

Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains one of the world’s deadliest infectious diseases and urgently requires new antibiotics to treat drug-resistant strains and to decrease the duration of therapy. During infection, Mtb encounters numerous stresses associated with host immunity, including hypoxia, reactive oxygen and nitrogen species, mild acidity, nutrient starvation, and metal sequestration and intoxication. The Mtb proteostasis network, composed of chaperones, proteases, and a eukaryotic-like proteasome, provides protection from stresses and chemistries of host immunity by maintaining the integrity of the mycobacterial proteome. In this Review, we explore the proteostasis network as a noncanonical target for antibacterial drug discovery.
Keywords: proteostasis, proteolysis, chaperones, proteasome, proteases, protein aggregation, protein folding, protein misfolding, proteostasis network, Mycobacterium tuberculosis, antibiotics, host immunity
Antibiotic-resistant bacterial pathogens, including Mycobacterium tuberculosis (Mtb), the etiological agent of tuberculosis (TB), present a serious health crisis. TB is now the leading cause of death from an infectious disease and at the forefront of diseases in dire need of new antibiotics. Treatment of drug-sensitive TB requires 6–9 months of therapy with up to four antibiotics, including the first-line drugs rifampicin (RIF), isoniazid (INH), pyrazinamide (PZA), and ethambutol (EMB). Multidrug resistant TB (MDR-TB), defined by resistance to RIF and INH, and extremely drug resistant TB (XDR-TB), defined by resistance to RIF, INH, a fluoroquinolone, and one of three injectable second-line drugs (amikacin, kanamycin, capreomycin), may require up to an additional 1.5 years of treatment (www.cdc.gov/tb).1 Other treatment options for TB often lack extensive data on efficacy and/or long-term safety, including bedaquiline (BDQ), delamid (DLM), linezolid (LZD), clofazimine (CFZ), and β-lactams paired with β-lactamase inhibitors, such as meropenem–amoxicillin–clavulanate.1,3 Bleak reports from the front lines of drug discovery indicate that the antibiotic pipeline is dwindling.4 To discover new antibiotics for the treatment of drug-resistant strains and to shorten therapy, we must consider adopting new drug discovery strategies. In this Review, we explore the proteostasis network and evaluate its suitability as a noncanonical target for the treatment of tuberculosis.
In Pursuit of Noncanonical Targets for Antibiotic Drug Discovery
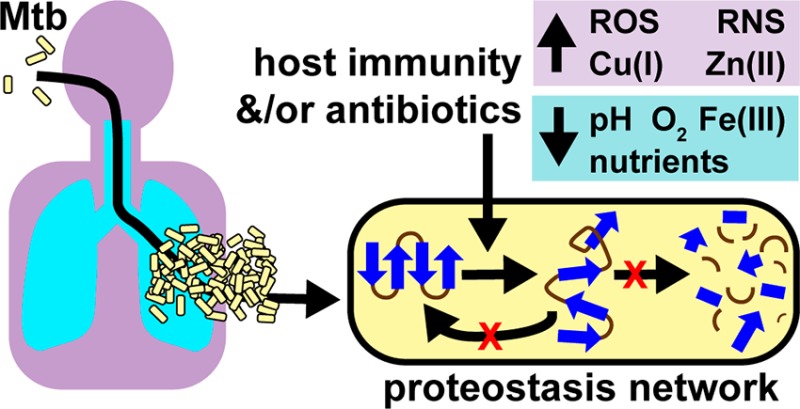
Most antibiotics were discovered by their ability to interfere with the synthesis of macromolecules such as DNA, RNA, lipids, proteins, and peptidoglycan and the vitamin folate, when bacteria are replicating in a synthetic growth medium in a laboratory.4−6 Antibiotics discovered using this strategy may fail to translate to a human host where conditions are often not conducive for Mtb replication.6 Mtb coevolved with the human host for tens to hundreds of thousands of years,7 and TB transpires from a complex interplay between Mtb, host immunity, host microenvironments, and host chemistries. Mtb can survive in a latent state for decades in a human.8,9 From the time Mtb is inhaled in an aerosolized droplet until the time the bacilli are eradicated by host immunity and/or with antibiotics, Mtb faces numerous cell types and host chemistries. Upon entering the respiratory tract, Mtb is ingested by alveolar macrophages and traffics to a phagosome. T-cells produce interferon-γ (IFNγ) that activates Mtb-infected macrophages, which is accompanied by a dramatic transcriptional change of both Mtb and the macrophage,10,11 and the Mtb-containing phagosome matures into a hostile environment that can slow or abrogate Mtb growth. Mtb can encounter suboptimal growth conditions in the caseum of a granuloma composed of necrotic macrophages, neutrophils, T cells, and B cells.12
Mtb has adapted to endure a diverse set of microenvironments in the host, including spatial compartments (lung, spleen, liver, brain, and adipose tissue), extracellular compartments (blood, granulomas, and caseum), and cell types (macrophages, dendritic cells, and neutrophils). Each microenvironment’s unique combination of carbon and nitrogen sources, micronutrients, pH, metal concentrations, gases, such as oxygen, carbon monoxide, carbon dioxide, and nitric oxide, and host immune effectors, encourages Mtb to proliferate, cease replicating and remain metabolically active, remain metabolically active and nonculturable, die by starvation for essential nutrients, or die after insult by host immune effectors.6
In many patients with TB, the foregoing immune mechanisms fail to eradicate Mtb without the assistance of antibiotics. TB is recalcitrant to antibiotic treatment, in part, due to populations of phenotypically tolerant Mtb (nongenetic drug resistance)6,13 generated by host immune chemistries and host microenvironments. Drug-tolerant Mtb persisters exist in animal models of tuberculosis14−16 and in humans.17 Phenotypic drug tolerance may result from arrest of replication for individual bacteria within a larger population (Class I persistence) or synchronous arrest of replication of a population of bacteria in response to an extrinsic stress (Class II persistence). While often associated with nonreplication, Class I drug tolerance may also result from phenomena unrelated to nonreplication,18,19 including stochastic expression of INH’s activating enzyme, catalase–peroxidase (KatG),14 or mistranslation of RIF’s target, RNA polymerase subunit RpoB.20,21 The mechanisms leading to Class II persistence are different from those leading to Class I persistence.19 For example, nonreplicating Class II persisters may have different uptake, retention, and metabolism of drugs.6,19,22 The Class II definition of persistence is further demarcated by the ability of bacteria, upon removal of the persistence-inducing stress, to recover as colonies on solid agar (Class IIa) or recover exclusively in liquid medium upon serial dilution (Class IIb).23,24 Bacteria exhibiting Class I phenotypic tolerance to one drug will often remain sensitive to other antibiotics,25 while bacteria exhibiting Class II phenotypic tolerance are broadly resistant to numerous antibiotics.19,26−29 Defining Class I and Class II persisters lays a foundation for designing rational strategies to treat human TB with antibiotics.13,19 Thus, in addition to targeting actively replicating Mtb, decreasing the time of TB treatment will require additional antibiotics that eradicate Class I, IIa, and IIb drug-tolerant Mtb persisters.6,13
TB drug discovery may benefit from embarking on a risky journey into target space enriched for noncanonical essential processes or pathways whose essentiality only becomes apparent during nonreplication and/or infection of a host.5,13 In fact, the first new TB drug in the last 40 years, BDQ, has a noncanonical target and kills both replicating and nonreplicating forms of Mtb by disrupting energy production via the C-subunit of ATP synthase.30,31 Other noncanonical antibiotic targets include redox homeostasis, cofactor/vitamin synthesis, metal homeostasis and uptake, signaling, and virulence factor production.5 Some examples of noncanonical target enzymes/pathways are thioredoxin reductase,32,33 dihydrolipoamide acyltransferase,34 depolarization of the bacterial membrane,35−37 and the proteolytic machinery.38−41 Of these noncanonical targets, the proteostasis network, consisting of chaperones that help fold and maintain client protein structure (proteostasis) and proteases that degrade client proteins (proteolysis), is particularly intriguing due to its potential to disrupt homeostatic levels of hundreds of client proteins.42
The Mycobacterial Proteostasis Network
The proteostasis network plays a critical role in Mtb survival against host immune stresses. In the absence of a functional proteostasis network to protect proteins, immune insults may irreversibly damage Mtb proteins and ultimately cause its death. For this critical role in protecting protein homeostasis, Mtb encodes over 100 proteins predicted or experimentally demonstrated to have a role in protein degradation (proteases, amidohydrolases, and the proteasome) or protein folding (chaperones and accessory proteins). The mycobacterial proteostasis network is summarized in Figure 1.
Figure 1.

Schematic of the mycobacterial proteostasis network. Mycobacteria possess numerous systems for maintaining protein structure and function via proteostasis (tan) or proteolysis (light blue). While proteolysis systems have become a rich source of antibiotic candidates, there are no described inhibitors of any members of mycobacterial chaperones or their accessory proteins. ClpP1P2 is associated with ClpC1 and ClpX, and Mtb20S is associated with Mpa and Paf. Lon is not found in pathogenic Mtb. The list of inhibitors targeting the Mtb proteolysis system is not comprehensive. HS proteins = heat shock proteins; poly-Pi = polyphosphate; LMW chaperones = low molecular weight chaperones; HspX = Acr1/α-crystallin 1; Hsp20 = Acr2/α-crystallin 2; TF = Trigger Factor; PTM = post-translational modification; ROS = reactive oxygen species.
Protein Maintenance by Chaperones and Their Cofactors
The molecular players in mycobacterial proteostasis are important in maintaining protein structure and function during normal cell growth and become essential in preventing damage and repairing proteins during periods of stress. DnaK (encoded by rv0350), the prokaryotic homologue of heat shock 70 kD proteins (Hsp70s), is an ATP-powered protein chaperone that is conserved from bacteria to humans. Several Hsp70-family members exist in eukaryotes, spanning different cellular compartments, with functions that include folding of nascent proteins, prevention and reactivation of protein aggregates, and intracellular trafficking.43−46 Along the same lines, Escherchia coli DnaK has been shown to be an important hub in the bacterial proteostasis network, as it is involved in protein folding, stabilizing non-native proteins for eventual folding by other chaperones, and mediating transfer of unfolded proteins to proteases for degradation.47,48 It has only recently been revealed that the specific roles of DnaK relative to other protein chaperones differ in mycobacteria compared to E. coli. In Mycobacterium smegmatis (Msm), a nonpathogenic, fast-growing mycobacterial saprophyte used as a model organism, DnaK is essential for cell growth and folding of nascent proteins following synthesis by the ribosome.49 Whole genome transposon-insertion mutant library analysis predicts that dnaK is also essential for viability of Mtb.50 However, DnaK is only conditionally essential in bacteria such as E. coli, in which deletion mutants can grow at low temperature (30 and 37 °C) but not at high temperature (42 °C).51 Additionally, the essential function of E. coli DnaK overlaps with that of the chaperone Trigger Factor (TF, encoded by rv246c/tig), as suggested by the observation that dnaK and tig form a synthetic lethal pair.52−54 Hence, the cellular roles of DnaK in mycobacteria cannot be extrapolated from studies in other organisms.
In mycobacteria, along with some Gram-negative and other high-GC-content Gram-positive bacteria, the repressor HspR (encoded by rv0353) regulates the expression of DnaK and a number of other heat shock-associated proteins.55,56 HspR is encoded on the same operon as dnaK and controls dnaK’s expression by binding an upstream HAIR (HspR-associated inverted repeat) sequence. DnaK modulates HspR binding to DNA through a proposed interaction with the unfolded C-terminal tail of HspR following denaturation by high temperatures.57,58 HspR also negatively regulates the expression of clpB, which encodes the protein disaggregase ClpB (encoded by rv0384c), and acr2 (encoded by rv0251c), which encodes a small heat shock protein called α-crystallin 2, or Hsp20. While hspR is under standard replicating conditions, infection of a mouse model with Mtb deficient in hspR revealed decreased viability compared to wild-type during the chronic phase of infection.59 One explanation for this observation is that the ΔhspR mutant’s lack of repression led to the overproduction of chaperones and increased protein secretion, which in turn stimulated the host’s immune response. Indeed, purified recombinant mycobacterial DnaK bound to peptide can act as an adjuvant in vitro.60−62
The dnaK operon also contains the protein cofactor-encoding genes dnaJ1(encoded by rv0352) and grpE (encoded by rv0351). In bacteria, DnaK’s protein folding activity is modulated by these cofactors, DnaJ (the homologue of eukaryotic Hsp40) and GrpE, a nucleotide exchange factor (NEF). DnaJ–proteins bind aggregated or non-native proteins prior to their delivery to DnaK in its ATP-bound state. Upon binding, the DnaJ–substrate complex activates ATP hydrolysis by DnaK, which is converted to an ADP-bound state. GrpE then mediates release of ADP by DnaK, which then binds ATP, allowing the reaction cycle to repeat until the substrate protein is folded to its native form or handed off to other chaperones in a semifolded state47 (Figure 2). The structural architecture of Hsp70s from all domains of life is highly conserved, with an N-terminal nucleotide binding domain (NBD) and a C-terminal substrate-binding domain (SBD) connected by a flexible linker.45 Cofactors predominantly interact with the N-terminal domain of Hsp70s.63 While eukaryotes possess many DnaJ–proteins, this is not the case in prokaryotes, where only GC-rich Gram-positive actinobacteria64 such as mycobacteria have more than one annotated dnaJ gene. Mtb has two copies of DnaJ, encoded by dnaJ1 and dnaJ2 (encoded by rv2373c). Studies on the overexpression of Mtb DnaJ1 and DnaJ2 suggest that each has a different cellular role65 and their encoding genes evolved separately. Genetic studies have recently shown that Msm dnaJ1 and dnaJ2 are individually dispensable but collectively required for cell growth.66grpE is essential in Msm49 and anticipated to be essential in Mtb, which has only a single annotated NEF in the genome.
Figure 2.

Function of mycobacterial protein chaperones includes both nascent protein folding and protein aggregate reactivation. Protein aggregates resulting from stress due to aging or antibiotic treatment are thought to be bound by small heat shock proteins (sHsps) prior to unfolding by the chaperone DnaK, its cofactor DnaJ, and the disaggregase ClpB in an ATP-dependent process that releases non-native proteins. These non-native proteins, which also result from ribosomal synthesis, are bound by DnaJ proteins, which then interact with DnaK in an ATP-bound state to stimulate ATP hydrolysis by DnaK. Once DnaK is converted to an ADP-bound state with attached protein substrate, the nucleotide-exchange factor GrpE binds to DnaK, causing release of ADP and binding of ATP to restart the catalytic cycle. The cofactor binding cycle repeats until the substrate is either released as a partially folded intermediate that is handled by other chaperones (such as Mtb GroEL/ES or HtpG) or released in a fully folded state. Mtb GroEL/ES may also act at earlier stages in the folding process, perhaps interacting with non-native proteins from the ribosome. Adapted with permission from Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Hartl, F. U. (2013) Annu. Rev. Biochem.82, 323–355.47 Copyright 2013 Annual Reviews.
The other conserved protein chaperone genes regulated by HspR, clpB and acr2, encode molecules that prevent and resolve protein aggregates in cooperation with the DnaK/cofactor system. Mtb acr2 regulation by HspR is modulated by interaction of the repressor with the response regulator PhoP.67 Bacterial ClpB, a homologue of eukaryotic Hsp104, is a hexameric disaggregase AAA+ ATPase (ATPase associated with various cellular activities) that hydrolyzes ATP while mediating protein unfolding through its central pore. DnaK and DnaJ–proteins deliver partially unraveled aggregated substrates to ClpB through direct interaction with the NBD of DnaK that competes for binding with GrpE.68 Small heat shock proteins (sHps) like mycobacterial α-crystallin 2 (Hsp20) and α-crystallin 1 (HspX; encoded by rv2031c) are typically annotated as HSPB (heat shock protein family B) in humans and Ibp (inclusion body protein) proteins in E. coli. Mtb lacks a third acr gene found in other mycobacteria such as Msm, M. marinum and M. leprae.69 sHps form oligomers that, in a nucleotide-independent manner, prevent aggregation by binding to proteins or assist other chaperones in protein disaggregation.70 Comparison of transcriptomes of Mtb under various growth and stress conditions shows that clpB expression is highly correlated with acr2 expression,71 suggesting that the encoded proteins cooperate in their cellular functions. While clpB is not essential under normal growth conditions, Mtb lacking clpB is defective for growth and virulence in a mouse model.72 Similarly, while neither acr1 nor acr2 is essential in Mtb under standard growth conditions, deletion of acr2 reduces the pathogenesis of Mtb in infected mice.69 Hence, these stress response genes are not required under standard laboratory conditions; however, under stresses that damage biomolecules, such as host infection, sublethal antibiotic treatment, or nonreplication, these genes encode proteins that constitute important pathways that repair aggregated or modified proteins.
DnaK, cofactor proteins, ClpB, and sHsps play important roles in reactivating damaged proteins. Microscopy studies in live mycobacteria have shown that ClpB and DnaK are recruited to a fluorescently tagged aggregating protein sequence (ELK16) and that tagged chaperones form foci that colocalize with these aggregates.49,72 Using aggregated luciferase as a model protein substrate, biochemical reconstitution of this proteostasis network consisting of Mtb DnaK, cofactors, ClpB, and Hsp20 showed that GrpE and at least one DnaJ–protein is required for luciferase refolding and that DnaJ1 and DnaJ2 exhibit nonredundant roles in protein folding.66 The in vitro requirements for these proteins in refolding reactions are consistent with grpE’s essentiality and dnaJ1 and dnaJ2’s synthetic lethality in Msm. Differences in the activity of mycobacterial DnaJ proteins were also evident in ATPase assays that showed DnaJ1 and DnaJ2 activate ATP hydrolysis by DnaK in the presence of GrpE to different degrees. Point mutations of a conserved three amino acid (histidine–proline–aspartate) motif73−75 in the N-terminal J-domain of DnaJ1 or DnaJ2 abrogate the ability of J-proteins to activate DnaK’s ATPase activity, inhibit aggregate reactivation when added to the reconstituted ClpB/DnaK/cofactor proteostasis system, and result in loss-of-function of DnaJs in mycobacterial cells. Since the DnaJ2 point mutant in this motif binds DnaK with affinity similar to the wild-type protein, these observations suggest that the native DnaK–DnaJ interaction can be disrupted. Targeting this interaction with selective small molecules could be lethal in mycobacteria,76 similar to what has been proposed for homologues in eukaryotes,77 in which the function of human Hsp70 proteins is essential in certain cell types, including some cancer cells.78
In bacteria, the GroE chaperone system often functions in parallel and downstream of the DnaK system. GroE consists of the chaperonin GroEL (Hsp60) and cochaperonin GroES (Hsp10) and is the only temperature-independent essential chaperone system in E. coli(79,80) (Table 1). The GroE system binds hydrophobic patches on polypeptides that are exposed during translation and helps stabilize folding intermediates by encasing them in a central channel.47,81 The GroE system also helps fold newly synthesized proteins that TF and DnaK cannot fold. GroE consists of two stacked heptameric rings, forming a barrel-like structure that is capped by two heptameric GroES subunits on either end.82 The canonical GroE system typically has strong ATPase activity and forms higher oligomeric structures.83 GroE from Mtb, however, is a dimer with weak ATPase activity that can partially refold proteins in vitro.83 Mycobacteria and other actinomycetes are unique in that they encode two copies of the chaperonin GroEL: GroEL1 (encoded by rv3417c) and GroEL2 (encoded by rv0440). groEL1 is in an operon with groES (encoded by rv3418c), and groEL2 is encoded elsewhere in the genome. Only GroEL2 is essential for in vitro survival of Mtb,84 suggesting its clients are essential proteins. Interestingly, the GroEL chaperonins are potent immunomodulators. GroEL2 is an immunodominant mycobacterial antigen,85 and both GroEL1 and GroEL2 play a role in cytokine induction and stimulating the host’s immune response to Mtb infection.84,86,87
Table 1. Select Chaperone Network Proteins and Their Essentiality for Optimal Growth in Vitro.
| protein | E. coli | M. smegmatis | M. tuberculosis |
|---|---|---|---|
| Trigger Factor | nonessential54 | nonessential49 | nonessentiala |
| DnaK | nonessential51 | essential49 | essentiala |
| GrpE | nonessential233 | essential49 | essentiala |
| DnaJ | nonessential75 | DnaJ1 and DnaJ2 collectively essential66 | DnaJ1 and DnaJ2 essentiala |
| ClpB | nonessential234 | nonessential72 | nonessential72 |
The ClpB/DnaK and GroE system in Mtb is important for the proper folding of proteins and polypeptides under standard growth conditions as well as under conditions of stress. The expression of dnaK, clpB, dnaJ1, grpE, acr2, groEL1, and groEL2 is upregulated at elevated temperature.55 Upon phagocytosis in macrophages, groEL1, groEL2, and acr are upregulated in mycobacteria.88
Other annotated chaperones present in the genome in Mtb have not been well-studied. For example, HtpG,89 the homologue of the eukaryotic protein chaperone Hsp90, is encoded by rv2299c in Mtb and is absent in Msm. While Hsp90 interactions in a larger protein network have been well-described,90 little is known of the interaction between HtpG and other proteostasis proteins in Mtb; hence, much remains to be determined in the network of proteins responsible for forming and maintaining protein structures in mycobacteria.
Protein Degradation by Proteases and the Proteasome
In addition to proteostasis systems that maintain proteins in their native conformation, the proteostasis network encompasses proteases that degrade damaged, aggregated, and overexpressed protein or proteins no longer needed by the cell.
Actinomycetes, including mycobacteria, encode four major cytosolic compartmentalized ATP-dependent proteases: ClpP, HslUV, Lon, and the proteasome. Neither HslUV nor Lon are present in Mtb. We will focus on the two cytosolic compartmentalized, two-component ATP-dependent proteases present in the pathogen Mtb: ClpP and the proteasome.
Under standard growth conditions, many Mtb proteins are degraded by the Clp (caseinolytic) protease.91 The Clp protease is comprised of a barrel shaped protease (ClpP) and an AAA+ ATPase (ClpC and ClpX in Mtb; see below).92 Clp proteases are present in eubacteria as well as in chloroplasts and mitochondria.93 In addition to misfolded or abnormal protein substrates, the Clp protease recognizes substrates that have been cotranslationally modified at the C-terminus with the 11 amino acid SsrA tag.92,94 Under standard growth conditions, in E. coli, Bacillus subtilis, and Staphylococcus aureus, the homotetradecamer Clp protease is nonessential; however, ClpP has a critical role for virulence (S. aureus) or under stresses.92,95 In Mtb, the Clp protease consists of a heterotetradecamer comprised of two subunits, ClpP1 and ClpP2, that possess chymotrypsin and caspase-like activities.96 The ClpP1P2 protease of Mtb is essential both for in vitro replication91 and in a mouse model of Mtb infection, where genetic knockdown of ClpP1P2 leads to decline in CFU in mouse lungs during the acute phase of infection and leads to the rapid elimination in mice by day 30.91,97,98 Under nonstress conditions, over 100 candidate substrates were identified in a regulated knockdown of ClpP1P2 in Mtb.99 This indicates that targeting ClpP1P2 with small molecules could cause dysregulation of numerous proteins under standard growth conditions and additionally lead to secondary effects by dysregulation of transcription factors, such as WhiB3, and their target genes.99
The Clp protease complex also has AAA+ ATPases, similar to ClpB, that interact with protein targets and use the energy from ATP hydrolysis to unfold protein substrates and feed them into the protease’s central pore. In Mtb, ClpC1 (encoded by rv3596c) interacts with ClpP1P2 to degrade a model substrate and has ATPase and chaperone activities (prevention and reactivation of luciferase aggregates).100,101 ClpX (encoded by rv2457c) is another AAA+ ATPase that interacts with and serves as a chaperone for ClpP1P2 in Mtb.100 While both ClpC1 and ClpX are predicted to be essential for Mtb survival in vitro, to date, no reports have confirmed this experimentally.102,103
The proteasome (PrcBA, herein Mtb20S), encoded by genes rv2110c and rv2109c (β and α subunits, respectively), was first identified in bacteria based on its sequence similarity to the eukaryotic proteasome.104 The eukaryotic proteasome degrades the majority of damaged, unfolded, or unwanted proteins in the cell and plays an essential role in cell survival.105 The eukaryotic core particle (CP) consists of heteroheptameric 14 α and 14 β subunits with peptidase activity afforded by the N-terminal threonine nucleophile of the β-subunit. In addition to the CP, the eukaryotic proteasome consists of a regulatory particle that flanks one or both ends of the CP and recognizes poly ubiquitin (Ub)-tagged substrates, removes Ub from tagged substrates, unfolds the substrates, and then delivers the substrates to the CP for degradation.106 The prokaryotic proteasome is present in archaea but limited to the bacterial orders Actinomycetales and Nitrosporales. The bacterial CP consists of homoheptameric 14 α and 14 β subunits with peptidase activity similar to that of eukaryotes.107 Unlike the eukaryotic proteasome, where the majority of the accessory factors have been characterized, the bacterial proteasome accessory factors are still being identified. Mpa (mycobacterial proteasome ATPase, encoded by rv2115c), an AAA+ ATPase, recognizes pupylated substrates, unfolds them, and delivers at least some for degradation by the CP.108,109 Pup (prokaryotic ubitiquitin-like protein, encoded by rv2111c) is a protein tag with function analogous to that of poly-Ub but without sequence or structural homology. Pup is intrinsically disordered110,111 and PafA (proteasome accessory factor A, encoded by rv2097c) attaches Pup to the ε-amino group of lysine residues on proteins112,113 after Pup activation by Dop (deamidase/depupylase of Pup, encoded by rv2112c).114,115 Dop also serves as a depupylase and removes Pup from protein substrates prior to entry into the proteasome. PafE (proteasome accessory factor E, encoded by rv3780), an ATP- and Pup-independent unfolding machine, associates with and activates proteolysis through the proteasome in Mtb.116,117 Similar to Mpa,118 PafE contains a C-terminal hydrophobic-tyrosine-X-motif (HbYX motif) that docks into the proteasome CP. In vitro, PafE enhances the CP-dependent degradation of unstructured proteins such as casein and is thought to deliver partially unfolded proteins (likely formed under conditions of stress) into the CP for degradation in the cell.116,117 Interestingly, PafE-deficient Mtb is unable to degrade HspR and is heat-sensitive, suggesting it is a regulator of the heat shock response in Mtb.116 Therefore, PafE may be both responsible for the degradation of misfolded proteins formed during heat stress and responsible for activating the chaperone response to heat stress. Under standard culture conditions, the Mtb proteasome degrades only a handful of proteins and is not essential.113 However, the proteasome and its accessory factors play a critical role in surviving host-relevant stress conditions such as nutrient starvation, prolonged stationary phase, reactive nitrogen species and heat.116,119−122 In the acute phase of a mouse model of infection, Mtb lacking the proteasome CP achieves bacterial burden equivalent to Mtb with a wild-type proteasome, but during the chronic phase of infection, the proteasome-deficient mutant was unable to sustain infection.120,121 It was recently proposed that the mycobacterial proteasome is important for amino acid recycling under nutrient starvation, in particular, nitrogen starvation in Msm.123,124
Role of the Proteostasis Network in Protecting Mtb from Host Immunity
In addition to general protein quality control, the Mtb proteostasis network is responsible for degrading damaged or prematurely terminated proteins, as well as supplying amino acids under conditions of stress. Mtb is reliant on the proteostasis network to survive host immunity and host chemistries.
To control Mtb growth, macrophages produce ROS and RNS via the phagocyte oxidase (phox)125 and the inducible isoform of nitric oxide synthase (iNOS),126 respectively. As demonstrated in Figure 3, numerous host stresses associated with phenotypic tolerance and treatment with antibiotics converge on the formation of ROS and RNS.72,127−130 RNS have been implicated in the antimycobacterial activity of the nitroimidazole drug, PA-824.131 RNS (including •NO, ONOO–, ONOOH, •NO2, N2O3, N2O5) and ROS (including •O2–, H2O2, ROOH, •OH,1O2, O3, HOCl, HOBr, HOI) may damage proteins by oxidation of cysteine and methionine, disruption of iron in heme and Fe–S clusters, oxidation of arginine, lysine, proline, and threonine, nitration of tyrosine, and S-nitrosylation of cysteine residues.6,132,133 Some protein damage inflicted by RNS/ROS, such as oxidation of methionine or cysteine, and S-nitrosylation of cysteine, is enzymatically reversible.134,135 Other damage to proteins, such as carbonylation or tyrosine nitration, is irreversible and may provoke protein misfolding, aggregation, degradation, or irreversible oxidation of proteins to protease resistant or protease inhibiting forms.46,136−138 Whole genome expression analysis of Mtb following addition of nitric oxide (NO) or hydrogen peroxide (H2O2) shows different expression patterns of proteostasis genes. In response to ROS, the dnaK operon, clpB, and acr2 are significantly upregulated; while in response to RNS, only acr2 and clpB show increased expression among the proteostasis and proteolysis genes described here.139 In particular, acr expression is induced in activated versus naïve macrophages following infection with Mtb over the course of 2 days, highlighting its role in responding to host stress environments.10
Figure 3.

Host microenvironments and immune chemistries, and antibiotic treatment, converge on the mycobacterial proteostasis network. In this example, antibiotics used to treat TB and/or host microenvironments and immune chemistries directly or indirectly generate ROS or RNS, which may damage proteins. Proteins damaged by ROS and/or RNS are often enzymatically inactivate and must be repaired or degraded. Killing of Mtb by the antibiotic PA-824 (pretomanid) is mediated in part via generation of RNS.
After stimulation by with IFNγ, the pH of the macrophage’s phagosomal compartment in which Mtb resides decreases to ∼4.5.140rv2224c, which encodes a protease involved in GroES processing, is important for survival in acidic conditions.141−144 The MarP protease plays a role in helping Mtb survive the acidic environment and maintain its intrabacterial pH.142,145 A member of a family of proteases involved in protecting proteins from acid stress, HtrA, was identified as the target of a benzoxazinone screening hit that arose from an HTS of over 300 000 small molecules for MarP protease inhibitors.146
Mtb faces varying degrees of hypoxia in a host.147 The nitroimidazole drug metronidazole kills Mtb under severe hypoxia (<0.1 ppm of O2) in vitro(148,149) and trends toward displaying higher activity in the animal models of TB with hypoxic lesions.6 Exposure to hypoxic conditions for 4 to 7 days increases expression of dnaK, clpB, and acr2 compared to nonstressed, log-phase cells.150
Mycobacterial metal homeostasis, in particular surviving copper or zinc intoxication and iron starvation, is critical during host infection.151−156 Iron starvation-induced growth arrest in Mtb leads to nonreplication and phenotypic tolerance to antibiotics. The chaperone ClpB plays a critical role in surviving stresses associated with nonreplication imposed by iron starvation.157
Mtb adapts to various degrees of nutrient starvation or changes to carbon and nitrogen availability or sources in the host. In one example of prolonged stationary phase (>100 days), DnaK protein was induced in Msm.158 In Mtb, the proteins GrpE, DnaK, and HspX (α-crystallin) were induced by 6-week starvation in phosphate-buffered saline (PBS).159 Nonreplication imposed by nutrient starvation or depletion of carbon and nitrogen sources can result in oxidative carbonylation of proteins.72 Thus, these data suggest that antibiotics targeting Mtb’s proteostasis network will sensitize it to proteotoxic stress from host chemistries and synergize with host immunity.
Targeting the Proteostasis Network: Chaperones and Their Cofactors
Chaperones have emerged as exciting targets for cancer chemotherapy, and numerous inhibitors of eukaryotic protein chaperones have entered clinical trials.160−162 Within their respective microenvironments, there are parallels to the stresses encountered by cancer cells in solid human tumors and Mtb in activated macrophages and/or granulomas. For example, solid tumors are nutrient deprived, hypoxic, and mildly acidic, and the degree of each stress is dependent on a cancerous cell’s location within the tumor and relative to the tumor’s vasculature.163−165 Cancer cells produce more ROS than wild-type cells.166,167 The microenvironmental conditions found in solid tumors lead to variable growth rates, and some or the majority of cancer cells may become tolerant to anticancer agents. Many of the predominant stresses, nutrient deprivation, hypoxia, acidity, and ROS, are associated with both drug resistance of cancer cells and phenotypic drug tolerance in Mtb.
Cells in solid tumors depend on the proteostasis network for survival. Chaperones may be highly induced on the transcriptional and protein level in cancer cells, and chaperones such as Hsp90 may exist in activation states and complexes that make them 100-fold more sensitive than wild-type cells to small-molecule inhibition.165,168 This parallels the upregulation of components of the proteostasis network in Mtb upon infection of macrophages or exposure to individual components of host immunity. Eukaryotic chaperones help malignant cells survive host stresses in solid tumors and help maintain pro-oncogenic proteins in a functional state that serves to keep cancer cells alive. Some examples of compounds with efficacy against Hsp70 and Hsp90 include geldanamycin (1), 17-allylamino-17-demethoxygeldanamycin (2), 15-desoxyspergualin (3), 2-phenylethynesulfonamide (4), gentamicin (5), and novobiocin (6) (Figure 4).
Figure 4.

Examples of compounds targeting human Hsp70 or Hsp90.
The strategy of treating cancer with chaperone inhibitors might translate to new strategies for bacterial drug discovery169 and, in particular, for TB. For example, the mycobacterial chaperone ClpC1 has become an exciting drug target and is discussed in further detail below. In other bacteria, the proteostasis network, including the GroE system, HtpG, and DnaK, has been disrupted by small molecules and peptides, many of which exert bactericidal activity. In light of a lack of understanding of which pharmocophores engage chaperones, we assembled structures of compounds targeting bacterial chaperones and their cofactors as a reference to serve as a starting point for mycobacterial chaperone inhibitors (structures and amino acid sequences are shown in Figure 5a,b). Compound EC3016 (7) is an inhibitor of both ATPase and chaperone activity of a modified E. coli GroEL/ES variant.170 Nontoxic inhibitors of GroEL/ES (compounds 8 (8) and 18 (9)) have bactericidal activity against E. coli.171 Large cyclic, lipopeptide antibiotics such as polymyxin B (10), polymyxin B nonapeptide (11), colistin (12), and gramicidin S (13) inhibit chaperone activity of HtpG in the model organism Synechococcus elongatus PCC7942 by binding its N-terminus and inducing oligomerization and/or aggregation.172 Compounds BI-88B3 (14) and BI-88D7 (15) bind the substrate-binding β-domain of the E. coli DnaK, and BI-88B3 (14) had weak antimicrobial activity against E. coli and Yersinia pseudotuberculosis.173 Nα-[Tetradecanoyl-(4-aminomethylbenzoyl)]-l-isoleucine (16) inhibits the secondary peptide cis–trans isomerase activity of the E. coli DnaK174 but has poor antimicrobial activity.174,175 The proline-rich antibacterial peptides of insect origin (Figure 5b), including drosocin (17), pyrrhocoricin (18), apidaecin 1a (19), bac-7 (20), and CP-105 (21), block the ATPase and chaperone activities of the E. coli DnaK.169,176−180 The proline-rich peptide CHP-105 (21) synergizes with levofloxacin to kill levofloxacin-sensitive and -resistant Klebsiella pneumoniae and E. coli.181 Pyrrhocoricin (18) and a stabilized pyrrhocoricin (Chex-pyrrhocoricin-Dap(Ac)) protected mice from E. coli septicemia.182 While the proline-rich antimicrobial peptides have weak to no activity against Gram positive pathogens,183 modifying peptide length and amino acid composition may improve anti-Mtb activity.184 Some proline-rich peptides, including pyrrhocoricin (18) and Bac-7 (20), were found to kill bacteria by blocking peptide exit from the 70S ribosome,185,186 indicating that at least some proline-rich peptides exert antimicrobial activity by inhibiting translation.
Figure 5.

Examples of compounds targeting bacterial chaperones. Representative structures are shown of (a) small molecules or (b) amino acid sequences of proline-rich peptides that inhibit bacterial GroEL/ES, HtpG, and DnaK. Some proline-rich peptides like Bac-7 and pyrrhocoricin may mediate antibacterial activity by inhibiting translation. To date, there are no examples of compounds or peptides targeting mycobacterial protein folding machinery.
While targeting the chaperone network is an active area of research for cancer therapy and has been explored for bacterial drug discovery, targeting chaperones for TB drug discovery is in its infancy. In fact, to our knowledge, there only has been one report of drug discovery efforts targeting protein-folding chaperones in Mtb.187 We propose further exploration of mycobacterial chaperones and their cofactors for TB drug discovery.
Targeting the Proteostasis Network: ClpC1, ClpP1P2, and Mtb20S
The majority of drug discovery work in the proteostasis field has focused on targeting barrel-shaped proteases and their Hsp100-family chaperone partners. Excellent reviews have extensively explored drug discovery for the bacterial proteolysis machinery.188−192 We have summarized information regarding inhibitors and activators of the Mtb proteolysis systems in Table 2. Targeting bacterial proteolysis provides a unique situation in which either inhibiting or activating the target pathway results in bacterial death.190,191 Inhibition or activation of proteases or their activators may alter the levels of client proteins (damaged, nascent, or “normal”), resulting in dysregulation, cell stress, and death.
Table 2. Summary of Select Inhibitors or Activators of ClpC1, ClpP1P2, or Mtb20Sa.
| compound class | compound | synthetic (S) or natural product (NP) | target | method of identification | activity on R Mtb | activity on NR Mtb | activity in mouse model of TB | FOR | references |
|---|---|---|---|---|---|---|---|---|---|
| peptide, cyclic | cyclomarin A | NP | ClpC1 | WCS against M. bovis BCG | yes | yes | NT | <10–9 | (40, 235) |
| peptide, cyclic | ecumicin | NP | ClpC1 | WCS vs actinomycetes extracts against Mtb | yes | yes | yes | 1 × 10–8 | (38) |
| peptide, cyclic | rufomycin | NP | ClpC1 | WCS vs actinomycetes extracts against Mtb | yes | NT | NT | NT | (197) |
| peptide, lasso | lassomycin | NP | ClpC1 | WCS of NP extracts against Mtb | yes | yes | NT | NT | (39) |
| acyldepsipeptide | ADEP-2 | NP | ClpP1P2 | N/A: previously shown to be active against S. aureus(42) | yes | NT | NT | 1 × 10–6(for S. aureus) | (42, 98, 196) |
| peptidyl boronate | examples: N-(2-(3,5-difluorophenyl)acetyl-Trp-boroMet); N-(Picolinoyl)-Ala-Lys-boroMet | S | ClpP1P2 | blocked tripeptide-AMC library to determine substrate preferences followed by rational design of inhibitors | yes | NT | NT | NT | (207) |
| peptidyl boronate | bortezomib (Velcade), MLN-273 | S | Mtb20S and ClpP1P2 (bortezomib); Mtb20S (MLN-273) | bortezomib: target-based whole-cell screen against M. smegmatis strain carrying SsrA-tagged GFP; MLN273: N/A | yes | yes | NT | NT | (107, 119, 203) |
| β-lactones | β-lactone 7 | S | ClpP1P2 | whole-cell screen of racemic mixture (14 scaffolds) of β-lactones against M. smegmatis | yes | NT | NT | NT | (201) |
| N, C-capped dipeptide | DPLG-2 | S | Mtb20S | purified recombinant Mtb20S-OG, hydrolysis of AMC-linked substrate | no | yes | NT | NT | (211) |
| oxathiazol-2-one | GL-5 | S | Mtb20S | purified recombinant Mtb20S-OG, hydrolysis of AMC-linked substrate | no | yes | NT | NT | (41) |
| styryl-oxathiazol-2-one | compound 17 | S | Mtb20S | purified recombinant Mtb20S-OG, hydrolysis of AMC-linked substrate | no | yes | NT | NT | (210) |
| macrolactam | syringolin analogues-compound 13 | S based on NP | Mtb20S | purified recombinant Mtb20S-OG, hydrolysis of AMC-linked substrate | NT | yes | NT | NT | (213) |
| epoxyketone peptide | epoxomicin | NP | Mtb20S | NA | NT | yes | NT | NT | (119) |
| lipopeptide aldehyde | fellutamide B | NP | Mtb20S | purified recombinant Mtb20S-OG, hydrolysis of AMC-linked substrate | NT | NT | NT | NT | (209, 236) |
| NA | MTBA, MTBB | S | Pup/Mpa interaction | in-cell STINT-NMR | NT | yes | NT | NT | (212) |
Abbreviations: NA = not applicable; NT = not tested or not found in the literature; S = synthetic; NP = natural product; FOR = frequency of resistance; Mtb20S = 20S proteasome (PrcBA); OG = open gate; AMC = aminomethyl coumarin; GFP = green fluorescent protein; R = replicating; NR = nonreplicating.
The discovery that ADEP-4 (22) (Figure 6a) activates proteolysis and kills both replicating and phenotypically drug tolerant S. aureus and pairs with RIF to eradicate a deep-seated thigh infection of S. aureus in mice indicates that proteolysis may be an exciting target pathway for latent diseases such as Mtb.42,193 In bacteria such as B. subtilis and E. coli, ADEPs bind ClpP and open the ClpP tetradecamer pore to permit unregulated degradation of nascent and flexible proteins.194 ADEP-bound ClpP subunits also fail to associate with their Hsp100 chaperones and subsequently lose tight regulation of ClpP substrates.194 Dysregulated degradation of the essential cell-division protein FtsZ plays a major role in ADEP-dependent bactericidal activity against B. subtilis and S. aureus.195 ADEPs have relatively weak activity against mycobacteria, with ADEP2 (23) having an MIC90 of 16–25 μg/mL against Mtb and M. bovis BCG.98,196 In the absence of ClpC1, ADEPs open the Mtb ClpP1P2 pore and increase protease activity, and in the presence of ClpC1, ADEPs interfere with the ClpC1–ClpP1P2 interaction.196 Dysregulated FtsZ proteolysis is not the cause of ADEP-dependent death in Mtb196 and the degradation of specific client proteins has not been elucidated. The activity of ADEPs against mycobacteria can be improved with efflux pump inhibitors or ClpP1P2 depletion,98,196 suggesting cell entry may be a limiting factor for activity. Thus, ADEPs appear to have a poor prognosis for further development for TB.
Figure 6.

Examples of compounds targeting mycobacterial protein degradation. Molecules targeting protein degradation in Mtb inhibit or activate (a) ClpC1 or (b) ClpP1P2.
There has been progress in the discovery of molecules that target ClpC1 and phenocopy treatment of other bacterial pathogens with ADEPs. In fact, experimental data generated with a handful of cyclic peptides support ClpC1’s candidacy as a high-priority target in Mtb (Figure 6a). The antimycobacterial peptides cyclomarin A (24),40 ecumicin (25),38 rufomycin (26),197 and lassomycin (27)39 target Mtb ClpC1 via different mechanisms. Cyclomarin A and rufomycin activate ClpC1-mediated, ATP-dependent proteolysis by ClpP1P2.40 Ecumicin and lassomycin uncouple ClpC1’s ATP hydrolysis,38,39 which ultimately prevents ClpC1’s ability to feed damaged and client proteins into ClpP1P2. Unregulated ATP hydrolysis can also deplete critical cellular ATP. In essence, ecumicin and lassomycin arrest ClpP1P2 protein degradation and function as ClpP1P2 inhibitors. The ClpC1-targeting peptides boast nanomolar to micromolar potency against replicating and nonreplicating Mtb, with limited toxicity to eukaryotic cells. Ecumicin and lassomycin were active against MDR and XDR Mtb,38,39 and ecumicin killed Mtb in both macrophages and mice.38 When tested, the microbial spectrum of activity was highly specific to Mtb. For example, lassomycin was inactive against Gram-positive bacteria such as S. aureus, Bacillus anthracis, and Klebsiella pneumoniae.39 The ClpC1-targeting peptides are natural products isolated from actinomycetes, including cyclomarin A (Streptomyces spp. CNB-982), ecumicin (Nonomuraea spp. MJM5123), lassomycin (Lentzea kentuckyensis spp. IO0009804), and rufomycin (Streptomyces atratus ATCC 14046). The biosynthesis of these peptides is complex; for example, the synthesis of cyclomarin A is mediated by over 23 genes (over 47 kb of DNA).40 The high prevalence of unnatural amino acids,192,198 including d-amino acids, methoxylated amino acids, N-methylleucine, N-methylhydroxyleucine, β-methoxyphenylalanine, 2-amino-3,5-dimethylhex-4-enoic acid, N-(1,1-dimethyl-2,3-epoxypropyl)-β-hydroxytryptophan, N-dimethylallyltryptophan, trans-2-crotylglycine, and 3-nitrotyrosine, indicated that it is unlikely that similar compounds or peptides exist in synthetic compound collections. A current and future challenge is in isolating and/or modifying complex natural products. Structure activity relationship studies may help identify critical pharmacophores that retain ClpC1 binding199 and permit identification of similar structures in screening decks. Recent developments in elucidating pathways encoding nonribosomally synthesized peptides200 may offer new avenues to discover and synthesize natural products targeting the ClpC1/ClpP1P2 in mycobacteria. To our knowledge, there are no described inhibitors or activators for the other ClpP1P2 accessory proteins, ClpC2 and ClpX.100
Compounds that directly inhibit or activate the ClpP1P2 tetradecamer include ADEPs, β-lactones, bortezomib, and peptidyl boronates (Figure 6b). Some β-lactones (28) inhibit ClpP1P2 and have antimycobacterial activity.201 Caution must be taken with β-lactones as ClpP1P2 inhibitors; as recently, a different member of the β-lactone class exerted antimycobacterial activity by inhibiting mycolic acid biosynthesis.202 While these β-lactones also associated with ClpP1P2 in pull-down experiments, further characterization demonstrated that they had poor inhibitory activity against recombinant ClpP1P2.202 A general concern of ClpP1P2-targeting β-lactones is that they lack structural features to generate target selectivity and risk indiscriminate reactivity with cellular nucleophiles. Another ClpP1P2 inhibitor, bortezomib (29), is a peptidyl boronate antineoplastic drug originally found to target the human 20S proteasome and was identified as an inhibitor of both ClpP1P2203 and Mtb20S.204 SAR campaigns have identified bortezomib analogues, including compound 58 (30), with improved selectivity toward ClpP1P2 and Mtb20S over the human proteasome.205 Other bortezomib analogues, in which the boronate was replaced with a chloromethyl ketone warhead, had negligible activity against the human proteasome.206 Numerous di- (example, N-2-(3,5-difluorophenyl)acetyl-Trp-boro-Met, 31) and tri- (example, n-(picolinoyl)-Ala-Lys-boro-Met, 32) peptidyl boronates with potent nM affinities for ClpP1P2 were inhibitory to Mtb at low μM concentrations.207 The stark difference in IC50 against the recombinant proteins and the MIC50 against Mtb indicates poor uptake into the bacillus or metabolism of the compound to inactive species. These peptidyl boronates inhibited human mitochondrial ClpP to a lesser extent than the Mtb ClpP1P2, and a select set were found to be nontoxic to human MM.1S myeloma cells.207 In summary, the majority of compounds directly binding ClpP1P2 can kill replicating Mtb in vitro, and to our knowledge, there is no evidence that they possess activity in animal models of TB. As stated above, one of the most promising cylic peptides targeting ClpC1, ecumicin, phenocopies ClpP1P2-inhibiting molecules and kills Mtb in murine models of TB.
The discovery of a eukaryotic-like proteasome in mycobacteria with an important role in defending Mtb against stresses of host immunity, such as RNI,119 spurred intense efforts to find species-selective Mtb20S inhibitors (Figure 7). A major concern pursuing the Mtb20S was the existence of the essential human proteasome. In fact, the first few characterized inhibitors of the Mtb proteasome were originally developed to target the human 26S proteasome, including bortezomib (29; described above), MLN-273 (33), and epoxomicin (34).119 While the cocrystal of MLN-273 and Mtb20S was solved, MLN-273 was not pursued due to lack of specificity for the mycobacterial proteasome.208 Similarly, fellutamide (35), the most potent Mtb proteasome inhibitor to date with a Ki under ∼7 nM, was not pursued as a lead candidate due to its potent inhibition of the human proteasome.209 The discovery of the oxathiazol-2-ones (36)41 importantly demonstrated the possibility of developing highly selective, irreversible Mtb proteasome inhibitors. Analogues of oxathiazol-2-ones, including styryl-oxathiazol-2-ones (37), were developed with improved antimycobacterial activity.210 The N,C-capped dipeptides, including DPLG-2 (38), are reversible inhibitors that gained selectivity for the Mtb proteasome by exploiting key differences revealed in the human and mycobacterial proteasome active sites.211 Recently, progress to identify novel pharmacophores for Mtb-selective proteasome inhibitors was inspired by syringolin, a natural product made by the plant pathogen Pseudomonas syringae.213 Compound 14 (39) was over 70-fold selective for the Mtb proteasome versus the human proteasome.213 Finally, the compounds MTBA (40) and MTBB (41) target the Pup/Mpa interface and prevent degradation of FabD, a physiological Mtb20S substrate. MTBA and MTBB kill M. bovis BCG under nitrosative stress conditions and were noncytotoxic.212 Unlike compounds targeting ClpC1 and/or ClpP1P2, in vitro the Mtb20S inhibitors require coadministration of additional stresses, usually RNS generated under mildly acidified conditions, to exert bactericidal activity.119,211 This corroborates genetic evidence that PrcBA (Mtb20S) is nonessential under standard replicating conditions and conditionally essential under nitrosative stress.119−121 To date, there is no evidence that compounds targeting Mtb20S have efficacy in TB animal models.
Figure 7.

Examples of compounds targeting Mtb’s eukaryotic-like proteasome, Mtb20S, and proteins serving the proteasome pathway, such as Pup and Mpa.
In addition to potent activity against replicating bacilli, many of the compounds targeting ClpC1, ClpP1P2, and Mtb20S also have activity against Mtb rendered nonreplicating by stresses that mimic components of host immunity described above. For example, Mtb rendered nonreplicating by acid/RNS stress is killed by proteasome inhibitors MLN-273, epoxomicin, GL-5, DPLG-2, and the syringolin-like compound 14 (39).41,119,211,213 Under nutrient starvation or stationary phase, Mtb is susceptible to ClpP1P2 inhibitors such as lassomycin and proteasome inhibitors such as compound 17 (37),39,210 presumably due to an increased demand on recycling proteins for amino acids. Under nonreplicating hypoxic conditions, Mtb is killed by ClpC1-targeting cyclic peptides, including cyclomarin A, ecumicin, and lassomycin.38−40
Although the frequencies of resistance (FOR) to ADEPs in nonmycobacterial organisms are borderline unacceptable for use as an antibiotic (∼1 × 10–6 in S. aureus(42)), molecules targeting the Mtb ClpC1 have attractive FORs on par with clinically used antibiotics such as RIF (cyclomarin A, <1 × 10–9;40 lassomycin, 3 × 10–7 39). At least one compound, bortezomib, targets both ClpP1P2 and the proteasome, an interesting feature that may ultimately lead to a lower frequency of resistance than targeting either the protease or proteasome individually.203,205 Standard methologies do not permit determination of the FORs for compounds that only kill when Mtb has been rendered nonreplicating with host-mimicking stresses such as RNI, nutrient starvation, or hypoxia.
Prospective: Could Disrupting the Proteostasis Network Serve as Adjunctive Therapy?
Targeting the proteostasis network may serve as an adjunctive therapy to help our current arsenal of TB drugs to kill Mtb that has survived stresses imposed by host immunity and/or to kill Mtb drug tolerant persisters. During the course of its infectious cycle, Mtb faces stresses that vary in nature and magnitude, and at all stages of infection, the proteostasis network contributes directly or indirectly to Mtb’s survival.
The proteostasis network plays an important role in allowing the majority of a population to adapt to stress conditions in a manner that permits their survival. However, how do cells handle host stresses or antibiotic stresses that arrive rapidly, with high intensity, and preclude bacterial adaptation? In these cases, pre-existing cell-to-cell heterogeneity permits survival of a minority of cells whose physiology differs from those of average cells in a normally distributed population (Figure 8). The proteostasis network bolsters diversity of phenotypic traits of individuals within a population. For example, it is possible that stochastic variations of antitoxin degradation of toxin–antitoxin systems by the Lon protease permit a minority of cells in a population, with higher toxin levels, to survive exposure to a stress such as antibiotics. In E. coli, mutants deficient in the Lon protease produce less drug tolerant persisters than a wild-type strain.214 Mtb does not have a Lon protease. Since some antitoxins are pupylated in Mtb,113 some of Mtb’s 80 toxin–antitoxins (TAs) are likely regulated by proteasomal degradation.214,215 Therefore, inhibition of the proteasome may kill some persister cells. Likewise, Mtb persisters surviving INH exposure had increased expression of dnaK, grpE, clpB, clpX, and groE.216 Cells expressing higher levels of dnaK represented the majority of the persister population and were more resistant to INH.
Figure 8.

Targeting the proteostasis network may sensitize Mtb to host immunity and antibiotics. In situations when host stresses or antibiotics reach Mtb slowly or at low intensity (left side), Mtb has ample time to adapt by inducing defense mechanisms. However, in situations in which stresses or antibiotics attain Mtb rapidly and/or with high intensity (right side), Mtb may not have time to adapt. The only surviving bacilli may be rare cells that are different from the general cell population that display phenotypic tolerance to stresses or antibiotics. The proteostasis network, via chaperones and the protein degradation machinery, contributes to adaptation (left side) or cell-to-cell heterogeneity (right side). Cell color key: alive, green; dead, light gray; rare outlier cells that are different from an average cell, red.
Prions are another example of the proteostasis network controlling cell-to-cell heterogeneity. Prions are self-propagating protein aggregates that confer protein-based phenotypic traits. The transcriptional terminator of Clostridium botulinum, Rho, is a native prion that can switch to a loss-of-function amyloidogenic form that causes genome-wide transcription.217 Propagation of the yeast Sup35 prion in E. coli required ClpB disaggregase activity.218 As prion transmission from parental to progeny cells requires the chaperone ClpB,218 inhibiting ClpB in prion-containing bacteria might decrease cell-to-cell heterogeneity, which has been associated with enhanced killing by antibiotics.219
TB patients requiring antibiotics represent a situation in which host immunity failed to kill and eradicate Mtb. In this case, Mtb must recover and adapt from damage by chaperone-mediated refolding of damaged proteins or degrading them with Clp proteases and the proteasome. If damaged proteins can neither be repaired nor degraded, Mtb deploys a last bastion of defense against proteotoxic stress, the ClpB-associated buffering, sequestration, and distribution between progeny cells of irreversibly oxidized proteins (IOP).72 Similar to the previous reasoning for prions, inhibiting ClpB may cripple the pathway by which Mtb eliminates damaged macromolecules. Interfering with metabolic processes might impact the ability of bacteria to sequester and asymmetrically distribute aggregates of damaged proteins or propagate prions. This could occur by impacting large particle (protein aggregates) diffusion in the cytosol or protein organization and compartmentalization by interfering with cytoplasmic fluid-to-glass or proteinaceous liquid–liquid phase transition.220,221 This raises the possibility that small molecules targeting metabolism may indirectly perturb proteostasis pathways.
During chronic infection, Mtb must adapt to its new environmental niche. A comparison of global transcriptomic data from four models of Mtb nonreplicating phenotypic tolerance, including persisters surviving d-cycloserine treatment, two models of hypoxia (the enduring hypoxic response and the Wayne model), and nutrient starvation, found acr2 was upregulated in all dormancy models, and clpB was upregulated in three of the four models.222 Mutations in genes encoding GrpE and TF were correlated with a high-persister phenotype in Mtb.17 Mtb deficient in α-crystallin (encoded by hspX) were cleared from mouse lungs more rapidly than the wild-type by a three-drug combination in the Cornell model of persistence (INH, RIF, PZA), and the hspX mutants had a decreased relapse rate upon treatment of mice with steroids.223 Targeting these pathways might affect Mtb’s ability to adapt to its host compartments.
Finally, in some instances, targeting the proteostasis network might have a detrimental impact by rendering Mtb more resistant to host stress or antibiotics. For example, knockdown of the Mtb proteasome121 or mutations in genes encoding accessory proteins Mpa and PafA119 led to hydrogen peroxide resistance. In another example, the proteostasis network may dictate the stability of target enzymes of antibiotics.224 PZA is a first-line drug that kills nonreplicating Mtb by inhibiting trans–translation and the synthesis of lipids, pantothenate, and coenzyme A,225−229 and two independent studies identified PZA resistance-conferring mutations in ClpC1.230,231 It is possible that the mutant ClpC1 has decreased affinity for client protein(s) that are the targets of PZA. This might contribute to PZA resistance by increasing the amount of PZA target enzyme in the cell. In another example, amikacin- and kanamycin-resistant Mtb had increased protein levels of chaperones TF (encoded by rv2462c) and α-crystallin (encoded by rv2031c) and proteolysis protein proteasome subunit alpha (encoded by rv2109).232
Conclusions
The proteostasis network repairs or degrades Mtb proteins damaged during infection. Some individual components of the proteostasis network, such as the proteasome, are vulnerable to selective inhibition over their eukaryotic counterparts,41 and we anticipate that species-selective inhibitors could be selected for most of the components of the pathway. Disrupting the Mtb proteostasis network as a noncanonical target pathway of small molecules is anticipated to have multiple effects on Mtb during human TB. First, targeting the proteostasis network will prevent Mtb from repairing damage to mycobacterial proteins that had already been inflicted by the host. Second, inhibiting or activating the proteostasis network may sensitize Mtb to stresses of host immunity, including ROS, RNS, hypoxia, mild acidity, nutrient starvation, and metal sequestration or intoxication, in part by preventing Mtb stress adaptation and/or generation of cell-to-cell heterogeneity. Finally, compounds inhibiting the proteostasis network may prevent the formation of drug-tolerant persister cells and serve as adjunctive therapy to boost the efficacy of conventional antibiotics. The ClpP1P2 accessory chaperone ClpC1 has emerged as an extremely promising noncanonical drug target.38−40,193 Of the compounds targeting mycobacterial proteolysis, only ecumicin has demonstrated efficacy in a murine model of TB.38 The next major hurdle will be to develop compounds with pharmacologic properties suitable for testing in animal models of TB, which will enable further testing of the hypothesis that targeting the proteostasis network renders Mtb hypersusceptible to host stresses during infection. To date, the majority of studies have focused on developing antibiotics targeting components of the Mtb proteolysis machinery, in particular ClpP1P2/ClpC1 and the eukaryotic-like proteasome, Mtb20S. However, the development of inhibitors of bacterial proteostasis in other bacteria, including GroEL/ES, GrpE, and DnaK, suggest that these proteins could be targeted in mycobacteria as well. Future studies will determine if molecules targeting the proteostasis network will impact Mtb pathogenesis in animal models or a human host.
Acknowledgments
We are grateful to Drs. Carl Nathan and Gang Lin (Weill Cornell Medicine) for critical discussions, chemistry guidance, and expert review of the manuscript. We thank Drs. Michael Glickman and Allison Fay (Memorial Sloan Kettering) and Drs. Nader Fotouhi, Khisi Mduli, Anna Upton, Christopher Cooper, and Takushi Kaneko (Global Alliance for TB Drug Development) for excellent ideas and discussions. We apologize to those authors whose references were omitted due to space constraints. This work was supported by the TB Drug Accelerator Program of the Bill and Melinda Gates Foundation, the Abby and Howard P. Milstein Program in Chemical Biology and Translational Medicine, and an NIH TB Research Unit (U19 AI111143). The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation. T.J.L. was supported by a Helen Hay Whitney and Simons Foundation Postdoctoral Fellowship.
Author Contributions
∥ T.J.L., J.V., and K.B.-H. contributed equally.
The authors declare no competing financial interest.
References
- WHO. (2014) Companion Handbook to the WHO Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis, WHO, Geneva. [PubMed] [Google Scholar]
- Diacon A. H.; van der Merwe L.; Barnard M.; von Groote-Bidlingmaier F.; Lange C.; Garcia-Basteiro A. L.; Sevene E.; Ballell L.; Barros-Aguirre D. (2016) beta-Lactams against Tuberculosis–New Trick for an Old Dog?. N. Engl. J. Med. 375 (4), 393–4. 10.1056/NEJMc1513236. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 6 (1), 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Nathan C. (2011) Making space for anti-infective drug discovery. Cell Host Microbe 9 (5), 343–8. 10.1016/j.chom.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Gold B.; Nathan C. (2017) Targeting Phenotypically Tolerant Mycobacterium tuberculosis. Tuberculosis and the Tubercle Bacillus 5 (1), 317–360. 10.1128/microbiolspec.TBTB2-0031-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S. (2012) Host-pathogen coevolution in human tuberculosis. Philos. Trans. R. Soc., B 367 (1590), 850–9. 10.1098/rstb.2011.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillebaek T.; Dirksen A.; Baess I.; Strunge B.; Thomsen V. O.; Andersen A. B. (2002) Molecular evidence of endogenous reactivation of Mycobacterium tuberculosis after 33 years of latent infection. J. Infect. Dis. 185 (3), 401–4. 10.1086/338342. [DOI] [PubMed] [Google Scholar]
- Lillebaek T.; Dirksen A.; Vynnycky E.; Baess I.; Thomsen V. O.; Andersen A. B. (2003) Stability of DNA patterns and evidence of Mycobacterium tuberculosis reactivation occurring decades after the initial infection. J. Infect. Dis. 188 (7), 1032–9. 10.1086/378240. [DOI] [PubMed] [Google Scholar]
- Schnappinger D.; Ehrt S.; Voskuil M. I.; Liu Y.; Mangan J. A.; Monahan I. M.; Dolganov G.; Efron B.; Butcher P. D.; Nathan C.; Schoolnik G. K. (2003) Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J. Exp. Med. 198 (5), 693–704. 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S.; Schnappinger D.; Bekiranov S.; Drenkow J.; Shi S.; Gingeras T. R.; Gaasterland T.; Schoolnik G.; Nathan C. (2001) Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med. 194 (8), 1123–40. 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois V. (2014) The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 12 (3), 159–67. 10.1038/nrmicro3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. (2012) Fresh approaches to anti-infective therapies. Sci. Transl. Med. 4 (140), 140sr2. 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamoto Y.; Dhar N.; Chait R.; Schneider K.; Signorino-Gelo F.; Leibler S.; McKinney J. D. (2013) Dynamic persistence of antibiotic-stressed mycobacteria. Science 339 (6115), 91–5. 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- McCune R. M.; Feldmann F. M.; Lambert H. P.; McDermott W. (1966) Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 123 (3), 445–468. 10.1084/jem.123.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manina G.; Dhar N.; McKinney J. D. (2015) Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17 (1), 32–46. 10.1016/j.chom.2014.11.016. [DOI] [PubMed] [Google Scholar]
- Torrey H. L.; Keren I.; Via L. E.; Lee J. S.; Lewis K. (2016) High Persister Mutants in Mycobacterium tuberculosis. PLoS One 11, e0155127. 10.1371/journal.pone.0155127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman M. A.; Brynildsen M. P. (2013) Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 57 (7), 3230–9. 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. (2017) Kunkel Lecture: Fundamental immunodeficiency and its correction. J. Exp. Med. 214 (8), 2175–2191. 10.1084/jem.20170637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javid B.; Sorrentino F.; Toosky M.; Zheng W.; Pinkham J. T.; Jain N.; Pan M.; Deighan P.; Rubin E. J. (2014) Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc. Natl. Acad. Sci. U. S. A. 111 (3), 1132–7. 10.1073/pnas.1317580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H. W.; Zhu J. H.; Li H.; Cai R. J.; Ealand C.; Wang X.; Chen Y. X.; Kayani M. U.; Zhu T. F.; Moradigaravand D.; Huang H.; Kana B. D.; Javid B. (2016) The essential mycobacterial amidotransferase GatCAB is a modulator of specific translational fidelity. Nature microbiology 1, 16147. 10.1038/nmicrobiol.2016.147. [DOI] [PubMed] [Google Scholar]
- Sarathy J.; Dartois V.; Dick T.; Gengenbacher M. (2013) Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating. Antimicrob. Agents Chemother. 57 (4), 1648–53. 10.1128/AAC.02202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K.; Warrier T.; Somersan-Karakaya S.; Kaminski L.; Mi J.; Jiang X.; Park S.; Shigyo K.; Gold B.; Roberts J.; Weber E.; Jacobs W. R. Jr.; Nathan C. F. (2017) Rifamycin action on RNA polymerase in antibiotic-tolerant Mycobacterium tuberculosis results in differentially detectable populations. Proc. Natl. Acad. Sci. U. S. A. 114 (24), E4832–E4840. 10.1073/pnas.1705385114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamolova G. V.; Turapov O.; Malkin J.; Woltmann G.; Barer M. R. (2010) Resuscitation-promoting factors reveal an occult population of tubercle Bacilli in Sputum. Am. J. Respir. Crit. Care Med. 181 (2), 174–80. 10.1164/rccm.200905-0661OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Barry C. E. 3rd; Boshoff H. I. (2010) The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J. Bacteriol. 192 (5), 1279–91. 10.1128/JB.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. S.; Kawate T.; Nag P. P.; Silvis M. R.; Gordon K.; Stanley S. A.; Kazyanskaya E.; Nietupski R.; Golas A.; Fitzgerald M.; Cho S.; Franzblau S. G.; Hung D. T. (2013) Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem. Biol. 8 (10), 2224–34. 10.1021/cb4004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzblau S. G.; DeGroote M. A.; Cho S. H.; Andries K.; Nuermberger E.; Orme I. M.; Mdluli K.; Angulo-Barturen I.; Dick T.; Dartois V.; Lenaerts A. J. (2012) Comprehensive analysis of methods used for the evaluation of compounds against. Tuberculosis (Oxford, U. K.) 92 (6), 453–88. 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Gold B.; Roberts J.; Ling Y.; Lopez Quezada L.; Glasheen J.; Ballinger E.; Somersan-Karakaya S.; Warrier T.; Warren J. D.; Nathan C. (2015) Rapid, semi-quantitative assay to discriminate among compounds with activity against replicating or non-replicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 6521. 10.1128/AAC.00803-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarayana S. B.; Huat T. B.; Ho P. C.; Manjunatha U. H.; Dartois V.; Dick T.; Rao S. P. (2015) Comprehensive physicochemical, pharmacokinetic and activity profiling of anti-TB agents. J. Antimicrob. Chemother. 70 (3), 857–67. 10.1093/jac/dku457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul A.; Dendouga N.; Vergauwen K.; Molenberghs B.; Vranckx L.; Willebrords R.; Ristic Z.; Lill H.; Dorange I.; Guillemont J.; Bald D.; Andries K. (2007) Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3 (6), 323–4. 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- Koul A.; Vranckx L.; Dendouga N.; Balemans W.; Van den Wyngaert I.; Vergauwen K.; Gohlmann H. W.; Willebrords R.; Poncelet A.; Guillemont J.; Bald D.; Andries K. (2008) Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 283 (37), 25273–80. 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- Harbut M. B.; Vilcheze C.; Luo X.; Hensler M. E.; Guo H.; Yang B.; Chatterjee A. K.; Nizet V.; Jacobs W. R. Jr.; Schultz P. G.; Wang F. (2015) Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. U. S. A. 112 (14), 4453–8. 10.1073/pnas.1504022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.; O’Brien K. M.; Trujillo C.; Wang R.; Wallach J. B.; Schnappinger D.; Ehrt S. (2016) Mycobacterium tuberculosis Thioredoxin Reductase Is Essential for Thiol Redox Homeostasis but Plays a Minor Role in Antioxidant Defense. PLoS Pathog. 12 (6), e1005675. 10.1371/journal.ppat.1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk R.; Gold B.; Venugopal A.; Singh J.; Samy R.; Pupek K.; Cao H.; Popescu C.; Gurney M.; Hotha S.; Cherian J.; Rhee K.; Ly L.; Converse P. J.; Ehrt S.; Vandal O.; Jiang X.; Schneider J.; Lin G.; Nathan C. (2008) Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3 (3), 137–45. 10.1016/j.chom.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Advani M. J.; Siddiqui I.; Sharma P.; Reddy H. (2012) Activity of trifluoperazine against replicating, non-replicating and drug resistant M. tuberculosis. PLoS One 7 (8), e44245. 10.1371/journal.pone.0044245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho L. P.; Darby C. M.; Rhee K. Y.; Nathan C. (2011) Nitazoxanide Disrupts Membrane Potential and Intrabacterial pH Homeostasis of Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2 (11), 849–854. 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Zhu W.; Schurig-Briccio L. A.; Lindert S.; Shoen C.; Hitchings R.; Li J.; Wang Y.; Baig N.; Zhou T.; Kim B. K.; Crick D. C.; Cynamon M.; McCammon J. A.; Gennis R. B.; Oldfield E. (2015) Antiinfectives targeting enzymes and the proton motive force. Proc. Natl. Acad. Sci. U. S. A. 112 (51), E7073–E7082. 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.; Kim J. Y.; Anderson J. R.; Akopian T.; Hong S.; Jin Y. Y.; Kandror O.; Kim J. W.; Lee I. A.; Lee S. Y.; McAlpine J. B.; Mulugeta S.; Sunoqrot S.; Wang Y.; Yang S. H.; Yoon T. M.; Goldberg A. L.; Pauli G. F.; Suh J. W.; Franzblau S. G.; Cho S. (2015) The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 59 (2), 880–9. 10.1128/AAC.04054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrish E.; Sit C. S.; Cao S.; Kandror O.; Spoering A.; Peoples A.; Ling L.; Fetterman A.; Hughes D.; Bissell A.; Torrey H.; Akopian T.; Mueller A.; Epstein S.; Goldberg A.; Clardy J.; Lewis K. (2014) Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 21 (4), 509–18. 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E. K.; Riwanto M.; Sambandamurthy V.; Roggo S.; Miault C.; Zwingelstein C.; Krastel P.; Noble C.; Beer D.; Rao S. P.; Au M.; Niyomrattanakit P.; Lim V.; Zheng J.; Jeffery D.; Pethe K.; Camacho L. R. (2011) The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem., Int. Ed. 50 (26), 5889–91. 10.1002/anie.201101740. [DOI] [PubMed] [Google Scholar]
- Lin G.; Li D.; de Carvalho L. P.; Deng H.; Tao H.; Vogt G.; Wu K.; Schneider J.; Chidawanyika T.; Warren J. D.; Li H.; Nathan C. (2009) Inhibitors selective for mycobacterial versus human proteasomes. Nature 461 (7264), 621–6. 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotz-Oesterhelt H.; Beyer D.; Kroll H. P.; Endermann R.; Ladel C.; Schroeder W.; Hinzen B.; Raddatz S.; Paulsen H.; Henninger K.; Bandow J. E.; Sahl H. G.; Labischinski H. (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11 (10), 1082–7. 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- Balchin D.; Hayer-Hartl M.; Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353 (6294), aac4354. 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Daugaard M.; Rohde M.; Jaattela M. (2007) The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 581 (19), 3702–10. 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Mayer M. P.; Bukau B. (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62 (6), 670–84. 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl F. U.; Bracher A.; Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475 (7356), 324–32. 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Kim Y. E.; Hipp M. S.; Bracher A.; Hayer-Hartl M.; Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Calloni G.; Chen T.; Schermann S. M.; Chang H. C.; Genevaux P.; Agostini F.; Tartaglia G. G.; Hayer-Hartl M.; Hartl F. U. (2012) DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 1 (3), 251–64. 10.1016/j.celrep.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Fay A.; Glickman M. S. (2014) An essential nonredundant role for mycobacterial DnaK in native protein folding. PLoS Genet. 10 (7), e1004516. 10.1371/journal.pgen.1004516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7 (9), e1002251. 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek K. H.; Walker G. C. (1987) Escherichia coli dnaK null mutants are inviable at high temperature. J. Bacteriol. 169 (1), 283–90. 10.1128/jb.169.1.283-290.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter S. A.; Houry W. A.; Ang D.; Tradler T.; Rockabrand D.; Fischer G.; Blum P.; Georgopoulos C.; Hartl F. U. (1999) Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell 97 (6), 755–65. 10.1016/S0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Genevaux P.; Keppel F.; Schwager F.; Langendijk-Genevaux P. S.; Hartl F. U.; Georgopoulos C. (2004) In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 5 (2), 195–200. 10.1038/sj.embor.7400067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E.; Schulze-Specking A.; Tomoyasu T.; Mogk A.; Bukau B. (1999) Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature 400 (6745), 693–6. 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- Stewart G. R.; Wernisch L.; Stabler R.; Mangan J. A.; Hinds J.; Laing K. G.; Young D. B.; Butcher P. D. (2002) Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 148 (Pt 10), 3129–3138. 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- Narberhaus F. (1999) Negative regulation of bacterial heat shock genes. Mol. Microbiol. 31 (1), 1–8. 10.1046/j.1365-2958.1999.01166.x. [DOI] [PubMed] [Google Scholar]
- Das Gupta T.; Bandyopadhyay B.; Das Gupta S. K. (2008) Modulation of DNA-binding activity of Mycobacterium tuberculosis HspR by chaperones. Microbiology 154 (Pt 2), 484–490. 10.1099/mic.0.2007/012294-0. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay B.; Das Gupta T.; Roy D.; Das Gupta S. K. (2012) DnaK dependence of the mycobacterial stress-responsive regulator HspR is mediated through its hydrophobic C-terminal tail. J. Bacteriol. 194 (17), 4688–97. 10.1128/JB.00415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G. R.; Snewin V. A.; Walzl G.; Hussell T.; Tormay P.; O’Gaora P.; Goyal M.; Betts J.; Brown I. N.; Young D. B. (2001) Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nat. Med. 7 (6), 732–7. 10.1038/89113. [DOI] [PubMed] [Google Scholar]
- MacAry P. A.; Javid B.; Floto R. A.; Smith K. G.; Oehlmann W.; Singh M.; Lehner P. J. (2004) HSP70 peptide binding mutants separate antigen delivery from dendritic cell stimulation. Immunity 20 (1), 95–106. 10.1016/S1074-7613(03)00357-1. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kelly C. G.; Karttunen J. T.; Whittall T.; Lehner P. J.; Duncan L.; MacAry P.; Younson J. S.; Singh M.; Oehlmann W.; Cheng G.; Bergmeier L.; Lehner T. (2001) CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity 15 (6), 971–83. 10.1016/S1074-7613(01)00242-4. [DOI] [PubMed] [Google Scholar]
- Lehner T.; Bergmeier L. A.; Wang Y.; Tao L.; Sing M.; Spallek R.; van der Zee R. (2000) Heat shock proteins generate beta-chemokines which function as innate adjuvants enhancing adaptive immunity. Eur. J. Immunol. 30 (2), 594–603. . [DOI] [PubMed] [Google Scholar]
- Gassler C. S.; Buchberger A.; Laufen T.; Mayer M. P.; Schroder H.; Valencia A.; Bukau B. (1998) Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc. Natl. Acad. Sci. U. S. A. 95 (26), 15229–34. 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M.; Canchaya C.; Bernini V.; Del Casale A.; Dellaglio F.; Neviani E.; Fitzgerald G. F.; van Sinderen D. (2005) Genetic characterization of the Bifidobacterium breve UCC 2003 hrcA locus. Appl. Environ. Microbiol. 71 (12), 8998–9007. 10.1128/AEM.71.12.8998-9007.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G. R.; Robertson B. D.; Young D. B. (2004) Analysis of the function of mycobacterial DnaJ proteins by overexpression and microarray profiling. Tuberculosis (Oxford, U. K.) 84 (3–4), 180–7. 10.1016/j.tube.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Lupoli T. J.; Fay A.; Adura C.; Glickman M. S.; Nathan C. F. (2016) Reconstitution of a Mycobacterium tuberculosis proteostasis network highlights essential cofactor interactions with chaperone DnaK. Proc. Natl. Acad. Sci. U. S. A. 113 (49), E7947–E7956. 10.1073/pnas.1617644113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Anil Kumar V.; Das A. K.; Bansal R.; Sarkar D. (2014) A transcriptional co-repressor regulatory circuit controlling the heat-shock response of. Mol. Microbiol. 94 (2), 450–65. 10.1111/mmi.12778. [DOI] [PubMed] [Google Scholar]
- Rosenzweig R.; Moradi S.; Zarrine-Afsar A.; Glover J. R.; Kay L. E. (2013) Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science 339 (6123), 1080–3. 10.1126/science.1233066. [DOI] [PubMed] [Google Scholar]
- Stewart G. R.; Newton S. M.; Wilkinson K. A.; Humphreys I. R.; Murphy H. N.; Robertson B. D.; Wilkinson R. J.; Young D. B. (2005) The stress-responsive chaperone alpha-Crystallin 2 is required for pathogenesis of Mycobacterium tuberculosis. Mol. Microbiol. 55 (4), 1127–37. 10.1111/j.1365-2958.2004.04450.x. [DOI] [PubMed] [Google Scholar]
- Reddy G. B.; Kumar P. A.; Kumar M. S. (2006) Chaperone-like activity and hydrophobicity of alpha-Crystallin. IUBMB Life 58 (11), 632–41. 10.1080/15216540601010096. [DOI] [PubMed] [Google Scholar]
- Wattam A. R.; Abraham D.; Dalay O.; Disz T. L.; Driscoll T.; Gabbard J. L.; Gillespie J. J.; Gough R.; Hix D.; Kenyon R.; Machi D.; Mao C.; Nordberg E. K.; Olson R.; Overbeek R.; Pusch G. D.; Shukla M.; Schulman J.; Stevens R. L.; Sullivan D. E.; Vonstein V.; Warren A.; Will R.; Wilson M. J.; Yoo H. S.; Zhang C.; Zhang Y.; Sobral B. W. (2014) PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42 (D1), D581–D591. 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaubourgeix J.; Lin G.; Dhar N.; Chenouard N.; Jiang X.; Botella H.; Lupoli T.; Mariani O.; Yang G.; Ouerfelli O.; Unser M.; Schnappinger D.; McKinney J.; Nathan C. (2015) Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe 17 (2), 178–90. 10.1016/j.chom.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall D.; Zylicz M.; Georgopoulos C. (1994) The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for lambda replication. J. Biol. Chem. 269 (7), 5446–5451. [PubMed] [Google Scholar]
- Tsai J.; Douglas M. G. (1996) A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J. Biol. Chem. 271 (16), 9347–54. 10.1074/jbc.271.16.9347. [DOI] [PubMed] [Google Scholar]
- Sell S. M.; Eisen C.; Ang D.; Zylicz M.; Georgopoulos C. (1990) Isolation and characterization of dnaJ null mutants of Escherichia coli. J. Bacteriol. 172 (9), 4827–35. 10.1128/jb.172.9.4827-4835.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C. G.; Chang L.; Gestwicki J. E. (2010) Heat shock protein 70 (Hsp70) as an emerging drug target. J. Med. Chem. 53 (12), 4585–602. 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L.; Miyata Y.; Ung P. M.; Bertelsen E. B.; McQuade T. J.; Carlson H. A.; Zuiderweg E. R.; Gestwicki J. E. (2011) Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chem. Biol. 18 (2), 210–21. 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylandsted J.; Brand K.; Jaattela M. (2000) Heat shock protein 70 is required for the survival of cancer cells. Ann. N. Y. Acad. Sci. 926, 122–5. 10.1111/j.1749-6632.2000.tb05605.x. [DOI] [PubMed] [Google Scholar]
- Chapman E.; Farr G. W.; Usaite R.; Furtak K.; Fenton W. A.; Chaudhuri T. K.; Hondorp E. R.; Matthews R. G.; Wolf S. G.; Yates J. R.; Pypaert M.; Horwich A. L. (2006) Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc. Natl. Acad. Sci. U. S. A. 103 (43), 15800–5. 10.1073/pnas.0607534103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayet O.; Ziegelhoffer T.; Georgopoulos C. (1989) The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J. Bacteriol. 171 (3), 1379–85. 10.1128/jb.171.3.1379-1385.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A.; Huber D.; Bukau B. (2011) Integrating protein homeostasis strategies in prokaryotes. Cold Spring Harbor Perspect. Biol. 3 (4), a004366. 10.1101/cshperspect.a004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Horwich A. L.; Sigler P. B. (1997) The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature 388 (6644), 741–50. 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- Qamra R.; Srinivas V.; Mande S. C. (2004) Mycobacterium tuberculosis GroEL homologues unusually exist as lower oligomers and retain the ability to suppress aggregation of substrate proteins. J. Mol. Biol. 342 (2), 605–17. 10.1016/j.jmb.2004.07.066. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Henderson B.; Lund P. A.; Tormay P.; Ahmed M. T.; Gurcha S. S.; Besra G. S.; Coates A. R. (2008) A Mycobacterium tuberculosis mutant lacking the groEL homologue cpn60.1 is viable but fails to induce an inflammatory response in animal models of infection. Infect. Immun. 76 (4), 1535–46. 10.1128/IAI.01078-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. B.; Ivanyi J.; Cox J. H.; Lamb J. R. (1987) The 65 kDa antigen of mycobacteria-a common bacterial protein?. Immunology today 8 (7–8), 215–9. 10.1016/0167-5699(87)90168-X. [DOI] [PubMed] [Google Scholar]
- Lewthwaite J. C.; Coates A. R.; Tormay P.; Singh M.; Mascagni P.; Poole S.; Roberts M.; Sharp L.; Henderson B. (2001) Mycobacterium tuberculosis chaperonin 60.1 is a more potent cytokine stimulator than chaperonin 60.2 (Hsp 65) and contains a CD14-binding domain. Infect. Immun. 69 (12), 7349–55. 10.1128/IAI.69.12.7349-7355.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cehovin A.; Coates A. R.; Hu Y.; Riffo-Vasquez Y.; Tormay P.; Botanch C.; Altare F.; Henderson B. (2010) Comparison of the moonlighting actions of the two highly homologous chaperonin 60 proteins of Mycobacterium tuberculosis. Infect. Immun. 78 (7), 3196–206. 10.1128/IAI.01379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan I. M.; Betts J.; Banerjee D. K.; Butcher P. D. (2001) Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology 147 (2), 459–71. 10.1099/00221287-147-2-459. [DOI] [PubMed] [Google Scholar]
- Genest O.; Hoskins J. R.; Camberg J. L.; Doyle S. M.; Wickner S. (2011) Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc. Natl. Acad. Sci. U. S. A. 108 (20), 8206–11. 10.1073/pnas.1104703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M.; Jarosz D. F.; Lindquist S. (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11 (7), 515–28. 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Raju R. M.; Unnikrishnan M.; Rubin D. H.; Krishnamoorthy V.; Kandror O.; Akopian T. N.; Goldberg A. L.; Rubin E. J. (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8 (2), e1002511. 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur E.; Ottofueling R.; Dougan D. A. (2013) Machines of destruction - AAA+ proteases and the adaptors that control them. Subcell. Biochem. 66, 3–33. 10.1007/978-94-007-5940-4_1. [DOI] [PubMed] [Google Scholar]
- Porankiewicz J.; Wang J.; Clarke A. K. (1999) New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol. Microbiol. 32 (3), 449–58. 10.1046/j.1365-2958.1999.01357.x. [DOI] [PubMed] [Google Scholar]
- Lies M.; Maurizi M. R. (2008) Turnover of endogenous SsrA-tagged proteins mediated by ATP-dependent proteases in. J. Biol. Chem. 283 (34), 22918–29. 10.1074/jbc.M801692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frees D.; Gerth U.; Ingmer H. (2014) Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of. Int. J. Med. Microbiol. 304 (2), 142–9. 10.1016/j.ijmm.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Akopian T.; Kandror O.; Raju R. M.; Unnikrishnan M.; Rubin E. J.; Goldberg A. L. (2012) The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 31 (6), 1529–41. 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personne Y.; Brown A. C.; Schuessler D. L.; Parish T. (2013) Mycobacterium tuberculosis ClpP proteases are co-transcribed but exhibit different substrate specificities. PLoS One 8 (4), e60228. 10.1371/journal.pone.0060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger J.; O’Malley T.; Kesicki E. A.; Odingo J.; Parish T. (2012) Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J. Bacteriol. 194 (3), 663–8. 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju R. M.; Jedrychowski M. P.; Wei J. R.; Pinkham J. T.; Park A. S.; O’Brien K.; Rehren G.; Schnappinger D.; Gygi S. P.; Rubin E. J. (2014) Post-translational regulation via Clp protease is critical for survival of Mycobacterium tuberculosis. PLoS Pathog. 10 (3), e1003994. 10.1371/journal.ppat.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz K. R.; Sauer R. T. (2014) Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol. Microbiol. 93 (4), 617–28. 10.1111/mmi.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar N. P.; Sikriwal D.; Rath P.; Choudhary R. K.; Batra J. K. (2008) Mycobacterium tuberculosis ClpC1: characterization and role of the N-terminal domain in its function. FEBS J. 275 (24), 6149–58. 10.1111/j.1742-4658.2008.06738.x. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48 (1), 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Rubin E. J. (2003) Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100 (22), 12989–94. 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A.; Zwickl P.; Baumeister W. (1994) Proteasome sequences in eubacteria. Trends Biochem. Sci. 19 (12), 533–4. 10.1016/0968-0004(94)90054-X. [DOI] [PubMed] [Google Scholar]
- Coux O.; Tanaka K.; Goldberg A. L. (1996) Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801–47. 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Voges D.; Zwickl P.; Baumeister W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–68. 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Lin G.; Hu G.; Tsu C.; Kunes Y. Z.; Li H.; Dick L.; Parsons T.; Li P.; Chen Z.; Zwickl P.; Weich N.; Nathan C. (2006) Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol. Microbiol. 59 (5), 1405–16. 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- Striebel F.; Hunkeler M.; Summer H.; Weber-Ban E. (2010) The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J. 29 (7), 1262–71. 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin K. H.; Lin G.; Chen Z.; Li H.; Nathan C. F. (2005) Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol. 55 (2), 561–71. 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- Liao S.; Shang Q.; Zhang X.; Zhang J.; Xu C.; Tu X. (2009) Pup, a prokaryotic ubiquitin-like protein, is an intrinsically disordered protein. Biochem. J. 422 (2), 207–15. 10.1042/BJ20090738. [DOI] [PubMed] [Google Scholar]
- Chen X.; Solomon W. C.; Kang Y.; Cerda-Maira F.; Darwin K. H.; Walters K. J. (2009) Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J. Mol. Biol. 392 (1), 208–17. 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous J.; Burns K.; Liu W. T.; Patel A.; Hook V.; Bafna V.; Barry C. E. 3rd; Bark S.; Dorrestein P. C. (2010) Expansion of the mycobacterial “PUPylome”. Mol. BioSyst. 6 (2), 376–85. 10.1039/B916104J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa R. A.; McAllister F.; Pearce M. J.; Mintseris J.; Burns K. E.; Gygi S. P.; Darwin K. H. (2010) Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected]. PLoS One 5 (1), e8589. 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imkamp F.; Striebel F.; Sutter M.; Ozcelik D.; Zimmermann N.; Sander P.; Weber-Ban E. (2010) Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep. 11 (10), 791–7. 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns K. E.; Cerda-Maira F. A.; Wang T.; Li H.; Bishai W. R.; Darwin K. H. (2010) “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol. Cell 39 (5), 821–7. 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastrab J. B.; Wang T.; Murphy J. P.; Bai L.; Hu K.; Merkx R.; Huang J.; Chatterjee C.; Ovaa H.; Gygi S. P.; Li H.; Darwin K. H. (2015) An adenosine triphosphate-independent proteasome activator contributes to the virulence of. Proc. Natl. Acad. Sci. U. S. A. 112 (14), E1763–72. 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delley C. L.; Laederach J.; Ziemski M.; Bolten M.; Boehringer D.; Weber-Ban E. (2014) Bacterial proteasome activator Bpa (Rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One 9 (12), e114348. 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Hu K.; Li D.; Bai L.; Yang S.; Jastrab J. B.; Xiao S.; Hu Y.; Zhang S.; Darwin K. H.; Wang T.; Li H. (2017) Mycobacterium tuberculosis proteasomal ATPase Mpa has a beta-grasp domain that hinders docking with the proteasome core protease. Mol. Microbiol. 105 (2), 227–241. 10.1111/mmi.13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin K. H.; Ehrt S.; Gutierrez-Ramos J. C.; Weich N.; Nathan C. F. (2003) The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302 (5652), 1963–6. 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- Gandotra S.; Lebron M. B.; Ehrt S. (2010) The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 6 (8), e1001040. 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandotra S.; Schnappinger D.; Monteleone M.; Hillen W.; Ehrt S. (2007) In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med. 13 (12), 1515–1520. 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda-Maira F. A.; Pearce M. J.; Fuortes M.; Bishai W. R.; Hubbard S. R.; Darwin K. H. (2010) Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol. Microbiol. 77 (5), 1123–35. 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fascellaro G.; Petrera A.; Lai Z. W.; Nanni P.; Grossmann J.; Burger S.; Biniossek M. L.; Gomez-Auli A.; Schilling O.; Imkamp F. (2016) Comprehensive Proteomic Analysis of Nitrogen-Starved Mycobacterium smegmatis Deltapup Reveals the Impact of Pupylation on Nitrogen Stress Response. J. Proteome Res. 15 (8), 2812–25. 10.1021/acs.jproteome.6b00378. [DOI] [PubMed] [Google Scholar]
- Elharar Y.; Roth Z.; Hermelin I.; Moon A.; Peretz G.; Shenkerman Y.; Vishkautzan M.; Khalaila I.; Gur E. (2014) Survival of mycobacteria depends on proteasome-mediated amino acid recycling under nutrient limitation. EMBO J. 33 (16), 1802–14. 10.15252/embj.201387076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng V. H.; Cox J. S.; Sousa A. O.; MacMicking J. D.; McKinney J. D. (2004) Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52 (5), 1291–302. 10.1111/j.1365-2958.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- MacMicking J. D.; North R. J.; LaCourse R.; Mudgett J. S.; Shah S. K.; Nathan C. F. (1997) Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94 (10), 5243–8. 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski M. A.; Dwyer D. J.; Hayete B.; Lawrence C. A.; Collins J. J. (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130 (5), 797–810. 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- McBee M. E.; Chionh Y. H.; Sharaf M. L.; Ho P.; Cai M. W.; Dedon P. C. (2017) Production of Superoxide in Bacteria Is Stress- and Cell State-Dependent: A Gating-Optimized Flow Cytometry Method that Minimizes ROS Measurement Artifacts with Fluorescent Dyes. Front. Microbiol. 8, 459. 10.3389/fmicb.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar M.; Nathan C.; Rhee K. Y. (2014) Isocitrate lyase mediates broad antibiotic tolerance in. Nat. Commun. 5, 4306. 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- Belenky P.; Ye J. D.; Porter C. B.; Cohen N. R.; Lobritz M. A.; Ferrante T.; Jain S.; Korry B. J.; Schwarz E. G.; Walker G. C.; Collins J. J. (2015) Bactericidal Antibiotics Induce Toxic Metabolic Perturbations that Lead to Cellular Damage. Cell Rep. 13 (5), 968–80. 10.1016/j.celrep.2015.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Manjunatha U.; Boshoff H. I.; Ha Y. H.; Niyomrattanakit P.; Ledwidge R.; Dowd C. S.; Lee I. Y.; Kim P.; Zhang L.; Kang S.; Keller T. H.; Jiricek J.; Barry C. E. 3rd (2008) PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322 (5906), 1392–5. 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee K. Y.; Erdjument-Bromage H.; Tempst P.; Nathan C. F. (2005) S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc. Natl. Acad. Sci. U. S. A. 102 (2), 467–72. 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C.; Cunningham-Bussel A. (2013) Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 13 (5), 349–61. 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. John G.; Brot N.; Ruan J.; Erdjument-Bromage H.; Tempst P.; Weissbach H.; Nathan C. (2001) Peptide methionine sulfoxide reductase from Escherichia coli and Mycobacterium tuberculosis protects bacteria against oxidative damage from reactive nitrogen intermediates. Proc. Natl. Acad. Sci. U. S. A. 98 (17), 9901–9906. 10.1073/pnas.161295398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. L.; Gold B.; Darby C.; Brot N.; Jiang X.; de Carvalho L. P.; Wellner D.; St. John G.; Jacobs W. R. Jr.; Nathan C. (2009) Mycobacterium tuberculosis expresses methionine sulfoxide reductases A and B that protect from killing by nitrite and hypochlorite. Mol. Microbiol. 71 (3), 583–593. 10.1111/j.1365-2958.2008.06548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlett B. S.; Stadtman E. R. (1997) Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272 (33), 20313–6. 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Grune T.; Jung T.; Merker K.; Davies K. J. (2004) Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ’aggresomes’ during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 36 (12), 2519–30. 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Dukan S.; Nystrom T. (1999) Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J. Biol. Chem. 274 (37), 26027–32. 10.1074/jbc.274.37.26027. [DOI] [PubMed] [Google Scholar]
- Voskuil M. I.; Bartek I. L.; Visconti K.; Schoolnik G. K. (2011) The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2, 105. 10.3389/fmicb.2011.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J. D.; Taylor G. A.; McKinney J. D. (2003) Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302 (5645), 654–659. 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- Vandal O. H.; Roberts J. A.; Odaira T.; Schnappinger D.; Nathan C. F.; Ehrt S. (2009) Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. J. Bacteriol. 191 (2), 625–631. 10.1128/JB.00932-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal O. H.; Pierini L. M.; Schnappinger D.; Nathan C. F.; Ehrt S. (2008) A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 14 (8), 849–854. 10.1038/nm.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal O. H.; Nathan C. F.; Ehrt S. (2009) Acid resistance in Mycobacterium tuberculosis. J. Bacteriol. 191 (15), 4714–4721. 10.1128/JB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J.; Murphy E.; Park A.; Krone C. L.; Hett E. C.; Bloom B. R.; Glimcher L. H.; Rubin E. J. (2008) Mycobacterium tuberculosis Rv2224c modulates innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 105 (1), 264–9. 10.1073/pnas.0710601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella H.; Vaubourgeix J.; Lee M. H.; Song N.; Xu W.; Makinoshima H.; Glickman M. S.; Ehrt S. (2017) Mycobacterium tuberculosis protease MarP activates a peptidoglycan hydrolase during acid stress. EMBO J. 36 (4), 536–548. 10.15252/embj.201695028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N.; Darby C. M.; Small J.; Bachovchin D. A.; Jiang X.; Burns-Huang K. E.; Botella H.; Ehrt S.; Boger D. L.; Anderson E. D.; Cravatt B. F.; Speers A. E.; Fernandez-Vega V.; Hodder P. S.; Eberhart C.; Rosen H.; Spicer T. P.; Nathan C. F. (2015) Target-based screen against a periplasmic serine protease that regulates intrabacterial pH homeostasis in. ACS Chem. Biol. 10 (2), 364–71. 10.1021/cb500746z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton M.; Brilha S.; Manavaki R.; Mauri F.; Nijran K.; Hong Y. T.; Patel N. H.; Dembek M.; Tezera L.; Green J.; Moores R.; Aigbirhio F.; Al-Nahhas A.; Fryer T. D.; Elkington P. T.; Friedland J. S. (2016) Hypoxia and tissue destruction in pulmonary TB. Thorax 71, 1145. 10.1136/thoraxjnl-2015-207402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne L. G.; Hayes L. G. (1996) An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64 (6), 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne L. G.; Sramek H. A. (1994) Metronidazole is bactericidal to dormant cells of. Antimicrob. Agents Chemother. 38 (9), 2054–8. 10.1128/AAC.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad T. R.; Harrell M. I.; Liao R.; Sherman D. R. (2008) The enduring hypoxic response of. PLoS One 3 (1), e1502. 10.1371/journal.pone.0001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez G. M.; Voskuil M. I.; Gold B.; Schoolnik G. K.; Smith I. (2002) An essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 70 (7), 3371–81. 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm J.; Post F. A.; Bekker L. G.; Walther G. B.; Wainwright H. C.; Manganelli R.; Chan W. T.; Tsenova L.; Gold B.; Smith I.; Kaplan G.; McKinney J. D. (2003) Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. U. S. A. 100 (24), 14321–6. 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B.; Deng H.; Bryk R.; Vargas D.; Eliezer D.; Roberts J.; Jiang X.; Nathan C. (2008) Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat. Chem. Biol. 4 (10), 609–16. 10.1038/nchembio.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland J. L.; Niederweis M. (2012) Resistance mechanisms of Mycobacterium tuberculosis against phagosomal copper overload. Tuberculosis (Oxford, U. K.) 92 (3), 202–10. 10.1016/j.tube.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolschendorf F.; Ackart D.; Shrestha T. B.; Hascall-Dove L.; Nolan S.; Lamichhane G.; Wang Y.; Bossmann S. H.; Basaraba R. J.; Niederweis M. (2010) Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U S A 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella H.; Peyron P.; Levillain F.; Poincloux R.; Poquet Y.; Brandli I.; Wang C.; Tailleux L.; Tilleul S.; Charriere G. M.; Waddell S. J.; Foti M.; Lugo-Villarino G.; Gao Q.; Maridonneau-Parini I.; Butcher P. D.; Castagnoli P. R.; Gicquel B.; de Chastellier C.; Neyrolles O. (2011) Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe 10 (3), 248–59. 10.1016/j.chom.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurthkoti K.; Amin H.; Marakalala M. J.; Ghanny S.; Subbian S.; Sakatos A.; Livny J.; Fortune S. M.; Berney M.; Rodriguez G. M. (2017) The Capacity of Mycobacterium tuberculosis To Survive Iron Starvation Might Enable It To Persist in Iron-Deprived Microenvironments of Human Granulomas. mBio 8 (4), e01092-17. 10.1128/mBio.01092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokpoel M. C.; Smeulders M. J.; Hubbard J. A.; Keer J.; Williams H. D. (2005) Global analysis of proteins synthesized by Mycobacterium smegmatis provides direct evidence for physiological heterogeneity in stationary-phase cultures. J. Bacteriol. 187 (19), 6691–700. 10.1128/JB.187.19.6691-6700.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts J. C.; Lukey P. T.; Robb L. C.; McAdam R. A.; Duncan K. (2002) Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43 (3), 717–31. 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- Taldone T.; Sun W.; Chiosis G. (2009) Discovery and development of heat shock protein 90 inhibitors. Bioorg. Med. Chem. 17 (6), 2225–35. 10.1016/j.bmc.2008.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T.; Patel H. J.; Bolaender A.; Patel M. R.; Chiosis G. (2014) Protein chaperones: a composition of matter review (2008–2013). Expert opinion on therapeutic patents. Expert Opin. Ther. Pat. 24 (5), 501–18. 10.1517/13543776.2014.887681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T.; Ochiana S. O.; Patel P. D.; Chiosis G. (2014) Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol. Sci. 35 (11), 592–603. 10.1016/j.tips.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D. D.; Morimoto R. I. (2004) Molecular chaperones and the stress of oncogenesis. Oncogene 23 (16), 2907–18. 10.1038/sj.onc.1207529. [DOI] [PubMed] [Google Scholar]
- Tredan O.; Galmarini C. M.; Patel K.; Tannock I. F. (2007) Drug resistance and the solid tumor microenvironment. Journal of the National Cancer Institute 99 (19), 1441–54. 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- Powers M. V.; Workman P. (2006) Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr.-Relat. Cancer 13 (Suppl 1), S125–35. 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- Trachootham D.; Zhou Y.; Zhang H.; Demizu Y.; Chen Z.; Pelicano H.; Chiao P. J.; Achanta G.; Arlinghaus R. B.; Liu J.; Huang P. (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10 (3), 241–52. 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Szatrowski T. P.; Nathan C. F. (1991) Production of large amounts of hydrogen peroxide by human tumor cells. Cancer research 51 (3), 794–798. [PubMed] [Google Scholar]
- Kamal A.; Thao L.; Sensintaffar J.; Zhang L.; Boehm M. F.; Fritz L. C.; Burrows F. J. (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425 (6956), 407–10. 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Alix J.-H. (2014) Targeting HSP70 to Fight Cancer and Bad Bugs: One and the Same Battle? In Antibiotics: Targets, Mechanisms and Resistance, Wiley-VCH Verlag GmbH & Co. KGaA, New York. [Google Scholar]
- Chapman E.; Farr G. W.; Furtak K.; Horwich A. L. (2009) A small molecule inhibitor selective for a variant ATP-binding site of the chaperonin GroEL. Bioorg. Med. Chem. Lett. 19 (3), 811–3. 10.1016/j.bmcl.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdeen S.; Salim N.; Mammadova N.; Summers C. M.; Frankson R.; Ambrose A. J.; Anderson G. G.; Schultz P. G.; Horwich A. L.; Chapman E.; Johnson S. M. (2016) GroEL/ES inhibitors as potential antibiotics. Bioorg. Med. Chem. Lett. 26 (13), 3127–3134. 10.1016/j.bmcl.2016.04.089. [DOI] [PubMed] [Google Scholar]
- Minagawa S.; Kondoh Y.; Sueoka K.; Osada H.; Nakamoto H. (2011) Cyclic lipopeptide antibiotics bind to the N-terminal domain of the prokaryotic Hsp90 to inhibit the chaperone activity. Biochem. J. 435 (1), 237–46. 10.1042/BJ20100743. [DOI] [PubMed] [Google Scholar]
- Cellitti J.; Zhang Z.; Wang S.; Wu B.; Yuan H.; Hasegawa P.; Guiney D. G.; Pellecchia M. (2009) Small molecule DnaK modulators targeting the beta-domain. Chem. Biol. Drug Des. 74 (4), 349–57. 10.1111/j.1747-0285.2009.00869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher M.; Jahreis G.; Lucke C.; Grabley S.; Raina S.; Schiene-Fischer C. (2007) Fatty acyl benzamido antibacterials based on inhibition of DnaK-catalyzed protein folding. J. Biol. Chem. 282 (7), 4437–46. 10.1074/jbc.M607667200. [DOI] [PubMed] [Google Scholar]
- Schiene-Fischer C.; Habazettl J.; Schmid F. X.; Fischer G. (2002) The hsp70 chaperone DnaK is a secondary amide peptide bond cis-trans isomerase. Nat. Struct. Biol. 9 (6), 419–24. 10.1038/nsb804. [DOI] [PubMed] [Google Scholar]
- Kragol G.; Lovas S.; Varadi G.; Condie B. A.; Hoffmann R.; Otvos L. Jr (2001) The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 40 (10), 3016–26. 10.1021/bi002656a. [DOI] [PubMed] [Google Scholar]
- Kragol G.; Hoffmann R.; Chattergoon M. A.; Lovas S.; Cudic M.; Bulet P.; Condie B. A.; Rosengren K. J.; Montaner L. J.; Otvos L. Jr (2002) Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin. Eur. J. Biochem. 269 (17), 4226–37. 10.1046/j.1432-1033.2002.03119.x. [DOI] [PubMed] [Google Scholar]
- Chesnokova L. S.; Slepenkov S. V.; Witt S. N. (2004) The insect antimicrobial peptide, L-pyrrhocoricin, binds to and stimulates the ATPase activity of both wild-type and lidless DnaK. FEBS Lett. 565 (1–3), 65–9. 10.1016/j.febslet.2004.03.075. [DOI] [PubMed] [Google Scholar]
- Scocchi M.; Tossi A.; Gennaro R. (2011) Proline-rich antimicrobial peptides: converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 68 (13), 2317–30. 10.1007/s00018-011-0721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scocchi M.; Luthy C.; Decarli P.; Mignogna G.; Christen P.; Gennaro R. (2009) The Proline-rich Antibacterial Peptide Bac7 Binds to and Inhibits in vitro the Molecular Chaperone DnaK. Int. J. Pept. Res. Ther. 15 (2), 147–155. 10.1007/s10989-009-9182-3. [DOI] [Google Scholar]
- Credito K.; Lin G.; Koeth L.; Sturgess M. A.; Appelbaum P. C. (2009) Activity of levofloxacin alone and in combination with a DnaK inhibitor against gram-negative rods, including levofloxacin-resistant strains. Antimicrob. Agents Chemother. 53 (2), 814–7. 10.1128/AAC.01132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L. Jr.; Bokonyi K.; Varga I.; Otvos B. I.; Hoffmann R.; Ertl H. C.; Wade J. D.; McManus A. M.; Craik D. J.; Bulet P. (2000) Insect peptides with improved protease-resistance protect mice against bacterial infection. Protein science: a publication of the Protein Society 9 (4), 742–9. 10.1110/ps.9.4.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L. Jr (2002) The short proline-rich antibacterial peptide family. Cell. Mol. Life Sci. 59 (7), 1138–1150. 10.1007/s00018-002-8493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon-Garcia S.; Mikut R.; Ng C.; Ruden S.; Volkmer R.; Reischl M.; Hilpert K.; Thompson C. J. (2013) Targeting Mycobacterium tuberculosis and other microbial pathogens using improved synthetic antibacterial peptides. Antimicrob. Agents Chemother. 57 (5), 2295–303. 10.1128/AAC.00175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon M. G.; Roy R. N.; Lomakin I. B.; Florin T.; Mankin A. S.; Steitz T. A. (2016) Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 44 (5), 2439–50. 10.1093/nar/gkw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizsan A.; Volke D.; Weinert S.; Strater N.; Knappe D.; Hoffmann R. (2014) Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew. Chem., Int. Ed. 53 (45), 12236–9. 10.1002/anie.201407145. [DOI] [PubMed] [Google Scholar]
- Mohammadi-Ostad-Kalayeh S.; Hrupins V.; Helmsen S.; Ahlbrecht C.; Stahl F.; Scheper T.; Preller M.; Surup F.; Stadler M.; Kirschning A.; Zeilinger C. (2017) Development of a microarray-based assay for efficient testing of new HSP70/DnaK inhibitors. Bioorg. Med. Chem. 25, 6345. 10.1016/j.bmc.2017.10.003. [DOI] [PubMed] [Google Scholar]
- Alhuwaider A. A. H.; Dougan D. A. (2017) AAA+ Machines of Protein Destruction in Mycobacteria. Frontiers in molecular biosciences 4, 49. 10.3389/fmolb.2017.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culp E.; Wright G. D. (2017) Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 70 (4), 366–377. 10.1038/ja.2016.138. [DOI] [PubMed] [Google Scholar]
- Roberts D. M.; Personne Y.; Ollinger J.; Parish T. (2013) Proteases in Mycobacterium tuberculosis pathogenesis: potential as drug targets. Future Microbiol. 8 (5), 621–31. 10.2217/fmb.13.25. [DOI] [PubMed] [Google Scholar]
- Raju R. M.; Goldberg A. L.; Rubin E. J. (2012) Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discovery 11 (10), 777–89. 10.1038/nrd3846. [DOI] [PubMed] [Google Scholar]
- Lee H.; Suh J. W. (2016) Anti-tuberculosis lead molecules from natural products targeting Mycobacterium tuberculosis ClpC1. J. Ind. Microbiol. Biotechnol. 43 (2–3), 205–12. 10.1007/s10295-015-1709-3. [DOI] [PubMed] [Google Scholar]
- Conlon B. P.; Nakayasu E. S.; Fleck L. E.; LaFleur M. D.; Isabella V. M.; Coleman K.; Leonard S. N.; Smith R. D.; Adkins J. N.; Lewis K. (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503 (7476), 365–70. 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J.; Hoffmann A.; Lilie H.; Schmidt R.; Rubsamen-Waigmann H.; Brotz-Oesterhelt H.; Mogk A.; Turgay K. (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO molecular medicine 1 (1), 37–49. 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass P.; Josten M.; Famulla K.; Schiffer G.; Sahl H. G.; Hamoen L.; Brotz-Oesterhelt H. (2011) Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc. Natl. Acad. Sci. U. S. A. 108 (42), 17474–9. 10.1073/pnas.1110385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulla K.; Sass P.; Malik I.; Akopian T.; Kandror O.; Alber M.; Hinzen B.; Ruebsamen-Schaeff H.; Kalscheuer R.; Goldberg A. L.; Brotz-Oesterhelt H. (2016) Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol. Microbiol. 101 (2), 194–209. 10.1111/mmi.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choules M.; Yu Y.; Cho S. H.; Anderson J.; Gao W.; Klein L.; Lankin D. C.; Kim J. Y.; Cheng J.; Yang S. H.; Lee H.; Suh J. W.; Franzblau S. G.; Pauli G. F. (2015) A rufomycin analogue is an anti-tuberculosis drug lead targeting CLPC1 with no cross resistance to ecumicin. Planta Med. 81 (11), 871–871. 10.1055/s-0035-1556165. [DOI] [Google Scholar]
- Tomita H.; Katsuyama Y.; Minami H.; Ohnishi Y. (2017) Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J. Biol. Chem. 292 (38), 15859–15869. 10.1074/jbc.M117.791269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie P.; Kazmaier U. (2016) Total synthesis of desoxycyclomarin C and the cyclomarazines A and B. Org. Biomol. Chem. 14 (25), 6055–64. 10.1039/C6OB00801A. [DOI] [PubMed] [Google Scholar]
- Vila-Farres X.; Chu J.; Inoyama D.; Ternei M. A.; Lemetre C.; Cohen L. J.; Cho W.; Reddy B. V.; Zebroski H. A.; Freundlich J. S.; Perlin D. S.; Brady S. F. (2017) Antimicrobials Inspired by Nonribosomal Peptide Synthetase Gene Clusters. J. Am. Chem. Soc. 139 (4), 1404–1407. 10.1021/jacs.6b11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton C. L.; Schmitz K. R.; Sauer R. T.; Sello J. K. (2013) Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS Chem. Biol. 8 (12), 2669–77. 10.1021/cb400577b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J.; Cheng T. Y.; Aggarwal A.; Park A. S.; Zeiler E.; Raju R. M.; Akopian T.; Kandror O.; Sacchettini J. C.; Moody D. B.; Rubin E. J.; Sieber S. A. (2018) An Antibacterial beta-Lactone Kills Mycobacterium tuberculosis by Disrupting Mycolic Acid Biosynthesis. Angew. Chem., Int. Ed. 57 (1), 348–353. 10.1002/anie.201709365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira W.; Ngan G. J.; Low J. L.; Poulsen A.; Chia B. C.; Ang M. J.; Yap A.; Fulwood J.; Lakshmanan U.; Lim J.; Khoo A. Y.; Flotow H.; Hill J.; Raju R. M.; Rubin E. J.; Dick T. (2015) Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 6 (3), e00253-15. 10.1128/mBio.00253-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Tsu C.; Dick L.; Zhou X. K.; Nathan C. (2008) Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem. 283 (49), 34423–31. 10.1074/jbc.M805324200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira W.; Santhanakrishnan S.; Ngan G. J. Y.; Low C. B.; Sangthongpitag K.; Poulsen A.; Dymock B. W.; Dick T. (2017) Towards Selective Mycobacterial ClpP1P2 Inhibitors with Reduced Activity against the Human Proteasome. Antimicrob. Agents Chemother. 61 (5), e02307-16. 10.1128/AAC.02307-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira W.; Santhanakrishnan S.; Dymock B. W.; Dick T. (2017) Bortezomib Warhead-Switch Confers Dual Activity against Mycobacterial Caseinolytic Protease and Proteasome and Selectivity against Human Proteasome. Front. Microbiol. 8, 746. 10.3389/fmicb.2017.00746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian T.; Kandror O.; Tsu C.; Lai J. H.; Wu W.; Liu Y.; Zhao P.; Park A.; Wolf L.; Dick L. R.; Rubin E. J.; Bachovchin W.; Goldberg A. L. (2015) Cleavage Specificity of Mycobacterium tuberculosis ClpP1P2 Protease and Identification of Novel Peptide Substrates and Boronate Inhibitors with Anti-bacterial Activity. J. Biol. Chem. 290 (17), 11008–20. 10.1074/jbc.M114.625640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.; Lin G.; Wang M.; Dick L.; Xu R. M.; Nathan C.; Li H. (2006) Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol. Microbiol. 59 (5), 1417–28. 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- Lin G.; Li D.; Chidawanyika T.; Nathan C.; Li H. (2010) Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch. Biochem. Biophys. 501 (2), 214–20. 10.1016/j.abb.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo F.; Gising J.; Akerbladh L.; Roos A. K.; Naworyta A.; Mowbray S. L.; Sokolowski A.; Henderson I.; Alling T.; Bailey M. A.; Files M.; Parish T.; Karlen A.; Larhed M. (2015) Optimization and Evaluation of 5-Styryl-Oxathiazol-2-one Mycobacterium tuberculosis Proteasome Inhibitors as Potential Antitubercular Agents. ChemistryOpen 4 (3), 342–62. 10.1002/open.201500001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Chidawanyika T.; Tsu C.; Warrier T.; Vaubourgeix J.; Blackburn C.; Gigstad K.; Sintchak M.; Dick L.; Nathan C. (2013) N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J. Am. Chem. Soc. 135 (27), 9968–71. 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMott C. M.; Girardin R.; Cobbert J.; Reverdatto S.; Burz D. S.; McDonough K.; Shekhtman A. (2018) Potent inhibitors of Mycobacterium tuberculosis growth identified by using in-cell NMR-based screening. ACS Chem. Biol. 10.1021/acschembio.7b00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro K. A.; Barthelme D.; Simpson P. T.; Jiang X.; Lin G.; Nathan C. F.; Sauer R. T.; Sello J. K. (2017) Rational Design of Selective and Bioactive Inhibitors of the Mycobacterium tuberculosis Proteasome. ACS Infect. Dis. 3 (2), 176–181. 10.1021/acsinfecdis.6b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A.; Fino C.; Sørensen M. A.; Semsey S.; Gerdes K. (2017) Nasty Prophages and the Dynamics of Antibiotic-Tolerant Persister Cells. bioRxiv 10.1101/200477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A.; Bordes P.; Genevaux P. (2014) Multiple toxin-antitoxin systems in. Toxins 6 (3), 1002–20. 10.3390/toxins6031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P.; Weinrick B. C.; Kalivoda E. J.; Yang H.; Munsamy V.; Vilcheze C.; Weisbrod T. R.; Larsen M. H.; O’Donnell M. R.; Pym A.; Jacobs W. R. (2016) Dual-Reporter Mycobacteriophages (Φ2DRMs) Reveal Preexisting Mycobacterium tuberculosis Persistent Cells in Human Sputum. mBio 7 (5), e01023-16. 10.1128/mBio.01023-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A. H.; Hochschild A. (2017) A bacterial global regulator forms a prion. Science 355 (6321), 198–201. 10.1126/science.aai7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A. H.; Garrity S. J.; Nako E.; Hochschild A. (2014) Prion propagation can occur in a prokaryote and requires the ClpB chaperone. eLife 3, e02949 10.7554/eLife.02949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego E. H.; Audette R. E.; Rubin E. J. (2017) Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546 (7656), 153–157. 10.1038/nature22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A. A.; Weber C. A.; Julicher F. (2014) Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 30, 39–58. 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Parry B. R.; Surovtsev I. V.; Cabeen M. T.; O’Hern C. S.; Dufresne E. R.; Jacobs-Wagner C. (2014) The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156 (1–2), 183–94. 10.1016/j.cell.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren I.; Minami S.; Rubin E.; Lewis K. (2011) Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2 (3), e00100-11. 10.1128/mBio.00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Liu A.; Menendez M. C.; Garcia M. J.; Oravcova K.; Gillespie S. H.; Davies G. R.; Mitchison D. A.; Coates A. R. (2015) HspX knock-out in Mycobacterium tuberculosis leads to shorter antibiotic treatment and lower relapse rate in a mouse model–a potential novel therapeutic target. Tuberculosis (Oxford, U. K.) 95 (1), 31–6. 10.1016/j.tube.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Rutherford S. L.; Henikoff S. (2003) Quantitative epigenetics. Nat. Genet. 33 (1), 6–8. 10.1038/ng0103-6. [DOI] [PubMed] [Google Scholar]
- Dillon N. A.; Peterson N. D.; Rosen B. C.; Baughn A. D. (2014) Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob. Agents Chemother. 58 (12), 7258–63. 10.1128/AAC.04028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W.; Chen J.; Feng J.; Cui P.; Zhang S.; Weng X.; Zhang W.; Zhang Y. (2014) Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerging Microbes Infect. 3 (8), e58. 10.1038/emi.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W.; Zhang X.; Jiang X.; Yuan H.; Lee J. S.; Barry C. E. 3rd; Wang H.; Zhang W.; Zhang Y. (2011) Pyrazinamide inhibits trans-translation in. Science 333 (6049), 1630–2. 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimhony O.; Cox J. S.; Welch J. T.; Vilcheze C.; Jacobs W. R. Jr (2000) Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of. Nat. Med. 6 (9), 1043–7. 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- Zimhony O.; Vilcheze C.; Arai M.; Welch J. T.; Jacobs W. R. Jr (2007) Pyrazinoic acid and its n-propyl ester inhibit fatty acid synthase type I in replicating tubercle bacilli. Antimicrob. Agents Chemother. 51 (2), 752–4. 10.1128/AAC.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee M.; Gopal P.; Dick T. (2017) Missense Mutations in the Unfoldase ClpC1 of the Caseinolytic Protease Complex Are Associated with Pyrazinamide Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 61 (2), AAC.02342-16. 10.1128/AAC.02342-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Tasneen R.; Yee M.; Lanoix J. P.; Sarathy J.; Rasic G.; Li L.; Dartois V.; Nuermberger E.; Dick T. (2017) Vivo-Selected Pyrazinoic Acid-Resistant Mycobacterium tuberculosis Strains Harbor Missense Mutations in the Aspartate Decarboxylase PanD and the Unfoldase ClpC1. ACS Infect. Dis. 3 (7), 492–501. 10.1021/acsinfecdis.7b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D.; Kumar B.; Lata M.; Joshi B.; Venkatesan K.; Shukla S.; Bisht D. (2015) Comparative Proteomic Analysis of Aminoglycosides Resistant and Susceptible Mycobacterium tuberculosis Clinical Isolates for Exploring Potential Drug Targets. PLoS One 10 (10), e0139414. 10.1371/journal.pone.0139414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang D.; Chandrasekhar G. N.; Zylicz M.; Georgopoulos C. (1986) Escherichia coligrpE gene codes for heat shock protein B25.3, essential for both lambda DNA replication at all temperatures and host growth at high temperature. J. Bacteriol. 167 (1), 25–9. 10.1128/jb.167.1.25-29.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires C. L.; Pedersen S.; Ross B. M.; Squires C. (1991) ClpB is the Escherichia coli heat shock protein F84.1. J. Bacteriol. 173 (14), 4254–62. 10.1128/jb.173.14.4254-4262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D.; Rao S. P.; Noble C. G. (2013) Structural basis of mycobacterial inhibition by cyclomarin A. J. Biol. Chem. 288 (43), 30883–91. 10.1074/jbc.M113.493767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H. C.; Singh P. K.; Fan H.; Wang R.; Sukenick G.; Nathan C.; Lin G.; Li H. (2017) Structural Basis for the Species-Selective Binding of N,C-Capped Dipeptides to the Mycobacterium tuberculosis Proteasome. Biochemistry 56 (1), 324–333. 10.1021/acs.biochem.6b01107. [DOI] [PMC free article] [PubMed] [Google Scholar]