

Abstract
Membrane proteins perform a host of vital cellular functions. Deciphering the molecular mechanisms whereby they fulfill these functions requires detailed biophysical and structural investigations. Detergents have proven pivotal to extract the protein from its native surroundings. Yet, they provide a milieu that departs significantly from that of the biological membrane, to the extent that the structure, the dynamics, and the interactions of membrane proteins in detergents may considerably vary, as compared to the native environment. Understanding the impact of detergents on membrane proteins is, therefore, crucial to assess the biological relevance of results obtained in detergents. Here, we review the strengths and weaknesses of alkyl phosphocholines (or foscholines), the most widely used detergent in solution-NMR studies of membrane proteins. While this class of detergents is often successful for membrane protein solubilization, a growing list of examples points to destabilizing and denaturing properties, in particular for α-helical membrane proteins. Our comprehensive analysis stresses the importance of stringent controls when working with this class of detergents and when analyzing the structure and dynamics of membrane proteins in alkyl phosphocholine detergents.
1. Introduction
Membrane proteins (MPs) are the gateways to the cell and to cellular compartments. In combination with their sophisticated environment, they perform a vast array of functions, such as signal transduction, transport of metabolites, or energy conversion.1 A significant portion of genomes, in humans about 15–25%, encodes for MPs, and MPs are the targets of the majority of drugs.2 Despite their number and importance for cellular processes, MPs are less well characterized than their soluble counterparts. The major bottleneck to studying MPs comes from the strong dependency of MP structure and stability on their lipid bilayer environment. Even though considerable technical progress has been made over the last years,3 the need to generate diffracting crystals from proteins reconstituted in detergent or lipidic cubic phase (LCP) for X-ray crystallography is still a major obstacle; often only ligand-inhibited states or mutants can be successfully crystallized, which limits the insight into the functional mechanisms. For solution-state NMR spectroscopy, the two-dimensional lipid bilayer generally needs to be abandoned to generate soluble particles, which also results in practical difficulties.4,5 Cryo-electron microscopy (cryoEM) can solve structures in situ by tomography,6 but for most applications MPs need to be solubilized and purified for electron crystallography of two-dimensional crystals or for imaging as single particles in nanodiscs or micelles.7 For solid-state NMR, the preparation of samples and the observation of high-resolution spectra for structural characterization remain difficult.3,8,9 Although this latter technology can characterize structure, interactions, and dynamics in lipid bilayers, all of the ex situ environments for MPs including lipid bilayers used by these technologies are membrane mimetics, while the native membrane is much more complex. Solid-state NMR spectroscopy of MPs in their native membrane environment is, in principle, possible,10−12 but suffers from limitations in resolution and sensitivity. Combined in situ solid-state NMR and electron cryotomography is being developed for integrative studies of atomic-level MP structure and dynamics in the context of the native membrane.13
Biochemical and biophysical studies of MPs require various preparative steps, such as extraction from native membranes, purification, and final reconstitution in a suitable membrane-mimicking environment, before they are subject to actual biophysical analyses. In a few cases, proteins are refolded from inclusion bodies. In the early days, detergents were the main molecules used to extract and stabilize MPs in a soluble form for functional, biophysical, and structural studies.14,15 In the past decade, different technologies have been proposed and are actively being developed for all of these steps, from extraction to final study, such as polymer-based native nanodiscs,16−19 nanolipoprotein particles (i.e., membrane-scaffold protein-based nanodiscs),20−24 bicelles,25−27 amphipols,28,29 fluorinated surfactants,30 lipidic cubic phase for crystallization,31 as well as crystallization from nanodiscs.32 Notwithstanding the range of different tools available, detergents remain to date by far the most commonly employed route for extraction, purification, and biophysical studies in solution or by crystallography. This importance is highlighted by the fact that from the 672 unique MP structures to date,33 about 80% have been obtained with detergents, either in solution by NMR, through electron microscopy, or by crystallization of detergent-solubilized protein (see statistics discussed further below). A large variety of detergents have been developed, and Figure 1 shows the chemical structures of some of the most frequently used ones.
Figure 1.

Chemical structures of some commonly used detergents: SDS, sodium dodecyl sulfate; LDAO, lauryldimethylamine N-oxide; LAPAO, 3-laurylamido-N,N″-dimethylpropylaminoxide; DPC, dodecylphosphocholine, also called Foscholine-12 (FC12); C8E4, tetraethylene glycol monooctyl ether; β-OG, β-octyl glucoside; DDM, dodecyl maltoside; 12MNG, 12-maltose neopentyl glycol, also called lauryl maltose neopentyl glycol, LMNG; and DHPC, 1,2-diheptanoyl-sn-glycero-3-phosphocholine. The focus of this Review is on the family of alkyl phosphocholine detergents, such as DPC. A list of further detergents and their chemical structures is shown in Table S1.
Detergents with specific and well-defined properties, suitable for crystallization, have been developed in the 1980s, in particular in the laboratory of J. Rosenbusch where the first well-diffracting crystal of a MP was obtained.34 In these early days, only proteins that are abundant in native membranes were studied. Therefore, a high solubilization yield was not necessarily a requirement, but conformational stability was mandatory to succeed in crystallization. This requirement restricted the nature of detergents to a limited number of classes. Despite the widespread use and frequent success of detergents for preparing and studying MPs, the properties of detergent micelles are significantly different from those of lipid bilayers, as discussed below, and the interactions that MPs form with these different surroundings also differ. This was the motivation for new developments such as the crystallization in lipidic cubic phase,35 which forms a three-dimensional bilayer matrix. The structure and dynamics of proteins result from a subtle balance of numerous weak interactions, and an altered environment is expected to induce structural changes. How exactly MP structures in detergents differ from those in lipid bilayers has been subject to debate and controversy for a long time. After several decades of structural biology with detergents, common trends can be identified.
The focus of this Review is on a particular class of detergents, termed alkyl phosphocholines. Throughout this Review, we will use the term alkyl phosphocholine for this group of detergents, or the appropriate names to refer to different alkyl chain lengths with 10 (decyl phosphocholine), 12 (dodecyl phosphocholine, abbreviated as DPC), 14 (tetradecyl phosphocholine), and 16 (hexadecyl phosphocholine) carbons. These are also known under their commercial name foscholine (FC), including FC10, FC12, FC14, and FC16. Forty years after the first applications of alkyl phosphocholine detergents in structural biology,36 a large number of MPs have been studied in these micelles. From the sheer statistics, alkyl phosphocholines have turned out to be very successful, particularly in solution-state NMR spectroscopy. Figure 2 shows the relative contributions of different techniques to solving MP structures, and the surfactants that have been used to determine these structures. Dodecyl phosphocholine has been used to obtain ca. 40% of the MP structures determined by solution-state NMR, making it the most frequently used detergent for this technique. Remarkably, however, it has been successful in generating only <1% of the MP structures determined by crystallography. The requirements for solution-state NMR and crystallography are quite different. For the former, the primary criterion for selecting a particular detergent is the solubility of the protein, and high resolution of the resulting NMR spectra. For the latter, restricting the conformational space in solution is important for crystallization. Highly flexible proteins may be very favorable for solution-state NMR and result in well-resolved spectra; yet, they likely will not crystallize. The strong bias toward alkyl phosphocholine in solution-state NMR and against this class of detergents in crystallography might possibly indicate some bias toward more dynamic proteins being studied by solution-state NMR, or it may suggest that DPC interferes with crystallization.
Figure 2.

Statistics on the use of membrane-mimicking environments for determining structures of MPs. (a) Surfactants used to determine MP crystal structures.37 (b) Surfactants used to determine structures of MPs from electron microscopy. (c) Surfactants used for solution-state NMR structures. These structures contain all integral MPs, peripheral MPs, and short membrane-inserted peptides, as compiled by Dror Warschawski38 and Stephen White.33 Besides a number of detergents, this list also contains structure solved in chloroform or DMSO (primarily of short peptides), isotropic bicelles (mostly formed by DHPC/DMPC), as well as one entry for a nanodisc-embedded protein. Panel (d) shows that in solution-state NMR the contribution of dodecyl phosphocholine (DPC) is about 40%, irrespective of whether the proteins are integral MPs, short peptides, β-barrels, or α-helical proteins. (Fluorinated alkyl phosphocholine in panel (b) is abbreviated as APC.)
In any study of MPs in artificial lipid-mimicking environments, one needs to address the question of the biological relevance of the sample. Are MPs in alkyl phosphocholine detergents in a conformation that resembles their state in a native membrane, or, conversely, do these detergents introduce systematic structural perturbations? Are MPs functional in alkyl phosphocholine detergents, and how do different detergents compare in this respect? Answering these questions in general terms is difficult, because MPs vastly differ in their topology (α-helical, β-barrel), size, and complexity. Nonetheless, from the large body of data collected over the last four decades, general trends emerge regarding the performance of this widely used class of detergents. The aim of this Review is to provide an overview of the properties, strengths, and weaknesses of alkyl phosphocholine detergents for MP studies. This Review is organized as follows. We first recapitulate the properties of lipid bilayer membranes and their interactions with MPs. We then discuss how detergents differ from lipids, and how the MP interactions are thereby altered. In section 3, we focus on available data for the functionality of MPs in alkyl phosphocholine detergents. Section 4 discusses in detail a number of examples of experimental studies of α-helical and β-barrel MPs and reveals how alkyl phosphocholines retain or distort the native structure, interactions, and dynamics. Section 5 discusses how molecular dynamics (MD) simulations contribute to our understanding of MP structure and dynamics, with a particular focus on effects of the membrane-mimicking environment. The general trends that are identified from this extensive literature survey are then summarized in section 6, and recommendations for useful and necessary control experiments are provided. We want to draw the reader’s attention also to existing reviews on the topics of detergents14,15,39−44 and the use of solution-NMR in MP studies.4,45,46
2. Membrane Protein Structure in Native and Artificial Environments
Protein structure is the result of molecular interactions within the protein and between the protein and its environment.47 However, getting a molecular description of MPs in their natural environment is a difficult task due to the heterogeneity of the environment.
Most MP purification protocols involve the solubilization of MPs from cellular membranes using a variety of detergents. Because detergent micelles form small molecular weight aggregates with MPs, they appear to be a good way for solution NMR spectroscopists to characterize MPs. LCPs were developed to reintroduce MPs into a lipidic bilayer during the crystallization process.35
The native environment for MPs is very heterogeneous ranging from the bulk aqueous environment through the membrane interfacial region to the very hydrophobic core of the cellular membrane. A detergent micelle provides a similar range of environments, and consequently it was not unreasonable to think that such detergent environments would be good models of a membrane environment as demonstrated with the first structures obtained by X-ray crystallography.48 Here, we will look carefully at the physical properties of a membrane and those properties provided by detergent micelles. In addition, an effort will be made to correlate the structural features observed for MPs in membrane mimetic environments with properties of those environments and also to attempt identification of key membrane environmental features that are important for stabilizing the native structure and dynamics of MPs.
Cellular membranes are indeed very heterogeneous, hosting many different proteins and many different lipids. Furthermore, the lipids are distributed asymmetrically between the two leaflets of the membrane. While a lot is known about the properties of the membrane interstices for transmembrane (TM) domains and a lot is known about the aqueous environment for water-soluble domains of MPs, much less is known about the bilayer interfacial region for the juxtamembrane domains of MPs where the heterogeneity and gradients in physical properties are very large.
Two classes of MPs are discussed here, α-helical proteins with either one TM helix or a bundle of helices, and β-barrels. Typically, TM helix proteins and β-barrel proteins have a fully hydrogen-bonded network of amide backbone sites. For the α-helix, there is i to i + 4 hydrogen bonding within each helix, and for β-barrel structures, the β-strands are completely hydrogen bonded between strands, such that the amide backbone, which dictates the secondary structure of these proteins and the tertiary structure of β-barrel proteins, is well-defined. This hydrogen bonding is assured by the low dielectric environment of the membrane interstices, where the strength of the hydrogen bonds is increased. In addition to the low dielectricity of the membrane interior, the lack of potentially competing hydrogen-bond donors and acceptors (i.e., water molecules) is another important factor that contributes to the effective/net strength of intraprotein hydrogen bonds.
For β-barrel proteins, an aqueous pore lined with hydrophilic side chains from the β-strand provides a dramatic dielectric gradient across the β-barrel from its interior to the interstices of the lipid environment. For both β-barrel and multihelix MPs, the tertiary structure can be sensitive to the membrane and membrane mimetic environment. For β-barrels, the shape of the pore, which appears to vary among structural characterizations, may reflect subtle differences in the membrane mimetic environment. For helical MPs, there is only rare hydrogen bonding between helices, and, therefore, the tertiary structure is sensitive to subtle changes in the protein’s environment. Like β-barrels, helical MPs may also have an aqueous pore, but only a portion of the helical backbone or other backbone structure, as in the selectivity filter of K+ channels, will have any significant exposure to the aqueous environment.49,50 In the early days of MP structural characterization, helical MPs were described as inside out as compared to water-soluble proteins51 with hydrophobic residues on the outside and hydrophilic residues on the interior contributing electrostatic interactions between helices. Later, a rule of thumb was that MP interiors were similar to the protein interior of water-soluble proteins,52 even though this appears to be an exaggeration of the electrophilicity of the MP interior. A recent study has shown that for helical MPs the hydrophilic amino acid composition is significantly less than for the typical water-soluble protein interior.53 It is reasonable to think that this may be necessary to avoid misfolding. Because hydrogen bonding is stronger in the membrane interstices,54 it would be important not to form incorrect hydrogen bonds or other strong electrostatic interactions as there is little, if any, catalyst (i.e., water) to rearrange the hydrogen bonding or electrostatic partners.55,56 Consequently, the interactions between TM helices are often weak, based largely on van der Waals interactions implying that the tertiary structure is stable only in the very low dielectric environment provided by the native membrane environment, whereas the hydrogen bonding that stabilizes β-barrel tertiary structure is not so easily disrupted. The structural situation in the interfacial region is different. Here, the dielectric constant is particularly large, as a result of the high density of charged groups. Consequently, the electrostatic interactions are even weaker than they are in a purely aqueous environment.57,58 For sure, this juxtamembrane region of MPs is where we know the least about the protein structure. It is also where the membrane mimetic environments for all of the structural technologies are the weakest. The two membrane surfaces of a plasma membrane have very different headgroup compositions, while the hydrocarbon interiors of the two leaflets are quite similar. Unfortunately, at this time debates still flourish about raft-like domains, further complicating our understanding of the interfacial region. Even characterizing the membrane interior remains an active arena for science. Below, we provide a summary of the model membrane mimetic environments used in structural studies of MPs including detergent micelles and lipid bilayers, and how the properties of native membranes may differ from these membrane mimetics.
2.1. Bilayer Properties
Both X-ray and neutron scattering technologies have been used to characterize liquid crystalline lipid bilayers, providing a glimpse into the heterogeneity of the physical properties of these environments.59 These environments are composed of two amphipathic monolayers with a mix of fatty acyl chains and sometimes sterols contributing to the hydrophobic interstices. The interfacial region between the aqueous environment and the hydrophobic interior is largely composed of phosphatidyl glycerols, although sterols and sphingomyelins contribute in many membranes. The two monolayers, as previously mentioned, have different compositions so the membranes are asymmetric. For their functional activities, most trans-membrane proteins exist in a unique orientation across their membrane environment, although a few dual-topology MPs were described.60 In addition to differing lipid compositions, membranes also have unique chemical and electrical potentials across the bilayer, resulting in unique environments for the aqueous portions of the protein on either side of the membrane.
While the hydrophobic interstices of membranes can vary in thickness as a result of varying fatty acyl chain composition, all membrane interiors have a very low dielectric constant that represents a barrier for the transit of hydrophilic compounds (see Figure 3). Because water is at a concentration of 55 molar, it is a bit of an exception in that it can pass across the cell membranes, albeit at such a low frequency that cells require aquaporins to transport significant quantities of water. The detailed mechanism by which water can pass through lipid bilayers is still debated. The result is that there is a water concentration gradient of many orders of magnitude between the membrane interior and membrane surfaces (see Figure 3). Some insight can be gained by considering the dielectric constant in the aqueous, membrane, and interfacial region. We note here, however, that the precise values of the dielectric constants are somewhat controversial, and the very concept of a dielectric constant is macroscopic in nature and has limited applicability at the molecular and submolecular levels. However, the trends help to rationalize some general properties, and we make use of available values below. The computational estimate of the dielectric constant in the interior of membranes is 1 over a broad span of ±12 Å from the bilayer center of 1-palmitoyl-2-oleoly-sn-glycero-3-phosphocholine (POPC) bilayers.57 Even if it is 2, this is a very low dielectric constant as compared to 80 for water, which dramatically alters the potential or energy associated with electrostatic interactions, because they are scaled by the inverse of the dielectric constant. Consequently, the energy associated with a hydrogen bond in the interstices of a lipid bilayer is going to be significantly strengthened by the dielectric constant of this medium. This has been clearly demonstrated by the enhanced uniformity of the transmembrane helical structures54,61,62 and the altered torsion angles of TM helices relative to water-soluble helices. The very low concentration of water in this region is also fundamentally important for the protein structure. Water and other protic solvents are known to be catalysts for hydrogen-bond exchange.56,63 Protic solvents were shown to have this catalytic effect when a mixture of four different double helical conformations of gramicidin in the nonprotic solvent, dioxane, interconvert very slowly with a half-life of 1000 h, but the addition of 1% water increases the interconversion rate by 3 orders of magnitude.56 In the TM domain of a protein, a misplaced hydrogen bond could be trapped and unable to rearrange, because of the lack of a catalytic solvent that could exchange the misplaced hydrogen bond correcting the misfolded state.64 Consequently, unsatisfied backbone hydrogen-bonding potential (i.e., exposed carbonyl oxygens and amide groups) in TM helices is not exposed to this low dielectric environment. In addition, side chains with hydrogen-bonding potential are also rarely exposed to these same lipid interstices. Interestingly, the side-chain hydroxyl of serine can hydrogen bond back to the polypeptide backbone, thus concealing this hydrogen-bonding potential. Small side chains, such as alanine and especially glycine that expose the polypeptide backbone more so than other residues to the low dielectric environment of the membrane interior, represent potential binding sites for other TM helices as they permit weak electrostatic interactions between helices including weak hydrogen bonds.65,66 In the TM domain of a protein, a misplaced hydrogen bond could be trapped and unable to rearrange, because of the lack of a catalytic solvent that could exchange a misplaced hydrogen bond with a correct hydrogen pairing, thereby correcting the misfolded state.64 Consequently, unsatisfied backbone hydrogen-bonding potential (i.e., exposed carbonyl oxygens and amide groups) in TM helices is not exposed to this low dielectric environment.
Figure 3.

Properties of lipid bilayers. (A) Distribution of moieties comprising lipids in a POPC bilayer along the bilayer normal (only one leaflet is illustrated), as obtained from MD simulations. The horizontal axis corresponds to the distance relative to the center of the bilayer. (B) Profile of the dielectric constant along the bilayer normal. Vertical lines correspond to confidence limits. As can be seen, alkyl chains possess a low dielectric constant, where it starts increasing at around 15 Å due to the presence of carbonyl groups. A large increase is observed at the phosphocholine head-groups, which cannot be accurately estimated; however, it is assumed to be several times larger than that of bulk water. Adapted with permission from ref (57). Copyright 2008 Elsevier.
The interfacial region of the membrane (between ±12 and ±17 Å from the bilayer center) has a slightly higher dielectric value that ranges upward of 3 or 4.57,58 This is the region where the first hydrogen bonds between the lipids and protein occur. Residues such as Trp and Tyr are known to be oriented so as to have their side-chain indole N–H and phenolic O–H groups oriented for hydrogen bonding to the lipid backbone ester groups tethering and orienting the protein with respect to the membrane surface.67,68 From within this region, but extending further to the phosphates of the membrane interface, are interactions between the phosphates and arginine and lysine side chains of the protein, known as snorkeling interactions with the lipids. Importantly, in this boundary between the hydrophilic and hydrophobic domains of the bilayer, a very significant pressure profile exists due to the free-energy cost of creating a hydrophobic/polar interface, which results in a tension (i.e., negative lateral pressure) in the interface region. At mechanical equilibrium, where the bilayer neither expands nor contracts, this tension is balanced by positive lateral pressure contributions from the headgroup and acyl-chain regions. In both of these regions, steric repulsion plays an important role, of course. In the headgroup region, another major contribution comes from electrostatic repulsion (monopoles, dipoles, etc.), while the acyl chains suffer from losses in conformational entropy upon compression. This lateral pressure at the hydrophobic/hydrophilic interface is thought to be on the order of several hundred atmospheres.69
Indeed, this contributes substantially to the dramatic barrier to water penetration into the bilayer interior. The pressure profile across the bilayer must be balanced, and indeed in the headgroup region a charge–charge repulsion appears to be responsible for a significant repulsive interaction, and potentially the high dynamics near the center of the bilayer may also contribute in a repulsive force to generate a net zero pressure profile. These repulsive forces occur over a much greater portion of the membrane profile and are not as dramatic as the narrow region associated with the profound attractive force that pinches off most of the water access to the membrane interior.
There is a dramatic demarcation between the interfacial and headgroup regions at 18 Å from the center of liquid crystalline POPC bilayers, based on the computed dielectric constant that jumps to above 200, well above the value for water. Hence, the transmembrane dielectric constant varies by more than a factor of 100. Not only does this influence the magnitude of the electrostatic interactions, but it also influences the distance range over which the interactions are significant. While long-range interactions are more significant in the membrane interior, the significant interactions in the headgroup region occur over a short distance. In this latter environment, the electrostatic interactions are reduced in strength to values that are significantly lower than those in the bulk aqueous solution.
The properties of the lipids in each monolayer of a membrane can include an inherent tendency to form a curved surface. In particular, the cross-sectional area in the headgroup region relative to the cross-sectional area in the fatty acyl region can lead to an inherent curvature for the monolayer if they are not equivalent.70,71 If the headgroup has a much larger cross-sectional area than the fatty acyl region, the result can be the formation of a micellar or hexagonal phase. If the headgroup has a much smaller cross-sectional area, the result can be for detergents, the formation of a reverse micelle or inverted hexagonal phase. When the differences in cross-sectional area are more subtle as for lipids, it is appropriate to think in terms of a tendency for a monolayer to curve. The curvature, of a lipid bilayer or membrane, is the result of the sum of these two tendencies that can lead to curvature frustration if the curvature tendencies are not complementary.72,73 Such curvature frustration may be alleviated by the MP through asymmetric contributions of juxtamembrane protein components, such as amphipathic helices or the packing of helices at one interface versus the other interface.74 In addition, a mismatch between the hydrophobic thickness of the membrane and that of the protein can alleviate or accentuate this frustration.75 In this context, it should be kept in mind that the relevant “effective” headgroup size takes into account not only the steric size but also electrostatic repulsion among headgroups. Particularly in the case of phosphocholine moieties, the effective headgroup size is considerably larger than it would be in the absence of a strong dipole moment. This is why DPC forms small, spherical micelles just above the CMC, whereas other C12 detergents with similarly sized or even larger but less polar headgroups form rodlike micelles.
Obviously, it is important for cells to maintain the integrity of their membranes, that is, the bilayer nature of the membrane. However, there are many processes that occur in cellular life that involve membrane fusion, vesicle budding, cell division, etc. These processes require bilayers to adapt to various nonbilayer structures, and consequently the lipid composition of cells is not achieved with lipids forming the most stable bilayers. For instance, phosphatidyl-ethanolamine, which has a much smaller cross-sectional area for its headgroup than for the fatty acyl chains, makes up approximately 75% of the lipid composition of the E. coli inner leaflet of the plasma membrane, while only a small fraction of the outer leaflet. Indeed, this lipid alone in aqueous preparations forms an inverted hexagonal phase. Such curvature frustration may be very appropriate for proteins with a greater hydrophobic thickness than that offered by the membrane. The result would be some relaxation of the curvature frustration surrounding each protein with the further result that protein oligomerization would be inhibited. This can also lead to stabilization of a particular functional state of a protein versus another, as in Rhodopsin.76,77 Shaping cell membranes, fusion, or fission of membranes were also modeled and described in line with experimental evidence by physical approaches.78
The dynamics and heterogeneity of the lipids are also important. NMR and ESR performed in nonfrozen samples can probe the dynamic disorder. For example, the analysis of the averaged quadrupolar coupling of 2H spins or dipolar couplings (e.g., between 1H and 13C) provides insight into the amplitude of motion experienced by a given bond averaged over time scales shorter than ca. microseconds.79 Experimental NMR 2H line shape analyses of lipids carrying specific 2H labels at different positions along the acyl chain show a gradient of dynamics:80,81 for approximately the first 6 carbons of the fatty acyl chains, the order parameter is approximately constant, that is, at values between 0.4 and 0.5 for both lipid chains, suggesting considerable residual order in pure lipid bilayers above the phase transition temperature. The site-specific order parameters for both chains decline steadily from the C6 to the bilayer center where the values are between 0.1 and 0.2. As alluded to above, ESR of spin labels at the C14 of stearic acid also reveal very low order parameters of the termini of the acyl chain.82 It is this high degree of fluidity at the bilayer center that is thought to contribute significantly to the repulsive lateral pressure at the bilayer center, making way for an attractive lateral pressure profile in the interfacial region.
2.2. Differences between Detergents and Lipids
In contrast to lipid membranes, which form two leaflets, detergent micelles have a single hydrophilic exterior surrounding a hydrophobic core. They are typically composed of detergents having a hydrophilic headgroup, but with only a single hydrocarbon chain or a pair of very short hydrocarbon chains. DPC micelles are the primary focus of this Review. This detergent molecule has a single 12 carbon saturated chain that in comparison with a diacyl lipid clearly results in a much smaller cross-sectional area for the alkyl chain than for the phosphocholine headgroup region. Such cone-shaped detergents lead to the formation of a micelle, which is not a bilayer. However, membrane proteins surrounded by a belt of detergent molecules can be maintained in solution. The protein–detergent complex is also called protein–detergent micelle, although it is not strictly a micelle.
Figure 4 shows results from MD simulations of detergent micelles formed by DPC, DDM, and SDS detergents. Such MD simulations reveal that the single alkyl chain does not have the extended structure of fatty acyl chains in lipid bilayers.84,85 Indeed, the hydrocarbon chain has considerable exposure to the aqueous environment on the surface of the micelle (estimated to be 20–30% of the surface), while it has essentially no exposure to the aqueous environment in a lipid bilayer.84−86 Consequently, the lateral pressure profile must be markedly reduced by the exposure of the hydrocarbon core to the aqueous environment, and therefore water penetration into the hydrocarbon core will be increased in comparison with lipid bilayers. Overall, this results in multiple differences between the environment formed by detergent micelles and that formed by lipid bilayers for membrane proteins. For one, the single thin interfacial region results in variable hydrophobic dimensions depending on the passage of the helix through the center Also, micelles have a weakened hydrophobicity for the micelle interior and a weakened hydrophilicity and thickness for the interfacial region. The dynamics of the detergent molecules is very different as compared to that of lipids. MD simulations showed that the order parameters for methylene segments of the lipid are roughly twice those of the surfactant,87 and that the order parameter of the detergent acyl-chains is closer to that of a liquid crystalline lipid bilayer than to that of a lipid bilayer in the gel phase.88 In lipids, as mentioned in section 2.1, the order parameters are small near the bilayer center, but for carbons 1–8 the order parameters are between 0.35 and 0.5. In micelles, it can be anticipated that the order parameters of carbons near the glycerol backbone are significantly reduced. Such increased dynamics may lead to void volumes facilitating the penetration of water molecules into the micelle interior, and because the hydrocarbon chains are exposed to the micelle surface this represents a likely mechanism for water permeation. Such water penetration can stabilize hydrophilic side chains and bent helical structures leading to non-native membrane protein structural features.
Figure 4.

MD simulations of detergent micelles formed of (A) 60 sodium docecyl sulfate (SDS), (B) 98 n-dodecyl β-d-maltoside (DDM), and (C) 65 DPC molecule. In (A), the sulfate group of SDS is represented by a yellow sphere, in (B) the two glucosides are shown by blue and turquoise spheres respectively, and in (C) the choline and phosphate groups are depicted as green and orange spheres, respectively, while the alky chains are represented as sticks. Atomic coordinates for SDS, DDM, and DPC micelles have been taken from https://www.tuhh.de/alt/v8/links/membranesmicelles.html,83http://micelle.icm.uu.se/example01.htm, and http://people.ucalgary.c/~tieleman/download.html, respectively. Part (D) shows the distribution of the different moieties of DPC as obtained from MD simulations.84
A major difference between lipids and detergents is the solubility in water as lipids, per definition, are quasi insoluble in water. Detergents are characterized by a critical micelle concentration (CMC), typically in the millimolar range (or less). Above the CMC the monomeric concentration is close to the CMC. In contrast, the concentration of monomeric lipid in the presence of lipid bilayers is nanomolar or less,89 that is, 106 times lower than typical detergent concentrations. The detergent molecules in the bulk aqueous environment can disrupt the quaternary structure, denature the tertiary structure, destabilize water-soluble domains, or interact with the aqueous pores of membrane proteins.90−95 In the case of cytochrome c oxidase, two different purification protocols led to crystals containing either 2 subunits96 or 4 subunits97 of the complex. The latter structure revealed that the two additional subunits are glued to the core structure by lipids that were displaced by detergents in the first structure. All of these features have the potential to influence the structure of membrane proteins in a way that is not native-like, not only for the TM region, but also for the juxtamembane and water-soluble domains of membrane proteins. Such interactions are likely to be present intermittently or consistently in detergent micelle solutions. The lifetime of interactions of membrane proteins with detergents and lipids may differ considerably. Conformational exchange dynamics in a G-protein coupled receptor has been shown to be directly related to the off-rate of detergent–protein interactions.98 It is important not to forget that ionic detergents are used to denature protein structures.
The micellar interfacial region is in sharp contrast with that of cellular membranes and many lipid bilayers, where this region is approximately 10 Å thick and has a dielectric constant that is considerably greater than that of the aqueous environment. Consequently, the lipid acyl chains are rejected from penetrating into this environment.
The single exterior of the micelle suggests that a hydrophilic side chain in the middle of a TM helix can “reach out” to the micelle surface without drawing the TM helical termini into the hydrophobic environment by forming a kink in the structure. This appears to be what happens in the GPGG motif in the middle of the TM part of protein Rv1761c, discussed in section 4.1. Gly and Pro are considered to be helix breakers.53 Even in membrane proteins, proline decreases the stability of a helix by forming a gap in the hydrogen-bonded helical structure, and glycine side chains expose the backbone of the helix to the hydrophobic environment. These residues have been referred to as “pro-kink” residues;62,85 in other words, they can form a “uniform” helical structure, or given the right conditions they can also induce a kink or bend in the helix as seen in mitochondrial carriers99 (see section 4.1.1). Glycine residues are also very important in allowing close approach of helices for enhancing electrostatic interactions between the helical backbones.66,100 Indeed, glycine residues do not appear to be conserved in TM helices unless they are used for helix–helix interactions or for kinking a helix. Yet the structure of the four-helix bundle protein KdpD has a helix with two glycine residues oriented toward the detergent environment.101 This structure also provides an example of hydrophilic side chains appearing to “reach out” to the micellar surface generating what appears to be an inside-out structure, instead of burying these residues in the interior of the helical bundle. In another example in the same publication, one of the two TM helices of ArcB has a distinct outward curvature of the helix that brings the hydrophilic helical backbone closer to the micelle surface, which is not possible in native membranes and in lipid bilayers. Furthermore, the hydrophilicity of the micellar interior is also demonstrated by extensive hydrogen/deuterium exchange for the amide sites in one of the helices of ArcB and three of the helices of KdpD.101 In fact, the very low dielectric environment of the lipid fatty acyl environment for TM helical bundles can induce the opposite, a slight hourglass shape, rather than the barrel shape as in the DPC micelle structure of DgkA102 (see section 4.1.2). A corollary to the single hydrophilic surface and the lack of a fixed hydrophobic dimension, as opposed to that in a lipid bilayer where a long α-helix is forced to tilt in the lipid bilayer, in a micelle the hydrophobic dimension can expand or contract to a certain extent to accommodate a long or short helix length.85 Indeed, different detergents prefer the formation of either oblate or prolate ellipsoids: DPC and dihexanoylphosphatidylcholine (DHPC) have tendencies to form prolate ellipsoids with the longer (major) axis being the symmetry axis, while DM and DDM form oblate ellipsoids103 with the shorter (minor) axis being the symmetry axis. For many helical membrane proteins, there is a considerable variation in the hydrophobic length of the helices, and the relatively rigid hydrophobic dimension of the lipid bilayer can be considered a restraint of the native helix packing.
In conclusion, the chemical nature of detergent molecules and detergent micelles explains how membrane proteins can be maintained in solution. However, lipids display a broad range of mesoscopic arrangements that convey plasticity and unique physical properties to the membrane that contribute to the structural and functional behavior of membrane proteins. Therefore, in the last decades, alternative approaches were developed. Nonetheless, the solubilization of membrane proteins with detergent molecules remains the most used approach, and a majority of structural studies currently rely on detergents, highlighting the importance of understanding the strengths and the limits of these molecules.
3. Alkyl Phosphocholines: History and MP Function
Alkyl phosphocholine detergents are classified in the category of lipid analogue detergents, which also comprises CHAPS and sodium cholate detergents. Indeed, alkyl phosphocholines and phosphatidylcholines both contain phosphocholine head groups and hydrocarbon tails. There are, however, two major differences, which explain why one is a detergent while the other is a lipid. Alkyl phosphocholine detergents contain a single alkyl chain and therefore are closer to lyso-lipids like 1-lauroyl-2-hydroxy-sn-glycero-3-phosphocholine (LDPC) than to diacyl phosphatidylcholine-type lipids. Indeed, DPC and LDPC have similar CMCs of 1 mM. The second major difference is the presence of a glycerol group between the headgroup and the carboxylic ester function of the acyl chain in phospholipids. In contrast, the alkyl chain of an alkyl phosphocholine is directly connected to the phosphate via a phosphoric ester function (see Figure 1 for two-dimensional structures of the aforementioned molecules).
3.1. Historical Background
One of the first publications reporting the use of DPC in membrane protein biophysics comes from Kurt Wüthrich’s laboratory. The authors first attempted to study a membrane peptide in phospholipid vesicles and failed to record the 1H NMR signal.36 Therefore, they asked whether lipids or detergent micellar systems were suitable for high-resolution NMR of membrane proteins. As a model membrane protein, they used melittin, an amphiphilic peptide of 26 amino acids that constitutes 50% of the dry weight of bee venom.104 It was known that at high concentrations, melittin lyses membranes and also activates phospholipase A, probably by modifying the local structural organization of the membrane. As micellar systems, they used short-chain detergents like 1,2-diheptanoyl-sn-glycero-3-phosphocholine (diC7PC), as well as LDAO and DPC, for which a chemical synthesis route had been previously established.105,106 Melittin was inserted either in detergent micelles or in a phospholipid bilayer, and secondary structure and conformational dynamics of the peptide were assessed by fluorescence spectroscopy, circular dichroism, analytical ultracentrifugation, quasi-elastic light scattering, and 1H NMR experiments. The main conclusions from this seminal paper are as follows:
(1) In the absence of lipids or detergent micelles, melittin changes its conformation to form a tetramer, which is soluble in solution. There was, therefore, a need to study melittin in micelles to understand its physiological function.
(2) Detergent micelles stabilize melittin in a single and homogeneous monomeric conformation easily detected by biophysical methods, especially by NMR.
(3) The conformation of melittin observed by NMR is independent of the type of detergent. However, detergents that form small-size micelles, like DPC (at a detergent/peptide ratio of 40/1), are more suitable for NMR analysis.
(4) Last, the author stated: In the systems studied here, the fluorescence and circular dichroism experiments provided direct evidence that the conformation of melittin bound to micelles or to phosphatidylcholine bilayers must be very similar.104
In the following years, several groups investigated the conformational dynamics of amphiphilic peptides in DPC. Mendz and colleagues identified by NMR peptide sequences of the myelin basic protein that interact with DPC micelles.107 The amino-terminus of the yeast mitochondrial cytochrome oxidase subunit IV precursor protein (p25) was also analyzed in DPC by NMR, and the authors showed that the N-terminal half of the peptide switched to an α-helical conformation upon binding to DPC micelles. Later, it was observed that addition of cardiolipins to p25 peptide/DPC micellar complexes stabilized the α-helix.108 In 2000 Anatrace added to its catalog fully deuterated DPC, which together with methodological and instrumental developments109 strongly stimulated the use of DPC for the study of larger membrane proteins by NMR. More recently, the Wüthrich laboratory, which initiated the use of DPC, extended the gamut of DPC derivative molecules in an unprecedented way. Using OmpX protein as a model β-barrel membrane protein, they screened detergents suitable for in vitro folding of this protein. Among 23 commercially available detergents, only the alkyl phosphocholine series (decyl, dodecyl, and tetradecyl phosphocholine) was able to support almost complete refolding of OmpX. For the case of OmpX where no functional assays can be performed, the refolding yield is a proxy, informing about the compatibility of the detergent with the folded state, even though direct conclusions on functionality should be treated with caution. In the case of OmpLA, DPC was only a moderately good refolding agent, but very good at preserving its enzymatic activity.110 From their observation on refolding yields with alkyl phosphocholines, the Wüthrich laboratory synthesized 42 new alkyl phosphocholine derivatives that more closely resemble lyso-phospholipids (Figure 5). To mimic lyso-phospholipids, which have been shown to preserve the activity of complex membrane proteins (LPG preserved the activity of the calcium ATPase for instance111), they added a polar spacer group, which mimics the glycerol motif between the phosphocholine headgroup and the alkyl chain (Figure 5). To approach the structure of short-chain phospholipids, which are usually considered as relatively mild detergents (like DHPC or diC7PC),112,113 they grafted short branches to the alkyl chain of DPC (Figure 5). All molecules were tested for their ability to refold efficiently OmpX. Five of them were further analyzed by NMR, and two of them, 138-fos and 179-fos, gave NMR spectra identical to that of the OmpX/DHPC complex.114 To conclude, the chemistry of alkyl phosphocholine detergents is a dynamic and productive field of research, mainly driven by chemists and biophysicists. In the next section, we examine the advantages and drawbacks of alkyl phosphocholine detergents from a biochemical and functional point of view.
Figure 5.

Alkyl phosphocholine derivatives from DPC (a) and DHPC (b) designed by the Wüthrich laboratory (figure reproduced with permission from Zhang et al.114). Copyright 2008 American Chemical Society.
3.2. Alkyl Phosphocholine Detergents: What Are They Good for in Membrane Protein Biochemistry?
There is a broad consensus on the ability of alkyl phosphocholine detergents to solubilize membrane proteins efficiently. In the frame of recombinant protein production of eukaryotic proteins, several laboratories have conducted systematic screening of detergents for the efficient solubilization of membrane proteins. For instance, Ren et al. have produced chemokine receptors in E. coli and have screened a large number of detergents and detergent mixtures.115 Human CCR5, CCR3, CXCR4, and CX3CR1 receptors were fused with thioredoxin in a pBAD vector and produced in the E. coli membrane (Top10 strain). The results of the detergent screen115 are shown in Table S2. The four chemokine receptors were readily solubilized by all alkyl phosphocholine detergents, including cyclofos detergents, while none of the other detergents tested in this study were able to solubilize the CX3CR1 receptor.115 The same group conducted a similar study with the formyl peptide receptors (FRP3) produced in the eukaryotic HEK293S stable cell line. Among 96 solubilization conditions with different detergents/detergent mixtures, three detergents, foscholine-unsat-11-10, Anapoe-X-100, and tetradecyl phosphoscholine (FC14), were found to be the most efficient for solubilizing FRP3 receptor. This detergent was chosen for its large-scale purification, low cost, and its structural similarity with phosphatidylcholine lipids, a major constituent of eukaryotic bilayers.116 Both examples illustrated the solubilization power of alkyl phosphocholine detergents for large-scale purification of recombinant proteins. However, the functionality of the receptors was not addressed in either study. In the next section, we review 21 membrane proteins of different origins and recombinant sources, which were purified in alkyl phosphocholine detergents and further characterized by functional tests.
3.3. Are Membrane Proteins Functional in Alkyl Phosphocholine Detergents?
It is not straightforward to answer in very general terms whether alkyl phosphocholines sustain the activity of membrane proteins. First, the impact of the detergent environment on activity certainly is different between different membrane proteins, making it difficult to derive general conclusions. Second, in the various available studies in the literature, the amount of copurified lipid differs, further complicating comparisons. Last, testing the activity of membrane proteins is, in some cases such as for transporters, only possible after reconstitution into lipid bilayers, not in the detergent-solubilized state. Detecting activity of reconstituted protein does not directly inform whether the initial state before reconstitution is active. With these limitations in mind, we summarize the results of 17 studies in Table S3.
There are few examples of successful recovery of membrane protein activity in the presence of alkyl phosphocholine detergents. The phosphate transporter from the plasma membrane of Saccharomyces cerevisiae was successfully produced in Pichia pastoris and purified in DPC detergent. Its activity was fully recovered after reconstitution in proteoliposomes with a similar substrate specificity as observed in an intact cell system.117 Conversely, opposite results were obtained with mitochondrial uncoupling proteins. The Chou laboratory reported proton-transport activity for both UCP1 and UCP2 proteins in DPC,118,119 while Zoonens and co-workers found that DPC fully inactivates both transporters.120 Asmar-Rovira and colleagues investigated how nine detergents influence the function of the nicotinic acetylcholine receptor (nAChR) of Torpedo electric rays.121 Below 45 mol of phospholipids per mole of nAChR, the receptor was rapidly inactivated. By carefully measuring the amount of residual lipids after solubilization of enriched Torpedo membranes, they could show that most detergents degraded the receptor during purification below the critical threshold to maintain its activity. For instance, Cymal-6, DDM, LDAO, and OG showed decreased stability and significant reduction or loss of ion-channel function. In contrast, CHAPS, DPC, and sodium cholate maintained stability and supported ion-channel function. Asmar-Rovira and colleagues concluded that in the case of nAChR, CHAPS, DPC, and sodium cholate mimic the lipids in the sense of being able to sustain lipid-dependent activity and stability.
The situation is even more complex with the human ABCG2 multidrug pump. MacDevitt et al. were able to solubilize the recombinant protein from sf9 insect cell membranes only with hexadecyl phosphocholine.122 After three purification steps in hexadecyl phosphocholine, the protein was still able to bind the substrate, but its ATPase activity in detergent was low, and the authors did not test ATPase activity after reconstitution of the protein in liposomes. They were nevertheless able to analyze single particles by cryoEM and obtained a low-resolution three-dimensional projection map showing a tetrameric structure, which was interpreted as four homodimers of ABCG2. A second study appeared a few years later, showing that the ABCG2 receptor purified in hexadecyl phosphocholine was irreversibly inactivated, while the same protein purified in DDM was active when reconstituted in liposomes containing an excess of cholesterol (40%).123 The authors concluded that the homodimers of ABCG2 were disrupted by hexadecyl phosphocholine, resulting in a complete inactivation of the receptor.124 Similar results were obtained for BmrA, a multidrug resistance efflux pump. The protein was inactivated by DPC, but in a reversible manner. Exchanging the alkyl phosphocholine detergent with DDM or anionic calix[4]arene-based detergents restored its activity. Reversible activation of pumps has also been seen with the human bile salt export pump, BSEP, produced in Pichia pastoris membranes and purified in phosphocholine detergents with linear or cyclic alkyl chains.125 Its activity was restored by exchanging the detergent with DDM.125 In the case of the multidrug resistance pump MDR3, addition of lipids to the alkyl phosphocholine-MDR3 complex resulted in a partial restoration of its activity.126
Aside from these examples of partial tolerance to DPC, there are numerous examples of membrane proteins that are fully inactivated by this detergent (see Table S2). For instance, diacylglycecol-kinase activity in the presence of DPC is extremely low as compared to a purification with the lysolipid 1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine, where the activity is 600 times higher.127 By performing NOE measurements in both conditions, Koehler and co-workers were able to evince the strong and non-native interactions of the indole rings of a tryptophan residue with the choline methyl protons at the end of the DPC headgroup, which could explain the loss of function. DPC has been also widely used for G-protein coupled receptor (GPCR) purification from recombinant eukaryotic cell membranes (see examples in Table S3). Receptors from this family are highly sensitive to the lipid environment,128 and their extraction from recombinant membranes is also cell-type dependent, as illustrated by the study of Thomas and Tate.129 These authors showed that the adenosine receptor is not functionally produced in sf9 cell, but rather in human iGnTI- cells. Accordingly, DDM detergent cannot extract the receptor from sf9 membranes, but the same receptor is fully extracted from iGnTI membranes and able to bind its ligand in DDM micelles. In contrast, DPC does not discriminate between folded and unfolded receptors. DPC was able to extract the adenosine receptor, regardless of the origin of the recombinant membranes, but ligand-binding assays revealed that the receptor was inactivated in that detergent solution.128 Similar results were obtained with the angiotensin II receptor, fully extracted with alkyl phosphocholine detergents, but showing no ligand-binding ability.128 Interestingly, a thermo-stabilized mutant of the same receptor was able to bind its ligand in alkyl phosphocholine micelles, but not in SDS micelles, thereby suggesting again that the use of alkyl phosphocholine detergents for functional studies is unpredictable and highly protein dependent.128 In another example, the Ste2p receptor produced in human BHK cells was fully extracted with DPC, and retained a significant ligand-binding capacity (Table S3), whereas the HCN2 voltage-gated cation channel produced and extracted from BHK membranes in the same conditions did not show any ligand-binding activity.130 Another interesting example is provided by the Ail protein, an outer membrane protein from Yersinia pestis bacteria. The Marassi laboratory showed that this protein is able to bind fibronectin or heparin in decyl phosphocholine detergents only at low detergent concentration, in this case, below its CMC.131
To conclude, it is apparent that alkyl phosphocholine detergents are powerful for solubilization and purification of membrane proteins. However, they do not discriminate between folded and unfolded proteins, and appear to maintain even unfolded membrane proteins in solution, possibly leading to heterogeneous samples, and representing a major limitation for most biophysical techniques. In addition, alkyl phosphocholine detergents have a pronounced tendency to inactivate the function of the protein, although some reports mention that the function can be restored by using lipids or exchanging the detergent.125 The use of alkyl phosphocholine detergents for functional studies of membrane proteins is, therefore, unpredictable and probably not recommended for fragile or complex membrane proteins, such as α-helical GPCR or transporters.
4. Studies of MPs in DPC Reveal Strengths and Weaknesses
The properties and stability of α-helical proteins differ considerably from those of β-barrels. While the tertiary structure of β-barrels is dictated by the hydrogen-bonded network, resulting in a stable tertiary arrangement, helix–helix contacts in the membrane involve weak packing interactions. Accordingly, these two types of proteins are very differently sensitive to the membrane/detergent environment, and are discussed separately in this section.
4.1. α-Helical Membrane Proteins
4.1.1. Mitochondrial Carriers
The mitochondrial carrier family (MCF) provides several examples that reveal effects of DPC on membrane protein structure and dynamics. Mitochondrial carriers (MCs) shuttle different classes of substrates, such as keto acids, amino acids, nucleotides, inorganic ions, and cofactors, across the inner mitochondrial membrane.132−134 The amino acid sequences of MCs comprise three homologous repeats of ca. 100 residues.135 In light of their sequence similarities, MCs are likely to have similar structures and transport mechanisms. Five decades of research on MCs has generated a large body of functional, biochemical, biophysical, and structural data,132,136−140 which can be compared to recent studies of MCs in DPC,118,141−146 thereby providing insights into the effects of the detergent environment on structural integrity and functional properties of MCs. The studies in DPC were carried out with MCs refolded from inclusion bodies produced in Escherichia coli, whereas the other studies used native MCs isolated from the inner membrane of mitochondria. MCs are among the most difficult membrane proteins to work with, as they are hydrophobic and highly dynamic.
The best characterized MC is the mitochondrial ADP/ATP carrier (AAC), which imports cytosolic ADP into the mitochondrion and exports ATP to the cytosol to replenish the cell with metabolic energy.136−138 Crystal structures of the bovine147 and yeast148 ADP/ATP carriers have been determined in LAPAO and maltoside detergents, respectively. In these structures, the presence of a high-affinity inhibitor, carboxyatractyloside (CATR), locks the transporter in an aborted cytoplasmic state in which the cavity is open to the intermembrane space/cytoplasm and closed to the mitochondrial matrix. Despite extensive efforts, no crystal structures of any state other than the CATR-inhibited state have been obtained, possibly due to the inherent dynamics of MCs. These aborted-state structures together with biochemical and computational data have allowed mechanisms of transport to be proposed, but many aspects are unresolved. In addition to AAC structures, a solution-state NMR backbone structure of uncoupling protein UCP2 in DPC has been determined.118 Uncoupling proteins dissipate the protein motive force in mitochondria to produce heat and are activated by fatty acids and inhibited by purine nucleotides, but the molecular mechanism is still debated.139,149,150 The structure was determined using a fragment-search approach with NMR residual-dipolar couplings (which provide information about the relative orientation of peptide planes) and paramagnetic relaxation-enhancement data (which probe distances of a given peptide plane to a spin label attached to a cysteine site). No NOEs were measured to provide short distance restraints. The structure of UCP2 in DPC has been solved in the presence of the inhibitor GDP, representing an aborted cytoplasmic state, similarly to the crystal structures of CATR-bound AAC. The comparison of these structures may thus provide insight into the effects of different detergents. (We note that structural differences might in part also be due to methodological differences, considering in particular that solution-state NMR with a protein of this size is challenging, and the obtained structure may, thus, also suffer from a lack of structural restraints. However, the trends revealed by these structure comparisons are also reflected in dynamics and interaction studies discussed further below.)
AACs and UCPs share ca. 25% identity over the entire length, despite having very different functions, indicating that they are likely to have a highly similar fold (Figure 6A). However, the structures of AAC1 in LAPAO and UCP2 in DPC are unexpectedly different, with a backbone RMSD of 9.52 Å (see Figure 6B,C). It is clear that the UCP2 structure is much wider and taller than the AAC structure, even though the overall sequence length is very similar. The distribution of the axial interhelical distances between TM α-helices shows that the average is 10 Å for the AAC1 structure, which is close to the norm, whereas it is 12.6 Å for the UCP2 structure (Figure 7A). Consequently, a large number of holes appear between the α-helices of the UCP2 structure,120 also in the membrane parts, which are highly improbable (red circles in Figure 7B and D). Importantly, the structure of UCP2 in the GDP-bound state in DPC features a large channel through the entire protein, which is big enough for protons and small molecules to pass through from the intermembrane space to the mitochondrial matrix, but this state is supposed to be proton-impermeable. In contrast, the cytoplasmic state of AAC has a ∼20 Å protein layer to prevent leak of protons147(Figure 7B, left). Mitochondrial carriers consist of three homologous domains, which is clearly reflected in the 3-fold pseudosymmetry of the structure. On the basis of sequence analyses, uncoupling proteins are among the most symmetrical, whereas ADP/ATP carriers are among the most asymmetrical of MCs.152 At odds, the structure of AAC1 has a high degree of symmetry in the structure of the domains and in the overall fold, whereas the structure of UCP2 does not (Figure 6B and C). The deviation in the symmetry of the fold can be best appreciated by comparing residue positions that belong to highly symmetrically conserved motifs. For instance, the glycine residues of the EG-motif are arranged in a symmetrical fashion in AAC1, whereas they are not at all in UCP2 (Figure 6B and C). Another striking difference between these structures is the angle of the TM α-helices with respect to the plane of the membrane, which is consistently 45° in the AAC structures,147,148 but a wider range of angles for the UCP2 structure are observed.118 Finally, in these aborted cytoplasmic states, residues of the matrix salt bridge network are supposed to be interacting, and they are in bonding distances in the AAC1 structure, but they are 11–14 Å apart in the UCP2 structure.
Figure 6.

Amino acid sequences and the structures of the mitochondrial ADP/ATP carrier AAC1 and uncoupling protein UCP2. (A) Aligned amino acid sequences of bovine AAC1 and mouse UCP2, shown in the ZAPPO color scheme using the program Jalview.151 Identical residues are shown in the consensus sequence and are indicated by black boxes. Also indicated are the positions of the matrix147 and cytoplasmic152 bridge networks. Mitochondrial carriers consist of three homologous sequence repeats, which are aligned beneath each other. (B) Cytoplasmic and (C) lateral views of the structures of bovine AAC1 (1OKC) determined by X-ray crystallography (left)147 and mouse UCP2 (2LCK) determined by solution NMR (right).118 The odd-numbered α-helices (H1, H3, H5), matrix α-helices (h12, h34, h56), and even-numbered α-helices (H2, H4, H6) are shown in green, blue, and red cartoon representations, respectively. Symmetry-related glycine residues of the EG-motif are shown in black spheres, whereas the residues of the matrix salt bridge network, which are interacting in these states (cyan dashes), are shown in yellow sticks. The 3-fold pseudosymmetrical axis is shown by a triangle.
Figure 7.

Structures of AAC (in DDM or LAPAO) and UCP2 (in DPC) have very different features. (A) Distribution of the axial interhelical distances of the bovine mitochondrial ADP/ATP carrier AAC147(wheat) and uncoupling protein UCP2118 (green). The dotted lines indicate the average values. (B) Cross-section through the middle of the bovine AAC1 (left) and mouse UCP2 (right) structures. AAC1 has a layer of about 20 Å to prevent the leak of protons, whereas UCP2 has a hole through the entire protein, which is large enough for small molecules and protons to pass through from the intermembrane space to the mitochondrial matrix and would short-circuit the mitochondrion. (C) Cross-sectional view of UCP2 in complex with GDP2– in MD simulations in explicit DPC.120 The detergent is organized in a bundle around the hydrophobic core, as well as in two extra micelles, assembled on the matrix and cytoplasmic sides around amphiphilic patches of amino acids. The internal cavity of the protein is fully opened on both sides of the protein and filled by a large number of water molecules. (D) Surface representation of UCP2 after 200 ns of MD simulation in explicit DPC, using the NMR structure as starting conformation. For clarity, ions, water molecules, and detergents are not shown. The lateral openings between helices can be clearly seen.
Given these unexpected features, it is likely that the structure of UCP2 in DPC does not represent a functionally relevant fold or conformation. It is interesting to note in this context that the AAC structures have held up in MD simulations, whereas UCP2 collapses,120 as discussed at the end of this section. In the following section, we investigate in more detail the possible origins of these structural differences, and reveal the effects of DPC on stability, secondary structure, interactions, and dynamics of several MCs.
4.1.1.1. Tertiary Structures of Mitochondrial Carriers Are Destabilized by DPC
Thermostability shift assays (TSA) can be used to assess membrane protein stability in different conditions153 and can provide information on the integrity and functionality of the protein in detergent solution.154−156 The thermostability of a population of purified MPs in detergent is monitored by a thiol-reactive coumarin maleimide probe, which forms a blue fluorescent adduct after reaction with exposed protein thiols.153
TSA studies have been carried out on two isoforms of the mitochondrial ADP/ATP carrier from yeast: AAC2157 and AAC3,158 which are produced by expression under aerobic and anaerobic conditions, respectively. When AAC3, purified from the yeast mitochondrial inner membrane, is diluted in dodecyl-maltoside (DDM), a typical unfolding curve is obtained with an apparent melting temperature of 48 °C. When the specific inhibitor CATR is added, a marked shift by 33 °C in thermostability occurs to 81 °C (Figure 8A). This shift is explained by the binding of CATR, which introduces a large number of polar interactions that stabilize the structure.148 However, when AAC3 is diluted in DPC, a high fluorescent baseline is observed at the start of the assay, indicating that all cysteines have become available for labeling prior to the temperature ramp and consequently no melting temperature could be assigned. In this case, the addition of CATR does not alter the stability, indicating that once AAC3 is in DPC, it is no longer competent to bind CATR (Figure 8B).
Figure 8.

Thermostability of the mitochondrial ADP/ATP carrier and uncoupling protein in different detergents. Carrier unfolding was monitored by the fluorescence of CPM-adduct formation at cysteine residues as they become solvent-exposed due to thermal denaturation.153,154 (A) Thermal denaturation profile (top) and corresponding first derivative (bottom) of native yeast ADP/ATP carrier AAC3 diluted into assay buffer in DDM in the absence (solid line) or presence (dashed line) of CATR. (B) Same as in (A), but with AAC3 diluted in DPC. (C) Apparent melting temperatures (TM) of native yeast ADP/ATP carrier AAC2 with or without bound CATR diluted in octyl to tridecyl maltoside (8M-13M), Cymal4–7, dodecyl and decyl maltose neopentyl glycol (12MNG and 10MNG), octyl glucose neopentyl glycol (8GNG), LAPAO, and DPC. (D) Thermal denaturation profile of native uncoupling protein UCP1 in decyl-maltose neopentyl glycol (10MNG) (top) and corresponding first derivative (bottom) in the absence (solid line) or presence (dashed line) of GDP. (E) Same as in (D), but with native UCP1 in DPC. The apparent melting temperatures are reported in the derivative profiles. Data are from refs (146) and (154).
The thermostability of the related AAC2 was assessed in a large number of different detergents. Some mild detergents with long hydrocarbon chains retain the folding of unliganded AAC2, whereas harsher detergents, such as octyl-maltoside, LAPAO, and DPC, do not (Figure 8C).154 When the native carrier is first inhibited by CATR before dilution into different detergents, the inhibitor provides some protection against unfolding by the increased number of inhibitor–protein interactions, as unfolding curves can be obtained for all detergents. As expected, the apparent melting temperatures for harsh detergents, like DPC, are much lower than for mild detergents. In retrospect, the structure of bovine AAC1 could be obtained because the carrier was inhibited by CATR prior to solubilization and because excess lipids were carried through by negative chromatography, providing further protection against unfolding.147 The observed melting temperatures (Figure 8C) correlate well with the size of the micelle154 and yields of purified carrier in these detergents.159,160
A similar study has been carried out with lamb uncoupling protein UCP1, isolated from the native mitochondrial membrane.154,155 Again, a typical unfolding curve is observed in decyl-maltose neopentyl glycol (10MNG), showing a melting temperature of 42 °C (Figure 8D). Addition of GDP, a specific inhibitor of UCP1, leads to an increase by 9 °C in thermostability to 51 °C, which was found to be pH-dependent,154 as observed in other studies.161,162 A detergent-dependent thermostability profile similar to that for AAC2 was obtained for UCP1,154 indicating that different members of the MC family have a comparable sensitivity to different detergents. However, when unliganded UCP1 is diluted in DPC, the protein loses its tertiary structure, whereas some protection against unfolding is observed when UCP1 is first inhibited by GDP (Figure 8E). These results show that the folded structure of native unliganded MCs cannot be maintained in DPC and that their ability to bind specific ligands is lost, whereas it is conserved in mild detergents.
4.1.1.2. Binding of Substrates and Inhibitors to MCs
Transport assays rely on membrane-separated compartments and substrate gradients, and thus the transport capability of membrane transporters cannot be studied with micelle-solubilized proteins. Instead, their binding affinity and specificity for ligands can be used to verify the functional state of these proteins in detergent. In lipid bilayers, MCs are highly specific; that is, they bind natural inhibitors and transport substrates at the exclusion of other solutes.
In the following, we will review the binding properties of specific natural inhibitors, and later substrate binding. AAC is a particularly relevant case, because two specific inhibitors are available, atractyloside (ATR) and CATR.163 The affinities of these two inhibitors have been reported multiple times,136 in isolated mitochondria, in solubilized and purified form, and after reconstitution into liposomes. AACs in the membrane bind ATR and CATR very strongly, with a dissociation constant in the range Kd = 5–12 nM (CATR),164−168 but the affinity is lower when AAC is solubilized in detergents. In isothermal calorimetry (ITC) measurements using native AAC3 from yeast mitochondria purified in DDM/tetraoleoyl cardiolipin, CATR binding has an average Kd of 72 nM; that is, the affinity is ca. 10-fold lower than in the membrane. In the zwitterionic detergent LAPAO, which is considered a relatively harsh detergent, the Kd of CATR binding to bovine AAC1 is 310 nM;164 that is, the affinity is ca. 45-fold lower than in membranes. In SDS, which is considered a very harsh detergent environment, CATR binding is abolished completely, suggesting that the protein is no longer in a folded state.169 When AAC3 is refolded from inclusion bodies in DPC, the CATR dissociation constants are 15 and 150 μM in ITC and NMR-observed titrations, respectively, which represent an ca. 1000–10 000-fold reduction in affinity as compared to AAC in lipid bilayers. This highly reduced affinity suggests that AAC3 in DPC does not retain key interactions required for inhibitor binding in agreement with the TSA data.
In addition, the residues that interact with CATR are very different in refolded AAC3 in DPC144 as compared to native AAC3 in decylmaltoside.148 NMR chemical-shift perturbations (CSPs) induced by different concentrations of CATR are found all over AAC3 in DPC,144 whereas in the crystal structure of AAC3 they are localized to a specific site in the central cavity,148 very similar to that in bovine AAC1147 and yeast AAC2.148 Out of the 14 residues known to interact with CATR,148 only one, R85, shows CSP, as well as some neighboring residues. However, about one-half of the residues showing CSPs are on structural elements that are not involved in CATR binding at all. One might argue that CSPs can be induced at remote sites via allosteric changes of structure and dynamics, and that the widespread CSPs in AAC3 do not necessarily point to a misfolding in DPC. This view is undermined by a recent study that uses the mitochondrial GDP/GTP carrier (GGC1), which does not bind CATR.170 Yet, the addition of CATR to GGC1 in DPC leads to CSPs of magnitude comparable to those in AAC in DPC146 (left panel of Figure 9d). Because GGC is not inhibited by CATR in lipid bilayers,170 the observed GGC1/CATR interactions in DPC must be nonspecific.146
Figure 9.

Loss of binding specificity of mitochondrial carriers (AAC3, GGC1) in DPC micelles. (a,b) Chemical-shift perturbations (CSP) observed in DPC-solubilized GGC1 upon addition of its substrate, GTP. Panel (a) shows residue-wise CSP values, which are plotted onto a structural model of GGC1 in panel (b). Panel (c) shows that the effects induced by addition of GTP and ATP are very similar, that is, that GGC1 interact with both nucleotides in a comparable manner, despite the fact that in lipid bilayers only GTP is bound, not ATP.146,170 (d) Chemical-shift perturbations upon addition of 5 mM CATR to GGC1 (left) or 7.5 mM CATR to AAC3 (right). Residues affected by inhibitor-binding are spread throughout large parts of the molecule, and the effects are similar in AAC3 (which is known to bind CATR physiologically) and GGC1 (which does not bind CATR in lipid bilayers). The data on GGC1 are from Kurauskas et al., and the panels were adapted with permission from ref (146). Copyright 2018 American Chemical Society. The AAC3/CATR interaction data are plotted using data reported by Brüschweiler et al.144
Inhibitor binding has also been studied in uncoupling proteins. In native UCP1 extracted from the mitochondrial membrane, the dissociation constant is 46 nM by ITC measurements.155 In contrast, Berardi et al. report a value of 5 μM118 for mouse UCP2 using a FRET assay. Zhao et al. report that for human UCP1 “titrating the NMR sample with GDP showed only small chemical-shift perturbation of the backbone amides even at very high GDP concentration (∼1 mM), which is inconsistent with the tight GDP binding reported for UCP1 reconstituted in a more native environment.”119
Substrate binding has been studied in several MCs in DPC by solution-state NMR, in AAC3 and GGC1143,144 as well as to the short Ca2+-binding mitochondrial carrier (SCaMC), which is another adenine nucleotide carrier, allowing a comparison to the properties of native proteins. Brüschweiler et al. have investigated ADP binding to AAC3 in DPC by NMR, and found a Kd value of 0.5 mM, approximately 85-fold higher than the published consensus values of the carrier in the mitochondrial membrane.136 Sounier et al. have investigated the binding of GTP, GDP, and AMP to GGC1 using CSPs.143 A range of different Kd values has been observed for different residues in GGC1 in DPC. The overall Kd for GTP was estimated to be 6.6 mM for GTP and 23 mM for GDP. These numbers are at least 3 orders of magnitude larger than the apparent KM values in transport assays (KmGTP = 1.2 μM and Km = 4.5 μM),170 which in turn must be larger than the Kd values for substrate binding. The Kd value for SCaMC in DPC was determined to be 1–2 mM for Mg-ATP,142 whereas the apparent KM value for ATP transport was 30 μM.171 Thus, in all cases where direct comparisons can be made, the affinities of the MC in DPC for the substrates and inhibitor (CATR) are several orders of magnitude lower than those for the native proteins in the membrane, suggesting the lack of interactions required for specific binding.
Mitochondrial carriers have been proposed to have a single substrate binding site in the central cavity,152,172,173 which has been corroborated by mutagenesis,174 photoaffinity labeling,175 and substrate specificity studies176 as well as MD simulations.177−179 Substrate interaction studies of MCs in DPC are not consistent with this site. ADP-induced chemical-shift perturbations (CSP) are found largely on the matrix side of AAC3,144 whereas they are found in multiple sites, rather than a single site, in GGC1. In SCaMC, the substrate interaction sites are found on the matrix and cytoplasmic side of the carrier and on transmembrane H4.142 Moreover, the nucleotide binding sites of AAC3 and ScaMC, which are closely related carriers, do not overlap, as one would expect. In conclusion, the nucleotide interaction sites highlighted by the studies in DPC are found all over the carriers rather than in a single substrate binding site in the central cavity, as proposed by the other studies.
Kurauskas et al. reasoned that the substrate and inhibitor interactions in DPC-solubilized MCs may be of electrostatic nature between the negatively charged substrates and the positively charged residues lining the cavity (pI values of MC are 10), and may not require a correctly arranged structural scaffold. To test this hypothesis, they performed titration experiments of AAC3 and GGC1 (in DPC) with both ATP and GTP to test the ability of these carriers to discriminate between different substrates.146 In lipid bilayers, GGC1 binds only GTP and AAC3 binds only ATP. However, in DPC, the two different nucleotides induce essentially identical CSPs in each of the proteins, showing that AAC3 and GGC1 in DPC lose their ability to discriminate between substrates of equal charge. This finding mirrors the unexpected similarity of the CATR interaction with GGC1 and AAC3, as discussed above.
Another important molecule that binds tightly to the mitochondrial ADP/ATP carrier is cardiolipin (CL), a major lipid constituent of the mitochondrial inner membrane.180 The structure of bovine AAC1 in LAPAO clearly showed that CL molecules were bound in three well-defined binding sites by hydrogen bonding.147,181 Very similar binding sites for CL were observed in the yeast AAC2 and AAC3, and it was postulated that the negatively charged CL molecules are also bound by electrostatic interactions with the positively charged helix dipole termini.148 Subsequently, it was shown that uncoupling protein UCP1 also binds CL in a 3:1 ratio, showing that it might be a universal property of mitochondrial carriers.155 The interactions between AAC extracted from the native membrane and CL molecules are very strong, as they remain attached to AAC even after extensive washing steps during purification.160
Recently, Zhao et al. have investigated CL binding to refolded AAC3 in DPC using solution NMR.145 They have shown that while the doubly charged CL produces clear chemical-shift perturbations, the uncharged POPE does not lead to spectral changes. NOESY and CSP data were used to identify the regions of AAC interaction with CL. The negatively charged head groups were found to bind largely at the same sites, which also contain positively charged residues, but some inconsistent and unusual features were also observed. First, the NMR titration data reveal that CL binding is in fast exchange; that is, CL molecules are not tightly attached to AAC3 in contrast to all previous studies that showed essentially irreversible binding. Second, the acyl chains of bound CLs traverse through the midpoint of the membrane to interact with the cytoplasmic side of AAC3. The resulting stretched conformation of the acyl chains is unprecedented. Third, NOE data show that the acyl chains are interacting with residues that are involved in binding of the head groups, again showing that they are not tightly bound in contrast to other studies. A likely explanation of the interaction data of Zhao et al. is that the interaction is primarily electrostatically driven, and that other important interactions are lacking. This interpretation would explain why the uncharged lipid does not produce detectable NMR spectral changes, and mirrors the situation of the electrostatically driven interactions of GTP and ATP to GGC1 and AAC3.
Fatty acid binding has also been investigated in uncoupling proteins, UCP2141 and UCP1119 in DPC. As UCPs are involved in fatty acid transport or flipping as part of the proton transport mechanism, studying these interactions is of direct functional importance. Both studies have used NMR titration experiments to identify a fatty-acid binding site at the interface between helices H1 and H6 on the matrix side of UCP1 and UCP2. Electrostatic interactions between the positively charged groups and the negatively charged carboxylic FA headgroup appear important for these interactions, as revealed by mutagenesis experiments.141 This is remarkable, however, because the fatty acid binding site overlaps with the highly conserved CL binding site.139,155 In fact, the residues interacting with the carboxylic headgroup are completely conserved between UCP1 and AAC1, even though the latter has no fatty acid flipping or transport activity. In the UCP2 study,141 the NMR sample contained CL; that is, the fatty acid has replaced CL in this sample, while in the UCP1 study119 no CL was present. The affinities in both cases were found to be very low (700 and 600 μM, respectively). The possible partitioning of fatty aids into micelles in the titration experiment makes these values an upper limit. Nonetheless, it is remarkable that the CL affinity in the UCP2/DPC sample is apparently very low, as it can be replaced by fatty acid readily. This is in contrast to the tight binding of CL to UCP1 extracted from the native membrane, which cannot be removed even after extensive washing with lipid-containing buffers.154,155 The unexpectedly low CL affinity mirrors the case of AAC145 discussed above, and might be explained by the loose structure (cf., Figure 7).
Taken together, the interactions of mitochondrial carriers in DPC show some expected features as well as several properties that are in contradiction to their behavior in lipid bilayers. The different carriers studied in DPC (GGC1, SCaMC1, AAC3, UCP2) interact with their respective substrates and with cardiolipin. However, these interactions appear to be nonspecific and likely driven by electrostatics; the binding affinities are greatly reduced and the specificities abolished. These observations point to a disrupted tertiary structure, as evidenced also by the TSA data (cf., Figure 8). We discuss below that signs of disrupted tertiary structure and high flexibility are visible in available NMR data.
4.1.1.3. Flexibility of Mitochondrial Carriers in DPC
NMR spectroscopy can directly report on dynamics on the level of individual residues. Several recent reports highlight that MCs in DPC are highly flexible on a wide range of kinetic time scales. Sounier et al. have reported an extensive analysis of NMR residual dipolar couplings (RDCs) of the GDP/GTP carrier (GGC1) in DPC.143 The authors reported that the RDC data from different parts of the structure, even on the same TM helix, cannot be fitted to a common molecular frame. In other words, the different parts of the molecule are highly flexible relative to each other, both in the presence and in the absence of substrates.
Further evidence that MCs in DPC deviate from the well-defined conformation observed in the aborted c-state crystal structures comes from an analysis of residue-wise backbone conformations. The NMR chemical shifts of backbone atoms depend on the local dihedral angles, and they thus provide direct access to the local secondary structure formation. Unlike the case of the crystal structures of AAC (Figure 6), there are extended parts of TM regions in DPC-solubilized MCs with nonhelical conformation, as reported for AAC3,144 GGC1,143 SCaMC1,142 and UCP1,119 discussed in ref (146). For the case of GGC1, these apparent nonhelical parts were shown directly to have increased flexibility, using NMR 15N R2 relaxation data143,146 (in particular, helix H4 for GGC1). These more flexible parts furthermore display increased solvent-accessibility, as probed by solvent-paramagnetic relaxation enhancement experiments;146 that is, they are not stably embedded in the micellar environment. Additional evidence for high flexibility comes from hydrogen–deuterium exchange experiments, which probe the local secondary structures. H/D exchange in GGC1 has been reported to be very fast, again supporting the view of highly mobile secondary structure elements. All of these observations indicate that MCs in DPC are significantly more flexible (on submillisecond time scales) than expected from the crystal structures.
A particularly interesting aspect of dynamics of MCs is the mobility on a time scale of hundreds of microseconds to a few milliseconds, because this time scale is comparable to the rate of solute transport.182 Brüschweiler et al.144 have studied microsecond–millisecond motions in yeast AAC3, and Kurauskas et al.146 studied additionally such motions in GGC1, ornithine carrier ORC1, and mutants of GGC1 and AAC3, in the presence of different substrates, inhibitors, and cardiolipin, probed by solution-state NMR relaxation-dispersion methods. All three proteins undergo extensive motions, on a time scale of ca. 1 ms, that involve about one-half of the protein in each case. The exchange rate constant in AAC3 is only slightly changed upon addition of inhibitor (CATR) and substrate (ADP), and the significance of this change has been questioned.183 Given the very strong abortive effect of CATR, the very modest (if not insignificant) effect on dynamics is surprising. Mutants of GGC1 and AAC3, which are nonfunctional, retain the same dynamics, further suggesting that the motion is not directly related to function, but that it might rather correspond to motions within a partly unfolded ensemble.146
In light of the highly flexible nature of MCs revealed by these NMR data, it is instructive to revisit the paramagnetic relaxation enhancement (PRE) data obtained with four different samples of UCP2 in DPC with nitroxide spin labels at four different positions, that is, at residues 68, 105, 205, and 255 of UCP2 (Figure 10). The PRE effect decreases proportionally to r–6, where r is the distance between the paramagnetic atom and the nuclear spin.185 Because the PRE data are correlated directly to the restraints imposed (deposited PDB data file LCK2), it is possible to verify whether the magnitude of the PRE effect correlates with the distance from the residue to the paramagnetic atom (Figure 10), and whether the observed PRE effects are in agreement with the known distance limits that this method can reliably detect. Of the 452 reported data for amide sites in the four differently labeled samples, 306 show no PRE effect, and therefore have no distance information. Of the remaining 146 PRE effects, 31 are on the same secondary-structural element, giving the strongest PRE as expected, but they provide no distance information with respect to the tertiary fold. Of the 115 that do, 56 PRE effects are observed at distances for amides that are more than 23 Å away from the paramagnetic atom (Figure 10). This distance, 23 Å, is to our knowledge the largest distance observed with MTSL-based PRE experiments of this kind and for a similar-size system,184,185 and is thus a reasonable upper limit for the observation of PRE effects. The fact that many PRE effects are observed up to ∼35 Å is, therefore, surprising. When the distances imposed by the restraints are plotted against the measured distances of the UCP2 model, the correlation has a slope of 2.5 rather than 1, meaning that PRE effects are observed at much greater distances than would be expected. This finding suggests that in DPC, UCP2 undergoes motions of significant amplitude, and in some of the temporarily populated states the respective amide site and paramagnetic labels are in close proximity, thus inducing paramagnetic bleaching. Some of these are found on the same α-helix as the paramagnetic atom, indicating that even the secondary structure is not stable. Moreover, for another five residues for which data are reported, one would have expected to see PRE effects, but none were observed (cyan arrow heads in Figure 10).
Figure 10.

Analysis of the paramagnetic relaxation enhancement data of the solution NMR structure of the mouse uncoupling protein UCP2. Panels A–D show the paramagnetic relaxation enhancement (PRE) data for the labeling with the nitroxide spin label MTSL (yellow spheres) attached to residues 68, 105, 202, and 255 of UCP2, respectively. The structure of UCP2 is shown in a tube representation in a rainbow color scheme from the N-terminus (blue) to the C-terminus (red). The structure and PRE restraints have been obtained from Protein Data Bank entry 2LCK. The size and color of the spheres, placed at amide proton sites, reflect the magnitude of the PRE effects, from large red (strongest PRE effects) to small pink spheres. The residues for which no PRE effects were reported are shown as black spheres. The numbers represent the distances from a given amide proton to the paramagnetic center (the nitrogen of the MTSL radical). The numbers are shown only for residues that are more than 23 Å from the paramagnetic center, which is the longest distance for which PRE effects have been reported.184,185 (E) Correlation between the imposed distances from the restraints and the measured distances in the structure of UCP2. The diagonal straight line is where the two distances are equal, and the red line is the linear correlation between the two sets of distances, which has a slope of 2.6. The dashed line indicates the 23 Å distance cutoff.
Taken together, the PRE data suggest that refolded UCP2 in DPC, inhibited by GDP, does not form a single defined structure, but a highly dynamic set of loose structures. Thus, it appears likely that UCP2 in DPC micelles has significantly more structural heterogeneity than represented in the well-defined bundle deposited in the Protein Data Bank.
4.1.1.4. Insights into Mitochondrial Carrier Structure and Dynamics from MD Simulations
Molecular simulations have proven a very valuable tool to rationalize the structure and dynamics of membrane carriers in both lipid bilayers and detergents.119,120,145,146,177−179,186−200
The vast majority of MD investigations were devoted to the ADP/ATP carrier embedded in a lipid bilayer, using either the bovine147 or the yeast AAC148 crystal structures as a starting point. All of the trajectories reported so far consistently described only a marginal evolution of the initial structure, suggesting that (i) CATR does not impose an incommensurable deformation on the protein c-state, and (ii) that the structures obtained in either LAPAO or DDM are compatible with a membrane environment. The importance of the electrostatic funnel formed by the cavity of the carrier has been studied further by simulations of the binding of ADP.177,178 The modeled binding motif has proven to be consistent with a body of biochemical data and sequence analyses.152,172,173,175
The UCP2 structure obtained by Berardi et al.118 was investigated through extensive MD simulations by Zoonens et al.120 From the onset, MD simulations of the protein restrained to its NMR structure and embedded in a lipid bilayer reveal that it allows a large number of water molecules to flow through it, reminiscent of α-hemolysin,201 which appears to be incompatible with its presumed biological function (Figure 7C). Furthermore, bereft of harmonic restraints, the membrane carrier collapsed in the lipid bilayer, suggesting that the structure obtained in DPC is not biologically representative. Additional molecular simulations in DPC micelles indicate that the unusual protein fold is stabilized by the spatial arrangement of the detergent molecules not only around the carrier, but also in its central pore and in the interstices separating TM segments (Figure 7C and D).
Molecular simulations were also employed to examine the propensity of the membrane carriers to bind in a specific fashion cardiolipins, an important component of the mitochondrial membrane.145,197,198,200 Comparing MD simulations in a POPC bilayer and NOE data recorded in DPC samples, Zhao et al.145 concluded that the detergent environment preserves the specific association of cardiolipins to AAC, at the headgroup binding sites highlighted by X-ray crystallography.147,148,181 Interestingly enough, in this work, the cardiolipins at play appear to be in an all-trans conformation, and remain so throughout the simulation. In stark contrast, a set of recent theoretical investigations underscore the significant flexibility of the cardiolipin acyl chains, which do not extend beyond the lipid leaflet on the matrix side.197,198,200 These studies suggest that, to fit the NOE data in DPC,145 the structure of the mitochondrial carrier must necessarily differ from the crystallographic conformation.147,148,181 Recently, Zhao et al. investigated the binding of a long-chain fatty acid to UCP1 with all-atom MD simulations.119 They built an homology model using the UCP2 structure as a template. Starting with three fatty-acids binding the surface of UCP1, they observed that only one remains associated after 50 ns, at a position that gave rise to a PRE signal. Yet, the conformational evolution of their homology model is not discussed and cannot be inferred solely from the binding property of the protein. Interestingly enough, Zoonens et al. have shown that in UCP2, the GDP inhibitor remains associated irrespective of the structure collapse.120
4.1.1.5. Conclusions about the Conformation of MCs in DPC
MCs have been extensively studied in DPC, and common trends emerge from these different structural, functional, and dynamic studies. In DPC, MCs retain a large part of their secondary structures, although some TM parts are disordered, and undergo motions on a picosecond–nanosecond time scale (as revealed by spin relaxation NMR measurements). Molecular-dynamics simulations highlighted the interplay between MCs and DPC and revealed how detergent molecules can diffuse between α-helical TM segments and maintain a distorted conformation, which collapses in a lipid environment. Thermostability shift assay experiments showed that MCs in DPC lack a cooperative unfolding transition, implying that the tertiary contacts are not stably formed. MD simulations revealed how DPC molecules penetrate between TM α-helices, stabilizing a distorted conformation that collapses in a model lipid bilayer. MCs undergo extensive dynamics on the microsecond–millisecond time scale, in a manner that is hardly affected by substrates, inhibitors, or severe mutations. The unexpectedly long-range PRE effects observed in UCP2 further support the view of a highly dynamic protein ensemble. While these data suggest that MCs in DPC are not correctly folded, interactions with substrates, inhibitors, and lipids have been reported, which suggest a functional fold. However, these interactions occur with much lower affinity, and lack the expected binding specificity. Unspecific electrostatic interactions are the likely reasons for these observations; such interactions do not rely on an intact tertiary fold, and may occur even in a loose ensemble of secondary structure elements.
4.1.2. Diacyl Glycerol Kinase (DgkA)
DgkA catalyzes the phosphorylation of diacylglycerol (DAG) by Mg-ATP to form phosphatidic acid.202 It was among the first integral membrane enzymes to be solubilized, purified, and mechanistically characterized.203 A solution-state NMR structure of the trimeric DgkA has been obtained in a DPC micelle environment,102 and three different X-ray crystal structures including a wild type (WT) and two thermally stabilized mutant structures were all obtained from a monoolein LCP.204 There is also limited Oriented Sample ssNMR data on DgkA in liquid crystalline lipid bilayers205 and MAS solid-state NMR investigations of its conformation.206
The solution NMR characterization was a heroic effort for such a large MP structure in 2009.102 The sample for structural study was shown to be functional at 37 °C, albeit with low affinity for substrate. The NMR experiments were collected at 45 °C. The result from a somewhat under-determined set of restraints, however, was a structure that was very different from that of the crystal structure determined in LCP (Figure 11).204 In the solution NMR structure, helices 1 and 3 are domain-swapped such that these helices primarily interact with helices from different monomers. Few examples of domain swapped TM proteins are present in the Protein Data Bank, including a solution NMR structure of the hepatitis C viral p7 protein,207 which is discussed further in this Review. Importantly, the TM helices of the solution DgkA NMR structure have an outward curvature giving rise to a barrel shaped structure that, as discussed earlier in this Review, is a potential artifact arising from the detergent micelle. This is in sharp contrast to the cylindrical nature of the crystal structure. Indeed, it appears that native-like MP structures may have a slight hourglass shape for TM helical bundles. This may result from the very low dielectric environment of the membrane interstices that strengthens and, consequently, shortens the helical hydrogen bonds that face the low dielectric fatty acyl environment. Moreover, these outward bowing helices could be induced by hydrophilic residues facing the fatty acyl environment (residues that should be oriented toward the interior of the helical bundle). Such residues could be “reaching” for the micellar hydrophilic surface that would not be accessible in a lipid bilayer.3 For the solution NMR structure, this outward curvature of the helices is therefore opposite to the natural tendency for the TM helices in a lipid bilayer environment. Here, in the DgkA solution NMR structure, helix 3 has no hydrophilic residues near the helical kink in the middle of the TM helix, and yet there is a broken hydrogen bond between Val101–Ile105 exposing the electrophilic carbonyl oxygen of Val101 to the micellar environment. This kinked helix resulted in a substantial tilt for both segments of this TM helix relative to the bilayer normal in conflict with the X-ray structure, which suggested a uniform helical structure and only a very small tilt relative to the bilayer normal.
Figure 11.
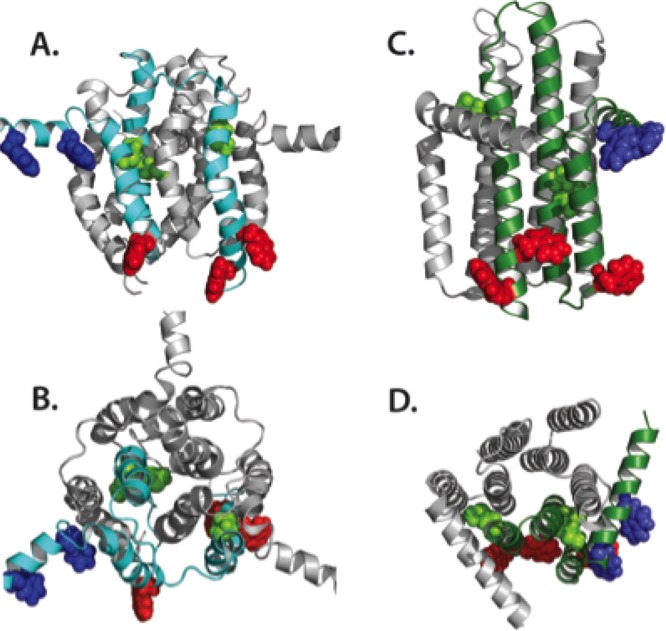
Structures of DgkA: cytoplasmic surface is at the top for the side views, and the end views are from the cytoplasmic surface. In each structure one monomer is highlighted with a colored backbone ribbon. (A and B) Views of the solution NMR structure in DPC micelles (PDB: 2KDC). (C and D) Views of the X-ray crystal structure in monoolein cubic phase (PDB: 3ZE4). TM helix tryptophan residues are in red, amphipathic helix tryptophan residues are in blue, and methionine residues are in green. (Reprinted with permission from ref (208). Copyright 2014 American Chemical Society.)
The wild-type DgkA structure obtained from X-ray diffraction is a triumph for the monoolein cubic phase sample preparation. Like the solution NMR structure, it is trimeric, but unlike the solution NMR structure there is no domain swapping of the TM helices that have a very uniform backbone structure, characteristic of most TM helices. For the WT crystal structure, the amphipathic helices (for two of the three monomers) are positioned approximately parallel to what would be the bilayer surface (defined via the bilayer normal that is assumed to be parallel to the trimeric axis), and the hydrophobic surface of the amphipathic helix faces appropriately toward the TM helix and fatty acyl environment. The packing of the amphipathic helix next to the trimeric helical bundle appears to be very reasonable as Ser17 of the amphipathic helix hydrogen bonds with the lipid facing Ser98 of helix 3.
An MAS ssNMR spectroscopic study of DgkA in liquid crystalline lipid bilayers (E. coli lipid extracts) assigned 80% of the backbone, a near complete assignment of the structured portion of the protein.206 The isotropic chemical shift data suggested that the residue makeup for the TM helices was nearly identical to that in the WT crystal structure. However, the positions of the nonhelical TM2-TM3 loop varied in the LCP environment for the WT (3ZE4) crystal structure from 82–90 to 86–91 for the mutant having 4 thermal stabilizing mutations (3ZE5), and to 82–87 for the mutant having 7 thermal stabilizing mutations (3ZE3), while the MAS ssNMR study found the nonhelical loop to be residues 81–85 for the WT. By contrast, the DPC micelle structure had the longest loop, between residues 80–90.
Limited OS ssNMR data were published prior to the solution NMR and X-ray crystal structures generating a fingerprint for residues in the amphipathic helix (Trp18 and Trp25), TM1 (Trp47), TM2 (Met63, Met66), and TM3 (Met96, Trp117).205 These observed resonances directly reflect the orientation of the backbone 15N–1H bonds with respect to the bilayer normal by correlating the 15N–1H dipolar interaction with the anisotropic 15N chemical shift. For α-helices, the N–H vector is tilted by approximately 17° with respect to the helix axis, and therefore helices that are parallel to the bilayer normal will have large 15N–1H dipolar coupling values of approximately 18 kHz along with large values of the anisotropic chemical shift values, while an amphipathic helix will be observed with half-maximal values of the dipolar interaction and minimal values of the anisotropic chemical shift. Because TM helical structures are remarkably uniform in structure,54,61 it is possible to predict the OS ssNMR anisotropic chemical shifts and dipolar couplings from PDB coordinates. Figure 12A,B shows the OS ssNMR experimental data (contours) as compared to the predictions (ovals) from the structures. Predictions from the solution NMR structure are shown in Figure 12A,B, and the predictions from the X-ray structures are shown in Figure 12C–H. Note that for the crystal structures there is more than one prediction for a residue due to differences between the monomers of a trimer arising from crystal contacts that perturb the 3-fold symmetry. While the calculated resonance frequencies from the solution NMR structure bear no resemblance to the observed spectra, the calculated frequencies from the WT crystal structure (3ZE4) are virtually identical to the observed values, supporting that the crystal structure, but not the solution-NMR structure, is indeed the conformation found in lipid bilayers.
Figure 12.

Comparisons of predicted OS ssNMR resonance frequencies from the DgkA structures with the 15N tryptophan and methionine labeled DgkA experimental data for methionine and tryptophan sites in a liquid crystalline lipid bilayer environment. Methionine resonance contours are green, TM tryptophan resonances are red, and amphipathic helix tryptophan resonances are blue. (A and B) Comparison with the solution NMR structure (PDB: 2KDC). M63 and M66 fit well with the experimental data, and W18 is not too far from one of the amphipathic helix experimental resonances, but the other resonances are not in agreement. (C,D) Comparison with the wild-type DgkA X-ray structure (PDB: 3ZE4). The A (green, red, blue) and C (black) monomers were used for the predictions. The amphipathic helix of monomer C did not diffract well enough for a structural characterization. Structure (PDB 3ZE5) using monomers A (green, red, blue) and B (black). (E,F) Comparison with the thermally stabilized (4 mutations) DgkA X-ray structure (PDB 3ZE5) using monomers A (green, red, blue) and B (black). One of the mutations is M96L, and therefore this resonance is not predicted. (G and H) Comparison with the thermally stabilized (7 mutations) DgkA structure (PDB 3ZE3) using monomers A (green, red, blue) and B (black). Two thermal stabilization mutations affect this spectrum, M96L as in 3ZE5, and A41C. (Reprinted with permission from ref (208). Copyright 2014 American Chemical Society.)
However, thermal stabilizing mutations that are often required for MP crystallizations did induce significant local distortions that caused dramatic deviations for the predicted resonances (Figure 12E–H). W47 and W117, which are located near the cytoplasmic termini of TM helices 1 and 3, are significantly influenced by these mutations. Most significantly, the indole N–H group of W47 in the WT structure is oriented toward what would be the bilayer surface as is typical of tryptophan residues that stabilize the orientation of MPs by hydrogen bonding from the TM helices to the interfacial region of the lipid bilayer. However, in monomer B of 3ZE3, which has 7 thermostabilizing mutations, the indole ring is rotated by ca. 180° so that the ring intercalates between helices 1 and 3 of the neighboring trimer in the crystal lattice and the indole N–H hydrogen bonds with the sulfhydral group of the hydrophobic to hydrophilic mutation, A41C. This emphasizes the hazards of thermostabilizing mutations that are used extensively in X-ray crystallography.
4.1.3. Tryptophan-Rich Translocator Protein (TSPO)
The 18 kDa-large translocator protein (TSPO), previously known as the peripheral benzodiazepine receptor, is a MP highly conserved from bacteria to mammals.208 In eukaryotes, TSPO is found primarily in the outer mitochondrial membrane and is thought to be involved in steroid transport to the inner mitochondrial membrane. TSPO also binds porphyrins and can catalyze porphyrin reactions.209−211 TSPO function in mammals remains poorly understood, but it is an important biomarker of brain and cardiac inflammation and a potential therapeutic target for several neurological disorders.212,213
Two NMR structures of mouse TSPO (MmTSPO) solubilized in DPC have been determined,214 one of wild-type214 and another of a A147T variant known to affect the binding of TSPO ligands.215,216 These structures can be compared to 10 X-ray crystallographic (XRC) structures in LCP or the detergent DDM. The XRC constructs were derived from the Gram-positive human pathogen Bacillus cereus (BcTSPO)211 or the purple bacteria Rhodobacter sphaeroides (RsTSPO)217 and crystallized in LCP or DDM in three different space groups.
The amino acid sequence of MmTSPO is 26% and 32% identical to that of BcTSPO and RsTSPO, respectively, whereas the bacterial TSPOs are 22% identical to each other. This sequence conservation predicts that there would not be large structural differences among the bacterial and eukaryotic TSPOs.218 Function also appears to be well conserved because rat TSPO can substitute for TSPO in R. sphaeroides,219 and BcTSPO and TSPO from humans and Xenopus tropicalis can catalyze degradation of protoporphyrin IX in vitro.211 In addition, RsTSPO retains cholesterol binding even though R. sphaeroides has only steroid-like hopanoids.220 Despite the conservation of sequence and function, large differences are seen in the structures of MmTSPO in DPC when compared to the bacterial TSPOs in DDM or LCP. The BcTSPO and RsTSPO structures were crystallized in several different space groups, in monomeric and oligomeric forms, and with and without bound PK11195, yet all of those structures are more similar to each other than to the MmTSPO structures (Figure 13). Whereas the largest Cα RMSD between the two bacterial proteins (BcTSPO versus the complete A139T RsTSPO structure) is 2.2 Å, the DPC-embedded structures of wild-type MmTSPO and the A147T variant of MmTSPO differ from the BcTSPO and RsTSPO structures by 4.1–4.6 and 3.7–3.9 Å, respectively. Although the greater dependence of NMR structure calculations on force fields may be a contributing factor, the quality of the structures determined in DPC is also dramatically lower than for those solved in LCP or DDM. The average Clashscore and overall MolProbity scores221 for the MmTSPO ensemble determined in DPC place it in the 12th and 7th percentiles, respectively, whereas the corresponding percentiles for the 10 structures solved by XRC are 95% and 97%.
Figure 13.
(A) Overlays of TSPO structures. Left: BcTSPO in DDM (4RYJ), overlaid with RsTSPO in LCP (4UC2). Middle: BcTSPO in DDM (4RYJ), overlaid with MmTSPO in DPC (2MGY). Right: A A147T variant of MmTSPO in DPC (2N02), overlaid with wild-type MmTSPO in DPC (2MGY). Whereas the structures in DDM and LCP are very similar (left), and the structures in DPC are very similar (right), the structures in DPC are different from those in LCP or DDM (middle). The Cα RMSDs indicated above the overlays were determined using the PyMol cealign function. (B) Cartoon representations of BcTSPO in LCP (left) and MmTSPO in DPC (right) showing a rotation of the fourth TM helix (TM4). Coloring is from blue (N-terminus) to red (C-terminus). To illustrate the rotation of TM4 by >120°, the side-chain atoms of three amino acids D111, L113, and L114 (MmTSPO residue numbering), which are conserved between MmTSPO and BcTSPO, are shown as spheres. D111, which packs against TM helix 5 (TM5) in BcTSPO in LCP, packs against TM helix 3 (TM3) in MmTSPO in DPC. Both structures were determined in the presence of bound ligand PK11195, which is shown in sticks.
There is a qualitative difference between the structures shown in Figure 13B that reflects a similar comparison in DgkA. The helices in the DPC environment are curved, reflecting a weak hydrophobic environment. In lipid bilayer environments, such helical bends in helices are atypical because of the low dielectric environment.62 One of the significant good qualities of the LPC environment is that it is highly hydrophobic. Consequently, as in the DgkA LPC structure, the LPC structure of BcTSPO has uniform helical structures reflecting strong hydrogen bonds in a hydrophobic environment.
A potential contribution to the structural discrepancies between the TSPO structures is the need to refold MmTSPO from E. coli inclusion bodies. Whereas BcTSPO and RsTSPO were inserted into membranes upon expression, the MmTSPO inclusion bodies were first solubilized in sodium dodecyl-sulfate (SDS), in which the protein is known to be unfolded and does not bind PK11195,222 and then exchanged into DPC. The protein prepared in this way further requires PK11195 to achieve stable tertiary protein interactions.214,223−225 In the absence of PK11195, MmTSPO adopts a molten globule-like structure with helices but no tertiary structure.223 By contrast, the apo form of a bacterial TSPO has been crystallized in both DDM and LPC.211 The differences in the stability of the apo forms have been taken to mean either that DPC destabilizes TSPO structure,217 or that MmTSPO structure and stability have diverged significantly from the bacterial homologues.216 The decreased stability and ligand binding affinity of the MmTSPO in DPC is reminiscent of the observations for the mitochondrial carriers discussed above, and the structural rearrangements of the TSPOs in the context of high sequence conservation point toward DPC-induced distortions of the MmTSPO structure. However, the refolding process for MmTSPO and the sequence differences between species are confounding factors, and it is necessary to determine structures of mammalian TSPO in milder detergents or LCP, or alternatively test the stabilities of unliganded bacterial TSPOs in DPC.
4.1.4. Hepatitis C p7 Channel Protein
Around 3% of the world’s population carries the hepatitis C virus (HCV), putting more than 200 million people at risk of developing liver disease. The HCV protein p7 is a viroporin that oligomerizes to form ion channels and is required for the assembly and secretion of infectious virus particles,226−228 making it an attractive drug target. Despite its therapeutic potential, the self-assembly of p7 into a functional viroporin and the molecular mechanisms that underlie ion channel activity remain poorly understood. A low resolution envelope of the p7 oligomer in the mild detergent diC7PC was calculated from negative-stain electron microscopy in 2009.229 In 2013, a high-resolution structure of p7 reportedly in an oligomeric form was published.207 This structure, determined by solution NMR spectroscopy of p7 (strain EUH1480) in DPC, failed to confirm several aspects of the known functional properties of the channel and was at odds with previous structural studies of the monomer and computational studies of the oligomer. The differences likely arise from the disruptive effects of DPC.
P7 is a relatively small protein of 63 amino acids, and several groups have investigated the structural properties of p7 in various membrane mimetics using NMR methods often combined with theoretical modeling.230−237 In one of the earliest studies, Patargias et al. elaborated a model based on secondary-structure prediction and protein–protein docking algorithms, resulting in an α-helical hairpin conformation of the TM domain.230 This monomeric structure served as a building block for construction of a putative pore-containing oligomer, which was validated by docking of the known inhibitor amantadine to residue His17 in the pore. Combining solution-state NMR and molecular dynamics simulations, Montserret et al. identified the secondary-structure elements of p7, and constructed a three-dimensional model of the monomer in a lipid bilayer.231 Remarkably, the resulting hairpin conformation of the protein was very similar to that inferred in silico by Patargias et al. The monomeric structure of p7 was subsequently utilized to build models of hexamers and heptamers, two likely oligomeric states found in the endoplasmic reticulum membrane, which were shown to function as ion channels in MD simulations.232
With the exception of the study of p7 in DPC, the large number of studies using wet-lab approaches and/or simulation are broadly consistent with each other in describing two hydrophobic TM regions that fold via a conserved basic loop region into hairpin-like structures (reviewed in ref (239)); for oligomeric models, the imidazole group of His17 is invariably placed into the channel pore.230−232,235,240,241 Instead of the expected hairpin conformation, the p7 subunits within the DPC-based oligomer adopt extended “horseshoe-like” conformations with each monomer making extensive intermolecular contacts and no long-range intramolecular contacts (Figure 14A). In vitro studies of p7 in liposomes have shown that monomers freely interchange between channels.242 However, the oligomer arrangement of OuYang et al., in which subunits crossover each other at about the midpoint of the peptide, results in a interwoven fold that raises questions as to how such a structure could exchange subunits within a membrane context, or indeed fold in the first place.239 Another controversial feature of the DPC-based p7 oligomer was the placement of His17, which pointed out and away from the oligomer rather than into the channel pore (Figure 14B), in contradiction with mutagenesis and Cu2+ inhibition studies indicating a key role for His17 in ion permeation.242,243 Perhaps the most striking aspect of the p7 oligomeric structure is the polarity of the lipid fatty-acyl facing protein surface, which would be energetically unfavorable in a lipid membrane, having three Arg side chains per monomer in what should be a hydrophobic environment (Figure 14A,C). As expected from its surface features, the p7 oligomer of OuYang et al. inserts poorly into simulated membranes and causes significant perturbations to the lipid bilayer244 (Figure 15A,B).
Figure 14.

(A,B) Cartoon representations of the p7 oligomer structure determined in DPC207 as viewed from within the membrane (A, left), from above the membrane (A, right), or from below the membrane (B). Two subunits are colored green and yellow to illustrate the extended, staple-like arrangement of the subunits, and to illustrate that adjacent subunits are arranged in a nearly parallel orientation with a single crossover point that results in an interwoven arrangement. In (A), Arg54, Arg57, Arg60, and the carboxyl containing C-terminal residue Ala63 are represented for two subunits as sticks to show charged groups that would be exposed to the hydrophobic region of the membrane. A double-arrowed vertical line corresponding to 30 Å is shown at the left. In (B), the residue H17, which is known to be involved in ion conduction but is not in the channel pore in the DPC-based structure, is represented as spheres. In (C), the electrostatic potential is mapped onto the surface of the p7 oligomer and indicates significant polarity on the surface region expected to be embedded in the hydrophobic interior of the membrane. The figure was generated using APBS,238 as implemented into PyMOL APBS Tools and displayed using a charge range from −4.0 to 4.0.
Figure 15.

Molecular-dynamics simulation of p7 oligomers embedded in a lipid bilayer. Membrane insertion of the hexameric structure of p7 reported by Chou and co-workers207 predicted from (A) MemProtMD195 and (B) a molecular-dynamics trajectory of 150 ns starting from the protein inserted in a thermalized lipid bilayer.236 Membrane insertion of the hexameric structures of p7 reported by (C) Foster et al.240 and (D) Chandler et al.232 The phosphate and choline moieties are depicted as yellow and ice blue spheres, respectively. The lipids tails are depicted by gray licorice. The protein is represented in cartoon with hydrophobic, polar, and basic residues colored white, green, and blue.
Small-molecule binding studies of p7 in DPC also support the hypothesis that the p7 structure in DPC is different in functionally important ways from that in membranes. Breitinger et al. measured the rimantadine IC50 values for four p7 constructs derived from a diverse set of HCV genotypes (1a–4a) and found that the IC50 values for proton conduction ranged from 0.7 to 24 nM.245 In contrast, the Kd values measured for rimantadine binding to p7 in DPC were 13 and 64 μM.207 Because an IC50 sets an upper limit for the corresponding Ki,246 there is a difference of 3 orders of magnitude or more between the rimantadine Kd in DPC and the Ki in membranes. Such a large discrepancy is unlikely to be accounted for by the small differences in solution conditions (salt, pH, etc.).
The unusual properties of the p7 oligomer structure prompted several computational studies. Using strains H77, J4, and EUH1480, Kalita et al. performed MD simulations in a lipid bilayer of the p7 protein in both its monomeric and its hexameric forms.235 These simulations revealed a partial collapse of the oligomeric architecture and full occlusion of the central pore over a 400 ns time scale. In an attempt to address the origin of the discrepant p7 oligomeric structures, discriminating between the effects of the strain and of the solubilizing agent, a series of molecular simulations was carried out in detergent and membrane environments, using strains J4 and EUH1480 projected onto α-helical hairpin231,240 and the horseshoe-like207 conformations, in conjunction with NMR spectroscopy236 (Figure 14B,C). The simulations underscored the crucial role played by the environment in shaping the monomeric structure of p7,247 with the lipid bilayer exhibiting a pronounced tendency to stabilize α-helical hairpin motifs, irrespective of the strain.
Assessing the biological relevance of the p7 oligomeric structure is important for the design of p7 inhibitors,237,248 but attempts to rationalize the unusual architecture of the OuYang et al. p7 oligomer in the context of the behavior of p7 in membranes have been unsuccessful thus far. Because DPC is known to weaken protein oligomerization,249,250 consideration of the procedure for determining the intermolecular contacts in the p7 oligomer may be relevant. The intermolecular NOE distance restraints that were used to determine the p7 hexamer were obtained from a sample in which 15N–2H-labeled and protonated subunits were mixed. The interpretation of NOE signals between amide protons and aliphatic protons as intermolecular contacts relies on the assumption that in the 15N–2H-labeled monomers the aliphatic hydrogens are completely replaced by deuterium, which has been questioned previously.251 More recently, a structure of the transmembrane domain of the HIV envelope spike (env) was reported to be trimeric on the basis of gel electrophoresis and weak intermolecular NOEs that were observed using the same mixed label sample approach as for p7.252 However, a more thorough evaluation came to the conclusion that the protein was predominantly monomeric under a range of conditions including those reported to provide data for the trimeric complex.253 The authors of that paper argued that the discrepancies can be reconciled if a small amount of dimeric or higher order oligomers were present in rapid exchange with a mostly monomer population. Furthermore, the apparent size observed on SDS-PAGE, which led Dev et al. to the proposition of a trimeric state, may be ascribed to the well-established observation that α-helical TM peptides exhibit anomalous migration on SDS-PAGE.254−256
Interestingly, the same NOE approach that has been used to determine intermolecular contacts in HIV env and p7 has also been employed for obtaining the pentamer structure of the calcium-uniporter in tetradecyl phosphocholine detergent.257 Although no data have been reported that challenge the calcium-uniporter structure, it is interesting to note that the affinity of this protein to the ligand Ru360 in this detergent (Kd = 24 μM) is 4 orders of magnitude lower than that in bilayers (Kd = 2 nM).258 Such low affinities may point to nonspecific interactions, as revealed for the case of mitochondrial carriers (cf., section 4.1.1).
4.1.5. Phospholamban
4.1.5.1. Importance of PLN in Cardiac Regulation
Phospholamban (PLN) is a single-pass TM protein that regulates Ca2+ uptake in the sarcoplasmic reticulum (SR) upon binding the sarcoplasmic reticulum Ca2+-ATPase (SERCA) (Figure 16).259,260 PLN’s primary sequence comprises 52 amino acids, and is highly conserved among different species.261 Sequence analysis, mutagenesis, and functional assays established that PLN comprises four structural domains: domain Ia (residues 1–18), a flexible loop (residues 19–23), domain Ib (residues 24–30), and domain II (31–52).261 While the TM domain II and the juxtamembrane domain Ib constitute the inhibitory region responsible for reducing the apparent Ca2+ affinity of SERCA,262 PLN’s cytoplasmic domain Ia has a regulatory role and harbors two phosphorylation sites: Ser16 and Thr17. Upon sympathetic simulation, PLN is phosphorylated at Ser16 by cAMP-dependent protein kinase A, which increases Ca2+ uptake in the SR (positive chronotropy).259 Protein phosphatase 1 (PP1) dephosphorylates PLN, re-establishing its basal inhibitory effect on SERCA. PLN is also phosphorylated at Thr17 by calmodulin-dependent protein kinase II (CaMKII) with functional effects similar to Ser16 phosphorylation. A recent work by Akaike at al.263 showed that Thr17 phosphorylated PLN is dephosphorylated by the PP2Ce phosphatase. While Ser16 phosphorylation is linked to physiological β-adrenergic stimulation, CaMKII-dependent PLN phosphorylation has been associated with cardioprotective action in response to pathogenic situations such as acidosis and ischemia/reperfusion.264 In the SR membrane, PLN oligomerizes and forms stable homopentamers that act as “storage” for active monomers, which are unleashed upon interaction with SERCA.260 Although the cysteine residues in PLN’s TM domain are not involved in disulfide bridges, they are responsible for maintaining the structural integrity of the pentamer.265 Removing one (Cys41) or all three (Cys36, Cys41, and Cys46) cysteine residues causes the formation of active monomers that bind and regulate SERCA with functional effects similar to those of PLN wild-type (PLNWT).265
Figure 16.

NMR structural models of PLN. (A) Average structure of the NMR ensemble in DPC micelles (PDB: 1ZLL) representing the “bellflower” model, with the cytoplasmic domains projected toward the bulk solvent. (B) Average structure of the “pinwheel” model (PDB: 2KYV) obtained with a hybrid solution/solid-state NMR method. The amphipathic cytoplasmic domain Ia is adsorbed on the surface of the lipid membrane. (C) Top view of the bellflower model. The pore of the pentameric assembly is quite large, suggesting a possible channel for chloride or calcium ions. (D) Top view of the pinwheel model, where the pore is less than 2 Å in diameter, preventing the passage of hydrated ions. Note that in the pinwheel ensemble several conformers have the Gln26 side chains pointing toward the center of the pore as supported by REDOR experiments.266
4.1.5.2. PLN Structure in DPC Micelles
Given its great biological importance and relatively small size, PLN has attracted the attention of several structural biology groups. However, attempts to crystallize PLN in detergents have failed, and NMR has represented the only viable technique to investigate its structure and dynamics. The first structure of PLN in DPC micelles was obtained using the PLNAFA monomeric mutant,267 where the cysteine residues C36, C41, and C46 in domain II were mutated into alanine, phenylalanine, and alanine, respectively. This functional mutant mimics the inhibitory potency of PLNWT and adopts an overall L-shaped topology similar to that of PLN in organic solvent.1000 However, the calculations for the conformers-based NMR restraints did not converge to a unique topology (i.e., arrangement of the secondary structure elements relative to the membrane bilayer). In fact, the interhelical angle obtained by the structural ensemble is essentially ill-defined, due to the high mobility of the interhelical loop,268 and concomitant lack of long-range NOE contacts between the helical domains. To define PLN’s topology in DPC micelles, a combination of paramagnetic quenching and H/D exchange experiments was used, which helped define the azimuthal angle for domain Ia.267 Following this work, a complete structure of pentameric PLNWT was obtained in DPC micelle by Chou and co-workers.269,270 This structure was determined using state-of-the-art solution NMR techniques. According to these authors, pentameric PLN adopts an unusual bellflower assembly, with a leucine/isoleucine zipper keeping the quaternary arrangement between the TM domains of PLN. The pentameric structural ensemble was obtained at remarkable resolution, with 0.61 Å rmsd for backbone atoms and 1.10 Å for all heavy atoms.269 In this structural ensemble, the interhelical loop adopts a well-defined β-turn like conformation similar to that found in monomeric PLNC41F and PLNAFA structures obtained in organic solvent and DPC micelles, respectively. The topological arrangement of the cytoplasmic domains Ia, however, is rather uncharacteristic, with all helical domains Ia pointing away from the surface of the micelle and projected toward the bulk water. No long-range distances (NOEs) were observed between the helical domains Ia, and their relative orientation was determined entirely from orientational restraints derived from residual dipolar couplings (RDCs).269 Importantly, the quaternary arrangement of the bellflower structure (PDB code: 2KYV) presents a large internal pore, suggestive of a possible role of PLN as a selective ion-channel for either Ca2+ or Cl– ions. The ion-channel hypothesis for pentameric PLN was first put forward by Kovacs et al.271 and more recently reexamined by Smeazzetto et al.272,273 However, electrochemical measurements and theoretical calculations suggest that pentameric PLN does not conduct ions due to the hydrophobic coating in the pore, which makes ion conduction energetically unfavorable.274 Interestingly, molecular dynamics (MD) simulation studies performed by several groups reported that the bellflower structure with a large central pore is stable for only about 1 ns, as discussed at the end of this section.275−277
4.1.5.3. PLN Structure in Lipid Membranes
Initial studies of PLN in lipid membranes were carried by Arkin et al.,278 who proposed a continuous helix model in which domains Ia of each monomer are completely helical and protrude toward the bulk water (reviewed in ref (279)). These results were further supported by site-specific solid-state NMR (ssNMR) measurements.280−282 While the helical nature of PLNWT was confirmed in lipid bicelles and mechanically oriented lipid membranes,283,284 oriented ssNMR experiments revealed the L-shaped topology for both monomeric and pentameric PLN. The complete structures of both the monomer and the pentamer in lipid membranes were accomplished using a combination of oriented and magic angle spinning (MAS) ssNMR methods.285−287 The structures confirmed the pinwheel topology of PLN in agreement with fluorescence measurements.288 The high-resolution structures obtained in lipid membranes showed that domain II forms an ideal α-helix, without the pronounced curvature reported for the bellflower model269 or the distortions observed in organic solvent.289 The ideal character of this TM segment is in agreement with both experimental and theoretical studies of MPs.54,61 The amphipathic domain Ia is adsorbed on the membrane surface in both the monomeric and the pentameric structures, with the hydrophobic face pointing toward the hydrocarbon region of the bilayer and the hydrophilic residues toward the bulk water in agreement with the amphipathic nature of domain Ia. PLN’s arginine residues (R9, R13, and R14) form electrostatic interactions with the lipid head groups and keep the helical domain anchored to the surface of the lipid membrane. Using ssNMR under similar experimental conditions, Lorigan and co-workers reached identical conclusions regarding the structural topology of pentameric PLN.290−294 In addition to the unusual topology of domains Ia, another significant difference between the bellflower and pinwheel structural models is the pore at the center of the pentamer assembly that crosses the membrane. In the bellflower, the size of the pore is on average 2.5 Å, changing from 5 to 2 Å across the membrane. In contrast, the pore in the pinwheel model is on average 2 Å, with a tight hydrophobic conduit that spans 25 Å in length, making it an unlikely path for hydrated ions to cross the membrane bilayer.
4.1.5.4. Effects of DPC Micelles on PLN Conformational Equilibrium and SERCA Regulation
NMR spin relaxation studies of monomeric PLNAFA in DPC micelles suggested that the cytoplasmic helical domain Ia is significantly more dynamic than the TM domain Ib and domain II.268 Importantly, combined NMR experiments and functional assays carried out on PLN phosphorylated at Ser16 or with single-point mutants in PLN’s cytoplasmic domain Ia and loop region showed that the structural dynamics of PLN can be tuned affecting PLN’s inhibition of SERCA.295−297 Although attempts to map the inhibitory interactions between PLN and SERCA in DPC gave useful insights,298 the functional effects of PLN are attenuated by this detergent in terms of both apparent Ca2+ affinity (pCa) and Vmax of the ATPase, suggesting that DPC interferes with the functional interactions between the two MPs.298 In particular, the Veglia group discovered that DPC drastically affects the conformational equilibrium of PLN. Both EPR and ssNMR in lipid membranes detected the presence of at least two discrete states of PLN: a more populated membrane associated T state, and a sparsely populated, membrane dissociated R state.265,283,299 The existence of the conformationally excited R state was also identified and characterized by Baldus and co-workers using ssNMR studies.300 These researchers discovered that the membrane-dissociated R state is quite dynamic and essentially unfolded. Further studies emphasized the importance of the T-to-R state equilibrium of PLN for SERCA regulation301 as well as recognition by protein kinase A.302 More importantly, using ssNMR in lipid membranes, it was found that the interactions between PLN and DPC micelles skewed the conformational equilibrium toward the folded T state, which caused to underestimate the population of the R state.303,304 These effects are even more apparent in the presence of SERCA, which shifts PLN’s equilibrium toward a SERCA-bound state (B state),305 which is only detectable by ssNMR in the presence of lipid membranes.
4.1.5.5. MD Simulations of Phospholamban
A handful of theoretical studies have focused on the conformational dynamics of the monomeric form of PLN.306−314 Models of monomeric PLN embedded in lipid bilayers have been put forth by coupling solid-state NMR in lipids to rigid-body MD simulations.306,311 Interestingly enough, Sayadi et al., resorting to an implicit representation of the membrane, demonstrated that the PLN conformational equilibrium depends on the thickness of lipid bilayer.313 On the basis of unrestrained simulations, Manna and Mukhopadhyay have proposed that the conformation of the cytoplasmic domain of PLN can be modulated by the presence of cholesterol in lipid bilayers.312 Molecular simulations have also been employed to address the conformational transition of PLN in response to phosphorylation.308−310,314 All of the aforementioned studies have highlighted the reorganization of the cytoplasmic domain upon phosphorylation of Ser16 and/or Thr17.
However, the pentameric forms of PLN have been the object of a lesser theoretical effort.275−277,315 Kim et al. have modeled the bellflower structure of Chou and co-workers in a POPC bilayer.275 Their simulations revealed an uncorrelated dynamics of the cytoplasmic domains undergoing large conformational changes. The authors reached the conclusion that in a biological membrane, the pinwheel structure is more likely to be observed. Beccucci et al. have investigated the putative ion-channel function of PLN.276 They computed the potentials of mean force associated with the transport of sodium and calcium ions through both the bellflower and the pinwheel models. They reported free-energy barriers to ion translocation as high as about 20 and 40 kcal/mol for Cl– and Ca2+, respectively, ruling out an ion-channel function for PLN. Their MD simulations further revealed a rapid collapse of the bellflower structure embedded in a POPC bilayer, associated with the expulsion of all water molecules initially inside the pore. Maffeo and Aksimentiev, using steered MD, equally reached the conclusion that transport of ions through PLN is thermodynamically unfavorable.277 They compared the dynamics of the bellflower and the pinwheel models in a lipid bilayer using 10–6 s-long coarse-grained simulations, supplemented by all-atom MD. Consistently with the work of Veglia and co-workers,276 their trajectories demonstrated unambiguously that the bellflower structure is not compatible with a membrane environment, contrasting markedly with the structural stability of the pinwheel model. Maffeo and Aksimentiev also performed coarse-grained and all-atom simulations of the bellflower conformation in DPC micelles. Noteworthily, they found that DPC stabilizes the pentameric fold by penetrating inside the pore of the protein, a behavior reminiscent of that observed by Zoonens et al. for UCP2 (see section 4.1.1).120 The phosphorylated states of both the bellflower and the pinwheel PLN have been studied by Lian et al., relying on molecular simulations.315 Their study suggests that, in response to phosphorylation, both structures are modified and evolve toward similar conformations.
Although PLN studies in DPC micelles represented a step ahead with respect to organic solvent mixtures, the effects of this detergent on the helical structure of this small MP are substantial. In particular, DPC introduced significant deviations from ideal helices producing “banana-shaped” helical domains that adapt to the curved surface of the detergent as was previously observed for other amphipathic polypeptides.316−318 Importantly, the unusual bellflower topology has misled scientists to consider pentameric PLN as a potential ion channel for either Cl– or Ca2+ ions. The latter is probably due to the sparse interhelical NOE structural restraints used in the calculations. The positioning of domains Ia in the pentamer is another significant concern. By using paramagnetic mapping of PLN’s topology, Shi et al. were able to lift the degeneracy of residual dipolar coupling and correct PLN’s topology in micelles;319 however, distortions in the helical domains caused by PLN’s interaction with DPC were observed. Interestingly enough, MD simulations276,277 pointed out that the structure obtained in DPC was not consistent with a physiological membrane environment. Significant improvement in resolving the reported distortions was achieved by combining solution NMR data in micelles describing PLN’s secondary structures with ssNMR distances and orientational restraints (i.e., hybrid NMR approach)285,286,320 obtained in lipid environments. Nonetheless, the most significant data regarding the structure–activity relationship in PLN have been obtained with ssNMR (oriented and/or MAS) using lipid mixtures that faithfully reproduce the inhibitory activity of PLN with SERCA.
4.1.6. Potassium Channel KcsA
Potassium channels are responsible for the selective conduction of K+ ions across cellular membranes, and are central to numerous biological function such as electrical signaling and neurotransmission.321−323 The macroscopic current behavior of the most prominent member of this family, KcsA, has been described by four stages,324−326 including a nonconductive resting state, an activated state with highest peak current, induced by a drop of the intracellular pH below 5, and an inactivation phase. The mechanisms leading to interconversions between activated, inactivated, and resting states have been studied extensively. Crystal structures of KcsA have provided the basis for molecular understanding of their function.50,322 Additionally, function, ion binding, activation, and inhibition have been extensively studied by various techniques, including electrophysiology measurements in lipid bilayers,326−328 solution-state NMR in nanodiscs329 and different detergents (including DPC,330 SDS,331,332 and DDM333), solid-state NMR spectroscopy in lipid bilayers,334−338 infrared spectroscopy,339,340 and MD simulations.341,342 The large body of functional, structural, and dynamical data in different environments makes KcsA an interesting case for the investigation of detergent effects.
KcsA is a highly robust α-helical protein, as highlighted by the fact that it retains its tetrameric structure even in SDS, exposed to 75 °C for 60 min, at pH 10 or 4 M urea.331 From this observation, one may deduce that it retains a functionally relevant conformation in a range of detergents. Nonetheless, structural differences have been reported between DPC-solublized and membrane-embedded KcsA for the N-terminus of TM1. In lipid bilayers, residues Ser22 to Gly30 show clear α-helical chemical shifts in solid-state NMR experiments. Helical structure is also seen in the crystal structure starting from residue 23.343 In contrast, this helix is shorter in SDS and DPC, where helix starts only at Gly30.335 Consequently, the functionally important gating residue His25 adopts a helical structure in lipid bilayers but a nonhelical structure in micelles. Interestingly, the secondary chemical shifts observed in SDS match those observed in lipid bilayers much better (Figure S6 of ref (335)).
Interestingly also, the effect of a pH-drop is sharply contrasted in lipid bilayers and DPC. Upon a pH change from neutral pH (6.9) to pH 3.5, only rather modest chemical-shift changes have been reported in DPC in the key structural parts of KcsA, involving Y78, G79, E71, and D80,330 while these changes are much more pronounced in lipid bilayers (Figure 2b of ref (334)). An important effect of pH drop is the appearance of a kink in TM helix 2, which has been observed in the crystal structure of the open state of KcsA,344,345 by chemical-shift changes in lipid bilayers (around Gly99334) and SDS micelles,332 but, strikingly, not in DPC.330 These observations might indicate that in DPC KcsA samples more conformations (more dynamics), such that the structural transition upon pH drop is “blurred”. This view of enhanced conformational flexibility in DPC would be in line with the behavior of mitochondrial carriers (see section 4.1.1), and might be related to the absence of lateral pressure or intercalation of detergent molecules between helices.
Another important consideration is the K+ affinity. A solid-state NMR study in DOPE/DOPS lipid bilayers showed a dissociation constant of ca. Kd = 7 μM.336 Using ITC measurements in DDM detergent, Lockless et al. have reported a value of 0.4 mM, that is, ca. 60-fold higher.346 Using solution-state NMR in SDS micelles, Chill et al. reported a Kd of 3 mM, and Shimada et al. reported a value of 6.1 mM in DDM.333 The value in DPC has, to our knowledge, not been reported, but these data make clear that affinities may strongly depend on the environment. It should be noted, however, that cellular K+ concentrations may be in the millimolar range, and thus millimolar affinities may well be of physiological relevance. More data in lipid bilayers are needed to draw definite conclusions.
Taken together, even though KcsA is a highly stable MP, differences in structure, dynamics, and interactions can clearly be seen between different membrane mimicking environments. DPC appears to retain the structure of KcsA largely, but the very different chemical-shift changes upon pH drop (as compared to lipid bilayers) show that it does not fully retain its function. From the available data, it is, however, not clear whether DPC performs better or worse than other detergents that have been employed.
4.1.7. Other α-Helical MPs in Alkyl Phosphocholine
Alkyl phosphocholines have been used to determine structures of a number of other α-helical MPs and single-span α-helices,347−355 some of which are briefly discussed here. A remarkable successful case of the use of alkyl phosphocholine is the crystal structure of the mechanosensitive channel MscC,355 determined by crystallography in tetradecyl phosphocholine, which is likely in the closed conformation. Booth and co-workers later solved its structure and in the open conformation,356 also in the same detergent. Rees and co-workers recently determined two structures in DDM, from E. coli and H. pylori, respectively, one in the open and one in the closed conformation. The structures determined in tetradecyl phosphocholine appear similar to those in DDM and reasonable in the light of their function. Call et al. solved the structure of the ζζ domain in mixed DPC:SDS (5:1) micelles,353 and it exhibits a highly regular coiled-coil arrangement similar to the GCN4 zipper357 with the Tyr and Thr side chains hydrogen bonded across the dimer interface protecting the hydrophilic hydroxyls from the hydrophobic interstices of the lipid bilayer. These cases may, thus, represent successful applications of alkyl phosphocholine detergents.
4.1.7.1. Rv1761c
Maybe one of the most dramatic examples of the difficulty in working with DPC micelles is the protein Rv1761c from Mycobacterium tuberculosis. Its structure has been determined by solution-state NMR based on 162 paramagnetic relaxation enhancement restraints, 36 hydrogen bonds, 210 dihedral restraints, and 218 residual dipolar couplings (PDB 2K3M),358 that is, >600 restraints, a generous number of restraints for this 127-residue protein. In this NMR structure, displayed in Figure 17, the Gly-Pro sequence in the middle of the TM sequence (RAVAAALVGPGGVALVVKV) is dramatically kinked in the middle of this very hydrophobic sequence, presumably bringing the kink site to the surface of the micelle. Many distance restraints between the C-terminal half of the TM helix and the amphipathic C-terminal region of the protein were obtained, indicating that the Gly-Pro sequence had been brought to the hydrophilic/hydrophobic interface, something that would not be possible in a TM topology in a lipid bilayer. In addition, over 55% of this 127 residue protein was characterized as α-helical with two adjacent helical sections being clearly amphipathic, and yet the structural data were not able to clearly define interfacial and aqueous regions of the protein, but were highly suggestive of being wrapped around the detergent micelle.
Figure 17.
Structure of Rv1761c (PDB entry 2K3M) in DPC micelles as determined by >600 NMR restraints, colored from blue (N-terminus) to red (C-terminus). The two representations are rotated by 90° relative to each other.
4.1.7.2. NccX
Another interesting case is the protein NccX,359 of which a crystal structure has been determined in DPC. Intriguingly, “the periplasmic domains of NccX appeared collapsed against the hydrophobic TM segments, leading to an aberrant topology incompatible with membrane insertion.” The authors explained this unusual topology with a “detergent-induced redistribution of the hydrophobic interactions among the TM helices and a pair of hydrophobic patches keeping the periplasmic domains together in the native dimer.” In this case, alkyl phosphocholine led to a very unusual architecture that does not seem to be of biological relevance.
4.1.7.3. CcdA
The bacterial electron transporter CcdA is another example of a MP that seems to undergo large-amplitude motions in DPC detergent.360 Williamson et al. have determined its structures from NOE restraints; in their approach, an initial set of only about 30 long-range NOEs was used to derive a first model, and with the aid of this model further NOE cross-peaks, which are not spectrally unambiguous, were then iteratively assigned; that is, the choice of assigning a given peak to a pair of atoms among several possibilities was done by rejecting possibilities that are not in agreement with the initial structure. This approach led to the assignment of 101 long-distance restraints, and a tightly defined bundle of structures with a pairwise backbone RMSD below 1 Å.
As this approach critically depends on the initial structure, derived here from only 30 NOEs, the authors used PREs to “independently validate” the structure. Interestingly, those PRE restraints point to large-scale motions, and are partly in disagreement with the structure. The authors have performed PRE experiments with MTSL spin labels attached at four different positions, in a manner similar to those discussed for UCP2 (see section 4.1.1 and Figure 10). As noted by the authors, the observed PRE effects, shown in supplementary Figure 3 of Williamson et al.,360 and highlighted for one of these cases in Figure 18, cannot be in agreement with a single structure. Unexpected PRE effects are observed for very long distances, whereas expected short-range PRE effects are absent. In the example of the spin label attached to residue 109 (Figure 18), strong PRE effects are found for residues in H5, located almost 30 Å from the spin label. For explaining the long-distance PRE effects, which are incompatible with a single structure, the authors invoke the existence of large-scale motions (cf., the discussion on PRE effects in UCP2, Figure 10).
Figure 18.

Analysis of paramagnetic relaxation enhancement (PRE) data of the bacterial CcdA protein in DPC micelles, exemplified by one of the spin-label experiments.360 (A–C) The peak intensity ratio of paramagnetic and diamagnetic samples, Ipara/Idia, obtained with a MTSL spin label attached to residue 109 (cyan) is plotted onto the structure determined by NMR in DPC (PDB 2N4X). Three different structural views are shown, corresponding to different orientations of CcdA. The graphs in (D) show the observed intensity ratios in HNCO experiments used for this color coding (top) and the distances of each amide hydrogen to the paramagnetic label (approximated by the CB position of M109; bottom). Three different regions of the protein are highlighted. (i) In the case of helix H3, which is in close proximity in the structure to the paramagnetic label (ca. 10–12 Å), strong PRE effects are found, leading to broadening of the resonance residues 48–68 beyond detection. This finding matches the close proximity in the structure. (ii) For helix H4, which is at the same distance to the spin label as H3, no PRE effects are observed, in striking contrast to the spatial proximity. (iii) Helix H5 is at a distance of ca. 30 Å from the spin label. Over such a distance, no PRE effects are expected, yet the peaks are largely broadened. An explanation put forward by the authors is the existence of large-scale motions, which transiently bring this part in close proximity to the spin label. Interestingly, however, the neighboring helix, which is closer to the spin label, does not experience sizable PRE effects. Taken together, these data show that at least CcdA must undergo large-scale motions, and additionally question the accuracy of the structure. The raw data were kindly provided by Prof. James J. Chou.
Even more intriguingly, many expected short-range distances are not found. For example, residues 85–100 in H4 and 170–180 in H6 are all much closer to M109 than 143–146 in H5, yet only for the latter long distances were PRE effects reported in the M109C-MTSL sample, not for the shorter distances. Even if there is a dynamic process that leads to transient contacts of the spin label position and H5 in a minor state, the major-state structure must fulfill all of the PRE restraints, which is clearly not the case of H4. Similarly, several other expected short-range distance are not found in the A126C-MTSL-labeled sample.360 Despite these unexpected features, the authors use the PRE restraints to “independently validate the NOE-derived structure”, and propose that the large-amplitude motions are functionally relevant, an assumption that is backed up by biochemical experiments. The fact that the presence of the alternative conformation(s) is not reflected in the very tightly defined bundle may be ascribed to the possibly low population of those states, which may not lead to detectable NOE restraints. Alternatively, in case some of the NOEs stem from the alternative states, the attempt to determine a single structure from restraints pertaining to several states may introduce artifacts in the structure. Irrespective of whether the apparent dynamics of CcdA is of relevance for function in a lipid bilayer, CcdA presents yet another example where large-amplitude motions are sustained by the alkyl phosphocholine environment, similarly to mitochondrial carriers (cf., section 4.1.1).
While this Review was in the final revision, an independent structure of CcdA from Thermus thermophilus was published, determined also in DPC detergent at a sample temperature of 70 °C. Interestingly, while the two proteins show significant sequence homology (29% identity, 60% similarity), they bear no structural resemblance. Although we are unable to conclude at this stage, there is a possibility that the differences might be due to lack of restraints in the structure calculation protocol.
4.1.7.4. Sigma-1 Receptor
The Sigma-1 Receptor (S1R) protein provides an example in which a alkyl phosphocholine can stabilize non-native secondary structure. S1R is a small membrane bound receptor that regulates the activity of a large number of cellular proteins and is itself regulated by small-molecule binding.361 S1R binds a large number of small molecules including cocaine, haloperidol, fluvoxamine, and steroid hormones such as progesterone, and dysfunction of S1R has been implicated in depression, addiction, and neuropathic pain.362,363 Earlier work had suggested that S1R contained an even number of TM domains,364−366 and sequence-based prediction algorithms had pointed to a TM helix in either residues 80–100 or 90–110, in addition to an N-terminal TM helix and signal peptide. A solution NMR study was carried out on a truncated form of S1R designed to exclude the N-terminal TM helix.367 The truncated construct could be produced and purified from E. coli membranes and was reconstituted into a mixture of DPC and DPPC to determine its secondary structure from chemical shift data. A putative TM helix was identified in residues 91–107, based on secondary chemical shifts and chemical shift perturbations induced by increasing the DPPC concentration. Subsequently, two structures of the full-length receptor produced in insect cells and crystallized in LCP were reported clearly showing just a single N-terminal TM helix.368 Remarkably, the region 91–107, which was helical in DPC, formed a β-hairpin conformation. The structures solved in LCP are consistent with the large number of mutagenesis studies of the receptor function in membranes, leaving little doubt about the absence of a second TM domain.368,369 Although the altered structure was observed on a truncated construct in which the native tertiary structure may have been compromised, the NMR studies of S1R are nonetheless a dramatic illustration that DPC is able to stabilize non-native secondary structure.
4.1.8. α-Helical MPs in DPC: Emerging Trends
The examples discussed above indicate that alkyl phosphocholine detergents can have a considerable impact on the structure, interaction, and dynamics of α-helical proteins. When analyzing structures obtained from solution-state NMR, one needs to keep in mind, however, the significant methodological challenge associated with the structural determination of proteins of tens of kilodaltons. Extensive broadening of NMR lines, the difficulty of correctly assigning intermolecular distance restraints, and the need for deuteration schemes, thus eliminating the possibility of using aliphatic protons as structural probes, make structure determination a heroic effort. Given that structures in particular of large MPs may, thus, contain some uncertainty related to the method, one needs to be cautious when ascribing unexpected structural features exclusively to the detergent. Nonetheless, the large body of structural information on α-helical proteins is also accompanied by data about dynamics, interactions, stability, and function, which allow us to draw general trends for MP/alkyl phosphocholine interactions.
One often observed tendency is the bowing of helices, to enable hydrophilic side chains to access the micelle exterior. Consequently, helices tend to be less straight than in lipid bilayers. This effect has been seen for the cases of DgkA and PLN and, more extreme, in Rv1761c (cf., discussions in sections 4.1.2, 4.1.5, and 4.1.7, respectively).
A common trend induced by detergents, in general, and by alkyl phosphocholines, in particular, is the loosening of helix–helix interactions. Examples of this effect are the loose ensembles of structures found in mitochondrial carriers, and in apo-TSPO, which becomes structured only upon inhibitor-binding (even though the crystal structure of apo-TSPO in lipidic cubic phase hardly deviates from the holo-state211). As a consequence, substrate interactions are generally weakened, and in some instances dramatically so; for example, in AAC or Ca-uniporter, the inhibitor binding affinity is reduced by over 3 orders of magnitude (see discussions in sections 4.1.1 and 4.1.4, respectively, and refs (146), (257), and (258)). The binding specificity may also be disrupted in the loose structures in alkyl phosphocholine, as exemplified with mitochondrial carriers.146 In line with such a loosened tertiary structure, the thermal stability has been observed to drop significantly in alkyl phosphocholines as compared to other detergents (cf., Figure 8). Alkyl phosphocholines have also been observed to lead to fraying of α-helices, such that the secondary structures are shorter in micelles than in lipid bilayers. Examples of such loosening of helices were reported for mitochondrial carriers146 and KcsA.335 These effects can be more or less pronounced, varying largely for different proteins. We have reported two cases, MscC356 and ζζ,353 which appear not to have structural distortions in alkyl phosphocholines. Monomeric single-span TM helices may not be impacted by these considerations, and in alkyl phosphocholine they may largely retain their structural properties (see the discussion on simulations of TM peptides in section 5 and references therein). This being said, the cases of NccX359 and Rv1761c358 show that also single-span helices may be significantly impacted in alkyl phosphocholine in terms of dimerization or local structure; the presence of hydrophilic or traditional helix breaking residues such as proline and glycine has led to an unphysiological structure in the latter case. Thus, even in single-span TM proteins, one needs to be cautious when interpreting structural data.
KcsA is another rather positive case: it forms its tetrameric structure in alkyl phosphocholines, but it does so even in SDS, known to be harsh. Disassembling the tetramer requires very harsh conditions of low pH, SDS, and heating.332 Even though KcsA is a very forgiving case, the helices in DPC are shortened as compared to lipid bilayers,335 and the pH-induced effects are very different in DPC and membranes.
Other proteins discussed in this Review, however, are highly sensitive to alkyl phosphocholines and appear to lose key structural and functional features in this environment. We have extensively investigated the case of mitochondrial carriers, which have only small helix–helix contact surfaces, such that their stability relies on the lateral pressure in the membrane. Accordingly, they appear to be easily destabilized in alkyl phosphocholine, likely because the small and flexible detergent molecules can compete with the intramolecular contacts and thus loosen the helix–helix interactions. They lose their substrate binding specificity, have very low affinity, and have dynamics that are not related to function (cf., section 4.1.1). The general trend of a very loose structure in DPC is also reflected by the TSPO case, which forms a molten globule in DPC unless it is locked by its inhibitor (which, however, binds at lower affinity than in bilayers). From these considerations, it is clear that one has to be extremely cautious especially when studying dynamics of α-helical MPs in detergents,144,224,360 as the motions of MPs in detergent are likely dictated by the environment and not representative of functional motions in bilayers.146,146
4.2. β-Barrel Membrane Proteins
Structures of several outer MPs (OMP) have been solved in different environments. In particular, a few OMP structures have been unraveled in DPC micelles. Interestingly, structures of the same proteins have been obtained in the presence of other detergents or even lipids (for a complete survey concerning OMP/DPC atomic structures, see Table 4 in the Supporting Information). Although most structural studies of OMP solubilized in DPC have been obtained by solution-state NMR spectroscopy, one of them, OmpF from Gram-negative bacteria, has been solved by X-ray crystallography (Table 4 in the Supporting Information).33,370,371 OmpF is one of the most studied OMP. Its trimeric structure has been determined by X-ray crystallography in the presence of several different detergents, including DPC, and a structure was also obtained from crystals grown in lipidic cubic phases.372 Different crystal packings were observed. The detergent arrangement in the trigonal and the tetragonal lattices was determined by low-resolution neutron diffraction,68,373 revealing a surprising detergent rearrangement from the solution to the trigonal crystal form, and an unexpected role of the detergent in the crystal contacts of the tetragonal form. Despite notable differences in chemical environment and crystal contacts, the backbones of all of the structures superimpose quite well, with an rmsd of 0.26 and 0.61 Å between the structure obtained in C8E4 with that in lipidic cubic phase and in DPC, respectively.
tOmpA is also an interesting example of an OMP bearing 8 β-strands, for which several NMR structures exist,374−376 including DPC,374 or in nondetergent solutions, that is, associated with amphipols377 or in nanodiscs.378 Overall, these structures are very similar. A notable feature is the observation of two sets of cross-peaks for the majority of residues in several detergents (DHPC, n-octyl glucoside or n-octyltetraoxyethylene).376 These two conformations were not in exchange, as no peak intensity change was observed by varying the temperature. The significance of these two sets of peaks remains elusive.
In the following subsections, we highlight the outer membrane proteins OmpX and PagP, two cases of interest because their structure and dynamics have been characterized in various media.
4.2.1. OmpX
OmpX is a particularly instructive case, because it has been studied extensively in several membrane-mimicking environments, and structures have been determined by solution-state NMR in DHPC,379 DPC,22 and phospholipid nanodiscs,22 as well as by crystallography in C8E4 detergent.380 In a comparative study, the structure and dynamics of OmpX in DPC and DMPC:DMPG (3:1) nanodiscs were determined by solution-state NMR at 45 °C,22 thus providing insight into the effects of DPC.
Focusing on the comparative study conducted in the presence of either DPC or lipid discs,22 important differences can be observed. First, each strand is, on average, up to two residues shorter in DPC solution.22 Similarly, differences in the length, but also sometimes in the orientation of the strands, have been observed with PagP discussed below. For OmpX, differences are particularly visible at the top of the strands 1, 3, and 8 and at the bottom of the strands 1, 2, 4, 5, and 8 (Figure 19). This is in accordance with hydrogen/deuterium exchange measurements performed after prolonged equilibration in D2O with OmpX in DHPC detergent micelles or associated with amphipols showing that residues belonging to the periplamic end of the barrel tend to exchange somewhat more in detergents than in amphipols.381 Most of the averaged 15N,1H chemical shift differences (Δδ [15N,1H]) between OmpX amino acid residues in DPC and nanodiscs are below 2 ppm (except eight residues, almost all located in the extracellular loops, with Δδ [15N,1H] above 3 ppm), suggesting that the differences observed in β-strand lengths may have some dynamic origins.
Figure 19.
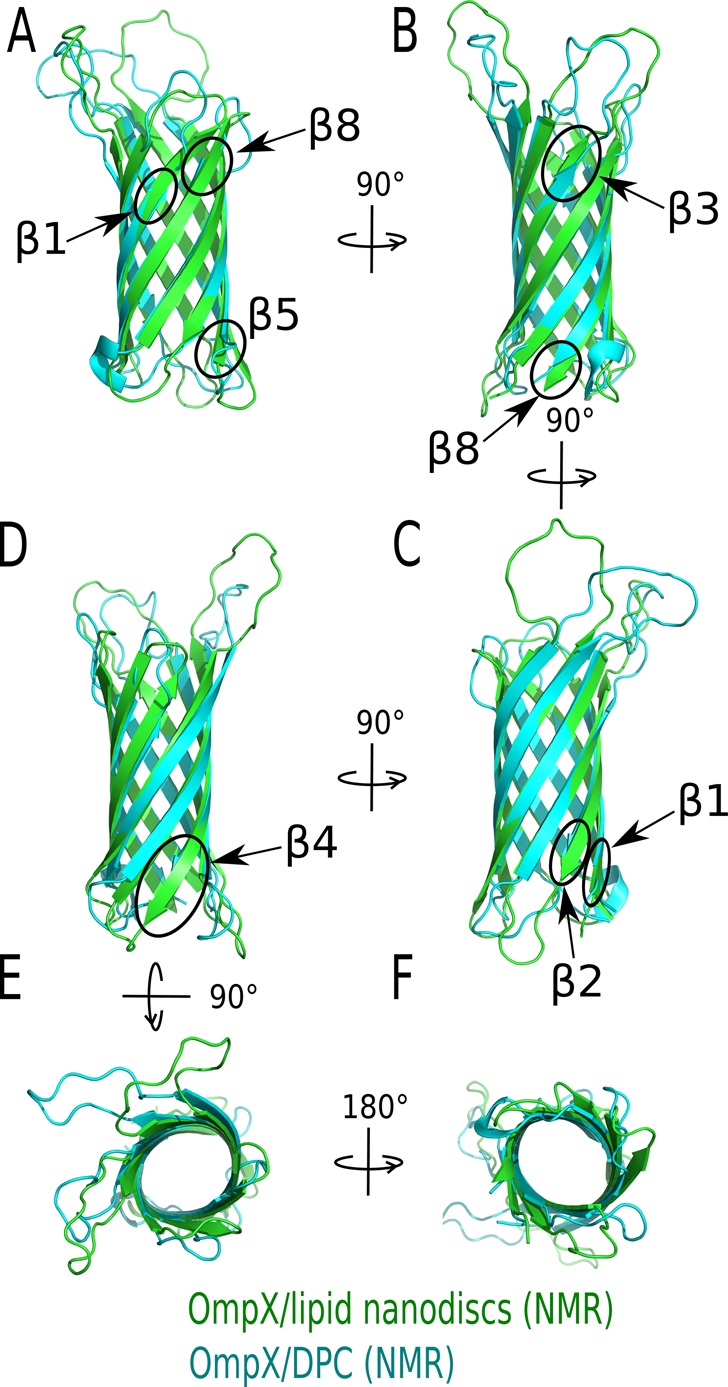
Comparison of NMR structures of OmpX in DPC micelles (in cyan; PDB code: 2M07)22 and in lipid nanodiscs (in green; PDB code: 2M06).22 Parts (A) to (D) correspond to lateral views, respectively, to the putative membrane plane, and (E) and (F) represent top and bottom views from the extracellular and periplasmic sides of the membrane, respectively. Ellipses in black indicate variations in length for β-strands 1, 2, 3, 4, 5, and 8 between the two structures.
Second, dynamics measurements by 1H–15N heteronuclear NOEs indicate that the first turn (following the nomenclature defined in reference Vogt and Schulz;382 residues Asp33 to Pro36; named loop L2 in ref (22)) and the loop L2 (residues ∼Glu47 to ∼Tyr62; named loop L3 in ref (22)) display marked motions at the picosecond-to-nanosecond time scale. Concerning L2, in DPC the dynamic behavior of this loop is split into two parts in contrast to observation in lipid discs where this loop seems entirely mobile. Indeed, in DPC solution, a rigid portion, from residues Glu47 to Ser54 (1H–15N heteronuclear NOEs > 0.7), precedes a more mobile part (Gly55 to Tyr62) with 1H–15N heteronuclear NOEs around 0.55, but associated with large error bars as compared to data in lipid discs in the same region of the protein. Overall, even if these measurements concern fast motions only, that is, in the picosecond-to-nanosecond time scale, they are in accordance with the generalized order parameter S2 calculated from chemical shift data, which indicate a larger flexibility or more ample motions in turn T1 and loop L2 in lipid discs. These large amplitude motions may involve much slower chemical exchanges as well, but not investigated in that study.
Frey et al. have also studied the dynamics of OmpX, and compared the motions in DPC, bicelles, and nanodiscs using 15N NMR spin-relaxation measurements.383 They report that the various β-strands have significant dynamic variability in lipid environment, but much less in DPC.
Another comparative study by NMR carried out in both DPC solution and lipid discs with Opa60 also indicates some variations in chemical shifts between the two media, and, as observed with OmpX, additional peaks are present with the protein in a lipid disc, which are restored in DPC solution when the long extracellular loops are removed by a proteolytic cleavage.384 This approach confirms that the dynamics of extracellular loops, but also periplamic turns like observed with OmpX, impact on the stability at the edges of the barrel, an impact that can be more or less important, depending on the protein and the media used to study the protein in solution or in a crystal.
4.2.2. PagP
The outer membrane palmitoyltransferase, or PagP, is an integral membrane enzyme of lipid metabolism, responsible for the incorporation into lipid A of a palmitate chain, resulting in the generation of a palmitoylated lipid A.385 The global fold of E. coli PagP was first determined by NMR spectroscopy from refolded material in DPC, and β-OG detergent solutions, respectively, at 45 °C386 and subsequently by X-ray crystallography in LDAO387 or SDS micelles.388 All of these structures described an eight-stranded antiparallel β-barrel associated with an N-terminal amphipathic α-helix. The global folds of the protein are very similar and essentially invariant to the different detergents used in these studies, with an average Cα rmsd of 1.8 Å between the crystal structure in LDAO and the average NMR backbone structure, excluding the leading α-helix and all connecting loops. A number of theoretical investigations aimed at elucidating the structural features of the integral membrane enzyme, and its relationship with its biological function.388−395
While the β-barrel part of PagP appears to be robust to different environments, including SDS, there are interesting differences in the dynamics and function. In particular, the long loop L1, which contains the greatest number of conserved polar residues (putatively involved in enzymatic activity), is highly dynamic. In the crystal structures, a large part of this loop is not resolved. Solution-state NMR relaxation measurements in DPC and β-OG directly show large-amplitude mobility,386 a finding that is also reflected in the conformational spread within the ensemble of NMR structures. In addition to these fast motions, NMR has also revealed slower (millisecond) motions in PagP.396 As a consequence of this conformational exchange process, many residues in loops L1, L3, and L4 and residues at the top of the connected β-strands could not been assigned because they are broadened beyond detection. Interestingly, the conformational dynamics depend on the employed detergent, and they appear to be related to function. In CYFOS-7, a alkyl phosphocholine with a cyclic extension at the acyl chain end in which PagP has been shown to be enzymatically active,397 this dynamic process has been studied in detail.396 A two-state exchange process was put forth, where the protein navigates between a state that the authors describe as a “closed” conformation, and a state where the β-barrel laterally opens. Arguably, the latter conformation may be important for the enzymatic activity, that is, for transfer of the sn-1 palmitate chain from phospholipid to lipopolysaccharide. In the case of PagP, conformational dynamics thus appears to be a hallmark of function. In DPC and β-OG, PagP has been reported not to be functional.397 In DPC, microsecond-to-millisecond dynamics are also observed for a large part of the protein, and the concerned residues partly coincide with those undergoing exchange in CYFOS-7; in β-OG only a few residues show dynamics (only residues 115–119 in the third loop were broadened by conformational exchange).
Taken together, PagP is a case where one alkyl phosphocholine, CYFOS-7, sustains enzymatic activity, in contrast to β-OG and DPC. The structures of PagP are very similar in the different detergents, highlighting again the robustness of β-barrel folds. The clearly different dynamics in different media, correlated to differences in enzymatic activity, highlight the importance that dynamics may have in particular for enzymes.
4.2.3. β-Barrel Proteins in DPC: Emerging Trends
Overall, structures of OMPs are quite well conserved regardless of the medium in which they are studied. Thus, from the structural data published so far for OMPs, alkyl phosphocholines can be considered as a possible detergent for structural studies. The alkyl phosphocholine CYFOS-7 has been shown to sustain activity of the PagP enzyme. The apparent robustness of β-barrel MPs is due to the constrained architecture involving a dense network of hydrogen bonds. Even under rather harsh experimental conditions (40–50 °C, hundreds of mM of alkyl phosphocholine detergent concentration), the global folds of the β-barrels are similar to available crystal structures or to other NMR structures conducted with different surfactants. However, as observed in the presence of other detergents, structures in DPC display a β-barrel that appears somewhat nibbled at edges. Through these studies briefly discussed here, this may be due to the presence of chemical exchanges at various time scales both in the trans-membrane β-barrel and in the extracellular loops, and to a lesser extent in the periplasmic turns. This includes large amplitude motions of the loops that seem to impact on the dynamic regime at the top of the barrel, that is, at the connection area with the loops, as observed with kpOmpA in a lipid bilayer.398 This has also been observed with other NMR studies of OMPs not developed here, like tOmpA,374,375,399,400 OmpG,401 Opa60,384,402 and OmpW (in 2-undecylphosphocholine, 30-Fos, detergent solution),123 but also with AAC3 and GGC1 α-helical MCs (see Flexibility of Mitochondrial Carriers in DPC in section 4.1.1).
As discussed in section 2, the physicochemical properties of the lipid bilayer/aqueous environment interface represent the most difficult part to reproduce with a non-native environment. This is due to important variations in fundamental variables like the dielectric constant and the pressure profile as compared to the membrane core. This is particularly true with DPC micelles where simulations indicate a considerable exposure of DPC hydrocarbon chains to water (cf., section 2.1). Among some hypotheses to explain the slow-to-intermediate chemical exchanges observed at the extracellular extremities of the β-strands and at the anchoring of the loops in DPC, it has been postulated that the polar headgroup of this detergent would be particularly flexible at the water interface and could increase internal motions toward the barrel edges.400 These motions could also be facilitated by the natural, large amplitude motions of the extracellular loops. In addition to some difficulties to reproduce membrane properties at the interface, the interaction with the girdles of aromatic residues that delineate the interface for the protein may be energetically unfavorable as compared to a lipid bilayer due to the inability for this detergent to reproduce the elastic properties of biological membranes.
4.3. Possibilities for Early Controls and a Posteriori Validation
Understanding the functional mechanisms of MPs at the atomic level requires the determination of high-resolution structures and investigations of their dynamics and interactions at the atomic scale. Obtaining samples that reflect the native behavior and are amenable to biophysical studies, for example, by crystallography or NMR, remains a daunting challenge. Although it is clear that detergents do not reflect the properties of the lipid bilayer, they often represent the only viable experimental approach to access structural information. The key question is whether the structural/dynamical/interaction data obtained in these environments can be interpreted as functionally relevant. Some of the examples shown here have highlighted that such interpretations must be made with caution, and it is crucial to use tools that allow one to decide, early in a study, whether a given experimental route should be pursued, or to validate a posteriori the relevance of the data. We briefly discuss here possible options.
Whenever possible, functional assays should be performed. In the case of transporters, where functional assays rely on compartments separated by a membrane and substrate gradients, which cannot be performed with solubilized protein, binding of ligands (substrates, inhibitors) can serve as a proxy. In such experiments, the binding specificity and affinity need to be carefully evaluated, as partially denatured proteins may still interact weakly/unspecifically, as revealed, for example, in mitochondrial carriers,146 TSPO, Ca-uniporter,257,258 and KcsA336 (cf., discussions in sections 4.1.1, 4.1.3,4.1.4, and 4.1.6, respectively). One possible route consists of performing titration experiments with a range of different substrates, for example, different nucleotides, or different amino acids in the case of a nucleotide-binding or amino-acid binding protein, respectively. MPs may be able to discriminate between these different solutes in lipid bilayers, but this ability may be lost in DPC (cf., the discussion about mitochondrial carriers above). A complementary route to assessing the relevance of structural/dynamical data is provided by studying the impact of mutations on function (in membranes) with their effects on structure/dynamics (in detergent). The function of the native conformation in the membrane may be critically dependent on defined residue–residue distances or electrostatic properties. In detergents, where the structure is loosened, those contacts may be less well-defined, and the effect of mutation on structure and dynamics may be negligible. The case of mitochondrial carriers is an example, where point-mutations lead to near-complete abolishment of functional turnover, but in DPC detergent the effects on structure and dynamics are very small.146
Alternatively, an investigation of thermal stability is a very efficient and cost-effective way to assess tertiary structures and function, and can, therefore, be performed at the early stages of a structural investigation; as highlighted with the example of mitochondrial carriers (section 4.1.1), such experiments readily revealed loss of specific binding and structural distortions that could later be detected with atomic-resolution techniques.
Many NMR parameters can also provide a detailed view of structure and may, thus, reveal possible unfolding. Secondary chemical shifts provide a view of the backbone structure, and nuclear Overhauser effects provide further views of intra- and intermolecular distances. Eichmann et al. have recently used exact NOEs to gain insight into detergent–protein proximities.403
Finally, molecular simulations have proven a powerful tool to assess the physiological meaning of the structures at hand by comparing their conformational dynamics and function in a native-like membrane environment and in detergent micelles. They have emerged as an indispensable tool and safeguard to guide structural biology investigations, especially when the protein fold is unusual. We propose the systematic application of such complementary experiments in structural investigations of MPs.
5. Simulation of Membrane Proteins in Native and Mimetic Lipid Environments
Molecular simulations have been employed traditionally to model MPs in native-like environments, and complement structural-biology experiments. Because the importance of detergents in structural investigations and their potential impact on membrane-protein structures, molecular simulations in detergent media, notably in DPC, have also been utilized, although more recently, to rationalize the effect of non-native-like environments on the structure, the dynamics, and the function of MPs. Outcomes of these theoretical studies have been presented for specific MPs, for example, AAC, UCP, p7, and PLN, in the previous section. In the following paragraphs, we complement the review by discussing in an exhaustive manner theoretical works addressing MP structures obtained in an alkyl phosphocholine environment.
As a preamble to focusing on the relationship between MPs and alkyl phosphocholine detergents, a rich literature of simulations of MPs in native-like media ought to be underscored. In a very systematic fashion, the growing number of MP structures that have been determined experimentally has inspired a host of molecular simulations performed in model-membrane environments, usually consisting of a single-lipid bilayer.404−420 In these simulations, the MP is generally embedded in a pre-equilibrated, fully hydrated patch of lipids organized in two leaflets. To eliminate edge effects, the simulation cell is replicated periodically in the three directions of Cartesian space, resulting effectively in a pseudo-infinite multilamellar molecular assembly.421,422 The bulk of the theoretical work devoted to MPs chiefly relies on a molecular mechanical description of the biological objects at play, and the use of MD simulations.423,424 The finite time step utilized to solve the Newton equations of motion, on the order of (1–2) × 10–15 s, imposed by a full atomistic representation of the molecular assembly, has severe limitations on both the size- and time-scales explored by the simulations, notwithstanding the continuous increase of the available computational resources. To circumvent these limitations, the granularity of the chemical description can be tuned to not only reduce the number of interactions to be evaluated, but also to dilate the time step used to propagate the motion. In such so-called coarse-grained simulations, a subset of atoms is represented by a single particle, allowing time steps as large as (30–40) × 10–15 s to be employed.425−427 Under these premises, very large biologically relevant assemblies of atoms have been examined over meaningful time scales, at the expense of preserving the fine atomic detail of the objects at play.428−430 Today, harnessing the considerable power of massively parallel architectures by means of highly scalable MD programs,431−435 the largest membrane assemblies have reached the level of small organelles formed by as many as 100 × 106 atoms,436 simulated over the 10–6 s time scale.
From the onset, the bulk of the theoretical effort to model MPs turned to native-like environments.437,438 Yet, the possibility that detergent media might render a different picture of the protein structure and dynamics led theorists to consider alternate surroundings in molecular simulations, chief among which are detergent micelles.439−443 In what follows, we will review as a preamble the models of DPC utilized in MD simulations. Next, we survey the simulations of MPs, the structure of which has been determined experimentally using DPC. For these different proteins, we will examine simulations performed in both lipid bilayers and alkyl phosphocholine micelles, emphasizing the role played by theory to highlight the differences and similarities in the structure and dynamics as a function of the environment.
5.1. Simulations of DPC Self-Organization
The first simulations of DPC micelles can be traced back to the late 1990s and relied on preformed self-organized objects.444 Despite the short simulations, on the 10–9 s time scale, the order parameters and correlation times extracted from the MD trajectories overall agreed with NMR relaxation data. Subsequent investigations explored the effect of the size of preformed micelles on the shape and dynamics of the latter.445 In a separate investigation, the detergent concentration was shown to modulate the shape of micelles, from worm-like at high concentration to spherical at low concentrations.446 On the basis of a 3.2 × 10–9 s simulation, the conformation, orientation, and dynamics of a 86-DPC-unit micelle were analyzed.447 Turning to a coarse-grained representation, Marrink et al. followed the self-aggregation of 400 DPC units, and observed on the 10–6 s time scale the formation of micelles of different sizes, compatible with experimental measurements.448 Using an implicit-solvent description, Lazaridis and co-workers investigated micelle formation, using a large number of 960 DPC units, and report aggregation numbers in close agreement with experiment.449 In addition, the effect of the interaction potential on detergent self-organization was also examined in a comparative study of academic macromolecular force fields.450
5.2. Early Simulations in DPC: Peptides, Glycophorin A, and Outer-Membrane Porins
Molecular simulations of membrane peptides and proteins in detergents appeared shortly after the first theoretical investigations of pure detergent self-aggregation. Aside from the noteworthy seminal work of Ceccarelli et al. in LDAO,440,451 of Braun et al. in SDS,441 of Khandelia and Kaznessis in SDS,452 of Böckmann and Caflisch in DHPC,443 and of Sansom and co-workers in DHPC and in OG,453,454 a large fraction of the simulations performed in a detergent environment followed the organization of DPC around a variety of integral α-helical and β-barrel proteins and peptides.439,442,455−463 Starting from the 310-helical form of adrenocotricotropin in DPC, Gao and Wong examined the binding mode of the peptide to the micelle, and showed that its interfacial behavior is similar to that observed in an SDS environment.455 In light of their comparative study in a preformed micelle of GM1 ganglioside and its isolated headgroup, Vasudevan and Balaji concluded that DPC packing modulates the conformation of the peptides, which follow a similar trend. Combining MD simulations and NMR spectroscopy, Dixon et al. have revealed the hairpin structure of a synthetic peptide containing the core sequence of an antibody-binding region of hemagglutinin A, and its location at the surface of the micelle.457 Using the outer-membrane protein OmpA, Bond and Sansom compared the dynamics of the latter embedded in a DPC micelle and in a lipid bilayer, and put forth that fluctuation of the protein structure is 1.5 times greater in the micellar surroundings, the loop region adopting moderately different conformations.439 In their 50 × 10–9 s, Bond and Sansom followed the formation of a DPC micelle around the paradigmatic glycophorin A α-helical dimer and the OmpA β-barrel, highlighting the preliminary self-organization of the detergents prior to association with the protein.442 The authors conclude that starting from a preformed micelle or from a polydisperse detergent solution, the resulting protein–detergent assemblies are remarkably similar, a conclusion also reached in light of their coarse-grained simulations.463 Khandelia and Kaznessis carried out a simulation of ovispirin-1, which was shown to partition to the surface of the micelle, adopting a conformation and orientation akin to that measured experimentally in the presence of a lipid bilayer.458 Similarly, in their theoretical investigation of the influenza hemagglutinin fusion peptide, Lagüe et al. observed a congruent structural behavior of the latter at both the micelle and the lipid bilayer surfaces, notwithstanding a significant increase of the overall solvation and rigidity in the micellar environment.459 In their series of simulations, Kaznessis and co-workers compared systematically insertion of α-helical and hairpin antimicrobial peptides in DPC and SDS micelles, and showed that the peptide–detergent interactions are different in zwitterionic and anionic micelles, underscoring the possibility of distinct modes of action at the surface of mammalian and bacterial membranes.460−462 These early simulations paved the way to subsequent investigations made possible by the concomitant improvement of force fields and ability to explore longer time scales afforded by massively parallel architectures.348,464−480 Aside from a rich literature investigating MPs in an explicit DPC environment, a number of studies have considered protein structures resolved experimentally in DPC and simulated in a lipid bilayer. Here, we focus on theoretical contributions comparing structures obtained from solution NMR and from high-resolution crystallography, which rely on distinct solubilizing agents. Kaznessis and co-workers pursued their systematic examination of environmental effects on antimicrobial peptides, comparing from experimental assays and molecular simulations SDS and DPC micelles, and confirm the previously observed distinct modes of action.464,467 To address the role of the peptide sequence on antimicrobial activity, these authors modeled a series of protegrin peptides and report different conformational arrangements and insertion depths in DPC micelles.465 They also showed that cation−π interactions stabilize indolicin in micelles formed by DPC, but not by SDS, resulting in a more ordered backbone with the former detergent.466 van der Spoel and co-workers have examined the effect of dehydration of a DPC micelle harboring OmpA, mimicking electrospray-ionization experiments, and revealed that while the protein remains structurally intact, the detergent molecules reorganize from a standard micelle to a spatial arrangement wherein the hydrocarbon tails are expelled outward, favoring headgroup–protein interactions.468 In their theoretical investigation of glycophorin A, Mineev et al. delved into the role played by the surroundings on α-helix dimerization, using DPC micelles and DMPC–DHPC bicelles, and showed that the overall architecture of the protein dimer remains unaltered, but its dynamics in a native membrane environment is better described in bicelles.470 Among the host of simulations of peptides in DPC micelles, several of them combined synergistically MD and NMR spectroscopy to render an enhanced picture of the interactions at play.348,469,471−473,475−477 In their simulations, Abel et al. compare the spatial arrangement of four membrane-spanning domains of an ABC transporter in DPC and DDM micelles, and report that these peptide chains migrate to the interfacial region, with a deeper penetration in the DDM detergents and a lesser tendency to unfold.474 Turning to an implicit-solvent description, Versace and Lazaridis examined a variety of interfacial peptides and β-barrel MPs in both DPC and SDS micelles, and noted little conformational deformation with respect to the reference, experimental structures.478 In their investigation of the N-terminal region of hemagglutinin in DPC micelles and in a DMPC bilayer, Victor et al. showed that this fusion peptide remains fully structured in the detergent medium, and adopts a membrane-spanning conformation in the bilayer, distorting locally the latter.479 Im and co-workers have designed a convenient tool for the construction of detergent micelles hosting proteins and peptides, and have applied it to the systematic study of a voltage-dependent potassium channel and the papiliocin peptide, showing an asymptotic limit of the protein–detergent interactions with the number of both DPC and DHPC detergent molecules.480
Molecular simulations are a versatile tool for studying the structure, dynamics, and ligand/lipid–interactions of MPs. Such simulations can furthermore not only be employed to investigate MPs near their equilibrium conformation, but also address the physiological relevance of structures obtained in non-native environments, and rationalize the interactions of detergents with MPs, as highlighted with several case studies presented in section 4.1.
6. Conclusions
MPs are a challenge from the standpoint of sample preparation and handling as well as for biophysical and structural techniques. Their size, heterogeneity, and intrinsic dynamics represent severe technical hurdles for structural and functional studies. The physiological relevance of MP structures has always been a matter of debate, at the theoretical as well as the experimental level. Every method has its particular requirements and may introduce specific artifacts. Crystallization selects a single conformation of the protein, the relevance of which has to be asserted by additional experiments. Not all conformations existing in a membrane may be prone to crystallization, making it difficult to decipher mechanistic details from a single frozen conformation. NMR spectroscopy, in its solution- and solid-state variants, is therefore complementary to crystallography, because the method can characterize proteins even if they coexist in multiple conformations, thereby providing access to systems that are not amenable to crystallography. However, as such measurements are almost always performed in non-native environments, the central question is to which extent the ensemble of conformations existing in a given membrane-mimicking environment reflects those present in membranes. In this Review, we have highlighted the effects of alkyl phosphocholines, and especially DPC, on MP structure, interactions, dynamics, and function. The fact that DPC is by far the most widely used detergent in solution-state NMR (Figure 2), and very powerful for solubilizing MPs (Section 3), raises the legitimate question of whether these solubilized proteins represent physiologically relevant conformations.
Even though the impact of detergents has to be evaluated for each protein individually, our survey reveals global trends. For most β-barrel proteins, alkyl phosphocholines seem to induce only very modest structural alterations as compared to other membrane-mimicking environments, although the proteins in alkyl phosphocholines appear more dynamic.
The situation appears to be different for MPs having transmembrane α-helices. An outward curvature that distorts single TM helices (e.g., Rv1761c) and disrupts tertiary helical interactions in multihelical proteins (e.g., DgkA) is often observed. The tertiary interactions in these proteins are weak, making them particularly sensitive to the small and flexible alkyl phosphocholine detergents. Furthermore, the ease with which a modestly hydrophilic site in the TM helix can reach the micelle surface can lead to distortions and bowing of TM helices. Albeit some rather successful cases of DPC-based studies of such proteins exist (such as KcsA), an increasing number of studies highlights that DPC weakens the tertiary contacts, enhances non-native dynamics, and may entail loss of binding specificity and activity.
Acknowledgments
We thank Dr. Vilius Kurauskas and Audrey Hessel for numerous discussions and for their groundbreaking work on mitochondrial carriers in DPC, which initiated this work. We thank Dr. Beate Bersch (IBS Grenoble, France), Dr. Robert Schneider (University of Lille, France), Dr. Stephanie Ravaud (IBCP Lyon, France), Dr. Karine Moncoq and Fabrice Giusti (IBPC Paris, France), and Dr. Phillip Stansfeld (University of Oxford, UK) for many insightful discussions and the three anonymous reviewers for their in-depth study of our manuscript and helpful suggestions. G.V. was supported by the National Institute of Health (GM 64742 and GM 72701). P.S. was supported by the European Research Council (ERC-StG-2012-311318). J.R.S. was supported by the UK Medical Research Council (M019152 and K018590). T.A.C. was supported in part by NIH grants AI023007 and AI-119178 and acknowledges the National High Magnetic Field Lab, supported by the NSF DMR-1157490 and the State of Florida. L.J.C. and B.M. were supported by the Centre National de la Recherche Scientifique, INSERM, by the “Initiative d’Excellence” program from the French State (Grant “DYNAMO”, ANR-11-LABEX-0011-01) and by the grant GHREDYN, ANR-13-BSV8-0006-01 from the Agence Nationale de la Recherche (ANR) to L.J.C. E.R.S.K. is supported by the UK Medical Research Council (MC_UU_00015/1). E.P.-P. was supported by the Institut Universitaire de France. The National High Magnetic Field Lab (NHMFL) is supported by the NSF Cooperative agreement DMR-1157490 and the State of Florida.
Biographies
Chris Chipot is a CNRS Research Director, with a joint appointment in the Physics Department of the University of Illinois at Urbana–Champaign. He obtained his Ph.D. in Theoretical Chemistry in 1994 from the University of Nancy with Bernard Maigret. He was a postdoctoral fellow in the groups of Peter Kollman at the University of California, San Francisco, and Andrew Pohorille at the NASA Ames Research Center. He was recruited by the CNRS in 1996, and obtained his habilitation in 2000. His research interests range from the structure and function of membrane proteins to the development of original strategies for calculating free energies and for exploring rare events by means of statistical simulations.
François Dehez is a CNRS Research Associate at the University of Lorraine, Nancy. He holds a M.Sc. in Physical Sciences from the University of Clermont-Ferrand. In 2002 he obtained a Ph.D. in Theoretical Chemistry from the University Henry Poincaré, Nancy, under the guidance of Pr. Claude Millot. He was a European Marie-Curie postdoctoral fellow with Pr. Francesco Zerbetto at the University of Bologna, where he investigated synthetic molecular switches and motors by means of statistical simulations. His research interests now focus essentially on modeling of membrane transport processes and DNA repair mechanisms.
Jason Schnell is an Associate Professor in the Department of Biochemistry at Oxford University. He received his Ph.D. in Biochemistry with Peter E. Wright from The Scripps Research Institute working on enzyme dynamics, and was a postdoctoral fellow at Harvard Medical School. The research interests of his lab are in structural biology, especially of proteins that interact with the membrane bilayer.
Nicole Zitzmann is Professor of Virology in the Department of Biochemistry at Oxford University. She received her Ph.D. in Biochemistry with Michael A. J. Ferguson, FRS, from Dundee University and was a postdoctoral fellow with Raymond A. Dwek, FRS, at the Oxford Glycobiology Institute. Her research interests are broad spectrum antiviral development, structural biology of host and viral targets, and mass spectrometry-based biomarker development.
Eva Pebay-Peyroula is Professor at University Grenoble Alpes and since 2016 adjunct Professor at Tromsø University. She received her Ph.D. in Physics. As a scientist at Institut Laue Langevin (ILL), she shifted her research field into biophysics and structural biology. She was then appointed by the University of Grenoble and joined the Institut de Biologie Structurale. In the frame of a long-term collaboration with J. Rosenbusch and E. Landau, she contributed to the developments of the crystallization in lipidic cubic phases. She studied bacterial rhodopsins and solved the first high-resolution structure of bacteriorhodopsin. Since 2000, her research interests are devoted to understanding the relationships between structure and function in membrane transporters. In this context, she solved the first structure of a mitochondrial carrier, the bovine ADP/ATP carrier.
Laurent J. Catoire is an Associate Research Scientist in the laboratory of Biology and Physico-Chemistry of Membrane Proteins at the Institut de Biologie Physico-Chimique (CNRS) in Paris. He received a Ph.D. in Molecular Biophysics (University Paris Diderot) and was a postdoctoral fellow at Rockefeller University. His research interest focuses on the energy landscape of membrane proteins and its modulation by allosteric regulators like lipids.
Bruno Miroux is the head of the Laboratory of Physical and Chemical Biology of Membrane Proteins in the Institute of Biological and Physical Chemistry in Paris, France. He obtained his Ph.D. in endocrinology and biochemistry in 1993. He has a strong interest in mitochondrial bioenergetics and was the first to experimentally demonstrate the topology of the mitochondrial uncoupling protein 1 and to initiate the genetic improvement of bacterial strains for the heterologous expression of membrane proteins.
Edmund Kunji holds a Ph.D. in Mathematics and Natural Sciences from the University of Groningen, The Netherlands. He was an EMBO Postdoctoral Fellow with Richard Henderson at the MRC Laboratory of Molecular Biology, Cambridge. Since 2000, he is a tenured Research Group Leader at the MRC Mitochondrial Biology Unit, University of Cambridge, where he works with his group on the structures of membrane proteins by X-ray crystallography and electron microscopy to elucidate the transport processes in mitochondria.
Gianluigi Veglia received both his M.S. and Ph.D. degrees in Chemistry from the University of Rome, La Sapienza, under the direction of Profs. M. R. Del Giudice and M. Delfini. In 1995, Dr. Veglia joined the laboratory of Prof. S. Opella at the University of Pennsylvania as a postdoctoral associate. In 2000, he joined the University of Minnesota, where he is now full Professor with a joint appointment between the Departments of Chemistry and Biochemistry, Molecular Biology & Biophysics. Dr. Veglia uses an interdisciplinary approach to integral and peripheral membrane proteins. He combines several biochemical assays with solution and solid-state NMR spectroscopy as well as computational methods to place the high-resolution structures and conformational dynamics of membrane proteins and protein complexes in the context of their biological function and disease associations.
Timothy A. Cross is Professor of Chemistry & Biochemistry at Florida State University and Director of the NMR & MRI User Program at the National High Magnetic Field Lab. He received his Ph.D. from the University of Pennsylvania with Prof. Stanley J. Opella, and following a postdoc with Opella he spent a final postdoctoral year in Basel, Switzerland, developing MRI/S technology in Prof. Joachim Seelig’s group at the Biozentrum before joining the faculty at FSU. His primary research interests are in the biophysics and solid-state NMR spectroscopy of membrane proteins.
Paul Schanda studied Chemistry at the University of Vienna (Austria) and received a Ph.D. in Physics from the University of Grenoble (France) in 2007, where he developed fast solution-state NMR methods for real-time investigation of protein folding. During his postdoctoral research at ETH Zürich (2008–2010) with Beat Meier and Matthias Ernst, he developed and applied solid-state NMR methods for protein dynamics studies. Since 2011 he works with his group at the Structural Biology Institute (IBS) in Grenoble, on various aspects of protein dynamics, ranging from fundamental processes and NMR methods development to applications in the field of membrane proteins, chaperones, and enzymes.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrev.7b00570.
Four tables showing the chemical structures of selected detergents, summaries of solubility and activity data, and available structures of β-barrels (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Coskun Ü.; Simons K. Cell membranes: the lipid perspective. Structure 2011, 19, 1543–1548. 10.1016/j.str.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Lunn C. A.Membrane Proteins as Drug Targets; Academic Press: Cambridge, MA, 2010; Vol. 91. [DOI] [PubMed] [Google Scholar]
- Zhou H.-X.; Cross T. A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013, 42, 361–92. 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J.; Howell S. C.; Van Horn W. D.; Jeon Y. H.; Sanders C. R. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 55, 335–360. 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T.; Hiller S.; Etzkorn M.; Wagner G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr. Opin. Struct. Biol. 2010, 20, 471–479. 10.1016/j.sbi.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum B.; Vonck J.; Bellack A.; Chaudhury P.; Reichelt R.; Albers S. V.; Rachel R.; Kuhlbrandt W. Structure and in situ organisation of the Pyrococcus furiosus archaellum machinery. eLife 2017, 6, 1. 10.7554/eLife.27470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldie K. N.; Abeyrathne P.; Kebbel F.; Chami M.; Ringler P.; Stahlberg H. In Electron Microscopy: Methods and Protocols; Kuo J., Ed.; Humana Press: Totowa, NJ, 2014; pp 325–341. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Yao Y.; Marassi F. M. Membrane protein structure determination in membrana. Acc. Chem. Res. 2013, 46, 2182–90. 10.1021/ar400041a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patching S. G. Solid-state NMR structures of integral membrane proteins. Mol. Membr. Biol. 2015, 32, 156–178. 10.3109/09687688.2016.1139754. [DOI] [PubMed] [Google Scholar]
- Renault M.; Cukkemane A.; Baldus M. Solid-state NMR spectroscopy on complex biomolecules. Angew. Chem., Int. Ed. 2010, 49, 8346–8357. 10.1002/anie.201002823. [DOI] [PubMed] [Google Scholar]
- Jacso T.; Franks W. T.; Rose H.; Fink U.; Broecker J.; Keller S.; Oschkinat H.; Reif B. Characterization of membrane proteins in isolated native cellular membranes by dynamic nuclear polarization solid-state NMR spectroscopy without purification and reconstitution. Angew. Chem., Int. Ed. 2012, 51, 432–435. 10.1002/anie.201104987. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Qin H.; Fu R.; Sharma M.; Can T. V.; Hung I.; Luca S.; Gor’Kov P. L.; Brey W. W.; Cross T. A. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew. Chem., Int. Ed. 2012, 51, 8383–8386. 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L. A.; Sinnige T.; Schellenberger P.; de Keyzer J.; Siebert C. A.; Driessen A. J.; Baldus M.; Grünewald K. Combined 1 H-Detected Solid-State NMR Spectroscopy and Electron Cryotomography to Study Membrane Proteins across Resolutions in Native Environments. Structure 2017, 26, 161. 10.1016/j.str.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privé G. G. Detergents for the stabilization and crystallization of membrane proteins. Methods 2007, 41, 388–397. 10.1016/j.ymeth.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Garavito R. M.; Ferguson-Miller S. Detergents as tools in membrane biochemistry. J. Biol. Chem. 2001, 276, 32403–32406. 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- Dörr J. M.; Scheidelaar S.; Koorengevel M. C.; Dominguez J. J.; Schäfer M.; van Walree C. A.; Killian J. A. The styrene maleic acid copolymer: a versatile tool in membrane research. Eur. Biophys. J. 2016, 45, 3–21. 10.1007/s00249-015-1093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postis V.; Rawson S.; Mitchell J. K.; Lee S. C.; Parslow R. a.; Dafforn T. R.; Baldwin S. a.; Muench S. P. The use of SMALPs as a novel membrane protein scaffold for structure study by negative stain electron microscopy. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 496–501. 10.1016/j.bbamem.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A. R.; O’Brien C. C.; Malhotra K.; Schwall C. T.; Albert A. D.; Watts A.; Alder N. N. A detergent-free strategy for the reconstitution of active enzyme complexes from native biological membranes into nanoscale discs. BMC Biotechnol. 2013, 13, 41. 10.1186/1472-6750-13-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersch B.; Dörr J. M.; Hessel A.; Killian J. A.; Schanda P. Proton-detected solid-state NMR spectroscopy of a zinc diffusion facilitator protein in native nanodiscs. Angew. Chem., Int. Ed. 2017, 56, 2508–2512. 10.1002/anie.201610441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I.; Grinkova Y.; Lazarides A.; Sligar S. Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 2004, 126, 3477–3487. 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- Nath A.; Atkins W. M.; Sligar S. G. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry 2007, 46, 2059–2069. 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- Hagn F.; Etzkorn M.; Raschle T.; Wagner G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135, 1919–1925. 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr M. L.; Baptista D.; Strauss M.; Sun Z.-Y. J.; Grigoriu S.; Huser S.; Plückthun A.; Hagn F.; Walz T.; Hogle J. M.; Wagner G. Covalently circularized nanodiscs for studying membrane proteins and viral entry. Nat. Methods 2017, 14, 49–52. 10.1038/nmeth.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I. G.; Sligar S. G. Nanodiscs in membrane biochemistry and biophysics. Chem. Rev. 2017, 117, 4669–4713. 10.1021/acs.chemrev.6b00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C. R.; Landis G. C. Reconstitution of membrane proteins into lipid-rich bilayered mixed micelles for NMR studies. Biochemistry 1995, 34, 4030–4040. 10.1021/bi00012a022. [DOI] [PubMed] [Google Scholar]
- Dürr U. H. N.; Soong R.; Ramamoorthy A. When detergent meets bilayer: birth and coming of age of lipid bicelles. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 69, 1–22. 10.1016/j.pnmrs.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerski L.; Sanders C. R. Functionality of a membrane protein in bicelles. Anal. Biochem. 2000, 284, 327–333. 10.1006/abio.2000.4720. [DOI] [PubMed] [Google Scholar]
- Popot J. L.; et al. Amphipols from A to Z. Annu. Rev. Biophys. 2011, 40, 379–408. 10.1146/annurev-biophys-042910-155219. [DOI] [PubMed] [Google Scholar]
- Tribet C.; Audebert R.; Popot J.-L. Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 15047–15050. 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyton C.; Pucci B.; Popot J.-L. Amphipols and fluorinated surfactants: two alternatives to detergents for studying membrane proteins in vitro. Methods Mol. Biol. 2010, 601, 17–38. 10.1007/978-1-60761-344-2_14. [DOI] [PubMed] [Google Scholar]
- Johansson L. C.; Wöhri A. B.; Katona G.; Engström S.; Neutze R. Membrane protein crystallization from lipidic phases. Curr. Opin. Struct. Biol. 2009, 19, 372–378. 10.1016/j.sbi.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Nikolaev M.; Round E.; Gushchin I.; Polovinkin V.; Balandin T.; Kuzmichev P.; Shevchenko V.; Borshchevskiy V.; Kuklin A.; Round A.; Bernhard F.; Willbold D.; G B.; V G. Integral membrane proteins can be crystallized directly from nanodiscs. Cryst. Growth Des. 2017, 17, 945–948. 10.1021/acs.cgd.6b01631. [DOI] [Google Scholar]
- White S. Membrane proteins of known 3D structure. 2017; http://blanco.biomol.uci.edu/mpstruc/.
- Garavito R. M.; Rosenbusch J. P. Three-dimensional crystals of an integral membrane protein: an initial x-ray analysis. J. Cell Biol. 1980, 86, 327–329. 10.1083/jcb.86.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau E. M.; Rosenbusch J. P. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 14532–14535. 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L.; Wüthrich K. NMR and ESR studies of the interactions of cytochrome c with mixed cardiolipin-phosphatidylcholine vesicles. Biochim. Biophys. Acta, Biomembr. 1977, 468, 389–410. 10.1016/0005-2736(77)90290-5. [DOI] [PubMed] [Google Scholar]
- Parker J. L.; Newstead S. Membrane protein crystallisation: current trends and future perspectives. Adv. Exp. Med. Biol. 2016, 922, 61–72. 10.1007/978-3-319-35072-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warschawski D. E. Membrane proteins of known structure by NMR (state: March 2017). 2017; http://www.drorlist.com/nmr/MPNMR.html.
- Seddon A. M.; Curnow P.; Booth P. J. Membrane proteins, lipids and detergents: not just a soap opera. Biochim. Biophys. Acta, Biomembr. 2004, 1666, 105–117. 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Bowie J. U. Stabilizing membrane proteins. Curr. Opin. Struct. Biol. 2001, 11, 397–402. 10.1016/S0959-440X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- Zhang X. Detergents: Friends not foes for high-performance membrane proteomics toward precision medicine. Proteomics 2017, 17, 1600209. 10.1002/pmic.201600209. [DOI] [PubMed] [Google Scholar]
- le Maire M.; Champeil P.; Møller J. V. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta, Biomembr. 2000, 1508, 86–111. 10.1016/S0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Gohon Y.; Popot J.-L. Membrane protein–surfactant complexes. Curr. Opin. Colloid Interface Sci. 2003, 8, 15–22. 10.1016/S1359-0294(03)00013-X. [DOI] [Google Scholar]
- Lichtenberg D.; Ahyayauch H.; Alonso A.; Goñi F. M. Detergent solubilization of lipid bilayers: a balance of driving forces. Trends Biochem. Sci. 2013, 38, 85–93. 10.1016/j.tibs.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Sim D.-W.; Lu Z.; Won H.-S.; Lee S.-N.; Seo M.-D.; Lee B.-J.; Kim J.-H. Application of solution NMR to structural studies on α-helical integral membrane proteins. Molecules 2017, 22, 1347. 10.3390/molecules22081347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm R.; Wagner G.; Hiller S.. Solution Nuclear Magnetic Resonance Spectroscopy of Integral Membrane Proteins. Reference Module in Life Sciences; Elsevier: New York, 2017. [Google Scholar]
- Anfinsen C. B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J.; Epp O.; Miki K.; Huber R.; Michel H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3Å resolution. Nature 1985, 318, 618–624. 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Yi M.; Dong H.; Qin H.; Peterson E.; Busath D. D.; Zhou H.-X.; Cross T. A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle D. A.; Cabral J. M.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Engelman D.; Zaccai G. Bacteriorhodopsin is an inside-out protein. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 5894–5898. 10.1073/pnas.77.10.5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M.; Patel A. B.; Liu W.; Smith S. O. Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys. J. 2002, 82, 2720–2736. 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X.; Cross T. A. Modeling the membrane environment has implications for membrane protein structure and function: influenza A M2 protein. Protein Sci. 2013, 22, 381–394. 10.1002/pro.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Cross T. A. Uniformity, ideality, and hydrogen bonds in transmembrane α-helices. Biophys. J. 2002, 83, 2084–2095. 10.1016/S0006-3495(02)73969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Wang A.; Vaughn J.; Cross T. A catalytic role for protic solvents in conformational interconversion. J. Am. Chem. Soc. 1996, 118, 9176–9177. 10.1021/ja9600892. [DOI] [Google Scholar]
- Xu F.; Cross T. Water: foldase activity in catalyzing polypeptide conformational rearrangements. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9057–9061. 10.1073/pnas.96.16.9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymeyer H.; Zhou H.-X. A method to determine dielectric constants in nonhomogeneous systems: application to biological membranes. Biophys. J. 2008, 94, 1185–1193. 10.1529/biophysj.107.117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern H. A.; Feller S. E. Calculation of the dielectric permittivity profile for a nonuniform system: application to a lipid bilayer simulation. J. Chem. Phys. 2003, 118, 3401–3412. 10.1063/1.1537244. [DOI] [Google Scholar]
- Wiener M. C.; White S. H. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 1992, 61, 434–447. 10.1016/S0006-3495(92)81849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluman N.; Tobiasson V.; von Heijne G. Stable membrane orientations of small dual-topology membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7987–7992. 10.1073/pnas.1706905114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R. C.; Kim S.; Cross T. A. Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure 2008, 16, 787–797. 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R. C.; Li C.; Hu J.; Gao F. P.; Cross T. A.. Lipid bilayers: an essential environment for the understanding of membrane proteins. Magn. Reson. Chem. 2007, 45.S2. 10.1002/mrc.2077 [DOI] [PubMed] [Google Scholar]
- Brooks C. L. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 1998, 8, 222–226. 10.1016/S0959-440X(98)80043-2. [DOI] [PubMed] [Google Scholar]
- Klibanov A. M. Enzymatic catalysis in anhydrous organic solvents. Trends Biochem. Sci. 1989, 14, 141–144. 10.1016/0968-0004(89)90146-1. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Flanagan J. M.; Treutlein H. R.; Zhang J.; Engelman D. M. Sequence specificity in the dimerization of transmembrane alpha-helixes. Biochemistry 1992, 31, 12719–12725. 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- Javadpour M. M.; Eilers M.; Groesbeek M.; Smith S. O. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys. J. 1999, 77, 1609–1618. 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian J. A.; von Heijne G. How proteins adapt to a membrane–water interface. Trends Biochem. Sci. 2000, 25, 429–434. 10.1016/S0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Garavito R.; Rosenbusch J.; Zulauf M.; Timmins P. Detergent structure in tetragonal crystals of OmpF porin. Structure 1995, 3, 1051–1059. 10.1016/S0969-2126(01)00241-6. [DOI] [PubMed] [Google Scholar]
- Cantor R. S. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 1999, 76, 2625–2639. 10.1016/S0006-3495(99)77415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Brink E.; Killian J. A.; de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim. Biophys. Acta, Biomembr. 2004, 1666, 275–288. 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- McMahon H. T.; Boucrot E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. 10.1242/jcs.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys. J. 2007, 93, 3884–3899. 10.1529/biophysj.107.107938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner S. M. Intrinsic curvature hypothesis for biomembrane lipid composition: a role for nonbilayer lipids. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 3665–3669. 10.1073/pnas.82.11.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.; Ursell T.; Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. Energetics of hydrophobic matching in lipid-protein interactions. Biophys. J. 2008, 94, 3996–4013. 10.1529/biophysj.107.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho A. V.; Huber T.; Sakmar T. P.; Brown M. F. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 2006, 91, 4464–4477. 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. F. Influence of nonlamellar-forming lipids on rhodopsin. Curr. Top. Membr. 1997, 44, 285–356. 10.1016/S0070-2161(08)60212-9. [DOI] [Google Scholar]
- Kozlov M. M.; Campelo F.; Liska N.; Chernomordik L. V.; Marrink S. J.; McMahon H. T. Mechanisms shaping cell membranes. Curr. Opin. Cell Biol. 2014, 29, 53–60. 10.1016/j.ceb.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P.; Ernst M. Studying dynamics by magic-angle spinning solid-state NMR spectroscopy: Principles and applications to biomolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 96, 1–46. 10.1016/j.pnmrs.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig A.; Seelig J. Dynamic structure of fatty acyl chains in a phospholipid bilayer measured by deuterium magnetic resonance. Biochemistry 1974, 13, 4839–4845. 10.1021/bi00720a024. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Marrink S.-J.; Berendsen H. J. A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta, Rev. Biomembr. 1997, 1331, 235–270. 10.1016/S0304-4157(97)00008-7. [DOI] [PubMed] [Google Scholar]
- Marsh D.; Watts A.; Pates R.; Uhl R.; Knowles P.; Esmann M. ESR spin-label studies of lipid-protein interactions in membranes. Biophys. J. 1982, 37, 265–274. 10.1016/S0006-3495(82)84675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter E.; Yordanova D.; Gerlach T.; Smirnova I.; Jakobtorweihen S. Molecular dynamics simulations of various micelles to predict micelle water partition equilibria with COSMOmic: influence of micelle size and structure. Fluid Phase Equilib. 2016, 422, 43–55. 10.1016/j.fluid.2016.03.006. [DOI] [Google Scholar]
- Abel S. .; Dupradeau F. .-Y.; Marchi M. Molecular dynamics simulations of a characteristic DPC micelle in water. J. Chem. Theory Comput. 2012, 8, 4610–4623. 10.1021/ct3003207. [DOI] [PubMed] [Google Scholar]
- Cross T. A.; Sharma M.; Yi M.; Zhou H.-X. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2011, 36, 117–125. 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faramarzi S.; Bonnett B.; Scaggs C. A.; Hoffmaster A.; Grodi D.; Harvey E.; Mertz B. Molecular Dynamics Simulations as a Tool for Accurate Determination of Surfactant Micelle Properties. Langmuir 2017, 33, 9934–9943. 10.1021/acs.langmuir.7b02666. [DOI] [PubMed] [Google Scholar]
- Schneider M. J.; Feller S. E. Molecular dynamics simulations of a phospholipid- detergent mixture. J. Phys. Chem. B 2001, 105, 1331–1337. 10.1021/jp003299g. [DOI] [Google Scholar]
- Tieleman D.; Van der Spoel D.; Berendsen H. Molecular dynamics simulations of dodecylphosphocholine micelles at three different aggregate sizes: micellar structure and chain relaxation. J. Phys. Chem. B 2000, 104, 6380–6388. 10.1021/jp001268f. [DOI] [Google Scholar]
- Cevc G.; Marsh D.. Phospholipid Bilayers: Physical Principles and Models; Wiley: New York, 1987. [Google Scholar]
- Dang S.; Sun L.; Huang Y.; Lu F.; Liu Y.; Gong H.; Wang J.; Yan N. Structure of a fucose transporter in an outward-open conformation. Nature 2010, 467, 734. 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- Liao J.; Li H.; Zeng W.; Sauer D. B.; Belmares R.; Jiang Y. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 2012, 335, 686–690. 10.1126/science.1215759. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Dahmane T.; Zhang Z.; Assur Z.; Brasch J.; Shapiro L.; Mancia F.; Hendrickson W. A. Structures from anomalous diffraction of native biological macromolecules. Science 2012, 336, 1033–1037. 10.1126/science.1218753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F.; Li S.; Jiang Y.; Jiang J.; Fan H.; Lu G.; Deng D.; Dang S.; Zhang X.; Wang J.; Yan N. Structure and mechanism of the uracil transporter UraA. Nature 2011, 472, 243–246. 10.1038/nature09885. [DOI] [PubMed] [Google Scholar]
- Stouffer A. L.; Acharya R.; Salom D.; Levine A. S.; Di Costanzo L.; Soto C. S.; Tereshko V.; Nanda V.; Stayrook S.; DeGrado W. F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales E. B.; Kawate T.; Gouaux E. Pore architecture and ion sites of acid sensing ion channels and P2X receptors. Biophys. J. 2010, 98, 610a. 10.1016/j.bpj.2009.12.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier C.; Harrenga A.; Ermler U.; Michel H. Structure at 2.7 Å resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10547–10553. 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrenga A.; Michel H. The cytochrome c oxidase from Paracoccus denitrificans does not change the metal center ligation upon reduction. J. Biol. Chem. 1999, 274, 33296–33299. 10.1074/jbc.274.47.33296. [DOI] [PubMed] [Google Scholar]
- Chung K. Y.; Kim T. H.; Manglik A.; Alvares R.; Kobilka B. K.; Prosser R. S. Role of detergents in conformational exchange of a G protein-coupled receptor. J. Biol. Chem. 2012, 287, 36305–36311. 10.1074/jbc.M112.406371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Brandolin G. Nucleotide exchange in mitochondria: insight at a molecular level. Curr. Opin. Struct. Biol. 2004, 14, 420–425. 10.1016/j.sbi.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Neil K. T.; DeGrado W. F. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 1990, 250, 646–651. 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- Maslennikov I.; Klammt C.; Hwang E.; Kefala G.; Okamura M.; Esquivies L.; Mörs K.; Glaubitz C.; Kwiatkowski W.; Jeon Y. H.; Choe S. Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 10902–10907. 10.1073/pnas.1001656107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Horn W. D.; Kim H.-J.; Ellis C. D.; Hadziselimovic A.; Sulistijo E. S.; Karra M. D.; Tian C.; Sönnichsen F. D.; Sanders C. R. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science 2009, 324, 1726–1729. 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J.; Columbus L.; Chu V. B.; Lesley S. A.; Doniach S. Size and shape of detergent micelles determined by small-angle X-ray scattering. J. Phys. Chem. B 2007, 111, 12427–12438. 10.1021/jp073016l. [DOI] [PubMed] [Google Scholar]
- Lauterwein J.; Bösch C.; Brown L. R.; Wüthrich K. Physicochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim. Biophys. Acta, Biomembr. 1979, 556, 244–264. 10.1016/0005-2736(79)90046-4. [DOI] [PubMed] [Google Scholar]
- Hirt R.; Berchtold R. Zur Synthese der Phosphatide. 2. Eine neue Synthese der Lecithine. Pharm. Acta Helv. 1958, 33, 349–356. [PubMed] [Google Scholar]
- Eibl H.; Arnold D.; Weltzien H.; Westphal O. On the synthesis of alpha and beta lecithins and their ether analogs. Justus Liebigs Ann. Chem. 1967, 709, 226–230. 10.1002/jlac.19677090124. [DOI] [PubMed] [Google Scholar]
- Mendz G. L.; Moore W. J.; Brown L. R.; Martenson R. E. Interaction of myelin basic protein with micelles of dodecylphosphocholine. Biochemistry 1984, 23, 6041–6046. 10.1021/bi00320a022. [DOI] [PubMed] [Google Scholar]
- Chupin V.; Leenhouts J. M.; de Kroon A. I.; de Kruijff B. Cardiolipin modulates the secondary structure of the presequence peptide of cytochrome oxidase subunit IV: a 2D 1H-NMR study. FEBS Lett. 1995, 373, 239–244. 10.1016/0014-5793(95)01054-I. [DOI] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 12366–12371. 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M.; Danielczak B.; Textor M.; Klement J.; Keller S. Modulating bilayer mechanical properties to promote the coupled folding and insertion of an integral membrane protein. Eur. Biophys. J. 2015, 44, 503–512. 10.1007/s00249-015-1032-y. [DOI] [PubMed] [Google Scholar]
- Huang P.; Liu Q.; Scarborough G. A. Lysophosphatidylglycerol: a novel effective detergent for solubilizing and purifying the cystic fibrosis transmembrane conductance regulator. Anal. Biochem. 1998, 259, 89–97. 10.1006/abio.1998.2633. [DOI] [PubMed] [Google Scholar]
- Shivanna B. D.; S R. E. Preservation of the native structure and function of Ca2+-ATPase from sarcoplasmic reticulum: solubilization and reconstitution by new short-chain phospholipid detergent 1, 2-diheptanoyl-sn-phosphatidylcholine. Biochem. J. 1997, 325, 533–542. 10.1042/bj3250533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser H. Short-chain phospholipids as detergents. Biochim. Biophys. Acta, Biomembr. 2000, 1508, 164–181. 10.1016/S0304-4157(00)00008-3. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Horst R.; Geralt M.; Ma X.; Hong W.; Finn M. G.; Stevens R. C.; Wüthrich K. Microscale NMR screening of new detergents for membrane protein structural biology. J. Am. Chem. Soc. 2008, 130, 7357–7363. 10.1021/ja077863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H.; Yu D.; Ge B.; Cook B.; Xu Z.; Zhang S. High-level production, solubilization and purification of synthetic human GPCR chemokine receptors CCR5, CCR3, CXCR4 and CX3CR1. PLoS One 2009, 4, e4509. 10.1371/journal.pone.0004509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhang S. Production of a bioengineered G-protein coupled receptor of human formyl peptide receptor 3. PLoS One 2011, 6, e23076. 10.1371/journal.pone.0023076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengottaiyan P.; Petrlova J.; Lagerstedt J. O.; Ruiz-Pavon L.; Budamagunta M. S.; Voss J. C.; Persson B. L. Characterization of the biochemical and biophysical properties of the Saccharomyces cerevisiae phosphate transporter Pho89. Biochem. Biophys. Res. Commun. 2013, 436, 551–556. 10.1016/j.bbrc.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Berardi M. J.; Shih W. M.; Harrison S. C.; Chou J. J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 2011, 476, 109–13. 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Wang S.; Zhu Q.; Wu B.; Liu Z.; OuYang B.; Chou J. J. Specific interaction of the human mitochondrial uncoupling protein 1 with free long-chain fatty acid. Structure 2017, 25, 1371–1379. 10.1016/j.str.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Zoonens M.; Masscheleyn S.; Comer J.; Pebay-Peyroula E.; Chipot C.; Miroux B.; Dehez F. Dangerous liaisons between detergents and membrane proteins. The case of mitochondrial uncoupling protein 2. J. Am. Chem. Soc. 2013, 135, 15174–15182. 10.1021/ja407424v. [DOI] [PubMed] [Google Scholar]
- Asmar-Rovira G. A.; Asseo-Garc a A. M.; Quesada O.; Hanson M. A.; Cheng A.; Nogueras C.; Lasalde-Dominicci J. A.; Stevens R. C. Biophysical and ion channel functional characterization of the Torpedo californica nicotinic acetylcholine receptor in varying detergent-lipid environments. J. Membr. Biol. 2008, 223, 13–26. 10.1007/s00232-008-9107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt C. A.; Collins R. F.; Conway M.; Modok S.; Storm J.; Kerr I. D.; Ford R. C.; Callaghan R. Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure 2006, 14, 1623–1632. 10.1016/j.str.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Horst R.; Stanczak P.; Wüthrich K. NMR polypeptide backbone conformation of the E. coli Outer membrane protein W. Structure 2014, 22, 1204–1209. 10.1016/j.str.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telbisz Á.; Özvegy-Laczka C.; Hegedus T.; Váradi A.; Sarkadi B. Effects of the lipid environment, cholesterol and bile acids on the function of the purified and reconstituted human ABCG2 protein. Biochem. J. 2013, 450, 387–395. 10.1042/BJ20121485. [DOI] [PubMed] [Google Scholar]
- Matar-Merheb R.; Rhimi M.; Leydier A.; Huché F.; Galián C.; Desuzinges-Mandon E.; Ficheux D.; Flot D.; Aghajari N.; Kahn R. Structuring detergents for extracting and stabilizing functional membrane proteins. PLoS One 2011, 6, e18036. 10.1371/journal.pone.0018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger P.; Kluth M.; Stindt J.; Smits S. H.; Schmitt L. Detergent screening and purification of the human liver ABC transporters BSEP (ABCB11) and MDR3 (ABCB4) expressed in the yeast Pichia pastoris. PLoS One 2013, 8, e60620. 10.1371/journal.pone.0060620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler J.; Sulistijo E. S.; Sakakura M.; Kim H. J.; Ellis C. D.; Sanders C. R. Lysophospholipid micelles sustain the stability and catalytic activity of diacylglycerol kinase in the absence of lipids. Biochemistry 2010, 49, 7089–7099. 10.1021/bi100575s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi M.; Damian M.; Lescop E.; Point E.; Moncoq K.; Morellet N.; Levy D.; Marie J.; Guittet E.; Banères J.-L.; Catoire L. J. Functional modulation of a G Protein-Coupled receptor conformational landscape in a lipid bilayer. J. Am. Chem. Soc. 2016, 138, 11170–11175. 10.1021/jacs.6b04432. [DOI] [PubMed] [Google Scholar]
- Thomas J. A.; Tate C. G. Quality control in eukaryotic membrane protein overproduction. J. Mol. Biol. 2014, 426, 4139–4154. 10.1016/j.jmb.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eifler N.; Duckely M.; Sumanovski L. T.; Egan T. M.; Oksche A.; Konopka J. B.; Lüthi A.; Engel A.; Werten P. J. Functional expression of mammalian receptors and membrane channels in different cells. J. Struct. Biol. 2007, 159, 179–193. 10.1016/j.jsb.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Fujimoto L. M.; Yao Y.; Marassi F. M. Solid-state NMR of the Yersinia pestis outer membrane protein Ail in lipid bilayer nanodiscs sedimented by ultracentrifugation. J. Biomol. NMR 2015, 61, 275. 10.1007/s10858-014-9893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunji E. In Comprehensive Biophysics; Egelman E. H., Ed.; Elsevier: Amsterdam, 2012; pp 174–205. [Google Scholar]
- Palmieri F.; Monné M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863, 2362–2378. 10.1016/j.bbamcr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Palmieri F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherited Metab. Dis. 2014, 37, 565–575. 10.1007/s10545-014-9708-5. [DOI] [PubMed] [Google Scholar]
- Saraste M.; Walker J. E. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982, 144, 250–254. 10.1016/0014-5793(82)80648-0. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 1978–2021. 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Nury H.; Dahout-Gonzalez C.; Trézéguet V.; Lauquin G. J. M.; Brandolin G.; Pebay-Peyroula E. Relations between structure and function of the mitochondrial ADP/ATP carrier. Annu. Rev. Biochem. 2006, 75, 713–741. 10.1146/annurev.biochem.75.103004.142747. [DOI] [PubMed] [Google Scholar]
- Kunji E. R.; Aleksandrova A.; King M. S.; Majd H.; Ashton V. L.; Cerson E.; Springett R.; Kibalchenko M.; Tavoulari S.; Crichton P. G.; Ruprecht J. J. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863, 2379–2393. 10.1016/j.bbamcr.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Crichton P. G.; Lee Y.; Kunji E. R. The molecular features of uncoupling protein 1 support a conventional mitochondrial carrier-like mechanism. Biochimie 2017, 134, 35–50. 10.1016/j.biochi.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M. UCP1-A sophisticated energy valve. Biochimie 2017, 134, 19–27. 10.1016/j.biochi.2016.10.012. [DOI] [PubMed] [Google Scholar]
- Berardi M. J.; Chou J. J. Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 2014, 20, 541–552. 10.1016/j.cmet.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Run C.; Yang Q.; Liu Z.; OuYang B.; Chou J. J. Molecular basis of MgATP selectivity of the mitochondrial SCaMC carrier. Structure 2015, 23, 1–10. 10.1016/j.str.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounier R.; Bellot G.; Chou J. J. Mapping conformational heterogeneity of mitochondrial nucleotide transporter in uninhibited states. Angew. Chem., Int. Ed. 2015, 54, 2436–2441. 10.1002/anie.201408417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüschweiler S.; Yang Q.; Run C.; Chou J. J. Substrate-modulated ADP/ATP-transporter dynamics revealed by NMR relaxation dispersion. Nat. Struct. Mol. Biol. 2015, 22, 636–641. 10.1038/nsmb.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Wang S.; Run C.; OuYang B.; Chou J. J. Specific lipid binding of membrane proteins in detergent micelles characterized by NMR and molecular dynamics. Biochemistry 2016, 55, 5317–5320. 10.1021/acs.biochem.6b00836. [DOI] [PubMed] [Google Scholar]
- Kurauskas V.; et al. How detergent impacts membrane proteins: atomic-level views of mitochondrial carriers in dodecylphosphocholine. J. Phys. Chem. Lett. 2017, 933. 10.1021/acs.jpclett.8b00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Dahout-Gonzalez C.; Kahn R.; Trezeguet V.; Lauquin G.; Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Ruprecht J. J.; Hellawell A. M.; Harding M.; Crichton P. G.; McCoy A. J.; Kunji E. R. S. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating–access transport mechanism. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E426–E434. 10.1073/pnas.1320692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M.; Winkler E.; Echtay K.. Uncoupling Protein, H+ Transport and Regulation; Portland Press Limited: UK, 2001. [DOI] [PubMed] [Google Scholar]
- Bertholet A. M.; Kirichok Y. UCP1: A transporter for H+ and fatty acid anions. Biochimie 2017, 134, 28–34. 10.1016/j.biochi.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A. M.; Procter J. B.; Martin D. M.; Clamp M.; Barton G. J. Jalview Version 2a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A. J.; Overy C.; Kunji E. R. S. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17766–17771. 10.1073/pnas.0809580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A. I.; Mileni M.; Chien E. Y. T.; Hanson M. A.; Stevens R. C. Microscale fluorescent thermal stability assay for membrane proteins. Structure 2008, 16, 351–359. 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Crichton P. G.; Lee Y.; Ruprecht J. J.; Cerson E.; Thangaratnarajah C.; King M. S.; Kunji E. R. S. Trends in thermostability provide information on the nature of substrate, inhibitor, and lipid interactions with mitochondrial carriers. J. Biol. Chem. 2015, 290, 8206–17. 10.1074/jbc.M114.616607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Willers C.; Kunji E. R.; Crichton P. G. Uncoupling protein 1 binds one nucleotide per monomer and is stabilized by tightly bound cardiolipin. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6973–6978. 10.1073/pnas.1503833112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. S.; Kerr M.; Crichton P. G.; Springett R.; Kunji E. R. Formation of a cytoplasmic salt bridge network in the matrix state is a fundamental step in the transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 14–22. 10.1016/j.bbabio.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson J.; Douglas M. G. Separate genes encode functionally equivalent ADP/ATP carrier proteins in Saccharomyces cerevisiae. Isolation and analysis of AAC2. J. Biol. Chem. 1988, 263, 14812–14818. [PubMed] [Google Scholar]
- Kolarov J.; Kolarova N.; Nelson N. A third ADP/ATP translocator gene in yeast. J. Biol. Chem. 1990, 265, 12711–12716. [PubMed] [Google Scholar]
- Kunji E. R.; Harding M.; Butler P. J. G.; Akamine P. Determination of the molecular mass and dimensions of membrane proteins by size exclusion chromatography. Methods 2008, 46, 62–72. 10.1016/j.ymeth.2008.10.020. [DOI] [PubMed] [Google Scholar]
- Bamber L.; Bamber L.; Harding M.; Harding M.; Butler P. J. G.; Butler P. J. G.; Kunji E. R. S.; Kunji E. R. S. Yeast mitochondrial ADP/ATP carriers are monomeric in detergents. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 16224–16229. 10.1073/pnas.0607640103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M. Nucleotide binding to uncoupling protein. Mechanism of control by protonation. Biochemistry 1988, 27, 781–791. 10.1021/bi00402a044. [DOI] [PubMed] [Google Scholar]
- Lin C. S.; Klingenberg M. Characteristics of the isolated purine nucleotide binding protein from brown fat mitochondria. Biochemistry 1982, 21, 2950–2956. 10.1021/bi00541a023. [DOI] [PubMed] [Google Scholar]
- Palmieri F.; Indiveri C.; Bisaccia F.; Iacobazzi V. Mitochondrial metabolite carrier proteins: purification, reconstitution, and transport studies. Methods Enzymol. 1995, 260, 349–369. 10.1016/0076-6879(95)60150-3. [DOI] [PubMed] [Google Scholar]
- Krämer R.; Klingenberg M. Reconstitution of inhibitor binding properties of the isolated adenosine 5′-diphosphate, adenosine 5′-triphosphate carrier-linked binding protein. Biochemistry 1977, 16, 4954–49561. 10.1021/bi00642a002. [DOI] [PubMed] [Google Scholar]
- Klingenberg M.; Grebe K.; Scherer B. The binding of atractylate and carboxy-atractylate to mitochondria. Eur. J. Biochem. 1975, 52, 351–363. 10.1111/j.1432-1033.1975.tb04003.x. [DOI] [PubMed] [Google Scholar]
- Vignais P. V.; Vignais P. M.; Defaye G. Adenosine diphosphate translocation in mitochondria. Nature of the receptor site for carboxyatractyloside (gummiferin). Biochemistry 1973, 12, 1508–1519. 10.1021/bi00732a007. [DOI] [PubMed] [Google Scholar]
- Krämer R. Interaction of membrane surface charges with the reconstituted ADP/ATP-carrier from mitochondria. Biochim. Biophys. Acta, Biomembr. 1983, 735, 145–159. 10.1016/0005-2736(83)90270-5. [DOI] [PubMed] [Google Scholar]
- Dupont Y.; Brandolin G.; Vignais P. V. Exploration of the nucleotide binding sites of the isolated ADP/ATP carrier protein from beef heart mitochondria. 1. Probing of the nucleotide sites by naphthoyl-ATP, a fluorescent nontransportable analog of ATP. Biochemistry 1982, 21, 6343–6347. 10.1021/bi00268a004. [DOI] [PubMed] [Google Scholar]
- Bojanovski D.; Schlimme E.; Wang C.-S.; Alaupovic P. Studies on the adenine nucleotide translocase from rat liver mitochondria. Eur. J. Biochem. 1976, 71, 539–548. 10.1111/j.1432-1033.1976.tb11143.x. [DOI] [PubMed] [Google Scholar]
- Vozza A.; Blanco E.; Palmieri L.; Palmieri F. Identification of the mitochondrial GTP/GDP transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 20850–20857. 10.1074/jbc.M313610200. [DOI] [PubMed] [Google Scholar]
- Harborne S. P.; King M. S.; Crichton P. G.; Kunji E. R. Calcium regulation of the human mitochondrial ATP-Mg/Pi carrier SLC25A24 uses a locking pin mechanism. Sci. Rep. 2017, 7, 45383. 10.1038/srep45383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A. J.; Kunji E. R. S. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2617–2622. 10.1073/pnas.0509994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunji E. R. S.; Robinson A. J. The conserved substrate binding site of mitochondrial carriers. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 1237–1248. 10.1016/j.bbabio.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Monné M.; Palmieri F.; Kunji E. R. The substrate specificity of mitochondrial carriers: mutagenesis revisited. Mol. Membr. Biol. 2013, 30, 149–159. 10.3109/09687688.2012.737936. [DOI] [PubMed] [Google Scholar]
- Dalbon P.; Brandolin G.; Boulay F.; Hoppe J.; Vignais P. V. Mapping of the nucleotide-binding sites in the ADP/ATP carrier of beef heart mitochondria by photolabeling with 2-azido[alpha-32P]adenosine diphosphate. Biochemistry 1988, 27, 5141–5149. 10.1021/bi00414a029. [DOI] [PubMed] [Google Scholar]
- Monné M.; Miniero D. V.; Bisaccia F.; Fiermonte G. The mitochondrial oxoglutarate carrier: from identification to mechanism. J. Bioenerg. Biomembr. 2013, 45, 1–13. 10.1007/s10863-012-9475-7. [DOI] [PubMed] [Google Scholar]
- Dehez F.; Pebay-Peyroula E.; Chipot C. Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J. Am. Chem. Soc. 2008, 130, 12725–33. 10.1021/ja8033087. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Tajkhorshid E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 9598–9603. 10.1073/pnas.0801786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud J.; Ravaud S.; Krammer E.-M.; Chipot C.; Kunji E. R. S.; Pebay-Peyroula E.; Dehez F. The substrate specificity of the human ADP/ATP carrier AAC1. Mol. Membr. Biol. 2013, 30, 160–8. 10.3109/09687688.2012.745175. [DOI] [PubMed] [Google Scholar]
- Beyer K.; Klingenberg M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by phosphorus-31 nuclear magnetic resonance. Biochemistry 1985, 24, 3821–3826. 10.1021/bi00336a001. [DOI] [PubMed] [Google Scholar]
- Nury H.; Dahout-Gonzalez C.; Trézéguet V.; Lauquin G.; Brandolin G.; Pebay-Peyroula E. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 2005, 579, 6031–6036. 10.1016/j.febslet.2005.09.061. [DOI] [PubMed] [Google Scholar]
- Gropp T.; Brustovetsky N.; Klingenberg M.; Muller V.; Fendler K.; Bamberg E. Kinetics of electrogenic transport by the ADP/ATP carrier. Biophys. J. 1999, 77, 714–726. 10.1016/S0006-3495(99)76926-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurauskas V.; Izmailov S. A.; Rogacheva O. N.; Hessel A.; Ayala I.; Woodhouse J.; Shilova A.; Xue Y.; Yuwen T.; Coquelle N.; Colletier J.-P.; Skrynnikov N. R.; Schanda P. Slow conformational exchange and overall rocking motion in ubiquitin protein crystals. Nat. Commun. 2017, 8, 145. 10.1038/s41467-017-00165-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste J. L.; Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 2000, 39, 5355–5365. 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Göbl C.; Madl T.; Simon B.; Sattler M. NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 80, 26–63. 10.1016/j.pnmrs.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Falconi M.; Chillemi G.; Di Marino D.; D’Annessa B. I.; Morozzo della Rocca; Palmieri L.; Desideri A. Structural dynamics of the mitochondrial ADP/ATP carrier revealed by molecular dynamics simulation studies. Proteins: Struct., Funct., Genet. 2006, 65, 681–691. 10.1002/prot.21102. [DOI] [PubMed] [Google Scholar]
- Johnston J. M.; Khalid S.; Sansom M. S. P. Conformational dynamics of the mitochondrial ADP/ATP carrier: A simulation study. Mol. Membr. Biol. 2008, 25, 506–517. 10.1080/09687680802459271. [DOI] [PubMed] [Google Scholar]
- Krammer E. M.; Ravaud S.; Dehez F.; Frelet-Barrand A.; Pebay-Peyroula E.; Chipot C. High-chloride concentrations abolish the binding of adenine nucleotides in the mitochondrial ADP/ATP carrier family. Biophys. J. 2009, 97, 25–27. 10.1016/j.bpj.2009.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marino D.; Oteri F.; Morozzo della Rocca B.; Chillemi G.; Falconi M. ADP/ATP mitochondrial carrier MD simulations to shed light on the structural-dynamical events that, after an additional mutation, restore the function in a pathological single mutant. J. Struct. Biol. 2010, 172, 225–232. 10.1016/j.jsb.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Feng J.; Lucchinetti E.; Enkavi G.; Wang Y.; Gehrig P.; Roschitzki B.; Schaub M. C.; Tajkhorshid E.; Zaugg K.; Zaugg M. Tyrosine phosphorylation by Src within the cavity of the adenine nucleotide translocase 1 regulates ADP/ATP exchange in mitochondria. Am. J. Phys. Cell Physiol. 2010, 298, C740–C748. 10.1152/ajpcell.00310.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravaud S.; Bidon-Chanal A.; Blesneac I.; Machillot P.; Juillan-Binard C.; Dehez F.; Chipot C.; Pebay-Peyroula E. Impaired transport of nucleotides in a mitochondrial transporter explains severe human genetic diseases. ACS Chem. Biol. 2012, 7, 1164–1169. 10.1021/cb300012j. [DOI] [PubMed] [Google Scholar]
- Di Marino D.; Oteri F.; della Rocca B.; D’Annessa I.; Falconi M. Mapping multiple potential ATP binding sites on the matrix side of the bovine ADP/ATP carrier by the combined use of MD simulation and docking. J. Mol. Model. 2012, 18, 2377–2386. 10.1007/s00894-011-1255-5. [DOI] [PubMed] [Google Scholar]
- Bidon-Chanal A.; Krammer E. M.; Blot D.; Pebay-Peyroula E.; Chipot C.; Ravaud S.; Dehez F. How do membrane transporters sense pH? The case of the mitochondrial ADP-ATP carrier. J. Phys. Chem. Lett. 2013, 4, 3787–3791. 10.1021/jz401847d. [DOI] [Google Scholar]
- Mielke C.; Lefort N.; McLean C. G.; Cordova J. M.; Langlais P. R.; Bordner A. J.; Te J. A.; Ozkan S. B.; Willis W. T.; Mandarino L. J. Adenine nucleotide translocase is acetylated in vivo in human muscle: Modeling predicts a decreased ADP affinity and altered control of oxidative phosphorylation. Biochemistry 2014, 53, 3817–3829. 10.1021/bi401651e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Goose J. E.; Caffrey M.; Carpenter E. P.; Parker J. L.; Newstead S.; Sansom M. S. P. MemProtMD: Automated insertion of membrane protein structures into explicit lipid membranes. Structure 2015, 23, 1350–1361. 10.1016/j.str.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietropaolo A.; Pierri C. L.; Palmieri F.; Klingenberg M. The switching mechanism of the mitochondrial ADP/ATP carrier explored by free-energy landscapes. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 772–781. 10.1016/j.bbabio.2016.02.006. [DOI] [PubMed] [Google Scholar]
- Hedger G.; Rouse S. L.; Domański J.; Chavent M.; Koldsø H.; Sansom M. S. P. Lipid-loving ANTs: Molecular simulations of cardiolipin interactions and the organization of the adenine nucleotide translocase in model mitochondrial membranes. Biochemistry 2016, 55, 6238–6249. 10.1021/acs.biochem.6b00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domański J.; Hedger G.; Best R. B.; Stansfeld P. J.; Sansom M. S. P. Convergence and sampling in determining free energy landscapes for membrane protein association. J. Phys. Chem. B 2017, 121, 3364–3375. 10.1021/acs.jpcb.6b08445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K.; Hayashi S. Atomistic modeling of alternating access of a mitochondrial ADP/ATP membrane transporter with molecular simulations. PLoS One 2017, 12, e0181489. 10.1371/journal.pone.0181489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehez F.; Schanda P.; King M. S.; Kunji E. R.; Chipot C. Mitochondrial ADP/ATP Carrier in Dodecylphosphocholine Binds Cardiolipins with Non-native Affinity. Biophys. J. 2017, 113, 2311–2315. 10.1016/j.bpj.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksimentiev A.; Schulten K. Imaging α–hemolysin with molecular dynamics: Ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005, 88, 3745–3761. 10.1529/biophysj.104.058727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérida I.; Ávila-Flores A.; Merino E. Diacylglycerol kinases: at the hub of cell signalling. Biochem. J. 2008, 409, 1–18. 10.1042/BJ20071040. [DOI] [PubMed] [Google Scholar]
- Walsh J. P.; Bell R. M. Diacylglycerol kinase from escherichia coli. Methods Enzymol. 1992, 209, 153–162. 10.1016/0076-6879(92)09019-Y. [DOI] [PubMed] [Google Scholar]
- Li D.; Lyons J. A.; Pye V. E.; Vogeley L.; Aragão D.; Kenyon C. P.; Shah S. T. A.; Doherty C.; Aherne M.; Caffrey M. Crystal structure of the integral membrane diacylglycerol kinase. Nature 2013, 497, 521–524. 10.1038/nature12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Berthold D. A.; Frericks H. L.; Gennis R. B.; Rienstra C. M. Partial 13C and 15N chemical-shift assignments of the disulfide-bond-forming enzyme DsbB by 3D magic-angle spinning NMR spectroscopy. ChemBioChem 2007, 8, 434–442. 10.1002/cbic.200600484. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhang Z.; Tang X.; Li J.; Glaubitz C.; Yang J. Conformation and topology of diacylglycerol Kinase in E.coli membranes revealed by solid-state NMR spectroscopy. Angew. Chem., Int. Ed. 2014, 53, 5624–5628. 10.1002/anie.201311203. [DOI] [PubMed] [Google Scholar]
- OuYang B.; Xie S.; Berardi M. J.; Zhao X.; Dev J.; Yu W.; Sun B.; Chou J. J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. 10.1038/nature12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavish M.; Bachman I.; Shoukrun R.; Katz Y.; Veenman L.; Weisinger G.; Weizman A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–50. [PubMed] [Google Scholar]
- Snyder S. H.; Verma A.; Trifiletti R. R. The peripheral-type benzodiazepine receptor: a protein of mitochondrial outer membranes utilizing porphyrins as endogenous ligands. FASEB J. 1987, 1, 282–288. 10.1096/fasebj.1.4.2820823. [DOI] [PubMed] [Google Scholar]
- Ginter C.; Kiburu I.; Boudker O. Chemical catalysis by the translocator protein (18 kDa). Biochemistry 2013, 52, 3609–3611. 10.1021/bi400364z. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Kalathur R. C.; Liu Q.; Kloss B.; Bruni R.; Ginter C.; Kloppmann E.; Rost B.; Hendrickson W. A. Structure and activity of tryptophan-rich TSPO proteins. Science 2015, 347, 551–555. 10.1126/science.aaa1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht R.; Papadopoulos V.; Rammes G.; Baghai T. C.; Fan J.; Akula N.; Groyer G.; Adams D.; Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discovery 2010, 9, 971–988. 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- Fairweather D.; Guilarte T. R.; Cooper J. L. T. Biomarker and more: can translocator protein 18 kDa predict recovery from brain injury and myocarditis?. Biomarkers Med. 2014, 8, 605–607. 10.2217/bmm.14.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaremko L.; Jaremko M.; Giller K.; Becker S.; Zweckstetter M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 2014, 343, 1363–1366. 10.1126/science.1248725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaremko M.; Jaremko L.; Giller K.; Becker S.; Zweckstetter M.; Jaremko L.; Jaremko M.; Giller K.; Becker S.; Zweckstetter M. Structural Integrity of the A147T Polymorph of Mammalian TSPO Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. ChemBioChem 2015, 16, 1483–9. 10.1002/cbic.201500217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Liu J.; Zheng Y.; Garavito R. M.; Ferguson-Miller S. Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–8. 10.1126/science.1260590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivella M.; Gonzalez A.; Pardo L.; Deupi X. Relation between sequence and structure in membrane proteins. Bioinformatics 2013, 29, 1589–92. 10.1093/bioinformatics/btt249. [DOI] [PubMed] [Google Scholar]
- Yeliseev A. A.; Krueger K. E.; Kaplan S. A mammalian mitochondrial drug receptor functions as a bacterial ”oxygen” sensor. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 5101–5106. 10.1073/pnas.94.10.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Xia Y.; Meiler J.; Ferguson-Miller S. Characterization and modeling of the oligomeric state and ligand binding behavior of purified translocator protein 18 kDa from Rhodobacter sphaeroides. Biochemistry 2013, 52, 5884–99. 10.1021/bi400431t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall r. W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacapère J. J.; Delavoie F.; Li H.; Peranzi G.; Maccario J.; Papadopoulos V.; Vidic B. Structural and functional study of reconstituted peripheral benzodiazepine receptor. Biochem. Biophys. Res. Commun. 2001, 284, 536–541. 10.1006/bbrc.2001.4975. [DOI] [PubMed] [Google Scholar]
- Murail S.; Robert J. C.; Coic Y. M.; Neumann J. M.; Ostuni M. A.; Yao Z. X.; Papadopoulos V.; Jamin N.; Lacapère J. J. Secondary and tertiary structures of the transmembrane domains of the translocator protein TSPO determined by NMR. Stabilization of the TSPO tertiary fold upon ligand binding. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 1375–1381. 10.1016/j.bbamem.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Jaremko L.; Jaremko M.; Giller K.; Becker S.; Zweckstetter M. Conformational Flexibility in the Transmembrane Protein TSPO. Chem. - Eur. J. 2015, 21, 16555–16563. 10.1002/chem.201502314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacapère J. J.; Iatmanen-Harbi S.; Senicort L.; Lequin O.; Tekely P.; Purusottam R. N.; Hellwig P.; Kriegel S.; Ravaud S.; Jullian-Binard C.; Pebay-Peyroula E.; Papadopoulos V.. Structural Studies of TSPO, a Mitochondrial Membrane Protein, 1st ed.; Membrane Protein Production for Structural Analysis; Springer-Verlag: New York, 2014; p 425. [Google Scholar]
- Jones C. T.; Murray C. L.; Eastman D. K.; Tassello J.; Rice C. M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann E.; Penin F.; Kallis S.; Patel A. H.; Bartenschlager R.; Pietschmann T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch J.; Brohm C.; Steinmann E.; Friesland M.; Menzel N.; Vieyres G.; Perin P. M.; Frentzen A.; Kaderali L.; Pietschmann T. hepatitis c Virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 2013, 9, e1003355. 10.1371/journal.ppat.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik P.; Chew C.; Aittoniemi J.; Chang J.; Wentworth P.; Dwek R. A.; Biggin P. C.; Vénien-Bryan C.; Zitzmann N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12712–12716. 10.1073/pnas.0905966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patargias G.; Zitzmann N.; Dwek R.; Fischer W. B. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J. Med. Chem. 2006, 49, 648–655. 10.1021/jm050721e. [DOI] [PubMed] [Google Scholar]
- Montserret R.; Saint N.; Vanbelle C.; Salvay A.; Simorre J. P.; Ebel C.; Sapay N.; Renisio J. G.; Böckmann A.; Steinmann E.; Pietschmann T.; Dubuisson J.; Chipot C.; Penin F. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J. Biol. Chem. 2010, 285, 31446–31461. 10.1074/jbc.M110.122895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler D.; Penin F.; Schulten K.; Chipot C. The p7 protein of Hepatitis C forms structurally plastic ion channels. PLoS Comput. Biol. 2012, 8, e1002702. 10.1371/journal.pcbi.1002702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-T.; Hsu H.-J.; Fischer W. B. Computational modeling of the p7 monomer from HCV and its interaction with small molecule drugs. SpringerPlus 2013, 2, 324. 10.1186/2193-1801-2-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-T.; Schilling R.; Fink R. H. A.; Fischer W. B. Ion-dynamics in hepatitis C virus p7 helical transmembrane domains – A molecular dynamics simulation study. Biophys. Chem. 2014, 192, 33–40. 10.1016/j.bpc.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Kalita M. M.; Griffin S.; Chou J. J.; Fischer W. B. Genotype-specific differences in structural features of hepatitis C virus (HCV) p7 membrane protein. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1383–1392. 10.1016/j.bbamem.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann N.; Chipot C.; Penin F.; Dehez F. Assessing the physiological relevance of alternate architectures of the p7 protein of hepatitis C virus in different environments. Bioorg. Med. Chem. 2016, 24, 4920–4927. 10.1016/j.bmc.2016.07.063. [DOI] [PubMed] [Google Scholar]
- Laasch N.; Kalita M. M.; Griffin S.; Fischer W. B. Small molecule ligand docking to genotype specific bundle structures of hepatitis C virus (HCV) p7 protein. Comput. Biol. Chem. 2016, 64, 56–63. 10.1016/j.compbiolchem.2016.04.010. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V.; Bartenschlager R. Structural and functional properties of the hepatitis C virus p7 viroporin. Viruses 2015, 7, 4461–4481. 10.3390/v7082826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster T. L.; et al. Structure-guided design affirms inhibitors of hepatitis C virus p7 as a viable class of antivirals targeting virion release. Hepatology 2014, 59, 408–422. 10.1002/hep.26685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. T.; Schilling R.; Fink R. H.; Fischer W. B. Ion-dynamics in hepatitis C virus p7 helical transmembrane domains–a molecular dynamics simulation study. Biophys. Chem. 2014, 192, 33–40. 10.1016/j.bpc.2014.06.001. [DOI] [PubMed] [Google Scholar]
- StGelais C.; Foster T. L.; Verow M.; Atkins E.; Fishwick C. W.; Rowlands D.; Harris M.; Griffin S. Determinants of hepatitis C virus p7 ion channel function and drug sensitivity identified in vitro. J. Virol. 2009, 83, 7970–779981. 10.1128/JVI.00521-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew C. F.; Vijayan R.; Chang J.; Zitzmann N.; Biggin P. C. Determination of pore-lining residues in the hepatitis C virus p7 protein. Biophys. J. 2009, 96, L10–2. 10.1016/j.bpj.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Goose J. E.; Caffrey M.; Carpenter E. P.; Parker J. L.; Newstead S.; Sansom M. S. MemProtMD: automated insertion of membrane protein structures into explicit lipid membranes. Structure 2015, 23, 1350–1361. 10.1016/j.str.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitinger U.; Farag N. S.; Ali N. K.; Breitinger H. G. Patch-clamp study of Hepatitis C p7 channels reveals genotype-specific sensitivity to inhibitors. Biophys. J. 2016, 110, 2419–2429. 10.1016/j.bpj.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Opella S. J. Relating structure and function of viral membrane-spanning miniproteins. Curr. Opin. Virol. 2015, 12, 121–125. 10.1016/j.coviro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichmann L.; Wang Y.-T.; Fischer W. B. Docking assay of small molecule antivirals to p7 of HCV. Comput. Biol. Chem. 2014, 53PB, 308–317. 10.1016/j.compbiolchem.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Cristian L.; Lear J. D.; DeGrado W. F. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14772–14777. 10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North B.; Cristian L.; Fu Stowell X.; Lear J. D.; Saven J. G.; Degrado W. F. Characterization of a membrane protein folding motif, the Ser zipper, using designed peptides. J. Mol. Biol. 2006, 359, 930–939. 10.1016/j.jmb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Mineev K. S.; Panova S. V.; Bocharova O. V.; Bocharov E. V.; Arseniev A. S. The membrane mimetic affects the spatial structure and mobility of EGFR transmembrane and juxtamembrane domains. Biochemistry 2015, 54, 6295–6298. 10.1021/acs.biochem.5b00851. [DOI] [PubMed] [Google Scholar]
- Dev J.; Park D.; Fu Q.; Chen J.; Ha H. J.; Ghantous F.; Herrmann T.; Chang W.; Liu Z.; Frey G.; Michael; Chen B.; Chou J. J. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 2016, 353, 172–175. 10.1126/science.aaf7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiliveri S. C.; Louis J. M.; Ghirlando R.; Baber J. L.; Bax A. Tilted, uninterrupted, monomeric HIV-1 gp41 transmembrane helix from residual dipolar couplings. J. Am. Chem. Soc. 2018, 140, 34–37. 10.1021/jacs.7b10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath A.; Glibowicka M.; Nadeau V. G.; Chen G.; Deber C. M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1760–1765. 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkenhorst W. F.; Merzlyakov M.; Hristova K.; Wimley W. C. Polar residues in transmembrane helices can decrease electrophoretic mobility in polyacrylamide gels without causing helix dimerization. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 1321–1331. 10.1016/j.bbamem.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian S. A.; Tamm L. K. Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin. Biochemistry 2000, 39, 496–507. 10.1021/bi991594p. [DOI] [PubMed] [Google Scholar]
- Oxenoid K.; Dong Y.; Cao C.; Cui T.; Sancak Y.; Markhard A. L.; Grabarek Z.; Kong L.; Liu Z.; Ouyang B.; Cong Y.; Mootha V. K.; Chou J. J. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. 10.1038/nature17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y.; Krapivinsky G.; Clapham D. E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Bers D. M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- MacLennan D. H.; Kranias E. G. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- Simmerman H. K.; Jones L. R. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998, 78, 921–947. 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Kurzydlowski K.; Tada M.; MacLennan D. H. Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J. Biol. Chem. 1996, 271, 21726–21731. 10.1074/jbc.271.36.21726. [DOI] [PubMed] [Google Scholar]
- Akaike T.; Du N.; Lu G.; Minamisawa S.; Wang Y.; Ruan H. A sarcoplasmic reticulum localized protein phosphatase regulates phospholamban phosphorylation and promotes ischemia reperfusion injury in the heart. JACC: Basic Transl. Sci. 2017, 2, 160–180. 10.1016/j.jacbts.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi A.; Kranias E. G.. The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 2014, 5. 10.3389/fphar.2014.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim C. B.; Kirby T. L.; Zhang Z.; Nesmelov Y.; Thomas D. D. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14437–14442. 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Fei J. Z.; Kawakami T.; Smith S. O. Structural constraints on the transmembrane and juxtamembrane regions of the phospholamban pentamer in membrane bilayers: Gln29 and Leu52. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 2971–2978. 10.1016/j.bbamem.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamoon J.; Mascioni A.; Thomas D. D.; Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys. J. 2003, 85, 2589–2598. 10.1016/S0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberth S.; Schmid H.; Muenchbach M.; Vorherr T.; Krebs J.; Carafoli E.; Griesinger C. NMR Structure of Phospholamban. Helv. Chim. Acta 2000, 83 (9), 2141–2152. . [DOI] [Google Scholar]
- Metcalfe E. E.; Zamoon J.; Thomas D. D.; Veglia G. 1 H/15 N heteronuclear NMR spectroscopy shows four dynamic domains for phospholamban reconstituted in dodecylphosphocholine micelles. Biophys. J. 2004, 87, 1205–1214. 10.1529/biophysj.103.038844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenoid K.; Chou J. J. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10870–10875. 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenoid K.; Rice A. J.; Chou J. J. Comparing the structure and dynamics of phospholamban pentamer in its unphosphorylated and pseudo-phosphorylated states. Protein Sci. 2007, 16, 1977–1983. 10.1110/ps.072975107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs R.; Nelson M.; Simmerman H.; Jones L. R. Phospholamban forms Ca2+-selective channels in lipid bilayers. J. Biol. Chem. 1988, 263, 18364–18368. [PubMed] [Google Scholar]
- Smeazzetto S.; Schröder I.; Thiel G.; Moncelli M. R. Phospholamban generates cation selective ion channels. Phys. Chem. Chem. Phys. 2011, 13, 12935–12939. 10.1039/c1cp20460b. [DOI] [PubMed] [Google Scholar]
- Smeazzetto S.; Saponaro A.; Young H. S.; Moncelli M. R.; Thiel G. Structure-function relation of phospholamban: modulation of channel activity as a potential regulator of SERCA activity. PLoS One 2013, 8, e52744. 10.1371/journal.pone.0052744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becucci L.; Cembran A.; Karim C. B.; Thomas D. D.; Guidelli R.; Gao J.; Veglia G. On the function of pentameric phospholamban: ion channel or storage form?. Biophys. J. 2009, 96, L60–L62. 10.1016/j.bpj.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. C.; Tang C.; Clore G. M.; Hummer G. Replica exchange simulations of transient encounter complexes in protein-protein association. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 12855–12860. 10.1073/pnas.0802460105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becucci L.; Cembran A.; Karim C. B.; Thomas D. D.; Guidelli R.; Gao J.; Veglia G. On the function of pentameric phospholamban: Ion channel or storage form?. Biophys. J. 2009, 96, L60–L62. 10.1016/j.bpj.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffeo C.; Aksimentiev A. Structure, dynamics, and ion conductance of the phospholamban pentamer. Biophys. J. 2009, 96, 4853–4865. 10.1016/j.bpj.2009.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin I. T.; Rothman M.; Ludlam C. F.; Aimoto S.; Engelman D. M.; Rothschild K. J.; Smith S. O. Structural model of the phospholamban ion channel complex in phospholipid membranes. J. Mol. Biol. 1995, 248, 824–834. 10.1006/jmbi.1995.0263. [DOI] [PubMed] [Google Scholar]
- Traaseth N. J.; Ha K. N.; Verardi R.; Shi L.; Buffy J. J.; Masterson L. R.; Veglia G. Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca2+-ATPase. Biochemistry 2008, 47, 3–13. 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z.; Reid D. G.; Watts A.; Middleton D. A. A solid-state NMR study of the phospholamban transmembrane domain: local structure and interactions with Ca 2+-ATPase. Biochim. Biophys. Acta, Biomembr. 2000, 1468, 187–198. 10.1016/S0005-2736(00)00257-1. [DOI] [PubMed] [Google Scholar]
- Middleton D. A.; Ahmed Z.; Glaubitz C.; Watts A. REDOR NMR on a hydrophobic peptide in oriented membranes. J. Magn. Reson. 2000, 147, 366–370. 10.1006/jmre.2000.2187. [DOI] [PubMed] [Google Scholar]
- Smith S. O.; Kawakami T.; Liu W.; Ziliox M.; Aimoto S. Helical structure of phospholamban in membrane bilayers. J. Mol. Biol. 2001, 313, 1139–1148. 10.1006/jmbi.2001.5101. [DOI] [PubMed] [Google Scholar]
- Traaseth N. J.; Verardi R.; Torgersen K. D.; Karim C. B.; Thomas D. D.; Veglia G. Spectroscopic validation of the pentameric structure of phospholamban. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14676–14681. 10.1073/pnas.0701016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascioni A.; Karim C.; Zamoon J.; Thomas D. D.; Veglia G. Solid-state NMR and rigid body molecular dynamics to determine domain orientations of monomeric phospholamban. J. Am. Chem. Soc. 2002, 124, 9392–9393. 10.1021/ja026507m. [DOI] [PubMed] [Google Scholar]
- Traaseth N. J.; Shi L.; Verardi R.; Mullen D. G.; Barany G.; Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10165–10170. 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verardi R.; Shi L.; Traaseth N. J.; Walsh N.; Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9101–9106. 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov V. V.; Mote K. R.; Verardi R.; Veglia G. Structural dynamics and topology of phosphorylated phospholamban homopentamer reveal its role in the regulation of calcium transport. Structure 2013, 21, 2119–2130. 10.1016/j.str.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robia S. L.; Flohr N. C.; Thomas D. D. Phospholamban pentamer quaternary conformation determined by in-gel fluorescence anisotropy. Biochemistry 2005, 44, 4302–4311. 10.1021/bi0478446. [DOI] [PubMed] [Google Scholar]
- Lamberth S.; Schmid H.; Muenchbach M.; Vorherr T.; Krebs J.; Carafoli E.; Griesinger C. NMR solution structure of phospholamban. Helv. Chim. Acta 2000, 83, 2141–2152. . [DOI] [Google Scholar]
- Tiburu E. K.; Karp E. S.; Dave P. C.; Damodaran K.; Lorigan G. A. Investigating the dynamic properties of the transmembrane segment of phospholamban incorporated into phospholipid bilayers utilizing 2H and 15N solid-state NMR spectroscopy. Biochemistry 2004, 43, 13899–13909. 10.1021/bi0490993. [DOI] [PubMed] [Google Scholar]
- Ghimire H.; Abu-Baker S.; Sahu I. D.; Zhou A.; Mayo D. J.; Lee R. T.; Lorigan G. A. Probing the helical tilt and dynamic properties of membrane-bound phospholamban in magnetically aligned bicelles using electron paramagnetic resonance spectroscopy. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 645–650. 10.1016/j.bbamem.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave P. C.; Tiburu E. K.; Damodaran K.; Lorigan G. A. Investigating structural changes in the lipid bilayer upon insertion of the transmembrane domain of the membrane-bound protein phospholamban utilizing 31 P and 2 H solid-state NMR spectroscopy. Biophys. J. 2004, 86, 1564–1573. 10.1016/S0006-3495(04)74224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Baker S.; Lu J.-X.; Chu S.; Shetty K. K.; Lorigan G. A. The structural topology of wild-type phospholamban in oriented lipid bilayers using 15N solid-state NMR spectroscopy. Protein Sci. 2007, 16, 2345–2349. 10.1110/ps.072977707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Baker S.; Lorigan G. A. Phospholamban and its phosphorylated form interact differently with lipid bilayers: a 31P, 2H, and 13C solid-state NMR spectroscopic study. Biochemistry 2006, 45, 13312–13322. 10.1021/bi0614028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe E. E.; Traaseth N. J.; Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry 2005, 44, 4386–4396. 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- Ha K. N.; Traaseth N. J.; Verardi R.; Zamoon J.; Cembran A.; Karim C. B.; Thomas D. D.; Veglia G. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. J. Biol. Chem. 2007, 282, 37205–37214. 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- Ha K. N.; Gustavsson M.; Veglia G. Tuning the structural coupling between the transmembrane and cytoplasmic domains of phospholamban to control sarcoplasmic reticulum Ca2+-ATPase (SERCA) function. J. Muscle Res. Cell Motil. 2012, 33, 485–492. 10.1007/s10974-012-9319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamoon J.; Nitu F.; Karim C.; Thomas D.; Veglia G. Mapping the interaction surface of a membrane protein: unveiling the conformational switch of phospholamban in calcium pump regulation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4747–4752. 10.1073/pnas.0406039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesmelov Y. E.; Karim C. B.; Song L.; Fajer P. G.; Thomas D. D. Rotational dynamics of phospholamban determined by multifrequency electron paramagnetic resonance. Biophys. J. 2007, 93, 2805–2812. 10.1529/biophysj.107.108910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronesi O. C.; Becker S.; Seidel K.; Heise H.; Young H. S.; Baldus M. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 2005, 127, 12965–12974. 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Karim C. B.; Lockamy E. L.; Thomas D. D.; Veglia G. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca 2+-ATPase. J. Mol. Biol. 2011, 408, 755–765. 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson L. R.; Yu T.; Shi L.; Wang Y.; Gustavsson M.; Mueller M. M.; Veglia G. cAMP-dependent protein kinase A selects the excited state of the membrane substrate phospholamban. J. Mol. Biol. 2011, 412, 155–164. 10.1016/j.jmb.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Veglia G. Activating and deactivating roles of lipid bilayers on the Ca2+-ATPase/phospholamban complex. Biochemistry 2011, 50, 10367–10374. 10.1021/bi200759y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Veglia G. Probing ground and excited states of phospholamban in model and native lipid membranes by magic angle spinning NMR spectroscopy. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 146–153. 10.1016/j.bbamem.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M.; Verardi R.; Mullen D. G.; Mote K. R.; Traaseth N. J.; Gopinath T.; Veglia G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17338–17343. 10.1073/pnas.1303006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascioni A.; Karim C.; Zamoon J.; Thomas D. D.; Veglia G. Solid-state NMR and rigid body molecular dynamics to determine domain orientations of monomeric phospholamban. J. Am. Chem. Soc. 2002, 124, 9392–9393. 10.1021/ja026507m. [DOI] [PubMed] [Google Scholar]
- Houndonougbo Y.; Kuczera K.; Jas G. S. Structure and dynamics of phospholamban in solution and in membrane bilayer: Computer simulations. Biochemistry 2005, 44, 1780–1792. 10.1021/bi0488404. [DOI] [PubMed] [Google Scholar]
- Paterlini M. G.; Thomas D. D. The alpha-helical propensity of the cytoplasmic domain of phospholamban: A molecular dynamics simulation of the effect of phosphorylation and mutation. Biophys. J. 2005, 88, 3243–3251. 10.1529/biophysj.104.054460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano S.; Carafoli E. The role of phosphorylation on the structure and dynamics of phospholamban: a model from molecular simulations. Proteins: Struct., Funct., Genet. 2007, 66, 930–940. 10.1002/prot.21239. [DOI] [PubMed] [Google Scholar]
- Sugita Y.; Miyashita N.; Yoda T.; Ikeguchi M.; Toyoshima C. Structural changes in the cytoplasmic domain of phospholamban by phosphorylation at Ser16: a molecular dynamics study. Biochemistry 2006, 45, 11752–11761. 10.1021/bi061071z. [DOI] [PubMed] [Google Scholar]
- Traaseth N. J.; Shi L.; Verardi R.; Mullen D. G.; Barany G.; Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10165–10170. 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna M.; Mukhopadhyay C. Cholesterol driven alteration of the conformation and dynamics of phospholamban in model membranes. Phys. Chem. Chem. Phys. 2011, 13, 20188–20198. 10.1039/c1cp21793c. [DOI] [PubMed] [Google Scholar]
- Sayadi M.; Tanizaki S.; Feig M. Effect of membrane thickness on conformational sampling of phospholamban from computer simulations. Biophys. J. 2010, 98, 805–814. 10.1016/j.bpj.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayadi M.; Feig M. Role of conformational sampling of Ser16 and Thr17-phosphorylated phospholamban in interactions with SERCA. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 577–585. 10.1016/j.bbamem.2012.08.017. [DOI] [PubMed] [Google Scholar]
- Lian P.; Wei D.-Q.; Wang J.-F.; Chou K.-C. An allosteric mechanism inferred from molecular dynamics simulations on phospholamban pentamer in lipid membranes. PLoS One 2011, 6, e18587. 10.1371/journal.pone.0018587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J. J.; Kaufman J. D.; Stahl S. J.; Wingfield P. T.; Bax A. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J. Am. Chem. Soc. 2002, 124, 2450–2451. 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- Ulmer T. S.; Bax A.; Cole N. B.; Nussbaum R. L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- Porcelli F.; Verardi R.; Shi L.; Henzler-Wildman K. A.; Ramamoorthy A.; Veglia G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. 10.1021/bi702036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Traaseth N. J.; Verardi R.; Gustavsson M.; Gao J.; Veglia G. Paramagnetic-based NMR restraints lift residual dipolar coupling degeneracy in multidomain detergent-solubilized membrane proteins. J. Am. Chem. Soc. 2011, 133, 2232–2241. 10.1021/ja109080t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Traaseth N. J.; Verardi R.; Cembran A.; Gao J.; Veglia G. A refinement protocol to determine structure, topology, and depth of insertion of membrane proteins using hybrid solution and solid-state NMR restraints. J. Biomol. NMR 2009, 44, 195–205. 10.1007/s10858-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B.Ion Channels of Excitable Membranes, 3rd ed.; Sinauer: Sunderland, MA, 2001. [Google Scholar]
- MacKinnon R. Potassium channels. FEBS Lett. 2003, 555, 62–65. 10.1016/S0014-5793(03)01104-9. [DOI] [PubMed] [Google Scholar]
- Gouaux E.; MacKinnon R. Principles of selective ion transport in channels and pumps. Science 2005, 310, 1461–1465. 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- Gao L.; Mi X.; Paajanen V.; Wang K.; Fan Z. Activation-coupled inactivation in the bacterial potassium channel KcsA. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17630–17635. 10.1073/pnas.0505158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales J. F.; Cuello L. G.; Zhao Y.; Jogini V.; Cortes D. M.; Roux B.; Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 2006, 13, 311–318. 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- Chakrapani S.; Cordero-Morales J. F.; Perozo E. A quantitative description of KcsA gating I: macroscopic currents. J. Gen. Physiol. 2007, 130, 465–478. 10.1085/jgp.200709843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ader C.; Schneider R.; Hornig S.; Velisetty P.; Vardanyan V.; Giller K.; Ohmert I.; Becker S.; Pongs O.; Baldus M. Coupling of activation and inactivation gate in a K+-channel: potassium and ligand sensitivity. EMBO J. 2009, 28, 2825–2834. 10.1038/emboj.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani S.; Cordero-Morales J. F.; Perozo E. A quantitative description of KcsA gating II: single-channel currents. J. Gen. Physiol. 2007, 130, 479–496. 10.1085/jgp.200709844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S.; Osawa M.; Mita K.; Toyonaga S.; Machiyama A.; Ueda T.; Takeuchi K.; Oiki S.; Shimada I. Functional equilibrium of the KcsA structure revealed by NMR. J. Biol. Chem. 2012, 287, 39634–39641. 10.1074/jbc.M112.401265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K. a.; Tzitzilonis C.; Kwiatkowski W.; Choe S.; Riek R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat. Struct. Mol. Biol. 2007, 14, 1089–1095. 10.1038/nsmb1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chill J. H.; Louis J. M.; Miller C.; Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Sci. 2006, 15, 684–698. 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chill J. H.; Louis J. M.; Delaglio F.; Bax A. Local and global structure of the monomeric subunit of the potassium channel KcsA probed by NMR. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 3260–3270. 10.1016/j.bbamem.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S.; Osawa M.; Takeuchi K.; Shimada I. Structural basis underlying the dual gate properties of KcsA. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6216–6221. 10.1073/pnas.0911270107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ader C.; Schneider R.; Hornig S.; Velisetty P.; Wilson E. M.; Lange A.; Giller K.; Ohmert I.; Martin-Eauclaire M.-F.; Trauner D.; Becker S.; Pongs O.; Baldus M. A structural link between inactivation and block of a K+ channel. Nat. Struct. Mol. Biol. 2008, 15, 605–612. 10.1038/nsmb.1430. [DOI] [PubMed] [Google Scholar]
- Schneider R.; Ader C.; Lange A.; Giller K.; Hornig S.; Pongs O.; Becker S.; Baldus M. Solid-state NMR spectroscopy applied to a chimeric potassium channel in lipid bilayers. J. Am. Chem. Soc. 2008, 130, 7427–7435. 10.1021/ja800190c. [DOI] [PubMed] [Google Scholar]
- Bhate M. P.; Wylie B. J.; Tian L.; McDermott A. E. Conformational dynamics in the selectivity filter of KcsA in response to potassium ion concentration. J. Mol. Biol. 2010, 401, 155–166. 10.1016/j.jmb.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhate M. P.; McDermott A. E. Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 15265–15270. 10.1073/pnas.1211900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhate M. P.; Wylie B. J.; Thompson A.; Tian L.; Nimigean C.; McDermott A. E. Preparation of uniformly isotope labeled KcsA for solid state NMR: Expression, purification, reconstitution into liposomes and functional assay. Protein Expression Purif. 2013, 91, 119–124. 10.1016/j.pep.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvil H. T.; Maj M.; Matulef K.; Annen A. W.; Ostmeyer J.; Perozo E.; Roux B.; Valiyaveetil F. I.; Zanni M. T. Probing the effects of gating on the ion occupancy of the K+ channel selectivity filter using 2D IR spectroscopy. J. Am. Chem. Soc. 2017, 139, 8837–8845. 10.1021/jacs.7b01594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvil H. T.; Carr J. K.; Matulef K.; Annen A. W.; Li H.; Maj M.; Ostmeyer J.; Serrano A. L.; Raghuraman H.; Moran S. D.; Skinner J. L.; Perozo E.; Roux B.; Valiyaveetil F. I.; Zanni M. T. Instantaneous ion configurations in the K+ ion channel selectivity filter revealed by 2D IR spectroscopy. Science 2016, 353, 1040–1044. 10.1126/science.aag1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneche S.; Roux B. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 2000, 78, 2900–2917. 10.1016/S0006-3495(00)76831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov S. Y.; Roux B. Ion selectivity in potassium channels. Biophys. Chem. 2006, 124, 279–291. 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Morais-Cabral J. H.; Kaufman A.; MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 angstrom resolution. Nature 2001, 414, 43–48. 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- Cuello L. G.; Jogini V.; Cortes D. M.; Perozo E. Structural mechanism of C-type inactivation in K+ channels. Nature 2010, 466, 203–208. 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal S.; Cuello L. G.; Cortes D. M.; Koide S.; Kossiakoff A. A.; Perozo E. Mechanism of activation gating in the full-length KcsA K+ channel. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11896–11899. 10.1073/pnas.1105112108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W.; Zhou M.; MacKinnon R. Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol. 2007, 5, e121. 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev K. S.; Efimov S. V.; Kolosova O.; Filippov A.; Klochkov V. V. High-resolution NMR structure of the antimicrobial peptide protegrin-2 in the presence of DPC micelles. J. Biomol. NMR 2015, 61, 227–234. 10.1007/s10858-014-9885-4. [DOI] [PubMed] [Google Scholar]
- Usachev K. S.; Efimov S. V.; Kolosova O. A.; Klochkova E. A.; Aganov A. V.; Klochkov V. V. Antimicrobial peptide protegrin-3 adopt an antiparallel dimer in the presence of DPC micelles: A high-resolution NMR study. J. Biomol. NMR 2015, 62, 71–79. 10.1007/s10858-015-9920-0. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Lesovoy D. M.; Goncharuk S. A.; Goncharuk M. V.; Hristova K.; Arseniev A. S. Structure of FGFR3 transmembrane domain dimer: implications for signaling and human pathologies. Structure 2013, 21, 2087–2093. 10.1016/j.str.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X.-M.; Ding Y.; Yu J.; Yao Y.; Marassi F. M. Structure of the Na, K-ATPase regulatory protein FXYD2b in micelles: Implications for membrane-water interfacial arginines. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 299–306. 10.1016/j.bbamem.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko V.; Xu Y.; Tang P. Structure of the first transmembrane domain of the neuronal acetylcholine receptor β 2 subunit. Biophys. J. 2007, 92, 1616–1622. 10.1529/biophysj.106.095364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie K. R.; Prestegard J. H.; Engelman D. M. A transmembrane helix dimer: structure and implications. Science 1997, 276, 131–133. 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- Call M. E.; Schnell J. R.; Xu C.; Lutz R. A.; Chou J. J.; Wucherpfennig K. W. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 2006, 127, 355–368. 10.1016/j.cell.2006.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi F. M.; Ding Y.; Schwieters C. D.; Tian Y.; Yao Y. Backbone structure of Yersinia pestis Ail determined in micelles by NMR-restrained simulated annealing with implicit membrane solvation. J. Biomol. NMR 2015, 63, 59–65. 10.1007/s10858-015-9963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass R. B.; Strop P.; Barclay M.; Rees D. C. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science 2002, 298, 1582–1587. 10.1126/science.1077945. [DOI] [PubMed] [Google Scholar]
- Wang W.; Black S. S.; Edwards M. D.; Miller S.; Morrison E. L.; Bartlett W.; Dong C.; Naismith J. H.; Booth I. R. The structure of an open form of an E. coli mechanosensitive channel at 3.45 Å resolution. Science 2008, 321, 1179–1183. 10.1126/science.1159262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. T.; Berger B. W.; DeGrado W. F. Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure 2008, 16, 991–1001. 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R. C.; Lee S.; Moore J. D.; Opella S. J.; Cross T. A. Backbone structure of a small helical integral membrane protein: A unique structural characterization. Protein Sci. 2009, 18, 134–146. 10.1002/pro.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziani W.; Maillard A. P.; Petit-Härtlein I.; Garnier N.; Crouzy S.; Girard E. F.; Covès J. The X-ray structure of NccX from cupriavidus metallidurans 31A illustrates potential dangers of detergent solubilization when generating and interpreting crystal structures of membrane proteins. J. Biol. Chem. 2014, 289, 31160–31172. 10.1074/jbc.M114.586537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. A.; Cho S.-H.; Ye J.; Collet J.-F.; Beckwith J. R.; Chou J. J. Structure and multistate function of the transmembrane electron transporter CcdA. Nat. Struct. Mol. Biol. 2015, 22, 809–814. 10.1038/nsmb.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T. P.; Su T. C.; Nakamura Y.; Tsai S. Y. The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol. Sci. 2016, 37, 262–78. 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S.; Su T. P.; Fujimoto M.; Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012, 35, 762–71. 10.1016/j.tins.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D.; Romero L.; Merlos M.; Vela J. M. Sigma 1 receptor: a new therapeutic target for pain. Eur. J. Pharmacol. 2013, 716, 78–93. 10.1016/j.ejphar.2013.01.068. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Su T. P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Aydar E.; Palmer C. P.; Klyachko V. A.; Jackson M. B. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 2002, 34, 399–410. 10.1016/S0896-6273(02)00677-3. [DOI] [PubMed] [Google Scholar]
- Balasuriya D.; Stewart A. P.; Edwardson J. M. The sigma-1 receptor interacts directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA receptor. J. Neurosci. 2013, 33, 18219–24. 10.1523/JNEUROSCI.3360-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Roldan J. L.; Ossa F.; Amin N. T.; Schnell J. R. Solution NMR studies reveal the location of the second transmembrane domain of the human sigma-1 receptor. FEBS Lett. 2015, 589, 659–65. 10.1016/j.febslet.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. R.; Zheng S.; Gurpinar E.; Koehl A.; Manglik A.; Kruse A. C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossa F.; Schnell J. R.; Ortega-Roldan J. L. A review of the human sigma-1 receptor structure. Adv. Exp. Med. Biol. 2017, 964, 15–29. 10.1007/978-3-319-50174-1_3. [DOI] [PubMed] [Google Scholar]
- Kefala G.; Ahn C.; Krupa M.; Esquivies L.; Maslennikov I.; Kwiatkowski W.; S C. Structures of the OmpF porin crystallized in the presence of foscholine-12. Protein Sci. 2002, 19, 1117–1125. 10.1002/pro.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaptal V.; Kilburg A.; Flot D.; Wiseman B.; Aghajari N.; Jault J. M.; Falson P. Two different centered monoclinic crystals of the E. coli outer-membrane protein OmpF originate from the same building block. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 326–332. 10.1016/j.bbamem.2015.11.021. [DOI] [PubMed] [Google Scholar]
- Efremov R. G.; Sazanov L. A. Structure of Escherichia coli OmpF porin from lipidic mesophase. J. Struct. Biol. 2012, 178, 311–318. 10.1016/j.jsb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Penel S.; Pebay-Peyroula E.; Rosenbusch J.; Rummel G.; Schirmer T.; Timmins P. Detergent binding in trigonal crystals of OmpF porin from Escherichia coli. Biochimie 1998, 80, 543546–551. 10.1016/S0300-9084(00)80019-X. [DOI] [PubMed] [Google Scholar]
- Arora A.; Abildgaard F.; Bushweller J. H.; Tamm L. K. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001, 8, 334–338. 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- Cierpicki T.; Liang B.; Tamm L. K.; Bushweller J. H. Increasing the accuracy of solution NMR structures of membrane proteins by application of residual dipolar couplings. High-resolution structure of outer membrane protein A. J. Am. Chem. Soc. 2006, 128, 6947–6951. 10.1021/ja0608343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández C.; Hilty C.; Bonjour S.; Adeishvili K.; Pervushin K.; Wüthrich K. Solution NMR studies of the integral membrane proteins OmpX and OmpA from Escherichia coli. FEBS Lett. 2001, 504, 173–178. 10.1016/S0014-5793(01)02742-9. [DOI] [PubMed] [Google Scholar]
- Zoonens M.; Catoire L. J.; Giusti F.; Popot J.-L. NMR study of a membrane protein in detergent-free aqueous solution. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 8893–8898. 10.1073/pnas.0503750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susac L.; Horst R.; Wüthrich K. Solution-NMR characterization of Outer-membrane protein A from E. coli in lipid bilayer nanodiscs and detergent micelles. ChemBioChem 2014, 15, 995–1000. 10.1002/cbic.201300729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández C.; Adeishvili K.; Wüthrich K. Transverse relaxation-optimized NMR spectroscopy with the outer membrane protein OmpX in dihexanoyl phosphatidylcholine micelles. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 2358–2363. 10.1073/pnas.051629298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautsch A.; Vogt J.; Model K.; Siebold C.; Schulz G. E. Strategy for membrane protein crystallization exemplified with OmpA and OmpX. Proteins: Struct., Funct., Genet. 1999, 34, 167–172. . [DOI] [PubMed] [Google Scholar]
- Catoire L. J.; Zoonens M.; van Heijenoort C.; Giusti F.; Guittet E.; Popot J.-L. Solution NMR mapping of water-accessible residues in the transmembrane beta-barrel of OmpX. Eur. Biophys. J. 2010, 39, 623–630. 10.1007/s00249-009-0513-2. [DOI] [PubMed] [Google Scholar]
- Vogt J.; Schulz G. E. The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure 1999, 7, 1301–1309. 10.1016/S0969-2126(00)80063-5. [DOI] [PubMed] [Google Scholar]
- Frey L.; Lakomek N.-A.; Riek R.; Bibow S. Micelles, bicelles, and nanodiscs: comparing the impact of membrane mimetics on membrane protein backbone dynamics. Angew. Chem., Int. Ed. 2017, 56, 380–383. 10.1002/anie.201608246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox D. A.; Larsson P.; Lo R. H.; Kroncke B. M.; Kasson P. M.; Columbus L. Structure of the Neisserial Outer membrane protein Opa 60: loop flexibility essential to receptor recognition and bacterial engulfment. J. Am. Chem. Soc. 2014, 136, 9938–9946. 10.1021/ja503093y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop R. E.; Gibbons H. S.; Guina T.; Trent M. S.; Miller S. I.; Raetz C. R. Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. EMBO J. 2000, 19, 5071–5080. 10.1093/emboj/19.19.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang P. M.; Choy W. Y.; Lo E. I.; Chen L.; Forman-Kay J. D.; Raetz C. R.; Privé G. G.; Bishop R. E.; Kay L. E. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 13560–13565. 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn V.; Lo E. I.; Engel C. K.; Chen L.; Hwang P. M.; Kay L. E.; Bishop R. E.; Privé G. G. A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO J. 2004, 23, 2931–2941. 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta-Seijo J. A.; Neale C.; Khan M. A.; Moktar J.; Tran C. D.; Bishop R. E.; Pomès R.; Privé G. G. PagP crystallized from SDS/cosolvent reveals the route for phospholipid access to the hydrocarbon ruler. Structure 2010, 18, 1210–1219. 10.1016/j.str.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai K.; Baaden M.; Murdock S.; Wu B.; Ng M. H.; Johnston S.; Boardman R.; Fangohr H.; Cox K.; Essex J. W.; Sansom M. S. P. Three hydrolases and a transferase: comparative analysis of active-site dynamics via the BioSimGrid database. J. Mol. Graphics Modell. 2007, 25, 896–902. 10.1016/j.jmgm.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Khan M. A.; Neale C.; Michaux C.; Pomès R.; Priv G. G.; Woody R. W.; Bishop R. E. Gauging a hydrocarbon ruler by an intrinsic exciton probe. Biochemistry 2007, 46, 4565–4579. 10.1021/bi602526k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K. A.; Bond P. J.; Ivetac A.; Chetwynd A. P.; Khalid S.; Sansom M. S. P. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure 2008, 16, 621–630. 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Cox K.; Bond P. J.; Grottesi A.; Baaden M.; Sansom M. S. P. Outer membrane proteins: Comparing X-ray and NMR structures by MD simulations in lipid bilayers. Eur. Biophys. J. 2008, 37, 131–141. 10.1007/s00249-007-0185-8. [DOI] [PubMed] [Google Scholar]
- Cox K.; Sansom M. S. P. One membrane protein, two structures and six environments: a comparative molecular dynamics simulation study of the bacterial outer membrane protein PagP. Mol. Membr. Biol. 2009, 26, 205–214. 10.1080/09687680902788967. [DOI] [PubMed] [Google Scholar]
- Neale C.; Madill C.; Rauscher S.; Pomès R. Accelerating convergence in molecular dynamics simulations of solutes in lipid membranes by conducting a random walk along the bilayer normal. J. Chem. Theory Comput. 2013, 9, 3686–3703. 10.1021/ct301005b. [DOI] [PubMed] [Google Scholar]
- Borysik A. J. Structure and dynamics of a protein-surfactant assembly studied by ion-mobility mass spectrometry and molecular dynamics simulations. Anal. Chem. 2015, 87, 8970–8976. 10.1021/acs.analchem.5b02172. [DOI] [PubMed] [Google Scholar]
- Hwang P. M.; Bishop R. E.; Kay L. E. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9618–9623. 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang P. M.; Kay L. E. Solution structure and dynamics of integral membrane proteins by NMR: a case study involving the enzyme PagP. Methods Enzymol. 2005, 394, 335–350. 10.1016/S0076-6879(05)94013-5. [DOI] [PubMed] [Google Scholar]
- Saurel O.; Iordanov I. O.; Nars G.; Demange P.; Le Marchand T.; Andreas L. B.; Pintacuda G.; Milon A. Local and global dynamics in Klebsiella pneumoniae outer membrane protein A in lipid bilayers probed at atomic resolution. J. Am. Chem. Soc. 2017, 139, 1590. 10.1021/jacs.6b11565. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; Abildgaard F.; Arora A.; Blad H.; Bushweller J. H. Structure, dynamics and function of the outer membrane protein A (OmpA) and influenza hemagglutinin fusion domain in detergent micelles by solution NMR. FEBS Lett. 2003, 555, 139–143. 10.1016/S0014-5793(03)01127-X. [DOI] [PubMed] [Google Scholar]
- Liang B.; Arora A.; Tamm L. K. Fast-time scale dynamics of outer membrane protein A by extended model-free analysis of NMR relaxation data. Biochim. Biophys. Acta, Biomembr. 2010, 1798, 68–76. 10.1016/j.bbamem.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B.; Tamm L. K. Structure of outer membrane protein G by solution NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16140–16145. 10.1073/pnas.0705466104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox D. A.; Columbus L. Solution NMR resonance assignment strategies for β-barrel membrane proteins. Protein Sci. 2013, 22, 1133–1140. 10.1002/pro.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichmann C.; Orts J.; Tzitzilonis C.; Vgeli B.; Smrt S.; Lorieau J.; Riek R. Intermolecular Detergent–Membrane Protein NOEs for the Characterization of the Dynamics of Membrane Protein–Detergent Complexes. J. Phys. Chem. B 2014, 118, 14288–14301. 10.1021/jp509137q. [DOI] [PubMed] [Google Scholar]
- Roux B.; Schulten K. Computational studies of membrane channels. Structure 2004, 12, 1343–1351. 10.1016/j.str.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Roux B.; Schulten K. Computational studies of membrane channels. Structure 2004, 12, 1343–1351. 10.1016/j.str.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Gumbart J.; Wang Y.; Aksimentiev A.; Tajkhorshid E.; Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr. Opin. Struct. Biol. 2005, 15, 423–431. 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond P. J.; Holyoake J.; Ivetac A.; Khalid S.; Sansom M. S. P. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 2007, 157, 593–605. 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Sansom M. S. P. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Khalili-Araghi F.; Gumbart J.; Wen P.-C.; Sotomayor M.; Tajkhorshid E.; Schulten K. Molecular dynamics simulations of membrane channels and transporters. Curr. Opin. Struct. Biol. 2009, 19, 128–137. 10.1016/j.sbi.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psachoulia E.; Marshall D. P.; Sansom M. S. P. Molecular dynamics simulations of the dimerization of transmembrane alpha-helices. Acc. Chem. Res. 2010, 43, 388–396. 10.1021/ar900211k. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. Molecular simulation approaches to membrane proteins. Structure 2011, 19, 1562–1572. 10.1016/j.str.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. From coarse grained to atomistic: a serial multiscale approach to membrane protein simulations. J. Chem. Theory Comput. 2011, 7, 1157–1166. 10.1021/ct100569y. [DOI] [PubMed] [Google Scholar]
- Maffeo C.; Bhattacharya S.; Yoo J.; Wells D.; Aksimentiev A. Modeling and simulation of ion channels. Chem. Rev. 2012, 112, 6250–6284. 10.1021/cr3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi N.; Winterhalter M.; Kleinekathöfer U. Computational modeling of ion transport through nanopores. Nanoscale 2012, 4, 6166–6180. 10.1039/c2nr31024d. [DOI] [PubMed] [Google Scholar]
- Baştuğ T.; Kuyucak S. Molecular dynamics simulations of membrane proteins. Biophys. Rev. 2012, 4, 271–282. 10.1007/s12551-012-0084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh S. A.; Li J.; Enkavi G.; Wen P.-C.; Huang Z.; Tajkhorshid E. Visualizing functional motions of membrane transporters with molecular dynamics simulations. Biochemistry 2013, 52, 569–587. 10.1021/bi301086x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluhackova K.; Wassenaar T. A.; Böckmann R. A. Molecular dynamics simulations of membrane proteins. Methods Mol. Biol. 2013, 1033, 85–101. 10.1007/978-1-62703-487-6_6. [DOI] [PubMed] [Google Scholar]
- Mondal S.; Khelashvili G.; Weinstein H. Not just an oil slick: how the energetics of protein-membrane interactions impacts the function and organization of transmembrane proteins. Biophys. J. 2014, 106, 2305–2316. 10.1016/j.bpj.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin P. C.; Bond P. J. Molecular dynamics simulations of membrane proteins. Methods Mol. Biol. 2015, 1215, 91–108. 10.1007/978-1-4939-1465-4_5. [DOI] [PubMed] [Google Scholar]
- Choong Y. S.; Yung-Hung R. L. A general overview on outer membrane protein (Omp) simulations. J. Comput. Sci. 2016, 17, 285–291. 10.1016/j.jocs.2016.03.008. [DOI] [Google Scholar]
- Chipot C.; Klein M. L.; Tarek M. In The Handbook of Materials Modeling. Methods and Models of Materials Modeling; Catlow R., Shercliff H., Yip S., Eds.; Kluwer Academic Publishers: Dordrecht, 2005; Vol. 1, pp 929–958. [Google Scholar]
- Kandt C.; Ash W. L.; Tieleman D. P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. 10.1016/j.ymeth.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Allen M. P.; Tildesley D. J.. Computer Simulation of Liquids; Clarendon Press: Oxford, 1987. [Google Scholar]
- Frenkel D.; Smit B.. Understanding Molecular Simulations: From Algorithms to Applications; Academic Press: San Diego, CA, 2002. [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Sansom M. S. P.; Scott K. A.; Bond P. J. Coarse-grained simulation: a high-throughput computational approach to membrane proteins. Biochem. Soc. Trans. 2008, 36, 27–32. 10.1042/BST0360027. [DOI] [PubMed] [Google Scholar]
- Takada S. Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol. 2012, 22, 130–137. 10.1016/j.sbi.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Arkhipov A.; Yin Y.; Schulten K. Membrane-bending mechanism of amphiphysin N-BAR domains. Biophys. J. 2009, 97, 2727–2735. 10.1016/j.bpj.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Schulten K. Membrane sculpting by F-BAR domains studied by molecular dynamics simulations. PLoS Comput. Biol. 2013, 9, e1002892. 10.1371/journal.pcbi.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Voth G. A. Membrane tension controls the assembly of curvature-generating proteins. Nat. Commun. 2015, 6, 7219. 10.1038/ncomms8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimpton S. Fast parallel algorithms for short–range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel L.; Kalé R. D.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; et al. Anton, a special-purpose machine for molecular dynamics simulation. SIGARCH Comput. Archit. News 2007, 35, 1–12. 10.1145/1273440.1250664. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Jung J.; Mori T.; Kobayashi C.; Matsunaga Y.; Yoda T.; Feig M.; Sugita Y. GENESIS: a hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2015, 5, 310–323. 10.1002/wcms.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sener M.; Strumpfer J.; Singharoy A.; Hunter C. N.; Schulten K. Overall energy conversion efficiency of a photosynthetic vesicle. eLife 2016, 5, 1. 10.7554/eLife.09541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf T. B.; Roux B. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11631–11635. 10.1073/pnas.91.24.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf T. B.; Roux B. Structure, energetics, and dynamics of lipid-protein interactions: A molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins: Struct., Funct., Genet. 1996, 24, 92–114. . [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Sansom M. S. P. Membrane protein dynamics versus environment: simulations of OmpA in a micelle and in a bilayer. J. Mol. Biol. 2003, 329, 1035–1053. 10.1016/S0022-2836(03)00408-X. [DOI] [PubMed] [Google Scholar]
- Ceccarelli M.; Marchi M. Simulation and modeling of the rhodobacter sphaeroides bacterial reaction center: structure and interactions. J. Phys. Chem. B 2003, 107, 1423–1431. 10.1021/jp0270001. [DOI] [Google Scholar]
- Braun R.; Engelman D. M.; Schulten K. Molecular dynamics simulation of micelle formation around dimeric glycophorin A transmembrane helices. Biophys. J. 2004, 87, 754–763. 10.1529/biophysj.104.040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond P. J.; Cuthbertson J. M.; Deol S. S.; Sansom M. S. P. MD simulations of spontaneous membrane protein/detergent micelle formation. J. Am. Chem. Soc. 2004, 126, 15948–15949. 10.1021/ja044819e. [DOI] [PubMed] [Google Scholar]
- Böckmann R. A.; Caflisch A. Spontaneous formation of detergent micelles around the outer membrane protein OmpX. Biophys. J. 2005, 88, 3191–3204. 10.1529/biophysj.105.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymore T.; Gao X.; Wong T. Molecular dynamics simulation of the structure and dynamics of a dodecylphosphocholine micelle in aqueous solution. J. Mol. Struct. 1999, 485, 195–210. 10.1016/S0022-2860(99)00090-3. [DOI] [Google Scholar]
- Tieleman D.; Van der Spoel D.; Berendsen H. Molecular dynamics simulations of dodecylphosphocholine micelles at three different aggregate sizes: micellar structure and chain relaxation. J. Phys. Chem. B 2000, 104, 6380–6388. 10.1021/jp001268f. [DOI] [Google Scholar]
- Marrink S.; Tieleman D.; Mark A. Molecular dynamics simulation of the kinetics of spontaneous micelle formation. J. Phys. Chem. B 2000, 104, 12165–12173. 10.1021/jp001898h. [DOI] [Google Scholar]
- Vasudevan S. V.; Balaji P. V. Conformation, orientation and dynamics of dodecylphosphocholine in micellar aggregate: a 3.2 ns molecular dynamics simulation study. Indian J. Biochem. Biophys. 2002, 39, 87–92. [PubMed] [Google Scholar]
- Marrink S. J.; De Vries A. H.; Mark A. E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 2004, 108, 750–760. 10.1021/jp036508g. [DOI] [Google Scholar]
- Lazaridis T.; Mallik B.; Chen Y. Implicit solvent simulations of DPC micelle formation. J. Phys. Chem. B 2005, 109, 15098–15106. 10.1021/jp0516801. [DOI] [PubMed] [Google Scholar]
- Abel S.; Dupradeau F.-Y.; Marchi M. Molecular dynamics simulations of a characteristic DPC micelle in water. J. Chem. Theory Comput. 2012, 8, 4610–4623. 10.1021/ct3003207. [DOI] [PubMed] [Google Scholar]
- Ceccarelli M.; Danelon C.; Laio A.; Parrinello M. Microscopic mechanism of antibiotics translocation through a porin. Biophys. J. 2004, 87, 58–64. 10.1529/biophysj.103.037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia H.; Kaznessis Y. N. Molecular dynamics simulations of helical antimicrobial peptides in SDS micelles: What do point mutations achieve?. Peptides 2005, 26, 2037–2049. 10.1016/j.peptides.2005.03.058. [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Cuthbertson J.; Sansom M. S. P. Simulation studies of the interactions between membrane proteins and detergents. Biochem. Soc. Trans. 2005, 33, 910–912. 10.1042/BST0330910. [DOI] [PubMed] [Google Scholar]
- Patargias G.; Bond P. J.; Deol S. S.; Sansom M. S. P. Molecular dynamics simulations of GlpF in a micelle vs in a bilayer: conformational dynamics of a membrane protein as a function of environment. J. Phys. Chem. B 2005, 109, 575–582. 10.1021/jp046727h. [DOI] [PubMed] [Google Scholar]
- Gao X.; Wong T. C. Molecular dynamics simulation of adrenocorticotropin (1–10) peptide in a solvated dodecylphosphocholine micelle. Biopolymers 2001, 58, 643–659. . [DOI] [PubMed] [Google Scholar]
- Vasudevan S. V.; Balaji P. V. Dynamics of ganglioside headgroup in lipid environment: molecular dynamics simulations of GM1 embedded in dodecylphosphocholine micelle. J. Phys. Chem. B 2001, 105, 7033–7041. 10.1021/jp0027952. [DOI] [Google Scholar]
- Dixon A. M.; Venable R. M.; Pastor R. W.; Bull T. E. Micelle-bound conformation of a hairpin-forming peptide: combined NMR and molecular dynamics study. Biopolymers 2002, 65, 284–298. 10.1002/bip.10249. [DOI] [PubMed] [Google Scholar]
- Khandelia H.; Kaznessis Y. N. Molecular dynamics simulations of the helical antimicrobial peptide ovispirin-1 in a zwitterionic dodecylphosphocholine micelle: Insights into host-cell toxicity. J. Phys. Chem. B 2005, 109, 12990–12996. 10.1021/jp050162n. [DOI] [PubMed] [Google Scholar]
- Lagüe P.; Roux B. t.; Pastor R. W. Molecular dynamics simulations of the influenza hemagglutinin fusion peptide in micelles and bilayers: conformational analysis of peptide and lipids. J. Mol. Biol. 2005, 354, 1129–1141. 10.1016/j.jmb.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Khandelia H.; Kaznessis Y. N. Molecular dynamics investigation of the influence of anionic and zwitterionic interfaces on antimicrobial peptides’ structure: implications for peptide toxicity and activity. Peptides 2006, 27, 1192–1200. 10.1016/j.peptides.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia H.; Langham A. A.; Kaznessis Y. N. Driving engineering of novel antimicrobial peptides from simulations of peptide-micelle interactions. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1224–1234. 10.1016/j.bbamem.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langham A. A.; Khandelia H.; Kaznessis Y. N. How can a beta-sheet peptide be both a potent antimicrobial and harmfully toxic? Molecular dynamics simulations of protegrin-1 in micelles. Biopolymers 2006, 84, 219–231. 10.1002/bip.20397. [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Sansom M. S. P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006, 128, 2697–2704. 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langham A. A.; Waring A. J.; Kaznessis Y. N. Comparison of interactions between beta-hairpin decapeptides and SDS/DPC micelles from experimental and simulation data. BMC Biochem. 2007, 8, 11. 10.1186/1471-2091-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolintineanu D. S.; Langham A. A.; Davis H. T.; Kaznessis Y. N. Molecular dynamics simulations of three protegrin–type antimicrobial peptides: interplay between charges at the termini, β–sheet structure and amphiphilic interactions. Mol. Simul. 2007, 33, 809–819. 10.1080/08927020701393481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia H.; Kaznessis Y. N. Cation-π interactions stabilize the structure of the antimicrobial peptide indolicidin near membranes: molecular dynamics simulations. J. Phys. Chem. B 2007, 111, 242–250. 10.1021/jp064776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyed-Ahmad A.; Khandelia H.; Kaznessis Y. N. Relative free energy of binding between antimicrobial peptides and SDS or DPC micelles. Mol. Simul. 2009, 35, 986–997. 10.1080/08927020902902742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friemann R.; Larsson D. S. D.; Wang Y.; van der Spoel D. Molecular dynamics simulations of a membrane protein-micelle complex in vacuo. J. Am. Chem. Soc. 2009, 131, 16606–16607. 10.1021/ja902962y. [DOI] [PubMed] [Google Scholar]
- Bourbigot S.; Dodd E.; Horwood C.; Cumby N.; Fardy L.; Welch W. H.; Ramjan Z.; Sharma S.; Waring A. J.; Yeaman M. R.; Booth V. Antimicrobial peptide RP-1 structure and interactions with anionic versus zwitterionic micelles. Biopolymers 2009, 91, 1–13. 10.1002/bip.21071. [DOI] [PubMed] [Google Scholar]
- Mineev K. S.; Bocharov E. V.; Volynsky P. E.; Goncharuk M. V.; Tkach E. N.; Ermolyuk Y. S.; Schulga A. A.; Chupin V. V.; Maslennikov I. V.; Efremov R. G.; Arseniev A. S. Dimeric structure of the transmembrane domain of glycophorin A in lipidic and detergent environments. Acta Naturae 2011, 3, 90–98. [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. A. M.; De Avila M.; Polverini E.; Bessonov K.; Bamm V. V.; Harauz G. Solution nuclear magnetic resonance structure and molecular dynamics simulations of a murine 18.5 kDa myelin basic protein segment (S72-S107) in association with dodecylphosphocholine micelles. Biochemistry 2012, 51, 7475–7487. 10.1021/bi300998x. [DOI] [PubMed] [Google Scholar]
- Cao C.; Mao J.; Li F.; Yang M.; He H.; Jiang L.; Liu M. Understanding the interaction between valsartan and detergents by NMR techniques and molecular dynamics simulation. J. Phys. Chem. B 2012, 116, 7470–7478. 10.1021/jp304304v. [DOI] [PubMed] [Google Scholar]
- Mugumbate G.; Jackson G. E.; van der Spoel D.; Kövér K. E.; Szilágyi L. Anopheles gambiae, Anoga-HrTH hormone, free and bound structure — A nuclear magnetic resonance experiment. Peptides 2013, 41, 94–100. 10.1016/j.peptides.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Abel S.; Lorieau A.; de Foresta B.; Dupradeau F.-Y.; Marchi M. Bindings of hMRP1 transmembrane peptides with dodecylphosphocholine and dodecyl-β-d-maltoside micelles: A molecular dynamics simulation study. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 493–509. 10.1016/j.bbamem.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Jackson G. E.; Gamieldien R.; Mugumbate G.; G de G. Structural studies of adipokinetic hormones in water and DPC micelle solution using NMR distance restrained molecular dynamics. Peptides 2014, 53, 270–277. 10.1016/j.peptides.2013.12.019. [DOI] [PubMed] [Google Scholar]
- Scrima M.; Di Marino S.; Grimaldi M.; Campana F.; Vitiello G.; Piotto S. P.; D’Errico G.; D’Ursi A. M. Structural features of the C8 antiviral peptide in a membrane-mimicking environment. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1010–1018. 10.1016/j.bbamem.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Weber D. K.; Yao S.; Rojko N.; Anderluh G.; Lybrand T. P.; Downton M. T.; Wagner J.; Separovic F. Characterization of the lipid-binding site of equinatoxin II by NMR and molecular dynamics simulation. Biophys. J. 2015, 108, 1987–1996. 10.1016/j.bpj.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versace R. E.; Lazaridis T. Modeling protein-micelle systems in implicit water. J. Phys. Chem. B 2015, 119, 8037–8047. 10.1021/acs.jpcb.5b00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor B. L.; Lousa D.; Antunes J. M.; Soares C. M. Self-assembly molecular dynamics simulations shed light into the interaction of the influenza fusion peptide with a membrane bilayer. J. Chem. Inf. Model. 2015, 55, 795–805. 10.1021/ci500756v. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Kim J.-K.; Kim Y.; Bowie J. U.; Im W. Molecular dynamics simulation strategies for protein-micelle complexes. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1566–1572. 10.1016/j.bbamem.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.