided additional variables to establish a potential SAR.
To test these extended-range azithromycin analogs, researchers in the Oyelere group subjected these to both in vitro and in vivo experiments. To bypass the issue of cell permeability, these analogs were tested directly against the E. coli ribosome in a cell-free context using a luciferase-based reporter to measure protein synthesis. Furthermore, these compounds were tested against S. aureus 29213, a susceptible strain. Only the analogs which maintained or improved upon azithromycin’s in vivo or in vitro activities are discussed in Table 3. Notably, the results do not correlate well. While several analogs outperformed azithromycin in the in vitro assay, those same molecules had considerably higher MIC50 values (121, 122, 124, 125, and 126). No analog was able to inhibit bacteria more efficiently than the known antibiotic, and each one matching azithromycin’s in vivo activity yielded inferior in vitro results. This discrepancy can only highlight a key difficulty in drug discovery—the sheer number of factors in drug efficacy besides affinity for the target.
Table 3.
Biological Evaluation of Extended-Reach Azithromycin Analogsa
| Linker | Indole | IC50 | MIC50 | |
|---|---|---|---|---|
| 119 | C3H8 |

|
0.54 | 0.78 |
| 120 | C3H8 |

|
0.88 | 0.78 |
| 121 | C3H8 |

|
0.227 | 1.56 |
| 122 | C3H8 |

|
0.128 | 1.56 |
| 123 | C3H8 |
|
0.49 | 0.78 |
| 124 | C3H8 |

|
0.147 | 1.56 |
| 125 | C6H4 |

|
0.12 | 3.13 |
| 126 | C6H4 |

|
0.197 | 1.56 |
| azithromycin (36) | 0.292 | 0.78 |
IC50 values in μM and tested against cell-free E. coli ribosomes; MIC50 values in μg/mL against a susceptible strain of S. aureus; green denotes an increased activity; orange, decreased; yellow, equipotent.
3.9. Summary
Despite the numerous obstacles encountered in semisynthesis, synthetic chemists have demonstrated that the exploration of unexplored chemical space surrounding these storied molecules can indeed be a fruitful endeavor. While semisynthetic approaches led to successful drugs such as the innovative azithromycin, a clear way forward relies on the application of total synthesis to develop compounds inaccessible by semisynthesis given the inherent limitations on chemistry imposed by Nature.
4. FROM BENCHTOP TO BEDSIDE—INNOVATIONS IN THE TETRACYCLINES
Tetracyclines have stood on the front lines in the battle against bacterial infections as powerful broad-spectrum antibiotics for nearly 70 years from the discovery of aureomycin in 1948 to recent efforts at Tetraphase Pharmaceuticals to develop and bring to market new totally synthetic tetracyclines.21,122
4.1. Mechanism of Action
Tetracyclines inhibit bacterial growth by reversibly binding to the bacterial ribosome, specifically the 30S subunit.123 As shown in Figure 14, the southern ridge of tetracycline forms an intricate hydrogen-bonding network with the sugar-ribose backbone of the rRNA. Such binding inhibits aminoacyl-tRNA (aa-tRNA) from entering the A-site of the ribosome, which is then unable to use aa-tRNA as building blocks for protein synthesis. Deactivating this critical process prevents bacteria from multiplying, which explains the tetracyclines’ bacteriostatic nature. Furthermore, the relatively nonspecific binding to the phosphate backbone suggests that the identity of the nucleotides is not critical to binding; this rationalizes the tetracyclines’ relatively broad spectrum of antimicrobial activity and explains the relative absence of ribosomal mutations as a mechanism of tetracycline resistance.
Figure 14.

Crystal structure of tetracycline (2) bound to E. coli ribosome; 50S subunit in orange, 30S subunit in yellow, ribosomal proteins omitted for clarity. Tetracycline shown in blue binding to the A site of the ribosome, which inhibits binding of acyl-amino-tRNA necessary for protein synthesis. On the right, 2 binding to selected residues demonstrating key hydrogen-bonding contacts between the southern ridge of 2, a magnesium ion, and the sugar−phosphate backbone. Images produced in UCSF Chimera, PDB ID 5J5B.
4.2. Resistance to Tetracyclines
4.2.1. Tetracycline Efflux
In the mid-1950s, scientists began to observe diminished activity of tetracycline against bacteria. These resistance factors, attributed to tet genes, are classified into two general modes of action. The first group identified, including the well-known TetA and TetB, among others, encode membrane-bound tetracycline efflux pumps, which actively remove tetracycline from the cytoplasm.124 These efflux pumps have been observed in both Gram-positive and Gram-negative species. In particular, TetA, which is only capable of exporting tetracycline proper, is observed in both families of pathogens; the more significant TetB is only observed in Gram-negative bacteria and confers resistance to more sophisticated tetracyclines, including minocycline, but not the more recently developed glycylcyclines.
4.2.2. Ribosomal Protection Proteins
A second mechanism of resistance features ribosomal protection proteins (RPPs), encoded by TetM, TetO, and others.125 These cytoplasmic proteins bear considerable sequence similarity to the elongation enzymes EF-G and EF-Tu.126 It was initially hypothesized that TetM and TetO were acting as Tc-resistant elongation factors and allowing protein synthesis to continue normally, but it was quickly shown that these enzymes do not exhibit such activity.127,128 It is, therefore, possible that the RPPs and elongation factors are related through evolution, though they do not show homologous activity. The RPPs bind to the ribosome in close proximity to the A (major) binding site of tetracycline, and it has been observed that the action of RPPs can reduce the apparent binding constant of tetracycline by a factor of 6.128,129 The mechanisms of action of both TetM and TetO have been studied by cryo-EM.130,131 Authors of both studies identify key loops near the C-terminus of the respective enzymes that perturb Tc binding, particularly near the D-ring. These hypotheses regarding the protective activities of TetM and TetO were further supported by experiments in which TetM and TetO knockouts, mutants, and truncations all restore susceptibility to tetracycline.
4.2.3. Other Resistance Phenotypes
Other species-specific resistance mechanisms have been characterized, including enzymatic oxidative decomposition of the drug in Bacteroides via TetX, which has not been observed in any other bacteria.132 A previously poorly understood TetU was thought to exhibit RPP-like activity based on reported low-level resistance and some sequence similarity with TetM, but expression of TetU alone in E. coli conferred no resistance to several common tetracyclines tested.133 Given that tetracyclines are intended as broad-spectrum agents, these singular instances of resistance are not of major concern to drug discovery efforts.
4.3. Semisynthetic Approaches to Tetracyclines
Given the rapid evolution of two mechanisms of resistance to tetracycline, the budding pharmaceutical industry fought to keep ahead of these pathogens by developing new tetracyclines that could overcome this resistance. The first tetracycline isolated from Nature was 7-chlorotetracycline (127), though Lloyd Conover at Pfizer later found that hydrogenolysis of the carbon–chlorine bond at C7 yielded another effective agent, tetracycline (2), which itself was later identified as a natural product (Figure 15). Shortly afterward, scientists at Pfizer identified oxytetracycline (129), which was marketed under the name terramycin; Woodward famously elucidated the structure of this antibiotic, which allowed chemists to start to tinker with the molecule. Later efforts focused on modifying a related natural product, 6-demethyltetracycline (128). In 1958, scientists at Pfizer found that treatment of 128 with hydrogen over palladium under acidic conditions also carries out deoxygenation at C6 to yield sancycline (130), the simplest tetracycline derivative that retains activity and is therefore a useful scaffold for semisynthetic drug discovery.134 Scientists at Lederle then found that nitration at C7 followed by a reduction/reductive amination with formaldehyde afforded another active species, minocycline (132).135,136 Decades later, in 1994, researchers at Wyeth repeated the nitration/reduction sequence at C9 and acylated the resultant amine with a glycine derivative to yield tigecycline (133), renowned as one of the few agents still capable of defeating multidrug-resistant bacteria.137 Computational studies suggest that tigecycline’s improved potency arises from an additional hydrogen-bond between the glycinyl side chain and C1054 in the ribosome.138 However, tigecycline received in 2013 an FDA “black box” warning for “all-cause mortality increase”, which renders the drug a medically useful option only as a last resort in life-threatening situations. More recently, researchers at Paratek Pharmaceuticals carried out a different semisynthesis from minocycline (132) arriving at a new class of tetracyclines termed aminomethylcyclines; the lead compound in this class, omadacycline (134),139 has recently met key Phase III clinical trial end points against community acquired bacterial pneumonia (CABP)140 and acute bacterial skin and skin-structure infections (ABSSIs).141 While greater than a half-century of semisynthesis has undoubtedly been fruitful, delivering many clinically useful agents, one might observe that the inherent chemical reactivity of the natural tetracycline scaffold is limited to modification at C7 and C9, which has been largely exhausted.
Figure 15.

Phylogenetic relationship of semisynthetic tetracycline derivatives and parent natural products with dates of FDA approval.90 Though tetracycline was first discovered following hydrogenolysis of chlorotetracycline, it was later identified as another streptomycete natural product.
4.4. Early Totally Synthetic Approaches
The semisynthetic efforts of the pharmaceutical industry did not go unnoticed; tetracyclines have always been popular targets for synthetic chemists (Figure 16). A relatively simplified anhydrotetracycline (135) was prepared by Shemyakin and coworkers at the former USSR Academy of Sciences in 1966 in a relatively short sequence,142 though the more complicated scaffolds required considerably more effort. Woodward’s synthesis of sancycline (130), beginning from the simple m-methoxybenzoic acid, established a trend of beginning with the D-ring and working to the east.143 Later syntheses followed this trend, borrowing Shemyakin’s starting material, juglone, as a precursor containing the D- and C-rings. Muxfeldt’s synthesis of the considerably more complex terramycin (129) avoids a number of synthetic pitfalls, namely various degradation pathways by retro-aldol-type mechanisms.144 While certainly an innovative work, their synthetic efforts are still not of significant use to the drug discovery field, given their 22-step sequence yielding only 0.06% of racemic terramycin. A more viable synthetic route to a tetracycline was not known until Stork’s 1996 synthesis of a 12a-deoxygenated tetracycline (136), which would require only C12 oxidation to yield the core structure, starting from juglone and requiring only 16 steps.145 Most impressive is the vast improvement in yield over previous work, rendering this approach attractive to industrial scale-up. Furthermore, the use of a benzyloxyisoxazole as a masked vinylogous carbamic acid on the A-ring was a key innovation, discovered in a previous effort,146 that will allow more efficient syntheses in the future. While limited by its racemic nature, the synthesis nonetheless demonstrated that total synthesis of tetracyclines could be industrially feasible, providing a realistic alternative to semisynthesis. The first asymmetric total synthesis of tetracycline147 followed Stork’s work by a few years, though its inefficiency did not provide a step forward in tetracycline drug discovery.
Figure 16.

Profiles of total syntheses of tetracyclines.
4.5. Modern Strategies in the Development of Totally Synthetic Tetracyclines
In their 1968 publication, Woodward and co-workers made key observations: that there existed a “need for a versatile method of synthesis which might be employed in exploring structure–activity relationships more deeply” and that there was a more fundamental need to prepare “tetracyclines which could not be obtained by partial synthesis or directly by fermentation.”143 Despite the recognized limitations of semisynthesis, total synthesis lagged behind its cousin for decades; it was, perhaps, itself limited by the state of existing methodology in constructing these formidable molecules. In order for tetracyclines to advance, chemists needed to develop synthetic methods to access these complex scaffolds with unnatural substituents in an efficient manner. In their landmark 2005 work, Myers and co-workers report precisely such a method: a convergent, efficient synthesis that allows for diversification to a variety of rationally designed tetracycline derivatives.148 From the prior half century of work, it is known that modifications on the D-ring of tetracyclines are the most fruitful. Charest et al. designed this synthesis to incorporate the D-ring at the last possible moment, allowing for late-stage diversification via varying D-ring donors without radically reengineering the synthesis (Figure 17). This approach can be classified as an exercise in diverted total synthesis, a scheme in which late-stage, reactive intermediates serve as a branching point to access more variation in chemical space.149
Figure 17.

Schematic comparison of a previous totally synthetic approach to the tetracyclines with Myers’ strategy.
4.5.1. First Generation of Novel Synthetic Tetracy-clines
Myers’ synthesis began with benzoic acid (137), which is dihydroxylated biosynthetically by the bacteria reported as A. eutrophus but known today as a member of the genus Cupriavides (Scheme 8). Hydroxyl-directed epoxidation and subsequent esterification with trimethylsilyldiazomethane followed. Treatment with tert-butyldimethylsilyl triflate forced transposition of the epoxide and formation of the less sterically hindered silyl ether 140. Lithiated isoxazole 141 added to the ester to yield epoxy ketone 142 (Scheme 8). Addition of the strongly Lewis-acidic lithium triflate mediated the addition of the amine to the epoxide, which tautomerized to the nitrogen ylide 143. Mechanistic studies performed by the Myers group indicated that this reaction followed a Stevens-like rearrangement, with the ylide dissociating to yield diradical intermediate 144, which then recombines to close the A-ring. Treatment with 2-nitro-benzenesulfonylhydrazine, a reagent developed by the Myers group, permitted deoxygenation with rearrangement to strategically position the alkene for enone formation.150 A final deprotection, oxidation, and reprotection sequence furnished key AB-enone 147 in 11 steps with 10% overall yield. Given that this enone is a critical building block in the construction of novel tetracyclines, the Myers group has reported a number of more efficient routes to this material,151,152 including several applicable to large-scale manufacturing.153,154
Scheme 8.

Myers’ First-Generation Synthetic Route to AB-Enone 147
Accomplishing what no previous synthetic campaign could, Myers and co-workers constructed a variety of structurally unprecedented tetracyclines in a highly convergent manner. The AB-enone was coupled with an ambiphilic D-ring donor bearing both nucleophilic and electrophilic functionalities to permit C-ring construction via a Michael–Claisen sequence. A benzylic nucleophile was generated by either deprotonation of a deactivated toluene or lithium-halogen exchange of the corresponding benzylic bromide. The resultant nucleophile added stereoselectively to the β-carbon of the enone in a Michael addition directed by the bulky silyl ether blocking the bottom face of the enone. An enolate was generated, which attacks the carbonyl of the nearby phenyl ester, which completes construction of the C-ring following collapse of the tetrahedral intermediate and tautomerization from the 1,3-diketone. All that remained was desilylation with hydrofluoric acid and hydro-genolysis to reveal the vinylogous carbamic acid from the 3-benzyloxyisoxazole. The sequence of these two steps differs among the various substrates for maximum efficiency. To further highlight the potential for structural diversity with this synthetic platform, Myers and co-workers were able to divert material designed for 6-deoxytetracyclines into a pathway to introduce C5 oxidation, which permitted access to other medically relevant tetracyclines such as doxycycline.
To summarize their first report, the Myers group constructed five new tetracycline compounds using this platform (Scheme 9); these featured somewhat unprecedented structural modifications including the incorporation of heterocycles as the D-ring and construction of a pentacycline with an additional fused ring (152). In the interest of brevity, we neglect the synthetic particulars of the late-stage construction and instead discuss the significance of this diverted total synthesis strategy.
Scheme 9.

Divergent yet Convergent Approach To Generating Diverse and Unprecedented Tetracycline Scaffolds
4.5.2. Evaluation of Novel Tetracyclines
While an impressive feat in the field of total synthesis, the Myers group did not construct these tetracyclines merely to admire their structures. A successful DTS campaign would yield new drug candidates that overcome resistance to the tetracyclines. These tetracyclines were assayed for antimicrobial inhibition against a number of species of pathogenic bacteria, both Gram-positive and −negative, with the natural tetracycline serving as a control. Selected results are summarized in Table 4. Two compounds, 6-deoxytetracycline (148) and pentacycline (152) performed as well as the natural tetracycline against a susceptible strain of S. aureus, but they had great success against a clinical multidrug-resistant MRSA strain, providing proof-of-concept that synthetic tetracyclines are a source of untapped potential. Results against the Gram-negative E. coli strains are less compelling, though this is not surprising, as combatting these bacteria is known to be challenging. While the pentacycline was more active against Gram-positive strains, its rather poor activity against E. coli limits its applicability as a broad-spectrum candidate. This DTS campaign was greatly successful as it provided two lead compounds with promising potencies. Given the nearly endless possibilities provided by this platform, these compounds, though far from clinical candidates, provide launching points in chemical space to explore finely tuned tetracyclines as ideal drug candidates.
Table 4.
Minimum Inhibitory Concentrations (MICs) in μg/mL of Compounds against Two Strains of S. aureus and Three Strains of E. colia
| Gram-positive | Gram-negative | ||||
|---|---|---|---|---|---|
|
| |||||
| Compound | S. aureus | E. coli | |||
| ATCC 29213 | ATCC 700699 | ATCC 25922 | ACH-0095 | PBR322 | |
| Tetracycline (2) | 1 | >64 | 1 | >64 | >64 |
| 148 | 1 | 2 | 4 | 16 | 16 |
| 149 | >64 | >64 | >64 | ND | ND |
| 150 | 8 | >64 | 2 | >64 | 64 |
| 151 | 16 | 64 | 32 | ND | ND |
| 152 | 1 | 1 | >64 | >64 | 64 |
![]()
In each category, the first strain listed is a susceptible QC strain; the second strain is a tetracycline-resistant clinical isolate. pBR322 is an engineered laboratory strain carrying a plasmid marker for tetracycline resistance. ND = not determined.
4.5.3. Later Generation Tetracyclines
Considering the promising leads gained from their first campaign, the Myers group continued their diverted total synthesis strategy toward developing new tetracyclines as clinical candidates. In their second report, which culminates in the construction of more than 50 new tetracyclines (including pentacycline derivatives), the approach is more finely tuned in developing analogs based around the two lead compounds from their previous work.155 Like in their prior work, key AB-enone 147 served as a common intermediate for a DTS effort (Scheme 10). Here, the D-ring donors are configured to place a 6-aryl substituent on the 6-deoxytetracycline scaffold, which is absolutely inaccessible by semisynthesis. While unlikely to enhance binding to the ribosome through π–π interactions, these aryl groups could modulate cell permeability or other pharmacological properties relevant to potency. To further explore the pentacycline scaffold, the naphthalene nucleophile can be decorated with a dimethylamino group at C7 and various aminomethyl groups at C10, perhaps inspired by minocycline and omadacycline, respectively. While the unsubstituted pentacycline prepared earlier had disappointing activity against Gram-negative species, further modification could yield a more potent broad-spectrum agent; taking inspiration from existing drugs is certainly a well-precedented strategy in medicinal chemistry.
Scheme 10.

Myers’ 2008 Extension of the DTS Approach To Expand on Leads Identified in 2005 Work
Thanks to the broad applicability of their drug discovery platform, Myers and co-workers rapidly constructed a variety of analogs in both scaffold families; the two best compounds are 153 and 154. While both retained the improvement against S. aureus seen in the prior work, the aryl tetracycline struggles against E. coli. However, the substituted pentacycline is very promising given its excellent broad-spectrum activity; this is a clear improvement over the previous compounds.
More recent efforts have also targeted specific novel tetracycline scaffolds, including the development of hexacycline derivatives, which exhibit promising results against Gram-negative pathogens including P. aeruginosa.156 More recently, the Myers group published a modified synthetic route to 5-oxytetracycline derivatives, which were hypothesized to enhance activity against Gram-negative strains thanks to an increase in polar surface area.157 However, these compounds did not show an increase in potency, which might be attributed to poor aqueous stability.
4.5.4. Translational Efforts from Tetraphase
With an efficient method to prepare highly diversified tetracyclines and promising results from preliminary synthetic work, it should be no surprise that Myers founded a pharmaceutical startup, Tetraphase, in the Boston area with the goal of applying this drug discovery platform to develop and market new tetracycline antibiotics. Despite Myers Group’s discovery of potent tetracycline derivatives, Tetraphase adopted a somewhat conservative approach in designing new compounds. From a fluorinated D-ring donor, researchers at Tetraphase rapidly constructed the tetracycline scaffold bearing C7 fluorination.158 A nitration/reduction sequence installs a C9 amino group while concurrently deprotecting the oxygens on the southern ridge of the molecule and cleaving the isoxazole. To generate unprecedented fluorinated glycylcyclines, the resulting amine was acylated either directly with a glycine derivative or with bromoacetyl bromide followed by substitution with the desired amine. Using this late-stage, narrowly focused diverted total synthesis approach, Tetraphase was able to accumulate a library of 7-fluoro-9-aminoacetamido-6-demethyl-6-deoxytetracyclines, hereafter referred to as fluorocyclines. Antimicrobial assays revealed that a pyrrolidine ring provided the highest potency against a very broad variety of pathogenic bacteria, including both Gram-positive and Gram-negative species. Most notably is that this compound (155) outperformed tigecycline, the most recent tetracycline to be approved for use, in many in vitro assays. The authors also explored substituted pyrrolidines, though the unsubstituted ring proved best. Further modification of this C9 side chain was also studied,159 though the pyrrolidine compound, first named TP-434, would be selected as the company’s lead compound (Figure 18). Chemists at Tetraphase quickly developed a synthetic route to prepare sufficient material for distribution.160 Eravacycline (155) was found to overcome the most prevalent tetracycline resistance mechanisms, including TetM, a ribosomal protection protein, and several efflux pumps, including TetA, TetB, and TetK.161 Only against the relatively rare tetX (degradation via oxidation) was tigecycline more active than eravacycline.
Figure 18.

Tetracycline derivatives under development by Tetraphase.
With a promising candidate in hand, Tetraphase began to explore the utility of eravacycline in the clinic. Despite promising early results, eravacycline encountered difficulties in September 2015; it was not able to show noninferiority compared to levofloxacin, a fluoroquinoline antibiotic, in treating complicated urinary tract infections (cUTIs). However, Tetraphase recently reported more promising results, in which eravacycline showed noninferiority compared to ertapenem in treating complicated intra-abdominal infections (cIAIs), achieving a cure rate within 1% of the existing treatment.162 Notably, eravacycline accomplished this end point at a lower dosing level (Eravacycline administered 1 mg/kg every 12 h, ertapenem 1 g every 24 h).
Tetraphase has expanded this technology to generate other clinical candidates, including those in Figure 18. An additional drug candidate, TP-6076, has entered Phase I trials, though to the best of our knowledge, its structure has not been publicly disclosed, presumably in accordance with patent proceedings. The promising outlook surrounding totally synthetic tetracyclines has very likely inspired the development of macrolides, as discussed in Section 3.6.
4.6. Viridicatumtoxin B – a Tetracycline in Disguise?
The viridicatumtoxin family of fungal natural products was first discovered in 1973;163 researchers identified viridicatumtoxin B (158) as a potent antibiotic in 2008 (Figure 19).164 A later screen suggested that the viridicatumtoxins and the related spirohexa-lines inhibit undecaprenyl phosphate (UPP) synthase,165 which is surprising given the considerable structural similarity to the tetracyclines, differing only in oxidation at C4a and C5, the spirocyclic ring system bridging C6 and C7, and the omission of the dimethylamino group at C4.
Figure 19.

Ring systems and numbering in viridicatumtoxin B (158).
4.6.1. Nicolaou’s Synthesis of Viridicatumtoxin B and Analogs
In 2013, Nicolaou reported a racemic synthesis of viridicatumtoxin B and offered a minor revision of the structure,166 which was followed closely by a series of racemic analogs to probe the SAR.167 Nicolaou very recently reported an asymmetric synthesis of the natural product and a small library of derivatives, which we discuss here in detail.168 While Myers’ innovations were helpful in developing this synthesis, there remained key synthetic challenges to overcome in this unique structure, including asymmetric formation of the spirocyclic system (rings E and F) and the unprecedented oxidation at C4a. Furthermore, preparation of both enantiomers was necessary to establish the absolute configuration of the natural product. Retrosynthetically, Nicolaou used a Myers-inspired Michael–Claisen reaction to form the A-ring bearing the familiar isoxazole (Scheme 11). A diastereoselective spirocyclization proceeded smoothly but required stereocontrol at C6, which was set by a catalytic asymmetric alkylation developed for this work.
Scheme 11.

Nicolaou’s Construction of the Viridicatumtoxin Skeleton
To effect the asymmetric alkylation, the Nicolaou group identified the suitable phase-transfer catalyst 161 and optimized a method to efficiently yield 162 with high enantioselectivity. Lewis acid-mediated spirocyclization set the E- and F-rings in the desired conformation; oxidation with phenyliodine diacetate (PIDA) was followed by treatment with acid to desaturate the E- ring. Subsequent oxidation with PIDA restored the benzoqui-none as the dimethoxyketal, which permitted addition of the deprotonated isoxazole derivative 166. This asymmetric Michael–Claisen reaction, inspired by the Myers group’s work, used phase-transfer catalyst 167 to define the stereochemistry at C4a. Following removal of the trimethylsilylethoxycarbonyl (Teoc) protecting group, oxidation at C12a resulted from the action of dimethyldioxirane and catalytic nickel (Scheme 12). 1,6-Reduction at C14 rendered the C-ring aromatic, while acid returned the ketal at C5 to the ketone. To achieve stereoselective oxidation at C4a, it was necessary to reduce and protect the ketone at C1 as the tert-butylsilyl ether to promote oxidation of the bottom face of the molecule with Davis’ oxaziridine. Straightforward chemistry restored the ketone at C1 and cleaved the isoxazole to provide (+)-viridicatumtoxin (158) as a single enantiomer.
Scheme 12.

Total Synthesis of Viridicatumtoxin B and Related Analogs
To establish a structure–activity relationship in this family of natural products, the Nicolaou group designed a series of simplified analogs diverted from intermediates near the end of the synthetic route, exploring the effect of inverting various stereocenters and altering oxidation and substitution at C4a and C5. Each analog with the proper stereochemistry at the EF spirocenter is given in Scheme 12, and the enantiomer of each was also evaluated; results against a small panel of pathogenic bacteria are provided in Table 5. Notably, removing oxidation at C4a had no major effect, though a ketone at C5 appeared beneficial, irrespective of the stereochemistry at the EF spirocenter.
Table 5.
Evaluation of Viridicatumtoxin B and Related Analogs against a Panel of Gram-Positive and −Negative Bacteriaa
|
|
||||
|---|---|---|---|---|
| VRE | MRSA | Gram-(−) | ||
|
|
||||
|
E. faecalis S613 |
E. faecium 105 |
S. aureus 371 |
A. baumannii AB210 |
|
|
|
||||
| minocycline (132) | 8 | 8 | 1 | 4 |
| tigecycline (133) | 0.5 | 0.25 | 2 | 0.5 |
| (+)-158 | 2 | 2 | 4 | 64 |
| (−)-158 | 4 | 4 | 8 | 64 |
| (+)-175 | 8 | 8 | 8 | 64 |
| (−)-175 | 16 | 16 | 32 | 64 |
| (+)-176 | 16 | 16 | 8 | 64 |
| (−)-176 | 8 | 16 | 64 | 64 |
| (−)-177 | 128 | 64 | 128 | 64 |
| (+)-177 | 1 | 2 | 64 | 64 |
| (−)-178 | 4 | 8 | 8 | 64 |
| (+)-178 | 2 | 8 | 8 | 64 |
| (+)-179 | 0.5 | 2 | 2 | 64 |
| (−)-179 | 1 | 1 | 2 | 64 |
| (−)-180 | 1 | 0.5 | 2 | 64 |
| (+)-180 | 1 | 1 | 2 | 64 |
MICs in μg/mL. Minocycline and tigecycline serve as positive controls. Analogs with enhanced activity against both VRE and MRSA relative to the natural product are highlighted in green.
Time-kill assays indicated that the viridicatumtoxins may share some mechanistic similarities with the tetracyclines, though a thorough biological investigation beyond in vitro assays and analog development is warranted.
5. VANCOMYCIN, GLYCOPEPTIDES, AND LIPOGLYCOPEPTIDES
Vancomycin (4), the prototypical glycopeptide, was isolated in 1955 from Actinobacter by McGuire and co-workers at Eli Lilly as a potent antibiotic,169 though its structure would remain unknown for another 25 years.170,171 Due in part to a lack of suitable treatment for serious staphylococcus infections, vancomycin was granted FDA approval in 1958. However, given concerns regarding side effects, including the infamous “red neck” syndrome and some reports of ototoxicity, vancomycin was relegated in favor of methicillin and used only as a last resort or in patients with a β-lactam allergy.172 Given the rise of MRSA over the last decades, vancomycin and related glycopeptides are enjoying a resurgence in usage, though this increased usage is accompanied by a rise in vancomycin-resistant bacteria.173
5.1. Mechanisms of Action and Resistance
5.1.1. Mechanism of Action
Vancomycin inhibits the critical process of cell wall biosynthesis, binding noncovalently to the D-Ala-D-Ala residues in the nascent peptidoglycan, inhibiting specifically the transpeptidation cross-linking.174 In the late stages of peptidoglycan biosynthesis, Lipid II is translocated from inside the cell membrane to the extracellular space by the recently discovered flippase, MurJ.175 Following linking of the lipid II monomers by transglycosylases, transpeptidases cross-link the pentaglycine side chain to the first D-alanine in the pentapeptide with loss of the terminal D-alanine. Vancomycin makes five key hydrogen bonds with the D-Ala-D-Ala dipeptide,176 as evidenced by crystallization with a model substrate (Figure 20).177
Figure 20.

Crystal structure of vancomycin bound to a Lipid II mimic. Below, computational studies demonstrating the decreased binding affinity associated with the VanA phenotype. PDB ID 1VFM.
5.1.2. Glycopeptide Resistance
Resistance to vancomycin and the glycopeptides is largely limited to two closely related mechanisms. Because vancomycin does not target protein or genetic material, mutations cannot confer resistance. Instead, bacteria have evolved to alter the D-Ala-D-Ala target in the nascent peptidoglycan via two closely related resistance phenotypes, VanA and VanB.178 Each comprising five genes, vanRSHA(or B)X, these inducible mechanisms detect vancomycin utilizing a two-component regulatory system comprising VanR, a response regulator, and VanS, a sensor kinase. This system then upregulates the remaining H, A, and X genes at the transcriptional level.179,180 VanH reduces pyruvate to D-lactate, and VanA and VanB, the namesakes of these resistance mechanisms, esterify D-lactate onto D-alanine (Figure 21). Finally, VanX cleaves the peptide bond between L-lysine and D-alanine in the nascent peptidoglycan; the D-Ala-D-Lac piece is ligated onto the cell wall precursor by the normal MurF.181
Figure 21.
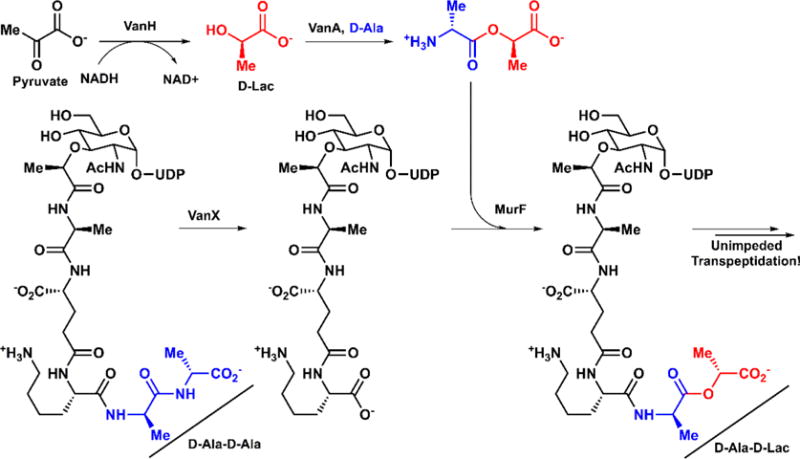
Biosynthesis of VanA-type Lipid II bearing a D-Lac substitution.
VanA and VanB effectively carry out a single atom substitution in the target of vancomycin. As shown in Figure 20, the amide carbonyl of the central amino acid in vancomycin makes a critical hydrogen bond with the amide nitrogen in D-Ala-D-Ala. If the amide nitrogen is replaced by oxygen, a hydrogen bond no longer exists, and the binding energy decreases dramatically.182 If vancomycin is unable to bind to the peptidoglycan, it cannot sterically inhibit transpeptidation, and cell wall biosynthesis proceeds normally.
5.2. Semisynthetic Glycopeptides and Lipoglycopeptides
Like other natural product antibiotics, semisynthesis spurred innovation in the development of new glycopeptides and lipoglycopeptides to overcome increasing resistance levels (Figure 22).
Figure 22.

Structures of approved glycopeptides and lipoglycopeptides with year of approval90 and sourcing. * denotes epivancosamine.
5.2.1. Telavancin
Telavancin (184) was reported as TD-6424 in 2003 by scientists at Theravance Pharmaceuticals as a semisynthetic lipoglycopeptide.183 Initially, it was found that alkylation of the vancosamine nitrogen with a decylaminoethyl group conferred activity against resistant strains. However, the increased hydrophobicity of this compound, THRX-689909, led to issues in absorption, distribution, metabolism, and excretion (ADME). To mediate these concerns, the group at Theravance sought to incorporate an additional hydrophilic moiety onto the vancomycin scaffold. Substitution of the resorcinol A-ring under Mannich conditions and further functionalization installed a phosphonic acid, giving rise to TD-6424 (184).184
Telavancin was selected for advancement into clinical trials as a compromise between slightly diminished antibacterial activity relative to THRX-689909 and a more favorable pharmacological profile, as it showed reduced accumulation in the liver and kidneys and improved excretion.185 Telavancin demonstrated clear superiority over vancomycin in treating Gram-positive skin infections, and it was approved by the FDA in 2009.186
5.2.2. Oritavancin
In the 1980s, scientists at Eli Lilly sought to identify new vancomycin-like natural products as leads for antibiotic development.187 Screening known glycopeptide producers yielded a closely related scaffold, chloroeremomycin, which differed from vancomycin in epimerization of vancos-amine as well as an additional L-4-epivancosamine on the sixth amino acid from the N-terminus. The further glycosylation appeared to cause increased potency, despite reduced solution binding to D-Ala-D-Ala substrates.
Reports of teicoplanin, the first lipoglycopeptide, encouraged medicinal chemists at Lilly to explore the functionalization of the epivancosamine nitrogen with various lipophilic groups. Preliminary work identified the addition of a p-chlorobenzyl-methyl group as beneficial; the p-chlorobiphenylmethyl enhanced activity considerably. This lead compound, known previously as LY333328, brought about a 2000-fold reduction in MIC90 against VanA-VRE over vancomycin. After changing hands several times in the early 21st century, oritavancin (183) was finally approved in 2014 and brought to market by The Medicines Company. It is indicated for Gram-positive acute bacterial skin and skin structure infections (ABSSSI) via IV administration.
5.2.3. Dalbavancin
Formerly known as BI 397, dalbavancin (181) is produced semisynthetically from the natural teicoplanin-like lipoglycopeptide A40926 from an actinomycete.188 Developed by Biosearch (Italy), this drug outperforms teicoplanin and vancomycin against Staphylococci and is equipotent with teicoplanin against Streptococci.189 Following successful murine models, dalbavancin performed well against Gram-positive skin infections in clinical trials.190 Furthermore, dalbavancin administered once-weekly demonstrated noninferiority to vancomycin and linezolid administered twice daily with fewer adverse effects.191 After changing hands numerous times, dalbavancin was brought to market by Durata Therapeutics, a subsidiary of Actavis, following its 2014 FDA approval.
5.3. Mechanistic Investigations of the Lipoglycopeptides
That lipoglycopeptides are able to overcome VanA-type resistance puzzled scientists for quite some time. Overcoming VanA resistance was thought to require a change in peptide binding, but these analogs retain the same peptide scaffold as vancomycin. Early hypotheses implicated membrane anchoring via the hydrophobic tails, which would render the peptide–peptide interaction pseudointramolecular and conceivably permit binding despite the disfavored hydrogen bond.192 However, there is insufficient evidence to suggest that this mechanism enhances binding to the extent that activity is restored. Another early hypothesis cited dimerization of these lipoglycopeptides, which has been observed in solution. While dimerized species were active against VanA resistance, there is no evidence that only dimerization (and not the attached groups to facilitate dimerization) restored activity.193 To elucidate this mechanism definitively, the Kahne laboratory designed experiments to identify precisely which step in peptidoglycan synthesis is inhibited by structural analogs of vancomycin.194 This can be determined using radiolabeled precursors and observing incorporation into the various peptidoglycan intermediates. For example, vancomycin is a transpeptidase inhibitor, and this is indicated by a decrease in mature (cross-linked) peptidoglycan and a buildup of the preceding immature peptidoglycan. If a chlorobiphenylmethyl (CBP) or n-decyl group is placed on vancosamine, the mechanism shifts to an upstream inhibition, with a decrease of both mature and immature peptidoglycan, indicating transglycosylase inhibition. Kahne postulates two mechanisms consistent with these results: (1) these lipophilic substituents anchor these molecules to the cell membrane where they interact with membrane-bound Lipid II instead of immature peptidoglycan and would therefore act as transglycosylase inhibitors, and (2) these lipoglycopeptides, instead of inhibiting the peptide substrate, target the enzymes responsible for peptidoglycan biosynthesis. These hypotheses, as Kahne would show, are easily distinguished by separating peptide binding from the carbohydrate moiety bearing a lipophilic substituent, and they do so by testing truncated analogs 189 and 190 (Figure 23). The unsubstituted disaccharide 185 had no activity, but the lipophilic group on 186 does confer activity, which is obviously not due to peptide binding! This result provides strong evidence that the lipoglycopeptides inhibit the transglycosylation enzymes directly instead of competitive binding for the peptide substrate.
Figure 23.

Truncated lipoglycopeptide analogs developed for mechanistic studies by Kahne.
5.4. Total Synthesis Campaigns
Given the immense structural complexity of the vancomycin scaffold, many of the top synthetic laboratories set out to construct this monolith. As is observed for numerous molecules, the chief synthetic target is the aglycon. We discuss here three synthetic campaigns toward the vancomycin aglycon, including one purely synthetic approach and two that began as synthetic efforts but evolved into analog development. The three synthetic campaigns were all completed within the span of about a year, the third of which remains underway seeking effective vancomycin derivatives. These syntheses must overcome a host of common difficulties, especially controlling the selective formation of atropisomers (rotational isomers). Synthetic campaigns toward vancomycin have been reviewed thoroughly;195,196 we therefore neglect some of the finer details and focus on the efforts in diversification of the glycopeptides.
5.5. Evans’ Total Synthesis of the Vancomycin Aglycon
Evans’ 1998 synthesis of the vancomycin aglycon197 proceeds from simple amino acid building blocks, beginning with the C-ring, which is elaborated into an amino acid derivative using Evans’ own asymmetric methodology (Scheme 13).198,199 The scaffold is rapidly elaborated to incorporate the A and B aryl groups, which are coupled using an oxidative technique developed by Evans.200 The resulting biaryl system 192 is coupled with a D-ring amino acid, which is then subjected to SNAr chemistry to form the C–D biaryl ether system with a 5:1 ratio of atropisomers. In order to permit construction of the scaffold, a number of protecting group manipulations are necessary, which lengthen this synthesis to a degree. Ultimately, Evans is able to construct ABCD precursor 195 with good control over locked rotational isomers in both the AB and CD systems. The remaining tripeptide bearing the E-ring is assembled rapidly beginning with Evans’ asymmetric methodology to develop the highly substituted phenylalanine and proceeding with standard peptide coupling (Scheme 14).201 The two precursors are united into the heptapeptide, and a second SNAr reaction completes the DE biaryl ether. Following seven protecting group manipulations, Evans and co-workers arrive at the completed vancomycin aglycon (201).
Scheme 13.

Evans’ Preparation of the Left-Hand Tetrapeptide 195
Scheme 14.

Preparation of Right-Hand Tripeptide 200, Union with 195, and Completion of the Synthesis
5.6. Nicolaou’s Total Synthesis of Vancomycin
The Nicolaou laboratory, formerly at The Scripps Research Institute, published their synthesis of the vancomycin aglycon immediately following Evans’ report in Angewandte Chemie. While they invoke similar disconnections as the Evans work, they constructed these precursors quite differently. Evans and coworkers used their venerable oxazolidinone chemistry; however, in contrast, Nicolaou turned to Sharpless’ asymmetric dihydroxylation (SAD)202 followed by Mitsunobu and Staudinger chemistries or Sharpless’ asymmetric aminohydroxylation (SAA)203 to construct these nonproteinogenic amino acids, four of which are necessary to construct the ABCD tetrapeptide precursor (Scheme 15).204 P-Aminobenzoic acid undergoes straightforward chemistry to arrive at amino alcohol 203. The diazonium salt is generated, which is trapped as a triazene by pyrrolidine under basic conditions. Following oxidation and Wittig methylenation, the resulting styrene undergoes SAD. Following monoprotection of the diol, Mitsunobu substitution with diphenylphosphanyl azide and Staudinger reduction yield the necessary amine, and standard manipulations yield building block 208 bearing a D-ring with a protected phenol. The C amino acid 213 was constructed similarly, using Horner–Emmons chemistry followed by SAA to generate the amino acid directly. A significant deviation from the Evans work is seen in the construction of the AB diphenyl system; the amino acids were coupled directly using Suzuki chemistry before integrating them into the scaffold. Both the A- and B-ring amino acids were constructed from simple aromatic molecules, which then undergo coupling to yield a mixture of atropisomers; the correct isomer was not identified until both were built into the scaffold and NOE data compared with natural material. Ultimately, Nicolaou and co-workers arrived at the AB biaryl intermediate 220 and were prepared to assemble the ABCD precursor.205
Scheme 15.

Nicolaou’s Preparation of Amino Acid Building Blocks
Meanwhile, the tripeptide bearing the E-ring was constructed in a straightforward fashion; styrene 211 was subjected to SAD, and the resulting diol was activated as the corresponding nosylate, permitting SN2 substitution with azide, which was reduced by tin (Scheme 16).206 Known asparagine derivative 224 was also modified. N-Protected leucine 221 was coupled with amino acid 222, and treatment with base revealed the C-terminal acid for the subsequent coupling step. With the tripeptide formed, simple protecting group manipulations and a chlorination completed the right-hand side of vancomycin.
Scheme 16.

Nicolaou’s Preparation of Right-Hand Tripeptide 227
The left-hand tetrapeptide 233 was constructed beginning with the peptide coupling of AB precursor 220 with 213, with a sequential deprotection and coupling to bring all building blocks into union (Scheme 17). The first macrocyclization was achieved through copper-mediated SNAr, yielding a mixture of atropisomers. Deprotections and a Staudinger reduction revealed the elements for peptide coupling nearest the C-terminus, which occurs in an intramolecular fashion upon treatment with pentafluorophenyl diphenylphosphinate (FDPP). The two fragments were united through a peptide coupling followed by the familiar SNAr cyclization to form the D–E biaryl ether, reporting a 3:1 ratio of atropisomers favoring the undesired product. However, Nicolaou reported that heating any mixture of atropisomers at 140 °C in o-dichlorobenzene yielded an equilibriated 1:1 mixture. Gratifyingly, the two were separable, and with enough patience, this material could be recycled almost completely.
Scheme 17.

Nicolaou’s Assembly of Left-Hand Tetrapeptide 233 and Construction of the Vancomycin Scaffold
With the completed scaffold 234 in hand, all that remained was to reveal the proper vancomycin aglycon. After much experimentation, Nicolaou arrived at a route to convert the triazine on the D-ring to the necessary phenol. Treatment with Raney nickel followed by hydrogenolysis generated the amine, while also debenzylating at the C-terminus. The amine was first oxidized to the diazonium species and then converted to the aryl iodide. Treatment with magnesium reagents rendered the aryl Grignard, which was trapped as the boronate; oxidation finally yielded the phenol. For efficient oxidation at the C-terminus, this much-maligned phenol was masked with a methyl group. Following the necessary oxidations, a global deprotection was performed with aluminum tribromide and ethanethiol, yielding the aglycon 201 (Scheme 18).207,208 Furthermore, Nicolaou later reported a glycosylation sequence to yield vancomycin proper.209
Scheme 18.

Endgame toward the Total Synthesis of 201
5.7. Preparation of Tethered Vancomycin Dimers
Studies have shown that vancomycin dimerizes in vitro, though the biological consequences of this activity remain unclear. To explore this phenomenon, the Nicolaou laboratory designed a series of analogs to explore the efficacy of tethered vancomycin dimers. This experimental design drew from the concept of target-accelerated synthesis, outlined briefly in Figure 24a.193 Considering a vancomycin modified with a functional handle, the ligand can dimerize (k1 ~ 7 × 102 M−1) or bind to the substrate (k2 ~ 104 M−1).210 However, it has been shown that dimerization is more favorable when bound to substrate (k3 ~ 106 M−1). For this reason, it is hypothesized that ligation would selectively tether bound complexes and reveal highly potent dimeric vancomycin analogs. Inspired by this investigative method in chemical biology, Nicolaou designed analogs to probe the optimal linker, varying both the composition (olefin or disulfide) and the length of this tether (Figure 24).210 The vancomycin scaffold was expanded semisynthetically at the vancosamine nitrogen, a known point of modification in the lipoglycopeptides. This benzyl group was modified with either a thiol or olefin tag, both of which dimerize efficiently through oxidation to the disulfide or olefin metathesis, respectively. Because the dimer must bind to two D-Ala-D-Ala peptides, the geometry must be optimized to ensure this binding was viable. As is evident in Figure 24c, there was an upper limit of 10 atoms between the aryl groups, and both the disulfide and olefin linkages retain activity. Compared to vancomycin, dimerization appeared to confer some activity in resistant strains, though it was not clear if this effect is due to dimerization or the introduction of a lipophilic group onto the vancosamine, effectively rendering it a lipoglycopeptide (refer to section 5.3). We should note that the olefin linkages were a ~1:1 mixture of E/Z isomers. With proof of concept in hand, Nicolaou expanded the scope of analog development to design vancomycin dimers with “mutations” at the first amino acid and an additional glycine at the C-terminus, which is feasible given the Nicolaou group’s work on solid-phase synthesis toward these scaffolds.211 Using a scaffold containing a short 6-atom olefin linkage, Nicolaou and co-workers generated a series of peptide analogs (Figure 25). The first analog was reminiscent of “broken” vancomycin, lacking the N-methylleucine residue, and it confirmed the inactivity. Generally, hydrophobic and charged side chains conferred activity against the VanA resistance phenotype, while hydrophobic residues did not. Despite these promising and intriguing results, the Nicolaou group appears to have ended this project, with no new results reported over the last 15 years.
Figure 24.

(a) Schematic representation and rationale of target-accelerated combinatorial synthesis. (b) General blueprint of the vancomycin dimer containing a tether through the vancosamine sugars. (c) Optimization of linker makeup and length. MICs in μg/mL.
Figure 25.

(a) Design of vancomycin analogs with N-terminal amino acid mutations and (b) biological evaluation. MICs in μg/mL. Gray shading indicates that no comparison can be made relative to the activity of vancomycin against a particular strain.
5.8. Boger’s Total Synthesis of Vancomycin Aglycon
In 1999, the Boger Laboratory at The Scripps Research Institute reported a third synthesis to the vancomycin aglycon. Notably, a key departure from the previous reports is a method to deal with the three axes of atropisomerism, the AB biphenyl system, and both the CD and DE biaryl ethers. Using model studies, Boger and co-workers determined the energies of atropisomeric interconversion for each structural element and conditions to equilibrate between both isomers to permit recycling of material. Upon determining the thermodynamic values at 26.6 kcal/mol for CD, 25.1 for AB, and 24.8 for DE, a logical sequence became apparent.212
Beginning with tripeptide 257, detailed in prior disclosures,213 Boger first closed the CD ether, yielding a 1:1 mixture of atropisomers, which were separated (Scheme 19). The undesired material 258 was recycled through equilibration, which permitted iterative enrichment. With the desired P-isomer in hand, reduction and oxidation to the diazonium facilitated Sandmeyer substitution to afford the aryl chloride 260. Wasting no time, the A-ring boronic acid 261 was incorporated using Suzuki chemistry. Though the undesired adduct was formed in a slight excess, this material was equilibrated very favorably to the needed S isomer. Following protecting group manipulations, intramolecular peptide coupling closed the macrocycle to afford left-hand tetrapeptide 265.
Scheme 19.

Boger’s Synthesis of the Left-Hand Tetrapeptide 265 Optimized for Atropisomeric Efficiency
The right-hand tripeptide 266, prepared in a straightforward manner, was coupled with its partner 265 to afford the heptapeptide, which was then treated with cesium fluoride to effect SNAr substitution. Notably, Boger and co-workers observed highly preferential formation of the desired P atropisomer 268, though recycling of 267 was also possible. A familiar sequence converted the D-ring nitro substituent to the aryl chloride, and 6 simple manipulations revealed the vancomycin aglycon 201 (Scheme 20).214,215
Scheme 20.

Assembly of Vancomycin Aglycon
5.9. Analogs with Affinity for D-Ala-D-Lac
The Boger group has designed analogs to overcome the critical VanA resistance mechanism, which replaces D-Ala-D-Ala to D-Ala-D-Lac. As discussed earlier, a single atom exchange removes a key hydrogen-bonding interaction between vancomycin and the cell wall precursor. Boger proposes “a complementary single atom exchange in the antibiotic to counter a corresponding single atom exchange in the cell wall precursors of resistant bacteria”. Specifically, Boger and co-workers plan to alter the amide carbonyl of the fourth amino acid to mitigate the disfavored hydrogen bond, or possibly establish a favorable interaction between this vancomycin analog and D-Ala-D-Lac.216 Planned single atom exchange analogs are described in Figure 26. For example, removing the carbonyl oxygen on the fourth amino acid might be predicted to tolerate the D-Lac substitution by removing the disfavored interaction between the lactate ester and the vancomycin amide.
Figure 26.

Boger’s vancomycin aglycon analogs with pocket modifications.
The simplest analog, the methylene 270, is constructed through reductive amination coupling between phenylglycine 273 and aminoaldehyde 274 (Scheme 21). The resulting amine is carried through the normal sequence following protection as the methyl carbamate. While SNAr cyclization is achieved with a higher selectivity of the desired atropisomer, Boger notes the difficulty of recycling undesired material through equilibration. Following coupling of the AB-system, the material is subjected to a similar sequence as described in Scheme 20 to furnish the deoxygenated vancomycin aglycon.
Scheme 21.

Boger’s Diverted Total Synthesis of Methylene Analog 270 via Intermediate 275
Boger and co-workers design a second analog to further probe this hydrogen bond as a contributor of resistance.217 Here, a thioamide is introduced via treatment of precursor 260 with Lawesson’s reagent. Due to difficulties in effecting Suzuki coupling, the thioamide must be masked as the methyl thioimidate 281. Upon successful union with the A-ring, the thioamide is again revealed, and the left-hand tetrapeptide is elaborated into the thioamide vancomycin analog. From this material, treatment with ammonia in methanol with silver acetate furnishes amidine analog 272, completing the set (Scheme 22).
Scheme 22.

Boger’s Synthetic Diversion of 260 to Thioamide 271 and Amidine 272
To establish proof-of-concept, Boger and co-workers subject these analogs to an in vitro binding assay against peptidoglycan precursor mimics 285 and 286, which mimic D-Ala-D-Ala and the resistant phenotype D-Ala-D-Lac. The disfavored hydrogen bonding in the lactate ester is apparent, giving rise to the ~1400-fold decrease in affinity for the vancomycin aglycon. As one might predict, the larger thioamide has decreased binding to both. The amidine, which can act as both a hydrogen bond donor and acceptor, exhibits slightly lower binding to the amide substrate but greatly improves upon binding to the ester, relative to the vancomycin aglycon. The consistency in binding to both model substrates is evident in the ratio of the binding affinities. A similar trend, albeit with binding constants an order of magnitude lower, is observed in the methylene analog.
These in vitro results translate well to in vivo assays, as the amidine was highly potent, followed by the methylene (Figure 27). These exciting results would provide the foundation for a more thorough investigation of vancomycin analogs.
Figure 27.

In vitro binding assays of aglycon analogues to peptidoglycan mimics 288 and 289. The ratio indicates x-fold decrease in binding affinities. In vivo MIC assay against E. faecalis BM 4166, expressing VanA, μg/mL.
With proof of concept in hand, Boger and co-workers set out to elaborate these substituted aglycons into full vancomycin analogs and their chlorobiphenylmethyl derivatives, inspired by the success of oritavancin.218,219 To effect glycosylation, Boger turns to Nature and uses recombinant glycosyltransferases from A. orientalis, a vancomycin-producing strain, leveraging work done by Kahne and Walsh.220 Using uridine diphosphate (UDP) sugars, glycosylation is carried out efficiently on all substrates except the amidine, which, due to its sensitivity, must be generated from the thioamide following glycosylation. This step also completes Boger’s total synthesis of vancomycin proper with a protecting group-free sequential glycosylation.
The approval of oritavancin, dalbavancin, and telavancin over the past decade clearly indicates that lipoglycopeptides are the present state of the art. To expand on these backbone-altered scaffolds, Boger introduces the chlorobiphenylmethyl (CBP) moiety onto the vancosamine sugar through reductive amination; no significant side reactions were observed, though the amidine, again, required diversion from the thioamide (Scheme 23).
Scheme 23.

Boger’s Elaboration of Aglycon Analogs to Glycosylated Vancomycin Analogs and CBP-Derivatives
These analogs are evaluated against a panel of relevant bacteria, including MSSA, MRSA, and VRE strains expressing VanA and VanB resistance mechanisms. The thioamide vancomycin, as one might predict from the binding assays of the aglycon, exhibits relatively poor inhibition, while the amidine excels. Addition of the CBP group increases activity nearly across the board; amidine CBP analog 293 boasts a 50,000-fold increase over vancomycin against VanA-expressing VRE strains. Given the enhanced activity against D-Ala-D-Lac-producing strains, Boger’s hypothesis regarding a single atom mutation restoring a critical hydrogen bond is clearly a valid one (Table 6).
Table 6.
MIC Assays of Analogsa
| S. aureus | E. faecalis | E. faecium | E. faecalis | |||
|---|---|---|---|---|---|---|
|
| ||||||
| ATCC 25923 | ATCC 43300 | BM 4166 | ATCC BAA-2317 | ATCC 51299 | ||
| QC | MRSA | VanA | VanA | VanB | ||
| Vancomycin (4) | 0.5 | 0.5 | 250 | 250 | 8 | |
|
| ||||||
| 287 | Y=S | >32 | >32 | >32 | >32 | >32 |
| 288 | Y=NH | nd | nd | 0.5 | 0.5 | nd |
| 289 | Y=H2 | nd | nd | 31 | 31 | nd |
|
| ||||||
| 291 | Y=O CBP | 0.03 | 0.03 | 2.5 | 2.5 | 0.03 |
| 292 | Y=S CBP | 2 | 2 | 4 | 4 | 2 |
| 293 | Y=NH CBP | 0.03 | 0.06 | 0.005 | 0.005 | 0.06 |
| 294 | Y=H2 CBP | 0.5 | 0.25 | 0.13 | 0.06 | 0.5 |
|
| ||||||
| 295 | Y=O CBP C0 | nd | nd | 5 | 5 | nd |
| 296 | Y=O CBP C1 | nd | nd | 0.25 | 0.5 | nd |
| 297 | Y=O CBP C5 | nd | nd | 2 | 2 | nd |
| 298 | Y=H2 CBP C1 | nd | nd | 0.01 | 0.005 | nd |
![]()
Color code indicates approximate x-fold change in activity relative to vancomycin. When considering “greater than” results, we assume the best-case scenario if a comparison can be made.
The Boger laboratory has also recently further modified the vancomycin scaffold at the C-terminus, installing a short hydrocarbon linkage tethering a tertiary amine or quaternary ammonium moiety (Figure 28).221 In particular, inclusion of the trimethylammonium increases the potency of the CBP-methylene scaffold by a factor of nearly ten (Table 6). Most intriguing is the discovery that compound 298 inhibits bacteria via five distinct mechanisms. In addition to the established binding to both D-Ala-D-Ala and D-Ala-D-Lac, this compound has also been shown to inhibit cell wall synthesis with or without ligand binding, likely assisted by the chlorobiphenylmethyl moiety, as suspected by Kahne years prior.194 Additionally, the quaternary ammonium moiety permits disruption of the cell wall. While Boger’s analogs have exhibited several of these mechanisms before, this analog alone demonstrates all five. Furthermore, a resistance assay showed that strains of VRE showed only a 0.4-fold increase in MIC over 50 cycles.
Figure 28.

Boger’s peripheral modifications to the CBP-vancomycin scaffold.
5.10. Summary
The sum of Boger’s results is particularly intriguing given that Kahne established clearly that the lipoglycopeptides’ mechanism of action is not consistent with peptide binding. Notably, Kahne postulated that “the complexity of the peptide portion of vancomycin makes it virtually impossible to reengineer the peptide backbone to include new contacts to the modified substrate”.194 In an elegant demonstration of the utility and robustness of chemical synthesis, Boger develops vancomycin derivatives and lipoglycopeptides that can do precisely what was perceived as impossible. It would be highly beneficial to the scientific community for the precise mechanism of action of these modified antibiotics to be established to inspire future drug discovery in the glycopeptide family.
6. TARGETING QUORUM SENSING AND BACTERIAL SIGNALING
6.1. Overview of Quorum Sensing and Virulence
In 1970, “Woody” Hastings at Harvard made an intriguing discovery. The bacteria Aliivibrio fischeri, famous as the bioluminescent commensal species in the tropical squid E. scolopes, only exhibits its trademark activity during a narrow window in the bacterial exponential growth phase.222 The Hastings laboratory, after careful experimentation, concluded that this was, by some unknown mechanism, a function of cell density, and regulated at the transcriptional level via a process they termed “autoinduction”. Later work identified the small molecule acyl homoserine lactone 301 as the A. fischeri luciferase autoinducer.223 Because of the apparent link to bacterial population density, this phenomenon was termed quorum sensing, which thanks to the efforts of Greenberg and Bassler, is widely known as the key method by which bacteria communicate; some seminal works are provided here.224–227
Quorum sensing has been implicated in both the formation of biofilms and the expression of virulence factors, two major mechanisms by which bacteria act as pathogens.228–230 Furthermore, this link is precisely what effectively “allows bacteria to function as multicellular organisms”.228 For example, quorum sensing machinery in Staphylococcus is known to regulate the production of hemolysins, which destroy erythrocytes to release the valuable iron-containing heme. As shown in Figure 29, quorum sensing systems in P. aeruginosa have been linked to the regulation of various virulence factors, including exotoxin A, various proteases, and siderophores such as pyoverdine and pyocyanin.231,232 These virulence factors are precisely what induce symptoms of infection.233,234 Contrasted with conventional approaches of inhibiting bacterial growth, inhibiting virulence offers a distinct advantage: inhibiting quorum sensing would present very little of the selection pressure that drives the evolution of resistant phenotypes.235 Controlling virulence could also, in theory, reduce aggregation into biofilms which might allow the immune system to fight off this more susceptible infection.
Figure 29.

Schematic representation of QS systems in P. aeruginosa. Color scheme: yellow/orange: LasR-mediated; blue: RhlR-mediated; green: promotors; red: repressors. Inspired from refs 225 and 226. Of particular interest is the regulation of toxins, as shown in the Venn diagram.
6.2. Acyl Homoserine Lactones
Since the identification of the acyl homoserine lactone (AHL) as the autoinducer of quorum sensing in many Gram-negative bacteria, scientists have leveraged this deceptively simple natural product scaffold to develop analogs with intriguing biological activity (Figure 30). Work toward developing unnatural AHL mimics has been reviewed extensively,236,237 including in this journal.238,239 We discuss here recent work by the Blackwell laboratory which seeks to better understand quorum sensing through chemical probing.
Figure 30.

(a) Natural and synthetic AHLs; (b) synthetic inhibitors; and (c) agonists of pyocyanin production in PAO1.
6.2.1. Blackwell’s Synthetic Campaign toward AHL Libraries
Having previously identified arylated AHLs as potent modulators of quorum sensing in Gram-negative bacteria, Blackwell and co-workers have constructed an extensive library of diverse AHLs to probe interactions with the P. aeruginosa quorum sensing enzymes LasR and RhlR and determine what effect, if any, is had on virulence regulation (Figure 30).240 Using straightforward assays in fluorescence reporter strains of P. aeruginosa and E. coli (which has had LasR and RhlR cloned in via plasmids), Blackwell and co-workers established an inverse relationship between pyocyanin inhibition and RhlR inhibition, noting that the strongest inhibitors of pyocyanin production tended to have agonistic effects on RhlR, expressed in both the native P. aeruginosa and nonnative E. coli (Table 7). Via transcriptomic experiments using the highly potent 303, Blackwell finds that the rhl cluster is up-regulated (~2×), while pqs is suppressed. Unsurprisingly, RhlR agonists up-regulate the production of rhamnolipids, another key signaling molecule in the Pseudomonads. The Blackwell laboratory has also successfully applied this library of AHL analogs to another Gram-negative pathogen, A. baumannii, which has an analogous AbaR receptor.241
Table 7.
Activities of Selected Analogs against QS in P. aeruginosa as % Activity at 100 μM Relative to Natural AHL Agonists and Antagonists
| PAO1 | PAO-JP2 | E. coli | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| pyocyanin inhibition |
LasR | RhlR | RhlR | ||||
| ant | ag | ant | ag | ant | ag | ||
| 303 | 88 | 31 | 8 | 12 | 105 | −110 | 91 |
| 304 | 84 | 15 | 10 | −9 | 86 | −95 | 81 |
| 305 | 83 | −61 | 99 | −40 | 102 | −122 | 83 |
| 306 | 79 | 33 | 7 | 15 | 46 | −52 | 79 |
| 307 | 67 | 3 | 7 | 41 | 48 | −64 | 84 |
| 308 | 60 | 60 | 7 | 78 | 32 | −47 | 71 |
|
| |||||||
| 309 | −25 | 31 | 9 | 56 | 2 | 60 | 11 |
| 310 | −38 | 47 | 5 | 85 | −1 | 64 | 5 |
| 311 | −44 | 60 | 2 | 92 | −1 | 74 | 7 |
| 312 | −49 | 33 | 7 | 54 | 2 | 84 | 1 |
6.3. Pseudomonas Quinolone Signal
The Pseudomonads possess an additional mechanism for quorum sensing and virulence utilizing quinolone 313, known as the Pseudomonas quinolone signal (PQS), which includes several related molecules (Figure 31).242 The PQS system is known to interact in a complex manner with both las and rhl, and it is also linked to the regulation of virulence factors including rhamnolipids, lectins, pyocyanin, and elastase.243
Figure 31.

PQS, due to its hydrophobicity, is transported between bacteria via membrane vesicles. When delivered to another P. aeruginosa cell (orange), PQS acts as a quorum sensing molecule. When delivered to a foreign species, such as S. aureus (green), PQS is an antibiotic, allowing P. aeruginosa to outcompete other species for valuable resources. Inspired from refs 227, 228, and 242. On the right, molecules implicated in the PQS system.
Because of the dual activities of these compounds, PQS is an attractive target for scientists seeking to develop analogs of these molecules to control both virulence and quorum sensing in the opportunistic pathogen P. aeruginosa as well as inhibit other bacteria (Figure 31).231
6.3.1. Huigens’ Synthesis of Phenazine-Inspired Analogs
Inspired by the antibacterial activity of the Pseudomonad virulence factor pyocyanin, Huigens designed a diverted total synthesis of a variety of phenazine analogs bearing simple modifications to establish a preliminary SAR, which demonstrated that a variety of bromophenazine derivatives were active against QS strains of S. aureus and S. epidermidis.244 This lead inspired further DTS work resulting in a variety of halogenated phenazine analogs (Figure 32), many of which exhibited low micromolar activities against various resistant Gram-positive bacteria.245
Figure 32.

(a) Selected halophenazine analogs by Huigens tested against Gram-positive bacteria with (b) MICs in μM.
Generally, these compounds exhibited pockets of activity across various strains, though compound 323 appears to have the best overall activity. These compounds were also shown to eradicate biofilms, and they were able to inhibit hemolysis, indicating that quorum sensing is interrupted. Unlike other autoinducer mimics, these compounds inhibit bacterial growth directly, suggesting that these drugs would have a fairly straightforward therapeutic application. Using a “scaffold-hopping” approach, Huigens sought to divert the success of the bromophenazine work to a simplified quinoline scaffold, generating a library of small molecules with comparable activities to the phenazine analogs, some of which were potent biofilm inhibitors.246,247
6.4. Autoinducer Peptides
Quorum sensing in Gram-positive bacteria is regulated by the interaction of short, cyclic autoinducer peptides (AIPs) with Agr enzymes.248 This pathway has been best characterized in Staphyloccus, though also known in Enterococcus, Lactobacillus, Listeria, and Clostridium.
6.4.1. Mechanism of AIP Regulation of QS
A schematic of the agr system is outlined in Figure 33.249 A region within the short peptide AgrD is excised, cyclized, and exported by the membrane-bound AgrB and SpsB. The resulting AIP can interact with the extracellular region of AgrC, which triggers kinase activity, phosphorylating AgrA. This enzyme then activates two promotors; P2 up-regulates the transcription of the agr cluster, resulting in increased production of AIP. P3, meanwhile, increases transcription of RNA III, which is implicated in the regulation of numerous virulence factors, including hemolysins, immunoglobulin G-binding protein, and staphylocoagulase, an enzyme which prevents the immune system from detecting this infection.250
Figure 33.

Schematic representation of Agr-mediated quorum sensing. Inspired from refs 239 and 249.
6.4.2. Muir’s Solid-Phase Synthesis of AIP Analogs
Given the rapid development of resistance so frequently observed in S. aureus, scientists quickly sought to better understand the dynamics of this pathogen with the eventual goal of targeting quorum sensing as an antibacterial strategy. In 1999 the Muir laboratory, formerly at Rockefeller and now at Princeton, established specificity within the agr subgroups, demonstrating that the AIP derived from AgrD-I activates only the corresponding AgrC-I, while inhibiting the enzyme from groups II and III (Figure 34, Table 8).251 Furthermore, small modifications to the AgrD-II AIP, including replacement of the thiolactone with a lactone or lactam, led to active compounds. Muir also pioneered the application of solid-phase peptide synthesis to the preparation of both naturally occurring and unnatural AIPs. To further highlight the therapeutic potential of AIP derivatives, Muir and co-workers show in a murine model that these AIPs can reduce the average lesion size resulting from a MRSA infection.
Figure 34.

AIPs from S. aureus.
Table 8.
Inhibition Assay of AIP Analogsa
| AgrC | Hemolysis Assay | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sequence | I | II | III | IV | I | II | III | IV | ||
| 329 | AIP-I | YST(CDFIM) | ag | 8.00 | 0.522 | nd | nd | 3.34 | 6.12 | 189 |
| 330 | AIP-II | GVNA(CSSLF) | 1.62 | ag | 0.532 | 0.396 | 0.89 | nd | 3.59 | 1.19 |
| 331 | AIP-III | IN(CDFLL) | 5.05 | 5.63 | ag | 8.53 | 8.07 | 0.456 | nd | 23.8 |
| 332 | AIP-IV | YST(CYFIM) | ND | 0.373 | 0.46 | ag | nd | 0.0897 | 1.49 | nd |
| 333 | I1A | AN(CDFLL) | 17.9 | 4.26 | 194 | 7.85 | 4.61 | 1.29 | nd | 12.5 |
| 334 | N2A | IA(CDFLL) | 3.6 | 0.732 | nd | 3.53 | 1.02 | 0.137 | nd | 2.64 |
| 335 | D4A | IN(CAFLL) | 0.485 | 0.429 | 0.0506 | 0.0349 | 0.082 | 0.0596 | 0.163 | 0.106 |
| 336 | I1A/N2A | AA(CDFLL) | 7.4 | 4.38 | 2.6 | 5.41 | nd | nd | nd | nd |
| 337 | I1A/D4A | AN(CAFLL) | 0.328 | 2.35 | 0.28 | 0.101 | 0.0103 | 0.793 | 0.551 | 0.284 |
| 338 | N2A/D4A | IA(CAFLL) | 0.331 | 0.204 | 0.0657 | 0.0221 | 0.0362 | 0.0661 | 0.216 | 0.122 |
| 339 | I1A/N2A/D4A | AA(CAFLL) | 0.304 | 0.604 | 0.0734 | 0.0161 | 0.0411 | 0.0606 | 0.243 | 0.14 |
| 340 | tD2A | Ac(CAFLL) | 0.257 | 0.9 | 0.329 | 0.0957 | 0.332 | 0.711 | 0.197 | 0.306 |
| 341 | tD2A/F3Y | Ac(CAYLL) | 0.279 | 1.15 | 0.387 | 0.0306 | 0.279 | 0.204 | 0.265 | 0.134 |
![]()
IC50 in nM. Parentheses indicate the cyclic portion within the sequence. ag = agonist; nd = not determined. x-fold change measured relative to APPEL.
Mechanistic work by the Muir group established the binding region within AgrC. By developing chimeric AgrC mutants, permutating two regions within the sensor domain established that specificity relies on hydrophobic interactions between the AIP and the distal subdomain within the receptor.252 The Muir laboratory’s efforts toward understanding the SAR of AIP-II have been thoroughly reviewed,253,254 including in this journal.247
6.4.3. Blackwell’s Synthesis of AIP Analogs
On the heels of their highly successful work in Gram-negative quorum sensing, Blackwell and co-workers established in 2013 a comprehensive SAR around the S. aureus AIP-III.255 As an initial screen, both alanine and D-amino acid scans indicated which sites in the peptide would be most receptive to alteration. After identifying positions 1, 2, and 4 as sites for potential improvement, these mutations were combined to arrive at an optimized AIP analog. These analogs were tested directly against AgrC using a fluorescent reporter assay and as overall inhibitors of quorum sensing via the down-regulation of hemolytic enzymes controlled by RNA III (Table 8). These assays reveal the D4A mutant as an especially potent pan-group AgrC inhibitor with picomolar activity.
Blackwell and co-workers further analyzed this SAR to gain mechanistic insight of the interaction between AIPs and AgrC.256 Using NMR techniques, the hydrophilic and hydrophobic regions of the AIP were deduced, and this structural information correlated with the biological activity. Blackwell was able to corroborate previous hypotheses which implicate hydrophobic residues around the macrocycle in AgrC recognition, independent of activation or inhibition, and a key hydrophobic residue in the N-terminal chain, which signals for activation of the AgrC kinase. Considering the experimental SAR, this is not surprising—each active compound has three large residues, often phenylalanine and two leucines. The N-terminal isoleucine in AIP-III is implicated as the activating residue. While D4A (335) retains this isoleucine, Blackwell postulates that the mutation within the macrocycle alters the conformation sufficiently to prevent this residue from activating the kinase.
While these AIP analogs are highly potent and excellent chemical tools for manipulating quorum sensing, their therapeutic potential is limited by the sensitivity of the thiolactone to hydrolysis. Analogous to Muir’s substitution of the thiolactone to the lactone and lactam, Blackwell applies this approach to their potent AgrC inhibitors derived from AIP-III (Table 9).257 While the amide analog of natural AIP-III exhibits poor activity, conversion to the amide renders analogs 343 and 344 only slightly less active than the original analogs. Furthermore, Blackwell shows that the lactam derivatives have enhanced stability to both degradation in alkaline conditions and tryptic proteolysis.
Table 9.
Biological Evaluation of AIP Analogs and Amide Derivatives Thereofa
| AgrC | ||||||
|---|---|---|---|---|---|---|
| Sequence | I | II | III* | IV | ||
| 331 | AIP-III | IN(CDFLL) | 5.05 | 5.63 | ag | 8.53 |
| 342 | AIP-III Amide | IN(XDFLL) | >1000 | >1000 | 75.2 | 299 |
| 335 | D4A | IN(CAFLL) | 0.485 | 0.429 | 0.0506 | 0.0349 |
| 343 | D4A Amide | IN(XAFLL) | 1.2 | 2.19 | 0.055 | 0.192 |
| 340 | tD2A | Ac(CAFLL) | 0.257 | 0.9 | 0.329 | 0.0957 |
| 344 | tD2A Amide | Ac(XAFLL) | 1.5 | 13.9 | 0.216 | 0.189 |
![]()
IC50 in nM, X = L-diaminopropionic acid, ag = agonist, t = truncated.
For the sake of comparison, x-fold improvement against AgrC-III is measured against the amide.
Recently, the Blackwell lab has attempted to develop AgrC inhibitors via a peptidomimetic approach in which portions of the peptide macrocycle were replaced with nonpeptidic portions, including methylene linkages, which allows ring expansion.258 None of these simplified analogs exhibited the same pan-group inhibition of AgrC observed in the AIP-III analogs. However, these peptidomimetic drugs exhibited far greater solubility.
Other efforts by the Blackwell laboratory include the application of this approach to develop pan-AgrC inhibitors in the related S. epidermidis, which, despite its evolutionary proximity to aureus, uses AIPs with distinct sequences (Figure 35).259 In an analogous fashion, Blackwell and co-workers identified sites for amino acid substitution and carry out the necessary solid-phase synthesis to generate a library of compounds, from which they identify analogs 348 and 349 as pan-group inhibitors with low nanomolar potencies. Because S. epidermidis is a clinical menace largely due to its propensity to form biofilms in indwelling medical devices, Blackwell and coworkers probed the effect of these AIPs and analogs on biofilm formation. It has been known for some time that inhibition of agr induces biofilm formation, while activation encourages dispersion, which is a significant quandary facing quorum quenching as a therapeutic method. Using a straightforward crystal violet assay to measure biofilm formation, Blackwell demonstrates that AIP-I, the native agonist, clearly reduces the biofilm. Likewise, a more potent AgrC-I agonist disperses biofilms to a greater extent. Surprisingly, preliminary results do not show increased biofilm formation following treatment with 349, a highly potent AgrC-I inhibitor, which Blackwell attributed in part to the highly complex (and poorly understood) relationship between quorum sensing networks and biofilm formation. For modulation of quorum sensing to be a viable therapeutic option, innovative chemical tools will be necessary to further our understanding of these intricate mechanisms and interactions.
Figure 35.

AIPs from S. epidermidis and analogs of 347 with IC50 values against AgrC I, II, and III.
6.5. Summary
While much has been learned about quorum sensing and virulence in a variety of pathogenic bacteria thanks to the use of small molecule probes inspired by natural autoinducing compounds, our understanding of these complex pathways must greatly increase for quorum sensing modulation to be a serious therapeutic option, especially the regulation of biofilms. Secondary to these issues are the many regulatory hurdles, as many of these molecules do not exhibit a straightforward overall inhibition of bacteria, and conventional clinical trials may be incompatible with these nonlethal agents.
7. NARROW-SPECTRUM SMALL MOLECULE NATURAL PRODUCTS
7.1. Consequences of Antibiotic Use on Microbiomes
While the interactions between conventional broad-spectrum antibiotics and pathogenic bacteria are relatively well-characterized, many more questions remain surrounding antibiotics’ effects on the nearly countless commensal bacteria strains with which we have a symbiotic relationship. Recent efforts by Martin Blaser at NYU Medical School to further understand how broad-spectrum antibiotics perturb the human microbiome, very frequently at birth, have led to insights regarding the critical role commensal species play in normal human development.260 It has been well established that microbiota imbalances and deficiencies are associated with numerous maladies, including obesity, irritable bowel disorder, and cancers,261 though Blaser intends to demonstrate that the increasing incidence of these conditions are in no small part due to our poor stewardship of antibiotics.260,262,263 The Blaser laboratory has shown in extensive animal modeling that disruptions within the micro-biome, especially those occurring early in life, are linked with metabolic issues, including obesity,264,265 though a recent medical study found that early antibiotic usage has devastating effects on the developing microbiome.266 Their work has suggested that strategies that target virulence or specific pathogenic bacteria may provide additional benefits over their commonly used broad-spectrum counterparts.
7.2. Fidaxomicin: A Narrow-Spectrum Macrolide Antibiotic
In 1972, scientists in Italy identified a new antibiotic from Actinoplanes,267 and because it was discovered on the 29th of February, it was cleverly named lipiarmycin. Active against a variety of Gram-positive species, no cross-resistance was observed against contemporary antibiotics.268 Furthermore, lipiarmycin was identified as an inhibitor of RNA synthesis.269 Sometime later, the structure was elucidated, and lipiarmycin (350) was shown to consist of a highly unsaturated 18-membered macrolactone bearing acylated rhamnose and noviose sugars,270 and the later discovered tiacumicin B was shown to have the same structure (Figure 36).271
Figure 36.
Structure of fidaxomicin, aka lipiarmycin, aka tiacumicin B, an 18-membered macrolide.
Lipiarmycin was identified to have especially potent activity (<1 μg/mL) against Clostridium difficile, a pathogen best known for debilitating diarrhea, often resulting in dehydration.272 This study, performed by scientists at Abbott, also demonstrated slow development of resistance and preliminary success in rodent models. Given the generic name fidaxomicin, this compound entered clinical trials, where it met the primary end point of noninferiority versus vancomycin against C. difficile infections, with an additional benefit of reduced infection reoccur-rence.273,274
Fidaxomicin was recently a popular target with synthetic chemists,275–278 though the only analog development known to us has been done via tailoring biosynthetic enzymes, which are beyond the defined scope of this review.279
7.3. Promysalin
In 2011, De Mot in Leuven, Belgium, reported a small molecule, dubbed promysalin (351), with intriguing biological activity within the pseudomonads.280 A secondary metabolite from the soil-dwelling Pseudomonas putida RW10S1 in the rhizosphere surrounding a Sri Lankan rice plant, promysalin, promoted swarming within the producing species but had potent antibacterial activity against other closely related strains, such as the highly pathogenic P. aeruginosa. At the time, little was known about its mechanism of action or the relative or absolute stereochemistry.
In 2015, Wuest and co-workers reported the total syntheses of four diastereomers of promysalin to identify the correct stereochemistry and further explore this compound’s exciting narrow spectrum activity (Scheme 24a).281 Genomic analysis of the biosynthetic cluster proposed by De Mot indicated that the natural L-proline is incorporated into the natural product; the remaining stereocenters were both in the myristate side chain. This synthetic approach featured a convergent strategy in which precursors each containing one stereocenter were joined through cross-metathesis, permitting straightforward preparation of the various diastereomers. The olefin is deleted through hydrogenation, and with Evans’ auxiliary removed, the side chain is obtained in five steps from known materials 352 and homoallylic alcohol 354. The left-hand fragment is derived from the amide coupling of trans-hydroxyproline and salicylate derivative 356. Following oxidation, ketone 357 is converted to the highly versatile enol triflate. To selectively deoxygenate, Wuest and coworkers found that a Stille reduction was highly effective. With acid 358 in hand, esterification with 355 under normal conditions followed by a global deprotection yields promysalin in an efficient, convergent route with 8 steps in the longest linear sequence. In the following year, the Musso laboratory in Milan reported a similar synthesis, with an improved route to the proline-salicylate precursor but a lengthier synthesis of the side chain.282
Scheme 24.

(a) Total Synthesis of Promysalin by Wuest; (b) Analogs of Promysalin Bearing Simple Modifications around the Pyrrolidine, Salicylate, and Myristate Portions; (c) Chimeric Promysalin Analogs Derived from the Fatty Acid Derivatives Lyngbic Acid and Hermitamides A and B
Wuest’s modular synthesis paved the way for a DTS campaign toward analogs to begin to probe the SAR of promysalin (Scheme 24b).283 Modifications to the dehydroproline ring were somewhat tolerated; fluorination and methylation at the γ-carbon still yielded active compounds within 10% of promysalin’s activity (Table 10). Saturated analog 359 was considerably less potent, and ring expansion to the piperidine (not shown) rendered an inactive compound. Methylation of either the salicylate phenol or the α-alcohol reduced potency as well. Deoxygenated compound 363 was nearly equipotent with the natural material, which is surprising given that the C2 epimer had considerably lower activity. This observation might indicate that the alcohol is not critical for activity, but incorrect stereo-chemistry results in a disfavored steric clash with whatever the target might be. An additional effort explored the possibility of constructing promysalin-inspired chimeric molecules, in which derivatives of the natural products lyngbic acid and hermitamides A and B replaced the normal side chain (Figure 24c).284 Unfortunately, these compounds showed no promising biological activity.
Table 10.
Activities of Selected Analogs (IC50 values, μM) against Strains of P. aeruginosa by Wuest
| P. aeruginosa | ||
|---|---|---|
| PAO1 | PA14 | |
| Promysalin (351) | 4.1 | 0.067 |
| C2 epi-351 | 33 | 4.3 |
| 359 | 111 | 28 |
| 360 | 7.7 | 0.019 |
| 361 | 32 | 12 |
| 362 | 57 | 6.7 |
| 363 | 5.8 | 0.035 |
| 364 | 38 | 11 |
| 365 | 8.3 | 0.067 |
Given some structural similarities with iron-binding molecules known in Pseudomonas, it was hypothesized that promysalin could act as a siderophore, though simple assays indicated only a qualitatively low affinity for iron. While not a bona fide siderophore, questions relating to iron uptake remained, given that promysalin inhibited pyoverdine production in P. putida strain KT2440.281 Biologically oriented investigations of the target are currently underway.
7.4. Carolacton
A collaborative group led by Kirschning reported in 2010 a polyketide macrolactone, carolacton (366), with antibiofilm properties.285 Isolated from S. cellulosum, this compound was found to radically alter cell wall structure, rendering the bacteria highly susceptible to acidic conditions.286 Furthermore, knockout assays established that a PknB mutant was not susceptible, though the mechanism of this interaction remains unclear. Later work established bactericidal activity against the related S. pneumoniae, another serious pathogen.287 Given the significance of S. mutans causing dental and heart issues including endocarditis, carolacton has received considerable attention as a potential narrow-spectrum antibiotic.
7.4.1. Kirschning’s Synthesis of Carolacton and Analogs
Kirschning and co-workers reported in 2012 the first total synthesis of carolacton, requiring 22 steps in the longest linear sequence (LLS) (Scheme 25).288 Toward the left-hand precursor, lactic acid was elaborated into alkyl bromide 371, which is converted to the organozinc reagent. Nucleophilic attack of racemic 372, guided by chiral ligand 373, afforded unsaturated ester 374, which, after conversion to the aldehyde, was attacked by the lithium enolate of 375. Cleavage of the acetal followed by hydrolysis revealed the diol functionality, which was protected as the acetonide. Finally, the venerable Swern oxidation yielded key aldehyde intermediate 378. The right-hand side chain was prepared through the coupling of alkyne 379 with aldehyde 380 via the Marshall reaction. A series of standard polyketide-type manipulations revealed an aldehyde from 383, which underwent Duthaler–Hafner chemistry to homologate by two carbons, capping the side chain with the tert-butyl ester. After methylation with Meerwein’s salt, the Schwarz reagent hydro-zirconates the alkyne, which was diverted to vinyl iodide 387. Aldehyde 378 was united with the side chain via the Nozaki–Hiyama–Kishi reaction, which gratifyingly furnished alcohol 389 in excellent yield with good diastereoselectivity. The macrocycle was formed using Shiina’s reagent, and standard manipulations provided carolacton (369).
Scheme 25.

Kirschning’s 2012 Synthesis of Carolacton (369): (a) Preparation of Macrocycle Precursor 378; (b) Synthesis of Side Chain Precursor 387 and NHK Coupling; (c) Macrocyclization and Endgame
Kirschning followed their synthetic route with an evaluation of late stage intermediates and semisynthetic derivatives against S. mutans biofilms and found that compounds 394 and 396 had micromolar activities, though with greatly diminished potency relative to carolacton (Figure 37).289 Given that each of these could be hydrolyzed to yield carolacton, it was hypothesized that these derivatives were merely acting as pro-drugs. Kirschning and co-workers did indeed detect small quantities of carolacton in the supernatant, indicating a weak pro-drug effect.
Figure 37.

Analogs of carolacton synthesized by Kirschning.
7.4.2. Phillips’ and Wuest’s Synthesis of Carolacton and Analogs
A collaborative effort by the Phillips lab at Yale and the Wuest lab developed a concise synthesis with just 14 steps in the LLS, and further work found that late-stage intermediates toward carolacton exhibit unique effects on S. mutans biofilm architecture (Scheme 26).290 To construct macrocycle precursor 400, gulonolactone derivative 397 was oxidized and olefinated to 398, and hydrolysis followed by Swern oxidation furnished lactone 399. A vinylogous (SN2′) substitution with a diallylcuprate provided acid 400 as a single diastereomer. The side chain was constructed through standard aldol and Wittig chemistry and culminated in a Leighton crotylation, yielding 406. The two were united through an efficient esterification, and the macrocycle was formed through ring-closing metathesis. A selective hydrogenation deleted the resultant olefin, and a final oxidation and deprotection yielded carolacton.
Scheme 26.

Phillips’ and Wuest’s Total Synthesis of Carolacton (369): (a) Preparation of Macrocycle 400; (b) Synthesis of Side Chain Precursor 406; (c) Esterification, Macrocycle, and Endgame; (d) Bioactive Simplified Analogs of Carolacton Accessed through Wuest’s DTS Approach
The Wuest group has recently reported the preparation of simplified analogs of carolacton, identifying that an aryl-containing side chain elicits various distinct phenotypic responses (Figure 38).291 Having drawn from Kirschning’s work, which indicated that the macrocycle does not tolerate modification, Wuest and co-workers hypothesized that replacement of the olefin in the side chain with an aryl moiety would both simplify the molecule and possibly retain activity. Additionally, truncated side chains were also explored. Of note, simplified carolacton analog 411 was found to inhibit S. mutans biofilms, with an MBIC50 of 63 μM. Furthermore, analog 412 elicited a carolacton-like phenotype at 500 nM. Notably, compounds 413 and 414 inhibited biofilm formation at the microcolony stage, indicating that carolacton and analogs thereof could serve as useful chemical tools for probing the biofilm life cycle.
A third complete synthesis of carolacton has also recently been reported, featuring a LLS of 13 steps and an impressive overall yield of 18.8%; though this work does not discuss analog development, an efficient and concise synthesis will certainly aid that process.292
7.5. Bromoageliferin and Simplified Analogs
The marine natural product bromoageliferin (415), a member of the oroidin (416) class, had been shown to disrupt biofilm formation in the Gram-negative marine bacteria R. salexigens (Scheme 27a). Considering the structural similarities within the oroidin alkaloids, which share a common 2-aminoimidazole (2-AI) core, the Melander laboratory at North Carolina State hypothesized that simplified analogs, derived from these intriguing natural products, could retain activity as antibiofilm compounds against persistent Gram-negative pathogens.293
Scheme 27.

(a) Related Natural Products Bromoageliferin (415) and Oroidin (416) and Analogs Derived Therefrom by Melander. (b) Representative Synthesis of 417; CAGE (418) Was Synthesized in an Analogous Manner from the Appropriate Epimer of 420
Melander and co-workers designed simplified analogs 417 and 418 which bear the 2-aminoimidazole core fused to the substituted cyclohexane ring, varying the stereochemistry.294
Both are prepared from diol 420 or its cis epimer through very straightforward transformations (Scheme 27). Both CAGE (418) and TAGE (417) inhibited biofilm formation in P. aeruginosa (IC50 100 μM vs PAO1, 180 μM and 190 μM, respectively, against PA14). A similar approach was applied to designing simple analogs of oroidin (416) through bromination at various sites around the ring, though the biological activities are comparable.295 These intriguing results led Melander and coworkers to further investigate 2-AI-bearing molecules as potential biofilm eradicators. 2-Aminobenzimidazoles such as 419 were shown to have potent activities against MRSA biofilms but were not lethal against planktonic cells.296 Furthermore, these derivatives were recently shown to act as adjuvants in sensitizing Mycobacteria to β-lactam antibiotics through a unique mechanism of action, as they were shown to not act as β-lactamase inhibitors.297
7.6. Summary
We have summarized here efforts to develop analogs of small molecule natural products with narrow-spectrum antibiotic activity. These molecules are of particular interest given recent insights into the importance of symbiotic relationships with bacteria in the gut microbiome. The clinical success of fidaxomicin in treating C. difficile should serve as a guiding principle that narrow-spectrum antibiotics may create a new paradigm in selectively targeting pathogenic bacteria while minimizing the impact on the human microbiome.
8. CONCLUSIONS
In most talks given by natural product chemists, it is commonly noted that while natural products can have outstanding biological activities, they are often poor choices for drugs, exemplified by erythromycin. While semisynthetic modifications have historically redirected this potent activity into a clinically useful drug, we have demonstrated that the need for new antibiotics to overcome resistant pathogens is so great as to require new generations of drugs occupying previously unexplored regions of chemical space. We have shown herein that the most direct, efficient, and fruitful method of generating drugs that can evade bacterial resistance mechanisms is through the power of total synthesis. We have outlined synthetic achievements toward many antibiotic scaffolds, both traditional and unexplored, and have discussed how these compounds fare against pathogenic bacteria. Traditional SAR studies can be undertaken to identify the key bioactive moieties, which can then be modified to generate more potent compounds. Additionally, synthetic approaches such as DOS, FOS, and CtD can be used to construct unprecedented scaffolds bearing the complexity of natural products, despite that these molecules may be foreign to Nature to the best of our knowledge. We hope that the examples discussed herein will spark further inspiration in the synthetic community to continue exploring innovative targets and methods to ensure a sustainable antibiotic supply.
Despite the potential for new antibiotic isolates and scaffolds, we must take care to preserve the efficacy of drugs currently prescribed (and overprescribed!). This requires improving upon our antibiotic stewardship by encouraging reduction in both the overprescription and misuse of these medicines. We must also continue to educate the public about the causes and persistence of antibiotic resistance, in part to drive public favor for a greater allocation of resources to address this crisis. Although the recognition of the term “antibiotic resistance” has increased, the understanding of how to avoid it and how it is caused has not been translated as effectively.15 Only by actively combining scientific innovation and communication can we avoid a postantibiotic era.
Acknowledgments
This work was supported by the National Science Foundation CHE-1454116 (W.M.W.), the National Institute of General Medical Studies R35 GM119426 (W.M.W.), and Temple University in the form of a Dissertation (M.H.F.) and a Presidential (S.E.R.) Fellowship. We are grateful to many members of the Wuest Group for a critical reading of this manuscript. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).298
ABBREVIATIONS
- ATCC
American Type Culture Collection
- Boc
t-butyloxy carbonyl
- CBP
p-chlorobiphenyl methyl
- Cbz
carboxy benzyl
- CDI
carbonyl diimidazole
- Cp
cyclopentyl
- CSA
10-camphorsulfonic acid
- CtD
complexity to diversity
- Cy
cyclohexyl
- dba
dibenzylideneacetone
- ddm
bis(4-methoxyphenyl)methyl
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DEAD
diethyl diazocarboxylate
- DEPBT
3-(diethoxyphosphoryloxy)-1,2,3- benzotriazin-4(3H)-one
- (DHQD)2AQN
hydroquinidine (anthraquinone-1,4-diyl) diether
- DIBAL-H
diisobutyl aluminum hydride
- DMAP
4-(N,N-dimethyl)aminopyridine
- DMDO
dimethyldioxirane
- DMP
Dess–Martin periodinane
- DPPA
diphenylphosphoryl azide
- DOS
diversity-oriented synthesis
- DTBMP
2,6-di-tert-butyl-4-methylpyridine
- DTS
diverted total synthesis
- EDC or EDCI
1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide
- FDPP
pentafluorophenyl diphenylphosphinate
- FOS
function-oriented synthesis
- GtfD
glucosyl transferase D
- GtfE
glucosyl transferase E
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HG-II
Hoveyda–Grubbs second generation catalyst
- HOAt
1-hydroxy-7-azabenzotriazole
- HOBt
hydroxybenzotriazole
- HMDS
hexamethyldisilazide
- IBX
2-iodoxybenzoic acid
- IC50
concentration inhibiting 50% of activity
- Lac
lactate
- LLS
longest linear sequence
- Lut
lutidine (2,6- unless otherwise noted)
- MBIC50
minimum inhibitory concentration of 50% of biofilms
- Mes
mesityl
- MIC
minimum inhibitory concentration
- MNBA
m-nitrobenzoic anhydride (Shiina’s reagent)
- MRSA
methicillin-resistant S. aureus
- MSSA
methicillin-susceptible S. aureus
- NBSH
2-nitrobenzenesulfonylhydrazine
- Oxaz
Davis’ N-sulfonyl oxaziridine
- PIDA
phenyliodine diacetate
- PMB
p-methoxybenzyl
- PMP
p-methoxyphenyl
- PPTS
pyridinium p-tolylsulfonate
- Pym
pyrimidyl
- Pyr
pyridyl
- SAR
structure–activity relationship
- SatA
and SatG streptogramin A acetyltransferases A and G
- SEM
[2-(trimethylsilyl)ethoxy]methyl
- TAS-F
tris(dimethylamino)sulfonium difluorotrimethylsilicate
- TBAF
tetrabutylammonium fluoride
- TBDPS
tert-butyldiphenylsilyl
- TEMPO
(2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical
- Teoc
trimethylsilylethoxycarbonyl
- TMSE
trimethylsilylethyl
- UDP
uridine diphosphate
- VISA
vancomycin-intermediate S. aureus
- VRE
vancomycin-resistant enterococcus
- VRSA
vancomycin-resistant S. aureus
- VSSA
vancomycin-susceptible S. aureus
Biographies
Sean E. Rossiter received his Bachelor of Science in Chemistry from Moravian College in Bethlehem, Pennsylvania, in 2015, graduating with honors for research in azulene chemistry under Carl O. Salter. He then matriculated into Temple University’s graduate program and began research under William Wuest toward the development of analogs of natural products with narrow-spectrum antibacterial activity. In 2017, he moved with the Wuest laboratory to Emory University.
Madison H. Fletcher received her Bachelor of Arts in Chemistry from Bard College in 2012 where she worked with Emily McLaughlin. She received her Ph.D. in chemistry from Temple University in the spring of 2017 with William Wuest developing analogs of cyclic dinucleotides for the investigation of bacterial signaling processes. She is excited to be working as a postdoctoral researcher at the University of California, Irvine with Gregory Weiss and looks forward to enjoying the beautiful weather that California has to offer year-round.
William M. Wuest obtained his B.S. degree in chemistry/business from the University of Notre Dame in 2003 and a Ph.D. in chemistry from the University of Pennsylvania under the tutelage of Amos. B. Smith, III in 2008. After an NIH NRSA Postdoctoral Fellowship with Christopher T. Walsh at Harvard Medical School, he joined the faculty in the department of chemistry at Temple University as an assistant professor in 2011 where he stayed until 2017 as the Daniel Swern Early Career Development Professor. In 2017, he moved his research group to Atlanta, GA, where he is currently a GRA Distinguished Investigator at Emory University. Research in the Wuest group focuses on the chemical biology of bacterial processes and, more specifically, the role that natural product-inspired analogs play in these events. For this, he and his group have received numerous awards including the NIH ESI Maximizing Investigators Research Award (MIRA), the NSF CAREER Award, the ACS Infectious Diseases Young Investigator Award, the Young Investigator Award from the Center for Biofilm Engineering at Montana State University, the New Investigator Award from the Charles E. Kaufman Foundation, and the Italia-Eire Foundation Distinguished Teacher of the Year Award at Temple University. Bill is an avid sports fan, with allegiances to the NY Yankees, NY Giants, and his alma mater, the Notre Dame Fighting Irish. Outside the lab he enjoys spending time with his wife, Liesl, and son, Max.
Footnotes
ORCID
William M. Wuest: 0000-0002-5198-7744
Notes
The authors declare no competing financial interest.
References
- 1.Patridge E, Gareiss P, Kinch MS, Hoyer D. An Analysis of FDA-Approved Drugs: Natural Products and Their Derivatives. Drug Discovery Today. 2016;21:204–207. doi: 10.1016/j.drudis.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Neu H. Crisis in Antibiotic Resistance. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 3.Spellberg B, Gilbert DN. The Future of Antibiotics and Resistance: A Tribute to a Career of Leadership by John Bartlett. Clin Infect Dis. 2014;59(Suppl 2):71–75. doi: 10.1093/cid/ciu392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viswanathan VK. Off-Label Abuse of Antibiotics by Bacteria. Gut Microbes. 2014;5:3–4. doi: 10.4161/gmic.28027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ventola CL. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. P T A peer-reviewed J Formul Manag. 2015;40:277–283. [PMC free article] [PubMed] [Google Scholar]
- 6.Goossens H, Ferech M, Coenen S, Stephens P, the European Sureillance of Antimicrobial Consumption Project Group Comparison of Outpatient Systemic Antibacterial Use in 2004 in the United States and 27 European Countries. Clin Infect Dis. 2007;44:1091–1095. doi: 10.1086/512810. [DOI] [PubMed] [Google Scholar]
- 7.Boeckel TPVan, Gandra S, Ashok A, Caudron Q, Grenfell BT, Levin SA, Laxminarayan R. Global Antibiotic Consumption 2000 to 2010: An Analysis of National Pharmaceutical Sales Data. Lancet Infect Dis. 2014;14:742–750. doi: 10.1016/S1473-3099(14)70780-7. [DOI] [PubMed] [Google Scholar]
- 8.Levy SB, Marshall B. Antibacterial Resistance Worldwide: Causes, Challenges and Responses. Nat Med. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 9.Barber M, Rozwadowska-dowzenko M. Infection by Penicillin Resistant Staphylococci. Lancet. 1948;252:641–644. doi: 10.1016/s0140-6736(48)92166-7. [DOI] [PubMed] [Google Scholar]
- 10.Levy SB. Microbial Resistance to Antibiotics: An Evolving and Persistant Problem. Lancet. 1982;320:83–88. doi: 10.1016/s0140-6736(82)91701-9. [DOI] [PubMed] [Google Scholar]
- 11.CDC. Antibiotic Resistance Threats in the United States. 2013 [Google Scholar]
- 12.White DG, Zhao S, Sudler R, Ayers S, Friedman S, Chen S, McDermott PF, McDermott S, Wagner DD, Meng J. The Isolation of Antibiotic-Resistant Salmonella from Retail Ground Meats. N Engl J Med. 2001;345:1147–1154. doi: 10.1056/NEJMoa010315. [DOI] [PubMed] [Google Scholar]
- 13.Chiu C-H, Wu T-L, Su L-H, Chu C, Chia J-H, Kuo A-J, Chien M-S, Lin T-Y. The Emergence in Taiwan of Fluoroquinolone Resistance in Salmonella Enterica Serotype Choleraesuis. N Engl J Med. 2002;346:413–419. doi: 10.1056/NEJMoa012261. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations the Review on Antimicrobial Resistance. 2016 [Google Scholar]
- 15.McCullough AR, Parekh S, Rathbone J, Del Mar CB, Hoffmann TC. A Systematic Review of the Public’s Knowledge and Beliefs about Antibiotic Resistance. J Antimicrob Chemother. 2016;71:27–33. doi: 10.1093/jac/dkv310. [DOI] [PubMed] [Google Scholar]
- 16.WHO. Antimicrobial Resistance. Bull World Health Organ. 2014;61:383–394. [PMC free article] [PubMed] [Google Scholar]
- 17.Johnning A, Kristiansson E, Angelin M, Marathe N, Shouche YS, Johansson A, Larsson DGJ. Quinolone Resistance Mutations in the Faecal Microbiota of Swedish Travellers to India. BMC Microbiol. 2015;15:235–242. doi: 10.1186/s12866-015-0574-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington, DC: 2003. [Google Scholar]
- 19.Walsh CT, Wencewicz TA. Antibiotics: Challenges, Mechanisms, Opportunities. ASM Press; Washington, DC: 2016. [Google Scholar]
- 20.Bourne CR. Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery. Antibiotics. 2014;3:1–28. doi: 10.3390/antibiotics3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wright PM, Seiple IB, Myers AG. The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew Chem, Int Ed. 2014;53:8840–8869. doi: 10.1002/anie.201310843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leech PN, Schoeffel EW, Kreider HR, Peterson JB, Geiling EMK, Coon JM, Cannon PR, Hagebusch OE. Elixir of Sulfanilamide-Massengill. J Am Med Assoc. 1937;109:1531–1539. [Google Scholar]
- 23.Ballentine C. Taste of Raspberries, Taste of Death: The 1937 Elixir Sulfanilamide Incident. FDA Consumer Magazine. 1981 Jun; [Google Scholar]
- 24.Coates A. In: Antibiotic Resistance. Coates A, editor. Springer; Heidelberg: 2012. [Google Scholar]
- 25.Dever LA. Mechanisms of Bacterial Resistance to Antibiotics. Arch Intern Med. 1991;151:886. [PubMed] [Google Scholar]
- 26.Crofts TS, Gasparrini AJ, Dantas G. Next-Generation Approaches to Understand and Combat the Antibiotic Resistome. Nat Rev Microbiol. 2017;15:422–434. doi: 10.1038/nrmicro.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munita JM, Arias CA, Unit AR, Santiago A De. Mechanisms of Antibiotic Resistance. Microb Spectr. 2016;4:1–37. doi: 10.1128/microbiolspec.VMBF-0016-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coates A. In: Antibiotic Resistance. Coates A, editor. Vol. 211 Springer; Heidelberg: 2012. [Google Scholar]
- 29.Munita JM, Arias CA, Unit AR, Santiago A De. Mechanisms of Antibiotic Resistance. Microb Spectr. 2016;4:1–37. doi: 10.1128/microbiolspec.VMBF-0016-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. Molecular Mechanisms of Antibiotic Resistance. Nat Rev Microbiol. 2015;13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 31.Langton KP, Henderson PJF, Herbert RB. Antibiotic Resistance: Multidrug Efflux Proteins, a Common Transport Mechanism? Nat Prod Rep. 2005;22:439–451. doi: 10.1039/b413734p. [DOI] [PubMed] [Google Scholar]
- 32.McMurry LM, Petrucci REJ, Levy SB. Active Efflux of Tetracycline Encoded by Four Genetically Different Tetracycline Resistance Determinants in Escherichia Coli. Proc Natl Acad Sci U S A. 1980;77:3974–3977. doi: 10.1073/pnas.77.7.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. Molecular Mechanisms of Antibiotic Resistance. Nat Rev Microbiol. 2015;13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 34.Nikaido H. Multidrug Resistance in Bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poole K. Efflux-Mediated Antimicrobial Resistance. J Antimicrob Chemother. 2005;56:20–51. doi: 10.1093/jac/dki171. [DOI] [PubMed] [Google Scholar]
- 36.Langton KP, Henderson PJF, Herbert RB. Antibiotic Resistance: Multidrug Efflux Proteins, a Common Transport Mechanism? Nat Prod Rep. 2005;22:439–451. doi: 10.1039/b413734p. [DOI] [PubMed] [Google Scholar]
- 37.Floss HG, Yu TW. Rifamycin-Mode of Action, Resistance, and Biosynthesis. Chem Rev. 2005;105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- 38.Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. Aminoglycosides: Activity and Resistance. Antimicrob Agents Chemother. 1999;43:727–737. doi: 10.1128/aac.43.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips I, Acar J, Bergan T, Degener J, Baquero F, Forsgren A, Schito G-C, Wiedemann B. Methods for the Determination of Susceptibility of Bacteria to Antimicrobial Agents. Terminology. Clin Microbiol Infect. 1998;4:291–296. [PubMed] [Google Scholar]
- 40.Mendelson M, Balasegaram M, Jinks T, Pulcini C, Sharland M. Antibiotic Resistance Has a Language Problem. Nature. 2017;545:23–25. doi: 10.1038/545023a. [DOI] [PubMed] [Google Scholar]
- 41.Repka LM, Chekan JR, Nair SK, van der Donk WA. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem Rev. 2017;117:5457–5520. doi: 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lovering F, Bikker J, Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 43.Lovering F. Escape from Flatland 2: Complexity and Promiscuity. MedChemComm. 2013;4:515–519. [Google Scholar]
- 44.Pawlowski AC, Johnson JW, Wright GD. Evolving Medicinal Chemistry Strategies in Antibiotic Discovery. Curr Opin Biotechnol. 2016;42:108–117. doi: 10.1016/j.copbio.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 45.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 46.Galloway WRJD, Bender A, Welch M, Spring DR. The Discovery of Antibacterial Agents Using Diversity-Oriented Synthesis. Chem Commun. 2009;18:2446–2462. doi: 10.1039/b816852k. [DOI] [PubMed] [Google Scholar]
- 47.Schreiber SL. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 48.Huigens RW, III, Morrison KC, Hicklin RW, Flood TA, Jr, Richter MF, Hergenrother PJ. A Ring-Distortion Strategy to Construct Stereochemically Complex and Structurally Diverse Compounds from Natural Products. Nat Chem. 2013;5:195–202. doi: 10.1038/nchem.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rafferty RJ, Hicklin RW, Maloof KA, Hergenrother PJ. Synthesis of Complex and Diverse Compounds through Ring Distortion of Abietic Acid. Angew Chem, Int Ed. 2014;53:220–224. doi: 10.1002/anie.201308743. [DOI] [PubMed] [Google Scholar]
- 50.Morrison KC, Hergenrother PJ. Natural Products as Starting Points for the Synthesis of Complex and Diverse Compounds. Nat Prod Rep. 2014;31:6–14. doi: 10.1039/c3np70063a. [DOI] [PubMed] [Google Scholar]
- 51.Allred TK, Manoni F, Harran PG. Exploring the Boundaries of “ Practical ”: De Novo Syntheses of Complex Natural Product-Based Drug Candidates. Chem Rev. 2017 doi: 10.1021/acs.chemrev.7b00126. [DOI] [PubMed] [Google Scholar]
- 52.Chou T-C, Dong H, Rivkin A, Yoshimura F, Gabarda AE, Cho YS, Tong WP, Danishefsky SJ. Design and Total Synthesis of a Superior Family of Epothilone Analogues which Eliminate Xenograft Tumors to a Nonrelapsable State. Angew Chem. 2003;115:4910–4915. doi: 10.1002/anie.200352361. [DOI] [PubMed] [Google Scholar]
- 53.Rivkin A, Chou T-C, Danishefsky SJ. On the Remarkable Antitumor Properties of Fludelone: How we Got There. Angew Chem, Int Ed. 2005;44:2838–2850. doi: 10.1002/anie.200461751. [DOI] [PubMed] [Google Scholar]
- 54.Danishefsky S. On the potential of natural products in the discovery of pharma leads: A case for reassessment. Nat Prod Rep. 2010;27:1114–1116. doi: 10.1039/c003211p. [DOI] [PubMed] [Google Scholar]
- 55.Wender PA, Quiroz RV, Stevens MC. Function through Synthesis-Informed Design. Acc Chem Res. 2015;48:752–760. doi: 10.1021/acs.accounts.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wender PA, Verma VA, Paxton TJ, Pillow TH. Function-Oriented Synthesis, Step Economy, and Drug Design. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 57.Cordier C, Morton D, Murrison S, Nelson A, O’Leary-Steele C. Natural Products as an Inspiration in the Diversity-Oriented Synthesis of Bioactive Compound Libraries. Nat Prod Rep. 2008;25:719–737. doi: 10.1039/b706296f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas GL, Spandl RJ, Glansdorp FG, Welch M, Bender A, Cockfield J, Lindsay JA, Bryant C, Brown DFJ, Loiseleur O, et al. Anti-MRSA Agent Discovery Using Diversity-Oriented Synthesis. Angew Chem, Int Ed. 2008;47(15):2808–2812. doi: 10.1002/anie.200705415. [DOI] [PubMed] [Google Scholar]
- 59.Socha AM, Tan NY, Laplante KL, Sello JK. Diversity-Oriented Synthesis of Cyclic Acyldepsipeptides Leads to the Discovery of a Potent Antibacterial Agent. Bioorg Med Chem. 2010;18:7193–7202. doi: 10.1016/j.bmc.2010.08.032. [DOI] [PubMed] [Google Scholar]
- 60.Paciaroni NG, Ratnayake R, Matthews JH, Norwood VM, IV, Arnold AC, Dang LH, Luesch H, Huigens RW., III A Tryptoline Ring-Distortion Strategy Leads to Complex and Diverse Biologically Active Molecules from the Indole Alkaloid Yohimbine. Chem – Eur J. 2017;23:4327–4335. doi: 10.1002/chem.201604795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McGuire JM, Bunch RL, Anderson RC, Boaz HE, Flynn EH, Powell HM, Smith JW. Ilotycin, a New Antibiotic. Antibiot Chemother. 1952;2:281–283. [PubMed] [Google Scholar]
- 62.Erythromycin: Another Antibiotic. Lancet. 1952;260:232–233. doi: 10.1016/S0140-6736(52)91557-2. [DOI] [PubMed] [Google Scholar]
- 63.Erythromycin, New and Promising Antibiotic. N Engl J Med. 1952;247:267–269. doi: 10.1056/NEJM195208142470710. [DOI] [PubMed] [Google Scholar]
- 64.Wiley PF, Gerzon K, Flynn EH, Sigal MV, Jr, Weaver O, Quarck UC, Chauvette RR, Monahan R. Erythromycin. X. Structure of Erythromycin. J Am Chem Soc. 1957;79:6062–6070. [Google Scholar]
- 65.Harris DR, McGeachin SG, Mills HH. The Structure and Stereochemistry of Erythromycin A. Tetrahedron Lett. 1965;6:679–685. doi: 10.1016/s0040-4039(00)90018-2. [DOI] [PubMed] [Google Scholar]
- 66.Celmer WD. Stereochemical Problems in Macrolide Antibiotics. Pure Appl Chem. 1971;28:413–453. doi: 10.1351/pac197128040413. [DOI] [PubMed] [Google Scholar]
- 67.Contreras A, Vázquez D. Cooperative and Antagonistic Interactions of Peptidyl-tRNA and Antibiotics with Bacterial Ribosomes. Eur J Biochem. 1977;74:539–547. doi: 10.1111/j.1432-1033.1977.tb11422.x. [DOI] [PubMed] [Google Scholar]
- 68.Andersson S, Kurland CG. Elongating Ribosomes in Vivo Are Refractory to Erythromycin. Biochimie. 1987;69:901–904. doi: 10.1016/0300-9084(87)90218-5. [DOI] [PubMed] [Google Scholar]
- 69.Dunkle JA, Xiong L, Mankin AS, Cate JHD. Structures of the Escherichia coli Ribosome with Antibiotics Bound near the Peptidyl Transferase Center Explain Spectra of Drug Action. Proc Natl Acad Sci U S A. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sutcliffe J, Tait-Kamradt A, Wondrack L. Streptococcus pneumoniae and Streptococcus pyogenes Resistant to Macrolides but Sensitive to Clindamycin: A Common Resistance Pattern Mediated by an Efflux System. Antimicrob Agents Chemother. 1996;40:1817–1824. doi: 10.1128/aac.40.8.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Felmingham D, Cantón R, Jenkins SG. Regional Trends in Beta-Lactam, Macrolide, Fluoroquinolone and Telithromycin Resistance among Streptococcus pneumoniae Isolates 2001–2004. J Infect. 2007;55:111–118. doi: 10.1016/j.jinf.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 72.Wierzbowski AK, Nichol K, Laing N, Hisanaga T, Nikulin A, Karlowsky JA, Hoban DJ, Zhanel GG. Macrolide Resistance Mechanisms among Streptococcus pneumoniae Isolated over 6 Years of Canadian Respiratory Organism Susceptibility Study (CROSS) (1998–2004) J Antimicrob Chemother. 2007;60:733–740. doi: 10.1093/jac/dkm273. [DOI] [PubMed] [Google Scholar]
- 73.Lubelski J, Konings WN, Driessen AJM. Distribution and Physiology of ABC-Type Transporters Contributing to Multidrug Resistance in Bacteria. Microbiol Mol Biol Rev. 2007;71:463–476. doi: 10.1128/MMBR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kobayashi N, Nishino K, Yamaguchi A. Novel Macrolide-Specific ABC-Type Efflux Transporter in Escherichia coli. J Bacteriol. 2001;183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ojo KK, Striplin MJ, Ulep CC, Close NS, Zittle J, Luis H, Bernardo M, Leitao J, Roberts MC. Staphylococcus Efflux msr(A) Gene Characterized in Streptococcus, Enterococcus, Corynebacterium, and Pseudomonas Isolates. Antimicrob Agents Chemother. 2006;50:1089–1091. doi: 10.1128/AAC.50.3.1089-1091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blair JMA, Piddock LJV. Structure, Function, and Inhibition of RND Efflux Pumps in Gram-Negative Bacteria: An Update. Curr Opin Microbiol. 2009;12:512–519. doi: 10.1016/j.mib.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 77.Chollet R, Chevalier J, Bryskier A, Pagès JM. The AcrAB-TolC Pump Is Involved in Macrolide Resistance but Not in Telithromycin Efflux in Enterobacter aerogenes and Escherichia coli. Antimicrob Agents Chemother. 2004;48:3621–3624. doi: 10.1128/AAC.48.9.3621-3624.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wehmeier C, Schuster S, Fähnrich E, Kern WV, Bohnert JA. Site-Directed Mutagenesis Reveals Amino Acid Residues in the Escherichia coli RND Efflux Pump AcrB That Confer Macrolide Resistance. Antimicrob Agents Chemother. 2009;53:329–330. doi: 10.1128/AAC.00921-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Canu A, Malbruny B, Coquemont M, Davies TA, Appelbaum PC, Leclercq R. Diversity of Ribosomal Mutations Conferring Resistance to Macrolides, Clindamycin, Streptogramin, and Telithromycin in Streptococcus pneumoniae. Antimicrob Agents Chemother. 2002;46:125–131. doi: 10.1128/AAC.46.1.125-131.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Björkholm B, Sjölund M, Falk PG, Berg OG, Engstrand L, Andersson DI. Mutation Frequency and Biological Cost of Antibiotic Resistance in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001;98:14607–14612. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allen NE. Macrolide Resistance in Staphylococcus aureus: Induction of Macrolide-Resistant Protein Synthesis. Antimicrob Agents Chemother. 1977;11:661–668. doi: 10.1128/aac.11.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allen NE. Macrolide Resistance in Staphylococcus aureus: Inducers of Macrolide Resistance. Antimicrob Agents Chemother. 1977;11:669–674. doi: 10.1128/aac.11.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pestka S, Vince R, Lemahieu R, Weiss F, Fern L, Unowsky J. Induction of Erythromycin Resistance in Staphylococcus aureus by Erythromycin Derivatives. Antimicrob Agents Chemother. 1976;9:128–130. doi: 10.1128/aac.9.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weisblum B. Insights into Erythromycin Action from Studies of Its Activity as Inducer of Resistance. Antimicrob Agents Chemother. 1995;39:797–805. doi: 10.1128/aac.39.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weisblum B, Siddhikol C, Lai CJ, Demohn V. Erythromycin-Inducible Resistane in Staphylococcus aureus: Requirements for Induction. J Bacteriol. 1971;106:835–847. doi: 10.1128/jb.106.3.835-847.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ounissi H, Courvalin P. Nucleotide Sequence of the Gene ereA Encoding the Erythromycin Esterase in Escherichia coli. Gene. 1985;35:271–278. doi: 10.1016/0378-1119(85)90005-8. [DOI] [PubMed] [Google Scholar]
- 87.Arthur M, Autissier D, Courvalin P. Analysis of the Nucleotide Sequence of the ereB Gene Encoding the Erythromycin Esterase Type II. Nucleic Acids Res. 1986;14:4987–4999. doi: 10.1093/nar/14.12.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR. Characterization of a New Metallo-β-Lactamase Gene, blaNDM-1, and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob Agents Chemother. 2009;53:5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morar M, Pengelly K, Koteva K, Wright GD. Mechanism and Diversity of the Erythromycin Esterase Family of Enzymes. Biochemistry. 2012;51:1740–1751. doi: 10.1021/bi201790u. [DOI] [PubMed] [Google Scholar]
- 90.U.S. Food and Drug Adminstration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf/
- 91.Kurath P, Jones PH, Egan RS, Perun TJ. Acid Degradation of Erythromycin A and Erythromycin B. Experientia. 1971;27:362. doi: 10.1007/BF02137246. [DOI] [PubMed] [Google Scholar]
- 92.Morimoto S, Takahashi T, Watanabe Y, Omura S. Chemical Modification of Erythromycins. I. Synthesis and Antibacterial Activity of 6-O-Methylerythromycins A. J Antibiot. 1984;37:187–189. doi: 10.7164/antibiotics.37.187. [DOI] [PubMed] [Google Scholar]
- 93.Djokic S, Kobrehel G, Lazarevski G, Lopotar N, Tamburasev Z, Kamenar B, Nagl A, Vickovic I. Erythromycin Series. Part 11. Ring Exansion of Erythromycin A Oxime by the Beckmann Rearrangement. J Chem Soc, Perkin Trans 1. 1986:1881–1890. [Google Scholar]
- 94.Farrell DJ, Flamm RK, Sader HS, Jones RN. Results from the Solithromycin International Surveillance Program (2014) Anti-microb Agents Chemother. 2016;60:3662–3668. doi: 10.1128/AAC.00185-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baker WR, Clark JD, Stephens RL, Kim KH. Modification of Macrolide Antibiotics. Synthesis of 11-Deoxy-11-(Carboxyamino)-6-O-Methylerythromycin A 11,12-(Cyclic Esters) via an Intramolecular Michael Reaction of O-Carbamates with an Alpha,beta-Unsaturated Ketone. J Org Chem. 1988;53:2340–2345. [Google Scholar]
- 96.Bertrand D, Bertrand S, Neveu E, Fernandes P. Molecular Characterization of Off-Target Activities of Telithroycin: A Potential Role for Nicotinic Acetycholine Receptors. Antimicrob Agents Chemo-ther. 2010;54:5399–5402. doi: 10.1128/AAC.00840-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.U.S. Food and Drug Adminstration. FDA Briefing Document: Solithromycin Oral Capsule and Injection. 2016 [Google Scholar]
- 98.Owens B. Solithromycin Rejection Chills Antibiotic Sector. Nat Biotechnol. 2017;35:187–188. doi: 10.1038/nbt0317-187. [DOI] [PubMed] [Google Scholar]
- 99.Corey EJ, Hopkins PB, Kim S, Yoo S-E, Nambiar KP, Falck JR. Total Synthesis of Erythromycins. 5. Total Synthesis of Erythronolide A. J Am Chem Soc. 1979;101:7131–7134. [Google Scholar]
- 100.Corey EJ, Kim S, Yoo S-E, Nicolaou KC, Melvin LS, Jr, Lett R, Sheldrake PW, Jr, Brunelle DJ, Falck JR, Trybulski EJ, et al. Total Synthesis of Erythromycins. 4. Total Synthesis of Erythronolide B. J Am Chem Soc. 1978;100:4620–4622. [Google Scholar]
- 101.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Auyeung BW, Balaram P, Browne LJ, Card PJ, Chen CH, et al. Asymmetric Total Synthesis of Erythromycin. 1. Synthesis of an Erythronolide A Seco Acid Derivative via Asymmetric Induction. J Am Chem Soc. 1981;103:3210–3213. [Google Scholar]
- 102.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung BW, Balaram P, Browne LJ, Card PJ, Chen CH, et al. Asymmetric Total Synthesis of Erythromycin. 2. Synthesis of an Erythronolide A Lactone System. J Am Chem Soc. 1981;103:3213–3215. [Google Scholar]
- 103.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung BW, Balaram P, Browne LJ, Card PJ, Chen CH. Asymmetric Total Synthesis of Erythromycin. 3. Total Synthesis of Erythromycin. J Am Chem Soc. 1981;103:3215–3217. [Google Scholar]
- 104.Martin SF, Hida T, Kym PR, Loft M, Hodgson A. The Asymmetric Synthesis of Erythromycin B. J Am Chem Soc. 1997;119:3193–3194. [Google Scholar]
- 105.Hergenrother PJ, Hodgson A, Judd AS, Lee W-C, Martin SF. An Abiotic Strategy for the Enantioselective Synthesis of Erythromycin B. Angew Chem, Int Ed. 2003;42:3278–3281. doi: 10.1002/anie.200351136. [DOI] [PubMed] [Google Scholar]
- 106.Breton P, Hergenrother PJ, Hida T, Hodgson A, Judd AS, Kraynack E, Kym PR, Lee W-C, Loft MS, Yamashita M, et al. Total Synthesis of Erythromycin B. Tetrahedron. 2007;63:5709–5729. [Google Scholar]
- 107.Kim HC, Kang SH. Total Synthesis of Azithromycin. Angew Chem, Int Ed. 2009;48:1827–1829. doi: 10.1002/anie.200805334. [DOI] [PubMed] [Google Scholar]
- 108.Seiple IB, Zhang Z, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, et al. A Platform for the Discovery of New Macrolide Antibiotics. Nature. 2016;533:338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Seiple IB, Hog DT, Myers AG. Practical Protocols for the Preparation of Highly Enantioenriched Silyl Ethers of (R)-3-Hydroxypentan-2-One, Building Blocks for the Synthesis of Macrolide Antibiotics. Synlett. 2016;27:57–60. [Google Scholar]
- 110.Morales MR, Mellem KT, Myers AG. Pseudoephenamine: A Practical Chiral Auxiliary for Asymmetric Synthesis. Angew Chem, Int Ed. 2012;51:4568–4571. doi: 10.1002/anie.201200370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Z, Kitamura Y, Myers AG. An Efficient Directed Claisen Reaction Allows for Rapid Construction of 5,6-Disubstituted 1,3-Dioxin-4-Ones. Synthesis. 2015;47:2709–2712. [Google Scholar]
- 112.Boeckman RK, Jr, Pruitt JR. A New, Highly Efficient, Selective Methodology for Formation of Medium-Ring and Macrocyclic Lactones via Intramolecular Ketene Trapping: An Application to a Convergent Synthesis of (−)-Kromycin. J Am Chem Soc. 1989;111:8286–8288. [Google Scholar]
- 113.Zhang Z, Fukuzaki T, Myers AG. Synthesis of -Desosamine and Analogs by Rapid Assembly of 3-Amino Sugars. Angew Chem, Int Ed. 2016;55:523–527. doi: 10.1002/anie.201507357. [DOI] [PubMed] [Google Scholar]
- 114.Tu D, Blaha G, Moore PB, Steitz TA. Structures of MLSBK Antibiotics Bound to Mutated Large Ribosomal Subunits Provide a Structural Explanation for Resistance. Cell. 2005;121:257–270. doi: 10.1016/j.cell.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 115.Andrade RB. Total Synthesis of Desmethyl Macrolide Antibiotics. Synlett. 2015;26:2199–2215. [Google Scholar]
- 116.Velvadapu V, Glassford I, Lee M, Paul T, Debrosse C, Klepacki D, Small MC, MacKerell AD, Andrade RB. Desmethyl Macrolides: Synthesis and Evaluation of 4,10-Didesmethyl Telithromycin. ACS Med Chem Lett. 2012;3:211–215. doi: 10.1021/ml200254h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Velvadapu V, Paul T, Wagh B, Klepacki D, Guvench O, MacKerell A, Andrade RB. Desmethyl Macrolides: Synthesis and Evaluation of 4,8,10-Tridesmethyl Telithromycin. ACS Med Chem Lett. 2011;2:68–72. doi: 10.1021/ml1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wagh B, Paul T, Glassford I, Debrosse C, Klepacki D, Small MC, MacKerell AD, Andrade RB. Desmethyl Macrolides: Synthesis and Evaluation of 4,8-Didesmethyl Telithromycin. ACS Med Chem Lett. 2012;3:1013–1018. doi: 10.1021/ml300230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Glassford I, Lee M, Wagh B, Velvadapu V, Paul T, Sandelin G, DeBrosse C, Klepacki D, Small MC, Mackerell AD, et al. Desmethyl Macrolides: Synthesis and Evaluation of 4-Desmethyl Telithromycin. ACS Med Chem Lett. 2014;5:1021–1026. doi: 10.1021/ml5002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gumbart J, Schreiner E, Wilson DN, Beckmann R, Schulten K. Mechanisms of SecM-Mediated Stalling in the Ribosome. Biophys J. 2012;103:331–341. doi: 10.1016/j.bpj.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Washington AZ, Tapadar S, George A, Oyelere AK. Exploiting Translational Stalling Peptides in an Effort to Extend Azithromycin Interaction Within the Prokaryotic Ribosome Nascent Peptide Exit Tunnel. Bioorg Med Chem. 2015;23:5198–5209. doi: 10.1016/j.bmc.2015.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu F, Myers AG. Development of a Platform for the Discovery and Practical Synthesis of New Tetracycline Antibiotics. Curr Opin Chem Biol. 2016;32:48–57. doi: 10.1016/j.cbpa.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 123.Brodersen DE, Clemons WM, Carter AP, Morgan-warren RJ, Wimberly BT, Ramakrishnan V. The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 124.Chopra I, Roberts M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol Mol Biol Rev. 2001;65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal Protection Proteins and Their Mechanism of Tetracycline Resistance. Antimicrob Agents Chemother. 2003;47:3675–3681. doi: 10.1128/AAC.47.12.3675-3681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sanchez-Pescador R, Brown JT, Roberts M, Urdea MS. Homology of the TetM with Translational Elongation Factors: Implications for Potential Modes of TetM-Conferred Tetracycline Resistance. Nucleic Acids Res. 1988;16:1218. doi: 10.1093/nar/16.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Burdett V. Purification and Characterization of Tet(M), a Protein That Renders Ribosomes Resistant to Tetracycline. J Biol Chem. 1991;266:2872–2877. [PubMed] [Google Scholar]
- 128.Burdett V. Tet(M)-Promoted Release of Tetracycline from Ribosomes Is GTP Dependent. J Bacteriol. 1996;178:3246–3251. doi: 10.1128/jb.178.11.3246-3251.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Trieber CA, Burkhardt N, Nierhaus KH, Taylor DE. Ribosomal Protection from Tetracycline Mediated by Tet(O): Tet(O) Interaction with Ribosomes Is GTP-Dependent. Biol Chem. 1998;379:847–856. doi: 10.1515/bchm.1998.379.7.847. [DOI] [PubMed] [Google Scholar]
- 130.Dönhöfer A, Franckenberg S, Wickles S, Berninghausen O, Beckmann R, Wilson DN. Structural Basis for TetM-Mediated Tetracycline Resistance. Proc Natl Acad Sci U S A. 2012;109:16900–16905. doi: 10.1073/pnas.1208037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li W, Atkinson GC, Thakor NS, Allas Ü, Lu C, Chan K-Y, Tenson T, Schulten K, Wilson KS, Hauryliuk V, et al. Mechanism of Tetracycline Resistance by Ribosomal Protection Protein Tet(O) Nat Commun. 2013;4:1477. doi: 10.1038/ncomms2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Speer BS, Bedzyk L, Salyers AA. Evidence That a Novel Tetracycline Resistance Gene Found on Two Bacteroides Transposons Encodes an NADP-Requiring Oxidoreductase. J Bacteriol. 1991;173:176–183. doi: 10.1128/jb.173.1.176-183.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Caryl JA, Cox G, Trimble S, O’Neill AJ. tet(U)” is Not a Tetracycline Resistance Determinant. Antimicrob Agents Chemother. 2012;56:3378–3379. doi: 10.1128/AAC.05957-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stephens CR, Murai K, Rennhard HH, Conover LH, Brunings KJ. Hydrogenolysis Studies in the Tetracycline Series – 6-Deoxytetracyclines. J Am Chem Soc. 1958;80:5324–5325. [Google Scholar]
- 135.Church FR, Weiss J. Synthesis of 7-Dimethylamino-6-Demethyl-6-Deoxytetracycline (Minocycline) via 9-Nitro-6-Demethyl-6-Deoxytetracycline. J Org Chem. 1971;36:723–725. doi: 10.1021/jo00804a025. [DOI] [PubMed] [Google Scholar]
- 136.Martell MJ, Boothe JH. The 6-Deoxytetracyclines. VII. Alkylated Aminotetracyclines Possessing Unique Antibacterial Activity. J Med Chem. 1967;10:44–46. doi: 10.1021/jm00313a009. [DOI] [PubMed] [Google Scholar]
- 137.Sum PE, Lee VJ, Testa RT, Hlavka JJ, Ellestad GA, Bloom JD, Gluzman Y, Tally FP. Glycylcyclines. 1. A New Generation of Potent Antibacterial Agents through Modification of 9-Aminotetracyclines. J Med Chem. 1994;37:184–188. doi: 10.1021/jm00027a023. [DOI] [PubMed] [Google Scholar]
- 138.Olson MW, Ruzin A, Feyfant E, Rush TS, III, O’Connell J, Bradford PA. Functional, Biophysical, and Structural Bases for Antibacterial Activity of Tigecycine. Antimicrob Agents Chemother. 2006;50:2156–2166. doi: 10.1128/AAC.01499-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Honeyman L, Ismail M, Nelson ML, Bhatia B, Bowser TE, Chen J, Mechiche R, Ohemeng K, Verma AK, Cannon EP, et al. Structure-Activity Relationship of the Aminomethylcyclines and the Discovery of Omadacycline. Antimicrob Agents Chemother. 2015;59:7044–7053. doi: 10.1128/AAC.01536-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paratek Announces Positive Phase 3 Study of Omadacycline in Community-Acquired Bacterial Pneumonia. 2017 Apr 3; [Google Scholar]
- 141.Paratek Announces Phase 3 Study of Oral-Only Dosing of Omadacycline Met All Primary and Secondary FDA and EMA Eficacy Endpoints in Acute Bacterial Skin Infections. 2017 Jul 17; [Google Scholar]
- 142.Gurevich AI, Karapetyan MG, Kolosov MN, Korobko V, Onoprienko VV, Popravko SA, Shemyakin M. Synthesis of 12a-Deoxy-5a, 6-Anhydrotetracycline. The First Total Synthesis of the Naturally Occurring Tetracycline. Tetrahedron Lett. 1967;8:131–134. doi: 10.1016/s0040-4039(00)90501-x. [DOI] [PubMed] [Google Scholar]
- 143.Korst JJ, Johnston JD, Butler K, Bianco EJ, Conover LH, Woodward RB. The Total Synthesis of Dl-6-Demethyl-6-Deoxytetracycline. J Am Chem Soc. 1968;90:439–457. [Google Scholar]
- 144.Muxfeldt H, Haas G, Hardtmann G, Kathawala F, Mooberry JB, Vedejs E. Tetracyclines. 9. Total Synthesis of Dl-Terramycin. J Am Chem Soc. 1979;101:689–701. doi: 10.1021/ja01025a063. [DOI] [PubMed] [Google Scholar]
- 145.Stork G, La Clair JJ, Spargo P, Nargund RP, Totah N. Stereocontrolled Synthesis of (±)-12a-Deoxytetracycline. J Am Chem Soc. 1996;118:5304–5305. [Google Scholar]
- 146.Stork G, Hagedorn AA., III 3-Benzyloxyisoxazole System in Construction of Tetracyclines. J Am Chem Soc. 1978;100:3609–3611. [Google Scholar]
- 147.Tatsuta K, Yoshimoto T, Gunji H, Okado Y, Takahashi M. The First Total Synthesis of Natural (−)-Tetracycline. Chem Lett. 2000;29:646–647. [Google Scholar]
- 148.Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. A Convergent Enantioselective Route to Structurally Diverse 6-Deoxytetracycline Antibiotics. Science. 2005;308:395–398. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]
- 149.Wilson RM, Danishefsky SJ. Small Molecule Natural Products in the Discovery of Therapeutic Agents: The Synthesis Connection. J Org Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- 150.Myers AG, Movassaghi M, Zheng B. Single-Step Process or the Reductive Deoxygenation of Unhindered Alcohols. J Am Chem Soc. 1997;119:8572–8573. [Google Scholar]
- 151.Brubaker JD, Myers AG. A Practical, Enantioselective Synthetic Route to a Key Precursor to the Tetracycline Antibiotics. Org Lett. 2007;9:3523–3525. doi: 10.1021/ol071377d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kummer DA, Li D, Dion A, Myers AG. A Practical, Convergent Route to the Key Precursor to the Tetracycline Antibiotics. Chem Sci. 2011;2:1710–1718. doi: 10.1039/C1SC00303H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang WY, Hogan PC, Chen CL, Niu J, Wang Z, Lafrance D, Gilicky O, Dunwoody N, Ronn M. Process Research and Development of an Enantiomerically Enriched Allyic Amine, One of the Key Intermediates for the Manufacture of Synthetic Tetracyclines. Org Process Res Dev. 2015;19:1784–1795. [Google Scholar]
- 154.Zhang W-Y, Chen C-L, HE M, Zhu Z, Hogan P, Gilicky O, Dunwoody N, Ronn M. Process Research and Development of TP-808: A Key Intermediate for the Manufacture of Synthetic Tetracyclines. Org Process Res Dev. 2017;21:377–386. [Google Scholar]
- 155.Sun C, Wang Q, Brubaker JD, Wright PM, Lerner CD, Noson K, Charest M, Siegel DR, Wang Y, Myers AG. A Robust Platform for the Synthesis of New Tetracycline Antibiotics. J Am Chem Soc. 2008;130:17913–17927. doi: 10.1021/ja806629e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sun C, Hunt DK, Chen CL, Deng Y, He M, Clark RB, Fyfe C, Grossman TH, Sutcliffe JA, Xiao XY. Design, Synthesis, and Biological Evaluation of Hexacyclic Tetracyclines as Potent, Broad Spectrum Antibacterial Agents. J Med Chem. 2015;58:4703–4712. doi: 10.1021/acs.jmedchem.5b00262. [DOI] [PubMed] [Google Scholar]
- 157.Liu F, Wright PM, Myers AG. Diastereoselective Michael–Claisen Cyclizations of γ-Oxa-A,β-Unsaturated Ketones En Route to 5-Oxatetracyclines. Org Lett. 2017;19:206–209. doi: 10.1021/acs.orglett.6b03491. [DOI] [PubMed] [Google Scholar]
- 158.Xiao X-Y, Hunt DK, He M, Achorn C, Chen CL, Deng Y, Fyfe C, Grossman TH, Hogan PC, O’Brien WJ, Clark RB, et al. Fluorocyclines. 1. 7-Fluoro-9-Pyrrolidinoacetamido-6-Demethyl-6- Deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent. J Med Chem. 2012;55:597. doi: 10.1021/jm201465w. [DOI] [PubMed] [Google Scholar]
- 159.Clark RB, Hunt DK, He M, Achorn C, Chen C-L, Deng Y, Fyfe C, Grossman TH, Hogan PC, O’Brien WJ, et al. Fluorocyclines. 2. Optimization of the C-9 Side-Chain for Antibacterial Activity and Oral Efficacy. J Med Chem. 2012;55:606–622. doi: 10.1021/jm201467r. [DOI] [PubMed] [Google Scholar]
- 160.Ronn M, Zhu Z, Hogan PC, Zhang WY, Niu J, Katz CE, Dunwoody N, Gilicky O, Deng Y, Hunt DK, et al. Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development. Org Process Res Dev. 2013;17:838–845. [Google Scholar]
- 161.Grossman TH, Starosta AL, Fyfe C, O’Brien W, Rothstein DM, Mikolajka A, Wilson DN, Sutcliffe JA. Target- and Resistance-Based Mechanistic Studies with TP-434, a Novel Fluorocycline Antibiotic. Antimicrob Agents Chemother. 2012;56:2559–2564. doi: 10.1128/AAC.06187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Solomkin J, Evans D, Slepavicius A, Lee P, Marsh A, Tsai L, Sutcliffe JA, Horn P. Assessing the Efficacy and Safety of Eravacycline vs Ertapenem in Complicated Intra-Abdominal Infections in the Investigating Gram-Negative Infections Treated With Eravacycline (IGNITE 1) Trial: A Randomized Clinical Trial. JAMA Surg. 2017;152:E1–E9. doi: 10.1001/jamasurg.2016.4237. [DOI] [PubMed] [Google Scholar]
- 163.Hutchison RD, Steyn PS, van Rensburg SJ. Viridicatumtoxin, a New Mycotoxin from Penicillium Viridicatum Westling. Toxicol Appl Pharmacol. 1973;24:507–509. doi: 10.1016/0041-008x(73)90057-4. [DOI] [PubMed] [Google Scholar]
- 164.Zheng C-J, Yu H-E, Kim E-H, Kim W-G. Viridicatumtoxin B, a New Anti-MRSA Agent from Penicillium Sp. FR11. J Antibiot. 2008;61:633–637. doi: 10.1038/ja.2008.84. [DOI] [PubMed] [Google Scholar]
- 165.Inokoshi J, Nakamura Y, Hongbin Z, Uchida R, Nonaka K, Masuma R, Tomoda H. Spirohexalines, New Inhibitors of Bacterial Undecaprenyl Pyrophosphate Synthase, Produced by Penicillium Brasilianum FKI-3368. J Antibiot. 2013;66:37–41. doi: 10.1038/ja.2012.83. [DOI] [PubMed] [Google Scholar]
- 166.Nicolaou KC, Nilewski C, Hale CRH, Ioannidou HA, Elmarrouni A, Koch LG. Total Synthesis and Structural Revision of Viridicatumtoxin B. Angew Chem, Int Ed. 2013;52:8736–8741. doi: 10.1002/anie.201304691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nicolaou KC, Hale CRH, Nilewski C, Ioannidou HA, Elmarrouni A, Nilewski LG, Beabout K, Wang TT, Shamoo Y. Total Synthesis of Viridicatumtoxin B and Analogues Thereof: Strategy Evolution, Structural Revision, and Biological Evaluation. J Am Chem Soc. 2014;136:12137–12160. doi: 10.1021/ja506472u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nicolaou KC, Liu G, Beabout K, McCurry MD, Shamoo Y. Asymmetric Alkylation of Anthrones, Enantioselective Total Synthesis of (−)- and (+)-Viridicatumtoxins B and Analogues Thereof: Absolute Configuration and Potent Antibacterial Agents. J Am Chem Soc. 2017;139:3736–3746. doi: 10.1021/jacs.6b12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.McCormick MH, Stark WM, Pittenger GE, Pittenger RC, McGuire JM. Vancomycin, a New Antibiotic. I. Chemical and Biologic Properties. Antibiot Annu. 1955;3:606–611. [PubMed] [Google Scholar]
- 170.Williamson MP, Williams DH. Structure Revision of the Antibiotic Vancomycin. The Use of Nuclear Overhauser Effect Difference Spectroscopy. J Am Chem Soc. 1981;103:6580–6585. [Google Scholar]
- 171.Harris CM, Kopecka H, Harris TM. Vancomycin: Structure and Transformation to CDP-I. J Am Chem Soc. 1983;105:6915–6922. [Google Scholar]
- 172.Levine DP. Vancomycin: A History. Clin Infect Dis. 2006;42:S5–12. doi: 10.1086/491709. [DOI] [PubMed] [Google Scholar]
- 173.Courvalin P. Vancomycin Resistance in Gram-Positive Cocci. Clin Infect Dis. 2006;42(Suppl 1):S25–34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 174.Anderson S. Biosynthesis of the Peptidoglycan of Bacterial Cell Walls. J Biol Chem. 1967;242:3180–3190. [PubMed] [Google Scholar]
- 175.Sham L-T, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. MurJ Is the Flippase of Lipid-Linked Precursors for Peptidoglycan Biogenesis. Science (Washington, DC, U S) 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Williams DH, Waltho JP. Molecular Basis of the Activity of Antibiotics of the Vancomycin Group. Biochem Pharmacol. 1988;37:133–141. doi: 10.1016/0006-2952(88)90765-4. [DOI] [PubMed] [Google Scholar]
- 177.Nitanai Y, Kikuchi T, Kakoi K, Hanamaki S, Fujisawa I, Aoki K. Crystal Structures of the Complexes between Vancomycin and Cell-Wall Precursor Analogs. J Mol Biol. 2009;385:1422–1432. doi: 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 178.Périchon B, Courvalin P. VanA-Type Vancomycin-Resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2009;53:4580–4587. doi: 10.1128/AAC.00346-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Arthur M, Molinas C, Courvalin P. The VanS-VanR Two-Component Regulatory System Controls Synthesis of Depsipeptide Peptidoglycan Precursors in Enterococcus faecium BM4147. J Bacteriol. 1992;174:2582–2591. doi: 10.1128/jb.174.8.2582-2591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wright GD, Holman TR, Walsh CT. Purification and Characterization of VanR and the Cytosolic Domain of VanS: A Two-Component Regulatory System Required for Vancomycin Resistance in Enterococcus faecium BM4147. Biochemistry. 1993;32:5057–5063. doi: 10.1021/bi00070a013. [DOI] [PubMed] [Google Scholar]
- 181.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and Lipoglycopeptide Antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 182.McComas CC, Crowley BM, Boger DL. Partitioning the Loss in Vancomycin Binding Affinity for D-Ala-D-Lac into Lost H-Bond and Repulsiv Lone Pair Contributions. J Am Chem Soc. 2003;125:9314–9315. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 183.Judice JK, Pace JL. Semi-Synthetic Glycopeptide Antibacterials. Bioorg Med Chem Lett. 2003;13:4165–4168. doi: 10.1016/j.bmcl.2003.08.067. [DOI] [PubMed] [Google Scholar]
- 184.Leadbetter MR, Adams SM, Bazzini B, Fatheree PR, Karr DE, Krause KM, Lam BMT, Linsell MS, Nodwell MB, Pace JL, et al. Hydrophobic Vancomycin Derivatives with Improved ADME Properties: Discovery of Telavancin. J Antibiot. 2004;57:326–336. doi: 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- 185.Krause KM, Renelli M, Difuntorum S, Wu TX, Debabov DV, Benton BM. In Vitro Activity of Telavancin against Resistant Gram-Positive Bacteria. Antimicrob Agents Chemother. 2008;52:2647–2652. doi: 10.1128/AAC.01398-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stryjewski ME, Graham DR, Wilson SE, O’Riordan W, Young D, Lentnek A, Ross DP, Fowler VG, Hopkins A, Friedland HD, et al. Telavancin Versus Vancomycin for the Treatment of Complicated Skin and Skin-Structure Infections Caused by Gram-Positive Organisms. Clin Infect Dis. 2008;46:1683–1693. doi: 10.1086/587896. [DOI] [PubMed] [Google Scholar]
- 187.Allen NE. From Vancomycin to Oritavancin: The Discovery and Development of a Novel Lipoglycopeptide Antibiotic. Anti-Infect Agents Med Chem. 2010;9:23–47. [Google Scholar]
- 188.Sosio M, Stinchi S, Beltrametti F, Lazzarini A, Donadio S. The Gene Cluster for the Biosynthesis of the Glycopeptide Antibiotic A40926 by Nonmuraea Species. Chem Biol. 2003;10:541–549. doi: 10.1016/s1074-5521(03)00120-0. [DOI] [PubMed] [Google Scholar]
- 189.Candiani G, Abbondi M, Borgonovi M, Romanò G, Parenti F. In-Vitro and in-Vivo Antibacterial Activity of BI 397, a New Semi-Synthetic Glycopeptide Antibiotic. J Antimicrob Chemother. 1999;44:179–192. doi: 10.1093/jac/44.2.179. [DOI] [PubMed] [Google Scholar]
- 190.Seltzer E, Dorr MB, Goldstein BP, Perry M, Dowell Ja, Henkel T. Once-Weekly Dalbavancin versus Standard-of-Care Antimicrobial Regimens for Treatment of Skin and Soft-Tissue Infections. Clin Infect Dis. 2003;37:1298–1303. doi: 10.1086/379015. [DOI] [PubMed] [Google Scholar]
- 191.Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-Weekly Dalbavancin versus Daily Conventional Therapy for Skin Infection. N Engl J Med. 2014;370:2169–2179. doi: 10.1056/NEJMoa1310480. [DOI] [PubMed] [Google Scholar]
- 192.Sharman GJ, Try AC, Dancer RJ, Cho YR, Staroske T, Bardsley B, Maguire AJ, Cooper MA, O’Brien DP, Williams DH. The Roles of Dimerization and Membrane Anchoring in Activity of Glycopeptide Antibiotics against Vancomycin-Resistant Bacteria. J Am Chem Soc. 1997;119:12041–12047. [Google Scholar]
- 193.Nicolaou KC, Hughes R, Cho SY, Winssinger N, Smethurst C, Labischinski H, Endermann R. Target-Accelerated Combinatorial Synthesis and Discovery of Highly Potent Antibiotics Effective Against Vancomycin-Resistant Bacteria. Angew Chem, Int Ed. 2000;39:3823–3828. doi: 10.1002/1521-3773(20001103)39:21<3823::AID-ANIE3823>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 194.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Vancomycin Derivatives That Inhibit Peptidoglycan Biosynthesis Without Binding D-Ala-D-Ala. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 195.Williams DH, Bardsley B. The Vancomycin Group of Antibiotics and the Fight against Resistant Bacteria. Angew Chem, Int Ed. 1999;38:1172–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 196.Okano A, Isley NA, Boger DL. Total Syntheses of Vancomycin-Related Glycopeptide Antibiotics and Key Analogues. Chem Rev. 2017 doi: 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Total Syntheses of Vancomcin and Eremomycin Aglycons. Angew Chem, Int Ed. 1998;37:2700–2704. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2700::AID-ANIE2700>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 198.Evans DA, Britton TC, Ellman JA, Dorow RL. The Asymmetric Synthesis of α-Amino Acids. Electrophilic Azidation of Chiral Imide Enolates, a Practical Approach to the Synthesis of (R)- and (S)-α-Azido Carboxylic Acids. J Am Chem Soc. 1990;112:4011–4030. [Google Scholar]
- 199.Evans DA, Evrad DA, Rychnovsky SD, Früh T, Whittingham WG, DeVries KM. A General Approach to the Asymmetric Synthesis of Vancomycin-Related Arylglycines by Enolate Azidation. Tetrahedron Lett. 1992;33:1189–1192. [Google Scholar]
- 200.Evans DA, Ellman JA, DeVries KM. The Oxidative Macrocyclization of Phenolic Peptides. A Biomimetic Approach to the Synthesis of the Vancomycin Family of Antibiotics. J Am Chem Soc. 1989;111:8912–8914. [Google Scholar]
- 201.Evans DA, Dinsmore CJ. Kinetic and Thermodynamic Atropdiastereoselection in the Synthesis of the M(5–7) Tripeptide Portion of Vancomycin. Tetrahedron Lett. 1993;34:6029–6032. [Google Scholar]
- 202.Kolb HC, VanNieuwenhze MS, Sharpless KB. Catalytic Asymmetric Dihydroxylation. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 203.Li G, Chang H, Sharpless KB. Catalytic Asymmetric Aminohydroxylation (AA) of Olefins. Angew Chem, Int Ed Engl. 1996;35:451–454. [Google Scholar]
- 204.Nicolaou KC, Natarajan S, Li H, Jain NF, Hughes R, Solomon ME, Ramanjulu JM, Boddy CNC, Takayanagi M. Total Synthesis of Vancomycin Aglycon-Part 1: Synthesis of Amino Acids 4–7 and Construction of the AB-COD Ring Skeleton. Angew Chem, Int Ed. 1998;37(19):2708–2714. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2708::AID-ANIE2708>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 205.Nicolaou KC, Li H, Boddy CNC, Ramanjulu JM, Yue TY, Natarajan S, Chu XJ, Brase S, Rübsam F. Total Synthesis of Vancomycin-Part 1: Design and Development of Methodology. Chem -Eur J. 1999;5:2584–2601. [Google Scholar]
- 206.Nicolaou KC, Jain NF, Natarajan S, Hughes R, Solomon ME, Li H, Ramanjulu JM, Takayanagi M, Koumbis AE, Bando T. Total Synthesis of Vancomycin Aglycon—Part 2: Synthesis of Amino Acids 1–3 and Construction of the AB-COD-DOE Ring Skeleton. Angew Chem, Int Ed. 1998;37:2714–2716. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2714::AID-ANIE2714>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 207.Nicolaou KC, Takayanagi M, Jain NF, Natarajan S, Koumbis AE, Bando T, Ramanjulu JM. Total Synthesis of Vancomycin Aglycon—Part 3: Final Stages. Angew Chem, Int Ed. 1998;37:2717–2719. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2717::AID-ANIE2717>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 208.Nicolaou KC, Koumbis AE, Takayanagi M, Natarajan S, Jain NF, Bando T, Li H, Hughes R. Total Synthesis of Vancomycin—Part 3: Synthesis of the Aglycon. Chem – Eur J. 1999;5:2622–2647. [Google Scholar]
- 209.Nicolaou KC, Mitchell HJ, Jain NF, Bando T, Hughes R, Winssinger N, Natarajan S, Koumbis AE. Total Synthesis of Vancomycin – Part 4: Attachment of the Sugar Moieties and Completion of the Synthesis. Chem – Eur J. 1999;5:2648–2667. [Google Scholar]
- 210.Gerhard U, Mackay JP, Maplestone RA, Williams DH. The Role of the Sugar and Chlorine Sustituents in the Dimerization of Vancomycin Antibiotics. J Am Chem Soc. 1993;115:232–237. [Google Scholar]
- 211.Nicolaou KC, Cho SY, Hughes R, Winssinger N, Smethurst C, Labischinski H, Endermann R. Solid- and Solution-Phase Synthesis of Vancomycin and Vancomycin Analogues with Activity against Vancomycin-Resistant Bacteria. Chem – Eur J. 2001;7:3798–3823. doi: 10.1002/1521-3765(20010903)7:17<3798::aid-chem3798>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 212.Boger DL, Castle SL, Miyazaki S, Wu JH, Beresis RT, Loiseleur O. Vancomycin CD and DE Macrocyclization and Atropisomerism Studies. J Org Chem. 1999;64:70–80. doi: 10.1021/jo980880o. [DOI] [PubMed] [Google Scholar]
- 213.Boger DL, Borzilleri RM, Nukui S, Beresis RT. Synthesis of the Vancomycin CD and DE Ring Systems. J Org Chem. 1997;62:4721–4736. [Google Scholar]
- 214.Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. Diastereoselective Total Synthesis of the Vancomycin Aglycon with Ordered Atropisomer Equilibrations. J Am Chem Soc. 1999;121:3226–3227. [Google Scholar]
- 215.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. Total Synthesis of the Vancomycin Aglycon. J Am Chem Soc. 1999;121:10004–10011. [Google Scholar]
- 216.Crowley BM, Boger DL. Total Synthesis and Evaluation of [Ψ[CH2NH]Tpg4]Vancomycin Aglycon: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J Am Chem Soc. 2006;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. Total Synthesis of [Ψ[C(=S)NH]Tpg4]-Vancomycin Aglycon, [Ψ[C(= NH)NH]Tpg4]Vancomycin Aglycon, and Related Key Compounds: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J Am Chem Soc. 2012;134:1284–1297. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Okano A, Nakayama A, Schammel AW, Boger DL. Total Synthesis of [Ψ[C(=NH)NH]Tpg4]Vancomycin and Its 4-Chlorobiphenyl)methyl Derivative: Impact of Peripheral Modifications on Vancomycin Analogues Redesigned for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding. J Am Chem Soc. 2014;136:13522–13525. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Okano A, Nakayama A, Wu K, Lindsey EA, Schammel AW, Feng Y, Collins KC, Boger DL. Total Syntheses and Initial Evaluation of [Ψ[C(=S)NH]Tpg4]vancomycin, [Ψ[=NH)NH]Tpg4]-vancomycin, [Ψ[CH2NH]Tpg4]vancomycin, and Their (4-Chlorobiphenyl)methyl Derivatives: Synergistic Binding Pocket and Peripheral Modificat. J Am Chem Soc. 2015;137:3693–3704. doi: 10.1021/jacs.5b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Losey HC, Jiang J, Biggins JB, Oberthur M, Ye XY, Dong SD, Kahne D, Thorson JS, Walsh CT. Incorporation of Glucose Analogs by GtfE and GtfD from the Vancomycin Biosynthetic Pathway to Generate Variant Glycopeptides. Chem Biol. 2002;9:1305–1314. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 221.Okano A, Isley NA, Boger DL. Peripheral Modifications of [Ψ[CH2NH]Tpg4]vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics. Proc Natl Acad Sci U S A. 2017:E5053–E5061. doi: 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nealson KH, Platt T, Hastings JW. Cellular Control of the Synthesis and Activity of the Bacterial Luminescent System. J Bacteriol. 1970;104:313–322. doi: 10.1128/jb.104.1.313-322.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Eberhard A, Burlingame AL, Eberhard C, Kenyon GL, Nealson KH, Oppenheimer NJ. Structural Identification of Autoinducer of Photobacterium Fischeri Luciferase. Biochemistry. 1981;20:2444–2449. doi: 10.1021/bi00512a013. [DOI] [PubMed] [Google Scholar]
- 224.Fuqua WC, Winans SC, Greenberg EP. Quorum Sensing in Bacteria: The LuxR-LuxI Family of Cell Density-Responsive Transcriptional Regulators. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fuqua C, Winans SC, Greenberg EP. CENSUS AND CONSENSUS IN BACTERIAL ECOSYSTEMS: The LuxR-LuxI Family of Quorum-Sensing Transcriptional Regulators. Annu Rev Microbiol. 1996;50:727–751. doi: 10.1146/annurev.micro.50.1.727. [DOI] [PubMed] [Google Scholar]
- 226.Surette MG, Miller MB, Bassler BL. Quorum Sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: A New Family of Genes Responsible for Autoinducer Production. Proc Natl Acad Sci U S A. 1999;96:1639–1644. doi: 10.1073/pnas.96.4.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bassler BL. How Bacteria Talk to Each Other: Regulation of Gene Expression by Quorum Sensing. Curr Opin Microbiol. 1999;2:582–587. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 228.Bassler BL, Losick R. Bacterially Speaking. Cell. 2006;125:237–246. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 229.Waters CM, Bassler BL. Quorum Sensing: Communication in Bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 230.Bassler BL. Small Talk: Cell-to-Cell Communication in Bacteria. Cell. 2002;109:421–424. doi: 10.1016/s0092-8674(02)00749-3. [DOI] [PubMed] [Google Scholar]
- 231.Diggle SP, Matthijs S, Wright VJ, Fletcher MP, Chhabra SR, Lamont IL, Kong X, Hider RC, Cornelis P, Cámara M, et al. The Pseudomonas aeruginosa 4-Quinolone Signal Molecules HHQ and PQS Play Multifunctional Roles in Quorum Sensing and Iron Entrapment. Chem Biol. 2007;14:87–96. doi: 10.1016/j.chembiol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 232.Withers H, Swift S, Williams P. Quorum Sensing as an Integral Component of Gene Regulatory Networks in Gram-Negative Bacteria. Curr Opin Microbiol. 2001;4:186–193. doi: 10.1016/s1369-5274(00)00187-9. [DOI] [PubMed] [Google Scholar]
- 233.Wilson JW, Schurr MJ, LeBlanc CL, Ramamurthy R, Buchanan KL, Nickerson CA. Mechanisms of Bacterial Pathogenicity. Postgrad Med J. 2002;78:216–224. doi: 10.1136/pmj.78.918.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Winzer K, Williams P. Quorum Sensing and the Regulation of Virulence Gene Expression in Pathogenic Bacteria. Int J Med Microbiol. 2001;291:131–143. doi: 10.1078/1438-4221-00110. [DOI] [PubMed] [Google Scholar]
- 235.Cámara M, Williams P, Hardman A. Controlling Infection by Tuning in and Turning down the Volume of Bacterial Small-Talk. Lancet Infect Dis. 2002;2:667–676. doi: 10.1016/s1473-3099(02)00447-4. [DOI] [PubMed] [Google Scholar]
- 236.Geske GD, O’Neill JC, Blackwell HE. Expanding Dialogues: From Natural Autoinducers to Non-Natural Analogues That Modulate Quorum Sensing in Gram-Negative Bacteria. Chem Soc Rev. 2008;37:1432–1447. doi: 10.1039/b703021p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Praneenararat T, Palmer AG, Blackwell HE. Chemical Methods to Interrogate Bacterial Quorum Sensing Pathways. Org Biomol Chem. 2012;10:8189–8199. doi: 10.1039/c2ob26353j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Quorum Sensing in Gram-Negative Bacteria: Small-Molecule Modulation of AHL and AI-2 Quorum Sensing Pathways. Chem Rev. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- 239.Stevens AM, Queneau Y, Soulère L, Bodman SVon, Doutheau A. Mechanisms and Synthetic Modulators of AHL-Dependent Gene Regulation. Chem Rev. 2011;111:4–27. doi: 10.1021/cr100064s. [DOI] [PubMed] [Google Scholar]
- 240.Welsh MA, Eibergen NR, Moore JD, Blackwell HE. Small Molecule Disruption of Quorum Sensing Cross-Regulation in Pseudomonas aeruginosa Causes Major and Unexpected Alterations to Virulence Phenotypes. J Am Chem Soc. 2015;137:1510–1519. doi: 10.1021/ja5110798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Stacy DM, Welsh Ma, Rather PN, Blackwell HE. Attenuation of Quorum Sensing in the Pathogen Acinetobacter baumannii Using Non-Native N-Acyl Homoserine Lactonres. ACS Chem Biol. 2012;7:1719–1728. doi: 10.1021/cb300351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Huse H, Whiteley M. 4-Quinolones: Smart Phones of the Microbial World. Chem Rev. 2011;111:152–159. doi: 10.1021/cr100063u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Williams P, Cámara M. Quorum Sensing and Environmental Adaptation in Pseudomonas aeruginosa: A Tale of Regulatory Networks and Multifunctional Signal Molecules. Curr Opin Microbiol. 2009;12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 244.Borrero NV, Bai F, Perez C, Duong BQ, Rocca JR, Jin S, Huigens RW. Phenazine Antibiotic Inspired Discovery of Potent Bromophenazine Antibacterial Agents against Staphylococcus aureus and Staphylococcus epidermidis. Org Biomol Chem. 2014;12:881–886. doi: 10.1039/c3ob42416b. [DOI] [PubMed] [Google Scholar]
- 245.Garrison AT, Abouelhassan Y, Norwood VM, Kallifidas D, Bai F, Nguyen MT, Rolfe M, Burch GM, Jin S, Luesch H, et al. Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines That Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis. J Med Chem. 2016;59:3808–3825. doi: 10.1021/acs.jmedchem.5b02004. [DOI] [PubMed] [Google Scholar]
- 246.Abouelhassan Y, Garrison AT, Burch GM, Wong W, Norwood VM, Huigens RW. Discovery of Quinoline Small Molecules with Potent Dispersal Activity against Methicillin-Resistant Staphylococcus aureus and Staphylococcus epidermidis Biofilms Using a Scaffold Hopping Strategy. Bioorg Med Chem Lett. 2014;24:5076–5080. doi: 10.1016/j.bmcl.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 247.Garrison AT, Abouelhassan Y, Yang H, Yousaf HH, Nguyen TJ, Huigens RW., III Microwave-Enhanced Friedländer Synthesis for the Rapid Assembly of Halogenated Quinolines with Antibacterial and Biofilm Eradication Activities against Drug Resistant and Tolerant Bacteria. MedChemComm. 2017;8:720–724. doi: 10.1039/c6md00381h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Peptide Signaling in the Staphylococci. Chem Rev. 2011;111:117–151. doi: 10.1021/cr100370n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Novick RP, Geisinger E. Quorum Sensing in Staphylococci. Annu Rev Genet. 2008;42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- 250.Gripenland J, Netterling S, Loh E, Tiensuu T, Toledo-Arana A, Johansson J. RNAs: Regulators of Bacterial Virulence. Nat Rev Microbiol. 2010;8:857–866. doi: 10.1038/nrmicro2457. [DOI] [PubMed] [Google Scholar]
- 251.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW. Structure-Activity Analysis of Synthetic Autoinducing Thiolactone Peptides from Staphylococcus aureus Responsible for Virulence. Proc Natl Acad Sci U S A. 1999;96:1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wright JS, Lyon GJ, George Ea, Muir TW, Novick RP. Hydrophobic Interactions Drive Ligand-Receptor Recognition for Activation and Inhibition of Staphylococcal Quorum Sensing. Proc Natl Acad Sci U S A. 2004;101:16168–16173. doi: 10.1073/pnas.0404039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.George EA, Muir TW. Molecular Mechanisms of Agr Quorum Sensing in Virulent. ChemBioChem. 2007;8:847–855. doi: 10.1002/cbic.200700023. [DOI] [PubMed] [Google Scholar]
- 254.Wang B, Muir TW. Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles. Cell Chem Biol. 2016;23:214–224. doi: 10.1016/j.chembiol.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. Highly Potent Inhibitors of Quorum Sensing in Staphylococcus aureus Revealed Through a Systematic Synthetic Study of the Group-III Autoinducing Peptide. J Am Chem Soc. 2013;135:7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]
- 256.Tal-Gan Y, Ivancic M, Cornilescu G, Cornilescu CC, Blackwell HE. Structural Characterization of Native Autoinducing Peptides and Abiotic Analogues Reveals Key Features Essential for Activation and Inhibition of an AgrC Quorum Sensing Receptor in Staphylococcus aureus. J Am Chem Soc. 2013;135:18436–18444. doi: 10.1021/ja407533e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Tal-Gan Y, Ivancic M, Cornilescu G, Yang T, Blackwell HE. Highly Stable, Amide-Bridged Autoinducing Peptide Analogues That Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus. Angew Chem, Int Ed. 2016;55:8913–8917. doi: 10.1002/anie.201602974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Vasquez JK, Tal-Gan Y, Cornilescu G, Tyler KA, Blackwell HE. Simplified AIP-II Peptidomimetics Are Potent Inhibitors of Staphylococcus aureus AgrC Quorum Sensing Receptors. ChemBioChem. 2017;18:413–423. doi: 10.1002/cbic.201600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yang T, Tal-Gan Y, Paharik AE, Horswill AR, Blackwell HE. Structure-Function Analyses of a Staphylococcus epidermidis Autoinducing Peptide Reveals Motifs Critical for AgrC-Type Receptor Modulation. ACS Chem Biol. 2016;11:1982–1991. doi: 10.1021/acschembio.6b00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Blaser MJ. Antibiotic Use and Its Consequences for the Normal Microbiome. Science. 2016;352:544–545. doi: 10.1126/science.aad9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Cho I, Blaser MJ. The Human Microbiome: At the Interface of Health and Disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Blaser M. Stop the Killing of Beneficial Bacteria. Nature. 2011;476:393–394. doi: 10.1038/476393a. [DOI] [PubMed] [Google Scholar]
- 263.Hicks LA, Blaser MJ. Variability in Antibiotic Prescribing: An Inconvenient Truth. J Pediatric Infect Dis Soc. 2015;4:e136–e138. doi: 10.1093/jpids/piu106. [DOI] [PubMed] [Google Scholar]
- 264.Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, et al. Altering the Intestinal Microbiota during a Critical Developmental Window Has Lasting Metabolic Consequences. Cell. 2014;158:705–721. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Hill C, Jain A, Takemoto H, Silver MD, Nagesh SVS, Ionita CN, Bednarek DR, Rudin S. Antibiotics in Early Life and Obesity. Nat Rev Endocrinol. 2015;73:389–400. [Google Scholar]
- 266.Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, A DL, Wu F, Perez-Perez GI, Chen Y, et al. Antibiotics, Birth Mode, and Diet Shape Microbiome Maturation during Early Life. Sci Transl Med. 2016;8:1–13. doi: 10.1126/scitranslmed.aad7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Parenti F, Pagani H, Beretta G. Lipiarmycin, a New Antibiotic from Actinoplanes I. Description of the Producer Strain and Fermentation Studies. J Antibiot. 1975;28:247–252. doi: 10.7164/antibiotics.28.247. [DOI] [PubMed] [Google Scholar]
- 268.Coronelli C, White RJ, Lancini GC, Parenti F. Lipiarmycin, a New Antibiotic from Actinoplanes. II. Isolation, Chemical, Biological and Biochemical Characterization. J Antibiot. 1975;28:253–259. doi: 10.7164/antibiotics.28.253. [DOI] [PubMed] [Google Scholar]
- 269.Sergio S, Pirali G, White R, Parenti F. Lipiarmycin, a New Antibiotic from Actinoplanes. III. Mechanism of Action. J Antibiot. 1975;28:543–549. doi: 10.7164/antibiotics.28.543. [DOI] [PubMed] [Google Scholar]
- 270.Cavalleri B, Arnone A, Di Modugno E, Nasini G, Goldstein BP. Structure and Biological Activity of Lipiarmycin B. J Antibiot. 1988;41:308–315. doi: 10.7164/antibiotics.41.308. [DOI] [PubMed] [Google Scholar]
- 271.Hochlowski JE, Swanson SJ, Ranfranz LM, Whittern DN, Buko AM, McAlpine JB. Tiacumicins, a Novel Complex of 18-Membered Macrolides. II. Isolation and Structure Determination. J Antibiot. 1987;40:575–588. doi: 10.7164/antibiotics.40.575. [DOI] [PubMed] [Google Scholar]
- 272.Swanson RN, Hardy DJ, Shipkowitz NL, Hanson CW, Ramer NC, Fernandes PB, Clement JJ. In Vitro and In Vivo Evaluation of Tiacumicins B and C against Clostridium difficile. Antimicrob Agents Chemother. 1991;35:1108–1111. doi: 10.1128/aac.35.6.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cornely OA, Crook DW, Esposito R, Poirier A, Somero MS, Weiss K, Sears P, Gorbach S. Fidaxomicin versus Vancomycin for Infection with Clostridium difficile in Europe, Canada, and the USA: A Double-Blind, Non-Inferiority, Randomised Controlled Trial. Lancet Infect Dis. 2012;12:281–289. doi: 10.1016/S1473-3099(11)70374-7. [DOI] [PubMed] [Google Scholar]
- 274.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, Gorbach S, Sears P, Shue Y-K. Fidaxomicin versus Vancomycin for Clostridium difficile Infection. N Engl J Med. 2011;364:422–431. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 275.Erb W, Grassot JM, Linder D, Neuville L, Zhu J. Enantioselective Synthesis of Putative Lipiarmycin Aglycon Related to Fidaxomicin/tiacumicin B. Angew Chem, Int Ed. 2015;54:1929–1932. doi: 10.1002/anie.201409475. [DOI] [PubMed] [Google Scholar]
- 276.Glaus F, Altmann KH. Total Synthesis of the Tiacumicin B (Lipiarmycin A3/fidaxomicin) Aglycone. Angew Chem, Int Ed. 2015;54:1937–1940. doi: 10.1002/anie.201409510. [DOI] [PubMed] [Google Scholar]
- 277.Miyatake-Ondozabal H, Kaufmann E, Gademann K. Total Synthesis of the Protected Aglycon of Fidaxomicin (Tiacumicin B, Lipiarmycin A3) Angew Chem, Int Ed. 2015;54:1933–1936. doi: 10.1002/anie.201409464. [DOI] [PubMed] [Google Scholar]
- 278.Kaufmann E, Hattori H, Miyatake-Ondozabal H, Gademann K. Total Synthesis of the Glycosylated Macrolide Antibiotic Fidaxomicin. Org Lett. 2015;17:3514–3517. doi: 10.1021/acs.orglett.5b01602. [DOI] [PubMed] [Google Scholar]
- 279.Xiao Y, Li S, Niu S, Ma L, Zhang G, Zhang H, Zhang G, Ju J, Zhang C. Characterization of Tiacumicin B Biosynthetic Gene Cluster Affording Diversified Tiacumicin Analogues and Revealing a Tailoring Dihalogenase. J Am Chem Soc. 2011;133:1092–1105. doi: 10.1021/ja109445q. [DOI] [PubMed] [Google Scholar]
- 280.Li W, Estrada-de los Santos P, Matthijs S, Xie G-L, Busson R, Cornelis P, Rozenski J, De Mot R. Promysalin, a Salicylate-Containing Pseudomonas putida Antibiotic, Promotes Surface Colonization and Selectively Targets Other Pseudomonas. Chem Biol. 2011;18:1320–1330. doi: 10.1016/j.chembiol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 281.Steele AD, Knouse KW, Keohane CE, Wuest WM. Total Synthesis and Biological Investigation of (−)-Promysalin. J Am Chem Soc. 2015;137:7314–7317. doi: 10.1021/jacs.5b04767. [DOI] [PubMed] [Google Scholar]
- 282.Kaduskar RD, Dhavan AA, Dallavalle S, Scaglioni L, Musso L. Total Synthesis of the Salicyldehydroproline-Containing Antibiotic Promysalin. Tetrahedron. 2016;72:2034–2041. [Google Scholar]
- 283.Steele AD, Keohane CE, Knouse KW, Rossiter SE, Williams SJ, Wuest WM. Diverted Total Synthesis of Promysalin Analogs Demonstrates That an Iron-Binding Motif Is Responsible for Its Narrow-Spectrum Antibacterial Activity. J Am Chem Soc. 2016;138:5833–5836. doi: 10.1021/jacs.6b03373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Knouse KW, Wuest WM. The Enantioselective Synthesis and Biological Evaluation of Chimeric Promysalin Analogs Facilitated by Diverted Total Synthesis. J Antibiot. 2016;69:337–339. doi: 10.1038/ja.2016.4. [DOI] [PubMed] [Google Scholar]
- 285.Jansen R, Irschik H, Huch V, Schummer D, Steinmetz H, Bock M, Schmidt T, Kirschning A, Müller R. Carolacton—A Macrolide Ketocarbonic Acid That Reduces Biofilm Formation by the Caries- and Endocarditis-Associated Bacterium Streptococcus mutans. Eur J Org Chem. 2010;2010:1284–1289. [Google Scholar]
- 286.Reck M, Rutz K, Kunze B, Tomasch J, Surapaneni SK, Schulz S, Wagner-Döbler I. The Biofilm Inhibitor Carolacton Disturbs Membrane Integrity and Cell Division of Streptococcus mutans through the Serine/Threonine Protein Kinase PknB. J Bacteriol. 2011;193:5692–5706. doi: 10.1128/JB.05424-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Donner J, Reck M, Bergmann S, Kirschning A, Müller R, Wagner-Döbler I. The Biofilm Inhibitor Carolacton Inhibits Planktonic Growth of Virulent Pneumococci via a Conserved Target. Sci Rep. 2016;6:29677. doi: 10.1038/srep29677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Schmidt T, Kirschning A. Total Synthesis of Carolacton, a Highly Potent Biofilm Inhibitor. Angew Chem, Int Ed. 2012;51:1063–1066. doi: 10.1002/anie.201106762. [DOI] [PubMed] [Google Scholar]
- 289.Stumpp N, Premnath P, Schmidt T, Ammermann J, Dräger G, Reck M, Jansen R, Stiesch M, Wagner-Döbler I, Kirschning A. Synthesis of New Carolacton Derivatives and their Activity Against Biofilms of Oral Bacteria. Org Biomol Chem. 2015;13:5765–5774. doi: 10.1039/c5ob00460h. [DOI] [PubMed] [Google Scholar]
- 290.Hallside MS, Brzozowski RS, Wuest WM, Phillips AJ. A Concise Synthesis of Carolacton. Org Lett. 2014;16:1148–1151. doi: 10.1021/ol500004k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Solinski AE, Koval AB, Brzozowski RS, Morrison KR, Carson CE, Eshraghi AR, Zhou G, Quivey RG, Voelz VA, Buttaro BA, et al. The Diverted Total Synthesis of Carolacton-Inspired Analogs Yields Three Distinct Phenotypes in Streptococcus mutans Biofilms. J Am Chem Soc. 2017;139:7188–7191. doi: 10.1021/jacs.7b03879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kuilya TK, Goswami RK. Stereoselective Total Synthesis of Carolacton. Org Lett. 2017;19:2366–2369. doi: 10.1021/acs.orglett.7b00903. [DOI] [PubMed] [Google Scholar]
- 293.Huigens RW, III, Ma L, Gambino C, Moeller PDR, Basso A, Cavanagh J, Wozniak DJ, Melander C. Control of Bacterial Biofilms with Marine Alkaloid Derivatives. Mol BioSyst. 2008;4:614–621. doi: 10.1039/b719989a. [DOI] [PubMed] [Google Scholar]
- 294.Huigens RW, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R, Melander C. Inhibition of Pseudomonas aeruginosa Biofilm Formation with Bromoageliferin Analogues. J Am Chem Soc. 2007;129:6966–6967. doi: 10.1021/ja069017t. [DOI] [PubMed] [Google Scholar]
- 295.Richards JJ, Ballard TE, Huigens RW, Melander C. Synthesis and Screening of an Oroidin Library against Pseudomonas aeruginosa Biofilms. ChemBioChem. 2008;9:1267–1279. doi: 10.1002/cbic.200700774. [DOI] [PubMed] [Google Scholar]
- 296.Lindsey EA, Brackett CM, Mullikin T, Alcaraz C, Melander C. The Discovery of N-1 Substituted 2-Aminobenzimida-zoles as Zinc-Dependent S. Aureus Biofilm Inhibitors. MedChemComm. 2012;3:1462. doi: 10.1039/C2MD20244A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Nguyen TV, Blackledge MS, Lindsey EA, Minrovic BM, Ackart DF, Jeon AB, Obregon-Henao A, Melander RJ, Basaraba RJ, Melander C. The Discovery of 2-Aminobenzimidazoles That Sensitize Mycobacterium smegmatis and M tuberculosis to β-Lactam Antibiotics in a Pattern Distinct from SS-Lactamase Inhibitors. Angew Chem, Int Ed. 2017;56:3940–3944. doi: 10.1002/anie.201612006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]