

ABSTRACT
Cyclic diguanylate (c-di-GMP) is a second messenger that regulates the transition from motile to sessile lifestyles in numerous bacteria and controls virulence factor production in a variety of pathogens. In Clostridium difficile, c-di-GMP negatively regulates flagellum biosynthesis and swimming motility and promotes the production of type IV pili (TFP), biofilm formation, and surface motility in vitro. Flagella have been identified as colonization factors in C. difficile, but the role of TFP in adherence to host cells and in colonization of the mammalian gut is unknown. Here we show that c-di-GMP promotes adherence to epithelial cells in vitro, which can be partly attributed to the loss of flagella. Using TFP-null mutants, we demonstrate that adherence to epithelial cells is partially mediated by TFP and that this TFP-mediated adherence requires c-di-GMP regulation. In a mouse model of colonization, the TFP-null mutants initially colonized the intestine as well as the parental strain but were cleared more quickly. Moreover, compared to the parent strain, C. difficile strains lacking TFP were particularly deficient in association with the cecal mucosa. Together these data indicate that TFP and their positive regulation by c-di-GMP promote attachment of C. difficile to the intestinal epithelium and contribute to persistence of C. difficile in the host intestine.
KEYWORDS: Clostridioides difficile, colonization, adhesin, type IV pili, c-di-GMP, riboswitch, TcdR, Clostridioides
INTRODUCTION
Clostridium difficile, recently reclassified as Clostridioides difficile (1), is a spore-forming, obligate anaerobe responsible for diarrheal diseases resulting in substantial morbidity and mortality (2). C. difficile infections (CDI) usually occur following antibiotic therapy; however, community-associated C. difficile infections in patients without recent antibiotic use are increasingly common (3). Moreover, ∼20 to 30% of CDI patients experience at least one recurrence of CDI after cessation of treatment, with additional recurrences becoming increasingly likely (4–6). Antibiotic treatment facilitates C. difficile infection by disrupting the normally protective microbiota, reducing competition from other bacteria, and altering the bile salt profile in the gut, allowing more-efficient germination and outgrowth of C. difficile (7–10). Actively growing C. difficile may produce one or both of the toxins TcdB and TcdA, which glucosylate Rho family GTPases in target cells, ultimately resulting in the disruption of the intestinal epithelium and the characteristic inflammation of CDI (11–13). Despite being essential for disease development, TcdA and TcdB are not required for colonization of humans or animal models of CDI (14–17). The factors involved in C. difficile colonization and persistence within the gut are largely unknown.
One of the best-studied colonization factors in C. difficile is the flagellum. In the epidemic-associated C. difficile strain R20291 (18), a mutation in fliC, which encodes flagellin, reduced adherence to Caco-2 epithelial cells in vitro (19). Additionally, fliC mutant bacteria were outcompeted by the parental strain in mouse coinfection experiments (19). In strain R20291, a point mutation in the flagellar motor gene motB, which results in bacteria that produce paralyzed flagella, did not influence colonization or attachment to the cecum in mice, indicating that the flagellum itself functions in adherence (19). However, in strain 630Δerm, an erythromycin-sensitive derivative of C. difficile strain 630 (20), mutations in fliC and fliD increased attachment to Caco-2 cells, increased virulence in hamster models of infection, and led to a modest defect in mouse cochallenge infections with the parental strain (19, 21, 22). These strain-specific phenotypes may be attributable to differences in the abilities of strains R20291 and 630Δerm to phase vary the production of flagella (23–26).
Other cell surface proteins have been implicated in adherence to epithelial cells in vitro, including SlpA, FbpA, and Cwp66 (27–31). While most of these proteins have not been shown to alter colonization of C. difficile in animal models, a mutant lacking the putative fructose binding protein FbpA displayed a modest defect in cecal colonization in a mouse model of C. difficile infection (30). Given these data and the lack of severe colonization defects reported for putative C. difficile colonization factors, it is likely that adhesion of C. difficile to the intestinal epithelium involves multiple, potentially redundant factors. Type IV pili (TFP) are involved in a number of bacterial behaviors in other species, including surface attachment, surface-based twitching motility, biofilm formation, and cell-cell interactions (32–34). In many Gram-negative pathogens, TFP contribute to adherence to both primary and transformed host cell lines (35–38). Accordingly, TFP are required for optimal colonization in animal models for a number of pathogens, such as Pseudomonas aeruginosa, Vibrio cholerae, Escherichia coli, and others (39–43). There are a number of mechanisms by which TFP machinery may enhance colonization, including direct adhesion to host cells (44), promotion of microcolony formation (45), and epithelial cell invasion (46). In Gram-positive bacteria, TFP have not been as well studied. Genes encoding TFP are rare in most bacilli but are nearly ubiquitous among clostridia (47). TFP are required for gliding motility in Clostridium perfringens (48). Ectopic expression of a pilin gene from C. perfringens in Neisseria gonorrhoeae increased attachment to mouse myoblasts, suggesting a role in adherence for TFP in C. perfringens (49).
TFP genes have been found in every sequenced strain of C. difficile, and a comparative phylogenomics analysis of C. difficile indicates that TFP genes are among the set of core genes for the species (50, 51). Work from our lab and others revealed that TFP in C. difficile are critical for a number of bacterial behaviors, particularly in response to increased levels of the second messenger cyclic diguanylate (c-di-GMP) (52–54). In many bacterial species, c-di-GMP regulates the switch between motile and nonmotile states (55, 56). Likewise in C. difficile, c-di-GMP negatively regulates flagellar swimming motility and positively regulates TFP biosynthesis and TFP-dependent behaviors (52, 53, 57). Regulation occurs via c-di-GMP-specific riboswitches upstream of the respective flagellum and TFP biosynthetic operons, where the flagellar riboswitch is an “off switch” and the TFP riboswitch is an “on switch” (53, 57–59). An insertional mutation in the major pilin gene pilA1 or in pilB1, which encodes the pilus assembly ATPase, results in bacteria that lack TFP (52, 53, 60). The pilA1 and pilB1 mutants display reduced autoaggregation, surface motility, and biofilm formation under conditions of high c-di-GMP (52, 53). While the contributions of biofilm formation and surface growth to C. difficile infection are unclear, bacterial mats have been observed in mouse models of C. difficile infection (61, 62).
Based on the known roles of TFP in host colonization by other pathogens and the importance of TFP to autoaggregation and surface behaviors of C. difficile, we hypothesized that TFP contribute to host cell attachment and colonization during C. difficile infection. Moreover, we postulated that c-di-GMP positively regulates TFP-mediated interactions with host tissues. In this study, we found that high c-di-GMP promotes early attachment to a variety of epithelial cell types in vitro and that TFP may be important for maintenance of adherence to epithelial cells. Using an antibiotic-treated mouse model, we further demonstrated that C. difficile mutants lacking TFP initially colonize as well as the parent strain but are eliminated more quickly from the intestine. This study provides evidence that TFP promote adherence of C. difficile to epithelial cells and are important for the persistence of C. difficile in the mammalian intestinal tract.
RESULTS
c-di-GMP promotes attachment of C. difficile to intestinal epithelial cells.
Work from our lab and others recently demonstrated that high intracellular c-di-GMP concentrations in C. difficile promote autoaggregation, biofilm formation, and surface motility (52, 53, 59). Because c-di-GMP increased both interbacterial interaction and attachment to a surface, we hypothesized that c-di-GMP also promotes attachment to epithelial cells. To test this hypothesis, we performed bacterial attachment assays under anaerobic conditions using monolayers of HT-29 and Caco-2 human intestinal epithelial cells. We used a previously described strategy to artificially increase intracellular concentrations of c-di-GMP in C. difficile, using the nisin-inducible expression of dccA, which encodes a C. difficile diguanylate cyclase (57). This concentration of nisin and level of dccA expression did not substantially inhibit growth, particularly at the mid-exponential phase, when the bacteria were collected (53). C. difficile strains grown with or without nisin to induce dccA expression and stimulate c-di-GMP production were incubated with HT-29 or Caco-2 cells. Incubations were limited to 1 h under strict anaerobic conditions (required by C. difficile), as the monolayers begin to lose integrity by 4 h (see Fig. S1 in the supplemental material). Expression of dccA in 630Δerm led to 12- and 96-fold increases in attachment to HT-29 and Caco-2 cell monolayers, respectively, compared to C. difficile bearing the control vector grown with nisin induction (Fig. 1A and B). In contrast, expression of dccAmut, which encodes a catalytically inactive variant of DccA, did not significantly alter attachment to either cell line. To confirm that the C. difficile isolates were associated with the epithelial cells in these assays, we visualized the bacteria attached to the epithelial cells using a Gram stain. While the bacteria harboring the vector and pDccAmut were sparsely and individually distributed on the surface of the epithelial cell monolayers, the bacteria harboring pDccA were present in much higher numbers and also clustered together (Fig. 1C, D, and E). Together these data indicate that c-di-GMP promotes attachment of C. difficile to intestinal epithelial cells.
FIG 1.

c-di-GMP promotes C. difficile attachment to epithelial cell monolayers in vitro. (A and B) C. difficile strain 630Δerm bacteria containing vector (pMC-Pcpr), pDccA, or pDccAmut were grown to mid-log phase in BHIS with 10 μg/ml thiamphenicol to maintain the plasmids and 1 μg/ml nisin to induce expression as indicated. Bacteria were added to HT-29 (A) or Caco-2 (B) epithelial cell monolayers, and the mixture was centrifuged briefly and incubated for 1 h. Serial dilutions of bacterial inoculums and outputs were plated, and CFU were enumerated to determine the percentage of bacteria that remained attached. The means and standard deviations for 3 biological replicates are shown. Data were analyzed using one-way analysis of variance (ANOVA) and Tukey's posttest. **, P < 0.01; ***, P < 0.001. (C to E) C. difficile containing the indicated plasmids was grown to mid-log phase as described for panels A and B and added to HT-29 cell monolayers grown on coverslips. After 1 h of incubation, coverslips were rinsed with PBS and stained with crystal violet and safranin counterstain. C. difficile cells are stained dark purple (indicated by white arrows), and the HT-29 cell monolayers are stained red. Bars, 10 μm. Images are representative of 4 biological replicates for each strain.
Negative regulation of flagellum biosynthesis by c-di-GMP contributes to increased attachment to epithelial cells in vitro.
Dingle et al. previously showed that C. difficile 630Δerm isolates with mutations in fliC or fliD, encoding flagellin or flagellar cap, respectively, are more adherent to Caco-2 monolayers (21). We thus tested whether inhibition of flagellum biosynthesis by c-di-GMP is responsible for the increased attachment. We used C. difficile 630Δerm with an intron insertion in sigD, which encodes the flagellar sigma factor (63); this mutant was previously shown to lack flagella and swimming motility (23, 53, 64). We compared the attachment rates of the sigD mutant bearing vector alone or complemented with a plasmid containing sigD under the control of a nisin-inducible cpr promoter to the attachment rates of the parental strain bearing vector. Consistent with the previous results showing that aflagellate C. difficile isolates are more adherent to tissue culture cells, the 630Δerm sigD mutant containing vector alone adhered to HT-29 cells at significantly higher levels than those of the parental vector-bearing strain (Fig. 2A). Complementation with the sigD gene under the control of the nisin-inducible cpr promoter restored adherence to parental levels (Fig. 2A). We next evaluated the effect of increasing c-di-GMP in the absence of flagella by stimulating c-di-GMP synthesis in the sigD mutant background. Expression of dccA in the sigD mutant strain resulted in a further 3.8-fold increase in attachment over the sigD mutant bearing vector or expressing the dccAmut allele (Fig. 2B). This result indicates that additional, sigD-independent factors mediate increased adherence to epithelial cells in response to c-di-GMP.
FIG 2.

Aflagellate C. difficile adheres better than flagellate C. difficile to HT-29 cell monolayers. C. difficile cultures were grown to mid-exponential phase in BHIS with 10 μg/ml thiamphenicol and 1 μg/ml nisin to induce gene expression as indicated. Bacteria were added to HT-29 epithelial cell monolayers, centrifuged briefly, and incubated for 1 h. Serial dilutions of bacterial inoculums and outputs were plated to enumerate CFU and determine the percentages of bacteria that remained attached. (A) Mutation of sigD increases attachment to HT-29 cells; (B) increasing c-di-GMP in the sigD mutant further promotes attachment to HT-29 cells. Strain backgrounds and plasmids are indicated. The means and standard deviations for 3 biological replicates are shown. Data were analyzed by one-way ANOVA and Tukey's posttest. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Type IV pili promote adherence of C. difficile to epithelial cells.
Because c-di-GMP promotes TFP production, we hypothesized that TFP contribute to the observed increase in adherence to epithelial cells. To test this, we examined the ability of 630Δerm with a mutation in pilA1 (encoding the major pilin) or pilB1 (encoding the pilus biosynthesis ATPase) to adhere to HT-29 and Caco-2 cells. These mutants were previously shown to lack the ability to produce TFP under either low or high c-di-GMP conditions and to be deficient in TFP-dependent behaviors (52, 53). The pilA1 and pilB1 mutants showed somewhat reduced adherence to HT-29 cells compared to the parental strain, but the differences were not statistically significant (Fig. 3A). C. difficile with basal c-di-GMP levels produce relatively low numbers of TFP during growth in BHIS medium (37 g/liter Bacto brain heart infusion, 5 g/liter yeast extract) (53). We reasoned that increasing intracellular c-di-GMP by overexpressing dccA, which stimulates TFP biosynthesis (53), might reveal a role for TFP. However, elevating c-di-GMP augmented adherence equally in the parental and TFP-null strains (Fig. 3B).
FIG 3.

Attachment to HT-29 cell monolayers at 1 h is not dependent on type IV pili. Mid-exponential-phase cultures of C. difficile 630Δerm, pilA1 mutant, or pilB1 mutant were added to HT-29 epithelial cell monolayers, and the mixture was centrifuged briefly and incubated for 1 h. Serial dilutions of bacterial inoculums and outputs were plated and enumerated to determine the percentages of bacteria that remained attached. (A) Attachment of 630Δerm and TFP-null mutants to HT-29 cells; (B) attachment of 630Δerm and TFP-null mutants containing pDccA, grown in the presence or absence of 1 μg/ml nisin, to HT-29 cells. Shown are the means and standard deviations from 3 biological replicates. No statistically significant differences were observed between the parental strain and either mutant strain grown under the same conditions.
While 1-hour adherence assays effectively measure the initial attachment of the bacteria to the epithelial cells, the relatively short incubation may miss factors that are important for maintained adherence or expansion of the attached bacteria. Indeed, many experiments evaluating bacterial adherence to epithelial cells use longer coincubation periods, such as 24 h (65, 66). However, Caco-2 and HT-29 cell monolayers lose integrity after prolonged incubation under the strict anaerobic conditions required by C. difficile (Fig. S1). To assess adherence of C. difficile to epithelial cells over a longer time frame, we decided to use another epithelial cell line that is more tolerant of anaerobic conditions, Madin-Darby canine kidney (MDCK) epithelial cells (67). Whereas monolayers of either Caco-2 or HT-29 cells were substantially disrupted after as few as 4 h in the anaerobic chamber, MDCK cells maintained their monolayers after 24 h of incubation in the anaerobic chamber (Fig. S1). As with HT-29 and Caco-2 cells, TFP were dispensable for attachment to MDCK cells at 1 h (Fig. 4A). After 24 h of incubation, the C. difficile pilA1 and pilB1 mutants showed significantly reduced attachment to MDCK cells (Fig. 4B). Growth of pilA1 and pilB1 mutant bacteria was equivalent to that of the parental strain in Dulbecco's modified Eagle medium (DMEM) plus 10% fetal bovine serum (FBS) (see Fig. S2 in the supplemental material). The mutations also did not affect flagellum-based swimming motility (see Fig. S3 in the supplemental material).
FIG 4.

Type IV pili promote adherence to MDCK cell monolayers at 24 h. (A and B) Attachment of 630Δerm and TFP-null mutants after 1 h (A) or 24 h (B) of incubation with MDCK cell monolayers. In panel A, data are expressed as the percentages of the original inoculum recovered following incubation and PBS washes, with 3 biological replicates for each strain. In panel B, data are expressed as the total CFU recovered per well after 24 h of incubation, with 6 biological replicates. Data were analyzed using a one-way ANOVA with Dunnett's test for multiple comparisons. (C) Complementation of the adherence defect of the pilA1 mutant after 24 h of incubation with MDCK cell monolayers. Symbols represent values from individual animals, and error bars indicate the standard deviations. Data were analyzed by one-way ANOVA using the Holm-Sidak method to correct for multiple comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To confirm that the reduced attachment to MDCK cells was due to the lack of TFP, we complemented the pilA1 mutation by ectopic expression of pilA1. For expression of pilA1, we used the native promoter and leader sequence (5′ untranslated region [UTR]) containing the c-di-GMP-sensing riboswitch. Ectopic expression of pilA1 from its native promoter (pPilA1) partially restored attachment of the mutant bacteria to near that of the parental strain (Fig. 4C). Incomplete complementation is likely due to polar effects of the intron insertion on the downstream TFP biosynthesis genes (53). However, expression of pilA1 under the control of the riboswitch containing a mutation (A70G) rendering it unable to bind and sense c-di-GMP (53) did not complement the effect of the pilA mutation on adherence to MDCK cells (Fig. 4C, pPilA1mut). Together these data indicate that TFP contribute to host cell colonization at later time points, and sensing of c-di-GMP by the riboswitch is important in this process.
Type IV pili are dispensable at early stages of infection but are important for persistence in the host intestine.
In many bacterial pathogens, TFP play a role in colonization of the host and/or virulence (68). We therefore examined the contribution of TFP to the ability of C. difficile to colonize and cause disease in an antibiotic-treated mouse model of CDI (69). After treatment with a cocktail of antibiotics, C57BL/6 mice were inoculated by oral gavage with 105 spores of the 630Δerm parental strain, the pilA1 mutant, or the pilB1 mutant. The intestinal burden of C. difficile was monitored by collecting feces from the mice daily and enumerating colonies on TCCFA (see Materials and Methods), a medium that allows outgrowth of C. difficile spores while inhibiting the growth of other bacteria (70). The numbers of spores recovered from the feces of mice infected with a mutant or parent strain were similar for the first 3 days of the infection, suggesting that TFP are dispensable for initial colonization of the mouse large intestine (Fig. 5). By days 4 and 5, mice infected with the pilB1 mutant showed a modest but statistically significant decline in CFU compared to those infected with 630Δerm bacteria. Both the pilA1 and pilB1 mutant-infected animals shed fewer CFU in feces on day 6, although the differences did not reach statistical significance due to high variability among mice infected with the pilA1 or pilB1 mutants. On day 7, spore counts for the TFP mutant infections were below the limit of detection in all mice, while all but one mouse infected with 630Δerm were still shedding spores above the limit of detection (Fig. 5). An independent mouse colonization experiment yielded similar results, but with slight changes in the timing of the decline in bacterial burden.
FIG 5.

Single-strain infections of mice by 630Δerm, pilA1 mutant, and pilB1 mutant. Mice that had been pretreated with antibiotics were inoculated with 105 spores. Feces were collected daily, and serial dilutions were plated on C. difficile selective medium with spore germinant (TCCFA) to monitor the burden of C. difficile. Symbols represent values from individual animals. Data were analyzed by two-way ANOVA using the Holm-Sidak method to correct for multiple comparisons. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To control for variations between mice and to better capture subtle differences between strains, we performed coinfections with 630Δerm and either the pilA1 or pilB1 mutant. The ermB gene used to generate the pilA1 and pilB1 mutations allowed differentiation between the Erm-resistant mutants and the Erm-sensitive parent strain. A competition between the parent strain and a tcdR mutant, which expresses the glucosylating toxin genes at significantly lower levels (63, 71) but should be competent for colonization (14, 15, 17), was included as a control. On day 1 postinoculation, the competition indices (CI) were ∼1, indicating equal abilities to establish colonization. Throughout the 14-day experiment, the pilA1 mutant was usually present in feces at numbers comparable to those of the parent strain; the exceptions are days 7 and 9, when significantly fewer pilA1 mutant bacteria were recovered (Fig. 6A). In contrast, the pilB1 mutant showed attenuated colonization after day 1, with CI significantly less than 1 on days 3, 5, 7, 9, and 13 (Fig. 6B). The control experiment using the tcdR mutant yielded CI equal to or somewhat greater than 1 (Fig. 6C), indicating that TcdR is dispensable for colonization in the mouse model and that the presence of the ermB cassette is not responsible for the fitness defect of the mutants.
FIG 6.

C. difficile pilus mutants are outcompeted by the parental strain in murine coinfections. Mice were inoculated with mixed inoculums containing ∼105 spores of each strain. Feces was collected every 2 days starting 1 day postinoculation and plated on TCCFA (total spores) and TCCFA with erythromycin (mutant spores only) to determine bacterial burdens. Competitive indices (mutant to parent) for the competition between 630Δerm and pilA1 mutant (A), 630Δerm and pilB1 mutant (B), and 630Δerm and tcdR mutant (C). Data were excluded if in total fewer than 10 spores were recovered. Symbols represent CI values from individual animals, and error bars indicate the standard deviations. Data were analyzed by the Wilcoxon rank sum test comparing values to a hypothetical CI of 1 indicating no difference. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To ensure that the observed defects in colonization for the TFP-null mutants are not specific to the 630Δerm background, we also examined the contribution of TFP to colonization and persistence in an epidemic-associated strain of C. difficile, the ribotype 027 strain R20291 (18). When inoculated as a coinfection with the parental strain, the R20291 pilB1 mutant was initially recovered from feces in numbers comparable to those of the parental strain but was outcompeted by the parental strain on days 3 and 5 postinoculation (see Fig. S4 in the supplemental material). These data indicate that TFP are dispensable for initial establishment of colonization by multiple strains of C. difficile but support a role for TFP in maintaining colonization of the mouse intestine.
Type IV pili promote association of C. difficile with the cecal epithelium.
We hypothesized that TFP enhance intestinal colonization by promoting attachment of the bacteria to the intestinal epithelium as observed in vitro. To test this, we compared the ratios of TFP-null and parent strain bacteria associated with the cecal mucosa and present in the lumen. Mice were coinoculated with the pilB1 mutant and 630Δerm strains, and ceca were harvested at day 3, when the mutant first showed decreased recovery from feces (Fig. 6B). The luminal fraction consisted of the cecal contents combined with the contents of a single PBS wash. The remaining cecal tissue comprised the tissue-associated fraction. Homogenates of each fraction were plated on selective medium, and the CI were calculated separately. In the cecal lumen, the pilB1 mutant bacteria were recovered at a lower rate (geometric mean of CI = 0.27), but in the tissue-associated fraction the CI was significantly lower (geometric mean of CI = 0.12) (Fig. 7). These data indicate that the pilB1 mutant bacteria are less likely to be found in close association with the epithelium, which may explain the defect in maintenance of colonization in the dual-strain infections and the lack of persistence in single-strain infections.
FIG 7.
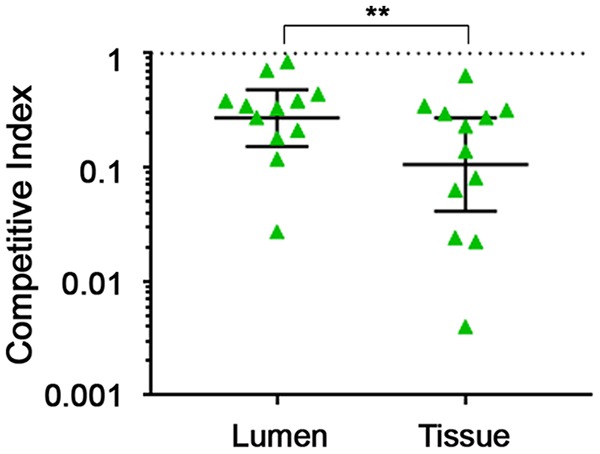
TFP promote association of C. difficile with the cecal epithelium. Mice were coinoculated with ∼105 spores each of 630Δerm and pilB1 mutant. Ceca were harvested anaerobically at day 3 postinoculation. Cecal contents and a 1-ml wash of PBS were combined into the luminal fraction. The tissue-associated fraction was obtained by homogenizing the remaining cecum. Serial dilutions of the luminal and tissue-associated fractions were plated on TCCFA (all C. difficile cells) and TCCFA with erythromycin (mutant C. difficile) to determine the bacterial burden of each strain. Symbols represent CI (mutant to parent) values from individual animals, and error bars indicate the standard deviations. Data were analyzed by the Wilcoxon rank sum test comparing the CI in the luminal fraction to the CI in the tissue-associated fraction. **, P < 0.01.
DISCUSSION
The mechanism by which C. difficile colonizes the host intestine is poorly understood, though multiple cell surface proteins and structures have been explored as possible adhesins. In C. difficile, c-di-GMP regulates the transition between motile and surface-associated states in part by negatively regulating flagellar gene expression and positively regulating TFP gene expression (52, 53). This study evaluated the contributions of c-di-GMP, flagella, and TFP to the ability of C. difficile to adhere to intestinal epithelial cells and the role of TFP in host colonization.
While previous work demonstrated a role for c-di-GMP in biofilm formation and autoaggregation of C. difficile, the effects of c-di-GMP signaling on the interaction between C. difficile and host cells had not been explored. In vitro adherence assays demonstrated that the second messenger c-di-GMP promotes attachment of C. difficile to HT-29 and Caco-2 colonic epithelial cells. Inhibition of flagellum biosynthesis by c-di-GMP is partially responsible for increased attachment, consistent with previous studies investigating the roles of flagellar proteins in C. difficile adherence (19, 21). It is possible that the presence of peritrichous flagella on the surface of C. difficile sterically interferes with the ability of adhesins on the bacterial surface to interact with the epithelial cells. In addition to removing the steric hindrance of the flagella, c-di-GMP may promote the presentation of other adhesins on the bacterial surface. Increasing intracellular c-di-GMP in a sigD mutant background led to a further increase in attachment, indicating that c-di-GMP regulates additional factors involved in attachment to epithelial cells. We hypothesized that TFP, which are positively regulated by c-di-GMP, function in this capacity. Our in vitro data indicate that TFP are not required for early attachment to epithelial cells but contribute to optimal prolonged adherence. Other putative adhesins regulated by c-di-GMP, such as CD2831 and CD3246, might promote early attachment of C. difficile to host cells (72, 73).
Our data also indicate that c-di-GMP signaling is required for promoting adherence by TFP. Restoration of adherence in the pilA1 mutant required a functional c-di-GMP riboswitch controlling pilA1 expression. Thus, c-di-GMP levels are high enough to support TFP production in these experiments. There is evidence that the interaction of C. difficile with a surface leads to increased intracellular c-di-GMP levels (52) and that stimulation of c-di-GMP production may occur during interactions with host cells as well. c-di-GMP may also posttranslationally regulate TFP biosynthesis in C. difficile. Some TFP are associated with PilB ATPases containing a MshEN domain, which binds c-di-GMP (74, 75). This c-di-GMP binding motif is conserved in the C. difficile MshE orthologue, PilB1, and potentially contributes to the positive regulation of TFP in C. difficile, as was recently shown for C. perfringens PilB2 (76). Future work will examine the potential roles of two distinct mechanisms of TFP regulation by c-di-GMP.
In a mouse model of CDI, both pilA1 and pilB1 mutant bacteria were deficient in long-term colonization of the intestine. The defect was more pronounced for the pilB1 mutant, suggesting that alternative pilins in addition to PilA1 are important for C. difficile persistence. The decreased recovery of the pilB1 mutant in the tissue-associated fraction of the cecum suggests a role for TFP in promoting close association with the intestinal epithelium. We speculate that C. difficile bacteria embedded more deeply in the mucus and/or in direct contact with epithelial cells are less likely to be excreted, serving as a reservoir for persistence and disease recurrence. While the importance of autoaggregation, microcolony formation, and biofilm development in the host for C. difficile disease development is unclear, mats of C. difficile associated with the intestinal epithelium of C. difficile-monoassociated mice (61, 62) and in mucus-associated mixed-species communities in the cecum and colon (77) have been observed. Given their role in autoaggregation, TFP may facilitate or stabilize the formation of single or multispecies microcolonies, and ultimately biofilms, on the epithelium, rather than function as adhesins per se.
Cell monolayers in general are not representative of the diverse range of cell types and structures found in the intestinal mucosa. Several groups have been working toward producing more physiologically relevant tissue culture models of the intestine that could improve the understanding of C. difficile interactions with host cells (78–81). For example, organoids and enteroids grown from primary stem or progenitor cells preserve some of the structure of intestinal villi and are also made up of a diverse array of cell types, unlike monocultures of transformed cell lines. While valuable for studying the disruption of the epithelial cell barrier by C. difficile toxins (82), these models may not be ideal for studying putative colonization factors because these are closed systems. Models using differentiated stem cells grown on scaffolds might enable coculture of C. difficile and epithelial cells by allowing oxygenation of the host cells at the basolateral surface while preserving an anaerobic environment in the apical space (81). Improvements to these models will allow for a much better understanding of intestinal colonization and host interaction of C. difficile and other anaerobes. Factors that influence C. difficile colonization and persistence may prove valuable in improving the treatment and prevention of CDI, especially in light of the high recurrence rate among patients treated for CDI (4–6).
MATERIALS AND METHODS
Bacterial growth conditions.
Strains and plasmids used in this study are listed in Table S1 in the supplemental material. Overnight cultures of C. difficile were grown in 2 ml of TY broth (30 g/liter Bacto tryptone, 20 g/liter yeast extract, 1 g/liter thioglycolate) in an anaerobic chamber (Coy Lab Products) with an atmosphere of 5% H2, 5% CO2, and 90% N2. For experiments, C. difficile was diluted 1:100 in BHIS (37 g/liter Bacto brain heart infusion, 5 g/liter yeast extract) for growth unless otherwise specified. E. coli cultures were grown in Luria-Bertani medium (Miller) with appropriate antibiotics as needed. Unless otherwise specified, antibiotics were used at the following concentrations: thiamphenicol (Tm), 10 μg/ml; chloramphenicol (Cm), 10 μg/ml; ampicillin (Amp), 100 μg/ml; and kanamycin (Kn), 100 μg/ml. Where appropriate, nisin was added at a final concentration of 1 μg/ml to induce transcription from the cpr promoter.
Construction of pilA1 complementation plasmids.
To generate pPilA1 (pMC123::PpilA1-RB-pilA1), the promoter, 5′ UTR, and coding sequence of pilA1 (CD630_35130) were amplified by PCR from 630Δerm genomic DNA using primers R1183 and R978 (see Table S2 in the supplemental material). This PCR product was digested with EcoRI and PstI, ligated into similarly digested pMC123, and transformed into DH5α. To generate pPilA1mut (pMC123::PpilA1-RBA70G-pilA1), we generated a PCR product containing the above fragment with an adenine-to-guanine substitution at position 70 of the riboswitch using splicing by overlap extension (SOE). The upstream region of homology was PCR amplified from 630Δerm genomic DNA using primers R1183 and R1184, and the downstream region was amplified using primers R1185 and R978. The two fragments were spliced together and amplified using R1183 and R978 and then cloned into pMC123 as above. Clones were confirmed by PCR and sequencing of the inserts. These pPilA1 and pPilA1mut plasmids were introduced into the pilA1 mutant via conjugation with HB101(pRK24) as described previously (57).
Attachment assays.
HT-29 or Caco-2 human intestinal epithelial cells were cultured in DMEM supplemented with FBS at 10% (HT-29) or 20% (Caco-2). Tissue culture-treated 24-well plates (Corning) were seeded with approximately ∼105 cells per well. Tissue culture cells were grown at 37°C in 5% CO2 for 5 to 7 days until a confluent monolayer was achieved, with fresh medium added as needed. To avoid oxygen toxicity to C. difficile, prior to inoculation the intestinal cells were transferred to the anaerobic chamber, and the medium was removed and replaced with anaerobic DMEM containing the appropriate concentration of FBS. For 24-h attachment assays, MDCK cells grown in DMEM with 10% FBS were seeded into 24-well plates with 105 cells per well. Cells were grown at 37°C for 3 days in 5% CO2 to allow the formation of confluent monolayers, with fresh medium added as needed.
C. difficile strains grown overnight (∼16 h) in TY with appropriate antibiotics were diluted 1:50 in filter-sterilized BHIS with antibiotics. Nisin was added if necessary for induction of gene expression. At mid-exponential phase, the bacteria were diluted 1:10 in DMEM with FBS and vortexed. We previously showed that C. difficile does not significantly aggregate at this stage of growth (53). Each diluted culture (50 μl) was added to 24-well plates seeded with HT-29, Caco-2, or MDCK cells and containing 450 μl DMEM with FBS. The plates were sealed with tape, removed from the anaerobic chamber, and centrifuged for 10 min at 1,000 × g to sediment the bacteria onto the tissue culture cells. This centrifugation step was performed because c-di-GMP inhibits flagellum biosynthesis and swimming motility, which might influence the ability of some strains tested to reach the cell monolayers. The plates were transferred back into the anaerobic chamber and incubated at 37°C for 1 h (HT-29 and Caco-2 cells) or 24 h (MDCK cells). During the incubation, dilutions of the bacterial inoculums were plated to enumerate the bacteria added to each well. Following incubation, the medium was removed, and the wells were washed 3 times with 1 ml Dulbecco's phosphate-buffered saline (DPBS; Gibco) to remove nonadherent bacteria. After the final wash, the epithelial cells and attached bacteria were scraped from the plate and suspended in 500 μl of DPBS by pipetting up and down until visible clumps were dispersed. Dilutions were plated on BHIS to enumerate the attached bacteria.
Microscopy.
HT-29 or MDCK cells were seeded at ∼105 cells per cm2 on Thermanox (ThermoFisher) coverslips and grown for 5 days (HT-29) or 3 days (MDCK) in DMEM–10% FBS. Bacteria were grown and added to the tissue culture cells, and the mixtures were incubated as described above for the attachment assays. Following incubation, the monolayers were washed twice with 500 μl PBS to remove unattached bacteria and then fixed by adding 500 μl of ice-cold methanol for 5 min. Methanol was removed, and samples were then stained with a Gram stain kit (Becton Dickinson), mounted on slides, and imaged with an Olympus BX61 v2 microscope at a magnification of ×600.
Animal experiments.
All animal studies were done in compliance with protocols approved by the UNC-CH Institutional Animal Care and Use Committee.
C. difficile spore inoculums were generated by streaking several colonies of C. difficile onto 70:30 agar (83) and incubating them at 37°C for 3 days in an anaerobic chamber. The growth was suspended in 10 ml DPBS, removed from the anaerobic chamber, and left overnight at room temperature to allow the release of mature spores. Spores were purified from the suspensions by sucrose gradient as previously described (84). Spores were enumerated by plating serial dilutions on BHIS agar containing the germinant 0.1% sodium taurocholate (85).
Groups of 8- to 10-week-old female C57BL/6 mice were obtained from Charles River Laboratories. Beginning 7 days prior to inoculation, the mice were given a cocktail of antibiotics in their drinking water, provided ab libitum for 3 days as described previously (69). The antibiotics were provided at the following concentrations: kanamycin (400 μg/ml), gentamicin (35 μg/ml), colistin (850 units/ml), vancomycin (45 μg/ml), and metronidazole (215 μg/ml) (69). Four days prior to inoculation, mice were switched back to regular water for the remainder of the experiment. A single intraperitoneal injection of clindamycin (10 μg/g body weight) was administered either 24 h prior to infection (single-strain infections) or 48 h prior to infection (competitions). The timing of the clindamycin injection was modified to 48 h prior to inoculation in the competition experiments to ensure that clindamycin concentrations did not favor growth of the mutants, which contain the ermB gene, which provides some resistance to clindamycin (86, 87). Mice were inoculated with 105 CFU of C. difficile spores (single-strain infections) or 2 × 105 total spores (coinfections) by oral gavage. Feces was collected in preweighed tubes every 24 or 48 h for 7 to 14 days, as indicated.
Fecal samples were weighed following collection and then suspended in 1 ml DPBS by vortexing. For single-strain infections, serial dilutions of fecal samples were plated on fructose agar containing 1 mg/ml sodium taurocholate, 16 μg/ml cefoxitin, and 250 μg/ml cycloserine (TCCFA). The burden of spores was calculated as CFU per gram of feces. For competition experiments, serial dilutions of fecal samples were plated on TCCFA to enumerate total spore burden, as well as TCCFA containing 2 μg/ml erythromycin (for 630Δerm strains) or 20 μg/ml lincomycin (for R20291 strains) to enumerate the CFU of mutant bacteria containing the ermB resistance gene. To calculate the competitive index (CI), the ratio of mutant to parental bacteria for each fecal sample, obtained with the formula (resistant CFU/[total CFU − resistant CFU])output, was divided by the ratio of mutant to parental bacteria in the initial spore inoculum, obtained with the formula (resistant CFU/[total CFU − resistant CFU])input. A CI of 1 indicates no difference in bacterial burden, and a CI of <1 indicates that the parental strain outnumbers the mutant, while a CI of >1 indicates that the mutant bacteria outnumber the parental strain bacteria.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kristen White at the UNC Microscopy Services Laboratory for help with microscopy and Brandon Anjuwon-Foster for critical reading of the manuscript.
This research was supported by NIH award R01-AI107029 to R.T. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00943-17.
REFERENCES
- 1.Lawson PA, Citron DM, Tyrrell KL, Finegold SM. 2016. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O'Toole 1935) Prevot 1938. Anaerobe 40:95–99. doi: 10.1016/j.anaerobe.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta A, Khanna S. 2014. Community-acquired Clostridium difficile infection: an increasing public health threat. Infect Drug Resist 7:63–72. doi: 10.2147/IDR.S46780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vardakas KZ, Polyzos KA, Patouni K, Rafailidis PI, Samonis G, Falagas ME. 2012. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents 40:1–8. doi: 10.1016/j.ijantimicag.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Johnson S. 2009. Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcomes. J Infect 58:403–410. doi: 10.1016/j.jinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 6.Surawicz CM, Alexander J. 2011. Treatment of refractory and recurrent Clostridium difficile infection. Nat Rev Gastroenterol Hepatol 8:330–339. doi: 10.1038/nrgastro.2011.59. [DOI] [PubMed] [Google Scholar]
- 7.Wilson KH, Perini F. 1988. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun 56:2610–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorg JA, Sonenshein AL. 2010. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol 192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. 2012. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, Li ZJ, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. 1995. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270:13932–13936. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 12.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 13.Abt MC, McKenney PT, Pamer EG. 2016. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol 14:609–620. doi: 10.1038/nrmicro.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 16.Gerding DN, Meyer T, Lee C, Cohen SH, Murthy UK, Poirier A, Van Schooneveld TC, Pardi DS, Ramos A, Barron MA, Chen H, Villano S. 2015. Administration of spores of nontoxigenic Clostridium difficile strain M3 for prevention of recurrent C. difficile infection: a randomized clinical trial. JAMA 313:1719–1727. doi: 10.1001/jama.2015.3725. [DOI] [PubMed] [Google Scholar]
- 17.Zhang K, Zhao S, Wang Y, Zhu X, Shen H, Chen Y, Sun X. 2015. The non-toxigenic Clostridium difficile CD37 protects mice against infection with a BI/NAP1/027 type of C. difficile strain. Anaerobe 36:49–52. doi: 10.1016/j.anaerobe.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baban ST, Kuehne SA, Barketi-Klai A, Cartman ST, Kelly ML, Hardie KR, Kansau I, Collignon A, Minton NP. 2013. The role of flagella in Clostridium difficile pathogenesis: comparison between a non-epidemic and an epidemic strain. PLoS One 8:e73026. doi: 10.1371/journal.pone.0073026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J Med Microbiol 54:137–141. doi: 10.1099/jmm.0.45790-0. [DOI] [PubMed] [Google Scholar]
- 21.Dingle TC, Mulvey GL, Armstrong GD. 2011. Mutagenic analysis of the Clostridium difficile flagellar proteins, FliC and FliD, and their contribution to virulence in hamsters. Infect Immun 79:4061–4067. doi: 10.1128/IAI.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aubry A, Hussack G, Chen W, Kuolee R, Twine SM, Fulton KM, Foote S, Carrillo CD, Tanha J, Logan SM. 2012. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect Immun 80:3521–3532. doi: 10.1128/IAI.00224-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anjuwon-Foster BR, Tamayo R. 2017. A genetic switch controls the production of flagella and toxins in Clostridium difficile. PLoS Genet 13:e1006701. doi: 10.1371/journal.pgen.1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anjuwon-Foster BR, Tamayo R. 2017. Phase variation of Clostridium difficile virulence factors. Gut Microbes 14:1–8. doi: 10.1080/19490976.2017.1362526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Twine SM, Reid CW, Aubry A, McMullin DR, Fulton KM, Austin J, Logan SM. 2009. Motility and flagellar glycosylation in Clostridium difficile. J Bacteriol 191:7050–7062. doi: 10.1128/JB.00861-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collery MM, Kuehne SA, McBride SM, Kelly ML, Monot M, Cockayne A, Dupuy B, Minton NP. 2016. What's a SNP between friends: the influence of single nucleotide polymorphisms on virulence and phenotypes of Clostridium difficile strain 630 and derivatives. Virulence 8:767–781. doi: 10.1080/21505594.2016.1237333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waligora AJ, Hennequin C, Mullany P, Bourlioux P, Collignon A, Karjalainen T. 2001. Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun 69:2144–2153. doi: 10.1128/IAI.69.4.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calabi E, Calabi F, Phillips AD, Fairweather NF. 2002. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun 70:5770–5778. doi: 10.1128/IAI.70.10.5770-5778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennequin C, Janoir C, Barc MC, Collignon A, Karjalainen T. 2003. Identification and characterization of a fibronectin-binding protein from Clostridium difficile. Microbiology 149:2779–2787. doi: 10.1099/mic.0.26145-0. [DOI] [PubMed] [Google Scholar]
- 30.Barketi-Klai A, Hoys S, Lambert-Bordes S, Collignon A, Kansau I. 2011. Role of fibronectin-binding protein A in Clostridium difficile intestinal colonization. J Med Microbiol 60:1155–1161. doi: 10.1099/jmm.0.029553-0. [DOI] [PubMed] [Google Scholar]
- 31.Merrigan MM, Venugopal A, Roxas JL, Anwar F, Mallozzi MJ, Roxas BA, Gerding DN, Viswanathan VK, Vedantam G. 2013. Surface-layer protein A (SlpA) is a major contributor to host-cell adherence of Clostridium difficile. PLoS One 8:e78404. doi: 10.1371/journal.pone.0078404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattick JS. 2002. Type IV pili and twitching motility. Annu Rev Microbiol 56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- 33.Anyan ME, Amiri A, Harvey CW, Tierra G, Morales-Soto N, Driscoll CM, Alber MS, Shrout JD. 2014. Type IV pili interactions promote intercellular association and moderate swarming of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 111:18013–18018. doi: 10.1073/pnas.1414661111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merz AJ, So M, Sheetz MP. 2000. Pilus retraction powers bacterial twitching motility. Nature 407:98–102. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- 35.Irvin RT, Doig P, Lee KK, Sastry PA, Paranchych W, Todd T, Hodges RS. 1989. Characterization of the Pseudomonas aeruginosa pilus adhesin: confirmation that the pilin structural protein subunit contains a human epithelial cell-binding domain. Infect Immun 57:3720–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.James-Holmquest AN, Swanson J, Buchanan TM, Wende RD, Williams RP. 1974. Differential attachment by piliated and nonpiliated Neisseria gonorrhoeae to human sperm. Infect Immun 9:897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woods DE, Straus DC, Johansen WG Jr, Berry VK, Bass JA. 1980. Role of pili in adherence of Pseudomonas aeruginosa to mammalian buccal epithelial cells. Infect Immun 29:1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knutton S, Shaw RK, Anantha RP, Donnenberg MS, Zorgani AA. 1999. The type IV bundle-forming pilus of enteropathogenic Escherichia coli undergoes dramatic alterations in structure associated with bacterial adherence, aggregation and dispersal. Mol Microbiol 33:499–509. doi: 10.1046/j.1365-2958.1999.01495.x. [DOI] [PubMed] [Google Scholar]
- 39.Bieber D, Ramer SW, Wu CY, Murray WJ, Tobe T, Fernandez R, Schoolnik GK. 1998. Type IV pili, transient bacterial aggregates, and virulence of enteropathogenic Escherichia coli. Science 280:2114–2118. doi: 10.1126/science.280.5372.2114. [DOI] [PubMed] [Google Scholar]
- 40.Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. 1999. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun 67:3625–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mundy R, Pickard D, Wilson RK, Simmons CP, Dougan G, Frankel G. 2003. Identification of a novel type IV pilus gene cluster required for gastrointestinal colonization of Citrobacter rodentium. Mol Microbiol 48:795–809. doi: 10.1046/j.1365-2958.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 43.Xicohtencatl-Cortes J, Monteiro-Neto V, Ledesma MA, Jordan DM, Francetic O, Kaper JB, Puente JL, Giron JA. 2007. Intestinal adherence associated with type IV pili of enterohemorrhagic Escherichia coli O157:H7. J Clin Invest 117:3519–3529. doi: 10.1172/JCI30727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farinha MA, Conway BD, Glasier LM, Ellert NW, Irvin RT, Sherburne R, Paranchych W. 1994. Alteration of the pilin adhesin of Pseudomonas aeruginosa PAO results in normal pilus biogenesis but a loss of adherence to human pneumocyte cells and decreased virulence in mice. Infect Immun 62:4118–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirn TJ, Lafferty MJ, Sandoe CM, Taylor RK. 2000. Delineation of pilin domains required for bacterial association into microcolonies and intestinal colonization by Vibrio cholerae. Mol Microbiol 35:896–910. doi: 10.1046/j.1365-2958.2000.01764.x. [DOI] [PubMed] [Google Scholar]
- 46.Zolfaghar I, Evans DJ, Fleiszig SM. 2003. Twitching motility contributes to the role of pili in corneal infection caused by Pseudomonas aeruginosa. Infect Immun 71:5389–5393. doi: 10.1128/IAI.71.9.5389-5393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melville S, Craig L. 2013. Type IV pili in Gram-positive bacteria. Microbiol Mol Biol Rev 77:323–341. doi: 10.1128/MMBR.00063-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varga JJ, Nguyen V, O'Brien DK, Rodgers K, Walker RA, Melville SB. 2006. Type IV pili-dependent gliding motility in the Gram-positive pathogen Clostridium perfringens and other Clostridia. Mol Microbiol 62:680–694. doi: 10.1111/j.1365-2958.2006.05414.x. [DOI] [PubMed] [Google Scholar]
- 49.Rodgers K, Arvidson CG, Melville S. 2011. Expression of a Clostridium perfringens type IV pilin by Neisseria gonorrhoeae mediates adherence to muscle cells. Infect Immun 79:3096–3105. doi: 10.1128/IAI.00909-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piepenbrink KH, Maldarelli GA, de la Pena CF, Mulvey GL, Snyder GA, De Masi L, von Rosenvinge EC, Gunther S, Armstrong GD, Donnenberg MS, Sundberg EJ. 2014. Structure of Clostridium difficile PilJ exhibits unprecedented divergence from known type IV pilins. J Biol Chem 289:4334–4345. doi: 10.1074/jbc.M113.534404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stabler RA, Gerding DN, Songer JG, Drudy D, Brazier JS, Trinh HT, Witney AA, Hinds J, Wren BW. 2006. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteriol 188:7297–7305. doi: 10.1128/JB.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purcell EB, McKee RW, Bordeleau E, Burrus V, Tamayo R. 2015. Regulation of type IV pili contributes to surface behaviors of historical and epidemic strains of Clostridium difficile. J Bacteriol 198:565–577. doi: 10.1128/JB.00816-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bordeleau E, Purcell EB, Lafontaine DA, Fortier LC, Tamayo R, Burrus V. 2015. Cyclic di-GMP riboswitch-regulated type IV pili contribute to aggregation of Clostridium difficile. J Bacteriol 197:819–832. doi: 10.1128/JB.02340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maldarelli GA, Piepenbrink KH, Scott AJ, Freiberg JA, Song Y, Achermann Y, Ernst RK, Shirtliff ME, Sundberg EJ, Donnenberg MS, von Rosenvinge EC. 2016. Type IV pili promote early biofilm formation by Clostridium difficile. Pathog Dis 74:ftw061. doi: 10.1093/femspd/ftw061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purcell EB, Tamayo R. 2016. Cyclic diguanylate signaling in Gram-positive bacteria. FEMS Microbiol Rev 40:753–773. doi: 10.1093/femsre/fuw013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell EB, McKee RW, McBride SM, Waters CM, Tamayo R. 2012. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J Bacteriol 194:3307–3316. doi: 10.1128/JB.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. 2008. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C, Sismeiro O, Semenova E, Severinov K, Le Bouguenec C, Coppee JY, Dupuy B, Martin-Verstraete I. 2013. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet 9:e1003493. doi: 10.1371/journal.pgen.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piepenbrink KH, Maldarelli GA, Martinez de la Pena CF, Dingle TC, Mulvey GL, Lee A, von Rosenvinge E, Armstrong GD, Donnenberg MS, Sundberg EJ. 2015. Structural and evolutionary analyses show unique stabilization strategies in the type IV pili of Clostridium difficile. Structure 23:385–396. doi: 10.1016/j.str.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lawley TD, Clare S, Walker AW, Goulding D, Stabler RA, Croucher N, Mastroeni P, Scott P, Raisen C, Mottram L, Fairweather NF, Wren BW, Parkhill J, Dougan G. 2009. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect Immun 77:3661–3669. doi: 10.1128/IAI.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soavelomandroso AP, Gaudin F, Hoys S, Nicolas V, Vedantam G, Janoir C, Bouttier S. 2017. Biofilm structures in a mono-associated mouse model of Clostridium difficile infection. Front Microbiol 8:2086. doi: 10.3389/fmicb.2017.02086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKee RW, Mangalea MR, Purcell EB, Borchardt EK, Tamayo R. 2013. The second messenger cyclic di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J Bacteriol 195:5174–5185. doi: 10.1128/JB.00501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.El Meouche I, Peltier J, Monot M, Soutourina O, Pestel-Caron M, Dupuy B, Pons JL. 2013. Characterization of the SigD regulon of C. difficile and its positive control of toxin production through the regulation of tcdR. PLoS One 8:e83748. doi: 10.1371/journal.pone.0083748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moreau-Marquis S, Redelman CV, Stanton BA, Anderson GG. 2010. Co-culture models of Pseudomonas aeruginosa biofilms grown on live human airway cells. J Vis Exp 2010(44):2186. doi: 10.3791/2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schiemann DA. 1995. Association with MDCK epithelial cells by Salmonella typhimurium is reduced during utilization of carbohydrates. Infect Immun 63:1462–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sheridan AM, Schwartz JH, Kroshian VM, Tercyak AM, Laraia J, Masino S, Lieberthal W. 1993. Renal mouse proximal tubular cells are more susceptible than MDCK cells to chemical anoxia. Am J Physiol 265:F342–F350. [DOI] [PubMed] [Google Scholar]
- 68.Craig L, Pique ME, Tainer JA. 2004. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- 69.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 70.Wilson KH, Kennedy MJ, Fekety FR. 1982. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J Clin Microbiol 15:443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mani N, Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci U S A 98:5844–5849. doi: 10.1073/pnas.101126598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hensbergen PJ, Klychnikov OI, Bakker D, van Winden VJ, Ras N, Kemp AC, Cordfunke RA, Dragan I, Deelder AM, Kuijper EJ, Corver J, Drijfhout JW, van Leeuwen HC. 2014. A novel secreted metalloprotease (CD2830) from Clostridium difficile cleaves specific proline sequences in LPXTG cell surface proteins. Mol Cell Proteomics 13:1231–1244. doi: 10.1074/mcp.M113.034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peltier J, Shaw HA, Couchman EC, Dawson LF, Yu L, Choudhary JS, Kaever V, Wren BW, Fairweather NF. 2015. Cyclic-di-GMP regulates production of sortase substrates of Clostridium difficile and their surface exposure through ZmpI protease-mediated cleavage. J Biol Chem 290:24453–24469. doi: 10.1074/jbc.M115.665091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones CJ, Utada A, Davis KR, Thongsomboon W, Sanchez DZ, Banakar V, Cegelski L, Wong GCL, Yildiz FH. 2015. C-di-GMP regulates motile to sessile transition by modulating MshA pili biogenesis and near-surface motility behavior in Vibrio cholerae. PLoS Pathog 11:e1005068. doi: 10.1371/journal.ppat.1005068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang YC, Chin KH, Tu ZL, He J, Jones CJ, Sanchez DZ, Yildiz FH, Galperin MY, Chou SH. 2016. Nucleotide binding by the widespread high-affinity cyclic di-GMP receptor MshEN domain. Nat Commun 7:12481. doi: 10.1038/ncomms12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hendrick WA, Orr MW, Murray SR, Lee VT, Melville SB. 2017. Cyclic di-GMP binding by an assembly ATPase (PilB2) and control of type IV pilin polymerization in the Gram-positive pathogen Clostridium perfringens. J Bacteriol 199:e00034-17. doi: 10.1128/JB.00034-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Semenyuk EG, Poroyko VA, Johnston PF, Jones SE, Knight KL, Gerding DN, Driks A. 2015. Analysis of bacterial communities during Clostridium difficile infection in the mouse. Infect Immun 83:4383–4391. doi: 10.1128/IAI.00145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. 2011. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 79.Pusch J, Votteler M, Gohler S, Engl Hampel JM, Walles H, Schenke-Layland K. 2011. The physiological performance of a three-dimensional model that mimics the microenvironment of the small intestine. Biomaterials 32:7469–7478. doi: 10.1016/j.biomaterials.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 80.Dedhia PH, Bertaux-Skeirik N, Zavros Y, Spence JR. 2016. Organoid models of human gastrointestinal development and disease. Gastroenterology 150:1098–1112. doi: 10.1053/j.gastro.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y, Gunasekara DB, Reed MI, DiSalvo M, Bultman SJ, Sims CE, Magness ST, Allbritton NL. 2017. A microengineered collagen scaffold for generating a polarized crypt-villus architecture of human small intestinal epithelium. Biomaterials 128:44–55. doi: 10.1016/j.biomaterials.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leslie JL, Huang S, Opp JS, Nagy MS, Kobayashi M, Young VB, Spence JR. 2015. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun 83:138–145. doi: 10.1128/IAI.02561-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Putnam EE, Nock AM, Lawley TD, Shen A. 2013. SpoIVA and SipL are Clostridium difficile spore morphogenetic proteins. J Bacteriol 195:1214–1225. doi: 10.1128/JB.02181-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Edwards AN, McBride SM. 2016. Isolating and purifying Clostridium difficile spores. Methods Mol Biol 1476:117–128. doi: 10.1007/978-1-4939-6361-4_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sorg JA, Sonenshein AL. 2008. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bakker D, Buckley AM, de Jong A, van Winden VJ, Verhoeks JP, Kuipers OP, Douce GR, Kuijper EJ, Smits WK, Corver J. 2014. The HtrA-like protease CD3284 modulates virulence of Clostridium difficile. Infect Immun 82:4222–4232. doi: 10.1128/IAI.02336-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kelly ML, Ng YK, Cartman ST, Collery MM, Cockayne A, Minton NP. 2016. Improving the reproducibility of the NAP1/B1/027 epidemic strain R20291 in the hamster model of infection. Anaerobe 39:51–53. doi: 10.1016/j.anaerobe.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.