

Abstract
Myelination by oligodendrocytes in the central nervous system permits high fidelity saltatory conduction from neuronal cell bodies to axon terminals. Dysmyelinating and demyelinating disorders impair normal nervous system functions. Consequently, an understanding of oligodendrocyte differentiation that moves beyond the genetic code into the field of epigenetics is essential. Chromatin reprogramming is critical for steering stage-specific differentiation processes during oligodendrocyte development. Fine temporal control of chromatin remodeling through ATP-dependent chromatin remodelers and sequential histone modifiers shapes a chromatin regulatory landscape conducive to oligodendrocyte fate specification, lineage differentiation, and maintenance of cell identity. In this Review, we will focus on the biological functions of ATP-dependent chromatin remodelers and histone deacetylases in myelinating oligodendrocyte development and implications for myelin regeneration in neurodegenerative diseases.
Keywords: Myelination, Histone Deacetylases, Chromatin Remodelers, Oligodendrocyte, Schwann cell, Temporal control, Lineage progression
I. Introduction
Oligodendrocytes are the specialized glial cells of the central nervous system that produce the myelin sheath that wraps around axons, the long-distance processes of neurons, providing insulation and ensuring saltatory nerve conduction to allow rapid communication over long distances. Defects in myelination or maintenance of the myelin sheath as well as regeneration after pathological insults could eventually lead to neurodegenerative diseases such as various leukodystrophies and multiple sclerosis (MS), respectively. MS often follows a relapsing-remitting course during early stages which underscores the importance of accelerating remyelination during periods of limited lesion progression to maximize the duration of remission. The understanding of myelination and remyelination processes may have an impact on promoting regeneration and functional nerve recovery in these devastating demyelinating diseases [1–4].
Oligodendrocytes trace their origin to multipotent neural progenitor cells (NPCs) which arise during embryonic development. The linage specification and progression from NPCs unfolds in distinct stages at different regions from primitive oligodendrocyte progenitor cells (pri-OPCs) also called uncommitted OPCs (Olig1/2+/A2B5+/PDGFRαlow) to committed OPCs (PDGFRαhigh/NG2+) to differentiating immature oligodendrocytes (O4+/CNP+) to differentiated oligodendrocytes (CC1+) and finally to mature myelinating oligodendrocytes (e.g. MBP+/PLP+/MOG+) (Figure 1). This step-wise differentiation process requires exquisitely precise coordination of a series of extracellular and intracellular cues including lineage-specific transcriptional regulators such as Olig1/2, Sox10, Yy1, Nkx2.2, Zeb2/Sip1, Zfp488, Smad7 and Myrf to regulate specification and differentiation programs, as well as epigenetic regulators that control gene expression by modifying the local state of chromatin [5–7].
Figure 1. Step-wise oligodendrocyte lineage differentiation process.
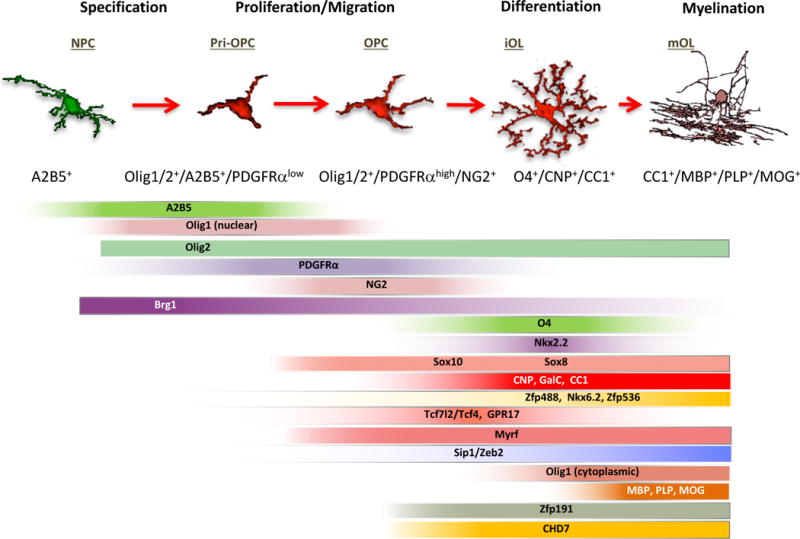
The linage specification and progression from NSCs unfolds in distinct stages at different regions from primitive oligodendrocyte progenitor cells (pri-OPCs) also called uncommitted OPCs (Olig1/2+/A2B5+/PDGFRαlow) to committed OPCs (PDGFRαhigh/NG2+) to differentiating immature oligodendrocytes (O4+/CNP+) to differentiated oligodendrocytes (CC1+) and finally to mature myelinating oligodendrocytes (e.g. MBP+/PLP+/MOG+).
How chromosomal regions are modified to fine-tune gene expression has not been fully understood [8,9]. The emerging picture is that distinct epigenetic codes, individually or in combination, are required for transcriptional programs during OPC specification, differentiation, and maturation in a stage- or region-specific manner. Epigenetic regulators, consisting primarily of enzymes which induce DNA modifications, histone modifications, and ATP-dependent chromatin remodeling, further fine tune gene expression via activity at the chromatin level [8][9].
Gene transcription is regulated by virtue of steric accessibility and dissociation of histones in the genome. When enzymes use energy from ATP hydrolysis to decrease histone-DNA contact, reposition nucleosomes, remove and replace histone subunit variants, or even disrupt and evict the nucleosome altogether, the process is termed ATP-dependent chromatin remodeling [9–11]. Thus, changes to chromatin structure serve to alter accessibility to certain genomic sequences, providing sites for further enzyme activity or transcriptional regulators to act upon and ultimately modulate gene expression.
At the histone level, histone-modifying enzymes may change chromatin structure or provide docking sites for recruitment of specific chromatin remodeling enzymes or transcriptional regulators for gene expression. A variety of modifications can occur along specific amino acid residues on the N-terminal tails of the protein subunits, including but not limited to acetylation, ADP-ribosylation, methylation, phosphorylation, ubiquitylation, and sumoylation, all of which can result in changes to gene expression [12]. The post-translational histone modifications are correlated with distinct chromosomal states that regulate access to DNA. A combination of distinct histone modifications likely forms a “histone code” to specify patterns of gene expression [13,14]. Histone acetylation has been studied extensively in the regulation of oligodendrocyte development. Histone acetyltransferases (HATs) [15] and histone deacetylases (HDACs) modulate histone acetylation states to tilt or maintain the balance between gene activation and repression, respectively. Histone acetylation status can be further recognized by bromo-, PHD-, Tudor-, or WD40-domain-containing activating regulators [16–18], which then further modulate target gene expression.
Cognizant that recent studies have revealed that properly operating epigenetic regulatory mechanisms are vital for oligodendrocyte development and function, this Review will provide an overview of current research into the impact of chromatin remodeling and histone acetylation states on the control of oligodendrocyte lineage specification, differentiation, myelination, and myelin repair in a variety of neurodegenerative diseases ending with a didactic comparison to the oligodendrocyte sibling, the Schwann cell, in the peripheral nervous system (PNS).
II. ATP-dependent remodelers in oligodendrocyte specification, lineage progression and myelination
Classes of chromatin remodelers
ATP-dependent chromatin remodelers utilize ATP as an energy source to reposition or evict nucleosomes, gating accessibility of chromatin to transcriptional regulators and thus acting as indirect effectors in gene expression [19,20]. They regulate various biological processes including cell growth, differentiation, and regeneration [21–23]. While united in their use of ATP, these remodeler complexes can be classified into four families depending on the makeup of their catalytic ATPase subunit: the SWI/SNF family includes ATPase subunits Brahma (Brm, also known as Smarca2) and Brahma-related 1 (Brg1, also known as Smarca4); the INO80 family includes ATPase subunits INO80, SRCAP, and P400; the ISWI family includes ATPase subunits SNF2L and SNF2H; and the CHD family includes the ATPase subunits CHD1-9 [20,24].
The SWI/SNF remodeler Brg1 is essential for specification of OPCs and impacts OPC differentiation
Brg1 (a.k.a. Samarca4), the helicase component of the mammalian SWI/SNF-related chromatin remodeling complex, has been of significant recent interest in neural development [25]. It is expressed in NPCs, OPCs, and differentiating oligodendrocytes during development, in addition to neurons, although it is downregulated in mature myelinating oligodendrocytes in adult mice [26,27]. Deletion of Brg1 in NPCs directed by nestin-Cre prevents oligodendrogenesis in the mouse embryonic cortex while leading to precocious neuronal differentiation of the ventricular zone progenitors [28]. Brg1 loss in these early NPCs results in reduced neurogenic Pax6 expression and ectopic Olig2 expression in the developing cerebral cortex. Olig2+ and Brg1− progenitors, however, fail to differentiate into oligodendrocytes in the developing brain [26], suggesting that Brg1 is vital for oligodendroglial fate specification from murine NPCs.
On the other hand, in cultured rat OPCs, when transient triiodothyronine (T3, a thyroid hormone) treatment was performed to induce OPC differentiation, Brg1 transcription was substantively increased, as illustrated by the extensive occupation of the Brg1 gene locus by RNA polymerase II across the promoter regulatory regions and entire gene body. Brg1 is further recruited by the pioneer oligodendrocyte lineage transcription factor Olig2 in cultured OPCs to achieve target binding specificity on oligodendrocyte-lineage genes and to activate oligodendrocyte differentiation-associated programs [27]. OPCs isolated from Brg1-deficient murine neocortices failed to differentiate into mature MBP+ oligodendrocytes in vitro, suggesting that Brg1 is critical for OPC differentiation.
In mice, deletion of Brg1 starting at the pri-OPC stage (Olig1+/A2B5+/PDGFRαlow) directed by an Olig1-Cre line results in a dysmyelinating phenotype with a defect in the formation of mature oligodendrocytes [27]. Despite the presence of normal levels of PDGFRα (an OPC marker) and overall numbers of OPCs in the developing CNS of Brg1-mutant animals [27], this phenotype persisted, suggesting that Brg1 is not necessarily required for OPC survival but is required for the initiation of OPC differentiation in vivo. Similarly, deletion of Brg1 in committed or postmitotic OPCs by CNP-Cre or NG2-Cre, which act later than Olig1-Cre, results in a decreased number of myelinating, differentiated oligodendrocytes in the developing murine brain [29]. Brg1 further regulates expression of pro-differentiation factor Sox10 during or immediately after OPC specification in vitro and in vivo [27,29]; however, it becomes less critical for Sox10 expression in committed or postmitotic NG2 cells and oligodendrocyte maturation [29]. Thus, Brg1 has distinct roles in OPC specification from NPCs, at the onset of differentiation in pri-OPCs, and during oligodendrocyte maturation.
The discrepancy in Brg1 functions observed across studies regarding OPC differentiation might be due to the difference in OPC temporal states between in vitro and in vivo conditions as cultured OPCs may represent early OPCs (Olig2+A2B5+ glial progenitors) under cell growth conditions with mitogens like PDGFAA and FGF. Alternatively, the distinct in vivo stages of OPCs (uncommitted pri-OPC, proliferative OPC or postmitotic OPC) [30,31] and/or differential recombination efficiency among Cre lines, such as NG2-Cre only recombining floxed alleles in a certain fraction of OPCs during murine CNS development, may account for disparate results on OPC differentiation. Further studies should be carried out by using region- and stage-specific Cre lines to better clarify the extent of the effect of Brg1 deletion during oligodendrocyte lineage development in the CNS. Nonetheless, deletion of Brg1 in PDGFRα+ OPCs in vivo has led to OPC differentiation defects (unpublished observations), suggesting that Brg1 is required for differentiation of PDGFRα+ OPCs, which appear prior to CNP+ immature oligodendrocytes, suggesting a stage-dependent Brg1 function in oligodendrocyte lineage progression. While Brg1 is necessary for NSC transition into OPCs and for the progression of Olig1+ pri-OPCs or PDGFRα+ OPCs into differentiating oligodendrocytes in vivo, the differentiation of CNP+- or NG2+-committed OPCs into full-fledged, mature oligodendrocytes during lineage progression seems to only partly require Brg1 activity [29]. Thus, chromatin remodeling mediated by Brg1-containing BAF complexes is critical for early events in OPC development including OPC specification and the initiation of OPC differentiation, but not critically required during postmitotic OPC or during oligodendrocyte differentiation, indicating a decline in lineage plasticity and a decreased requirement for Brg1-mediated nuclear reorganization at the later stage of oligodendrocyte maturation.
Vertebrate SWI/SNF complexes contain at least 15 subunits including a catalytic subunit, Brg1 or its paralog Brm, which are mutually exclusive [23] despite interchangeability in some contexts. Brm deletion does not appear to cause obvious neural phenotypes [32], whereas Brg1 is critical for neural development [25]. Although oligodendroglial lineage cells co-express Brg1 and Brm at all stages of development, Brm-deficient mice are viable and fertile, while Brg1-deficient mice are not. shRNA-mediated knockdown of Brm does not lead to a significant reduction of MBP expression in transfected oligodendroglial cells in vitro [29]. Further, in Brg1-deficient mice, Brm is not upregulated in any compensatory role [29]. These observations suggest that Brg1 and Brm have non-overlapping functions in oligodendrocyte development, while the in vivo role of Brm in CNS myelination remains to be further defined. Although other subunits of SWI/SNF complexes have a cell-type specific function during development [25,33], their functions remain to be determined during oligodendrocyte lineage progression and myelination.
OPCs can be reprogrammed to a more developmentally plastic state called a multipotent neural stem/progenitor cell-like (NSC-like) in vitro [34]. This reprogramming is mediated in part through the recruitment of the chromatin-remodeler Brm to the Sox2 promoter, promoting Sox2 expression [35]. Reversing OPCs into a primitive multipotent NPC state is at odds with the established effects of Brg1 in OPC specification, suggesting an opposing role of Brm antagonistic towards Brg1.
SWI/SNF chromatin-remodeling factors Brg1 and Brm have been shown to regulate through altering the state of DNA methylation in a context-dependent manner [36–38]. DNA methylation is critical for efficient OPC expansion and early onset of differentiation, but not OPC survival [39], and is also important for efficient remyelination [40]. Whether and how SWI/SNF chromatin-remodeling links to DNA methylation to control oligodendrocyte lineage development and remyelination remain to be investigated in the future.
SWI/SNF complexes have also been implicated in recruitment of histone methyltransferases [41]. Histone methyltransfereases (HMT) like Ezh2 in the PRC2 complex can cause chromatin compaction by trimethylation of histone K27, which leads to a repressive state [42]. Ezh2 has been shown to promote murine oligodendrocyte proliferation from NSCs in vitro [Sher et al., 2008], while deletion of the murine Ezh2 in Olig1+ early progenitors led to dramatic reduction in mature oligodendrocytes and myelination in mice [43].
CHD chromatin remodelers like CHD7 are critical to oligodendrocyte development and regeneration
Brg1 and Olig2 work cooperatively in oligodendrocyte lineage progression regulation, targeting distinct subsets of genes [27,44], one of which is the gene encoding chromatin remodeler CHD7, an ATP-dependent nucleosome remodeling factor in the chromodomain helicase DNA-binding (CHD) family. Brg1 and Olig2 target and activate Chd7 in the differentiating phase of oligodendrocyte development.
Mutations in CHD7 are the major cause of human CHARGE syndrome, an autosomal dominant disorder characterized by a non-random association of multiple birth defects including impaired white matter development and myelination [45,46]. Unexpectedly, CHD7 expression is highly enriched in oligodendrocyte lineage cells in the CNS, with a peak of expression in differentiating oligodendrocytes. Inactivation of CHD7 in mice causes a delay in oligodendrocyte differentiation and myelination since CHD7 cooperates with Sox10 to activate myelin-associated gene expression, however, CHD7 inactivation does not seem to affect OPC formation in vivo [44], despite a defect in OPC proliferation [47].
Furthermore, CHD7 is analogously critical for remyelination after demyelinating injury [44], consistent with the notion that oligodendrocyte lineage development is recapitulated in regeneration. Notably, CHD7 can cooperate with Sox2 to activate OPC proliferation after spinal cord injury in mice [47]. Despite being unnecessary for OPC formation, CHD7 ablation leads to impaired OPC differentiation during remyelination after lysolecithin-induced demyelination [44] as well as reduced OPC recruitment and differentiation after spinal cord injury by laminectomy [47]. In addition, CHD7 appears to collaborate with Sox10 and Sox2 to regulate OPC differentiation and activation, respectively [44,47] and is therefore an important determinant of effective myelin repair. CHD7 function in OPC recruitment and proliferation after injury appears to exhibit a different extent between lysolecithin-induced demyelination model versus spinal cord transection injury model, which remains to be further defined. The difference in Chd7 floxed alleles may also account for the discrepancy. CHD7 targets the enhancers of Myrf and Olig1 and promotes their expression to induce oligodendrocyte maturation [44]. Besides its role in OPC differentiation, CHD7 appears to regulate OPC proliferation through activating expression of PKCΘ and Rgcc (regulator of cell cycle) [47]. In addition, Chd7 can target and regulate other oligodendrocyte maturation-associated factors such as Osterix/Sp7 and Creb3l2, which are also critical for bone formation [44]. Thus, Chd7 can act as a regulatory nexus that control the development of diverse lineages including oligodendrocyte lineage cells and osteoblasts.
It is worth noting that the chromatin remodelers CHD7 and Brg1 appear to target a distinct set of genes pertaining to oligodendrocyte lineage differentiation [44], suggesting that each chromatin remodeler has distinct targets at different stages during oligodendrocyte lineage progression (Figure 1).
CHD7 belongs to the group III CHD family of ATP-dependent chromatin remodeling enzymes, comprising CHD6, CHD7, CHD8, and CHD9 [48]. CHD7 has been shown to interact with its close homolog CHD8 [49]. Mutations in CHD8 manifest in and define a specific subtype of autism spectrum disorders (ASDs) [50–53] and cause developmental white matter abnormalities in the brain [54–57]. CHD8 is also highly expressed in OPC and oligodendrocyte lineage cells during development (unpublished observations). One would suggest that, based on the homologies, the group III CHD family enzymes have limited functional redundancy, which potentially compensates for defective CHD7 mutants in terms of myelination. At present, the roles of other ATP-dependent chromatin remodelers in oligodendrocyte lineage development remain to be determined. The distinct chromatin remodelers may act independently or cooperate to regulate chromatin accessibility of lineage-specific genes in a temporally distinct manner, possibly as a sequential cascade, to control oligodendrocyte lineage progression (Figure 2).
Figure 2. Sequential Chromatin remodeling cascade drives oligodendrocyte lineage progression.
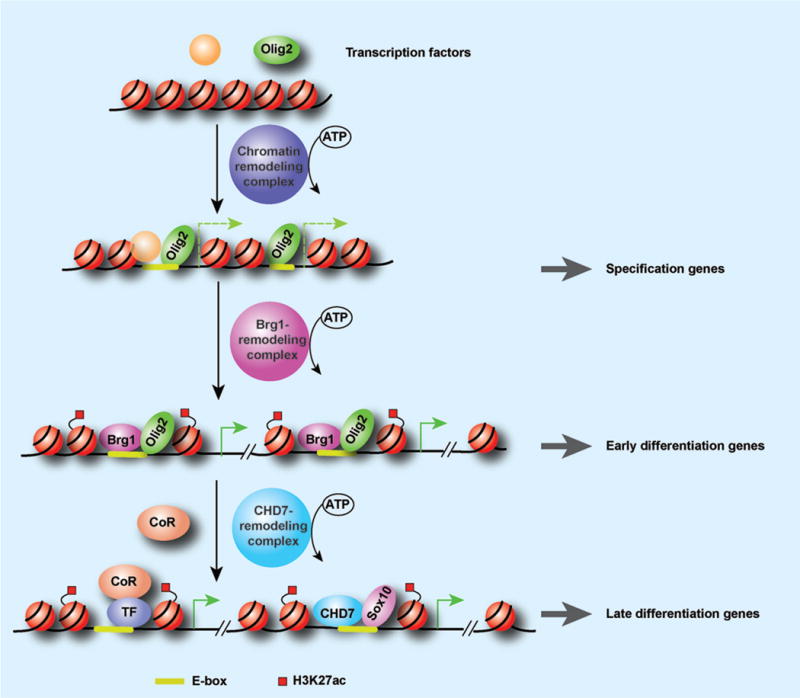
Chromatin remodelers depend on ATP to change the chromatin structure to allow oligodendrocyte lineage pioneer transcription factors such as Olig2 to activate lineage-specification gene expression. The Brg1-containing SWI/SNF complex can be recruited by Olig2 to activate Sox10 expression, and cooperates with Olig2 to promote OPC specification from NSCs and OPC differentiation from pri-OPC progenitors. CHD7, a member of another family of ATP-dependent chromatin remodelers, interacts with Sox10 to regulate the timing of oligodendrocyte differentiation. Thus, successive chromatin remodeling cascades are required to drive proper oligodendrocyte lineage progression.
At present, how expression of multiple chromatin remodeling genes is regulated during oligodendrocyte development or myelin regeneration is unknown, making the field an exciting new frontier. Each chromatin remodeler may act independently or cooperatively to regulate chromatin accessibility of lineage-specific genes in a temporally distinct manner (as seen with Brg1) to control oligodendrocyte lineage progression. To date, little is known regarding regulation of multiple chromatin remodeling genes during oligodendrocyte development or myelin regeneration. Moreover, current research is limited on how the various chromatin remodeling enzymes coordinate with either each other or, for that matter, with other chromatin modifying machinery to regulate the transcriptional output for oligodendrocyte differentiation. This field offers exciting opportunities for multi-enzyme knockout studies and enzyme structure modeling.
III. The function of histone deacetylase protein family protein in oligodendrocyte lineage development
Histone deacetylases (HDACs), as a class of enzymes, remove acetyl groups from an ε-N-acetyl lysine amino acid on a histone, promoting compact chromatin conformation. In mammals, there are four classes of HDACs, assigned based on their similarity to their yeast homologs: class I contains HDAC1, −2, −3, and −8; class II contains HDAC4, −5, −6, −7, −9, and −10; SIRTs 1-7 (also known as sirtuins) are NAD-dependent and belong to class III HDAC; and HDAC11 represents the sole member of class IV [58,59]. Individual HDACs exhibit a dynamic expression pattern during oligodendrocyte lineage progression (Figure 3A).
Figure 3. Histone acetylation in the regulation of oligodendrocyte development.
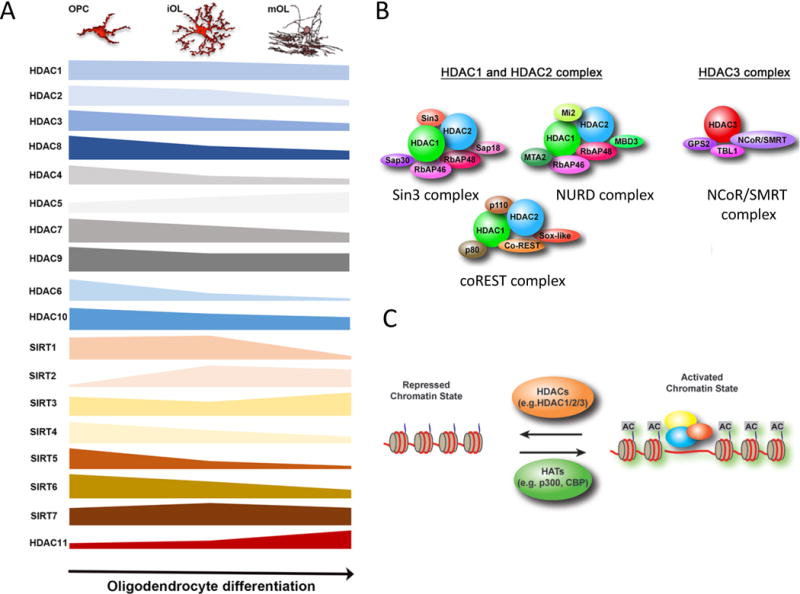
A) Dynamic expression of class I-IV HDACs during oligodendrocyte lineage differentiation based on the transcriptome database [124].
B) Diverse multiprotein complexes containing HDAC1-HDAC2 and HDAC3.
C) A balance of gene transcription regulated by histone acetyltransferases (HATs), which establishes a more relaxed chromatin state resulting in transcriptional activation (activated state), and HDACs, which reverses the activating chromatin state to a “repressed” chromatin state by deacetylation to inhibit gene transcription.
Effects of pharmacological pan-HDAC inhibitors on OPC differentiation
A series of studies have utilized different pharmacological HDAC inhibitors (HDACi) to elucidate the potential role of HDACs in OPC differentiation [60–62]. Treatment with the pan-HDAC inhibitor valproic acid (VPA) inhibits differentiation of rat NPC cells (or HCN cells) into OPCs, while enhancing NPC differentiation towards a neuronal fate, suggesting that HDAC activity modulates OPC and neuronal determination in NPCs [61]. Inhibition of HDACs appears to activate expression of NeuroD, a neurogenic transcription factor, to promote neurogenesis while inhibiting oligodendrogenesis [61]. Similarly, blocking HDAC activity with another pan-HDACi trichostatin A (TSA) prevents the progression of OPCs into mature oligodendrocytes [62] and reverses the lineage-restricted OPCs in vitro to multipotent NPC-like cells, thereby inducing developmental plasticity [60]. TSA decreases differentiation of OPCs by disinhibiting expression of differentiation inhibitors Id2, Egr1, and Sox11 in rats and ID4 and SOX2 in human cells [63,64]. These studies suggest a requirement of HDAC activity for OPC differentiation.
Such studies illustrate the necessity of HDAC activity through multiple stages of oligodendrocyte lineage progression, yet while VPA inhibits OPC differentiation when used early in oligodendrocyte development, the effect of VPA on inhibition of oligodendrocyte differentiation is transient, only taking place during the first postnatal week. The HDACi does not have a substantial effect on oligodendrocyte maturation when utilized after the onset of myelination in vivo [65], suggesting a critical timing window for HDAC-mediated chromatin modification to impact OPC differentiation and myelination. Utilization of HDAC class-specific inhibitors in future studies will continue to provide additional insight into the specific roles of these differentiation inhibitors.
Regulation of OPC differentiation by genetic ablation of HDACs
Studies using a variety of genetic models indicate that HDAC1 and HDAC2 exert both overlapping and non-redundant functions in different cell types at specific developmental stages [66,67]. HDAC functions are critical for oligodendrocyte differentiation in the developing vertebrate brain. HDAC1 is vital for OPC specification in zebrafish through promoting Olig2 expression [68]. In mice, ablation of both HDAC1 and HDAC2, but not of either individual gene alone, in oligodendrocyte lineage cells blocks OPC proliferation and differentiation in the CNS, suggesting functional redundancy between HDAC1/2 in mammalian OPC development [66]. A key difference in these two studies is the use of zebrafish and mammalian models, which may indicate functional redundancy of HDAC1/2 that is phylogenetically distinct.
Exemplifying the unique function of certain HDACs, HDAC3 deletion in early Olig1-expressing pri-OPCs leads to increased numbers of astrocytes with a proportional decrease in oligodendrocyte lineage cells [69], a phenomenon that does not occur in HDAC1/2-deficient OPCs in vivo. These results suggest that HDAC3 functions as a molecular switch for oligodendrocyte and astrocyte lineage fate determination in the developing brain.
In NPC culture, siRNA knockdown of HDAC2 alone in conjunction with T3 treatment in NPCs increases expression of oligodendrocyte genes through disinhibition of Sox8 and Sox10 expression [70]. Similarly, knockdown of HDAC3 initiates a neuronal differentiation pathway in cultured NSCs [70], as opposed to the astrocytic pathway in HDAC3-ablated pri-OPCs in vivo. How can these differing roles for HDAC2 and 3 be reconciled? The explanation may lie in a stage- and context-dependent functional model for individual HDACs. At present, the function of the class I HDACs in oligodendrocyte remyelination after demyelinating injury remains elusive.
Regulation of non-histone substrates by HDACs in oligodendrocyte development
While HDACs are primarily associated with histone modifications, HDACs have been shown to exhibit non-histone-dependent functions in oligodendrocyte development. Both class I and II HDACs (specifically, HDAC1, HDAC3, HDAC10) can deacetylate the Olig1 transcription factor, increasing its likelihood of nuclear translocation and ultimate promotion of OPC differentiation [71]. This process can be inversely regulated by the CREB-binding protein (CBP), a p300-related acetyltransferase, which enhances Olig1 acetylation and interaction with ID2 to facilitate its retention in the cytoplasm of mature oligodendrocytes in vitro [71].
HDAC3 deacetylase activity can inhibit STAT3 acetylation to antagonize JAK-STAT3 mediated astrogliogenesis in vivo [69], which may play a role in the previously discussed capacity of HDAC3 to specify whether an NSC would progress to an astrocyte or oligodendrocyte [69,70].
Genome-wide mapping study reveals that HDACs bind to chromatin at the loci of active, not silent, genes, where they reset chromatin by removing acetylation [72]. Specifically, HDAC1 and HDAC3 are mainly associated with promoters of active genes, whereas HDAC2 can be localized to both promoter and gene body coding regions [72]. Further research would need to determine whether this action of individual HDACs links to specific gene expression or programs for oligodendrocyte development and myelinogenesis.
HDACs function as transcriptional co-regulators independent of deacetylase activity
Two class I HDACs, HDAC1 and HDAC2, can associate with co-repressors to assemble the following repressive complexes: SIN3, nucleosome remodeling and deacetylating (NuRD), and Co-REST complexes (Figure 3B) [73,74]. HDAC1/2 are also often recruited and interact with transcription factors to target specific genes. The transcription factor YY1 recruits HDAC1 to the promoter regions of inhibitory genes such as Id4 to repress their expression during oligodendrocyte differentiation in vitro [75]. In addition, the HDAC1/2-mediated repressor complex can regulate WNT and p53 pathways [76] and compete with β-catenin to interact with TCF7L2 (TCF4), a member of the TCF transcription factor family [66]. Interaction between β-catenin with TCF7L2 can inhibit oligodendrocyte differentiation by activating downstream inhibitory effectors. However, an HDAC1/2 co-repressor complex competing for TCF7L2 binding with β-catenin would block β-catenin signaling and permit oligodendrocyte differentiation to continue unimpeded [66].
Unlike HDAC1/2, HDAC3 is mainly associated with NCoR and SMRT co-repressor complexes (Figure 3B), which stimulate HDAC3 enzymatic activity [77]. NCoR inhibits astroglial differentiation [78], and the loss of NCoR leads to activation of astrocytic pathways in NPCs [78]. Thus, the HDAC3/NCoR complex not only suppresses astrogliogenic programs but also inhibits STAT3 activation through deacetylation of STAT3, an effector of astrogliogenesis, to antagonize astrogliogenesis mediated by JAK-STAT signaling [69].
HDAC3, on the other hand, cooperates with acetyltransferase p300 (also known as EP300) to promote expression of pro-oligodendrocyte genes such as Olig2 and thereby establishes OPC identity [69]. While the coordination of HDAC3 and p300 may seem paradoxical or even a recipe for a futile cycle as the former is a deacetylase and the latter an acetyltransferase, HDACs paired with EP300 were shown to positively regulate gene transcription possibly due to activation of increased net HAT activity, which has been shown to promote transcriptional events leading to OPC specification [69]. HDAC3 has long be known to function not only as a transcriptional co-repressor through interaction with NCoR/SMRT [79] but also as a transcriptional co-activator such as in activating retinoic acid response elements [80,81], so its role in disparate processes is not unprecedented.
Although HDAC3 is tightly associated with NCoR1 and SMRT, it may not be entirely dependent upon the overall complex for all of its function. In mice, deletion of DAD domains in both NCoR1 and SMRT results in non-functional histone deacetylase activity in the overall complex [82], but doing so does not derail oligodendrocyte development or result in a dysmyelinating phenotype [69], indicating that HDAC3 functionality remains unaffected even in the absence of NCoR1/SMRT-mediated histone deacetylation during oligodendrocyte development. This suggests that HDAC3 function in oligodendrogenesis is likely independent of NCoR1/SMRT-mediated histone deacetylation activity and that HDAC3 transcriptional activity per se, HDAC3/NCoR-mediated transcriptional co-repressor complexes, or modification of non-histone regulators may control oligodendrocyte-astrocyte fate switching, which remains to be further defined.
Another layer of regulation of HDAC3 activity is modulation of its phosphorylation state. Protein kinase CK2 phosphorylates HDAC3, activating its deacetylase activity, while protein phosphatase 4 removes the phosphate group and deactivates HDAC3 [74]. In an interesting coincidence of homologous substrate sites, CK2 also phosphorylates and activates Olig2 in vitro [83]. Interaction between HDAC3 and p300 also activates Olig2 and initiates OPC specification programs. From three seemingly disparate proteins, a potential regulatory circuitry emerges that connects the CK2, HDAC3 and Olig2 pathways. Future studies regarding the specific amino acid residues involved and conservation between rodent and human oligodendrocyte development would serve to better tease out this connection.
IV. Functions of other classes of HDACs in oligodendrocyte maturation
Class II HDACs show minimal deacetylation capacity alone, but when recruited with class I HDACs or a multiprotein co-repressor complex containing HDAC3 and SMRT/NCoR, they possess deacetylation capabilities [84]. As yet, their functions in oligodendrocyte development and myelination have not been fully explored, a gap that future research may be able to fill. In rat oligodendrocytes, HDAC6 has been observed to downregulate the acetylation of the microtubule-associated protein tau as well as α-tubulin. Further, HDAC6 can modulate tau phosphorylation and degradation in oligodendrocytes [85]. Tau is necessary for cellular process outgrowth and the transport of MBP mRNA to the cell periphery, suggesting that HDAC6 may have a role in tau and protein aggregate formation during oligodendrocyte maturation [86].
Class III HDACs including Sirtuin 1-7 (SIRT1-7), catalyze the deacetylation of protein targets such as K16 of histone 4 (H4K16) by hydrolyzing NAD, thereby generating a deacetylated protein and nicotinamide molecule [87,88]. SIRT1 and SIRT2 have redundant functions in specification of NPCs to adopt the oligodendrocyte fate through regulation of NAD+ levels and activity of nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in mammalian NAD+ biosynthesis [89]. SIRT1 has been demonstrated to be a regulator of OPC proliferation and regeneration in white matter after neonatal brain injury [90].
Over the course of oligodendrocyte lineage progression, SIRT2 levels increase [91]. Besides acting on histones, SIRT2 also deacetylates α-tubulin by interacting with HDAC6 in the myelin proteome [92,93]. SIRT2-mediated deacetylation of α-tubulin leads to curtailment of oligodendrocyte arborization in culture [91,94]. Intriguingly, SIRT2 can also promotes differentiation of CG4 oligodendroglial cells [95]. SIRT2 mRNA is stabilized by the RNA-binding protein Quaking, which causes persistent SIRT2 levels and the promotion of oligodendrocyte lineage progression [96]. It is likely that this seemingly contradictory role of SIRT2 as a factor at once promoting and inhibiting differentiation may be resolved by future study into stage- and temporally-specific function. The precise function of SIRT2 in the regulation of oligodendrocyte differentiation and morphogenesis in vivo remains to be defined.
HDAC11, the class IV HDAC, regulates H3K9 and H3K14 acetylation states near the regulatory elements of the Mbp and Plp genes, enhancing maturation of an oligodendrocyte cell line (OL-1) in vitro [97,98], suggesting a role of HDAC11 in regulating myelin gene expression.
V. HDAC inhibitors in neural protection and regeneration after pathological insults
Although HDAC activity is essential for oligodendrocyte differentiation and normal nerve function in the developing CNS, transient HDAC inhibition has been shown to have a neuroprotective function after brain injury. After a permanent middle cerebral artery occlusion (MCAO), a rat stroke model, the number of NG2+ OPCs expressing nuclear HDAC1 and HDAC2 increases substantially [99]. When HDACi like sodium butyrate and TSA were administered immediately following an MCAO, some protection against losing oligodendrocytes seemed to be conferred [100]. Interestingly, even delayed treatment with the HDAC inhibitor VPA after MCAO increased oligodendrocyte differentiation and survival [101]. HDACi-mediated protection has been suggested to act through promoting OPC proliferation and differentiation, upregulating VEGF, suppressing inflammation, and decreasing caspase-3-mediated apoptosis [100,101]. In support of the inflammation suppression hypothesis, treatment with scriptaid, a class I and II HDACi, after a traumatic brain injury in mice promoted conversion of microglia/macrophages to the beneficial M2 phenotype, thereby decreasing inflammatory cytokine secretion [102].
The class III HDAC SIRT1 and its modulation by NAD+ levels have also been shown to offer some neuroprotection in the event of ischemic stroke in animal models [103–107]. Inhibition of SIRT1 promotes OPC differentiation after neonatal hypoxia by increasing the cell-cycle exit of proliferative OPCs [90]. Intriguingly, inactivation of SIRT1 in NPCs results in OPC proliferation through activating p38 MAPK and AKT pathways, and in turn improves remyelination in animal models of demyelinating injury including lysolecithin-induced demyelination and chronic experimental autoimmune encephalomyelitis (EAE) [108]. The seemingly opposite effects of SIRT1 inactivation illustrate a context-dependent complexity of epigenetic regulation of OPC proliferation and differentiation in different injury models.
In the frontal lobe of MS patients, a steady decline in histone deacetylation is observed following an early period of functional HDACs [109], yet in EAE, a common animal model of MS, TSA treatment has been documented to improve EAE clinical symptoms, likely through regulating immune cell properties [110]. Combined treatment with HDACi and T3 have potential in improving remyelination and targeting inflammation. Finally, treatment with resveratrol (SRT501), which activates SIRT1, can prevent neuronal damage and long-term neurologic dysfunction in EAE [111,112], suggesting a neuroprotective effect of SIRT1 through both immunomodulatory and neuroprotective mechanisms.
Because most HDACi exhibit broad-spectrum effects targeting many HDACs, outcomes can be mixed and in some cases entirely counterproductive. Suberoylanilide hydroxamic acid (SAHA), an FDA-approved HDACi, has been shown to decrease mouse cortical OPC survival and, accordingly, may decrease CNS myelination rates [113]. Development of specific HDACi with isoform selectivity should minimize side effects and enhance treatment efficacy.
VI. Chromatin modifications with convergent or divergent functions in CNS and PNS myelination
Given that diseases like MS do not solely target the myelinating cells of the CNS but extend to the PNS as well, the roles of HDACs have also been studied in Schwann cell development and peripheral myelination. Inhibition of HDACs by TSA promotes Schwann cell proliferation, while inhibiting their maturation, a phenomenon also seen in oligodendrocytes [62,114], where oligodendrocyte chromatin generally becomes more dense and likely deacetylated as cells differentiate. Similarly, deletion of both HDAC1 and 2, but not either gene in isolation, causes Schwann cell apoptosis and hypomyelinating phenotype, suggesting overlapping functions in HDAC1 and 2 in regard to Schwann cell development and myelination [115,116]. Intriguingly, although blocking Schwann cell remyelination, short-term HDAC1/2 inhibition can improve recovery after nerve injury. This is likely due to a capacity Schwann cells do not share with oligodendrocytes, namely conversion to repair cells, which increase the rate axon regrowth at the expense of remyelination [117].
In contrast to HDAC1/2, HDAC3 appears to exert unique functions distinct from other class I HDACs. Inhibition of HDAC3 seems to promote Schwann cell maturation and enhance myelin sheath growth [4], suggesting, as in oligodendrocytes, specific functions unique to individual HDAC family members in Schwann cell myelinogenesis. In contrast to its early role in oligodendrocyte specification during development [69], HDAC3 appears to have a later function as an inhibitor of Schwann cell maturation and myelinogenesis [4]. Thus, HDAC3 may have a distinct role in myelinating cell development in the central and peripheral nervous systems, although the potential functions of HDAC3 in myelin growth and repair in the CNS remain to be fully determined. If these divergent functions among class I HDACs in a temporal specific manner are supported in future research, it indicates the caution required in therapeutic administration of broad spectrum HDACi and the even greater need for highly specific therapies for myelinating cell dysfunction.
ATP-dependent chromatin remodelers have also been shown to regulate Schwann cell development and PNS myelination. In a subtle contrast to its early role in OPC specification and differentiation [27,29], Brg1 is also required for immature Schwann cells to differentiate and myelin formation in vivo [118,119]. As it does for OPCs in the CNS, Brg1 activates Sox10, though in Schwann cells this occurs at the onset of myelination and in cooperation with the transcription factor NF-κB [118]. Brg1 then also interacts with Sox10 to activate Schwann cell maturation and myelination via the activation of myelin gene expression [119,120].
Although oligodendrocytes and Schwann cells arise from separate progenitors in the neural tube and neural crest, respectively, and utilize anatomically distinct strategies to enwrap target axons (multiple axons for oligodendrocytes and single axons for Schwann cells), chromatin remodelers serve as critical regulators of the myelination program in both cell types. However, the effects of chromatin remodeling may be accomplished through distinct mechanisms, suggesting common and divergent features of chromatin remodeling behind regulation of central and peripheral myelination.
VII. Conclusion and perspectives
Nuclear reorganization through chromatin remodeling and histone modifications has vital roles in oligodendrocyte development, myelination and regeneration after injury or pathological insults. A growing understanding of the critical role of chromatin modifications facilitates dissecting the intricate molecular network underpinning myelinogenesis and provides strategies to enhance neuroprotection and myelin repair.
Given that many of the chromatin modifiers discussed in this Review are druggable enzymes, further research will create new opportunities to modulate chromatin remodeling activity in combination with other cellular signaling pathways to promote myelin repair after injury. Fully understanding the chromatin modification mechanisms mediated by individual epigenetic enzymes will facilitate the identification of drug targets for myelin repair. Given that HDAC inhibitors are generally well-tolerated and have been clinically approved for treating certain cancers [121,122], their neuroprotective and regenerative actions indicate that they may be considered as therapeutic agents to treat brain injury. Due to the intricate, distinct and nuanced functions of individual HDAC family members, their effects should be taken into account in light of the stage- and context-specific expression of individual HDACs. Specifically, given that HDAC ablation reveals highly specific functions of individual HDACs, such complexity calls for careful interpretation of study outcomes that employ pan-HDAC inhibitors. It is also worth underlining the potential impact of HDAC inhibitors on psychological functions such as cognition, learning, memory and mood in young, healthy animals [123].
Additionally, a better understanding of the molecular framework underlying chromatin remodelers Brg1 and CHD7 in oligodendrocyte lineage development may help to identify signaling pathways and molecules as therapeutic targets for promoting myelin recovery in patients with BRG1-associated diseases, CHARGE syndrome or CHD8-associated autisms, respectively. These chromatin remodelers also play a critical role in oligodendrocyte lineage development, and will likely have some part to play in the future treatment of MS. Indeed, identification and development of selective small molecule compounds that modulate chromatin modifying enzyme activity such that the myelinogenic program is activated will be essential for treating the patients afflicted by nerve injury or demyelinating diseases, such as MS.
Acknowledgments
The authors thank Jiajia Wang and Xanyao Zhou for drawing figures, and Dr. Ed Hurlock for critical reading. This study was funded in part by grants from the US National Institutes of Health R01NS072427 and R01NS075243 to QRL, the National Multiple Sclerosis Society (NMSS-4727) to QRL
Footnotes
MR. ALEXANDER GREGATH (Orcid ID : 0000-0002-3956-1249)
References
- 1.Franklin RJ, Gallo V. The translational biology of remyelination: past, present, and future. Glia. 2014;62:1905–15. doi: 10.1002/glia.22622. [DOI] [PubMed] [Google Scholar]
- 2.Bei F, et al. Restoration of Visual Function by Enhancing Conduction in Regenerated Axons. Cell. 2016;164:219–32. doi: 10.1016/j.cell.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toosy AT, Mason DF, Miller DH. Optic neuritis. Lancet Neurol. 2014;13:83–99. doi: 10.1016/S1474-4422(13)70259-X. [DOI] [PubMed] [Google Scholar]
- 4.He X, et al. A Histone Deacetylase 3-Dependent Pathway Delimits Peripheral Myelin Growth and Functional Regeneration. Nature Medicine. 2018;25 doi: 10.1038/nm.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuchero JB, Barres BA. Intrinsic and extrinsic control of oligodendrocyte development. Curr Opin Neurobiol. 2013;23:914–20. doi: 10.1016/j.conb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emery B, Lu QR. Transcriptional and Epigenetic Regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harb Perspect Biol. 2015;7:a020461. doi: 10.1101/cshperspect.a020461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wegner M. A matter of identity: transcriptional control in oligodendrocytes. J Mol Neurosci. 2008;35:3–12. doi: 10.1007/s12031-007-9008-8. [DOI] [PubMed] [Google Scholar]
- 8.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 9.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Kanagaratham C, Radzioch D. Chromatin Remodelling. Chromatin Remodelling 2013 [Google Scholar]
- 11.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–87. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 12.Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Couture JF, Trievel RC. Histone-modifying enzymes: encrypting an enigmatic epigenetic code. Curr Opin Struct Biol. 2006;16:753–60. doi: 10.1016/j.sbi.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Prakash K, Fournier D. Evidence for the implication of the histone code in building the genome structure. Biosystems. 2017 doi: 10.1016/j.biosystems.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Marmorstein R, Zhou MM. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harbor Perspectives in Biology. 2014;6 doi: 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Research. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiang CM. Nonequivalent response to bromodomain-targeting BET inhibitors in oligodendrocyte cell fate decision. Chem Biol. 2014;21:804–6. doi: 10.1016/j.chembiol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gacias M, et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem Biol. 2014;21:841–854. doi: 10.1016/j.chembiol.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hota SK, Bruneau BG. ATP-dependent chromatin remodeling during mammalian development. Development. 2016;143:2882–97. doi: 10.1242/dev.128892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Runge JS, Raab JR, Magnuson T. Epigenetic Regulation by ATP-Dependent Chromatin-Remodeling Enzymes: SNF-ing Out Crosstalk. Curr Top Dev Biol. 2016;117:1–13. doi: 10.1016/bs.ctdb.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yodh J. ATP-Dependent Chromatin Remodeling. Adv Exp Med Biol. 2013;767:263–95. doi: 10.1007/978-1-4614-5037-5_13. [DOI] [PubMed] [Google Scholar]
- 22.Mayes K, Qiu Z, Alhazmi A, Landry JW. ATP-dependent chromatin remodeling complexes as novel targets for cancer therapy. Adv Cancer Res. 2014;121:183–233. doi: 10.1016/B978-0-12-800249-0.00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–6. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Serna IL, Ohkawa Y, Imbalzano AN. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nature Reviews Genetics. 2006;7:461–473. doi: 10.1038/nrg1882. [DOI] [PubMed] [Google Scholar]
- 25.Yoo AS, Crabtree GR. ATP-dependent chromatin remodeling in neural development. Curr Opin Neurobiol. 2009;19:120–6. doi: 10.1016/j.conb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto S, Banine F, Feistel K, Foster S, Xing R, Struve J, Sherman LS. Brg1 directly regulates Olig2 transcription and is required for oligodendrocyte progenitor cell specification. Dev Biol. 2016;413:173–87. doi: 10.1016/j.ydbio.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Y, et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013;152:248–61. doi: 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto S, et al. Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev Biol. 2006;289:372–83. doi: 10.1016/j.ydbio.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 29.Bischof M, Weider M, Kuspert M, Nave KA, Wegner M. Brg1-dependent chromatin remodelling is not essentially required during oligodendroglial differentiation. J Neurosci. 2015;35:21–35. doi: 10.1523/JNEUROSCI.1468-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303:390–6. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- 31.Goldman SA, Kuypers NJ. How to make an oligodendrocyte. Development. 2015;142:3983–95. doi: 10.1242/dev.126409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) EMBO J. 1998;17:6979–91. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–84. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells [see comments] Science. 2000;289:1754–7. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- 35.Kondo T, Raff M. Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev. 2004;18:2963–72. doi: 10.1101/gad.309404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han P, et al. Epigenetic response to environmental stress: Assembly of BRG1-G9a/GLP-DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta. 2016;1863:1772–81. doi: 10.1016/j.bbamcr.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao C, et al. Chromatin-remodelling factor Brg1 regulates myocardial proliferation and regeneration in zebrafish. Nat Commun. 2016;7:13787. doi: 10.1038/ncomms13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, Weissman BE, Sherman LS. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res. 2005;65:3542–7. doi: 10.1158/0008-5472.CAN-04-3554. [DOI] [PubMed] [Google Scholar]
- 39.Moyon S, et al. Functional Characterization of DNA Methylation in the Oligodendrocyte Lineage. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moyon S, Ma D, Huynh JL, Coutts DJC, Zhao C, Casaccia P, Franklin RJM. Efficient Remyelination Requires DNA Methylation. eNeuro. 2017;4 doi: 10.1523/ENEURO.0336-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokpor G, Xie Y, Rosenbusch J, Tuoc T. Chromatin Remodeling BAF (SWI/SNF) Complexes in Neural Development and Disorders. Front Mol Neurosci. 2017;10:243. doi: 10.3389/fnmol.2017.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He D, et al. lncRNA Functional Networks in Oligodendrocytes Reveal Stage-Specific Myelination Control by an lncOL1/Suz12 Complex in the CNS. Neuron. 2017;93:362–378. doi: 10.1016/j.neuron.2016.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He D, Marie C, Zhao C, Kim B, Wang J, Deng Y. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nature. 2016 doi: 10.1038/nn.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin DM. Chromatin remodeling in development and disease: focus on CHD7. PLoS Genet. 2010;6:e1001010. doi: 10.1371/journal.pgen.1001010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2014;48:334–42. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- 47.Doi T, Ogata T, Yamauchi J, Sawada Y, Tanaka S, Nagao M. Chd7 Collaborates with Sox2 to Regulate Activation of Oligodendrocyte Precursor Cells after Spinal Cord Injury. J Neurosci. 2017;37:10290–10309. doi: 10.1523/JNEUROSCI.1109-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manning BJ, Yusufzai T. The ATP-dependent chromatin remodeling enzymes CHD6, CHD7, and CHD8 exhibit distinct nucleosome binding and remodeling activities. J Biol Chem. 2017;292:11927–11936. doi: 10.1074/jbc.M117.779470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batsukh T, et al. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum Mol Genet. 2010;19:2858–66. doi: 10.1093/hmg/ddq189. [DOI] [PubMed] [Google Scholar]
- 50.Stolerman ES, Smith B, Chaubey A, Jones JR. CHD8 intragenic deletion associated with autism spectrum disorder. Eur J Med Genet. 2016;59:189–94. doi: 10.1016/j.ejmg.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Cotney J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katayama Y, et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature. 2016;537:675–679. doi: 10.1038/nature19357. [DOI] [PubMed] [Google Scholar]
- 53.Bernier R, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158:263–76. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deoni SC, Zinkstok JR, Daly E, Ecker C, Consortium M.A. Williams SC, Murphy DG. White-matter relaxation time and myelin water fraction differences in young adults with autism. Psychol Med. 2015;45:795–805. doi: 10.1017/S0033291714001858. [DOI] [PubMed] [Google Scholar]
- 55.Hardan AY, Fung LK, Frazier T, Berquist SW, Minshew NJ, Keshavan MS, Stanley JA. A proton spectroscopy study of white matter in children with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2016;66:48–53. doi: 10.1016/j.pnpbp.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Casanova MF. Neuropathological and genetic findings in autism: the significance of a putative minicolumnopathy. Neuroscientist. 2006;12:435–41. doi: 10.1177/1073858406290375. [DOI] [PubMed] [Google Scholar]
- 57.Boddaert N, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e4415. doi: 10.1371/journal.pone.0004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thiagalingam S, Cheng KHH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Annals of the New York Academy of Sciences. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 59.Glaser KB. HDAC inhibitors: Clinical update and mechanism-based potential. Biochemical Pharmacology. 2007;74:659–671. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 60.Lyssiotis CA, Walker J, Wu C, Kondo T, Schultz PG, Wu X. Inhibition of histone deacetylase activity induces developmental plasticity in oligodendrocyte precursor cells. Proceedings of the National Academy of Sciences. 2007;104:14982–14987. doi: 10.1073/pnas.0707044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:10333–10345. doi: 10.1523/JNEUROSCI.22-23-10333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conway GD, O’Bara MA, Vedia BH, Pol SU, Sim FJ. Histone deacetylase activity is required for human oligodendrocyte progenitor differentiation. Glia. 2012;60:1944–53. doi: 10.1002/glia.22410. [DOI] [PubMed] [Google Scholar]
- 64.Swiss VA, Nguyen T, Dugas J, Ibrahim A, Barres B, Androulakis IP, Casaccia P. Identification of a Gene Regulatory Network Necessary for the Initiation of Oligodendrocyte Differentiation. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0018088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. The Journal of Cell Biology. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye F, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the β-catenin–TCF interaction. Nature Neuroscience. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, Morrisey EE, Millar SE. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell. 2010;19:807–18. doi: 10.1016/j.devcel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cunliffe VT, Casaccia-Bonnefil P. Histone deacetylase 1 is essential for oligodendrocyte specification in the zebrafish CNS. Mechanisms of Development. 2006;123:24–30. doi: 10.1016/j.mod.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Zhang L, et al. Hdac3 Interaction with p300 Histone Acetyltransferase Regulates the Oligodendrocyte and Astrocyte Lineage Fate Switch. Dev Cell. 2016;36:316–30. doi: 10.1016/j.devcel.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castelo-Branco G, et al. Neural stem cell differentiation is dictated by distinct actions of nuclear receptor corepressors and histone deacetylases. Stem Cell Reports. 2014;3:502–15. doi: 10.1016/j.stemcr.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dai J, Bercury KK, Jin W, Macklin WB. Olig1 Acetylation and Nuclear Export Mediate Oligodendrocyte Development. J Neurosci. 2015;35:15875–93. doi: 10.1523/JNEUROSCI.0882-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Ruijter A, van Gennip AH, Caron HN, Kemp S, van Kuilenburg A. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochemical Journal. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He Y, et al. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen S, Yao X, Li Y, Saifudeen Z, Bachvarov D, El-Dahr SS. Histone deacetylase 1 and 2 regulate Wnt and p53 pathways in the ureteric bud epithelium. Development. 2015;142:1180–92. doi: 10.1242/dev.113506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hermanson O, Jepsen K, Rosenfeld MG. N-CoR controls differentiation of neural stem cells into astrocytes. Nature. 2002;419:934–9. doi: 10.1038/nature01156. [DOI] [PubMed] [Google Scholar]
- 79.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89:349–56. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 80.Jepsen K, et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell. 2000;102:753–63. doi: 10.1016/s0092-8674(00)00064-7. [DOI] [PubMed] [Google Scholar]
- 81.Greer CB, Tanaka Y, Kim YJ, Xie P, Zhang MQ, Park IH, Kim TH. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015;13:1444–55. doi: 10.1016/j.celrep.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–7. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou J, et al. A Sequentially Priming Phosphorylation Cascade Activates the Gliomagenic Transcription Factor Olig2. Cell Rep. 2017;18:3167–3177. doi: 10.1016/j.celrep.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic Activity Associated with Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Molecular Cell. 2000;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 85.Noack M, Leyk J, Richter-Landsberg C. HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia. 2014;62:535–547. doi: 10.1002/glia.22624. [DOI] [PubMed] [Google Scholar]
- 86.Richter-Landsberg C. Protein aggregate formation in oligodendrocytes: tau and the cytoskeleton at the intersection of neuroprotection and neurodegeneration. Biol Chem. 2016;397:185–94. doi: 10.1515/hsz-2015-0157. [DOI] [PubMed] [Google Scholar]
- 87.Revollo JR, Li X. The ways and means that fine tune Sirt1 activity. Trends Biochem Sci. 2013;38:160–7. doi: 10.1016/j.tibs.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vaquero A, Scher M, Erdjument-Bromage H, Tempst P, Serrano L, Reinberg D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450:440–444. doi: 10.1038/nature06268. [DOI] [PubMed] [Google Scholar]
- 89.Stein LR, Imai Si. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO Journal. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jablonska B, et al. Sirt1 regulates glial progenitor proliferation and regeneration in white matter after neonatal brain injury. Nat Commun. 2016;7:13866. doi: 10.1038/ncomms13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li W, et al. Sirtuin 2, a Mammalian Homolog of Yeast Silent Information Regulator-2 Longevity Regulator, Is an Oligodendroglial Protein That Decelerates Cell Differentiation through Deacetylating α-Tubulin. The Journal of Neuroscience. 2007;27:2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Molecular Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 93.Roth AD, Ivanova A, Colman DR. New observations on the compact myelin proteome. Neuron Glia Biology. 2005;2:15. doi: 10.1017/S1740925X06000068. [DOI] [PubMed] [Google Scholar]
- 94.Tang B, Chua C. SIRT2, tubulin deacetylation, and oligodendroglia differentiation. Cell Motility and the Cytoskeleton. 2008;65:179–182. doi: 10.1002/cm.20253. [DOI] [PubMed] [Google Scholar]
- 95.Ji S, Doucette RJ, Nazarali AJ. Sirt2 is a novel in vivo downstream target of Nkx2.2 and enhances oligodendroglial cell differentiation. Journal of Molecular Cell Biology. 2011;3:351–359. doi: 10.1093/jmcb/mjr009. [DOI] [PubMed] [Google Scholar]
- 96.Thangaraj MP, Furber KL, Gan JK, Ji S, Sobchishin L, Doucette JR, Nazarali AJ. RNA-binding Protein Quaking Stabilizes Sirt2 mRNA during Oligodendroglial Differentiation. J Biol Chem. 2017;292:5166–5182. doi: 10.1074/jbc.M117.775544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu H, Hu Q, D’Ercole JA, Ye P. Histone deacetylase 11 regulates oligodendrocyte-specific gene expression and cell development in OL-1 oligodendroglia cells. Glia. 2009;57:1–12. doi: 10.1002/glia.20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Douvaras P, Rusielewicz T, Kim K, Haines JD, Casaccia P, Fossati V. Epigenetic Modulation of Human Induced Pluripotent Stem Cell Differentiation to Oligodendrocytes. International Journal of Molecular Sciences. 2016;17:614. doi: 10.3390/ijms17040614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kassis H, Chopp M, Liu XS, Shehadah A, Roberts C, Zhang ZG. Histone deacetylase expression in white matter oligodendrocytes after stroke. Neurochem Int. 2014;77:17–23. doi: 10.1016/j.neuint.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim HJ, Chuang DM. HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am J Transl Res. 2014;6:206–23. [PMC free article] [PubMed] [Google Scholar]
- 101.Liu XS, et al. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience. 2012;220:313–21. doi: 10.1016/j.neuroscience.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang G, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A. 2015;112:2853–8. doi: 10.1073/pnas.1501441112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1–uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hernández-Jiménez M, Hurtado O, Cuartero MI. Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke. 2013 doi: 10.1161/STROKEAHA.113.001715. [DOI] [PubMed] [Google Scholar]
- 105.Wang P, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate–activated kinase pathway. Annals of Neurology. 2011;69:360–374. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- 106.Wang S, et al. Cellular NAD Replenishment Confers Marked Neuroprotection Against Ischemic Cell Death. Stroke. 2008;39:2587–2595. doi: 10.1161/STROKEAHA.107.509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ying W, Wei G, Wang D, Wang Q, Tang X, Shi J, Zhang P, Lu H. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front Biosci. 2007;12:2728–34. doi: 10.2741/2267. [DOI] [PubMed] [Google Scholar]
- 108.Rafalski VA, et al. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nature cell biology. 2013;15:614–624. doi: 10.1038/ncb2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pedre X, Mastronardi F, Bruck W, López-Rodas G, Kuhlmann T, Casaccia P. Changed Histone Acetylation Patterns in Normal-Appearing White Matter and Early Multiple Sclerosis Lesions. The Journal of Neuroscience. 2011;31:3435–3445. doi: 10.1523/JNEUROSCI.4507-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Camelo S, et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 111.Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:3602–9. doi: 10.1167/iovs.07-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shindler KS, Ventura E, Dutt M, Elliott P, Fitzgerald DC, Rostami A. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J Neuroophthalmol. 2010;30:328–39. doi: 10.1097/WNO.0b013e3181f7f833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dincman TA, Beare JE, Ohri SS, Gallo V, Hetman M, Whittemore SR. Histone deacetylase inhibition is cytotoxic to oligodendrocyte precursor cells in vitro and in vivo. Int J Dev Neurosci. 2016;54:53–61. doi: 10.1016/j.ijdevneu.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 114.Wang Y, Wu X, Zhong Y, Shen J, Wu X, Ju S, Wang X. Effects of histone deacetylase inhibition on the survival, proliferation and migration of Schwann cells, as well as on the expression of neurotrophic factors and genes associated with myelination. Int J Mol Med. 2014;34:599–605. doi: 10.3892/ijmm.2014.1792. [DOI] [PubMed] [Google Scholar]
- 115.Jacob C, et al. HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat Neurosci. 2011;14:429–36. doi: 10.1038/nn.2762. [DOI] [PubMed] [Google Scholar]
- 116.Chen Y, et al. HDAC-mediated deacetylation of NF-κB is critical for Schwann cell myelination. Nature Neuroscience. 2011;14:437–441. doi: 10.1038/nn.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brugger V, Duman M, Bochud M, Munger E, Heller M, Ruff S, Jacob C. Delaying histone deacetylase response to injury accelerates conversion into repair Schwann cells and nerve regeneration. Nat Commun. 2017;8:14272. doi: 10.1038/ncomms14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Limpert AS, Bai S, Narayan M, Wu J, Yoon SO, Carter BD, Lu QR. NF-kappaB forms a complex with the chromatin remodeler BRG1 to regulate Schwann cell differentiation. J Neurosci. 2013;33:2388–97. doi: 10.1523/JNEUROSCI.3223-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weider M, et al. Chromatin-remodeling factor Brg1 is required for Schwann cell differentiation and myelination. Dev Cell. 2012;23:193–201. doi: 10.1016/j.devcel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 120.Marathe HG, Mehta G, Zhang X, Datar I, Mehrotra A, Yeung KC, de la Serna IL. SWI/SNF enzymes promote SOX10- mediated activation of myelin gene expression. PLoS One. 2013;8:e69037. doi: 10.1371/journal.pone.0069037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bickhart DM, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22:778–90. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]