

Abstract
During evolution, individuals whose brains and bodies functioned well in a fasted state were successful in acquiring food, enabling their survival and reproduction. With fasting and extended exercise, liver glycogen stores are depleted and ketones are produced from adipose-cell-derived fatty acids. This metabolic switch in cellular fuel source is accompanied by cellular and molecular adaptations of neural networks in the brain that enhance their functionality and bolster their resistance to stress, injury and disease. Here, we consider how intermittent metabolic switching, repeating cycles of a metabolic challenge that induces ketosis (fasting and/or exercise) followed by a recovery period (eating, resting and sleeping), may optimize brain function and resilience throughout the lifespan, with a focus on the neuronal circuits involved in cognition and mood. Such metabolic switching impacts multiple signalling pathways that promote neuroplasticity and resistance of the brain to injury and disease.
Food scarcity has been a driving force for the evolution of nervous systems; indeed, even the most advanced capabilities of the human brain (imagination, creativity and language) arose via selection for individuals who were adept at cooperating to acquire and share food1–3. However, people in modern societies typically consume food three or more times each day. Every time they eat, the glycogen stores in their liver are replenished; liver glycogen provides 700–900 calories of glucose or energy, an amount that will last 10–14 hours in individuals who are not exercising. Subsequently, liver energy stores are depleted, circulating glucose levels remain low and adipose cells release fatty acids, which are converted in the liver to the ketone bodies β-hydroxybutyrate (BHB) and acetoacetate (AcAc), which are released into the blood and are used as energy substrates by neurons4,5 (FIG. 1). The transition from utilization of carbohydrates and glucose to fatty acids and ketones as the major cellular fuel source can be referred to as the ‘G-to-K switch’. Upon consumption of food after a fast, the major energy source for cells switches back to glucose (‘K-to-G switch’). Because exercise accelerates the depletion of liver glycogen stores, it hastens the onset of the G-to-K switch. For example, the metabolic switch may occur in someone who runs for 1 hour (during which time they use approximately 600 calories) beginning 4 hours after their most recent meal.
Figure 1. Biochemical pathways involved in the metabolic switch.

During fasting and sustained exercise, liver glycogen stores are depleted and lipolysis of triacylglycerols and diacylglycerols in adipocytes generates free fatty acids (FFAs), which are then released into the blood. The FFAs are transported into hepatocytes, where they are metabolized via β-oxidation to acetyl CoA, which is then used to generate the ketones acetone, acetoacetate (AcAc) and β-hydroxybutyrate (BHB). BHB and AcAc are transported from the blood into the brain and then into neurons via monocarboxylic acid transporters (MCTs) in the membranes of vascular endothelial cells and neurons. Within neurons, BHB and AcAc are metabolized to acetyl CoA, which then enters the tricarboxylic acid (TCA) cycle in mitochondria, resulting in the production of ATP and the reducing agents that transfer electrons to the electron transport chain. BHB can also upregulate the expression of brain-derived neurotrophic factor (BDNF) and may thereby promote mitochondrial biogenesis, synaptic plasticity and cellular stress resistance. In addition to blood-borne ketones, astrocytes are capable of ketogenesis, which may provide an important local source of BHB for neurons. Intermittent metabolic switching also increases insulin sensitivity, thereby enhancing uptake and utilization of glucose by neurons. Upon refeeding, ingested carbohydrates (CHO) and glucose stimulate release into the blood of the incretin hormone glucagon-like peptide 1 (GLP1) from enteroendocrine cells in the gut. GLP1 enhances clearance of glucose from the blood by stimulating insulin release from the pancreas and increases the insulin sensitivity of cells. GLP1 crosses the blood–brain barrier and can act directly on neurons to promote synaptic plasticity, enhance cognition and bolster cellular stress resistance. HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; GT, glucose transporter.
Beyond the switch in the major cellular fuel source, emerging findings are revealing remarkably complex and coordinated adaptations of the brain and body that enable the individual to maintain and even enhance their cognitive and physical performance for extended time periods in the fasted state6,7. In general, signalling pathways engaged when the G-to-K switch occurs serve to bolster cellular stress resistance and set the stage for subsequent cellular structural and functional plasticity that occurs after the K-to-G switch. We define intermittent metabolic switching (IMS) as scenarios in which an individual’s eating and exercise patterns result in periodic G-to-K switches. The most commonly employed IMS protocols in animals involve intermittent fasting (IF), including alternate-day fasting (ADF) and daily time-restricted feeding (TRF), and are described in BOX 1. Studies of animals and humans are revealing beneficial effects of IF on health beyond those resulting from caloric restriction (CR) without IMS8–11. Here, we review the molecular and cellular adaptations of neurons to IMS from the perspectives of brain evolution, neuroplasticity and resistance of the nervous system to injury and disease. We focus on brain regions involved in cognition, mood regulation and motor control; for those interested in the hypothalamus and neuroendocrine regulation of feeding behaviour and circadian rhythms, we refer readers to recent review articles12,13.
Box 1. Animal models of intermittent metabolic switching.
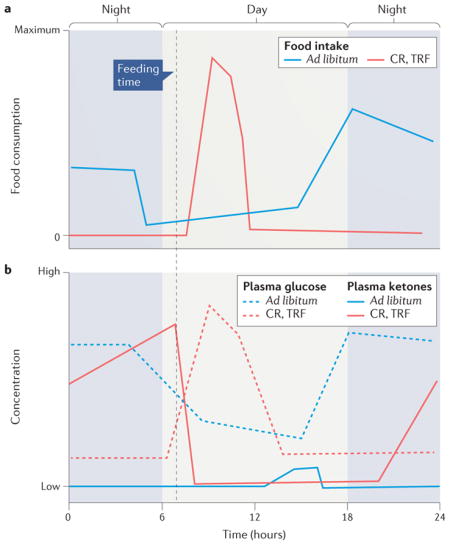
Alternate-day fasting (ADF) and daily time-restricted feeding (TRF) are the most widely employed intermittent fasting (IF) approaches for controlled frequent intermittent metabolic switching (IMS) in mice and rats. In ADF, the animals are deprived of food for 24 hours every other day8. For daily TRF, there are two different protocols: 1) animals are deprived of food for a specific time period each day (for example, 16 hours)9, and 2) animals are provided a daily allocation of an amount of food that is 30–40% less than their ad libitum consumption (that is, caloric restriction (CR)), in which case they typically consume all their food within 2 4 hours of their feeding time184–186. The figure shows depictions of food intake in rats or mice on 30–40% CR (TRF) during a 24-hour period (panel a) and the corresponding changes in plasma ketone and glucose concentrations (panel b; solid lines are ketone concentrations and dashed lines are glucose concentration). Animals on the CR diets consume all their daily food allotment within a 4 hour time window and therefore fast for 20 hours; during the last half of the fasting period, plasma ketone concentrations are elevated and the plasma glucose concentration remains low. The illustrations are based on data in several different studies, including those in REFS 184–187. Although plasma β-hydroxybutyrate levels are rarely measured in exercise studies, there are data showing that bouts of vigorous exercise (running or swimming) do elevate circulating ketone levels in rodents188,189.
We suggest that switching between time periods of negative energy balance (short fasts and/or exercise) and positive energy balance (eating and resting) can optimize general health and brain health. Illustrative of the sustained health benefits of IMS are enhanced insulin sensitivity, reduced abdominal fat, maintenance of muscle mass and reduced resting heart rate and blood pressure7. It is important to note that the short-term IF discussed herein is in contrast with long-term fasts of a week or more, which may have detrimental effects on muscle mass. Because sedentary, overindulgent lifestyles can increase the risk of several neurodegenerative and psychiatric disorders (see section on IMS and neurological disorders below), a better understanding of the neurobiology of IMS may lead to new approaches for improving brain health throughout the life course.
Neuronal adaptations to IMS
Behavioural adaptations
Experimental evidence supports the evolutionary principle that the brain and body perform at high levels in environments that demand IMS. Because many studies that have evaluated the effects of IF and exercise on cognition and mood did not include measurements of circulating ketones, we describe here only a few examples that either included ketone measurements or employed IF protocols known to involve IMS. Thirty years ago, Ingram et al. reported that mice maintained on a daily TRF schedule (40% CR; see BOX 1) beginning at weaning age exhibit no age-related learning and memory impairment and improved locomotor performance in middle and old age compared with control mice fed ad libitum14. When fasting was initiated in 14-month-old mice, their spatial navigation, working memory, strength and coordination at 22–25 months of age were superior to those of control mice fed ad libitum. Whereas mice fed ad libitum develop age-related hippocampus-dependent spatial learning and memory deficits, mice maintained on daily TRF (40% CR) during their adult life do not experience a decrement in cognition15. Daily TRF also ameliorates age-related anxiety-like behaviours in mice16. Middle-aged mice fed a very-low-calorie diet for 4 consecutive days every other week (during which ketones were elevated) for 7 months exhibited significantly better spatial learning and memory in the Barnes maze, and significantly higher recognition and working memory in the novel object recognition task and Y-maze, than mice continuously fed ad libitum17.
Evidence from human and animal studies has established that regular aerobic exercise can improve cognitive performance and can protect against anxiety and depression18,19. Cognitive challenges performed during exercise result in levels of enhancement of synaptic plasticity, as well as learning and memory, greater than either challenge alone20. However, in most studies of effects of exercise on the brain, ketone levels are not measured; therefore, it is not known whether IMS occurs. Nevertheless, from an evolutionary perspective, it is noteworthy that precise navigational decision-making (cognitive challenge) while rapidly traversing the landscape (running) in a food-deprived and/or fasted state would be critical for survival. Indeed, combining exercise with cognitive challenges and/or IF results in improvements in neuroplasticity and cognitive performance superior to those from the individual challenges alone21,22.
Neuronal network activity, synaptic plasticity and neurogenesis
Hippocrates wrote more than 2,400 years ago that fasting can prevent or lessen the severity of epilepsy, a disorder he called “the sacred disease” (REF. 23). During the past 50 years, it was established that ketones can constrain neuronal network activity and ketogenic diets can benefit many patients with epilepsy24. More recently, it was shown that rats maintained on ADF exhibit resistance to seizure-induced hippocampal damage and associated cognitive impairment25 and that IF can reduce seizure frequency in children who do not fully respond to a ketogenic diet26.
Although IMS elicited by IF and/or regular vigorous exercise can protect neuronal circuits against aberrant hyperexcitability, IMS also enhances structural and functional synaptic plasticity. Long-term potentiation (LTP) is an enduring activity-dependent increase in synaptic strength that occurs in response to repetitive stimulation of synapses and is believed to be a common cellular manifestation of learning and memory. Rats or mice that run or are fasted intermittently exhibit enhanced LTP at hippocampal synapses compared with sedentary animals fed ad libitum27–29. Daily TRF (22 hours of fasting every day) for 3 weeks improves performance in the Barnes maze test and increases dendritic spine density and LTP in CA1 neurons in rats30. IMS can also prevent age-related deficits in LTP31,32. IMS regimens involving daily TRF (30% CR) alone, or in combination with running-wheel exercise, result in increased dendritic spine density in hippocampal dentate granule neurons in wild-type mice and in hyperphagic diabetic mice21. The latter study demonstrated additive effects of dietary energy restriction and running on dentate neuron dendritic spine density, consistent with the notion that synaptic plasticity can be optimized by combining exercise with energy restriction.
New neurons are born continuously throughout adult life in the hippocampal dentate gyrus of mammals, including rodents, monkeys and humans. The newly generated neurons differentiate into granule neurons and receive synaptic inputs from other granule neurons, interneurons and neurons in the entorhinal cortex, basal forebrain (including the septum) and mammillary bodies33. Both running and IF can enhance neurogenesis, with running stimulating the proliferation of the stem cells33 and IF increasing the survival of the newly generated neurons34. Running also strengthens inputs of newly generated neurons from brain regions critical for spatial learning and memory, including the entorhinal cortex and basal forebrain (including the septum)35. However, the roles of the G-to-K switch in the effects of running and IF on neurogenesis remain to be determined.
Signalling pathways impacted by IMS
Neurotransmitters and neurotrophic factors
Glutamate is the major excitatory neurotransmitter in the CNS and, as such, is essential for all behaviours, including learning and memory, emotional responses, sensory input and integration and motor system activity. Glutamate receptor activation triggers Ca2+ influx into the post-synaptic neuron, which engages downstream pathways that mediate adaptive structural (dendritic spine growth and synaptogenesis) and functional (LTP and long-term depression) responses of neuronal circuits to environmental challenges. The downstream pathways involve Ca2+-responsive kinases such as calcium/calmodulin-dependent kinases and protein kinase C as well as transcription factors such as cAMP-responsive element-binding protein (CREB) and nuclear factor-κB (NF-κB)36,37. In rats, 18 hours of fasting results in CREB activation in hippocampal and entorhinal cortex neurons38. Running-wheel exercise also triggers upregulation of CREB in hippocampal and cerebral cortical cells in mice39. This pathway is highly conserved because fasting enhances long-term memory formation in multiple tasks in Drosophila melanogaster by a mechanism requiring CREB activation in mushroom body neurons40. Ketones, particularly BHB, upregulate GABAergic tone, which can protect against seizures41,42, and may mediate adaptive responses of neuronal circuits to IMS. The monoamine neurotransmitters serotonin, noradrenaline and dopamine may also play important roles in the effects of IMS on neuronal network activity43,44.
A robust and readily discernible effect of IMS on a neurotransmitter system involves brainstem cholinergic neurons that innervate the heart. Endurance athletes exhibit low resting heart rates and blood pressure and high heart rate variability (HRV; the variation in time intervals between individual heartbeats) as a result of increased activity of the brainstem cardiovagal neurons45. Similar enhancement of parasympathetic tone and consequent reductions in resting heart rate and blood pressure, and increased HRV, occur in response to ADF or daily TRF46,47. Recordings of cardiovagal neuronal activity in brain-derived neurotrophic factor (BDNF) heterozygous knockout mice suggest that BDNF mediates the enhancement of parasympathetic tone in response to IMS48.
BDNF plays pivotal roles in synaptic plasticity (synaptogenesis and LTP), neurogenesis and neuronal stress resistance49. BDNF likely plays important roles in behavioural and neuronal network adaptations to IMS because BDNF expression in cells in multiple regions is induced by running and IF49, and ablation of TrkB tyrosine kinase (also known as BDNF/NT3 growth factor receptor (NTRK2)) in hippocampal neural stem cells abolishes the ability of IMS to enhance neuroplasticity50. Insulin-like growth factor 1 (IGF1) signalling is also upregulated in response to IMS as demonstrated in studies of the effects of ketogenic diets, exercise and CR and/or IF on brain IGF1 signalling51–53. Two recent studies showed that BHB induces BDNF expression in hippocampal and cortical neurons in cell culture and in vivo54,55, suggesting that BHB is itself a signal that ‘alerts’ neurons in the brain that the metabolic switch has occurred.
Fibroblast growth factor 2 (FGF2) plays important roles in stimulating neural stem cell proliferation and regulating neurite outgrowth and cell survival during brain development56. FGF2 can also protect neurons against excitotoxic, metabolic and oxidative stress by mechanisms involving enhancement of cellular Ca2+ homeostasis and upregulation of antioxidant enzymes57,58. FGF2 levels increase in the cerebral cortex and striatum of mice in response to ADF59 and in the hippocampus in response to running-wheel exercise60. The formation and extinction of memories of fearful events have been reported to be mediated, in part, by FGF2, suggesting a role for FGF2 in the critical synaptic plasticity required for survival during periods of food scarcity61. However, whether and to what extent FGF2 signalling is critical for the enhancement of cognition, synaptic plasticity and neurogenesis by IMS remains to be determined.
Mitochondrial biogenesis and cellular stress-resistance pathways
In response to intermittent bouts of exercise, the number of well-functioning mitochondria in muscle cells increases in a process called mitochondrial biogenesis, which is mediated by reactive oxygen species (ROS), Ca2+/calmodulin-dependent protein kinase type IIγ (CaMKII), AMP kinase (AMPK; also known as 5′-activated protein kinase (PRKAA)) and NAD-dependent lysine deacetylase sirtuin 1 (SIRT1) and the transcription factor peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α; also known as PPARGC1A)62–64 (FIG. 2). Emerging findings suggest that IMS stimulates mitochondrial biogenesis in neurons and that such mitochondrial biogenesis enables synaptic plasticity. When mitochondrial biogenesis is inhibited by knockdown of PGC-1α in developing hippocampal neurons, basal synapse formation is reduced and the ability of BDNF to induce synaptogenesis is abolished65. PGC1α is also essential for the long-term maintenance of dendritic spines of hippocampal dentate granule neurons in adult mice65. Because BDNF expression is upreg-ulated in response to the G-to-K switch and BHB54,55, BDNF signalling may stimulate mitochondrial biogenesis to provide the increased numbers of mitochondria required to support the function of newly formed and potentiated synapses (FIG. 2). Interestingly, PGC1α can enhance BDNF expression66, suggesting a positive feedback signalling mechanism whereby BDNF induces PGC1α and vice versa. The increased activity in neuronal circuits that occurs during exercise and fasting may also contribute to mitochondrial biogenesis via a Ca2+–CaMKII–CREB–PGC1α pathway6,67.
Figure 2. Signalling pathways by which neurons respond to the metabolic switch during fasting and exercise.

Increased excitatory synaptic activity triggers Ca2+ influx through plasma membrane channels, resulting in the activation of multiple kinases, including calcium/calmodulin-dependent kinases (Ca2+/CaMK) and mitogen-activated protein kinases (MAPKs), nitric oxide synthase (NOS) and the protein phosphatase calcineurin. Neurotrophic factor signalling pathways are also engaged in response to fasting and exercise, resulting in the activation of signalling pathways involving phosphatidylinositol 3-kinase (PI3K), RACα serine/threonine-protein kinase (AKT) and MAPKs. These activity-dependent and neurotrophic-factor-dependent signalling pathways converge on transcription factors, including cAMP-responsive element-binding protein (CREB), nuclear regulatory factor 2 (NRF2; also known as NFE2L2), nuclear factor-κB (NF-κB) and myocyte-specific enhancer factor 2 (MEF2), which induce the expression of many different genes that encode proteins involved in cellular stress adaptation. Three genes that may be particularly important in the enhancement of neuroplasticity and stress resistance in response to intermittent metabolic switching are those encoding brain-derived neurotrophic factor (BDNF), NAD-dependent protein deacetylase sirtuin 3 (SIRT3) and peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α). SIRT3 localizes to the mitochondria, where it deacetylates proteins involved in antioxidant defence (superoxide dismutase [Mn], mitochondrial (SOD2)), energy production (isocitrate dehydrogenase [NADP], mitochondrial (IDH2); acetyl CoA synthase 2 (AceCS2; also known as ACSS1); and electron transport chain (ETC) proteins) and protection against apoptosis (cyclophilin D). PGC1 α is a key transcriptional regulator of mitochondrial biogenesis that acts, in part, by upregulating NRF1 and mitochondrial transcription factor A (TFAM), which, in turn, upregulate oxidative phosphorylation, mitochondrial DNA (mtDNA) replication and transcription and mitochondrial protein import. CREB, NRF2 and NF-κB induce the expression of antioxidant enzymes, DNA repair enzymes, anti-apoptotic proteins and protein chaperones and Ca2+-handling proteins. The ketone β-hydroxybutyrate (BHB), which is elevated during fasting and extended exercise, provides an energy substrate for neurons and induces the expression of BDNF. The reduction in availability of glucose and amino acids during fasting and exercise results in a reduction in the AMP:ATP ratio, which activates AMP kinase (AMPK) and, in turn, stimulates autophagy. The reduction in glucose and amino acid availability also reduces activation of mammalian target of rapamycin (mTOR) and further enhances the AMPK-driven autophagy pathway. AMPK activates the serine/threonine-protein kinase ULK1 (ULK1) complex, which stimulates engulfment of damaged proteins and organelles in autophagosomes, which, in turn, fuse with lysosomes. Collectively, activation of these different pathways in response to the metabolic switch bolsters neuronal bioenergetics, improves Ca2+ handling and protects against oxidative, excitotoxic and proteotoxic stress. AC, acetyl group; LC3-PE, light chain 3– phosphatidylethanolamine; NAM, nicotinamide adenine mononucleotide; OXPHOS, ETC complexes involved in oxidative phosphorylation.
The metabolic efficiency and stress tolerance of neuronal mitochondria can be enhanced by IMS. To illustrate this point, we highlight the mitochondrial NAD+-dependent protein deacetylase SIRT3, which is emerging as a key mediator of adaptive responses of neurons to bioenergetic challenges68. SIRT3 expression is upregulated in the cerebral cortical and hippocampal neurons of mice in response to running-wheel exercise by a mechanism requiring activation of NMDA glutamate receptors69. Deletion of SIRT3 from neurons renders them vulnerable to excitotoxicity and mitochondrial stress, whereas overexpression of SIRT3 is neuroprotective69. SIRT3-mediated deacetylation of proteins in the electron transport chain and tricarboxylic acid (TCA) cycle may enhance the efficiency of mitochondrial ATP production, whereas deacetylation of superoxide dismutase 2 reduces mitochondrial oxidative stress and deacetylation of cyclophilin D protects neurons against apoptosis69,70 (FIG. 2).
Mammalian target of rapamycin and autophagy
A highly conserved pathway by which cells regulate protein synthesis in response to fluctuations in nutrient availability (principally glucose and amino acids) involves a protein called mechanistic target of rapamycin (mTOR; also known as serine/threonine-protein kinase mTOR (MTOR) and mammalian target of rapamycin)71,72. In the fed state, activation of mTOR by upstream nutrient-sensing proteins activates ribosomal protein S6 kinase to increase general protein synthesis and activates sterol regulatory element-binding protein (SREBP1; also known as SREBF1) to increase lipid synthesis. Conversely, during fasting and extended exercise, when the metabolic switch is on, mTOR activity decreases. In essence, cells are in a ‘growth mode’ when mTOR is active and in a ‘conserve-resources mode’ when the mTOR pathway is inactive. During fasting and sustained exercise, the mTOR pathway is inhibited and autophagy is stimulated in a wide range of tissues, including those in the nervous system73,74. The metabolic shift to ketogenesis not only is associated with the conserve-resources mode of cells but may also be causally involved because BHB can stimulate autophagy75. Indeed, fasting or treatment of animals with the mTOR inhibitor rapamycin can extend lifespan and healthspan in several different organisms, including mice7,76. Conversely, lack of IMS may impair autophagy and increase the risk of neurodegenerative disorders77,78.
Autophagy is upregulated in cerebral cortical and cerebellar neurons in response to fasting73, and genetic and pharmacological manipulations of mTOR and autophagy alter synaptic plasticity and neurogenesis in ways consistent with the involvement of these pathways in neuronal network adaptations to IMS73. mTOR mediates the local synthesis of proteins in dendrites that is required for long-term changes in synaptic strength that are believed to be critical for learning and memory79–81, and mTOR is also involved in some forms of synaptic plasticity82–84. Because IF and exercise enhance synaptic plasticity and cognition, it will be important to establish the specific contributions of the regulation of mTOR and autophagy pathways to the underlying structural and functional adaptations of neuronal networks to IMS.
AMPK is a cellular energy status responsive enzyme (activated when the ratio of ATP to AMP and/or ADPdecreases) that mediates the downregulation of mTOR and upregulation of autophagy in response to fasting and vigorous exercise in muscle cells85,86. Recent findings suggest that AMPK also mediates adaptive responses of neuronal networks to IMS. Daily treadmill training in rats results in activation of AMPK in hippocampal cells87, and pharmacological inhibition of AMPK enhances contextual fear memory in mice88. AMPK activity is increased in response to fasting and is required for fasting-induced dendritic spine formation and synaptic functional plasticity in hypothalamic arcuate nucleus neurons89. Mice treated with an AMPK agonist exhibit enhanced cognition and motor system function, consistent with roles for AMPK in the beneficial effects of IMS on neuroplasticity90. Although intermittent AMPK activation resulting from physiological bioenergetic challenges can enhance neuroplasticity, sustained AMPK activation can impair axonal and dendritic plasticity91, consistent with the importance of a recovery period for optimal neuroplasticity (FIG. 3).
Figure 3. Model for how intermittent metabolic switching may optimize brain performance and increase resistance to injury and disease.

Intermittent metabolic switching (IMS) involves repeating time periods of a bioenergetic challenge (fasting and/or exercise) when the metabolic switch is on (that is, liver glycogen stores are depleted and ketones are produced) and recovery periods (eating, resting and sleeping) when the metabolic switch is off. When the metabolic switch is on, levels of circulating ketones, ghrelin and myokines are elevated, whereas levels of glucose, leptin, insulin and pro-inflammatory cytokines are maintained at low levels. Adaptive responses of neurons to fasting and exercise include utilization of ketones as an energy substrate; increased expression of brain-derived neurotrophic factor (BDNF) and fibroblast growth factor 2 (FGF2); activation of the transcription factors cAMP-responsive element-binding protein (CREB) and peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α), which induce the expression of genes involved in synaptic plasticity, neurogenesis and mitochondrial biogenesis; and upregulation of autophagy and DNA repair pathways. During the bioenergetic challenge, activity of the mammalian target of rapamycin (mTOR; also known as serine/threonine-protein kinase mTOR (MTOR) and mechanistic target of rapamycin) pathway and global protein synthesis are reduced in neurons, and levels of pro-inflammatory cytokines in the brain are reduced. Collectively, the pathways activated when the metabolic switch is on enhance neuronal stress resistance and bolster repair and recycling of damaged molecules. When the metabolic switch is off, circulating levels of glucose, leptin, insulin and pro-inflammatory cytokines increase, whereas levels of ketones, ghrelin and myokines decrease. In brain cells, the mTOR pathway is active, protein synthesis increases and mitochondrial biogenesis occurs during the recovery period. The activation state of neurotrophic signalling and adaptive cellular stress-response pathways may decrease when the metabolic switch is off. Cycles of IMS may optimize brain health by promoting synaptic plasticity and neurogenesis, improving cognition, mood, motor performance and autonomic nervous system (ANS) function and bolstering resistance of neurons to injury and neurodegenerative disease. SIRT, NAD-dependent protein deacetylase sirtuin.
IMS-associated peripheral signals
Multiple neuroactive signalling molecules are released into the blood from peripheral organs in response to the G-to-K metabolic switch. The ‘hunger hormone’ ghrelin is among the most intensively studied of such signals because of its effects on hypothalamic regulation of food intake. Ghrelin is produced in a subpopulation of cells in the gut from which it is released into the blood in response to fasting, whereas food intake inhibits its release. Ghrelin and the additional peripheral signals generated when the metabolic switch is on also affect the plasticity and resilience of neuronal circuits throughout the brain, including those involved in motivation and cognition92,93. Ghrelin enhances hippocampal synaptic plasticity, neurogenesis and hippocampus-dependent learning and memory by direct actions on hippocampal cells and by stimulating serotonergic neurons in the brainstem raphe nucleus, which innervate the hippocampus94. Ghrelin can also have an anxiolytic effect on mice95. However, it is unclear whether ghrelin mediates the beneficial effects of IMS on neuroplasticity and behaviour.
Several proteins released from skeletal muscle during exercise, so-called myokines, may contribute to the CNS adaptations associated with IMS27. Interleukin 6 (IL-6) is best known as a mediator of immune cell responses to infectious agents and tissue damage, but IL-6 is also produced in and released from muscle cells in response to vigorous exercise and fasting and increases the insulin sensitivity of muscle cells96,97. Studies of IL-6-knockout mice have revealed important roles for IL-6 in CNS neuroplasticity, including regulation of hippocampus-dependent learning and memory98. Insulin-like growth factor I (IGF1) and fibroblast growth factor 2 (FGF2) are two neurotrophic factors that can enhance neuroplasticity and protect neurons against metabolic and oxidative stress99,100. IGF1 and FGF2 are produced by neurons and astrocytes but are also myokines produced in response to fasting and exercise101,102. Although the effects of circulating FGF2 on neuroplasticity are unclear, studies in which circulating IGF1 is selectively neutralized suggest an important role for peripheral-organ-derived IGF1 in the beneficial effects of IMS on brain neuroplasticity. Running stimulates IGF1 transport from the blood into neurons in the brain, and this uptake is associated with running-induced increases of neuronal network activity and BDNF expression103. Neutralization of circulating IGF1 blocks the abilities of exercise to modulate hippocampal neuronal network activity and protect neurons against excito-toxicity104. Nevertheless, the involvement of peripheral-organ-derived IGF1 in the beneficial effects of metabolic switching on the brain is complex because although exercise increases circulating IGF1 levels, fasting reduces circulating IGF1 levels by reducing production of IGF1 in the liver. However, similar to the prominent decrease in circulating insulin levels and the associated increase in sensitivity of insulin receptors to insulin in response to IMS, it is possible that fasting increases the sensitivity of the IGF1 receptor signalling pathway to IGF1.
Originally touted for its ability to ‘brown’ white fat, thereby increasing its thermogenic potential, the protein irisin is released from muscle cells in response to exercise and has been reported to enter the brain and upregulate BDNF expression66. However, it remains to be determined whether irisin is particularly important in the enhancement of neuroplasticity and stress resistance conferred by IMS. A recent study provided evidence that the enzyme cathepsin B is an exercise-induced peripheral signal that contributes to the cognitive benefits of exercise105. Best known for its role as a lysosomal protease, plasma cathepsin B levels are increased in response to exercise in mice, monkeys and humans, and circulating cathepsin B levels are positively correlated with aerobic fitness and recall memory in humans105. Hippocampus-dependent spatial memory is impaired in cathepsin-B-deficient mice, and these mice also lack the normal exercise-induced enhancement of hippocampal neurogenesis and spatial memory105. The mechanism by which exercise induces cathepsin B release from muscle cells is unknown, but it might involve stimulation of autophagic flux.
Emerging findings are revealing that BHB regulates neuronal gene transcription in ways that promote synaptic plasticity and cellular stress resistance. BHB induces the expression of BDNF in cultured cerebral cortical neurons, an effect that occurs only when glucose levels are fairly low62. Running-wheel exercise increases circulating BHB levels in mice, and intraventricular infusion of BHB induces Bdnf gene transcription in hippocampal cells55. BHB may induce Bdnf gene transcription by inhibiting histone deacetylases (HDACs) that normally repress Bdnf expression55,106. Given that CR, fasting and exogenous ketone supplementation increase histone acetylation in peripheral mouse tissue and that exercise decreases histone deacetylation in the hippocampus, it is likely that the inhibitory effects of ketones on HDACs are amplified when exercise occurs in the fasted state107. In addition, via actions in the mitochondria, BHB triggers the activation of the cytoplasmic transcription factor NF-κB in neurons, which then translocates to the nucleus and induces Bdnf expression54. Such findings suggest that BHB is not only a biomarker of the metabolic switch and an energy source for neurons but is also a peripheral signal that activates neuronal signalling pathways that enhance synaptic plasticity, cognition and neuronal stress resistance. Indeed, it was reported that oral administration of a BHB ester to rats for 5 days improves their spatial learning and memory and enhances their endurance in a treadmill test108. In addition to their roles as energy substrates and signalling molecules, BHB and AcAc are also precursors to membrane lipids in brain cells, including neurons and, notably, the oligodendrocytes109, suggesting a benefit of IMS for axonal myelination.
IMS and acute CNS injury
Epilepsy, stroke and traumatic brain and spinal cord injury are major causes of morbidity throughout the world. Excitotoxicity, metabolic failure and oxidative stress play prominent roles in these acute neurological insults110. Rats maintained on IF for several months before exposure to the seizure-inducing excitotoxin kainate exhibit reduced loss of hippocampal pyramidal neurons and improved performance in a hippocampus-dependent water maze test of spatial learning and memory compared with rats fed ad libitum25. A daily IF and/or CR diet that elevates circulating BHB levels also suppresses seizures in a genetic mouse model of epilepsy111. In models of focal ischaemic stroke, rats or mice maintained on IF (ADF) prior to cerebral vessel occlusion exhibit reduced death of cerebral cortical neurons and improved functional outcomes59,112. Compared with rats fed ad libitum, rats maintained on daily TRF (40% CR) beginning 3 months before, and continuing for 70 days after, global cerebral ischaemia exhibited recovery of spatial memory deficits113. IF initiated either before or after a contusion injury to the thoracic spinal cord significantly improves recovery of motor function in rats114, and similar beneficial effects of IF following injury are evident in a rat model of incomplete cervical spinal cord injury115. A single 24 hour fast following injury lessens brain damage and ameliorates cognitive deficits in a rat model of traumatic brain injury116. These findings demonstrate that IMS can protect neurons and enhance resilience following injury. In light of the lack of any effective drugs for acute CNS injury, it would seem prudent to evaluate IF interventions in randomized controlled trials in human patients who have suffered a stroke or traumatic CNS injury.
With regards to cellular and molecular mechanisms, IF upregulates pathways involved in resistance to proteotoxic stress, neurotrophic factor signalling, DNA repair, mitochondrial metabolism and bioenergetics, antioxidant defences and Ca2+ buffering and downregulates pro-inflammatory cytokines117–123. Targeted analyses of proteins in pathways of interest have revealed that IF upregulates expression of the protein chaperones heat shock 70-kilodalton protein (HSP70; also known as HSPA) and 78-kilodalton glucose-regulated protein (HSPA5; also known as GRP78), the antioxidant enzyme haem oxygenase 1 and the neurotrophic factors BDNF and FGF2. IF reduces levels of pro-inflammatory cytokines (tumour necrosis factor (TNF), IL-1β and IL-6) in the uninjured brain and in ischaemic brain tissue in a mouse model of focal ischaemic stroke59. Similar to IF, exercise upregulates neurotrophic signalling and suppresses neuroinflammation120. The contribution of ketosis to the excitoprotective effects of IMS has not been established but could be tested in animal models in which ketone production or transport into neurons is genetically or pharmacologically disabled. Ketones may also mediate neuronal resistance to ischaemic and traumatic insults, as demonstrated in animal models in which BHB treatment lessened brain damage by mechanisms involving enhanced mitochondrial bioenergetics and stress resistance as well as suppression of neuroinflammation124–126. Indeed, BHB exerts anti-inflammatory actions by activating hydroxy-carboxylic acid receptor 2 and inhibiting the NRLP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome127.
IMS and neurological disorders
Alzheimer disease (AD) and Parkinson disease (PD) are the two most common neurodegenerative disorders128,129. The major risk factor for both diseases is age, with symptom onset typically occurring in people older than 70 years of age. The number of elderly individuals afflicted with AD or PD is rising rapidly because life expectancy has increased as a result of medical advances that enable those who would have previously died at a younger age from cardiovascular disease, cancer or diabetes to live beyond the age of 70 years130. Because there are no treatments that impact the disease progression in patients with AD or PD, lifelong diet and lifestyle interventions that retard ageing processes and thereby reduce disease risk may be of great value. In laboratory animals, IF (daily TRF or ADF) extends the average lifespan by up to 40% and protects against major chronic diseases, including cancer, diabetes and kidney disease7. Increasing evidence suggests that individuals with sedentary, overindulgent lifestyles are at increased risk of developing AD and PD, and animal studies support the notion that a chronic positive energy balance devoid of IMS renders the brain prone to AD and PD131,132. Conversely, exercise and moderation of energy intake reduce the risk of AD and PD133,134, and exercise intervention trials further suggest that IMS reduces clinical symptoms in many patients with AD and PD135,136.
The identification of genetic mutations that cause early-onset AD enabled the generation of transgenic mouse models that manifest some features of AD, including β-amyloid (Aβ) plaque and neurofibrillary tangle-like pathologies and cognitive impairment. Several studies have demonstrated that IMS regimens can counteract neuropathological and behavioural features in AD mouse models. For example, daily TRF (30% or 40% CR) reduced the accumulation of Aβ plaques in App-mutant mice137,138, and long-term (1-year) IF (ADF) and daily TRF prevented the development of cognitive impairment in 3xTg-AD mice that express beta-amyloid precursor protein (APP), presenilin 1 and tau mutations139. Interestingly, in the latter study, daily TRF lessened Aβ and tau pathologies, whereas ADF did not, suggesting that ADF protects neurons against synaptic dysfunction and cognitive deficits despite the presence of pathological Aβ and tau. As reviewed recently, aerobic exercise regimens counteract Aβ pathology and improve cognition in AD mouse models140.
Mechanisms by which IMS may lessen neuropathology and ameliorate cognitive deficits in AD mice include a reduction of amyloidogenic enzymatic processing of APP, suppression of inflammation, constraint of neuronal hyperexcitability and upregulation of adaptive neuronal stress-resistance pathways (for example, neurotrophic factors, antioxidant defences and nuclear and mitochondrial sirtuins)6,141,142. Ketones may counteract AD pathogenesis because a ketone ester diet that elevates BHB more than tenfold lessens Aβ and tau pathologies and ameliorates behavioural abnormalities in 3xTg-AD mice143. Moreover, Castellano et al. recently reported that a 3-month aerobic training (brisk walking) programme increased brain cell ketone utilization in patients with AD threefold without affecting cerebral glucose utilization144, which may be one reason that aerobic exercise can protect the brain against AD. That IF may be another approach for subjects at risk of or with AD is suggested by a study using 11C-acetoacetate positron emission tomography (PET) imaging that showed that within 48 hours of the onset of fasting, there is a sevenfold to eightfold increase in brain uptake of ketones4,145. Moreover, whereas brain cell uptake of glucose is severely impaired in patients with AD, the cells remain capable of utilizing ketones146.
Selective degeneration of nigrostriatal dopaminergic neurons, similar to that occurring in PD, can be induced by administration of mitochondrial toxins that selectively accumulate in dopaminergic neurons. When mice are maintained on an ADF IMS regimen before neurotoxin administration, their dopaminergic neurons are relatively resistant to degeneration and their motor deficits are reduced compared with mice fed ad libitum147. Daily TRF (30% CR) was also effective in reducing motor deficits and attenuating dopamine depletion in a unilateral 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) lesion rhesus macaque PD model148. Intermittent treadmill exercise is also beneficial in protecting dopaminergic neurons and promoting functional recovery in MPTP PD models149. Compared with neurotoxin models, the impact of IMS on neuropathology and functional deficits in transgenic PD animal models is largely unexplored. Transgenic mice expressing a mutant form of α-synuclein (A53T) that causes familial PD exhibit age-dependent accumulation of α-synuclein inclusions in neurons throughout the neuraxis develop progressive autonomic nervous system and motor dysfunction and die before they reach 1 year of age. Autonomic dysfunction in these mice is ameliorated by ADF and exacerbated by excessive caloric intake150. Enhancing neurotrophic support (BDNF and glial-cell-line-derived neurotrophic factor), induction of protein chaperones, ketogenesis and ghrelin signalling are among the neuroprotective mechanisms of action of IMS suggested from the experiments with PD animal models151–153.
Depression and anxiety disorders are major causes of morbidity, and their incidence has increased in parallel with increases in obesity and diabetes in many countries154. Data from human and animal studies suggest that sedentary, overindulgent lifestyles increase the risk of anxiety disorders and depression155 and that IMS effected by exercise or IF can improve mood and ameliorate anxiety and depression156–159. The ability of exercise to ameliorate symptoms of depression and anxiety is well known, and the underlying mechanisms have been elucidated, with upregulation of BDNF expression in neuronal circuits that regulate mood playing a role160–162. Increased activity within hippocampal and frontal cortical networks and enhanced noradrenergic input to those neurons may mediate upregulation of BDNF expression via activation of CREB by Ca2+-dependent and cAMP-dependent pathways163,164. Considerable evidence also suggests that BDNF signalling mediates the anxiolytic and antidepressant actions of the most commonly prescribed antidepressant drugs (serotonin and noradrenaline reuptake inhibitors). Interestingly, selective ablation of the BDNF receptor TrkB in hippocampal neural stem cells abolishes the abilities of wheel running and anti-depressant drugs to ameliorate anxiety and depression in mice165. The recent finding that BHB induces BDNF expression in hippocampal and cortical neurons54,55 suggests that ketones mediate the antidepressant effects of exercise and IF. Anxiolytic and antidepressant effects of IGF1 are suggested by data showing that peripheral administration of IGF1 exerts antidepressant actions, whereas peripheral IGF1-neutralizing antibodies attenuate the antidepressant effects of exercise in mice166,167. Similar experiments have not yet been performed to determine whether IGF1 also plays a role in the anxiolytic and antidepressant effects of IF.
During the past 3 decades, there has been a rapid increase in the number of children diagnosed with an autism spectrum disorder (ASD), which manifests, to varying extents, in social communication and language deficits, anxiety and repetitive behaviours168,169. The increase in autism tracks remarkably closely with the increase in childhood overweight or obesity during the same time period (data from autismspeaks.org and the US Centers for Disease Control), suggesting a causal link between lack of metabolic switching and autistic behaviours. In support of the latter possibility are data showing that nearly twice as many children diagnosed with ASD are overweight or obese as control children170. The underlying neurobiological mechanisms may include reduced BDNF expression and excessive mTOR pathway activation. Indeed, human adolescents with BDNF haploinsufficiency score higher on an autism clinical rating scale than age-matched control subjects171, and mTOR is hyperactivated in animal models of ASDs, which may result in aberrant dendritic spinogenesis and dysregulation of neuronal network activity172. Consistent with a potential benefit of IMS in ASD, exercise is effective in reducing behavioural issues in many children with ASD173. In the BTBR mouse model of ASD, a ketogenic diet improved symptoms by increasing sociability and decreasing self-directed repetitive behaviour174. It was recently reported that a ketogenic diet also improves symptoms in children with ASD175. As yet, trials of IF in ASD have not been performed.
IMS and optimization of brain health
It has become clear that sedentary overindulgent lifestyles in which IMS is lacking preclude optimal brain health. The rapid increase in overweight and obese individuals, including children, during the past 40 years is contributing to an emerging ‘epidemic’ of cognitive impairment and dementia that will intensify in coming decades. As reviewed elsewhere, evidence supporting the latter two statements is strong, and the mechanisms by which lack of IMS adversely affects neuroplasticity and risk of major brain disorders are becoming understood176,177. When rodents are maintained on an obesogenic diet (high fat and sugar) or are genetically defective in leptin signalling and therefore eat excessively, they perform more poorly on learning and memory tests than nonobese control animals178–180. Studies of the hippocampus have shown that deleterious effects of obesity on cognition involve impaired synaptic plasticity and neurogenesis, reduced expression of BDNF and local neuroinflammation176–180. IMS, triggered with running-wheel exercise and/or IF, can counteract the adverse effects of excessive energy intake on hippocampal plasticity6. Although a chronic positive energy balance is not optimal for brain health and may increase the risk of neurological disorders, including anxiety, depression, AD and PD, a chronic negative energy balance (starvation and/or excessive exercise) is also detrimental for health and survival. It therefore becomes important to establish parameters of IMS (frequency, duration and ‘intensity’) that promote optimal brain function and disease resistance.
Thus far, we have considered the metabolic and cell-signalling-mediated adaptations of the brain to IMS, with a focus on the events occurring during the period of metabolic challenge (during fasting and exercise). However, events occurring in the brain during the recovery period (eating after fasting, resting after exercise and sleep) are also important to understand when considering IMS and neuroplasticity (FIG. 3). In this regard, knowledge of how muscle cells respond to exercise and recovery cycles provides a general framework for understanding how IMS affects brain cells. The cellular stress that muscle cells experience during exercise activates several key pathways of the conserve-resources mode, including inhibition of mTOR and overall protein synthesis, upregulation of autophagy and upregulation of genes encoding antioxidant enzymes, sirtuins, protein chaperones and lipolytic enzymes181. The latter genes are induced by Ca2+, ROS and an increase in the AMP:ATP ratio, which activate kinases (CaMKII and AMPK) and downstream transcription factors (nuclear regulatory factor 2 (NRF2; also known as NFE2L2), NRF1, peroxisome proliferator-activated receptors (PPARs) and forkhead box protein O1 (FOXO1)). PGC1α is also rapidly upregulated to facilitate the mitochondrial biogenesis that occurs during the subsequent recovery period after exercise; muscle cells grow not during the exercise period but rather during the recovery period. The sequential upregulation of autophagy and stress-resistance pathways during the exercise period and mitochondrial biogenesis and cell growth during the recovery period are both critical for the improvements in endurance and strength that result from intermittent exercise181.
We suggest that a generally similar scenario occurs at the cellular and molecular levels in response to IMS in many neuronal networks in the brain (FIG. 3). Neurons respond to the G-to-K switch by downregulating mTOR and global protein synthesis while concomitantly upregulating cellular stress-resistance pathways and autophagy. Upstream triggers of the latter pathways include BHB, Ca2+ (CaMKII) and ROS, which activate transcription factors including CREB, NF-κB and PGC1α182. The latter pathways not only bolster cellular stress resistance but also set the stage for the structural and functional neuroplasticity that occur during the K-to-G recovery phase of IMS cycles. In this view, outgrowth of dendrites and axons, synaptogenesis and neurogenesis occur during the recovery phase, when the cells are in growth mode, with high levels of mTOR activity, protein synthesis and mitochondrial biogenesis. We envision that IMS involves alternating cycles of upregulation of neuronal autophagy and stress-response pathways when the switch is on and the expansion or adaptive remodelling of neuronal circuits when the switch is off (FIG. 3). This model does not preclude structural and functional adaptations (for example, dendritic spine formation or retraction, LTP and long-term depression) during periods of time when the switch is on. With regards to IMS and cognition, this hypothesis could be tested in experiments in which individual neurons are interrogated by time-lapse imaging and electrophysiological recordings during cycles of fasting and feeding. Experiments in which mTOR and PGC-1α are (genetically or pharmacologically) inhibited or upregulated during on and off phases of IMS cycles and synaptic plasticity, neurogenesis and cognition are evaluated would provide cause–effect data to support (or not) our working model.
Although IMS can enhance brain function and stress resistance, the question of what specific IMS regimen(s) is ideal for brain health throughout the life course remains unanswered. This could first be addressed in animal studies by systematically varying the duration of fasting and feeding periods (for example, ADF versus 16 hours daily TRF versus fasting 2 days per week) and evaluating behavioural, and the related electrophysiological and neurochemical, end points. Ideally, such experiments would be done in several strains of mice and/or rats and would include both males and females and ageing cohorts. To increase relevance to animals in the wild and human diet and lifestyle interventions, different exercise regimens could be incorporated into IF studies. Although an increasing number of studies have demonstrated improvements in a range of peripheral health indicators when individuals who had previously eaten three or more meals every day change their eating pattern to an IF diet182, data on the impact of IMS on the human brain are sparse. However, a recent study showed that a 2-year multidomain intervention (diet, exercise, cognitive training and vascular risk monitoring) results in improved scores on cognitive tests in elderly subjects compared with a nonintervention control group183. Randomized controlled trials of IF and exercise regimens with neurological end points (psychological test batteries, functional brain imaging and molecular interrogation of cerebrospinal fluid) at baseline and at designated time points during the IMS period will be of great value. On the basis of the evidence from the animal studies reviewed in this article, one might predict that IMS will improve mood and cognition, change resting-state and task-related neuronal network activity, increase levels of neurotrophic factors and reduce markers of inflammation in cerebrospinal fluid. Performance of such IMS trials in subject populations at risk of a neurological disorder (for example, subjects with metabolic syndrome), in subjects who have suffered an acute neurological insult and in patients in the early stages of an age-related neurodegenerative disorder may eventually lead to prescriptions for IMS instead of or in conjunction with drugs that target a specific pathway.
Conclusions and future directions
The evidence reviewed in this article leads to several general conclusions regarding IMS and neuroplasticity. First, cognition, sensory–motor function and physical performance can be enhanced by IMS protocols involving IF and/or vigorous exercise. Second, by providing an alternative energy source and activating signalling pathways involved in neuroplasticity and cellular stress resistance, the ketone BHB plays a particularly important role in neuronal adaptations to fasting and exercise. Third, neurons respond to the G-to-K switch by engaging a ‘cell-preservation mode’ and adopt a ‘cell-growth mode’ by activation of certain signalling pathways when the switch is off (food, rest and sleep). Fourth, fasting and exercise upregulate neurotrophic factor signalling, antioxidant and DNA repair enzymes, protein deacetylases and autophagy, which protects neurons against stress and sets the stage for mitochondrial biogenesis and cell growth and plasticity during recovery periods (FIG. 3). Fifth, lifestyles characterized by little or no IMS (three meals per day plus snacks and negligible exercise) result in suboptimal brain functionality and increase the risk of major neurodegenerative and psychiatric disorders. Sixth, many different IMS regimens are likely to improve brain health such that individuals may choose an approach that suits their particular daily and weekly schedules.
There are many gaps in our knowledge of the neurobiology of IMS and the application of such knowledge to the prevention and treatment of neurological disorders. Among the outstanding questions, we find the following particularly pressing. First, what adaptive responses of the nervous system to metabolic switching are mediated by and/or require ketones? This might be answered by studies in which the ketone-generating enzyme 3-hydroxy-3-methylglutaryl-CoA lyase (HMG-CoA lyase) or neuronal monocarboxylate transporter 2 (MCT2; also known as SLC16A7) is knocked down or out in mice and then neurological responses to fasting and exercise are evaluated. Because of the criticality of ketones for survival during fasting, such experiments will require ‘titration’ of circulating ketone levels by administration of exogenous ketones. Second, what are the contributions of intrinsic neuronal network signalling pathways and peripheral signals entering the brain from the circulation to IMS-induced neuroplasticity? Third, which pathways are common to the mechanisms of action of exercise, fasting and cognitive challenges, and which are unique to a specific metabolic challenge? Fourth, are there retrograde transneuronal signals from the peripheral nervous system to the CNS that mediate effects of IMS on CNS neuroplasticity and resistance to injury and disease? Fifth, are there roles for glial cells in adaptations of the CNS to IMS? Although IF and exercise can suppress microglia-mediated neuroinflammation, the underlying mechanisms remain to be established. The possible effects of IMS on astrocytes are also unexplored. Do IF and/or exercise stimulate neurotrophic factor production in astrocytes, and are ketones involved? Does IMS affect axonal myelination? Sixth, are there roles for cerebrovascular remodelling in the beneficial effects of IMS on cognition and stress resistance, and what are the underlying mechanisms? Seventh, does IF improve cognition in humans, and which cognitive domains are affected? Eigth, will there be clinical applications of IMS ‘prescriptions’ for a range of neurological disorders? Several randomized controlled trials of IF in subjects with or at high risk of a neurological disorder are in progress, and results will be forthcoming.
Acknowledgments
This work was supported by the Intramural Research Program of the US National Institute on Ageing.
Glossary
- β-Hydroxybutyrate(BHB)
A ketone, generated from fatty acids during fasting and extended exercise, that functions as a cellular energy source and as a signalling molecule that induces the expression of brain-derived neurotrophic factor.
- Intermittent metabolic switching (IMS)
Repeating cycles of a metabolic challenge (fasting and/or exercise) sufficient to deplete liver glycogen stores and elevate circulating ketone levels, followed by a recovery period (eating, resting and sleeping).
- Brain-derived neurotrophic factor (BDNF)
A protein produced and released from neurons in response to synaptic activity, exercise and fasting that acts to enhance synaptic plasticity and cellular stress resistance.
- Mitochondrial biogenesis
The proliferation of mitochondria in neurons in response to metabolic challenges and neurotrophic factors to produce new mitochondria that promote synaptic plasticity and cellular stress resistance.
- Deacetylase
An enzyme that removes an acetyl group from lysine residues of substrate proteins; the sirtuins SIRT1 and SIRT3 are deacetylases that play particularly important roles in adaptive responses of neurons to metabolic challenges.
- Mechanistic target of rapamycin
(mTOR; also known as serine/threonine-protein kinase mTOR (MTOR) and mammalian target of rapamycin). A kinase that plays a pivotal role in stimulating cellular protein synthesis and suppressing autophagy when nutrients (glucose and amino acids) are plentiful.
- Autophagy
A complex process by which cells recognize damaged dysfunctional proteins and organelles, engulf them in a membrane and target them for enzymatic degradation in lysosomes to generate recyclable undamaged components (for example, amino acids and lipids).
- Ketogenesis
The process by which spillover of acetyl CoA results from β-oxidation of fatty acids in the liver during fasting and extended exercise.
- Myokines
Proteins and peptides released from muscle cells during exercise that can enter the brain and affect neuroplasticity; examples include interleukin-6, cathepsin B and irisin.
Footnotes
Author contributions
M.P.M., K.M., N.G., M.S. and A.C. researched data for the article, made a substantial contribution to the discussion of content and contributed to the writing, review and editing of the manuscript before submission’
Competing interests statement
The authors declare no competing interests.
Reviewer information
Nature Reviews Neuroscience thanks S. Cunnane, J. Rho and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Mattson MP. Lifelong brain health is a lifelong challenge: from evolutionary principles to empirical evidence. Ageing Res Rev. 2015;20:37–45. doi: 10.1016/j.arr.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bramble DM, Lieberman DE. Endurance running and the evolution of Homo. Nature. 2004;432:345–352. doi: 10.1038/nature03052. [DOI] [PubMed] [Google Scholar]
- 3.Mattson MP. Superior pattern processing is the essence of the evolved human brain. Front Neurosci. 2014;8:265. doi: 10.3389/fnins.2014.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Courchesne-Loyer A, et al. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J Cereb Blood Flow Metab. 2017;37:2485–2493. doi: 10.1177/0271678X16669366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017;36:1474–1492. doi: 10.15252/embj.201695810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mattson MP. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 2012;16:706–722. doi: 10.1016/j.cmet.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anson RM, et al. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci USA. 2003;100:6216–6220. doi: 10.1073/pnas.1035720100. The data in this study provide the first evidence that IMS exerts beneficial effects on the brain that are independent of overall energy intake. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatori M, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. This study finds that 16 hours of fasting each day can prevent obesity and related metabolic morbidities in mice fed a high-fat diet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harvie MN, et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int J Obes (Lond) 2011;35:714–727. doi: 10.1038/ijo.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harvie M, et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. Br J Nutr. 2013;110:1534–1547. doi: 10.1017/S0007114513000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanevski F, Xu B. Molecular and neural bases underlying roles of BDNF in the control of body weight. Front Neurosci. 2013;7:37. doi: 10.3389/fnins.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bechtold DA, Loudon AS. Hypothalamic clocks and rhythms in feeding behaviour. Trends Neurosci. 2013;36:74–82. doi: 10.1016/j.tins.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Ingram DK, Weindruch R, Spangler EL, Freeman JR, Walford RL. Dietary restriction benefits learning and motor performance of aged mice. J Gerontol. 1987;42:78–81. doi: 10.1093/geronj/42.1.78. [DOI] [PubMed] [Google Scholar]
- 15.Means LW, Higgins JL, Fernandez TJ. Mid-life onset of dietary restriction extends life and prolongs cognitive functioning. Physiol Behav. 1993;54:503–508. doi: 10.1016/0031-9384(93)90243-9. [DOI] [PubMed] [Google Scholar]
- 16.Parikh I, et al. Caloric restriction preserves memory and reduces anxiety of aging mice with early enhancement of neurovascular functions. Aging. 2016;8:2814–2826. doi: 10.18632/aging.101094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandhorst S, et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015;22:86–99. doi: 10.1016/j.cmet.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stillman CM, Cohen J, Lehman ME, Erickson KI. Mediators of physical activity on neurocognitive function: a review at multiple levels of analysis. Front Hum Neurosci. 2016;10:626. doi: 10.3389/fnhum.2016.00626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper C, Moon HY, van Praag H. On the run for hippocampal plasticity. Cold Spring Harb Perspect Med. 2017 doi: 10.1101/cshperspect.a029736. http://dx.doi.org/10.1101/cshperspect.a029736. [DOI] [PMC free article] [PubMed]
- 20.Raichlen DA, Alexander GE. Adaptive capacity: an evolutionary neuroscience model linking exercise, cognition, and brain health. Trends Neurosci. 2017;40:408–421. doi: 10.1016/j.tins.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stranahan AM, et al. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X, Yin S, Lang M, He R, Li J. The more the better? A meta-analysis on effects of combined cognitive and physical intervention on cognition in healthy older adults. Ageing Res Rev. 2016;31:67–79. doi: 10.1016/j.arr.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Hippocrates. On the Sacred Disease. (400 BCE). English translation by F. Adams: http://classics.mit.edu/Hippocrates/sacred.html.
- 24.Ruskin DN, Masino SA. The nervous system and metabolic dysregulation: emerging evidence converges on ketogenic diet therapy. Front Neurosci. 2012;6:33. doi: 10.3389/fnins.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann Neurol. 1999;45:8–15. [PubMed] [Google Scholar]
- 26.Hartman AL, Rubenstein JE, Kossoff EH. Intermittent fasting: a “new” historical strategy for controlling seizures? Epilepsy Res. 2013;104:275–279. doi: 10.1016/j.eplepsyres.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA. 1999;96:13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farmer J, et al. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague–Dawley rats in vivo. Neuroscience. 2004;124:71–79. doi: 10.1016/j.neuroscience.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 29.O’Callaghan RM, Ohle R, Kelly AM. The effects of forced exercise on hippocampal plasticity in the rat: a comparison of LTP, spatial- and non-spatial learning. Behav Brain Res. 2007;176:362–366. doi: 10.1016/j.bbr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Talani G, et al. Enhanced glutamatergic synaptic plasticity in the hippocampal CA1 field of food-restricted rats: involvement of CB1 receptors. Neuropsychopharmacology. 2016;41:1308–1318. doi: 10.1038/npp.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckles-Smith K, Clayton D, Bickford P, Browning MD. Caloric restriction prevents age-related deficits in LTP and in NMDA receptor expression. Mol Brain Res. 2000;78:154–162. doi: 10.1016/s0169-328x(00)00088-7. [DOI] [PubMed] [Google Scholar]
- 32.Hori N, Hirotsu I, Davis PJ, Carpenter DO. Long-term potentiation is lost in aged rats but preserved by calorie restriction. Neuroreport. 1992;3:1085–1088. doi: 10.1097/00001756-199212000-00013. [DOI] [PubMed] [Google Scholar]
- 33.Vivar C, et al. Monosynaptic inputs to new neurons in the dentate gyrus. Nat Commun. 2012;3:1107. doi: 10.1038/ncomms2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- 35.Vivar C, Peterson BD, van Praag H. Running rewires the neuronal network of adult-born dentate granule cells. Neuroimage. 2016;131:29–41. doi: 10.1016/j.neuroimage.2015.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen SM, Li B, Tsien RW, Ma H. Evolutionary and functional perspectives on signaling from neuronal surface to nucleus. Biochem Biophys Res Commun. 2015;460:88–99. doi: 10.1016/j.bbrc.2015.02.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 38.Estrada NM, Isokawa M. Metabolic demand stimulates CREB signaling in the limbic cortex: implication for the induction of hippocampal synaptic plasticity by intrinsic stimulus for survival. Front Syst Neurosci. 2009;3:5. doi: 10.3389/neuro.06.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med. 2014;16:161–174. doi: 10.1007/s12017-013-8270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirano Y, et al. Fasting launches CRTC to facilitate long-term memory formation in Drosophila. Science. 2013;339:443–446. doi: 10.1126/science.1227170. [DOI] [PubMed] [Google Scholar]
- 41.Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. 2016;6:a022780. doi: 10.1101/cshperspect.a022780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, O’Leary EI, Tanner GR. The ketogenic diet metabolite beta-hydroxybutyrate (β-HB) reduces incidence of seizure-like activity (SLA) in a Katp- and GABAb-dependent manner in a whole-animal Drosophila melanogaster model. Epilepsy Res. 2017;133:6–9. doi: 10.1016/j.eplepsyres.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Blier P, El Mansari M. Serotonin and beyond: therapeutics for major depression. Phil Trans R Soc B Biol Sci. 2013;368:20120536. doi: 10.1098/rstb.2012.0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kondo M, Nakamura Y, Ishida Y, Shimada S. The 5-HT3 receptor is essential for exercise-induced hippocampal neurogenesis and antidepressant effects. Mol Psychiatry. 2015;20:1428–1437. doi: 10.1038/mp.2014.153. [DOI] [PubMed] [Google Scholar]
- 45.Billman GE, et al. Exercise training-induced bradycardia: evidence for enhanced parasympathetic regulation without changes in intrinsic sinoatrial node function. J Appl Physiol. 2015;118:1344–1355. doi: 10.1152/japplphysiol.01111.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wan R, Camandola S, Mattson MP. Intermittent food deprivation improves cardiovascular and neuroendocrine responses to stress in rats. J Nutr. 2003;133:1921–1929. doi: 10.1093/jn/133.6.1921. [DOI] [PubMed] [Google Scholar]
- 47.Mager DE, et al. Caloric restriction and intermittent fasting alter spectral measures of heart rate and blood pressure variability in rats. FASEB J. 2006;20:631–637. doi: 10.1096/fj.05-5263com. This study establishes that IF lowers blood pressure and heart rate and increases heart rate variability by a mechanism involving enhancement of parasympathetic tone. [DOI] [PubMed] [Google Scholar]
- 48.Wan R, et al. Evidence that BDNF regulates heart rate by a mechanism involving increased brainstem parasympathetic neuron excitability. J Neurochem. 2014;129:573–580. doi: 10.1111/jnc.12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marosi K, Mattson MP. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol Metab. 2014;25:89–98. doi: 10.1016/j.tem.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng CM, et al. A ketogenic diet increases brain insulin-like growth factor receptor and glucose transporter gene expression. Endocrinology. 2003;144:2676–2682. doi: 10.1210/en.2002-0057. [DOI] [PubMed] [Google Scholar]
- 52.Bohannon NJ, et al. Characterization of insulin-like growth factor I receptors in the median eminence of the brain and their modulation by food restriction. Endocrinology. 1988;122:1940–1947. doi: 10.1210/endo-122-5-1940. [DOI] [PubMed] [Google Scholar]
- 53.Llorens-Martín M, Torres-Alemán I, Trejo JL. Mechanisms mediating brain plasticity: IGF1 and adult hippocampal neurogenesis. Neuroscientist. 2009;15:134–148. doi: 10.1177/1073858408331371. [DOI] [PubMed] [Google Scholar]
- 54.Marosi K, et al. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem. 2016;139:769–781. doi: 10.1111/jnc.13868. This study shows that the ketone BHB can act directly on neurons to induce transcription of the gene encoding BDNF by a mechanism involving NF-κB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sleiman SF, et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. eLife. 2016;5:e1509. doi: 10.7554/eLife.15092. This study finds that the ketone BHB can induce Bdnf gene expression in the hippocampus in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zechel S, Werner S, Unsicker K, von Bohlen und Halbach O. Expression and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation. Neuroscientist. 2010;16:357–373. doi: 10.1177/1073858410371513. [DOI] [PubMed] [Google Scholar]
- 57.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3740. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mark RJ, Keller JN, Kruman I, Mattson MP. Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997;756:205–214. doi: 10.1016/s0006-8993(97)00196-0. [DOI] [PubMed] [Google Scholar]
- 59.Arumugam TV, et al. Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann Neurol. 2010;67:41–52. doi: 10.1002/ana.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gómez-Pinilla F, Dao L, So V. Physical exercise induces FGF-2 and its mRNA in the hippocampus. Brain Res. 1997;764:1–8. doi: 10.1016/s0006-8993(97)00375-2. [DOI] [PubMed] [Google Scholar]
- 61.Graham BM, Richardson R. Memory of fearful events; the role of fibroblast growth factor-2 in fear acquisition and extinction. Neuroscience. 2011;189:156–169. doi: 10.1016/j.neuroscience.2011.05.041. [DOI] [PubMed] [Google Scholar]
- 62.Hood DA, Tryon LD, Carter HN, Kim Y, Chen CC. Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem J. 2016;473:2295–2314. doi: 10.1042/BCJ20160009. [DOI] [PubMed] [Google Scholar]
- 63.Kerr JS, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40:151–166. doi: 10.1016/j.tins.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hepple RT. Why eating less keeps mitochondria working in aged skeletal muscle. Exerc Sport Sci Rev. 2009;37:23–28. doi: 10.1097/JES.0b013e3181877dc5. [DOI] [PubMed] [Google Scholar]
- 65.Cheng A, et al. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250. doi: 10.1038/ncomms2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wrann CD, et al. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013;18:649–659. doi: 10.1016/j.cmet.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fusco S, et al. A role for neuronal cAMP responsive-element binding (CREB)-1 in brain responses to calorie restriction. Proc Natl Acad Sci USA. 2012;109:621–626. doi: 10.1073/pnas.1109237109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van de Ven RAH, Santos D, Haigis MC. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. 2017;23:320–331. doi: 10.1016/j.molmed.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng A, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23:128–142. doi: 10.1016/j.cmet.2015.10.013. This study shows that the mitochondrial deacetylase SIRT3 is upregulated in response to exercise and excitatory synaptic activity and that SIRT3 protects neurons against excitotoxic and metabolic stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang X, et al. PGC-1α/ERRα-Sirt3 pathway regulates DAergic neuronal death by directly deacetylating SOD2 and ATP synthase β. Antioxid Redox Signal. 2016;24:312–328. doi: 10.1089/ars.2015.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bockaert J, Marin P. mTOR in brain physiology and pathologies. Physiol Rev. 2015;95:1157–1187. doi: 10.1152/physrev.00038.2014. [DOI] [PubMed] [Google Scholar]
- 73.Alirezaei M, et al. Short-term fasting induces profound neuronal autophagy. Autophagy. 2010;6:702–710. doi: 10.4161/auto.6.6.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.He C, Sumpter R, Jr, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. 2012;8:1548–1551. doi: 10.4161/auto.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005;280:25864–25870. doi: 10.1074/jbc.M502456200. [DOI] [PubMed] [Google Scholar]
- 76.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 78.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang SJ, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–472. doi: 10.1073/pnas.012605299. This study demonstrates a role for the nutrient-sensing mTOR pathway in hippocampal synaptic plasticity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cammalleri M, et al. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci USA. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoeffer CA, et al. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weston MC, Chen H, Swann JW. Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J Neurosci. 2012;32:11441–11452. doi: 10.1523/JNEUROSCI.1283-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci. 2012;32:10413–10422. doi: 10.1523/JNEUROSCI.4533-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hernandez D, et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron. 2012;74:277–284. doi: 10.1016/j.neuron.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Palacios OM, et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging. 2009;1:771–783. doi: 10.18632/aging.100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mounier R, Théret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab. 2015;26:275–286. doi: 10.1016/j.tem.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 87.Marosi K, et al. Long-term exercise treatment reduces oxidative stress in the hippocampus of aging rats. Neuroscience. 2012;226:21–28. doi: 10.1016/j.neuroscience.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 88.Han Y, et al. AMPK signaling in the dorsal hippocampus negatively regulates contextual fear memory formation. Neuropsychopharmacology. 2016;41:1849–1864. doi: 10.1038/npp.2015.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kong D, et al. A postsynaptic AMPK — p21-activated kinase pathway drives fasting-induced synaptic plasticity in AgRP neurons. Neuron. 2016;91:25–33. doi: 10.1016/j.neuron.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kobilo T, et al. AMPK agonist AICAR improves cognition and motor coordination in young and aged mice. Learn Mem. 2014;21:119–126. doi: 10.1101/lm.033332.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramamurthy S, Chang E, Cao Y, Zhu J, Ronnett GV. AMPK activation regulates neuronal structure in developing hippocampal neurons. Neuroscience. 2014;259:13–24. doi: 10.1016/j.neuroscience.2013.11.048. [DOI] [PubMed] [Google Scholar]
- 92.Ferrario CR, et al. Homeostasis meets motivation in the battle to control food intake. J Neurosci. 2016;36:11469–11481. doi: 10.1523/JNEUROSCI.2338-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Andrews ZB. The extra-hypothalamic actions of ghrelin on neuronal function. Trends Neurosci. 2011;34:31–40. doi: 10.1016/j.tins.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 94.Kim Y, et al. Ghrelin is required for dietary restriction-induced enhancement of hippocampal neurogenesis: lessons from ghrelin knockout mice. Endocr J. 2015;62:269–275. doi: 10.1507/endocrj.EJ14-0436. [DOI] [PubMed] [Google Scholar]
- 95.Jensen M, et al. Anxiolytic-like effects of increased ghrelin receptor signaling in the amygdala. Int J Neuropsychopharmacol. 2016;19:pyv123. doi: 10.1093/ijnp/pyv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wallenius V, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–79. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 97.Wueest S, et al. Interleukin-6 contributes to early fasting-induced free fatty acid mobilization in mice. Am J Physiol Regul Integr Comp Physiol. 2014;306:R861–R867. doi: 10.1152/ajpregu.00533.2013. [DOI] [PubMed] [Google Scholar]
- 98.Baier PC, May U, Scheller J, Rose-John S, Schiffelholz T. Impaired hippocampus-dependent and -independent learning in IL-6 deficient mice. Behav Brain Res. 2009;200:192–196. doi: 10.1016/j.bbr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 99.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3740. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mattson MP, Maudsley S, Martin B. A neural signaling triumvirate that influences ageing and age-related disease: insulin/IGF-1, BDNF and serotonin. Ageing Res Rev. 2004;3:445–464. doi: 10.1016/j.arr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 101.Vendelbo MH, et al. Exercise and fasting activate growth hormone-dependent myocellular signal transducer and activator of transcription-5b phosphorylation and insulin-like growth factor-I messenger ribonucleic acid expression in humans. J Clin Endocrinol Metab. 2010;95:E64–68. doi: 10.1210/jc.2010-0689. [DOI] [PubMed] [Google Scholar]
- 102.Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–465. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- 103.Carro E, Nuñez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carro E, Trejo JL, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J Neurosci. 2001;21:5678–5684. doi: 10.1523/JNEUROSCI.21-15-05678.2001. The authors of this study use an IGF1-blocking antibody to demonstrate a role for circulating IGF1 in the neuroprotective actions of exercise in mouse models of excitotoxic neuronal degeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moon HY, et al. Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab. 2016;24:332–340. doi: 10.1016/j.cmet.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shimazu T, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murray AJ, et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016;30:4021–4032. doi: 10.1096/fj.201600773R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Webber RJ, Edmond J. The in vivo utilization of acetoacetate, D-(–)-3-hydroxybutyrate, and glucose for lipid synthesis in brain in the 18-day-old rat. Evidence for an acetyl-CoA bypass for sterol synthesis. J Biol Chem. 1979;254:3912–3920. [PubMed] [Google Scholar]
- 110.Chamorro Á, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016;15:869–881. doi: 10.1016/S1474-4422(16)00114-9. [DOI] [PubMed] [Google Scholar]
- 111.Greene AE, Todorova MT, McGowan R, Seyfried TN. Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia. 2001;42:1371–1378. doi: 10.1046/j.1528-1157.2001.17601.x. [DOI] [PubMed] [Google Scholar]
- 112.Yu ZF, Mattson MP. Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: evidence for a preconditioning mechanism. J Neurosci Res. 1999;57:830–839. [PubMed] [Google Scholar]
- 113.Roberge MC, Messier C, Staines WA, Plamondon H. Food restriction induces long-lasting recovery of spatial memory deficits following global ischemia in delayed matching and non-matching-to-sample radial arm maze tasks. Neuroscience. 2008;156:11–29. doi: 10.1016/j.neuroscience.2008.05.062. [DOI] [PubMed] [Google Scholar]
- 114.Jeong MA, et al. Intermittent fasting improves functional recovery after rat thoracic contusion spinal cord injury. J Neurotrauma. 2011;28:479–492. doi: 10.1089/neu.2010.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Plunet WT, et al. Dietary restriction started after spinal cord injury improves functional recovery. Exp Neurol. 2008;213:28–35. doi: 10.1016/j.expneurol.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 116.Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008;86:1812–1822. doi: 10.1002/jnr.21628. This study shows that when initiated after traumatic brain injury, fasting and ketone administration reduce neuronal loss in rats. [DOI] [PubMed] [Google Scholar]
- 117.Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- 118.Xu X, et al. Gene expression atlas of the mouse central nervous system: impact and interactions of age, energy intake and gender. Genome Biol. 2007;8:R234. doi: 10.1186/gb-2007-8-11-r234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schafer MJ, Dolgalev I, Alldred MJ, Heguy A, Ginsberg SD. Calorie restriction suppresses age-dependent hippocampal transcriptional signatures. PLoS ONE. 2015;10:e0133923. doi: 10.1371/journal.pone.0133923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Di Benedetto S, Müller L, Wenger E, Düzel S, Pawelec G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci Biobehav Rev. 2017;75:114–128. doi: 10.1016/j.neubiorev.2017.01.044. [DOI] [PubMed] [Google Scholar]
- 121.Kiprianova I, et al. Enlarged infarct volume and loss of BDNF mRNA induction following brain ischemia in mice lacking FGF-2. Exp Neurol. 2004;189:252–260. doi: 10.1016/j.expneurol.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 122.Bo H, et al. Exercise-induced neuroprotection of hippocampus in APP/PS1 transgenic mice via upregulation of mitochondrial 8-oxoguanine DNA glycosylase. Oxid Med Cell Longev. 2014;2014:834502. doi: 10.1155/2014/834502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med. 2014;16:161–174. doi: 10.1007/s12017-013-8270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Prins ML, Lee SM, Fujima LS, Hovda DA. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J Neurochem. 2004;90:666–672. doi: 10.1111/j.1471-4159.2004.02542.x. [DOI] [PubMed] [Google Scholar]
- 125.Rahman M, et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun. 2014;5:3944. doi: 10.1038/ncomms4944. [DOI] [PubMed] [Google Scholar]
- 126.Yin J, Han P, Tang Z, Liu Q, Shi J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J Cereb Blood Flow Metab. 2015;35:1783–1789. doi: 10.1038/jcbfm.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Simeone TA, Simeone KA, Rho JM. Ketone bodies as anti-seizure agents. Neurochem Res. 2017;42:2011–2018. doi: 10.1007/s11064-017-2253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Poewe W, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 130.Kontis V, et al. Future life expectancy in 35 industrialised countries: projections with a Bayesian model ensemble. Lancet. 2017;389:1323–1335. doi: 10.1016/S0140-6736(16)32381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Frisardi V, et al. Metabolic-cognitive syndrome: a crosstalk between metabolic syndrome and Alzheimer’s disease. Ageing Res Rev. 2010;9:399–417. doi: 10.1016/j.arr.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 132.Lee EB, Mattson MP. The neuropathology of obesity: insights from human disease. Acta Neuropathol. 2014;127:3–28. doi: 10.1007/s00401-013-1190-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Baumgart M, et al. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement. 2015;11:718–726. doi: 10.1016/j.jalz.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 134.LaHue SC, Comella CL, Tanner CM. The best medicine? The influence of physical activity and inactivity on Parkinson’s disease. Mov Disord. 2016;31:1444–1454. doi: 10.1002/mds.26728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Groot C, et al. The effect of physical activity on cognitive function in patients with dementia: a meta-analysis of randomized control trials. Ageing Res Rev. 2016;25:13–23. doi: 10.1016/j.arr.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 136.Uhrbrand A, Stenager E, Pedersen MS, Dalgas U. Parkinson’s disease and intensive exercise therapy — a systematic review and meta-analysis of randomized controlled trials. J Neurol Sci. 2015;353:9–19. doi: 10.1016/j.jns.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 137.Wang J, et al. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2005;19:659–661. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- 138.Patel NV, et al. Caloric restriction attenuates a beta-deposition in Alzheimer transgenic models. Neurobiol Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 139.Halagappa VK, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 140.Ryan SM, Kelly ÁM. Exercise as a pro-cognitive, pro-neurogenic and anti inflammatory intervention in transgenic mouse models of Alzheimer’s disease. Ageing Res Rev. 2016;27:77–92. doi: 10.1016/j.arr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 141.Qin W, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 142.Zilberter M, et al. Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer’s disease. J Neurochem. 2013;125:157–171. doi: 10.1111/jnc.12127. [DOI] [PubMed] [Google Scholar]
- 143.Kashiwaya Y, et al. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2013;34:1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023. This study shows that dietary supplementation with a ketone (BHB) ester ameliorates behavioural deficits and lessens amyloid and tau pathologies in a triple transgenic mouse model of AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Castellano CA, et al. A 3-month aerobic training program improves brain energy metabolism in mild Alzheimer’s disease: preliminary results from a neuroimaging study. J Alzheimers Dis. 2017;56:1459–1468. doi: 10.3233/JAD-161163. [DOI] [PubMed] [Google Scholar]
- 145.Bentourkia M, et al. PET study of 11C-acetoacetate kinetics in rat brain during dietary treatments affecting ketosis. Am J Physiol Endocrinol Metab. 2009;296:E796–E801. doi: 10.1152/ajpendo.90644.2008. [DOI] [PubMed] [Google Scholar]
- 146.Croteau E, et al. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp Gerontol. 2017 doi: 10.1016/j.exger.2017.07.004. http://dx.doi.org/10.1016/j.exger.2017.07.004. Employing PET imaging with radiotracers for glucose and the ketone AcAc, the authors of this study provide evidence that although cerebral glucose utilization is impaired in patients with AD, their neurons remain capable of utilizing ketones. [DOI] [PubMed]
- 147.Duan W, Mattson MP. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J Neurosci Res. 1999;57:195–206. doi: 10.1002/(SICI)1097-4547(19990715)57:2<195::AID-JNR5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 148.Maswood N, et al. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. This study demonstrates that daily CR lessens motor deficits and dopamine depletion and elevates striatal levels of glial-cell-line-derived neurotrophic factor in a nonhuman primate model of PD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Petzinger GM, et al. Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2 ,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. J Neurosci. 2007;27:5291–5300. doi: 10.1523/JNEUROSCI.1069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Griffioen KJ, et al. Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant α-synuclein. Neurobiol Aging. 2013;34:928–935. doi: 10.1016/j.neurobiolaging.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gerecke KM, Jiao Y, Pagala V, Smeyne RJ. Exercise does not protect against MPTP-induced neurotoxicity in BDNF haploinsufficient mice. PLoS ONE. 2012;7:e43250. doi: 10.1371/journal.pone.0043250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bayliss JA, et al. Ghrelin-AMPK signaling mediates the neuroprotective effects of calorie restriction in Parkinson’s disease. J Neurosci. 2016;36:3049–3063. doi: 10.1523/JNEUROSCI.4373-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tieu K, et al. D-Beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ferrari AJ. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10:e1001547. doi: 10.1371/journal.pmed.1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hryhorczuk C, Sharma S, Fulton SE. Metabolic disturbances connecting obesity and depression. Front Neurosci. 2013;7:177. doi: 10.3389/fnins.2013.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hallgren M, et al. Exercise, physical activity, and sedentary behavior in the treatment of depression: broadening the scientific perspectives and clinical opportunities. Front Psychiatry. 2016;7:36. doi: 10.3389/fpsyt.2016.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Johnson JB, et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med. 2007;42:665–674. doi: 10.1016/j.freeradbiomed.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Archer T, Josefsson T, Lindwall M. Effects of physical exercise on depressive symptoms and biomarkers in depression. CNS Neurol Disord Drug Targets. 2014;13:1640–1653. doi: 10.2174/1871527313666141130203245. [DOI] [PubMed] [Google Scholar]
- 159.Patki G, et al. Novel mechanistic insights into treadmill exercise based rescue of social defeat-induced anxiety-like behavior and memory impairment in rats. Physiol Behav. 2014;130:135–144. doi: 10.1016/j.physbeh.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Adlard PA, Cotman CW. Voluntary exercise protects against stress-induced decreases in brain-derived neurotrophic factor protein expression. Neuroscience. 2004;124:985–992. doi: 10.1016/j.neuroscience.2003.12.039. [DOI] [PubMed] [Google Scholar]
- 161.Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF) Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 162.Taliaz D, et al. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J Neurosci. 2011;31:4475–4483. doi: 10.1523/JNEUROSCI.5725-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Garcia C, Chen MJ, Garza AA, Cotman CW, Russo-Neustadt A. The influence of specific noradrenergic and serotonergic lesions on the expression of hippocampal brain-derived neurotrophic factortranscripts following voluntary physical activity. Neuroscience. 2003;119:721–732. doi: 10.1016/s0306-4522(03)00192-1. [DOI] [PubMed] [Google Scholar]
- 164.Aguiar AS, et al. Effects of exercise on mitochondrial function, neuroplasticity and anxio-depressive behavior of mice. Neuroscience. 2014;271:56–63. doi: 10.1016/j.neuroscience.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 165.Li Y, et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Glasper ER, Llorens-Martin MV, Leuner B, Gould E, Trejo JL. Blockade of insulin-like growth factor-I has complex effects on structural plasticity in the hippocampus. Hippocampus. 2010;20:706–712. doi: 10.1002/hipo.20672. [DOI] [PubMed] [Google Scholar]
- 167.Duman CH, et al. Peripheral insulin-like growth factor-I produces antidepressant-like behavior and contributes to the effect of exercise. Behav Brain Res. 2009;198:366–371. doi: 10.1016/j.bbr.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zipfel S, Giel KE, Bulik CM, Hay P, Schmidt U. Anorexia nervosa: aetiology, assessment, and treatment. Lancet Psychiatry. 2015;2:1099–1111. doi: 10.1016/S2215-0366(15)00356-9. [DOI] [PubMed] [Google Scholar]
- 169.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 170.Criado KK, et al. Overweight and obese status in children with autism spectrum disorder and disruptive behavior. Autism. 2017 doi: 10.1177/1362361316683888. http://dx.doi.org/10.1177/1362361316683888. [DOI] [PMC free article] [PubMed]
- 171.Han JC, et al. Association of brain-derived neurotrophic factor (BDNF) haploinsufficiency with lower adaptive behaviour and reduced cognitive functioning in WAGR/11p13 deletion syndrome. Cortex. 2013;49:2700–2710. doi: 10.1016/j.cortex.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Huber KM, Klann E, Costa-Mattioli M, Zukin RS. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci. 2015;35:13836–13842. doi: 10.1523/JNEUROSCI.2656-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bremer E, Crozier M, Lloyd M. A systematic review of the behavioral outcomes following exercise interventions for children and youth with autism spectrum disorder. Autism. 2016;20:899–915. doi: 10.1177/1362361315616002. [DOI] [PubMed] [Google Scholar]
- 174.Ruskin DN, et al. Ketogenic diet improves core symptoms of autism in BTBR mice. PLoS ONE. 2013;8:e65021. doi: 10.1371/journal.pone.0065021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.El-Rashidy O, et al. Ketogenic diet versus gluten free casein free diet in autistic children: a case-control study. Metab Brain Dis. 2017;32:1935–1941. doi: 10.1007/s11011-017-0088-z. [DOI] [PubMed] [Google Scholar]
- 176.Stranahan AM. Models and mechanisms for hippocampal dysfunction in obesity and diabetes. Neuroscience. 2015;309:125–139. doi: 10.1016/j.neuroscience.2015.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.O’Brien PD, Hinder LM, Callaghan BC, Feldman EL. Neurological consequences of obesity. Lancet Neurol. 2017;16:465–477. doi: 10.1016/S1474-4422(17)30084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lindqvist A, et al. High-fat diet impairs hippocampal neurogenesis in male rats. Eur J Neurol. 2006;13:1385–1388. doi: 10.1111/j.1468-1331.2006.01500.x. [DOI] [PubMed] [Google Scholar]
- 179.Stranahan AM, et al. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stranahan AM, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.MacInnis MJ, Gibala MJ. Physiological adaptations to interval training and the role of exercise intensity. J Physiol. 2017;595:2915–2930. doi: 10.1113/JP273196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev. 2017;39:46–58. doi: 10.1016/j.arr.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ngandu T, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385:2255–2263. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- 184.Duffy PH, Feuers R, Nakamura KD, Leakey J, Hart J. Effect of chronic caloric restriction on the synchronization of various physiological measures in old female Fischer 344 rats. Chronobiol Int. 1990;7:113–124. doi: 10.3109/07420529009056963. [DOI] [PubMed] [Google Scholar]
- 185.Duffy PH, Feuers RJ, Hart RW. Effect of chronic caloric restriction on the circadian regulation of physiological and behavioral variables in old male B6C3F1 mice. Chronobiol Int. 1990;7:291–303. doi: 10.1080/07420529009064635. [DOI] [PubMed] [Google Scholar]
- 186.Acosta-Rodríguez VA, de Groot MHM, Rijo-Ferreira F, Green CB, Takahashi JS. Mice under caloric restriction self-impose a temporal restriction of food intake as revealed by an automated feeder system. Cell Metab. 2017;26:267–277. doi: 10.1016/j.cmet.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Schupp M, et al. Metabolite and transcriptome analysis during fasting suggest a role for the p53-Ddit4 axis in major metabolic tissues. BMC Genomics. 2013;14:758. doi: 10.1186/1471-2164-14-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Askew EW, Dohm GL, Huston RL. Fatty acid and ketone body metabolism in the rat: response to diet and exercise. J Nutr. 1975;105:1422–1432. doi: 10.1093/jn/105.11.1422. [DOI] [PubMed] [Google Scholar]
- 189.Raja G, Bräu L, Palmer TN, Fournier PA. Repeated bouts of high-intensity exercise and muscle glycogen sparing in the rat. J Exp Biol. 2003;206:2159–2166. doi: 10.1242/jeb.00416. [DOI] [PubMed] [Google Scholar]