

Abstract
Plasmodium falciparum parasites are purine auxotrophs that rely exclusively on the salvage of preformed purines from their human hosts to supply the requirement for purine nucleotides. Hypoxanthine-guanine-xanthine phosphor-ibosyltransferase (HGXPRT) catalyzes the freely reversible Mg2+-dependent conversion of 6-oxopurine bases to their respective nucleotides and inorganic pyrophosphate. The phosphoribosyl group is derived from 5-phospho-α-D-ribosyl 1-pyrophosphate (PRPP). The enzyme from malaria parasites (PfHGXPRT) is essential as hypoxanthine is the major precursor in purine metabolism. We used specific heavy atom labels in PRPP and hypoxanthine to measure primary (1-14C and 9-15N) and secondary (1-3H and 7-15N) intrinsic kinetic isotope effect (KIE) values for PfHGXPRT. Intrinsic isotope effects contain information for understanding enzymatic transition state properties. The transition state of PfHGXPRT was explored by matching KIE values predicted from quantum mechanical calculations to the intrinsic values determined experimentally. This approach provides information about PfHGXPRT transition state bond lengths, geometry, and atomic charge distribution. The transition state structure of PfHGXPRT was determined in the physiological direction of addition of ribose 5-phosphate to hypoxanthine by overcoming the chemical instability of PRPP. The transition state for PfHGXPRT forms nucleotides through a well-developed and near-symmetrical DN*AN, SN1-like transition state.
Graphical abstract
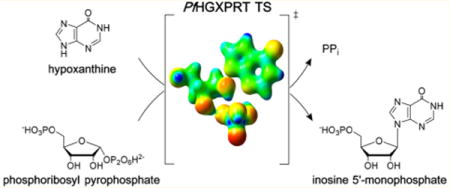
Extensive nucleic acid production occurs during the blood and liver stages of Plasmodium falciparum when there is a high demand for purine nucleotides. Like most parasitic protozoa, P. falciparum lacks a de novo purine nucleotide biosynthetic pathway1 and relies exclusively on the salvage of preformed purines from its human host. Thus, enzymes of the salvage pathway are essential to purine cofactor and nucleic acid synthetic pathways.2 Hypoxanthine is a key precursor salvaged for purine nucleotide synthesis in P. falciparum,3 and the most critical enzyme is hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRT; EC 2.4.2.8). This enzyme catalyzes the freely reversible Mg2+-dependent conversion of 6-oxopurine bases to their respective nucleoside monophosphates and inorganic pyrophosphate (PPi), with the phosphoribosyl group being derived from 5-phospho-α-D-ribosyl 1-pyrophosphate (PRPP) (Figure 1).
Figure 1.
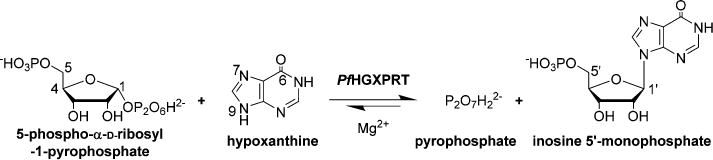
Physiological reaction for nucleotide synthesis by PfHGXPRT. The reaction is a freely reversible Mg2+-dependent conversion of 6-oxopurine bases to their respective nucleoside monophosphates and inorganic pyrophosphate (PPi), with the phosphoribosyl group being derived from 5-phospho-α-D-ribosyl 1-pyrophosphate (PRPP).
P. falciparum HGXPRT (PfHGXPRT) is similar to human HGPRT, whose genetic deficiency or insufficiency is related to Lesch-Nyhan syndrome or gouty arthritis, respectively.4,5 The human and P. falciparum phosphoribosyltransferases share a sequence identity of 44% and a similarity of 76%. The structures of both enzymes have been determined in complex with transition state analogues, PPi, and two Mg2+ ions.6,7 Their catalytic sites consist of a cleft between core and hood subdomains. Catalytic site residues that make contact with the reactants are identical. Kinetic mechanisms for the human,8 schistosomal,9 Trypanosoma cruzi,10 and more recently P. falciparum11 enzymes are reported to be ordered bi-bi substrate addition and product release, in which PRPP binds first to the enzyme, followed by 6-oxopurine binding. PPi dissociates first from the enzyme, followed by nucleotide release.
The considerable similarity between human and malarial enzymes is in contrast to their substrate specificity and enzyme activity. Only PfHGXPRT is capable of catalyzing the phosphoribosylation of xanthine.12 A comparison of kinetic constants of human HGPRT with PfHGXPRT indicates similar efficiencies of hypoxanthine salvage. Thus, the kcat/Km values are within a factor of 4 for the human and P. falciparum enzymes with hypoxanthine.13 Despite the similar catalytic efficiencies, the differences in substrate specificity for these enzymes make it possible to generate species-specific inhibitors, similar to the case for other systems.14 Several acyclic immucillin phosphonates have been described as nanomolar inhibitors of PfHGXPRT with specificity values that are 500–600 times greater for the parasite than for the human enzyme.15 The enzymes of purine salvage pathways have been targeted for the development of antimalarials since the discovery that Plasmodium parasites are purine auxotrophs.16 As PfHGXPRT is essential for P. falciparum survival,17 this enzyme continues to be of interest as a candidate for drug design.
Human malaria has been a leading cause of death in tropical nations,18 and the gradual development of P. falciparum drug resistant strains has created a need for new and selective chemotherapeutic agents. The coupling of kinetic isotope effects (KIEs) to computational chemistry has the potential to provide details about enzymatic transition states to guide the design of transition state analogues. When transition state features can be approximated into chemically stable mimics, inhibitors with femtomolar dissociation constants have been obtained.19
Transition state analysis of phosphoribosyltransferases has been reported for Salmonella typhimurium, human, and P. falciparum orotate phosphoribosyltransferases (OPRTs) and for human nicotinamide phosphoribosyltransferase.20–22 In every case, these transition states were studied from the direction of nucleoside monophosphate pyrophosphorolytic cleavage from the base and phosphorylated ribosyl groups. For orotate phosphoribosyltransferases, insignificant isotope effects were observed with pyrophosphate as the substrate, but use of phosphonoacetic acid, a slow-reacting analogue of pyrophosphate, permitted measurement of the KIE values. Nicotinamide phosphoribosyltransferase gave significant KIE values for the pyrophosphorolysis of nicotinamide mononucleotide; thus, its transition state structure was determined with the natural reactants but in the reverse physiological direction. No transition state structure of a phosphoribosyltransferase has been determined in the physiological direction using isotopically labeled PRPP as the substrate. The chemical instability of PRPP and the need for high-purity labeled reactants when measuring KIE values have contributed to this gap in the literature. For example, the half-life of PRPP in dilute formic acid (a chromatography eluent) has been reported to be from 1.8 to 10 min, depending on the conditions.23 We used enzymatic synthesis of labeled PRPPs followed by rapid purification under mild conditions to permit analysis of the transition state structure of PfHGXPRT using natural substrates in the physiological direction.
METHODS
Recombinant Enzyme Expression and Purification
The codon-optimized open reading frame, purchased from DNA2.0 and using the amino acid sequence from PfHGXPRT (Gene ID PF3D7_1012400, http://plasmodb.org/plasmo/), was inserted into pDEST-14, an inducible high-level expression plasmid. An N-terminal thrombin cleavable six-histidine tag was added to assist subsequent protein purification steps. Nucleotide sequencing validated the DNA sequence for PfHGXPRT. The pDEST-14-PfHGXPRT construct was transformed into Escherichia coli One Shot BL21-AI chemically competent cells (Invitrogen) and plated. A single overnight colony was grown in LB Carbenicillin (50 μg mL−1) medium at 37 °C and 200 rpm to an OD600 of 0.3–0.6, when the temperature was decreased to 18 °C. After 45 min, cells were induced by addition of L-arabinose (final concentration of 0.2%), grown for an additional 12–18 h, harvested by centrifugation (5000g for 20 min), and stored at −80 °C. All subsequent steps were performed at 4 °C, unless stated otherwise.
Cells were suspended in 20 mM Tris-HCl, 5 mM imidazole, 500 mM NaCl, 1 mM DTT (pH 7.9) (5 mL/g of cell pellet), with a spatula tip of both lysozyme and DNase I (Sigma), and protease inhibitor complete Mini EDTA-free (one tablet per 20 g of cell pellet; Roche). After being stirred for 30 min, the cells were disrupted by sonication (15 s on, 15 s off, at 30% amplitude for 30 min) and centrifuged (20000g for 20 min) to remove cell debris. The supernatant was incubated with Ni-NTA agarose (1.3 mL of slurry/g of cell pellet; Qiagen) for 45 min with rocking, and the mixture was poured into a column and washed with 12 column volumes of the cell suspension buffer. The collection of 4 column volume fractions from a 50 to 500 mM imidazole stepwise elution gradient was followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (200 V and 185 mA for 60 min in MOPS running buffer) analysis, and the fractions containing the target protein were pooled and dialyzed against 50 mM KH2PO4-KOH, 5 mM imidazole, 150 mM KCl, 1 mM DTT, and 10% glycerol (pH 7.5) using 10 kDa dialysis cassettes (Thermo Scientific). The enzyme solution was concentrated to approximately 300 μM (8 mg mL−1), and aliquots were flash-frozen in liquid nitrogen and stored at −80 °C.
Synthesis of Isotopically Labeled PRPP
Commercially available radiolabeled D-[6-14C,6-3H2]glucose and D-[1-14C,1-3H]ribose (American Radiolabeled Chemicals) were converted to [5-14C,5-3H2,1-14C,1-3H]PRPP by a sequence of coupled enzymatic reactions (Figure 2).
Figure 2.
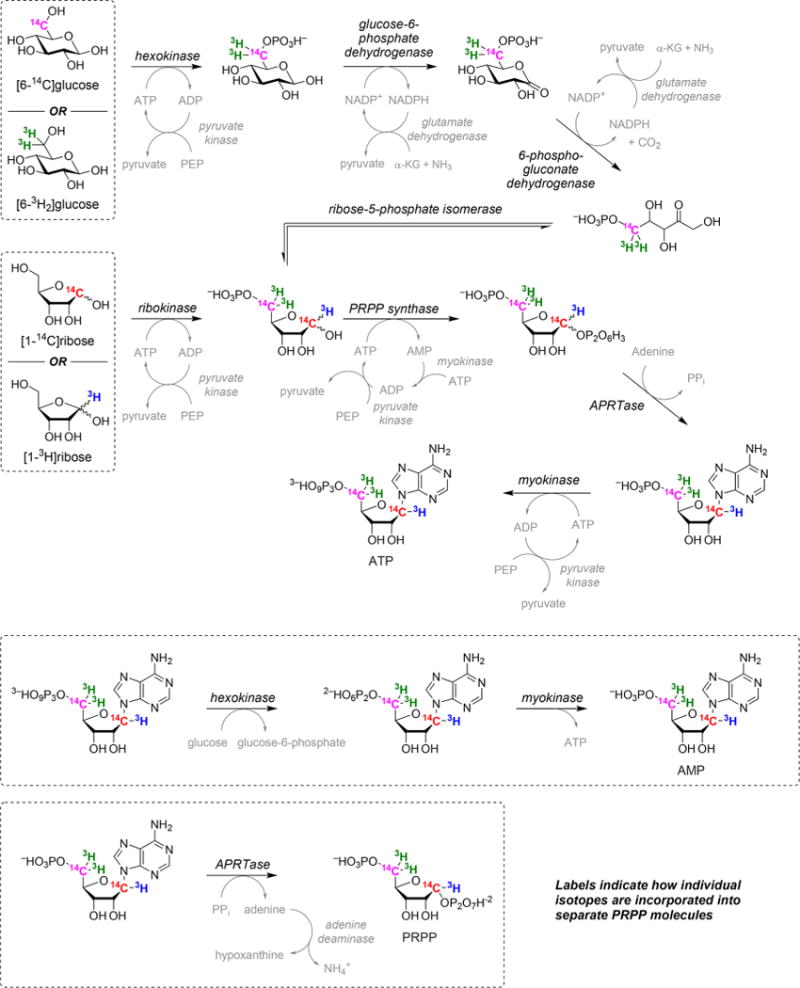
Enzymatic synthesis of isotopically labeled PRPP from glucose or ribose. The final product of the first synthesis reaction is ATP, which is subsequently converted to AMP. Isotopically labeled PRPP is produced in the final synthesis step and used as soon as possible following isolation.
PRPP with 5-14C and 5-3H2 labels was synthesized from D-[6-14C,6-3H2]glucose by preparing 1 mL reaction mixtures containing 2 mM D-glucose, 20 mM phosphoenolpyruvate, 200 μM adenosine 5′-triphosphate (ATP), 200 μM β-NADP+, 20 mM α-ketoglutaric acid, 5.5 mM ammonium chloride, 50 mM magnesium chloride, 2.5 mM adenine, 50 mM potassium chloride, 2.5 mM DTT, and 140 mM potassium phosphate, pH of 7.4. Reactions were initiated by adding 1 unit each of hexokinase, L-glutamic dehydrogenase, phosphoriboisomerase, 6-phosphogluconic dehydrogenase, glucose-6-phosphate dehydrogenase, adenine phosphoribosyltransferase (APRTase), myokinase, pyruvate kinase, and PRPP synthase. PRPP with 1-14C and 1-3H labels was synthesized from D-[1-14C,1-3H]-ribose by preparing 1 mL reaction mixtures containing 1 mM D-ribose, 20 mM phosphoenolpyruvate, 200 μM ATP, 50 mM magnesium chloride, 1.2 mM adenine, 50 mM potassium chloride, 1 mM DTT, and 100 mM potassium phosphate, pH of 7.4. Reactions were initiated by adding 1 unit each of ribokinase, APRTase, myokinase, pyruvate kinase, and PRPP synthase. After 10 h at room temperature, 25 units of hexokinase, 200 units of myokinase, and 6.25 mM glucose were added to each quenched reaction mixture (boiled and filtered) to produce the corresponding radiolabeled AMP from ATP (final product of the first synthesis reaction; 30 min at 37 °C). Radiolabeled AMPs were purified by high-performance liquid chromatography (HPLC) using a Delta Pak C18 15 μm, 300 Å, 300 mm × 3.9 mm column (Waters) in 50 mM potassium phosphate and 8 mM tetrabutylammonium bisulfate (pH 6.0) in an acetonitrile gradient. A subsequent acetonitrile gradient desalted the radiolabeled AMP for storage at −80 °C.
Each isotopically labeled PRPP was generated from its corresponding radiolabeled AMP and used immediately to minimize hydrolysis. The final synthesis step was performed in 330 μL reaction mixtures that contained 50 μM radiolabeled AMP, 1.4 mM PPi, 1.4 mM magnesium chloride, 100 μM DTT, and 50 mM potassium phosphate, pH of 7.4. Reactions were initiated by adding 1 unit each of APRTase and adenine deaminase. After 10 min at room temperature, each reaction was quenched with 5 mM EDTA (pH 8.0), and each mixture was filtered (10000 NMWL) at 4 °C and flash-frozen in liquid nitrogen. Each radiolabeled PRPP was purified by HPLC using a HiTrap QFF 5 mL column (GE Healthcare) on a 0 to 1.5 M linear gradient of ammonium acetate (pH 7.0) by tracking the radioactive counts (Figure S1), lyophilized (Figure S2), and used directly.
Synthesis of Isotopically Labeled Hypoxanthine
[7-15N]- and [9-15N]Adenine, synthesized from 4,6-diamino-5-[15N]nitrosopyrimidine and [4-15N]diamino-6-chloropyrimidine, respectively, as previously described,24 were used to synthesize [7-15N]- and [9-15N]hypoxanthine (Figure S3). [7-15N]- or [9-15N]Adenine was added to 400 μL reaction mixtures containing 500 μM labeled adenine, 0.1 mM DTT, and 50 mM potassium phosphate buffered to a pH of 7.4, and reactions were initiated by adding 1 unit of adenine deaminase to form [7-15N]- or [9-15N]hypoxanthine, respectively. Each synthesis reaction mixture was filtered (10000 NMWL) after 40 min at room temperature, and each labeled adenine was purified by HPLC using a Delta Pak C18 15 μm, 300 Å, 300 mm × 3.9 mm column (Waters) in 50 mM triethylammonium acetate (pH 5.0) in an acetonitrile gradient.
Measurement of Substrate-Based KIEs
Competitive Radiolabeled Approach
KIEs for the nucleotide synthesis reaction of PfHGXPRT were measured using the competitive radiolabeled approach. PRPP substrates with the appropriate isotopic label, deemed “heavy” substrates, 1-3H or 1-14C, were mixed with the corresponding “light” remote labeled PRPP containing a 5-14C or 5-3H2 label, respectively. Heavy and light PRPPs were mixed in a 3:1 3H:14C ratio of specific radioactivity, and HGXPRT reaction mixtures contained 10 μM PRPP, 2.5 mM hypoxanthine, 1.4 mM magnesium chloride, 0.1 mM DTT, and 50 mM potassium phosphate buffered to a pH of 7.4. KIE reactions were initiated by the addition of 114 μM PfHGXPRT and quenched after 20 min by the addition of 5 mM EDTA (pH 8.0) at fractions of conversion (f) between 0.1 and 0.7, and mixtures were flash-frozen in liquid nitrogen. Unreacted substrate (PRPP) and product (IMP) were purified by HPLC using a Luna C18(2) 15 μm, 100 Å, 250 mm × 4.6 mm column (Phenomenex) in 25 mM potassium phosphate and 4 mM tetrabutylammonium bisulfate (pH 6.0) in an acetonitrile gradient. This protocol resolved R5P, a PRPP breakdown product. R5P, IMP, and PRPP eluted from the column with retention times of 3.5, 13.5, and 25.5 min, respectively. These samples were dried by vacuum centrifugation using a Savant SC210A SpeedVac Concentrator (ThemoFisher Scientific), dissolved in 500 μL of H2O, and mixed with 10 mL of Ultima Gold scintillation fluid (PerkinElmer) for radiolabeled analysis in a Tri-Carb 2910 TR Liquid Scintillation Analyzer (PerkinElmer). Scintillation counting used a dual-channel analysis in which counts from tritium isotopes appeared in channel A while counts from 14C isotopes appeared in channels A and B. The counts per minute for each sample for tritium and 14C were used to measure sample specific radioactivity and were taken as an average of 10 count cycles of 10 min each. The counts per minute for 3H and 14C were determined using eqs 1 and 2
| (1) |
| (2) |
where r represents the fraction of 14C counts appearing in channel A.
Kinetic isotope effects were measured using eq 3,
| (3) |
where V/K is the observed KIE, f is the reaction fraction of conversion, R0 is the ratio of heavy substrate to light substrate when the reaction was initiated, and Rf is the ratio of heavy substrate to light substrate at fraction of conversion f. The f value was calculated by comparing the CPM of IMP to the total CPM of R5P, PRPP, and IMP. All reactions and procedures were performed at room temperature (25 °C).
1-14C KIEs were corrected for the remote 5-3H2 KIE using eq 4. Because remote 3H KIEs can report on distortional binding effects, they must be corrected for to obtain the V/K KIE values.
| (4) |
Mass Spectrometry
Kinetic isotope effects for PfHGXPRT were measured for hypoxanthine using 15N-labeled substrate as the “heavy” substrate, with unlabeled hypoxanthine as the “light” substrate. Heavy and light hypoxanthine were mixed at equal concentrations, and HGXPRT reaction mixtures contained 190 μM PRPP, 140 μM hypoxanthine, 1.4 mM magnesium chloride, 0.1 mM DTT, and 50 mM potassium phosphate buffered to a pH of 7.4. KIE reactions were initiated by the addition of 109 μM PfHGXPRT and quenched after 20 min by the addition of 5 mM EDTA (pH 8.0) at fractions of conversion (f) between 0.4 and 0.5, and mixtures were flash-frozen in liquid nitrogen. Unreacted hypoxanthine was purified from the remaining reaction contents by HPLC using a Luna C18(2) 15 μm, 100 Å, 250 mm × 4.6 mm column (Phenomenex) in 50 mM triethylammonium acetate (pH 5.0) in an acetonitrile gradient.
Purified hypoxanthine samples were dried by vacuum centrifugation and dissolved in 50 μL of MeOH, and the isotopic ratio of the contents of each hypoxanthine sample was assessed via mass spectrometry using an LTQ Orbitrap Velos Mass Spectrometer (ThermoFisher Scientific). The flow rate was maintained at 0.2 μL/min on 90% MeOH. The ratio of 15N-containing hypoxanthine to unlabeled hypoxanthine was then used to calculate the V/K KIE values for the 7-15N and 9-15N positions of hypoxanthine using eq 3. All reactions and procedures were performed at room temperature (22 °C).
Measurement of Forward Commitment Factor
The forward commitment factor (Cf) was measured for PfHGXPRT using the isotope trapping method developed by Rose.25 To estimate the Cf value for PRPP in the PfHGXPRT-catalyzed reaction, a 320 μL mixture contained 10 μM [5-14C]PRPP, 114 μM PfHGXPRT, 1.4 mM magnesium chloride, 0.1 mM DTT, and 50 mM potassium phosphate, pH of 7.4. Following preincubation, a 180 μL “chase” solution containing 2.5 mM hypoxanthine, 2 mM (200-fold) unlabeled PRPP, 1.4 mM magnesium chloride, 0.1 mM DTT, and 50 mM potassium phosphate, pH of 7.4 was added. Following the addition of the chase solution, 120 μL aliquots of the reaction mixture were quenched at 30, 60, 90, and 120 s by the addition of 5 mM EDTA (pH 8.0) and flash-frozen in liquid nitrogen. The product (IMP) from each time point was purified by HPLC using a Luna C18(2) 15 μm, 100 Å, 250 mm × 4.6 mm column (Phenomenex) in 25 mM potassium phosphate and 4 mM tetrabutylammonium bisulfate (pH 6.0) in an acetonitrile gradient. Samples were dried by vacuum centrifugation, dissolved in 500 μL of H2O, and mixed with 10 mL of scintillation fluid for radiolabeled analysis (as described above) to estimate product ([5′-14C]IMP) from the initial solution. The ratio of radiolabeled IMP to the initial concentration of the enzyme–substrate complex was plotted as a function of time to extrapolate to zero (Y), based on eq 5. Cf was calculated using eq 6. Finally, intrinsic KIEs for the PfHGXPRT-catalyzed reaction were obtained by correcting the experimental KIE values for Cf using eq 7.
| (5) |
| (6) |
| (7) |
Computational Methods
The transition state structure of PfHGXPRT was determined by comparing intrinsic KIE values to calculated KIE values for theoretical transition states generated in Gaussian 09 using the B3LYP level of theory and the 6-31g(d) basis set and ISOEFF98.26,27 The starting coordinates for the reaction were taken from the crystal structure of malarial purine phosphoribosyltransferase bound to (1S)-1-(9-deazahypoxanthin-9-yl)-1,4-dideoxy-1,4-imino-D-ribi-tol-5-phosphate and PPi [Protein Data Bank (PDB) entry 1CJB].7
The structure of PRPP was simplified to α-D-ribose 1-phosphate [to serve as a truncated PRPP (see Figure S4)] to simplify QM calculations. The full hypoxanthine structure was used with both a protonated and unprotonated N-7 position. Ground state structures were taken from the same crystal structure. All calculations were performed using water as an implicit solvent, and all structures were assigned a multiplicity of 1. Transition state structures were assigned a formal charge of 0; ground state α-D-ribose 1-phosphate (to serve as a truncated PRPP) was assigned a formal charge of 0, and ground state hypoxanthine was assigned a formal charge of 0 (monoprotonated N-7 or N-9) or +1 (diprotonated N-7 and N-9). Initial transition state calculations were performed by fixing the 1′-C–1-O(PPi) and 1′-C–N-9 bonds and varying their bond lengths from 1.6 to 3.0 Å in 0.2 Å increments. Transition state structures were refined from the results of the first calculation by varying the C–N bond length from 2.30 to 2.50 Å in 0.05 Å increments and by varying the C–O bond length from 2.50 to 2.70 Å in 0.05 Å increments. In a final refinement step, the C–O bond length was fixed at 2.60 Å and the C–N bond length was varied from 2.41 to 2.44 Å in 0.01 Å increments.
Calculated kinetic isotope effects (see Table 1) were found using ISOEFF98 from the scaled vibrational frequencies (SCFACT = 0.958) of transition state structures and ground state structures. Calculations were conducted using water as an implicit solvent and a temperature of 298.15 K. Calculated KIEs were then compared to experimentally measured intrinsic KIEs to determine theoretical transition state structures. The final transition state structure had a single imaginary vibrational frequency.
Table 1.
V/K KIEs, Intrinsic KIEs, and Calculated KIEs for PfHGXPRT at Four Atomic Positions
| heavy substrate | starting material | light substrate | type of KIE | V/K KIE | intrinsic KIE | calculated KIEe |
|---|---|---|---|---|---|---|
| 1-14C | [1-14C]ribose | 5-3H2 | PRPP primary | 1.025 ± 0.004b | 1.025 ± 0.004c | 1.031 |
| 1-3H | [1-3H]ribose | 5-14Ca | α-secondary Hypoxanthine | 1.205 ± 0.006 | 1.205 ± 0.006c | 1.212 |
| 7-15N | [7-15N]adenine | hypoxanthine | β-secondary | 0.988 ± 0.009 | N/Ad | 0.984 |
| 9-15N | [9-15N]adenine | hypoxanthine | primary | 0.997 ± 0.005 | N/Ad | 1.014 |
The KIE value from the remote 5-14C label is assumed to be unity.
This experimental V/K KIE value is dependent on the remote 5-3H2 KIE according to the expression KIEV/K = KIEobserved × KIEremote. In this system, no correction is needed.
The intrinsic KIE values were determined by correcting V/K KIE values using eq 7 assuming Cf = 0.001.
N/A, not applicable; the forward commitment factor was not measured for this substrate.
Bond orders for the final transition state structure were determined from the Pauling bond order correlation. Reference C–O and C–N bond lengths were taken from structures of PRPP and IMP from the Coot database and used to calculate the transition state bond order using eq 8
| (8) |
where nx represents the transition state bond order, n0 represents the ground state bond order (assigned a value of 1.0), r0 is the reference bond length, rx is the transition state bond length, and c is a proportionality constant equal to 0.6 for >2.0 Å bonds and 0.3 for <2.0 Å bonds.28,29 Electrostatic potential maps were generated from the cube function in Gaussian 09 from the checkpoint files of optimized transition state and ground state structures.
RESULTS AND DISCUSSION
Recombinant PfHGXPRT Expression and Purification
PfHGXPRT was heterologously overexpressed in E. coli with an N-terminal six-histidine tag. Samples deemed to be homogeneous based on SDS–PAGE analysis yielded approximately 14 mg of enzyme. Nucleotide sequencing of the pDEST-14-PfHGXPRT construct validated its DNA sequence to be encoding PfHGXPRT.
Synthesis of Isotopically Labeled PRPP
Although PRPP is the natural substrate for KIE measurements on PfHGXPRT, its chemical instability leads to a significant solvolysis to ribose 5-phosphate (R5P) and PPi. Given the level of precision necessary for KIE measurements, substrate integrity is essential to the analysis of isotope effects. Synthesis and long-term storage of radiolabeled PRPP were not practical. Our approach was to synthesize adenosine 5′-monophosphate (AMP) carrying the isotope labels of interest. Conversion of labeled AMP molecules to labeled PRPPs (Figure 2) was accomplished in a single rapid step by adenine phosphoribosyltransferase for immediate use in KIE experimental protocols.
Forward Commitment Factor
Intrinsic KIEs are essential for establishing the transition state structure, but the experimental values may be masked by commitment factors. Determination of commitment factors permits intrinsic KIE values to be calculated. The forward commitment factor (Cf) is defined as the likelihood that the enzyme–substrate complex (the Michaelis complex) will cross the barrier to product formation rather than dissociate into unreacted substrates and the free enzyme. We have determined the forward commitment factor using the isotope trapping method developed by Rose.25 From a preformed PfHGXPRT–[5-14C]PRPP complex, a dilution into excess unlabeled PRPP and hypoxanthine allows radiolabeled inosine 5′-monophosphate (IMP) product formation ([5′-14C]IMP) to be monitored under conditions where [5′-14C]IMP released from the Michaelis complex cannot rebind. The extrapolation to time zero in eq 5 allows the determination of the fraction of bound PRPP converted to IMP (product). The experimental data were plotted as a function of time (Figure 3), and Cf was calculated using eq 6.
Figure 3.
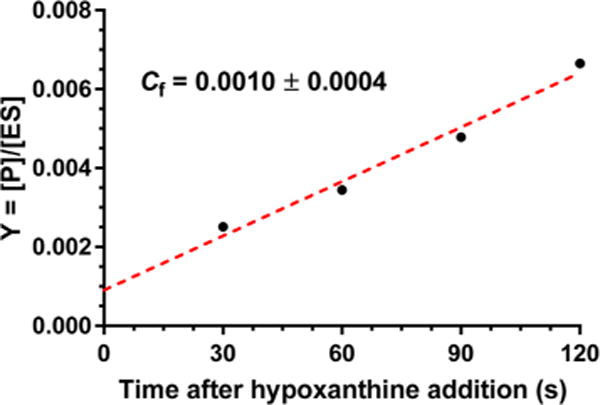
Measurement of the forward commitment factor (Cf) by isotope trapping from Michaelis complexes of PfHGXPRT.
The Cf for the PfHGXPRT-catalyzed reaction was estimated to be approximately 0.001, and an insignificant fraction of the Michaelis complex is converted to product without equilibration with unreacted substrate. This forward commitment is negligible relative to experimental error and does not contribute to the intrinsic KIE value. Low forward commitments are expected when catalysis is slow. The kcat for PfHGXPRT is approximately 0.4 s−1 under optimal conditions with hypoxanthine,13,15 sufficient time for the Michaelis complex to equilibrate with unbound reactants. Under conditions used here, the kcat is even slower.
Intrinsic KIEs Determined by the Competitive Radio-labeled Approach and Mass Spectrometry
KIEs for the synthesis of IMP by PfHGXPRT were measured using the competitive radiolabeled approach and by mass spectrometry. The individual isotopic substitutions and the experimental KIEs are included in Table 1 and Figure 4.
Figure 4.
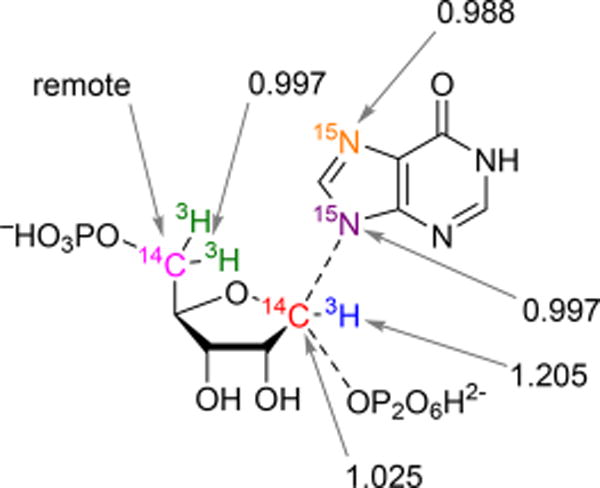
Intrinsic KIE values by atom position after correction for forward commitment factors.
In the double-label approach to KIE values, a control label remote from the bonds broken in catalysis is required. Here, [5-3H2]PRPP served as the remote label. Although remote [5-3H2]PRPP KIEs can occur in cases of distortional binding effects, the near-unity KIE values obtained for the 5-3H2 remote (0.997 ± 0.002) indicate no correction is required for this remote isotope label. Furthermore, on the basis of the low Cf obtained (0.1%), experimental KIEs required no correction to generate their corresponding intrinsic values. The relatively large errors for the 7-15N and 9-15N KIEs measured by mass spectrometry are caused by the decreased sensitivity of this assay when compared to that of the competitive radiolabeled method used for the PRPP KIEs.
An α-secondary [1-3H]PRPP KIE of 1.205 indicates an increased degree of bond vibrational freedom for the 1-3H label of PRPP at the transition state. In SN2 reactions, partial bonding to both the incoming nucleophile and departing leaving group constrains the out-of-plane motion to result in small or no α-secondary tritium isotope effects, but in an SN1 transition state, rehybridization of the anomeric carbon from sp3 to sp2 gives an increased degree of freedom to the out-of-plane mode, causing large secondary isotope effects. The tritium result indicates that PfHGXPRT proceeds by an SN1-like transition state. In SN2 reactions, partial bonding to both the incoming nucleophile and departing leaving group makes the anomeric carbon in [1-14C]PRPP the center of reaction coordinate motion. Kinetic isotope effects for 1-14C in SN2 reactions can be as large as 1.14.30 Isotope effects at this position decrease as the reaction becomes more dissociated with a limiting KIE of unity or slightly inverse when the transition state is a fully dissociated ribocation. The small but intermediate value of 1.025 ± 0.004 for the primary [1-14C]PRPP KIE is evidence of a mostly ribocation-dissociated transition state but with weak participation of one of the reaction coordinate nucleophiles.
Transition State Structure of PfHGXPRT
The transition state structure of PfHGXPRT was determined by using experimentally measured intrinsic KIEs as constraints for QM computational analysis. A simplified model of the HGXPRT reaction was generated from the coordinates of the crystal structure of PfHGXPRT bound to (1S)-1-(9-deazahypoxanthin-9-yl)-1,4-dideoxy-1,4-imino-D-ribitol-5-phosphate and PPi (PDB entry 1CJB) (see Figure S4). Calculations used Gaussian 09 with the B3LYP level of theory and the 6-31g(d) basis set. The lengths of the leaving group bond [1-C–1-O(PPi)] and the incoming nucleophile bond (9-N–1′-C) were varied at fixed lengths from 1.6 to 3.0 Å to determine theoretical transition states with calculated KIEs (through ISOEFF98) that best match the experimental intrinsic KIE values (Figure 5).
Figure 5.
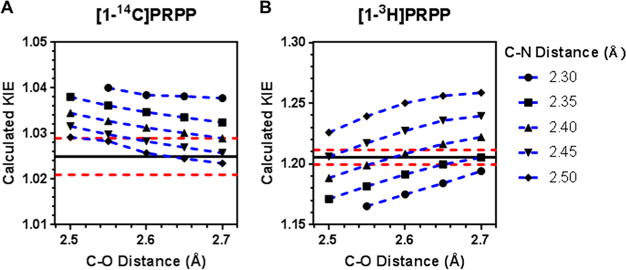
Calculated KIEs at specific C–O and C–N bond lengths generated from Gaussian 09 and ISOEFF98 for (A) 1-14C and (B) 1-3H. The black line represents the experimentally measured intrinsic KIEs, and the dashed red lines represent the upper and lower margins of error, respectively, for the experimentally measured values.
The structure with C–O and C–N bond lengths of 2.60 and 2.41 Å, respectively (compared to 1.43 and 1.46 Å, respectively, in the reactant state), gave calculated KIE values that most closely matched the experimentally measured values (Table 1).
The bond orders for the leaving group C–O bond and the nucleophilic C–N bond were estimated using the Pauling bond correlation and were determined to be 0.142 and 0.205, respectively. The net bond order of 0.347 indicates a loose SN1-like transition state but with an asymmetric bond contribution to the leaving pyrophosphate and the incoming purine N-9 nucleophile. The bond to the pyrophosphate leaving group is weaker than that to the incoming nitrogen, making this more product-like (IMP-like) than reactant-like along the reaction coordinate.
Forming the PfHGXPRT transition state requires generation of the ribocation and chemical activation of both the leaving group pyrophosphate and attacking nucleophilic N-9 of the hypoxanthine group. Hypoxanthine in solution is neutral with an N-7/N-9 tautomeric proton. Bond formation from N-9 of the purine ring requires the proton to be at N-7. Conversion from a tautomeric proton in the solution reactant state to the N-7 protonated state of the transition state adds bond order to N-7 and stiffens its bond vibrational environment. This is reflected in the inverse 7-15N KIE of 0.988 ± 0.009 (Table 1). The interpretation of the 7-15N KIE is also supported by the crystal structure with immucillin-H 5′-phosphate and PPi at the catalytic site of the enzyme. Asp148 donates a proton to N-7 with a favorable 2.8 Å hydrogen bond.7 A downfield 1H NMR signal at 14.3 ppm has been assigned to the N-7 proton using isotope editing with [7-15N]immucillin-H 5′-phosphate.31
Activation of the pyrophosphate to become a leaving group from PRPP is most favorable if the departing group is neutral. An anionic leaving group in this SN1 mechanism creates a chemically unfavorable anion–cation reactive pair at the transition state. Pyrophosphate in the complex of PfHGXPRT with immucillin-H 5′-phosphate is bound between two magnesium ions, each with a bidentate interaction with two oxygens of the pyrophosphate. A rotation of the attacking phosphoryl group by only 10° places the departing oxygen in position to be neutralized by the catalytic site Mg2+ ions.7 The Mg2+–pyrophosphate complex in human HGPRT is similar to that for the PfHGXPRT enzyme.6 In that case, isotope-edited difference Raman and FTIR studies demonstrated that PPi is fully ionized to P2O74− and in the di-Mg2+–pyrophosphate complex when bound with immucillin-H 5′-phosphate. Extrapolation to the present transition state of PfHGXPRT proposes coordination of the leaving group pyrophosphate oxygen to one of the active site Mg2+ ions to achieve the neutral leaving group.
Transition State Characteristics
PRPP phosphoribosyl-transferases share common features of the pyrophosphate leaving group and the aromatic nitrogen nucleophiles.20–22 However, because of different experimental approaches, the PfHGXPRT transition state is the only one determined in the physiological direction with natural substrates. In principle, transition state structure should be independent of the direction in which it is characterized if the reaction is characterized by a single transition state. However, we expect that the relative bond orders to the leaving group and the attacking nucleophiles of the transition states will differ because of the altered reactivity of the purine, pyrimidine, or pyridine heterocycles and the difference between the pyrophosphate and phosphonoacetic acid groups used as nucleophiles in the reverse reactions for the OPRTs. A convenient method for comparing transition states is a More O’Farrell–Jencks reaction diagram of bond orders at the transition states.32–34 Phosphoribosyltransferase transition states cluster in the DN + AN region of the plot (Figure 6), with a low but significant level of participation of the leaving group and the nucleophile. The S. typhimurium OPRT is an exception as it lies on the leaving group ordinate, an early dissociative transition state.20
Figure 6.
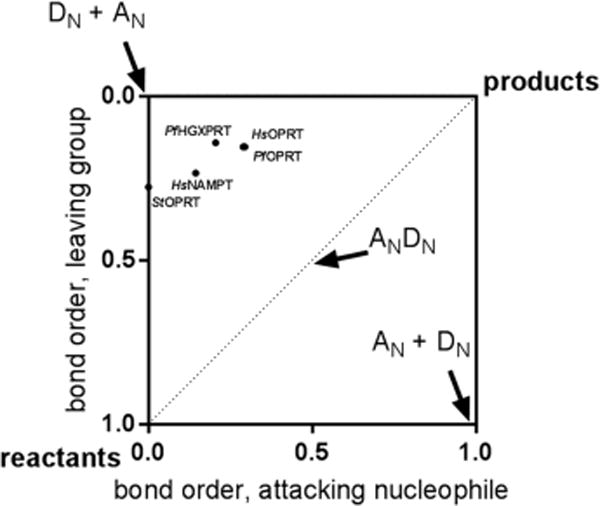
More O’Ferrall–Jencks plot for the transition states of phosphoribosyltransferases. PfHGXPRT shows an almost-symmetrical leaving group (pyrophosphate) and attacking nucleophile (hypoxanthine). The PfHGXPRT transition state is closer to the DN + AN corner than other phosphoribosyltransferase transition states. Pauling bond order used a proportionality constant of 0.6 when the bond order is low (>2.0 Å) and 0.3 when the bond is <2.0 Å.29 Organisms: Hs, Homo sapiens; Pf, P. falciparum; St, S. typhimurium. Enzymes: HGXPRT, hypoxanthine-guanine-xanthine phosphoribosyltransferase; NAMPT, nicotinamide phosphoribosyltransferase; OPRT, orotate phosphoribosyltransferase.
Electrostatic Potential of Reactants and the Transition State
Electrostatic potential surface (EPS) maps of the transition state structure (Figure 7) show the relative size and charge distribution of central portions of the transition state structure. Upon comparison to the EPS maps for the ground state structures for the α-D-ribose 1-phosphate (to serve as a truncated PRPP) and hypoxanthine substrates, a significant positive charge is present on the anomeric carbon at the transition state. The partial charge results from the bond deficiency at the transition state, where the covalent bond of the reactant has decreased to 0.347 bond order. Thus, the transition state is a partial carbocation occurring in this hybrid SN1 mechanism. This positive charge also serves to neutralize the negatively charged functional groups of PRPP relative to the ground state structure. Additionally, an increased negative charge is observed at the N-9 position on the hypoxanthine nucleophile at the transition state. In contrast to the ground state of hypoxanthine, negative charge appears at N-7, and N-9 is relatively positively charged. The development of negative charge at N-9 as hypoxanthine approaches the transition state is needed to perform a nucleophilic attack on the anomeric carbon of PRPP following carbocation formation.
Figure 7.
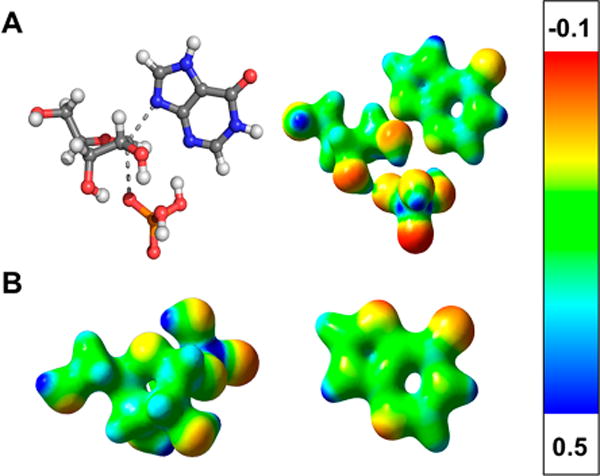
Electrostatic potential surface map for (A) the transition state of PfHGXPRT and (B) the PfHGXPRT substrates, α-D-ribose 1-phosphate (to serve as a truncated PRPP) and hypoxanthine. Hypoxanthine is shown as the neutral N-9-protonated structure, the favored ground state. During the reaction coordinate, N-7-protonated hypoxanthine forms and is the reactive species at the transition state.
Supplementary Material
Acknowledgments
The authors thank E. Nieves from the laboratory of R. H. Angeletti for assistance with mass spectrometry analysis. The authors also thank Drs. C. F. Stratton, S. A. Cameron, M. B. Poulin, and E. S. Burgos for insightful discussions about kinetic isotope effects and forward commitment factor measurement and analysis.
Funding: This work was supported by Grants GM041916 and AI127807 from the National Institutes of Health. The mass spectrometer system was provided by NIH SIG RR029389.
ABBREVIATIONS
- AMP
adenosine 5′-monophosphate
- APRTase
adenine phosphoribosyltransferase
- ATP
adenosine 5′-triphosphate
- Cf
forward commitment factor
- EPS
electrostatic potential surface
- HGXPRT
hypoxanthine-guanine-xanthine phosphoribosyltransferase
- IMP
inosine 5′-monophosphate
- KIE
kinetic isotope effect
- OPRT
orotate phosphoribosyltransferase
- PfHGXPRT
P. falciparum HGXPRT
- PPi
inorganic pyrophosphate
- PRPP
5-phospho-α-D-ribosyl 1-pyrophosphate
- QM
quantum mechanical
- R5P
ribose 5-phosphate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b01027.
Figures S1–S7 (PDF)
ORCID
Rodrigo G. Ducati: 0000-0002-8783-8847
Vern L. Schramm: 0000-0002-8056-1929
Notes
The authors declare no competing financial interest.
References
- 1.Sherman IW. Biochemistry of Plasmodium (malarial parasites) Microbiol Rev. 1979;43:453–495. doi: 10.1128/mr.43.4.453-495.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ducati RG, Namanja-Magliano HA, Schramm VL. Transition-state inhibitors of purine salvage and other prospective enzyme targets in malaria. Future Med Chem. 2013;5:1341–1360. doi: 10.4155/fmc.13.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman PA, Human L, Freese JA. Xanthine oxidase inhibits growth of Plasmodium falciparum in human erythrocytes in vitro. J Clin Invest. 1991;88:1848–1855. doi: 10.1172/JCI115506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesch M, Nyhan WL. A Familial Disorder of Uric Acid Metabolism and Central Nervous System Function. Am J Med. 1964;36:561–570. doi: 10.1016/0002-9343(64)90104-4. [DOI] [PubMed] [Google Scholar]
- 5.Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, O’Neill JP. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res, Rev Mutat Res. 2000;463:309–326. doi: 10.1016/s1383-5742(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 6.Shi W, Li CM, Tyler PC, Furneaux RH, Grubmeyer C, Schramm VL, Almo SC. The 2.0 A structure of human hypoxanthine-guanine phosphoribosyltransferase in complex with a transition-state analog inhibitor. Nat Struct Biol. 1999;6:588–593. doi: 10.1038/9376. [DOI] [PubMed] [Google Scholar]
- 7.Shi W, Li CM, Tyler PC, Furneaux RH, Cahill SM, Girvin ME, Grubmeyer C, Schramm VL, Almo SC. The 2.0 A structure of malarial purine phosphoribosyltransferase in complex with a transition-state analogue inhibitor. Biochemistry. 1999;38:9872–9880. doi: 10.1021/bi990664p. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Grubmeyer C. Catalysis in human hypoxanthine-guanine phosphoribosyltransferase: Asp 137 acts as a general acid/base. Biochemistry. 1998;37:4114–4124. doi: 10.1021/bi972519m. [DOI] [PubMed] [Google Scholar]
- 9.Yuan L, Craig SP, 3rd, McKerrow JH, Wang CC. Steady-state kinetics of the schistosomal hypoxanthine-guanine phosphoribosyltransferase. Biochemistry. 1992;31:806–810. doi: 10.1021/bi00118a024. [DOI] [PubMed] [Google Scholar]
- 10.Wenck MA, Medrano FJ, Eakin AE, Craig SP. Steady-state kinetics of the hypoxanthine phosphoribosyltransferase from Trypanosoma cruzi. Biochim Biophys Acta, Proteins Proteomics. 2004;1700:11–18. doi: 10.1016/j.bbapap.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Roy S, Nagappa LK, Prahladarao VS, Balaram H. Kinetic mechanism of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase. Mol Biochem Parasitol. 2015;204:111–120. doi: 10.1016/j.molbiopara.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Queen SA, Vander Jagt D, Reyes P. Properties and substrate specificity of a purine phosphoribosyltransferase from the human malaria parasite. Mol Biochem Parasitol. 1988;30:123–133. doi: 10.1016/0166-6851(88)90105-3. [DOI] [PubMed] [Google Scholar]
- 13.Keough DT, Ng AL, Winzor DJ, Emmerson BT, de Jersey J. Purification and characterization of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase and comparison with the human enzyme. Mol Biochem Parasitol. 1999;98:29–41. doi: 10.1016/s0166-6851(98)00139-x. [DOI] [PubMed] [Google Scholar]
- 14.Taylor Ringia EA, Tyler PC, Evans GB, Furneaux RH, Murkin AS, Schramm VL. Transition state analogue discrimination by related purine nucleoside phosphorylases. J Am Chem Soc. 2006;128:7126–7127. doi: 10.1021/ja061403n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hazleton KZ, Ho MC, Cassera MB, Clinch K, Crump DR, Rosario I, Jr, Merino EF, Almo SC, Tyler PC, Schramm VL. Acyclic immucillin phosphonates: second-generation inhibitors of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase. Chem Biol. 2012;19:721–730. doi: 10.1016/j.chembiol.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Büngener W, Nielsen G. Nucleic acid metabolism in experimental malaria. 2. Incorporation of adenosine and hypoxanthine into the nucleic acids of malaria parasites (Plasmodium berghei and Plasmodium vinckei) Z Tropenmed Parasitol. 1968;19:185–197. [PubMed] [Google Scholar]
- 17.Dawson PA, Cochran DA, Emmerson BT, Gordon RB. Inhibition of Plasmodium falciparum hypoxanthine-guanine phosphoribosyltransferase mRNA by antisense oligodeoxynucleotide sequence. Mol Biochem Parasitol. 1993;60:153–156. doi: 10.1016/0166-6851(93)90039-z. [DOI] [PubMed] [Google Scholar]
- 18.Cox FE. History of human parasitology. Clin Microbiol Rev. 2002;15:595–612. doi: 10.1128/CMR.15.4.595-612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schramm VL. Enzymatic transition states and transition state analogues. Curr Opin Struct Biol. 2005;15:604–613. doi: 10.1016/j.sbi.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Tao W, Grubmeyer C, Blanchard JS. Transition state structure of Salmonella typhimurium orotate phosphoribosyltransferase. Biochemistry. 1996;35:14–21. doi: 10.1021/bi951898l. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Luo M, Schramm VL. Transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J Am Chem Soc. 2009;131:4685–4694. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgos ES, Vetticatt MJ, Schramm VL. Recycling nicotinamide. The transition-state structure of human nicotinamide phosphoribosyltransferase. J Am Chem Soc. 2013;135:3485–3493. doi: 10.1021/ja310180c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen KF, Houlberg U, Nygaard P. Thin-layer chromatographic methods to isolate 32P-labeled 5-phosphoribosyl-alpha-1-pyrophosphate (PRPP): determination of cellular PRPP pools and assay of PRPP synthetase activity. Anal Biochem. 1979;98:254–263. doi: 10.1016/0003-2697(79)90138-6. [DOI] [PubMed] [Google Scholar]
- 24.Yuan H, Stratton CF, Schramm VL. Transition State Structure of RNA Depurination by Saporin L3. ACS Chem Biol. 2016;11:1383–1390. doi: 10.1021/acschembio.5b01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose IA. The isotope trapping method: desorption rates of productive E.S complexes. Methods Enzymol. 1980;64:47–59. doi: 10.1016/s0076-6879(80)64004-x. [DOI] [PubMed] [Google Scholar]
- 26.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima TE, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc.; Wallingford, CT: 2009. [Google Scholar]
- 27.Anisimov V, Paneth P. ISOEFF98. A program for studies of isotope effects using Hessian modifications. J Math Chem. 1999;26:75–86. [Google Scholar]
- 28.Pauling L. Atomic Radii and Interatomic Distances in Metals. J Am Chem Soc. 1947;69:542–553. [Google Scholar]
- 29.Houk KN, Gustafson SM, Black KA. Theoretical secondary kinetic isotope effects and the interpretation of transition state geometries. 1. The Cope rearrangement. J Am Chem Soc. 1992;114:8565–8572. [Google Scholar]
- 30.Schramm VL. Enzymatic transition states and transition state analog design. Annu Rev Biochem. 1998;67:693–720. doi: 10.1146/annurev.biochem.67.1.693. [DOI] [PubMed] [Google Scholar]
- 31.Li CM, Tyler PC, Furneaux RH, Kicska G, Xu Y, Grubmeyer C, Girvin ME, Schramm VL. Transition-state analogs as inhibitors of human and malarial hypoxanthine-guanine phosphoribosyltransferases. Nat Struct Biol. 1999;6:582–587. doi: 10.1038/9367. [DOI] [PubMed] [Google Scholar]
- 32.O’Ferrall M. Relationships between E2 and E1cB mechanisms of β-elimination. J Chem Soc B. 1970;0:274–277. [Google Scholar]
- 33.Jencks WP. General acid-base catalysis of complex reactions in water. Chem Rev. 1972;72:705–718. [Google Scholar]
- 34.Schramm VL. Enzymatic transition state poise and transition state analogues. Acc Chem Res. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.