

Summary
Respiratory syncytial virus (RSV) is the most common cause of hospitalization due to bronchiolitis in infants. Although the mechanisms behind this association are not completely elucidated, they appear to involve an excessive immune response causing lung pathology. Understanding the host response to RSV infection may help in the identification of targets for therapeutic intervention. We infected in‐vitro human monocyte‐derived dendritic cells (DCs) with RSV and analysed various aspects of the cellular response. We found that RSV induces in DCs the expression of CD38, an ectoenzyme that catalyses the synthesis of cyclic ADPR (cADPR). Remarkably, CD38 was under the transcriptional control of RSV‐induced type I interferon (IFN). CD38 and a set of IFN‐stimulated genes (ISGs) were inhibited by the anti‐oxidant N‐acetyl cysteine. When CD38‐generated cADPR was restrained by 8‐Br‐cADPR or kuromanin, a flavonoid known to inhibit CD38 enzymatic activity, RSV‐induced type I/III IFNs and ISGs were markedly reduced. Taken together, these results suggest a key role of CD38 in the regulation of anti‐viral responses. Inhibition of CD38 enzymatic activity may represent an encouraging approach to reduce RSV‐induced hyperinflammation and a novel therapeutic option to treat bronchiolitis.
Keywords: inflammation, interferons, monocyte‐derived dendritic cells, respiratory syncytial virus
Abbreviations
- 8‐Br‐cADPR
8‐Bromo‐cADP ribose
- cADPR
cyclic adenosine diphosphate ribose
- IFN
interferon
- ISGs
IFN‐stimulated genes
- LRTI
lower respiratory tract infection
- MDDC
monocyte‐derived dendritic cells
- NAC
N‐acetyl‐cysteine
- NAD+
nicotinamide adenine dinucleotide
- RSV
respiratory syncytial virus
- SOD2
superoxide dismutase 2
Introduction
Human respiratory syncytial virus (RSV) is a negative single‐stranded RNA virus of the Pneumoviridae family, previously classified in the Paramyxoviridae family.1 RSV is the commonest cause of lower respiratory tract infection (LRTI) in children and the major cause of infantile bronchiolitis: up to 70% of newborn infants become infected during their first year of life, and virtually all children by 2 years of age have been infected by RSV.2 In most cases the infection does not cause serious damage; however, some individuals, such as preterm infants, are at higher risk for developing severe bronchiolitis with viral spreading to the lower respiratory tract. High morbidity, high hospitalization rates and significant mortality rates are features of RSV‐associated disease.3 Multiple epidemiological studies have clearly demonstrated that RSV bronchiolitis is frequently associated with subsequent persistent wheezing, childhood asthma or both.4, 5 Neither a safe vaccine nor effective therapy are yet available.6, 7
The causes of severe RSV disease are not fully understood, but might involve both magnitude and quality of the host immune response. RSV infection initiates a cascade of events leading to the recruitment and activation of immune effectors, including monocyte/macrophages and dendritic cells (DCs) which, in turn, trigger the production of cytokines and chemokines, which orchestrate and potentiate the immune response.8 However, when exacerbated, the immune response to RSV may lead to tissue damage and to the establishment of bronchiolitis.9
In this scenario, a crucial role is played by DCs that act as a portal for virus invasion as well as potent antigen‐presenting cells, positioned to link innate to adaptive immune responses.10 Indeed, some studies suggest that myeloid DCs, along with macrophages, are the major source of type I interferons (IFNs) during pulmonary viral infections.11 There is also evidence that DCs, along with epithelial cells, produce IFN lambda (i.e. type III IFN) in response to RSV.12 The induction of types I and III IFNs, of IFN‐stimulated genes (ISGs) and of proinflammatory cytokines and chemokines are hallmarks of anti‐RSV immunity.13, 14 It has been shown that type I IFN production in response to RSV infection not only induces an anti‐viral state, but also operates to amplify proinflammatory responses in the respiratory tract. This process is a natural component of the early inflammatory response to RSV infection, designed to keep the virus under control until adaptive immunity is activated to clear the virus.15 However, type I IFN activation must be tightly regulated in order to reduce viral load but not allow excessive inflammatory cytokine and chemokine production. It is possible to hypothesize that the onset of RSV‐induced bronchiolitis in some human patients is related to genetically or environmentally driven alterations to this balance.15
As well as the inflammatory response, RSV induces oxidative stress, defined as a disruption of the pro‐oxidant/antioxidant balance,16 with cell death, loss of immune function, increased viral replication and inflammatory response, thereby contributing to pathogenesis.
In a previous study, we have shown that RSV triggers type I IFNs expression in monocyte‐derived DCs (MDDCs).17 We also showed that RSV infection induces up‐regulation of surface CD38, a marker of DCs activation induced by multiple inflammatory stimuli.18 CD38 is a multifunctional ectoenzyme that catalyses the synthesis of adenosine diphosphate ribose (ADPR) and cyclic ADPR (cADPR), second messengers involved in the regulation of cytoplasmic Ca2+ fluxes.19, 20, 21 CD38 also influences both innate and adaptive immune responses by regulating the trafficking of cells (e.g. neutrophils, DCs) to the sites of inflammation 22 and multiple aspects related to DCs maturation, such as chemotaxis and transendothelial migration, longevity, interleukin (IL)‐12 production and T helper type 1 (Th1) polarization.18
Here, we investigated the role of CD38 in the regulation of cellular responses to RSV in infected MDDCs with the aim of identifying possible lines of clinical intervention to ameliorate RSV‐associated LRTI treatment.
Materials and methods
Ethics statement
This study was conducted according to the principles expressed in the Declaration of Helsinki. Blood donors provided written informed consent.
Chemicals
8‐Br‐cADPR was from Santa Cruz Biotechnology (Dallas, TX); nicotinamide adenine dinucleotide (NAD)+, potassium dihydrogen phosphate (KH2PO4), acetonitrile [high‐performance liquid chromatography (HPLC)‐grade reagent], kuromanin, N‐acetylcysteine (NAC), erythro‐9‐(2‐hydroxy‐3‐nonyl) adenine (EHNA; adenosine deaminase inhibitor), dypiridamole (phosphodiesterase inhibitor) and levamisole (alkaline phosphatase inhibitor) were all from Sigma‐Aldrich (St Louis, MO). All the chemical reagents used were of analytical grade.
Purification and culture of MDDCs
Human monocytes were purified from peripheral blood of healthy blood donors (courtesy of Dr Girelli, ‘Centro Trasfusionale Policlinico Umberto I’, University La Sapienza, Rome, Italy) using Ficoll gradients (lympholyte‐H; Cedarlane, Burlington, ON). CD14 cells were purified by anti‐CD14 monoclonal antibody (mAb)‐conjugated magnetic microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and were cultured in the presence of human recombinant granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and IL‐4 (50 ng/ml each; Immunological Sciences, Rome, Italy); after 6 days immature MDDCs were washed and analysed using cytofluorimetry for the expression of the surface markers CD11c, CD14, CD83 and CD38 employing the fluorochrome‐conjugated mAbs (BD Biosciences, San Jose, CA). MDDCs were used in the experiments if > 90% CD11c and < 5% CD14. All the experiments were performed with MDDCs obtained from different donors.17
Preparation of RSV stocks and MDDCs infection
Plaque‐purified human RSV [type A2 strain from the American Type Culture Collection (ATCC)] was grown in human epithelial type 2 (HEp‐2) cells.23 Mock‐infected HEp‐2 cells were similarly processed to generate the mock control stock preparation. Viral titres were determined by plaque assay.
MDDCs (106 cell/ml) were incubated with RSV at multiplicity of infection (MOI) equal to 1. After 2 hr at 37°C cells were extensively washed and incubated in 1 ml of complete medium at 37°C, 5% CO2 for 20 hr post‐infection (p.i.). As negative control, an equal volume of mock control medium was added to MDDCs.
In some experiments, cells were pretreated for 20 min with kuromanin (100 μ m), 8‐Br‐cADPR (5 μg/ml), NAC (2 mm) or a combination of human anti‐human IFN (IFN)‐α and IFN‐β antibody (National Institutes of Health, Bethesda, ND). The inhibitors were re‐added at the same concentration to the culture medium after the infection and the washing steps.
Apoptosis assay
Apoptosis was measured in RSV‐infected MDDCs after 20 hr in the presence of NAC or 8 Br‐cADPR using an annexin V apoptosis detection kit (BD Biosciences). The assay was carried out following the manufacturer's protocol. Briefly, infected cells were incubated with different dilutions of NAC or 8‐Br‐cADPR for 20 hr. Cells were harvested, washed in phosphate‐buffered saline (PBS), pelleted and resuspended in binding buffer; 100 μl of the cell suspension (105 cells) was then mixed with 5 μl of fluorescein isothiocyanate (FITC) annexin V and 5 μl propidium iodide (PI). The samples were kept in the dark and incubated for 15 min at room temperature prior to the addition of binding buffer. Samples were analysed by cytofluorimetry to detect apoptotic cells and dead cells.
TaqMan‐based real‐time reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA was extracted at 4 and 20 hr p.i. from RSV‐infected and mock‐infected MDDCs using the total RNA Purification Kit (Norgen Biotek Corp., Thorold, ON) and was retrotranscribed as previously described.17 Quantitative real‐time PCR was carried out with a Viia7 real‐time PCR system (Applied Biosystems, Foster City, CA). Primers and probes for each gene were added to the Probes Master Mix at 500 and 250 nm, respectively, in a final volume of 20 μl.17, 24 The housekeeping gene β‐glucuronidase (or human β‐actin) was used as internal control. All the determinations were performed in duplicate. Gene expression values were calculated by the comparative Ct (ΔΔCT) method. Fold changes in gene expression levels were calculated by comparison with the gene expression in mock samples, which were assigned an arbitrary value of 1.
RSV amplification by RT–PCR
Total RNA was extracted from RSV‐infected and mock‐infected MDDCs and reverse transcription was carried out as described by Loebbermann and colleagues25 using 1 μg of total RNA for cDNA synthesis. Quantitative PCR (qPCR) specific for the RSV L gene was performed using 900 nm forward primer (5′‐GAACTCAGTGTAGGTAGAATGTTTGCA‐3′), 300 nm reverse primer (5′‐TTCAGCTATCATTTTCTCTGCCAAT‐3′) and 175 nm probe (5′‐6‐carboxyfluorescein‐TTTGAACCTGTCTGAACATTCCCGGTT‐6‐carboxytetramethylrhodamine‐3′). Copy numbers were determined from standard curves of pCDNA3 containing a fragment of the RSV L‐gene.
Adenosine generation assay
MDDCs were infected with RSV in the presence or absence of kuromanin, as described above. After the infection, cells (1 × 106/ml) were suspended in AIM V serum‐free medium (Invitrogen, Carlsbad, CA) containing a mix of EHNA, dipyridamole and levamisole and incubated for 15 min at 37°C, 5% CO2. NAD+ (100 m) was then added and cells were incubated for 30 min at 37°C. Following centrifugation (700 g; 5 min at 4°C) supernatants were collected in tubes containing 1 ml of ice‐cold acetonitrile to stabilize adenosine. Supernatants were evaporated by speed‐vacuum, reconstituted in mobile‐phase buffer, and assayed by HPLC.
Chromatographic analysis was performed with an HPLC system (Beckman Gold 126/166NM; Beckman Coulter, Brea, CA) equipped with a reverse‐phase column (Hamilton C18, 5 μm; 250 × 4·5 mm). Separation of nucleotides and nucleosides was performed using a mobile‐phase buffer (0·125 m citric acid and 0·025 m KH2PO4), pH 5·1 with 8% acetonitrile fot 10 min at a flow rate of 0·8 ml/min. UV absorption spectra were measured at 254 nm. HPLC‐grade standards used to calibrate the signals were dissolved in AIM V serum‐free medium, pH 7·4, 0·2 μm sterile‐filtered and injected in a buffer volume of 20 μl.
The retention time (R t, in min) of standard ADPR was: 3·2. Peak integration was performed using karat software (Beckman Coulter). Quantitative measurements were inferred by comparing the peak area of samples with calibration curve for peak area of standard compound.
Statistical analysis
All results are expressed as mean ± standard error of the mean (SEM). Statistical significance was determined using a two‐tailed, unpaired Student's t‐test (*P < 0·05, **P < 0·01). Values of P < 0·05 and P < 0·01 were considered significant (prism software; Graph‐Pad Software Inc., San Diego, CA).
Results
CD38 expression is regulated by type‐I IFNs in RSV‐infected MDDCs
In a previous study we showed that RSV infection induces a significant up‐regulation of CD38 surface expression as well as IFN‐β production in MDDCs.17 Here, we first analysed whether the increased expression of CD38 at the cell surface is paralleled by increased transcription of the CD38 gene. Infection of MDDCs by RSV was checked by measuring RSV L gene transcripts, as an index of viral copies (data not shown). As shown in Fig. 1a, RSV induced a significant increase of CD38 transcription compared to mock treated cells, mostly at 20 hr p.i. The levels of CD38 surface expression, checked in parallel by flow cytometry, confirmed the gene expression data (Fig. 1b). As type I IFNs activate CD38 expression,26 we then asked whether the observed activation of CD38 was dependent upon type I IFNs produced by MDDCs in response to RSV infection. With this aim, we performed in‐vitro infection experiments in the presence of a combination of neutralizing antibodies directed against type I IFNs (e.g. IFN‐α and ‐β). As shown in Fig. 1c, blocking of type I IFNs signalling significantly reduced RSV‐induced CD38 gene expression at 20 hr p.i., confirming our hypothesis.
Figure 1.
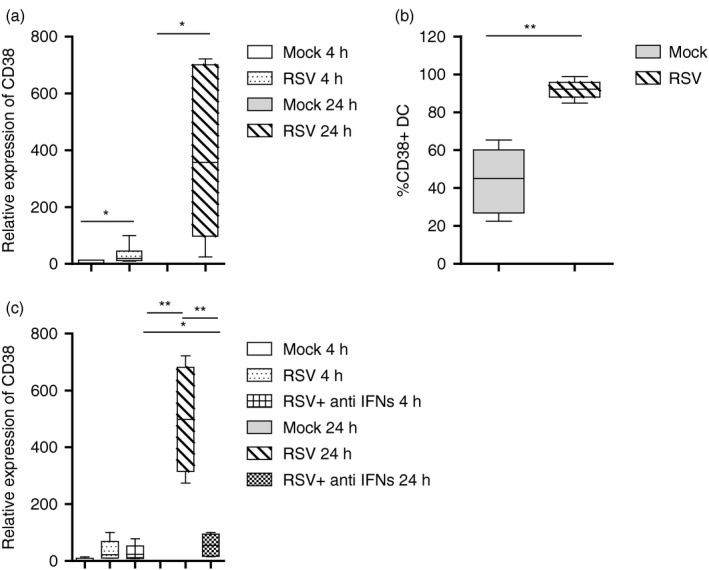
Respiratory syncytial virus (RSV)‐induced CD38 expression in monocyte‐derived dendritic cells (MDDCs). MDDCs were infected with RSV [multiplicity of infection (MOI) of 1] and in (c), cells were also treated with a combination of human anti‐human interferon (IFN)‐α and ‐β antibody. Mock‐infected cells were added as control. In (a) and (c), total RNA was extracted at the indicated time‐points, and the kinetics of mRNA expression for CD38 was evaluated by real‐time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). The mRNA transcripts were normalized with respect to the endogenous reference (human β‐actin) sample. Data were expressed as fold increase versus mock‐treated cells at 4 hr [mean ± standard error of the mean (SEM) of seven or five experiments in (a) and (c), respectively]. (b) RSV‐infected MDDCs, after 20 hr, were analysed for the CD38 surface marker. Fluorescence data are as percentage of positive cells. Values are expressed as mean ± SEM of five independent experiments. For all, statistical significance was determined using a two‐tailed, unpaired Student's t‐test (*P < 0·05, **P < 0·01).
CD38 and IFNs‐dependent response induced by RSV are linked to oxidative stress
CD38 has been described to play a role in the regulation of oxidative stress and accumulation of reactive oxygen species in eukaryotic cells,27, 28 a process that is also a hallmark of RSV infection.26 The observation that, in airway epithelial cells, RSV infection modulates the oxidative balance through up‐regulation of superoxide dismutase 2 (SOD2),29 prompted us to investigate whether the same event occurs in MDDCs. A strong induction of SOD2 expression was recorded in RSV‐infected MDDCs, at either 4 or 20 hr p.i. Remarkably, in the presence of NAC, SOD2 gene expression decreased, reaching a significant reduction at 20 hr p.i. (Fig. 2a).
Figure 2.
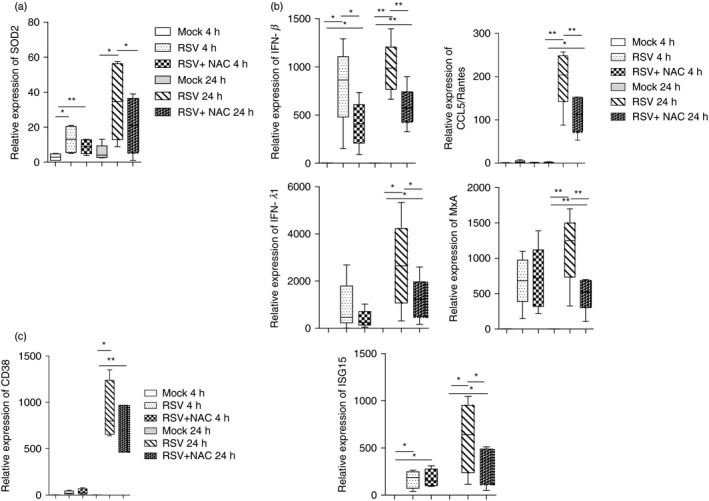
NAC treatment and genes expression in respiratory syncytial virus (RSV)‐infected monocyte‐derived dendritic cells (MDDCs). Cells were infected with RSV [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, some samples were pretreated with N‐acetyl‐cysteine (NAC) (2 mm) for 20 min. Mock‐infected cells were added as control. After infection, cells were washed, incubated in fresh medium and, where indicated, NAC was re‐added. At the indicated time‐points, total RNA was extracted and kinetics of mRNA expression for all genes analysed was evaluated by real‐time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). The mRNA transcripts were normalized with respect to the endogenous reference sample. Data were expressed as fold increase versus mock‐treated cells at 4 hr [mean ± standard error of the mean (SEM) of five experiments in (a) and (b), mean ± standard error of the mean (SEM) of four experiments in (c)]. Statistical significance was determined using a two‐tailed, unpaired Student's t‐test (*P < 0·05, **P < 0·01).
We measured the expression of an array of IFNs and ISGs that are key markers of the anti‐RSV innate immune response, such as IFN‐β, IFN‐λ1/IL‐29, chemokine (C‐C motif) ligand 5/regulated on activation, normal T cell expressed and secreted (CCL5/RANTES), myxovirus resistance A (MxA) and IFN‐stimulated gene 15 (ISG15)26, 30, 31, 32, 33 in MDDCs infected in the presence or absence of NAC. As shown in Fig. 2b, all the genes analysed were up‐regulated in infected MDDCs; in particular we noticed an early expression of IFN‐β, while IFN‐λ1 and ISGs peaked at a later time‐point p.i., suggesting two separate waves of transcription.
We found that NAC statistically reduced IFN‐β induction at 4 and 20 hr p.i., while the expression of all ISGs was reduced at the later time‐point in RSV‐infected MDDCs. Finally, down‐regulation of CD38 expression was recorded in NAC‐treated MDCCs at 20 hr p.i. (Fig. 2c). The fact that ISGs are modulated by RSV and NAC is proof of IFN protein expression. Such evidence is important, as mRNA levels may not necessarily be correlated with the protein levels.
A cADPR antagonist inhibits RSV‐driven proinflammatory responses in human MDDCs
Once established that either oxidative stress or type‐I IFN responses drive the up‐regulation of CD38 in RSV‐infected MDDCs, we sought to determine whether the enzymatic functions of CD38 are involved in the activation of anti‐viral and proinflammatory responses.
To this aim, MDDCs were infected with RSV in the presence of 8‐Bromo‐cADP ribose (8‐Br‐cADPR), a cell‐permeant cADPR antagonist34 capable of inhibiting the release of intracellular Ca2+ mediated by CD38/cADPR.35 In order to verify that the concentrations of NAC and 8‐Br‐cADPR used in our experiments did not affect cell viability, we performed two apoptosis assays. RSV‐infected MDDCs were treated with three increasing doses of NAC (0·2, 2 and 10 mm) or 8‐Br‐cADPR (2, 5 and 20 μg/ml) and, after 20 hr, we assessed the levels of early apoptosis (annexin V‐positive cells) and late apoptosis/dead (annexin V/PI‐positive cells) of the cells. Our results have shown that the NAC and 8‐Br‐cADPR doses utilized did not affect the cell viability (Supporting information, Fig. S1). As shown in Fig. 3, we found that 8‐Br‐cADPR significantly reduced the expression of all the genes analysed, at 20 hr p.i. The evidence that gene expression inhibition occurred at the later time‐point corroborates the notion that CD38 might be induced during the second wave of RSV‐induced gene transcription through the action of early produced IFN‐β. Overall, these findings identify a putative functional axis linking oxidative stress, IFN/ISGs production and CD38, which might be involved in a positive feedback loop that enhances proinflammatory responses during RSV infection.
Figure 3.
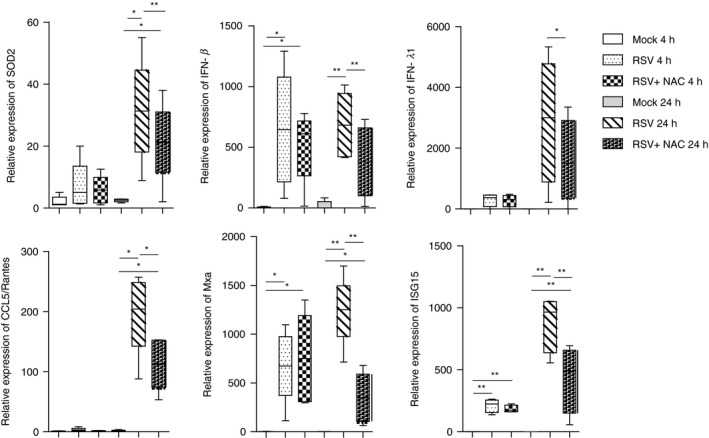
8‐Bromo‐cADP ribose (8‐Br‐cADPR) treatment and genes expression in respiratory syncytial virus (RSV) ‐infected monocyte‐derived dendritic cells (MDDCs). Cells were infected with RSV [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, some samples were pretreated with 8‐Br‐cADPR (5 μg/ml) for 20 min. Mock‐infected cells were added as control. After infection, cells were washed, incubated in fresh medium and, where indicated, 8‐Br‐cADPR was re‐added. At the indicated time‐points, total RNA was extracted and kinetics of mRNA expression for all genes analysed was evaluated by real‐time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). The mRNA transcripts were normalized with respect to the endogenous reference sample. Data were expressed as fold increase versus mock‐treated cells at 4 hr [mean ± standard error of the mean (SEM) of five experiments]. For all, statistical significance was determined using a two‐tailed, unpaired Student's t test (*P < 0·05, **P < 0·01).
Inhibition of CD38 activity by kuromanin decreases RSV‐induced responses in MDDCs
To demonstrate that RSV‐induced CD38 is catalytically active in MDDCs, we performed a set of experiments to directly measure its enzymatic activity. To this end, we evaluated the accumulation of ADPR, a product of the CD38 catalytic reaction, in the supernatants of RSV‐infected cells. The experiments were performed in the presence of the natural anthocyanin kuromanin, a CD38 ADP‐ribosylation cyclase inhibitor, which has been shown to inhibit CD38 enzymatic activity.36 After the addition of exogenous nicotinamide adenine dinucleotide (NAD+) to RSV‐infected MDDCs as substrate for CD38, we observed an ADPR accumulation. Remarkably, the presence of kuromanin decreased ADPR levels, with about 50% inhibition (Fig. 4a). The inhibitory effect of kuromanin treatment was not caused by decreased CD38 expression (Supporting information, Fig. S2). As previously reported with other flavonoids with similar activity,37 no cell toxicity was observed when MDDCs were exposed to 100 μ m kuromanin for up to 20 hr (not shown). Additionally, to exclude that the inhibitory effect of kuromanin on ADPR accumulation in MDCCs could be related to its interference on RSV entry or replication, the levels of RSV, expressed as number of viral copies/μg RNA, in both kuromanin‐treated or ‐untreated RSV‐infected cells were analysed. As shown in Fig. 4c, the amount of RSV copies/μg RNA had the same order of magnitude, independently of kuromanin addition to the MDCCs cultures.
Figure 4.
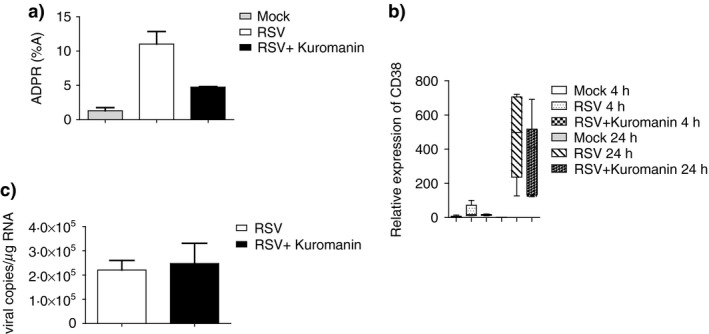
Inhibition of CD38 enzymatic activity by a natural anthocyanin. Cells were infected with respiratory syncytial virus (RSV) [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, some samples were pretreated with kuromanin (100 μ m) for 20 min. Mock‐infected cells were added as control. After infection, cells were washed, incubated in fresh medium and, where indicated, kuromanin was re‐added. (a) RSV‐infected monocyte‐derived dendritic cells (MDDCs), at 20 hr, were harvested and incubated with 0·1 mm NGD +. Generation of ADPR was measured by high‐performance liquid chromatography (HPLC). Quantitative measurements were inferred by comparing the peak area of the samples with the calibration curve for peak area of standard compound. Values were expressed as the percentage of the peak area and are the mean ± standard error of the mean (SEM) of three independent experiments. (b) RSV‐infected MDDCs were harvested at the indicated time‐points, and total RNA was extracted. The kinetics of mRNA expression for all genes analysed was evaluated by real‐time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). The mRNA transcripts were normalized with respect to the endogenous reference sample. Data were expressed as fold increase versus mock‐treated cells at 4 hr and are mean ± SEM of five experiments. (c) RSV‐infected MDDCs were collected at 20 hr p.i. and viral copies for each sample were determined byRT –PCR (mean ± SEM of two experiments).
Given the efficient inhibition on CD38 enzymatic activity exerted by kuromanin, we examined the effects of kuromanin on RSV‐induced oxidative and IFN responses. Results are summarized in Fig. 5. Expression of all the genes tested was reduced, and only IFN‐λ1 reduction failed to reach statistical significance. These results confirm that CD38 plays a central role in fostering anti‐viral responses.
Figure 5.
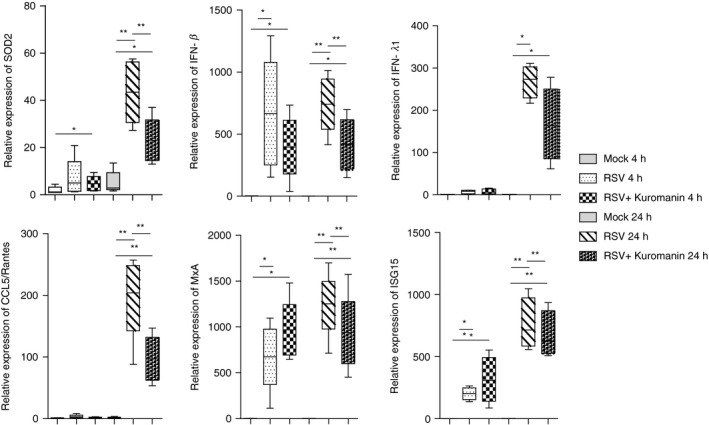
Kuromanin treatment and genes expression in respiratory syncytial virus (RSV)‐infected monocyte‐derived dendritic cells (MDDCs). Cells were infected with RSV [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, some samples were pretreated with kuromanin (100 μ m) for 20 min. Mock‐infected cells were added as control. After infection, cells were washed, incubated in fresh medium and, where indicated, kuromanin was re‐added. At the indicated time‐points, total RNA was extracted and kinetics of mRNA expression for all genes analysed was evaluated by real‐time quantitative reverse transcription–polymerase chain reaction (qRT–PCR). The mRNA transcripts were normalized with respect to the endogenous reference (human β‐actin) sample. Data were expressed as fold increase versus mock‐treated cells at 4 hr and are the mean ± standard error of the mean (SEM) of five experiments. For all, statistical significance was determined using a two‐tailed, unpaired Student's t‐test (*P < 0·05, **P < 0·01).
Discussion
Despite extensive attempts, an RSV vaccine is not yet available.6 Analogously, a safe and effective cure for RSV remains elusive: treatment consists of supportive care including supplemental oxygen and mechanical ventilation, while bronchodilators, corticosteroids and ribavirin have failed to show clear benefit.7 Immune‐prophylaxis with the neutralizing monoclonal antibody palivizumab is used to prevent RSV disease in extremely premature infants or those with congenital heart disease.38 Thus, new intervention strategies are urgently needed.
The current study explored our hypothesis that CD38 is at the centre of a functional axis linking the IFNs response and the oxidative stress induced by RSV. Taking advantage of a well‐established preclinical human MDDCs model, we analysed the role of CD38 in the context of RSV infection.
Type I IFNs are classically elicited in response to viral infection; they induce a vast array of ISGs whose activity impairs viral replication in infected cells.39 This cell‐intrinsic action plays a crucial role in protecting the lungs from spread of respiratory viruses.40 However, type I IFNs also play a key role in the initiation of lung inflammatory responses by inducing recruitment and activation of immune cells, a process that can cause detrimental immunopathology and contribute to disease severity.40 Here, we showed that IFN‐β is induced as early as 4 hr p.i. in RSV‐infected MDDCs; gene expression analysis also demonstrates that a set of typical ISGs is induced in a second wave of transcription. These genes include MxA and ISG15, both endowed with broad anti‐viral activity and markers of RSV infection,32, 33 and RANTES/CCL5, an inflammatory chemokine associated with increased likelihood of severe asthma after RSV lung disease.41 In addition, as the emerging role of type III IFNs during in RSV infection,24 the production of IFN‐λ1/IL‐29 was also measured. We also found that the CD38 gene is expressed in the second wave of RSV‐induced transcription. The association between RSV‐induced type I IFNs and CD38 de‐novo transcription, is witnessed by the finding that blocking type I IFNs significantly reduces RSV‐induced CD38 gene expression.
A key aspect of the present study is the finding that the enzymatic activity of CD38 is involved in the anti‐viral and proinflammatory responses induced by RSV. cADPR is one of the main products of CD38 catalytic activity and has been recognized as a principal second messenger involved in Ca2+ mobilization from intracellular stores.42 Moreover, the CD38/cADPR pathway plays an important role in several inflammatory processes.43, 44 Here, we found that the expression of RSV‐induced IFN‐β, IFN‐λ1/IL‐29, CCL5/RANTES, MxA and ISG15 is reduced significantly in MDDCs by the cADPR analogue 8‐Br‐cADPR.
Based on reports on the key role of oxidative stress in RSV pathology16,29 and on the activity of CD38 in other cellular settings, it is tempting to speculate that the IFN/CD38 axis activated by RSV in MDDCs could also be implicated in the regulation of oxidative responses. It is known that CD38 is involved in reactive oxygen species (ROS) production through cADPR.27, 45 Our results were in agreement with those reported by Hosakote et al.29 indeed, NAC treatment significantly decreased SOD2 gene expression, confirming that increased SOD2 expression is part of an unbalanced oxidative response caused by RSV infection. Notably, when RSV‐infected MDDCs were treated with 8‐Br‐cADPR, SOD2 expression was also markedly and significantly reduced. Moreover, when RSV‐infected MDDCs were treated with the anti‐oxidant NAC the reduction of RSV‐induced IFN‐β, as well as ISGs, was verified. Taken together, our results suggest the existence of a functional axis that, when unbalanced during RSV infection, may favour the onset of lung immunopathology; the CD38/cADPR pathway appears at the centre of a functional axis set at the crossroad between type I IFN‐dependent anti‐viral and proinflammatory responses and the oxidative burst. We hypothesize that, in RSV‐infected MDDCs, CD38‐induced cADPR opens ryanodine receptor Ca2+ channels, as shown in different settings.46 Elevations in Ca2+ may induce local production of ROS,47 fostering the activation of inflammatory and anti‐viral processes (Fig. 6). A directed demonstration that RSV‐induced CD38 is enzymatically active derives from the use of NAD+ as CD38 substrate. Indeed, after the addition of exogenous NAD+ to RSV‐infected MDDCs we observed an accumulation of ADPR. Further studies are needed to directly correlate CD38‐mediated cADPR with increased levels of intracellular Ca2+ and generation of ROS.
Figure 6.
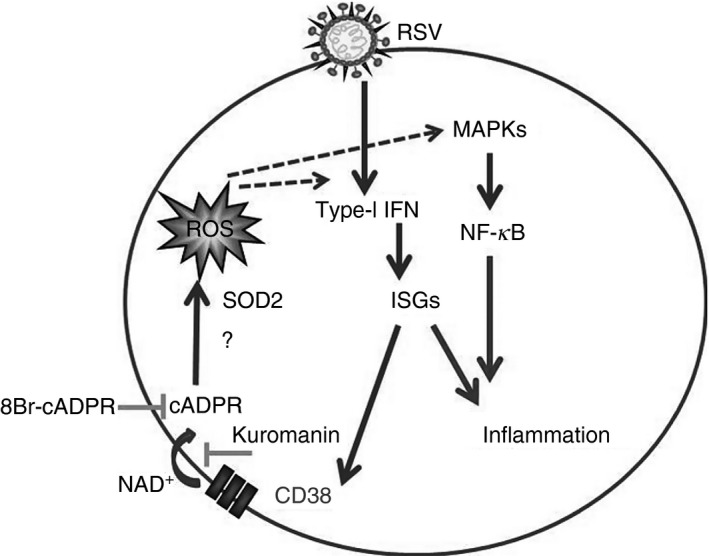
Schematic representation of the proposed functional axis between the CD38/cyclic adenosine diphosphate ribose (cADPR) pathway, type‐I interferon (IFN) response and the oxidative burst induced by respiratory syncytial virus (RSV) in monocyte‐derived dendritic cells (MDDCs). ROS: reactive oxygen species.
A consequence of the present findings is that inhibitors of CD38 enzymatic activities may prove therapeutically useful: an ideal candidate for this purpose is kuromanin.48, 49 Flavonoids, such as kuromanin, have been shown to inhibit CD38 enzymatic activities.36
Among the available molecules, kuromanin is active in the low micromolar range and has been recently proved to be a potent CD38 inhibitor in chronic lymphocytic leukaemia cells.50 At the same time, kuromanin is a natural product and its prospective use for human health appears to be safe. In RSV‐infected MDDCs, kuromanin inhibited CD38 enzymatic activity with approximately 50% reduction, restraining RSV‐induced proinflammatory and pro‐oxidant responses in MDDCs.
Similarly to what was observed with 8‐Br‐cADPR, addition of kuromanin decreased the expression of IFN‐β, as well as of ISGs and SOD2. In this regard it is important to note that kuromanin is endowed with anti‐oxidant activity,51 thus it is possible to envisage a twofold mechanism employed to prevent the onset of an exaggerated inflammatory response, a specific inhibition of the enzymatic activity of CD38 with consequent reduction in cADPR production; a more general anti‐oxidant effect by scavenging free radicals.
Other hints on the mechanisms through which kuromanin may exert its effects come from recent studies. An example comes from a recent study showing that kuromanin inhibits the production of proinflammatory cytokines in intestinal Caco‐2 cells through activation of the Nrf‐2 pathway.52 In this respect, it is known that RSV infection induces a reduction in nuclear and total cellular levels of the NRF2 protein, thus fostering oxidative damage.53 Further studies are required to investigate whether activation of Nrf2 by kuromanin also occurs in RSV‐infected MDDCs and whether it could be correlated with inhibition of the CD38/cADPR pathway.
In conclusion, the present study, for the first time, identified CD38 and cADPR as key players in the orchestration of cellular responses to RSV by infected MDDCs and possible targets for interventions aimed at controlling exacerbated lung responses. Kuromanin appears a promising candidate for its intrinsic features.
Author contributions
I.S. and G.F. designed the study, performed the experiments and wrote the paper; C.S., A.P., P.L. and A.L.H. performed the experiments; F.M. and C.M.A. contributed to study design. The authors thank Simonetta Pietrangeli for excellent technical assistance.
Disclosures
All the authors declare no commercial or financial conflicts of interest.
Supporting information
Figure S1. Cells were infected with respiratory syncytial virus (RSV) [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, samples were pretreated with the indicated concentrations of N‐acetyl‐cysteine (NAC) and 8‐Bromo‐cADP ribose (8Br) for 20 min.
Figure S2. Cells were infected with respiratory syncytial virus (RSV) [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, samples were pretreated with Kuromanin (100 μ m) for 20 min.
Acknowledgements
This work was supported by grants from faculty research funds of Sapienza University (2015/2016) to C.S. and A.P.
References
- 1. Afonso CL, Amarasinghe GK, Bányai K, Bào Y, Basler CF, Bavari S et al Taxonomy of the order Mononegavirales: update 2016. Arch Virol 2016; 161:2351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hall CB. The burgeoning burden of respiratory syncytial virus among children. Infect Disord Drug Targets 2012; 12:92–7. [DOI] [PubMed] [Google Scholar]
- 3. Meissner HC. Viral bronchiolitis in children. N Engl J Med 2016; 374:62–72. [DOI] [PubMed] [Google Scholar]
- 4. Koponen P, Helminen M, Paassilta M, Luukkaala T, Korppi M. Preschool asthma after bronchiolitis in infancy. Eur Respir J 2012; 39:76–80. [DOI] [PubMed] [Google Scholar]
- 5. Voraphani N, Stern DA, Wright AL, Guerra S, Morgan WJ, Martinez FD. Risk of current asthma among adult smokers with respiratory syncytial virus illnesses in early life. Am J Respir Crit Care Med 2014; 190:392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jorquera PA, Anderson L, Tripp RA. Understanding respiratory syncytial virus (RSV) vaccine development and aspects of disease pathogenesis. Expert Rev Vaccines 2016; 15:173–87. [DOI] [PubMed] [Google Scholar]
- 7. Ralston SL, Lieberthal AS, Meissner HC, Alverson BK, Baley JE, Gadomski AM et al American Academy of Pediatrics. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics 2014; 134:e1474–502 [Erratum in: Pediatrics 2015; 136:782]. [DOI] [PubMed] [Google Scholar]
- 8. González PA, Bueno SM, Carreño LJ, Riedel CA, Kalergis AM. Respiratory syncytial virus infection and immunity. Rev Med Virol 2012; 22:230–44. [DOI] [PubMed] [Google Scholar]
- 9. Farrag MA, Almajhdi FN. Human respiratory syncytial virus: role of innate immunity in clearance and disease progression. Viral Immunol 2016; 29:1–26. [DOI] [PubMed] [Google Scholar]
- 10. Neyt K, Lambrecht BN. The role of lung dendritic cell subsets in immunity to respiratory viruses. Immunol Rev 2013; 255:57–67. [DOI] [PubMed] [Google Scholar]
- 11. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E et al Alveolar macrophages are the primary interferon‐alpha producer in pulmonary infection with RNA viruses. Immunity 2007; 27:240–52. [DOI] [PubMed] [Google Scholar]
- 12. Hillyer P, Mane VP, Chen A, Dos Santos MB, Schramm LM, Shepard RE et al Respiratory syncytial virus infection induces a subset of types I and III interferons in human dendritic cells. Virology 2017; 504:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McNamara PS, Flanagan BF, Hart CA, Smyth RL. Production of chemokines in the lungs of infants with severe respiratory syncytial virus bronchiolitis. J Infect Dis 2005; 191:225–32. [DOI] [PubMed] [Google Scholar]
- 14. Mukherjee S, Lukacs NW. Innate immune responses to respiratory syncytial virus infection. Curr Top Microbiol Immunol 2013; 372:139–54. [DOI] [PubMed] [Google Scholar]
- 15. Goritzka M, Durant LR, Pereira C, Salek‐Ardakani S, Openshaw PJ, Johansson C. Alpha/beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J Virol 2014; 88:6128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garofalo RP, Kolli D, Casola A. Respiratory syncytial virus infection: mechanisms of redox control and novel therapeutic opportunities. Antioxid Redox Signal 2013; 18:186–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schiavoni I, Fedele G, Quattrini A, Bianco M, Schnoeller C, Openshaw PJ et al Live attenuated B. Pertussis BPZE1 rescues the immune functions of respiratory syncytial virus infected human dendritic cells by promoting Th1/Th17 responses. PLOS ONE 2014; 9:e100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frasca L, Fedele G, Deaglio S, Capuano C, Palazzo R, Vaisitti T et al CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood 2006; 107:2392–9. [DOI] [PubMed] [Google Scholar]
- 19. Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos‐Argumedo L, Parkhouse RM et al Formation and hydrolysis of cyclic ADP‐ribose catalyzed by lymphocyte antigen CD38. Science 1993; 262:1056–9. [DOI] [PubMed] [Google Scholar]
- 20. Guse AH, da Silva CP, Berg I, Skapenko AL, Weber K, Heyer P et al Regulation of calcium signaling in T lymphocytes by the second messenger cyclic ADP‐ribose. Nature 1999; 398:70–3. [DOI] [PubMed] [Google Scholar]
- 21. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E et al Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 2008; 88:841–86. [DOI] [PubMed] [Google Scholar]
- 22. Partida‐Sanchez S, Gasser A, Fliegert R, Siebrands CC, Dammermann W, Shi G et al Chemotaxis of mouse bone marrow neutrophils and dendritic cells is controlled by adp‐ribose, the major product generated by the CD38 enzyme reaction. J Immunol 2007; 179:827–39. [DOI] [PubMed] [Google Scholar]
- 23. Lee DC, Harker JA, Tregoning JS, Atabani SF, Johansson C, Schwarze J et al CD25+ natural regulatory T cells are critical in limiting innate and adaptive immunity and resolving disease following respiratory syncytial virus infection. J Virol 2010; 84:8790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Selvaggi C, Pierangeli A, Fabiani M, Spano L, Nicolai A, Papoff P et al Interferon lambda 1–3 expression in infants hospitalized for RSV or HRV associated bronchiolitis. J Infect 2014; 68:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loebbermann J, Schnoeller C, Thornton H, Thornton H, Durant L, Sweeney NP et al IL‐10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLOS ONE 2012; 7:e32371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bauvois B, Durant L, Laboureau J, Barthélémy E, Rouillard D, Boulla G et al Upregulation of CD38 gene expression in leukemic B cells by interferon types I and II. J Interferon Cytokine Res 1999; 19:1059–66. [DOI] [PubMed] [Google Scholar]
- 27. Lee S, Paudel O, Jiang Y, Yang XR, Sham JS. CD38 mediates angiotensin II‐induced intracellular Ca(2 + ) release in rat pulmonary arterial smooth muscle cells. Am J Respir Cell Mol Biol 2015; 52:332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guan XH, Liu XH, Hong X, Zhao N, Xiao YF, Wang LF et al CD38 deficiency protects the heart from ischemia/reperfusion injury through activating SIRT1/FOXOs‐mediated antioxidative stress pathway. Oxid Med Cell Longev 2016; 2016:7410257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hosakote YM, Liu T, Castro SM, Garofalo RP, Casola A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am J Respir Cell Mol Biol 2009; 41:348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS Jr, Bakaletz LO et al Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo . J Virol 2007; 81:9790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saito T, Deskin RW, Casola A, Häeberle H, Olszewska B, Ernst PB et al Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J Infect 1997; 175:497–504. [DOI] [PubMed] [Google Scholar]
- 32. González‐Sanz R, Mata M, Bermejo‐Martín J, Álvarez A, Cortijo J, Melero JA et al ISG15 is upregulated in respiratory syncytial virus infection and reduces virus growth through protein ISGylation. J Virol 2016; 90:3428–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engelmann I, Dubos F, Lobert PE, Houssin C, Degas V, Sardet A et al Diagnosis of viral infections using myxovirus resistance protein A (MxA). Pediatrics 2015; 135:e985–93. [DOI] [PubMed] [Google Scholar]
- 34. Walseth TF, Lee HC. Synthesis and characterization of antagonists of cyclic‐ADP‐ribose‐induced Ca2+ release. Biochim Biophys Acta 1993; 1178:235–42. [DOI] [PubMed] [Google Scholar]
- 35. White TA, Kannan MS, Walseth TF. Intracellular calcium signaling through the cADPR pathway is agonist specific in porcine airway smooth muscle. FASEB J 2003; 17:482–4. [DOI] [PubMed] [Google Scholar]
- 36. Kellenberger E, Kuhn I, Schuber F, Muller‐Steffner H. Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett 2011; 21:3939–42. [DOI] [PubMed] [Google Scholar]
- 37. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT et al Flavonoid apigenin is an inhibitor of the NAD+ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013; 62:1084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. American Academy of Pediatrics Committee on Infectious Diseases . American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 2014; 134:415–20. [DOI] [PubMed] [Google Scholar]
- 39. Reid E, Charleston B. Type I and III interferon production in response to RNA viruses. J Interferon Cytokine Res 2014; 34:649–58. [DOI] [PubMed] [Google Scholar]
- 40. Makris S, Pulsen M, Johansson C. Type I Interferons as regulators of lung inflammation. Front Immunol 2017; 8:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bacharier LB, Cohen R, Schweiger T, Yin‐Declue H, Christie C, Zheng J et al Determinants of asthma after severe respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol 2012; 130:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wei W, Graeff R, Yue J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca/2+ signaling pathway. World J Biol Chem 2014; 5:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choe CU, Lardong K, Gelderblom M, Ludewig P, Leypoldt F, Koch‐Nolte F et al CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLOS ONE 2011; 6:e19046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deaglio S, Robson SC. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol 2011; 61:301–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wei W, Lu Y, Hao B, Zhang K, Wang Q, Miller AL et al CD38 is required for neural differentiation of mouse embryonic stem cells by modulating reactive oxygen species. Stem Cells 2015; 33:2664–73. [DOI] [PubMed] [Google Scholar]
- 46. Partida‐Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B et al Cyclic ADP‐ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo . Nat Med 2001; 7:1209–16. [DOI] [PubMed] [Google Scholar]
- 47. Zhang F, Jin S, Yi F, Xia M, Dewey WL, Li PL. Local production of O2‐ by NAD(P)H oxidase in the sarcoplasmic reticulum of coronary arterial myocytes: cADPR‐mediated Ca2+ regulation. Cell Signal 2008; 20:637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Um MY, Ahn J, Ha TY. Hypolipidaemic effects of cyanidin 3‐glucoside rich extract from black rice through regulating hepatic lipogenic enzyme activities. J Sci Food Agric 2013; 93:3126–8. [DOI] [PubMed] [Google Scholar]
- 49. Francis FJ. Food colorants: anthocyanins. Crit Rev Food Sci Nutr 1989; 28:273–314. [DOI] [PubMed] [Google Scholar]
- 50. Vaisitti T, Audrito V, Serra S, Buonincontri R, Sociali G, Mannino E et al The enzymatic activities of CD38 enhance CLL growth and trafficking: implications for therapeutic targeting. Leukemia 2015; 29:356–68. [DOI] [PubMed] [Google Scholar]
- 51. Ciocoiu M, Badescu L, Miron A, Badescu M. The Involvement of a polyphenol‐rich extract of black chokeberry in oxidative stress on experimental arterial hypertension. Evid Based Complement Alternat Med 2013; 2013:912769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ferrari D, Speciale A, Cristani M, Fratantonio D, Molonia MS, Ranaldi G et al Cyanidin‐3‐O‐glucoside inhibits NF‐kB signaling in intestinal epithelial cells exposed to TNF‐α and exerts protective effects via Nrf2 pathway activation. Toxicol Lett 2016; 264:51–8. [DOI] [PubMed] [Google Scholar]
- 53. Komaravelli N, Tian B, Ivanciuc T, Mautemps N, Brasier AR, Garofalo RP et al Respiratory syncytial virus infection down‐regulates antioxidant enzyme expression by triggering deacetylation‐proteasomal degradation of Nrf2. Free Radic Biol Med 2015; 88:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cells were infected with respiratory syncytial virus (RSV) [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, samples were pretreated with the indicated concentrations of N‐acetyl‐cysteine (NAC) and 8‐Bromo‐cADP ribose (8Br) for 20 min.
Figure S2. Cells were infected with respiratory syncytial virus (RSV) [multiplicity of infection (MOI) of 1] for 2 hr. Where indicated, samples were pretreated with Kuromanin (100 μ m) for 20 min.