

Abstract
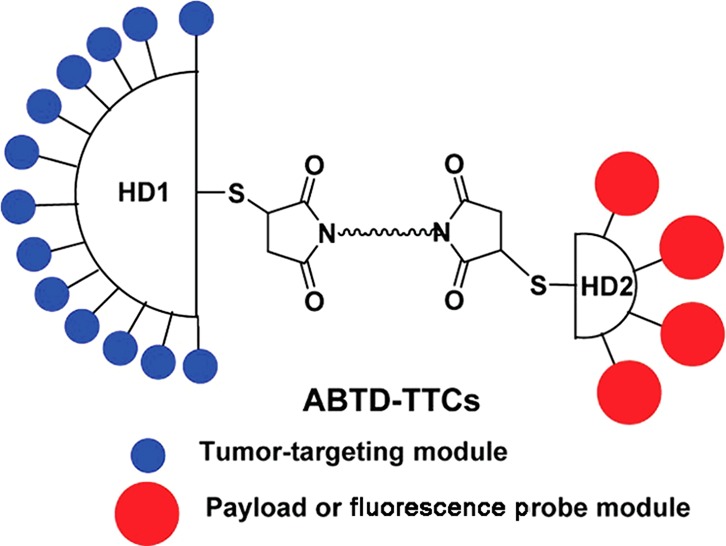
A unique asymmetric bow-tie poly(amidoamine) (PAMAM) dendrimer (ABTD) scaffold was designed and developed as a well-defined macromolecular carrier for tumor-targeted drug delivery. The ABTD scaffold in this study consists of a G3-half-dendron (G3-HD) unit and a G1-half-dendron (G1-HD) unit, bearing thiol moiety in each unit and a bis(maleimide) linker unit, which undergo sequential thiol–maleimide coupling to assemble the scaffold. This assembly methodology is applicable to all other combinations of different generations of PAMAM dendrimers. In the prototype ABTD in this study, 16 biotin moieties were tethered to the G3-HD unit and 4 payloads (new-generation taxoid) to the G1-HD via a self-immolative linker to form an ABTD-tumor-targeting conjugate (ABTD-TTC-1). Two other ABTD-TTCs were synthesized, wherein the G1-HD unit was tethered to a fluorescence-labeled taxoid or to a fluorescent probe. These three ABTD-TTCs were constructed by using a common key ABTD 6 bearing a terminal acetylene group in the G1-HD unit, which was fully characterized as a single molecule by high-resolution mass spectrometry and NMR despite its high molecular weight (Mw: 12 876). Then, the click reaction was employed to couple ABTD 6 with a small-molecule payload or fluorescence probe unit bearing a terminal azide moiety. ABTD-TTC-3, as a surrogate of ABTD-TTC-2, showed substantially enhanced internalization into two cancer cell lines via receptor-mediated endocytosis, attributed to multibinding effect. ABTD-TTC-1 exhibited a remarkable selectivity to cancer cells (1400–7500 times) compared to human normal cells, which demonstrates the salient feature and bright prospect of the ABTD-based tumor-targeted drug-delivery system.
Introduction
Cancer is the second leading cause of death, as well as the number 1 cause of death under the age of 85, in the United States.1 It has been estimated that approximately 1.7 million new cancer cases will occur and over half a million deaths will be attributed to cancer.2 Despite the significant progress in the development of cancer detection, prevention, surgery, chemotherapy, and radiation therapy, no common cure has been fully established for patients with this malignant disease. Traditional chemotherapy relies on the premise that the rapidly proliferating tumor cells are more likely to be killed by cytotoxic agents than normal cells. In reality, however, due to the little or no specificity of these cytotoxic agents, severe systemic toxicity and dose-limiting side effects may occur and result in anemia, hair loss, and damage to the liver, kidney, and bone marrow. Accordingly, extensive efforts have been made for developing tumor-specific delivery systems to overcome the shortcomings of conventional chemotherapy in the last few decades.3,4
In general, a tumor-targeted drug conjugate consists of a tumor recognition moiety and a cytotoxic agent connected directly or through a suitable linker. The tumor-targeted drug conjugate should be systemically nontoxic, which means the linker must be stable during the blood circulation, but readily cleaved to release the cytotoxic agent efficiently upon internalization into the tumor cells. In addition, the linker should be bifunctional so that both cytotoxic agent and tumor-targeting moiety can be connected to the linker.
Rapidly growing cancer cells require various nutrients and vitamins so that certain cancer-specific receptors are overexpressed to enhance the uptake. These receptors can be used as targets to deliver the cytotoxic drugs specifically to the cancer cells via receptor-mediated endocytosis (RME). Various ligands, including mAbs,5−9 PUFAs,10,11 folic acid,12,13 transferrin,14 oligopeptides,15 aptamers,16 and hyaluronic acid,17 have been studied as TTMs to construct the tumor-targeted drug conjugates.
Biotin, well known as vitamin H or B7, is essential for cell growth, production of fatty acids, and metabolism of fats and amino acids. Considerable evidence has shown that various cancer cells overexpress biotin receptors (BRs) significantly more than normal cells.18 Accordingly, we selected biotin receptors on cancer cell surface as the molecular target and employed biotin as the primary tumor-targeting moiety to construct our drug conjugate with cytotoxic agents.
Paclitaxel and docetaxel have made significant impacts on cancer chemotherapy.19 However, these drugs seriously suffer from the lack of tumor specificity and MDR. For solid tumors, such as breast, ovarian, and lung cancers, paclitaxel and docetaxel show efficacy, but these taxane drugs are not effective against colon, pancreatic, melanoma, and renal cancers. For example, human colon carcinoma is inherently multidrug-resistant because of the overexpression of P-glycoprotein (P-gp), which is an effective ABC transporter. Overexpression of P-gp can lead to multidrug resistance as the result of pumping hydrophobic anticancer agents, such as paclitaxel and docetaxel, out of cancer cells.20 On the basis of our extensive SAR study of taxoids, we have developed a series of highly potent second- and third-generation taxoids.21−29 Most of these new-generation taxoids exhibited 2–3 orders of magnitude higher potency than paclitaxel and docetaxel against MDR cell lines expressing P-gp.30 Accordingly, we chose a highly potent second-generation taxoid, SB-T-1214, as the cytotoxic payload.
Tumor-targeted drug-delivery strategies are based on “active” or “passive” targeting. Active targeting relies on the difference in the expression level of cell surface biomarkers, such as antigen or receptor, between cancer and normal tissue. Extensive preclinical and clinical studies have been performed on antibody–drug conjugates (ADCs) for tumor-targeted drug delivery in the last two decades, wherein monoclonal antibodies (mAbs) targeting cancer surface-specific antigens, e.g., EGFR, Her-2, CD22, CD30, CD33, etc., are used for “active” targeting.4,9,31 Small-molecule drug conjugates (SMDCs), especially targeting vitamin receptors in tumor and cancer cells, have also been actively studied.30,32−34 Another strategy termed as “passive targeting” aims at employing macromolecules, including polymers or nanoparticles as vehicles. These macromolecules do not directly interact with tumor cells, but strongly influence the accumulation, transportation, and biodistribution of their drug conjugates in tumor tissues due to the EPR effect. The EPR effect was first elucidated by Maeda and co-workers in the 1980s.35 In principle, tumor cell clusters induce angiogenesis for their increasing demands of oxygen and nutrition. The new blood vessels generated by the tumor often have irregular shape and large size pore compared to the healthy tissue. The leaky and defective vessels make the vasculature of tumor tissue permeable to macromolecules, which hardly pass the endothelial barrier of the blood vessels of healthy tissue. Besides, the enhanced uptake of macromolecules in tumor tissue is also related to its increased retention in the tumor. If the molecular weight of a macromolecule reaches 40 kDa, it is difficult to be cleared from the tumor due to the lack of its lymphatic drainage system.36
Dendrimer is a class of polymers that are also often called starburst polymers because of their shapes. In general, a dendrimer has a treelike molecular architecture with an interior core, interior layers (also called generations), which consist of repeating units regularly attached to the core, as well as an exterior surface of functional terminal groups attached to the outermost generation. Generation number (usually abbreviated to G#) is determined by the number of focal points going from the core toward the surface.37 The molecular weight of dendrimer itself and the number of terminal functional group can be easily determined by the generation. Dendrimer could be divided into various classes, such as poly(amidoamine-organosilicon), poly(propyleneimine), amphiphilic, chiral, multilingual, and micellar dendrimers, as well as Tecto-dendrimers and Frechet-type dendrimers.38 One of the commonly used dendrimers as carriers in medicinal chemistry is poly(amidoamine) (PAMAM) dendrimer. PAMAM dendrimers are commercially available, usually in a methanol solution, which can reach generation 10 with molecular weight over 930 000 g/mol.39
Dendrimer has been used in drug delivery as a macromolecular carrier to target and deliver a wide range of therapeutic agents.40−48 Compared to the small molecular drug conjugates, which only have limited number of targeting moiety in each conjugate, dendrimer-based drug conjugates can largely increase the loading capacity of the targeting moiety to achieve high concentration at tumor sites. Recently, Baker and his co-workers have used PAMAM dendrimers as the platform for functionalizing it with different types of biological molecules for targeted drug delivery and have shown efficient receptor-mediated cellular internalization in vitro and improved chemotherapeutic activity in vivo.49−53 For example, the surface amino groups of a G5 PAMAM dendrimer were partially acetylated (up to 85%), and the remaining amino groups were sequentially modified with different functionalities, including folic acid, paclitaxel, and fluorescein isothiocyanate (FITC).49
However, it is impossible to modify the surface of a Gn dendrimer with two different modifiers, e.g., tumor-targeting module and payload module, with an exact ratio, because these modifications will produce modified Gn dendrimers with statistical distributions of the two modifiers. Thus, this type of traditional approach cannot produce reproducible materials with exact loading of payloads or tumor-targeting molecules, which may seriously limit its clinical applications.47
To solve this highly challenging problem, we came up with an idea of coupling two fully functionalized half-dendrons, modified with two different modifiers, through a bifunctional linker (Figure 1). A fully functionalized Gn PAMAM dendrimer can be readily prepared by modifying all amino terminals of the dendrimer with a cystamine core, PAMAM(cystamine), using excess modifying reagents, e.g., an activated ester of a tumor-targeting module or that of a cytotoxic drug module. Then, the fully functionalized Gn PAMAM(cystamine) dendrimer can be reduced to exactly the same half-dendrons, e.g., Gn-HD1-SH and Gn-HD2-SH. Subsequent stepwise coupling of Gn1-HD-SH and Gn2-HD-SH with a bis-maleimide linker should give a novel asymmetric bow-tie PAMAM dendrimer (ABTD) (Figure 1). A salient feature of this methodology is that it is applicable to the same- or different-generation dendrimers; hence, it provides a highly robust and flexible platform for a variety of applications.
Figure 1.

Construction of a well-defined asymmetric bow-tie dendrimer scaffold using two differently functionalized half-dendrons.
While our work on ABTDs was in progress,54,55 Hartley and co-workers reported a G3 PAMAM dendrimer-based platform for vaccine delivery.56 A peptide/protein array, as well as a mannose array, was fully functionalized on two different G3 PAMAM(cystamine) dendrimers. Then, these two G3 dendrimers were reduced to the corresponding half-dendrons to construct G3 heterodendrimers through orthogonal reattachment using a diblock linker bearing a fluorescein probe for imaging purpose.56 Although the basic idea is similar to ours, their work was limited to the same-generation (G3) dendrimers and not relevant for the construction of tumor-targeting drug conjugates, in which only a small number of cytotoxic payloads need to be introduced as compared to the tumor-targeting modules. In contrast, our work deals with the construction of ABTDs bearing two different generations of half-dendrons, which can accommodate the loadings of functionally different modules. Although a number of asymmetric bow-tie dendrimers consisting of two dendrimeric wedges and terminated by two different functionalities, i.e., “Janus dendrimers”, have been reported57−59 and some of them have shown promising results on the drug delivery in vivo,60,61 our approach is unique and unprecedented.
We describe here the design, synthesis, and biological evaluations of a novel ABTD-based tumor-targeting drug conjugate (ABTD-TTC-1, 1), which exhibited extremely high selectivity (>1000 times) to cancer cells compared to normal cells, and its fluorescent congeners as mechanistic probes for tumor-targeting and efficient receptor-mediated endocytosis (RME).
Results and Discussion
Design and Strategy for Construction of Tumor-Targeted ABTD Conjugates with Payloads
We have designed a novel ABTD-based tumor-targeted drug delivery system (DDS) platform, consisting of 16 tumor-targeting modules on a G3 half-dendron (G3-HD1) unit and 4 payloads (or probes) with self-immolative linkers on a G1 half-dendron (G1-HD2) unit, which are connected by a bis-thiosuccinimide linker unit (Figure 2). To increase the solubility of this macromolecule structure, a PEG short chain (PEG3) is inserted into each dendrimer arm and the linker unit. The payload/probe can be a cytotoxic drug, a fluorescent dye, or a drug with a fluorescent dye.
Figure 2.

Schematic representation of asymmetric bow-tie dendrimer-based conjugates.
Building upon the ABTD-DDS platform illustrated in Figure 2, we further designed three novel tumor-targeting ABTD conjugates 1–3 with a payload or a probe, which are shown in Figure 3. Biotin (vitamin B7) was selected as the tumor-targeting molecule since we have been successfully using this vitamin in our SMDCs,32,62−65 as well as single-walled carbon nanotube (SWNT)-based drug conjugates.30,66,67 Also, a highly potent second-generation taxoid, SB-T-1214 (4), was selected as the cytotoxic payload. As fluorescent probes, fluorescein and SB-T-1214-fluorescein (5) were selected. As Figure 3 shows, all three conjugates (ABTD-TTCs) share the same platform except for the payload or probe component. ABTD-TTC-1 (1) bears taxoid 4 as the payload with self-immolative disulfide linker unit, which has been successfully used in our previous SMDCs and a SWNT-TTDDS.32,62−66 ABTD-TTC-2 (2) has fluorescein isobutyl ester with a short tether as a fluorescent probe and ABTD-TTC-3 (3) is a fluorescent probe of 1, wherein fluorescein isobutyl ester with a short tether is attached to 4. These three ABTD-TTCs are planned to be constructed from the common key construct bearing alkyne termini and three azide-bearing components using the click reaction.
Figure 3.

ABTD-based tumor-targeted conjugates 1–3.
Synthesis of Biotin–ABTD–Alkyne Construct 6, the Key Intermediate for ABTD-TTCs
As Figure 4 illustrates our retrosynthetic analysis, biotin–ABTD–alkyne construct 6 was synthesized following the strategy outlined in Figure 1. First, G3-PAMAM(cystamine) dendrimer was fully functionalized by attaching biotin-NH-PEG3-(CH2)2CO units to give dendrimer 7. In a similar manner, G1-PAMAM(cystamine) dendrimer was converted to fully functionalized dendrimer 8, bearing 4-pentynoyl-NH-PEG3-(CH2)2CO units. Then, ω,ω′-bis-[6-maleimidohexanoyl-NH(CH2)2]2-PEG3 (9) was prepared as the linker unit. Next, both dendrimers, 7 and 8, were converted to the corresponding half-dendrons, 10 and 11, bearing sulfhydryl groups, through reductive cleavage of the cystamine disulfide cores. Then, bismaleimide linker 9 was reacted with a large excess of G1-half-dendron-SH 11 to afford the corresponding monoaddition product 12. Finally, G1-half-dendron-S-linker-maleimide 12 was reacted with G3-half-dendron-SH 10 to give the key common intermediate, biotin-ABTD-alkyne intermediate 6. The syntheses of fully functionalized dendrimers 7 and 8 from G3- and G1-PAMAM(cystamine) dendrimers are shown in Schemes 1 and 2, respectively.
Figure 4.

Retrosynthetic analysis for the construction of key intermediate 6.
Scheme 1. Synthesis of Fully ω-Biotinylated G3-PAMAM Dendrimer 7.

Reaction conditions: (i) N,N′-diisopropylcarbodiimide (DIC) (2 equiv), N-hydroxysuccinimide (NHS) (3 equiv), dimethylformamide (DMF) (0.2 M), 45 °C, 18 h; (ii) CH2Cl2, 25 °C, 23 h; (iii) trifluoroacetic acid (TFA) (1.0 equiv), CH2Cl2, 25 °C, 3 h; (iv) DIC (2.0 equiv), NHS (3.0 equiv), CH2Cl2, 25 °C, 48 h; (v) MeOH, 25 °C, 24 h.
Scheme 2. Synthesis of Fully Functionalized ω-Alkynoyl-G1-PAMAM Dendrimer 3.

Reaction conditions: (i) 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) (1.1 equiv), 4-N,N-dimethylaminopyridine (DMAP) (0.5 equiv), CH2Cl2 (0.02 M), rt, 24 h; (ii) TFA (1.0 equiv), CH2Cl2, rt, 4 h (98%); (iii) NHS (2 equiv), EDC·HCl (2.0 equiv), CH2Cl2, rt, 17 h (70%); (iv) MeOH, rt, 48 h.
Fully ω-Biotinylated G3-PAMAM Dendrimer 7
G3-PAMAM dendrimer bearing a cystamine core (DNT-296) was fully functionalized with tumor-targeting modules (32 in total) through reaction with an N-hydroxysuccinimide activated ester of the biotin-PEG module, biotin-NH-PEG3-(CH2)2-CO-NHS 14 (Scheme 1). Activated ester 14 was readily prepared in good overall yield from biotin and tert-butyl 12-amino-4,7,10-trioxadodecanoate (13) through amide coupling, followed by deprotection of the tert-butyl moiety by TFA,68 and subsequent esterification with NHS.
Then, activated ester 14 (2 equiv/amine terminus) was reacted with G3-dendrimer (DNT-296) in methanol at ambient temperature for 48 h to give fully ω-biotinylated G3-dendrimer 7. Due to limited solubility, the purification of the product was performed by dialysis in methanol using the molecular weight cut-off (MWCO) 6000–8000 tubing membrane to remove unreacted excess 14. The structure and purity of isolated G3-dendrimer 7 was confirmed by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) using 2,5-dihydroxybenzoic acid (DHB) as matrix (Figure S35). The m/z peak at 20 748.3 was consistent with the expected fully ω-biotinylated G3-dendrimer 7. The highest m/z peak 10 378.6 corresponds to the half-dendron of 7, generated by interior disulfide bond cleavage under the MALDI measurement conditions.
Fully Functionalized G1-PAMAM Dendrimer with ω-Alkanoyl Groups 8
In a manner similar to the synthesis of G3-dendrimer 7, 4-pentynoic acid (15) was coupled with 13, followed by deprotection of tert-butyl group by TFA, and subsequent esterification with NHS to give activated ester 16 in good overall yield. Then, G1-PAMAM(cystamine) dendrimer (DNT-294) was reacted with 16 (3 equiv/amino terminus) in methanol at ambient temperature to give the corresponding fully functionalized G1-dendrimer 8 as the product (Scheme 2). The dendrimer 8 was isolated by preparative high-performance liquid chromatography (HPLC) using a C18 column. The structure and purity of ω-alkynoyl G1-dendrimer 8 was confirmed by MALDI analysis (Figure S36) and LC-UV-TOF (Figures S37 and S38). The highest m/z peak 1895.205 corresponds to the half-dendron of 8 via cleavage of the interior disulfide linkage, generated under the MALDI-TOF measurement conditions. The 3788.202 peak corresponds to the molecular ion. The LC-UV/TOF analysis clearly showed a major peak with retention time at 21.8 min (UV at 215 nm). The LC-TOF analysis showed 1895.0713 (M + 2H+)2+/2, 1263.7156 (M + 3H+)3+/3, 948.0384 (M + 4H+)4+/4, and 758.6320 (M + 5H+)5+/5 ion peaks (Figure S37), which were deconvoluted to the mass number of the molecular ion, 3788.125 (Figure S38).
Fluorescence-Labeled Constructs, 19 and 20
In our previous fluorescence imaging studies, we used fluorescein methyl ester.32,62,66 However, under the conditions of the present study, it was found that fluorescein methyl ester was prone to be converted to the corresponding nonfluorescent spirolactam.69−71 Thus, we used more stable fluorescein isobutyl ester as the fluorescent label for taxoid 4. 6-Hydroxycarbonylpropylfluorescein isobutyl ester 17 was prepared using the method previously reported by our laboratory.64 Amide coupling of 17 with 11-azido-3,6,9-trioxaundecan-1-amine (18)72,73 using EDC·HCl gave a click-ready fluorescent label construct 6 in good yield (Scheme 3). Amide coupling-ready fluorescent label 20 was also prepared from 17 and NHS in excellent yield (Scheme 3), which would be used to synthesize fluorescence-labeled taxoid 5 (Scheme 4).
Scheme 3. Preparation of Click-Ready and Coupling-Ready Fluorescent Constructs.

Reaction conditions: (i) 18 (1.0 equiv), DMAP (0.5 equiv), EDC·HCl (1.2 equiv), DMF/CH2Cl2, rt, 21 h; (ii) NHS (1.5 equiv), EDC (1.5 equiv), DMAP (0.5 equiv), CH2Cl2, rt, 17 h.
Scheme 4. Synthesis of a Fluorescein-Labeled Taxoid 5.

Reaction conditions: (i) TBDMS-Cl (5 equiv), imidazole (10 equiv), DMF, rt, 4 h; (ii) 20 (2.0 equiv), EDC·HCl (2.0 equiv), DMAP (1.0 equiv), CH2Cl2/DMF, rt, 9 d; (iii) HF/Py, CH3CN/Py, 0 °C to >rt, 24 h.
Click-Ready Fluorescence-Labeled Drug-Linker and Drug-Linker Conjugates62
The C2′-hydroxy group of taxoid 4 was protected as a tert-butyldimethylsilyl (TBDMS) ether, which was coupled with 20 under EDC·HCl/DMAP conditions, followed by deprotection with HF/pyridine to afford 7-fluorescein-O-butanoyl-taxoid 5 (Scheme 4).
Fluorescein-labeled taxoid 5 was coupled with 2-(triisopropylsiloxycarbonylbutan-2-yl)disulfanylphenylacetic acid (21)63−65 under EDC·HCl/DMAP conditions to give the corresponding taxoid-2′-ester, followed by deprotection with HF/pyridine and subsequent esterification with NHS/EDC·HCl to afford activated ester 22 in good overall yield (Scheme 5). Then, activated ester 22 was coupled with 11-azido-3,6,9-trioxaundecan-1-amine (18) to give click-ready azido-SS-linker-taxoid-fluorescein conjugate 24 in 70% yield (Scheme 5). The corresponding azido-SS-linker-taxoid conjugate 25 was also synthesized from activated ester 23 in a similar manner from taxoid 4, which was previously reported by our laboratory (Scheme 5).64 It should be noted that the disulfide linker unit incorporated into the conjugates 24 and 25 is a second-generation self-immolative linker developed in our laboratory, which is proven to be stable in blood plasma, but readily cleaved in cancer cells to release the taxoid payload.30,63−65,74
Scheme 5. Synthesis of Click-Ready Fluorescence-Labeled Drug-Linker (24) and Drug-Linker (25) Components.

Reaction conditions and yields: (i) 21 (1.0 equiv), EDC·HCl (1.5 equiv), DMAP (0.25 equiv), CH2Cl2 (0.01 M), 0 °C to >rt, 16 h, 50–93%; (ii) HF/Py, CH3CN/Py, 0 °C to >rt, 25 h, 71%; (iii) NHS (1.2 equiv), EDC·HCl (1.2 equiv), CH2Cl2 (0.01 M), rt, 17 h, 86%; (iv) 18 (1.5 equiv), CH2Cl2, rt, 17 h: 24, 70%; 25, 85%.
PEGylated Bis(maleimide) Linker 9
Two equivalents of 6-maleimidohexanoic acid (26) were coupled with 4,7,10-trioxa-1,13-tridecanediamine (27) through activated ester method using isobutyl chloroformate and N-methylmorphorine to give PEGylated bis(maleimide) linker 9 in moderate yield (Scheme 6).
Scheme 6. Synthesis of Bis(maleimide)-PEG3-Linker 9.

Reaction conditions and yields: (i) ClCO2Bu-i (1.0 equiv), N-methylmorpholine (NMM) (1.0 equiv), THF, 0 to >25 °C, 3 h; (ii) 4,7,10-trioxa-1,13-tridecanediamine (27) (0.5 equiv), THF (0.1 M), 0 to >25 °C, 22 h; 50% for two steps.
“Click-Ready” ABTD Construct 6
As illustrated in the retrosynthetic analysis (Figure 4), to construct the “click-ready” ABTD construct 6 bearing 4 terminal alkyne arms and 16 terminal biotin arms, fully functionalized G3 dendrimer 7 and G1 dendrimer 8 should be cleaved to form the corresponding G3- and G1-half dendrons, 10 and 11, respectively, which should be cross-coupled through bis(maleimido) linker 9 stepwise. We selected G1 dendrimer 8 over G3 dendrimer 7 to first couple with 9 because (i) 8 has better solubility than 7, hence purification with preparative HPLC is feasible and (ii) difference in molecular weight is very large in the transformation of G1-half dendron-linker 12 to ABTD construct 6 through cross-coupling of 12 with 10, due to the dominant contribution of 10 to the molecule size so that dialysis can be used as the most relevant purification method after the cross-coupling (Scheme 7).
Scheme 7. Click-Ready ABTD Construct 6.

Reaction conditions and yields: (i) 9 (10 equiv), tris(2-carboxyethyl)phosphine (TCEP) (3 equiv), MeOH, 25 °C, 24 h, 49%; (ii) 10 (0.5 equiv), TCEP (3 equiv), MeOH/H2O (1/1), 25 °C, 24 h, 49%.
Purification of 12 was performed by preparative HPLC using Jupiter C18 column. Solvents were water with 0.1% TFA (solvent A) and acetonitrile with 0.1% TFA (solvent B). We were able to remove the impurities and defect derivatives to solely isolate the desired product as single component. The purity of 12 was confirmed by MALDI-TOF (Figure S39) and LC-UV-TOF (Figures S40 and S41). The formation of 6 (Mw: 12 876) was confirmed by the LC-UV-TOF analysis. Construct 6 was purified by dialysis with MWCO 6000–8000 dialysis tubing membrane, and the structure of 6 was confirmed by MALDI-TOF (Figure S8) and LC-UV-TOF analyses (Figures S9–S11).
ABTD-Based Tumor-Targeting Drug/Probe Conjugates 1–3
Click-ready ABTD 6 was reacted with azido-SS-linker-taxoid conjugate 25(75) in the presence of a catalytic amount of copper sulfate pentahydrate and sodium ascorbate in methanol/distilled water (4/1) at room temperature for 24 h. The progress of the reaction was monitored by FIA-MS. Upon completion, the reaction mixture was subjected to dialysis using an MWCO 6000–8000 tubing membrane for 48 h against methanol. The resulting solution was filtrated to remove insoluble solid and then lyophilized to give tumor-targeting asymmetric bow-tie dendrimer 1 (ABTD-TTC-1) in 72% yield (Scheme 8). In the same manner, click-ready ABTD 6 was reacted with azido-SS-linker-taxoid(fluorescein) probe 24 and azido-linker-fluorescein 19 to give 2 (ABTD-TTC-2) and 3 (ABTD-TTC-3) in 69 and 76% yields, respectively (Scheme 8). Attempted characterizations of these ABTD-TTCs 1–3 by MALDI-TOF or LC-TOF were unfortunately not successful, due to the lack of sufficient ionization. Thus, these compounds were characterized by 1H and 13C NMR, as well as gel permeation chromatography (GPC) analyses, which showed >98% purity for all compounds (Figures S1–S6, S45–S47 in the Supporting Information).
Scheme 8. Synthesis of ABTD-TTCs 1–3.

See Figure 3 for the structures of payload or probe moieties.
Biological Evaluations
Internalization of ABTD-TTC-2 (2) and ABTD-TTC-3 (3) Fluorescent Probes into Cancer Cells via Receptor-Mediated Endocytosis (RME)
Internalization of 2 via RME was confirmed by confocal fluorescence microscopy (CFM) and flow cytometry (see Figure S48 in the Supporting Information). Internalization of 3 was monitored by CFM, and the fluorescence was quantified by flow cytometry, using ca. 10 000 treated live ID-8 (ovarian) and MX-1 (breast) cancer cells, overexpressing biotin receptors (Figure 5). CFM images in Figure 5 clearly show the efficient internalization of 3 via RME. Flow cytometry also indicates that at 20 μM concentration, the fluorescein signal intensity increases from 1 to 3 h incubation period.
Figure 5.

CFM images and flow cytometry analysis of different types of cells after incubation with 3 at the final concentration of 20 μM at 37 °C for different periods. (A) Flow cytometry analysis of 3 in ID-8 at 0 h (red, control), 1 h (green), and 3 h (blue). (B, C) CFM images and flow cytometry analysis in ID8 at 1 and 3 h, respectively. (D) Flow cytometry analysis of 3 in MX-1 at 0 h (red, control), 1 h (green), and 3 h (blue). (E, F) CFM images and flow cytometry analysis in MX-1 at 1 and 3 h, respectively. (For full internalization study results using ID8 and MX-1 cancer cells, as well as WI-38 normal human lung fibroblast cells, see Figure S49 in the Supporting Information.).
Multibinding Effect
The multiple TTMs (16 biotin modules in this case) on a single ABTD macromolecular drug conjugate are expected to increase its targeting efficiency to get internalized through RME via multibinding to the biotin receptors that are overexpressed on the surface of cancer cells. To prove this hypothesis, we synthesized a small-molecule fluorescent probe 28 (Figure 6), which only has one biotin moiety for one fluorescent moiety (TTM/fluorescein ratio = 1), for comparison with 3, bearing 16 biotin moieties for 4 fluorescent moiety (TTM/fluorescein ratio = 4).
Figure 6.

Biotin-PEG3-fluorescein probe 28 and biotin-SS-linker-taxoid conjugate 29 (BLT-S).
Figure 7 clearly shows that at 10 μM concentration of fluorescent probes, with increasing incubation time, both 28 and 3 were internalized via RME into two different cancer cell lines (ID-8 and MX-1) overexpressing biotin receptors. It also reveals that at the initial stage the RME rate of 3 is higher than 28, which indicates that the multiple TTM can increase the targeting efficiency to the cancer cells. In addition, for each time point, in terms of the different number of inherited fluorescein of 3 and 28, the fluorescein signal intensity of 3 is more than 4 times of 28 against ID-8 cell line, suggesting that the multiple TTM can enhance the cellular uptake of the whole conjugate via RME, resulting in more probes internalized at the end.
Figure 7.

Time-dependent RME rate comparison between a small fluorescent probe 28 (blue) and 3 (red). Curves were drawn based on the flow cytometry results by incubation of 3 at the concentration of 10 μM at 37 °C for 0 h (control), 1, 2, 4, 6, and 16 h, in ID-8 (left) and MX-1 (right) (for the flow cytometry data to produce these graphs, see Figures S50 and S51).
Cancer Cell Selective Cytotoxicity
Cytotoxicity assessment of ABTD conjugates 1–3 using MTT assay against ID-8 (murine ovarian cancer), MX-1 (human breast cancer), and WI-38 (human lung fibroblast, normal) cell lines were performed. For comparison purpose, paclitaxel, free taxoid 4, its fluorescein conjugate 5, and small-molecule biotin-SS-linker-taxoid conjugate, BLT-S 29 (Figure 5),64 were assayed as well. The results are summarized in Table 1.
Table 1. Cytotoxicity (IC50, nM) of Paclitaxel, 4, 5, 29, and ABTD-TTCs 1–3b.
| entry | compound | ID-8 | MX-1 | WI-38 |
|---|---|---|---|---|
| 1 | paclitaxel | 21.2 ± 4.30 | 3.83 ± 0.59 | 175 ± 51.6 |
| 2 | 4 (SB-T-1214) | 1.89 ± 0.30 | 2.90 ± 0.47 | 4.14 ± 0.82 |
| 3 | 5 (SB-T-1214-fluorescein) | 846 ± 112 | N.D. | 519 ± 156 |
| 4 | 29 (BLT-S) | 7.84 ± 1.85 | 26.7 ± 3.44 | 519 ± 90.3 |
| 5 | 29 + GSH-OEta | 5.91 ± 0.32 | 1.52 ± 0.34 | N.D. |
| 6 | 1 (ABTD-TTC-1) | 0.66 ± 0.03 | 2.05 ± 0.91 | 582 ± 48.8 |
| 7 | 1 + GSH-OEta | 0.62 ± 0.07 | 0.12 ± 0.05 | 903 ± 15 |
| 8 | 2 (ABTD-TTC-2) | >5000 | 3630 ± 1300 | N.D. |
| 9 | 2 + GSH-OEta | 651 ± 169 | 411 ± 80.9 | N.D. |
| 10 | 3 (ABTD-TTC-3) | >5000 | >5000 | N.D. |
The concentration of cytotoxic compounds that inhibit 50% (IC50, nM) of different types of cells after 72 h of drug exposure at 37 °C under 5% CO2 was examined. N.D. = not determined.
24 h of drug exposure, followed by thorough washing with Dulbecco's phosphate-buffered saline (DPBS), and 48 h incubation with 6 equiv of glutathione ethyl ester (GSH-OEt) at 37 °C under 5% CO2.
As Table 1 shows, the IC50 values of second-generation taxoid 4 against ID-8, MX-1, and WI-38 are 1.89, 2.90, and 4.14 nM, respectively, which are 1–2 orders of magnitude more potent than those of paclitaxel (entries 1 and 2). Taxoid 4 exhibited similar IC50 values against these three cell lines (entry 2). Paclitaxel showed small differences in their IC50 values against the three cell lines (entry 1). Thus, paclitaxel possesses appreciable selectivity difference between cancer cells and normal cells, but 4 shows little selectivity between cancer cells and normal cells, as anticipated. Table 1 also shows that fluorescence-labeled taxoid 5 with modification at the C7 position exhibits 2–3 orders of magnitude less potency than 4 (entry 3). Similar results were also observed from the comparison between 1 and 2 against ID-8 (IC50 0.66 nM, >5000 nM, respectively) and MX-1 (IC50 2.05, 3630 nM, respectively) (entries 6–10).
ABTD-TTC-1 1 exhibits remarkable potency (IC50 0.66 nM), 3 times higher than that of free taxoid 4 (IC50 1.89 nM) against ID-8 (entry 6), indicating excellent efficiency of this ABTD-based drug-delivery system, which is consistent with the CFM and flow cytometry results shown in Figures 5 and 6. When 6 equiv of glutathione ethyl ester were added to fully cleave the linker and release all taxoid payload, the IC50 did not change much (IC50 0.62 nM), indicating that endogenous glutathione level (and possibly some other thiols) was sufficient to cleave all of the internalized ABTD conjugate in 72 h incubation (entry 7). Drug conjugate 1 shows similar potency to free taxoid 4 against MX-1 (IC50 2.05, 2.90 nM, respectively) without addition of glutathione ethyl ester (entry 6). However, as anticipated, 1 exhibited 1 order of magnitude higher potency (IC50 0.12 nM) once 6 equiv of glutathione ethyl ester were added after 24 h and incubated for another 48 h, indicating the insufficient endogenous glutathione level in MX-1 cells (entry 7).
ABTD-TTC-1 (1) and small-molecule conjugate 29 exhibit a weak cytotoxicity (IC50 519 and 582 nM, respectively) against WI-38 normal cell line, while showing strong cytotoxicity against cancer cells (entries 4 and 6). The results indicate that excellent tumor-targeting efficiency was achieved by these conjugates using the biotin receptor as tumor-specific target. Moreover, 1 exhibited 2–3 orders of magnitude higher cytotoxicity against ID8 and MX-1 compared to normal cell line WI-38 (entry 6), which indicates that 1 has more than 10 times higher cancer cell selectivity than 29. Same trend was also observed on addition of 6 equiv of glutathione ethyl ester (entry 7). Therefore, the results indicate that a higher intracellular concentration of taxoid payload was achieved by utilizing the ABTD-DDS with multiple payloads, resulting in a higher potency of drug conjugate than the small-molecule conjugate 29 bearing single TTM with single taxoid payload. To assess a possible cytotoxicity of dendrimer itself as macromolecular vehicle, cytotoxicity of 3, which only has TTM and fluorescein moiety without cytotoxic payload, was examined. As Table 1 clearly shows, the IC50 values of 3 against ID-8 and MX-1 are both over 5000 nM, indicating that the cytotoxicity of ABTD scaffold itself is nontoxic and biocompatible (entry 10).
Conclusions
Asymmetric bow-tie PAMAM dendrimer-based (ABTD) platform as a novel tumor-targeting drug-delivery system has been designed, and three tumor-targeting ABTD conjugates (ABTD-TTC-1∼3) have been successfully synthesized. The ABTD-TTCs were obtained in good purity and biological evaluations were reproducible. By means of flow cytometry and confocal fluorescence microscopy (CFM), we confirmed that these macromolecular drug conjugates were internalized into cancer cells through RME. We also disclosed the multibinding effect of the tumor-targeting moieties by comparing the RME internalization rate and the fluorescein signal intensity between 3 and a small fluorescent probe 28. The IC50 values observed for 1 clearly showed the high efficiency of this dendrimer-based drug-delivery system compared to the drug itself and a small drug conjugate 29. The IC50 values observed for 3 indicated the nontoxicity and biocompatibility of this drug-delivery vehicle. Further studies along this line include different tumor-targeting molecules as well as newer-generation taxoids with higher potency. The present synthetic protocol is applicable to higher-generation dendrimers. In place of attaching a fluorescent probe at the C7 position of a taxoid, which resulted in significant loss of cytotoxicity, a branched bis(maleimide) linker that can accommodate a fluorescent probe or a radionuclide for PET, SPECT, or magnetic resonance imaging for diagnostic and theranostic studies has been designed. Further studies along this line are actively underway in our laboratory.
Experimental Section
General Methods
1H NMR and 13C NMR spectra were measured on a Varian 300 or 500 MHz spectrometer or a Bruker 400, 500, or 700 MHz NMR spectrometer. Melting points were measured on a Thomas–Hoover capillary melting point apparatus and are uncorrected. Thin-layer chromatography (TLC) analyses were performed on aluminum-backed Silica G TLC plates (Sorbent Technologies, 200 μm, 20 cm × 20 cm), visualized with UV light, and stained with sulfuric acid-EtOH, 10% PMA-EtOH, 10% vanillin-EtOH with 1% sulfuric acid or ninhydrin-butanol with 10% AcOH. Column chromatography was carried out on silica gel 60 (Merck, 230–400 mesh ASTM). Purification were also performed using Jupiter C18 semipreparative HPLC column (5 μm, 300 Å, 250 mm × 10 mm) or Jupiter C18 preparative HPLC column (5 μm, 300 Å, 250 mm × 21.2 mm) on a Shimadzu CBM-10AW VP communications bus module, Shimadzu SPD-10A VP UV–vis detector, and Shimadzu LC-6AD liquid chromatography assembly. Chemical purity was determined with a Shimadzu L-2010A HPLC HT series HPLC assembly, using a Kinetex PFP column (4.6 mm × 100 mm, 2.6 μm), employing CH3CN/water as the solvent system with a flow rate of 1 mL/min. GPC analysis was performed with a Shimadzu L-2010A HPLC HT series HPLC assembly, using a Ultrahydrogel 500 column (7.8 mm × 300 mm, 10 μm), employing CH3CN/water (20/80) with a flow rate of 0.5 mL/min. High-resolution mass spectrometry analysis was carried out on an Agilent LC-UV-TOF mass spectrometer using Jupiter C18 analytical column with 2.6 μm, 100 Å, 2.1 mm × 100 mm, with 0.1% TFA in water (optima grade) as solvent A and 0.1% TFA in CH3CN (optima grade) as solvent B, running temperature 25 °C, and flow rate 0.5 mL/min, at the Institute of Chemical Biology and Drug Discovery (ICB&DD), Stony Brook, NY, or at the Mass Spectrometry Laboratory, University of Illinois at Urbana–Champaign, Urbana, IL. MALDI-TOF analysis for the determination of molecular weight was carried out using DHB as matrix at the Institute of Chemical Biology and Drug Discovery (ICB&DD), Stony Brook, NY.
Materials
The chemicals were purchased from Sigma-Aldrich, Fisher Scientific, or VWR International and used as received or purified before use by standard methods. Dichloromethane and methanol were dried before use by distillation over calcium hydride under nitrogen. Ether and tetrahydrofuran were dried before use by distillation over sodium benzophenone kept under nitrogen. 10-Deacetylbaccatin III was obtained from Indena SpA, Italy. Reaction flasks were dried in a 100 °C oven and allowed to cool to room temperature in a desiccator over calcium sulfate and assembled under an inert nitrogen gas atmosphere. 4-(9-(2-Isobutoxycarbonylphenyl)-3-oxo-3H-xanthen-6-yloxy)butanoic acid (17),64 11-azido-3,6,9-trioxaundecan-1-amine (18),72 SB-T-1214 (4),28 2′-TBDMS-4,62 2-(5-oxo-triisopropylsiloxypent-2-yl)disulfanylphenylacetic acid (21),75 SB-T-1214-linker-SS-PEG3-azide 25,75tert-butyl 12-amino-4,7,10-trioxadodecanoate (13),76,77 12-biotynylamino-4,7,10-trioxadodecanoic acid,55,78 and biotin-PEG3-tether-SS-linker-SB-T-1214 (BLT-S) 29(64) were prepared by the literature methods. The PAMAM dendrimers bearing cystamine cores were purchased as methanol solution from Dendritic Nanotechnologies, Inc. G3 PAPAM(cystamine) dendrimer, DNT-296 (MW: 7001 g/mol) (10.1% in methanol) and G1 PAPAM(cystamine) dendrimer, and DNT-294 (MW: 1522 g/mol) (20.1% in methanol) were used as received. Biochemical materials, including RPMI-1640 cell culture media, DPBS buffer, fetal bovine serum (FBS), PenStrep, and TrypLE, were obtained from Gibco and VWR International and used as received for cell-based assays.
Synthetic Procedures
7-FOB-Taxoid 5
A solution of 2′-TBDMS-462 (184 mg, 0.19 mmol), DMAP (23 mg, 0.19 mmol), 20 (180 mg, 0.38 mmol), and EDC·HCl (72.8 mg, 0.38 mmol) in DCM (10 mL) and DMF (1 mL) was stirred for 9 days at rt. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated in vacuo and the crude product was purified on a silica gel column (DCM/MeOH with 99/1 to 9/1 gradient) to give 7-FOB-2′-TBDMS-taxoid (260 mg, 95% yield) as an orange solid: 1H NMR (500 MHz, CDCl3) δ 0.07 (s, 3H), 0.10 (s, 3H), 0.67 (d, J = 6.7 Hz, 3H), 0.69 (d, J = 6.7 Hz, 3H), 0.87 (m, 2H), 0.93 (s, 9H), 0.98 (m, 2H), 1.05 (m, 1H), 1.16 (s, 3H), 1.23 (s, 3H), 1.34 (s, 9H), 1.59 (m, 1H), 1.67 (m, 1H), 1.74 (s, 3H), 1.77 (s, 3H), 1.79 (s, 3H), 1.93 (s, 3H), 2.03 (d, J = 4.8 Hz, 2H), 2.09 (m, 2H), 2.29 (m, 1H), 2.40 (s, 3H), 2.46 (m, 2H), 3.71 (m, 2H), 3.96 (d, J = 6.8 Hz, 1H), 4.05 (m, 2H), 4.17 (d, J = 8.4 Hz, 1H), 4.23 (d, J = 3.2 Hz, 1H), 4.30 (d, J = 8.4 Hz, 1H), 4.75 (m, 1H), 4.82 (d, J = 9.7 Hz, 1H), 4.94 (d, J = 8.8 Hz, 1H), 5.22 (d, J = 7.4 Hz, 1H), 5.59 (m, 1H), 5.66 (d, J = 7.4 Hz, 1H), 6.12 (t, J = 8.8 Hz, 1H), 6.29 (s, 1H), 6.46 (s, 1H), 6.54 (m, 1H), 6.71 (m, 1H), 6.86 (t, J = 9.7 Hz, 2H), 6.95 (m, 1H), 7.27 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 7.4 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.64 (t, J = 7.6 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 8.00 (s, 1H), 8.08 (d, J = 7.4 Hz, 2H), 8.24 (d, J = 7.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ −5.2, −4.8, 8.8, 10.9, 12.8, 13.7, 14.6, 18.4, 18.6, 18.8, 18.9, 19.1, 20.7, 21.4, 22.6, 23.7, 25.7, 25.8, 26.1, 27.5, 28.3, 30.4, 30.6, 31.4, 33.3, 35.4, 36.5, 43.2, 46.8, 56.0, 60.4, 64.4, 67.9, 71.2, 71.4, 71.9, 74.6, 74.9, 75.0, 78.8, 80.7, 84.0, 100.7, 105.7, 113.9, 114.7, 117.5, 121.8, 128.6, 128.9, 129.2, 129.6, 129.8, 130.1, 130.3, 130.5, 130.8, 131.3, 132.3, 132.5, 133.7, 134.2, 136.5, 141.6, 150.6, 154.4, 155.1, 159.0, 162.6, 163.6, 165.5, 166.8, 169.9, 171.7, 171.9, 172.9, 173.8, 185.7, 202.4. HRMS (TOF) [M + H]+ calcd for C79H98NO21Si+ 1424.6395, found 1424.6398 (Δ = 0.2 ppm).
To an ice-cooled solution of 7-FOB-2′-TBDMS-taxoid (243 mg, 0.17 mmol) in a 1:1 mixture of acetonitrile/pyridine (9 mL, 0.02 M) was slowly added 2.4 mL of HF/pyridine under nitrogen. The reaction mixture was stirred at rt for 24 h and the progress was monitored by TLC. Upon completion, the reaction was quenched with 0.2 M aqueous citric acid (5 mL) and extracted with EtOAc (30 mL × 3). The organic layer was collected and washed with CuSO4 and distilled water until the organic layer became clear. Then, the organic layer was washed with brine (30 mL × 3), dried over anhydrous MgSO4, and concentrated in vacuo. The resulting crud product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 5 (115 mg, 53% yield) as an orange solid: mp 170–172 °C; 1H NMR (500 MHz, CDCl3) δ 0.67 (d, J = 6.7 Hz, 3H), 0.69 (d, J = 6.7 Hz, 3H), 0.85 (m, 2H), 0.95 (m, 2H), 1.00 (m, 1H), 1.07 (m, 1H), 1.18 (s, 3H), 1.23 (s, 3H), 1.35 (s, 9H), 1.59 (m, 1H), 1.68 (m, 1H), 1.76 (s, 3H), 1.79 (s, 3H), 1.80 (s, 3H), 2.01 (s, 3H), 2.08 (m, 2H), 2.37 (s, 3H), 2.41 (m, 2H), 2.56 (m, 1H), 3.73 (m, 2H), 3.96 (d, J = 6.8 Hz, 1H), 4.13 (m, 2H), 4.17 (d, J = 8.5 Hz, 1H), 4.26 (s, 1H), 4.31 (d, J = 8.5 Hz, 1H), 4.75 (t, J = 8.5 Hz, 1H), 4.95 (m, 2H), 5.35 (d, J = 7.4 Hz, 1H), 5.65 (t, J = 7.4 Hz, 2H), 6.11 (t, J = 8.5 Hz, 1H), 6.31 (s, 1H), 6.43 (s, 1H), 6.53 (m, 1H), 6.72 (d, J = 8.5 Hz, 1H), 6.86 (t, J = 9.2 Hz, 2H), 7.11 (s, 1H), 7.28 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.4 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 8.09 (d, J = 7.4 Hz, 2H), 8.24 (d, J = 7.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 8.8, 8.9, 10.8, 12.9, 14.1, 14.7, 18.6, 18.8, 18.9, 21.1, 22.3, 22.6, 23.4, 25.7, 26.2, 27.5, 28.2, 29.9, 30.0, 31.6, 33.4, 35.5, 43.2, 47.0, 51.5, 56.1, 67.9, 71.5, 71.9, 72.0, 73.6, 74.4, 75.1, 78.6, 79.7, 80.8, 83.9, 100.8, 105.7, 114.3, 114.7, 117.4, 120.9, 128.7, 128.8, 128.9, 129.1, 129.6, 129.8, 130.1, 130.3, 130.5, 130.8, 130.9, 131.3, 132.5, 133.8, 134.2, 137.5, 141.2, 150.8, 154.5, 155.4, 159.1, 163.7, 165.5, 166.8, 170.1, 172.1, 172.1, 172.9, 173.1, 185.7, 202.4. HRMS (TOF) [M + H]+ calcd for C73H84NO21+ 1310.5530, found 1310.5540 (Δ = 0.7 ppm).
(G1 Half-Dendron)-Linker-(G3 Half-Dendron) Construct 6
To a solution of (G1 half-dendron)-linker construct 12 (21 mg, 8.4 μmol) and biotin-PEG3-(G3 half-dendron) 10 (87 mg, 4.2 μmol) in MeOH/distilled water (1/1, 2.0 mL) was added 0.5 M TCEP (25.2 μmol, 50 μL) at rt. The resulting solution was stirred overnight. The progress of the reaction was monitored by FIA-MS until the starting material was totally consumed. Then, the solution was subjected to dialysis using an MWCO 6000–8000 tubing membrane against MeOH for 48 h. The solution was lyophilized to give asymmetric bow-tie dendrimer 6 as (53 mg, 49% yield) a yellow solid: 1H NMR (700 MHz, CD3OD) δ 0.87 (m, 32H), 1.27 (m, 32H), 1.43 (m, 32H), 1.59 (m, 32H), 1.64 (m, 32H), 1.73 (m, 32H), 1.80 (m, 8H), 2.07 (m, 72H), 2.16 (m, 8H), 2.20 (m, 32H), 2.27 (m, 8H), 2.35 (m, 48H), 2.38 (m, 32H), 2.44 (m, 120H), 2.49 (m, 16H), 2.57 (m, 32H), 2.62 (m, 8H), 2.70 (m, 8H), 2.78 (m, 64H), 2.84 (s, 32H), 2.91 (m, 16H), 2.98 (s, 32H), 3.09 (m, 16H), 3.19 (m, 32H), 3.23 (s, 8H), 3.33 (s, 16H), 3.35 (m, 32H), 3.46 (m, 8H), 3.49 (m, 8H), 3.52 (m, 48H), 3.60 (m, 96H), 3.62 (m, 80H), 3.65 (s, 8H), 3.66 (m, 24H), 3,68 (m, 16H), 3.72 (m, 42H), 4.30 (m, 16H), 4.48 (m, 16H). 13C NMR (176 MHz, CD3OD) δ 11.8, 14.3, 23.5, 23.9, 24.8, 25.4, 25.5, 28.0, 28.1, 28.3, 28.4, 30.2, 33.2, 34.3, 34.5, 35.3, 35.4, 35.5, 36.3, 37.2, 38.6, 38.7, 38.9, 39.0, 39.1, 39.6, 39.7, 39.8, 42.2, 48.4, 49.6, 49.7, 50.7, 54.2, 55.5, 55.6, 60.2, 61.9, 66.2, 66.8, 68.5, 69.0, 69.1, 69.2, 69.7, 69.8, 69.9, 70.0, 70.1, 70.2, 82.2, 99.9, 163.4, 164.6, 172.6, 172.7, 173.6, 174.7, 177.6, 177.7. MS (LC-TOF) [M + H]+ calcd for C574H1001N130O161S18+ 12 877.1505, found 1840.4098, 2146.9901, deconvolution value (neutral form) 12876.2067.
Fully Biotinylated G3 PAMAM Dendrimer 7
To a solution of G3 dendrimer DNT-296 in MeOH (1.88 g, 27 μmol) was added a solution of 14 (937 mg, 1.7 mmol, 2 equiv per amino group in DNT-296) in MeOH (19 mL). The resulting solution was stirred for 2 days at rt. The resulting product was purified by dialysis against MeOH for 24 h, followed by removal of solvent by lyophilizer at −49 °C to give 7 (435 mg, 64% yield) as a white solid: 1H NMR (700 MHz, CD3OD) δ 1.44 (m, 64H), 1.61 (m, 32H), 1.65 (m, 64H), 1.74 (m, 32H), 2.21 (t, J = 7.4 Hz, 64H), 2.37 (m, 113H), 2.45 (t, J = 6.3 Hz, 64H), 2.61 (m, 64H), 2.70 (d, J = 12.6 Hz, 32H), 2.81 (m, 124H), 2.91 (m, 42H), 3.21 (m, 32H), 3.34 (t, J = 5.3 Hz, 64H), 3.52 (t, J = 5.3 Hz, 64H), 3.61 (m, 256H), 3.72 (t, J = 6.3 Hz, 64H), 4.30 (m, 32H), 4.49 (m, 32H), 4.55 (s, 32H). 13C NMR (176 MHz, CD3OD) δ 20.0, 21.3, 25.4, 25.5, 28.1, 28.2, 28.3, 28.4, 33.3, 34.3, 35.3, 35.4, 36.3, 37.2, 38.7, 38.9, 39.0, 39.6, 39.8, 48.4, 49.6, 49.7, 50.7, 52.1, 53.4, 55.5, 55.7, 60.2, 61.9, 66.2, 66.9, 69.1, 69.2, 69.8, 69.9, 70.0, 70.1, 70.2, 164.6, 172.4, 172.6, 173.2, 173.6, 174.5, 174.7. MALDI-TOF-MS [M + H]+ calcd for C912H1605N218O252S34+ 20 747.1855, found 20 748.3, 10 378.6.
Fully Alkynylated G1 PAMAM Dendrimer 8
To a solution of G1 cystamine-PAMAM dendrimer (Dendritic Nanotechnologies Inc. Item # DNT-294, 20.1% solid in MeOH, 271 mg, 0.18 mmol, MW: 1522 g/mol) in MeOH (1.36 g, 0.18 mmol) was added 16 (1.14 g, 2.86 mmol, 3 equiv per amino group on G1 dendrimer) in MeOH (2.0 mL), and the resulting solution was stirred at rt for 2 days. The reaction mixture was purified by dialysis against MeOH for 2 days, and the resulting solution was concentrated by lyophilization to afford fully functionalized G1 dendrimer 8 as a sticky yellow solid (435 mg, 64% yield). For analytical purpose, further purification was performed by semipreparative HPLC using a Jupiter C18 column (5 μm, 300 Å, 250 mm × 21.2 mm) (conditions: solvent A = H2O + 0.1% TFA, solvent B = acetonitrile + 0.1% TFA with gradient from 20% B to 35% B; low flow rate: 10 mL/min): 1H NMR (700 MHz, CD3OD) δ 2.33 (m, 8H), 2.37 (t, J = 6.4 Hz, 16H), 2.41 (t, J = 6.4 Hz, 16H), 2.48 (m, 32H), 2.61 (m, 8H), 2.81 (m, 16H), 2.84 (m, 16H), 3.32 (m, 48H), 3.36 (m, 48H), 3.38 (t, J = 5.5 Hz, 16H), 3.55 (t, J = 5.5 Hz, 16H), 3.62 (m, 32H), 3.65 (m, 24H), 3.74 (t, J = 6.4 Hz, 16H). 13C NMR (176 MHz, CD3OD) δ 14.3, 33.2, 34.5, 36.3, 37.2, 38.6, 39.0, 48.4, 49.4, 49.6, 52.0, 66.8, 69.1, 69.2, 69.8, 69.9, 70.0, 70.1, 82.2, 172.6, 172.7, 173.2, 173.6. MS (MALDI-TOF) [M + H]+ calcd for C176H301N34O52S2+ 3789.6495, found 1895.205, 3788.202.
N,N′-Bis[6-(maleimido)hexanoylamino]-4,7,10-trioxatridecane (9)
To an ice-cooled solution of N-maleimidohexanoic acid (26) (1.1 g, 5.4 mmol) and NMM (546 mg, 5.4 mmol, 0.6 mL) in THF (20 mL, 0.25 M) was slowly added isobutyl chloroformate (738 mg, 5.4 mmol) with stirring. A white precipitate was immediately generated. The reaction mixture was allowed to warm to rt for 3 h, and the progress of the reaction was monitored by TLC. Upon completion, the precipitate was filtered out and the resulting THF solution was immediately taken to the next step. A small portion of the solution was concentrated in vacuo for 1H NMR analysis, which confirmed the formation of the corresponding activate ester, 6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoic propionic anhydride: 1H NMR (500 MHz, CDCl3) δ 0.96 (d, J = 6.7 Hz, 6H), 1.35 (m, 2H), 1.58 (m, 2H), 1.66 (m, 2H), 1.99 (m, 1H), 2.44 (t, J = 7.4 Hz, 2H), 3.50 (t, J = 7.4 Hz, 2H), 4.03 (d, J = 6.7 Hz, 2H), 6.68 (s, 2H). 13C NMR (125 MHz, CDCl3) δ 18.8, 23.6, 25.9, 27.6, 28.1, 33.9, 37.5, 75.5, 134.0, 149.2, 167.7, 170.8.
To an ice-cooled solution of the activated ester of 26 in THF was slowly added 4,7,10-trioxa-1,13-tridecanediamine (27) (595 mg, 2.7 mmol) with stirring. The resulting solution was allowed to warm to rt for 22 h, and the progress of the reaction was monitored by TLC. Upon completion, the solvent was removed in vacuo and the crude product was purified by column chromatography on silica gel (DCM/MeOH with gradient 99/1 to 9/1) to give 9 (814 mg, 50% yield for two steps) as a white foam-like solid: 1H NMR (500 MHz, CDCl3) δ 1.30 (quint, J = 7.4 Hz, 4H), 1.61 (quint, J = 7.4 Hz, 4H), 1.65 (quint, J = 7.4 Hz, 4H), 1.77 (quint, J = 7.4 Hz, 4H), 2.15 (t, J = 7.4 Hz, 4H), 3.35 (q, J = 6.4 Hz, 4H), 3.52 (t, J = 7.4 Hz, 4H), 3.57 (t, J = 6.4 Hz, 4H), 3.61 (m, 4H), 3.66 (m, 4H), 6.22 (m, 2H), 6.71 (s, 4H). 13C NMR (125 MHz, CDCl3) δ 25.2, 26.4, 28.3, 29.1, 36.5, 37.7, 37.8, 70.0, 70.1, 70.4, 134.0, 170.8, 172.7. HRMS (TOF) [M + H]+ calcd for C30H47N4O9+ 607.3338, found 607.3334 (Δ = −0.6 ppm).
PEGylated “Click-Ready” G1 PAMAM Half-Dendron Linker Construct 12
To a solution of 8 (66 mg, 17 μmol) and bis(maleimido) linker 9 (103 mg, 170 μmol) in MeOH (3.0 mL) was slowly added 0.5 M TCEP (51 μmol, 0.1 mL) at rt with stirring. Upon addition, the solution turned cloudy. The resulting mixture was stirred at rt for 6 h, and the progress of the reaction was monitored by FIA-MS. Upon completion, the reaction mixture was diluted with distilled water (1.0 mL) and purified by semipreparative chromatography using a Jupiter C18 column (5 μm, 300 Å, 250 mm × 21.2 mm; acidic solvent A: H2O + 0.1% TFA; solvent B: acetonitrile + 0.1% TFA; gradient: 45% B to 55% B; flow rate: 10 mL/min). The fractions containing product were combined and concentrated by lyophilization to give 12 (21 mg, 25% yield) as a yellow oil: 1H NMR (700 MHz, CD3OD) δ 1.28 (m, 4H), 1.58 (m, 4H), 1.62 (m, 4H), 1.74 (m, 4H), 2.17 (q, J = 7.2 Hz, 4H), 2.31 (t, J = 2.6 Hz, 4H), 2.41 (t, J = 7.2 Hz, 8H), 2.47 (t, J = 5.7 Hz, 16H), 2.76 (t, J = 6.2 Hz, 8H), 2.94 (m, 4H), 3.26 (m, 6H), 3.30 (m, 8H), 3.35 (m, 8H), 3.37 (t, J = 5.7 Hz, 8H), 3.43 (t, J = 5.7 Hz, 4H), 3.50 (m, 8H), 3.54 (m, 16H), 3.60 (m, 6H), 3.65 (m, 36H), 3.73 (m, 4H), 3.75 (t, J = 6.2 Hz, 8H), 5.50 (s, 4H), 6.82 (s, 2H). 13C NMR (176 MHz, CD3OD) δ 14.3, 25.1, 25.9, 26.0, 26.9, 27.8, 28.0, 28.3, 29.0, 29.1, 34.2, 34.5, 35.2, 35.4, 35.5, 36.2, 36.4, 36.5, 37.0, 38.0, 38.4, 38.9, 39.0, 39.5, 50.0, 50.1, 52.3, 52.6, 53.4, 66.8, 68.5, 68.6, 69.0, 69.1, 69.8, 69.9, 70.0, 70.1, 82.2, 133.9, 171.1, 171.3, 172.5, 172.6, 172.9, 174.3, 174.4, 175.0, 177.6. MS (LC-TOF) [M + H]+ calcd for C118H198N21O35S+ 2503.0535, found 657.3406, 835.4750, 1252.7086; deconvolution value: 2503.40.
1-Biotynyl-1-amino-3,6,9-trioxadodecanoyl-OSu 14
Activated ester 14 was prepared by the condensation of NHS with 12-biotynylamino-4,7,10-trioxadodecanoic acid,55,78 which was readily prepared from biotin through coupling of biotin with tert-butyl 12-amino-4,7,10-trioxadodecanoate (13),76,77 followed by deprotection of the tert-butyl ester moiety with HF/pyridine in accordance with the patent literature method.55,78 A solution of 12-biotynylamino-4,7,10-trioxadodecanoic acid (950 mg, 2.12 mmol), EDC·HCl (487 mg, 2.54 mmol), and NHS (293 mg, 2.54 mmol) in DCM (21 mL, 0.1 M) was stirred at rt for 48 h. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated in vacuo and the resulting crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 14 (977 mg, 86% yield) as a white sticky solid: 1H NMR (500 MHz, CDCl3) δ 1.24 (m, 2H), 1.40 (m, 2H), 1.64 (m, 2H), 1.71 (m, 2H), 2.19 (t, J = 7.4 Hz, 2H), 2.56 (t, J = 6.4 Hz, 1H), 2.68 (s, 1H), 2.71 (d, J = 12.8 Hz, 1H), 2.82 (s, 4H), 2.86 (t, J = 6.3 Hz, 2H), 3.10 (m, 1H), 3.40 (m, 2H), 3.53 (t, J = 5.0 Hz, 2H), 3.60 (m, 8H), 3.66 (s, 1H), 3.81 (t, J = 6.3 Hz, 2H), 4.28 (m, 1H), 4.47 (m, 1H). 13C NMR (125 MHz, CDCl3) δ 14.1, 22.6, 25.6, 28.2, 31.6, 32.2, 36.0, 39.1, 40.5, 55.6, 60.3, 61.8, 65.7, 66.6, 69.9, 70.1, 70.3, 70.4, 70.7, 164.3, 166.8, 169.1, 173.4. HRMS (TOF) [M + H]+ calcd for C23H37N4O9S+ 545.2276, found 545.2276 (Δ = 0 ppm).
12-(Pent-4-ynoylamino)-4,7,10-trioxadodecanoyl-OSu 16
ω-Alkynyl acid-activated ester 16 was prepared from 4-pentynoic acid (15) and 13, followed by deprotection of the tert-butyl moiety and esterification with NHS, as described below.
To a solution of 4-pentynoic acid (15) (1.04 g, 5.35 mmol) in DCM (27 mL, 0.2 M) was added tert-butyl 12-amino-4,7,10-trioxadodecanoate (13)76,77 (1.63 g, 5.88 mmol) and EDC·HCl salt (1.12 g, 5.88 mmol), and the solution was stirred for 35 h under rt. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated in vacuo and purified by column chromatography on silica gel (hexanes/EtOAc with 10/90-70/30 gradient) to give tert-butyl 12-(pent-4-ynoylamino)-4,7,10-trioxadodecanoate (1.64 g, 86% yield) as a light yellow oil: 1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 2.02 (m, 1H), 2.41 (m, 2H), 2.50 (m, 4H), 3.45 (m, 2H), 3.56 (m, 2H), 3.63 (m, 8H), 3.72 (m, 2H), 6.34 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 14.8, 28.0, 35.2, 36.2, 39.2, 66.9, 69.2, 69.8, 70.2, 70.3, 70.4, 70.5, 80.5, 83.0, 170.8, 170.9. HRMS (TOF) [M + H]+ calcd for C18H32NO6+ 358.2224, found 358.2231 (Δ = 1.9 ppm).
To a solution of tert-butyl 12-(pent-4-ynoylamino)-4,7,10-trioxadodecanoate (1.56 g, 4.36 mmol) in DCM (22 mL, 0.2 M) was slowly added TFA (4.4 mL) and the resulting solution was stirred for 4 h at rt. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated in vacuo and purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to afford 12-(pent-4-ynoylamino)-4,7,10-trioxadodecanoic acid (950 mg, 96% yield.) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 2.05 (m, 1H), 2.47 (m, 2H), 2.53 (m, 2H), 2.64 (m, 2H), 3.49 (m, 2H), 3.59 (m, 2H), 3.65 (m, 8H), 3.78 (m, 2H), 6.79 (s, 1H), 10.78 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 14.9, 34.8, 35.0, 39.5, 66.4, 69.6, 69.7, 70.2, 70.3, 70.4, 70.5, 82.6, 172.3, 175.2. HRMS (TOF) [M – H]− calcd for C14H22NO6– 300.1453, found 300.1465 (Δ = 4.0 ppm).
A solution of 12-(pent-4-ynoylamino)-4,7,10-trioxadodecanoic acid (1.35 g, 4.48 mmol), NHS (618 mg, 5.37 mmol), and EDC·HCl (1.03 g, 5.37 mmol) in DCM (22 mL, 0.2 M) was stirred for 19 h at rt. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was concentrated in vacuo and purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give activated ester 16 (1.22 g, 70% yield) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 2.00 (m, 1H), 2.35 (m, 2H), 2.46 (m, 2H), 2.80 (s, 4H), 2.86 (m, 2H), 3.40 (m, 2H), 3.51 (m, 2H), 3.60 (m, 8H), 3.80 (m, 2H), 5.27 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 14.8, 25.4, 25.5, 32.1, 35.1, 39.2, 65.6, 69.2, 69.7, 70.1, 70.3, 70.5, 70.6, 83.1, 166.8, 169.2, 171.0. HRMS (TOF) [M + H]+ calcd for C18H27N2O8+ 399.1762, found 399.1783 (Δ = 5.2 ppm).
Fluorescein-O-butanoyl-NH-PEG3-(CH2)2-N319
To a solution of 4-(9-(2-isobutoxycarbonylphenyl)-3-oxo-3H-xanthen-6-yloxy)butanoic acid (17)64 (116 mg, 0.24 mmol), 1.2 equiv EDC·HCl salt (55 mg, 0.29 mmol), and 0.5 equiv DMAP (14.7 mg, 0.29 mmol) in DCM (12 mL) was slowly added a solution of 11-azido-3,6,9-trioxaundecan-1-amine (18)72 (63 mg, 0.29 mmol) in DCM (2 mL), and the resulting solution was stirred under rt for 24 h. The progress of the reaction was monitored by TLC. Upon completion, the solvent was evaporated and the resulting crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 19 (136 mg, 0.20 mmol, 82% yield) as a sticky orange solid: 1H NMR (500 MHz, CD3OD) δ 0.70 (d, J = 6.7 Hz, 3H), 0.71 (d, J = 6.7 Hz, 3H), 1.09 (m, J = 6.7 Hz, 1H), 2.14 (quint, J = 6.7 Hz, 2H), 2.43 (t, J = 6.7 Hz, 2H), 3.36 (t, J = 6.7 Hz, 2H), 3.38 (t, J = 6.7 Hz, 2H), 3.54 (t, J = 5.4 Hz, 2H), 3.60 (m, 2H), 3.64–3.67 (m, 8H), 3.73–3.82 (m, 2H), 4.21 (t, J = 6.3 Hz, 2H), 6.51 (s, 1H), 6.59 (d, J = 9.6 Hz, 1H), 6.96 (d, J = 9.6 Hz, 1H), 7.06 (d, J = 4.0 Hz, 1H), 7.08 (d, J = 4.0 Hz, 1H), 7.25 (s, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.85 (t, J = 7.6 Hz, 1H), 8.32 (d, J = 7.6 Hz, 1H). 13C NMR (125 MHz, CD3OD) δ 17.8, 17.9, 24.8, 27.4, 31.8, 39.0, 50.3, 68.1, 69.1, 69.7, 69.8, 70.1, 70.2, 70.3, 71.4, 100.6, 104.1, 114.6, 114.8, 116.8, 128.0, 129.5, 129.9, 130.3, 130.5, 130.9, 131.2, 132.6, 133.6, 154.7, 155.0, 160.1, 164.8, 165.5, 173.8, 185.8. HRMS (TOF) [M + H]+ calcd for C36H43N4O9+ 675.3025, found 675.3064 (Δ = 5.7 ppm).
Fluorescein-O-butanoyl-OSu 20
A dark orange solution of 17 (100 mg, 0.21 mmol), NHS (36.8 mg, 0.32 mmol), DMAP (12.8 mg, 0.11 mmol), and EDC·HCl (61.3 mg, 0.32 mmol) in DCM (10 mL, 0.02 M) was stirred at rt for 17 h. The progress of the reaction was monitored by TLC. Upon completion, the solvent was removed in vacuo and the resulting crude product was directly purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give FOB-OSu 20 (114 mg, 95% yield) as an orange solid: 1H NMR (500 MHz, CDCl3) δ 0.64 (d, J = 6.7 Hz, 3H), 0.66 (d, J = 6.7 Hz, 3H), 1.54 (m, J = 6.7 Hz, 1H), 2.20 (quint, J = 6.7 Hz, 2H), 2.79 (s, 4H), 2.80 (m, 2H), 3.67 (m, 2H), 4.11 (t, J = 6.7 Hz, 2H), 6.37 (s, 1H), 6.46 (d, J = 7.8 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 6.82 (d, J = 4.0 Hz, 2H), 6.91 (d, J = 4.0 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.66 (t, J = 7.6 Hz, 1H), 8.20 (d, J = 7.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 18.8, 18.9, 24.1, 25.6, 27.4, 27.6, 66.7, 71.8, 100.8, 105.7, 113.6, 115.0, 117.6, 129.0, 129.7, 129.8, 130.3, 130.5, 130.7, 131.2, 132.6, 134.1, 150.3, 154.2, 158.9, 163.0, 165.5, 168.1, 169.1, 185.6. HRMS (TOF) [M + H]+ calcd for C32H30NO9+ 572.1915, found 572.2016 (Δ = 17 ppm).
7-FOB-2′-(2-SuO-tether-SS-phenylacetyl)taxoid 22
A solution of 2-(5-oxo-triisopropylsiloxypent-2-yl)disulfanylphenylacetic acid (21)75 (38 mg, 0.084 mmol), DMAP (2.6 mg, 0.02 mmol), EDC·HCl (24 mg, 0.126 mmol), and 7-FOB-taxoid 5 (115 mg, 0.09 mmol) in DCM (9 mL, 0.01 M) was prepared at 0 °C, kept at the same temperature for 10–15 min, and then allowed to warm to rt with stirring. The progress of the reaction was monitored by TLC. Upon completion, the solvent was evaporated in vacuo and the resulting crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 7-FOB-2′-(2-TIPSO-tether-SS-phenylacetyl)taxoid (101 mg, 73% yield) as an orange solid: 1H NMR (500 MHz, CDCl3) δ 0.66 (d, J = 6.7 Hz, 3H), 0.67 (d, J = 6.7 Hz, 3H), 0.82 (m, 2H), 0.86 (m, 2H), 0.90 (m, 2H), 0.94 (m, 2H), 1.00 (m, 1H), 1.03 (d, J = 7.4 Hz, 18H), 1.07 (m, 1H), 1.16 (s, 3H), 1.22 (s, 3H), 1.24 (m, 2H), 1.28 (m, 2H), 1.33 (s, 9H), 1.36 (s, 1H), 1.58 (m, 1H), 1.68 (s, 3H), 1.70 (s, 3H), 1.78 (s, 3H), 1.84 (m, 2H), 1.92 (m, 2H), 1.96 (s, 3H), 2.06 (m, 2H), 2.35 (s, 3H), 2.61 (m, 1H), 2.91 (m, 1H), 3.70 (m, 2H), 3.91 (m, 2H), 4.06 (m, 3H), 4.15 (d, J = 8.5 Hz, 1H), 4.29 (d, J = 8.5 Hz, 1H), 4.84 (m, 1H), 4.93 (m, 3H), 5.05 (d, J = 7.4 Hz, 1H), 5.58 (t, J = 3.0 Hz, 1H), 6.11 (t, J = 8.5 Hz, 1H), 6.28 (s, 1H), 6.31 (t, J = 8.5 Hz, 1H), 6.43 (s, 1H), 6.51 (d, J = 11.0 Hz, 1H), 6.72 (d, J = 8.5 Hz, 1H), 6.84 (m, 2H), 6.94 (m, 1H), 7.20 (m, 1H), 7.22 (d, J = 7.8 Hz, 1H), 7.27 (m, 2H), 7.28 (m, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.57 (t, J = 7.6 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 7.6 Hz, 2H), 8.24 (d, J = 7.5 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 8.7, 8.8, 10.9, 11.4, 11.8, 12.8, 14.1, 14.5, 17.8, 18.5, 18.8, 18.9, 20.5, 20.6, 20.7, 21.4, 22.3, 22.6, 23.7, 25.2, 25.7, 26.2, 27.5, 29.0, 29.7, 30.3, 30.4, 30.9, 31.0, 31.6, 32.9, 33.0, 33.3, 34.5, 34.6, 35.3, 36.0, 38.7, 43.2, 45.9, 46.8, 56.0, 68.0, 71.5, 71.7, 71.8, 74.6, 74.8, 75.0, 76.3, 78.7, 79.7, 80.6, 83.9, 100.7, 105.7, 113.9, 114.7, 117.5, 119.9, 127.7, 128.3, 128.6, 128.9, 129.2, 129.6, 129.8, 130.0, 130.1, 130.2, 130.5, 130.8, 130.9, 131.3, 132.1, 132.5, 133.1, 133.6, 134.2, 137.3, 137.7, 150.4, 154.3, 155.0, 159.0, 163.6, 165.5, 166.9, 168.1, 169.5, 169.6, 169.9, 171.7, 172.8, 173.0, 185.6. HRMS (TOF) [M + H]+ calcd for C95H118NO24S2Si+ 1749.7283, found 1749.7279 (Δ = −0.2 ppm).
To an ice-cooled solution of 7-FOB-2′-(2-TIPSO-tether-SS-phenylacetyl)taxoid (120 mg, 0.07 mmol) in a 1:1 mixture of acetonitrile/pyridine (1.4 mL, 0.05 M) was slowly added HF/pyridine (1.2 mL) under nitrogen. The reaction mixture was allowed to warm to rt, and the progress was monitored by TLC. Upon completion, the reaction was quenched with 0.2 M aqueous citric acid solution (10 mL) and extracted with EtOAc (10 mL × 3). The organic layer was collected and washed with CuSO4 and distilled water until the organic layer became clear. Then, the organic layer was washed with brine (10 mL × 3), dried over anhydrous MgSO4, and concentrated in vacuo to afford a crude product. The crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 7-FOB-2′-(2-carbohydroxy-tether-SS-phenylacetyl)taxoid (79 mg, 71% yield) as an orange solid: 1H NMR (500 MHz, CDCl3) δ 0.66 (d, J = 6.7 Hz, 3H), 0.67 (d, J = 6.7 Hz, 3H), 0.82 (m, 2H), 0.87 (m, 2H), 0.90 (m, 2H), 0.94 (m, 2H), 0.97 (m, 2H), 1.02 (m, 1H), 1.16 (s, 3H), 1.22 (s, 3H), 1.24 (m, 2H), 1.26 (m, 2H), 1.34 (s, 9H), 1.58 (m, 1H), 1.64 (m, 3H), 1.70 (s, 3H), 1.78 (s, 3H), 1.84 (m, 2H), 1.92 (m, 2H), 1.96 (s, 3H), 2.03 (m, 2H), 2.37 (m, 3H), 2.60 (m, 1H), 2.95 (m, 1H), 3.70 (m, 2H), 3.92 (m, 2H), 4.07 (m, 3H), 4.30 (d, J = 8.5 Hz, 1H), 4.94 (t, J = 8.5 Hz, 3H), 5.57 (t, J = 7.7 Hz, 1H), 5.66 (d, J = 6.9 Hz, 1H), 6.15 (q, J = 7.5 Hz, 1H), 6.28 (s, 1H), 6.56 (s, 1H), 6.59 (d, J = 9.7 Hz, 1H), 6.74 (d, J = 8.5 Hz, 1H), 6.88 (t, J = 10 Hz, 2H), 6.99 (s, 1H), 7.20 (m, 1H), 7.22 (d, J = 7.8 Hz, 1H), 7.27 (m, 2H), 7.28 (m, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.58 (t, J = 7.6 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.70 (t, J = 7.5 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 7.6 Hz, 2H), 8.25 (d, J = 7.5 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 8.7, 10.8, 12.8, 14.1, 14.6, 18.5, 18.8, 18.9, 20.3, 20.4, 21.3, 22.3, 22.7, 22.9, 23.6, 23.7, 25.7, 27.2, 27.5, 28.1, 28.2, 29.3, 29.7, 30.1, 30.4, 30.7, 30.8, 31.1, 31.9, 33.3, 35.3, 38.7, 43.2, 45.7, 46.0, 46.9, 49.0, 55.9, 68.0, 71.5, 71.8, 74.6, 74.8, 75.0, 76.3, 78.7, 79.8, 80.7, 83.9, 100.6, 105.4, 114.6, 114.7, 117.3, 119.9, 127.7, 128.3, 128.6, 129.0, 129.1, 129.5, 129.7, 130.1, 130.5, 130.7, 130.8, 131.0, 131.3, 132.1, 132.5, 133.2, 133.7, 134.1, 141.9, 151.8, 154.6, 159.3, 164.0, 165.4, 166.9, 168.3, 170.1, 171.8, 172.8, 185.7, 202.5. HRMS (TOF) [M + H]+ calcd for C86H98NO24S2+ 1592.5915, found 1592.5934 (Δ = 1.2 ppm).
A solution of 7-FOB-2′-(2-carbohydroxy-tether-SS-phenylacetyl)taxoid (79 mg, 0.05 mmol), N-hydroxysuccinimide (6.3 mg, 0.055 mmol), and EDC·HCl (10.5 mg, 0.055 mmol) in DCM (5 mL) was stirred at rt for 18 h, and the reaction was monitored by TLC. Upon completion, the solvent was evaporated in vacuo and the resulting crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 22 (73 mg, 86% yield) as an orange solid: 1H NMR (500 MHz, CDCl3) δ 0.69 (d, J = 6.7 Hz, 3H), 0.72 (d, J = 6.7 Hz, 3H), 0.88 (m, 2H), 0.92 (m, 2H), 1.00 (m, 2H), 1.08 (m, 2H), 1.19 (s, 3H), 1.24 (s, 3H), 1.27 (m, 3H), 1.31 (m, 3H), 1.36 (s, 9H), 1.60 (m, 1H), 1.69 (m, 2H), 1.72 (s, 3H), 1.80 (s, 3H), 1.87 (m, 1H), 1.98 (s, 3H), 2.05 (s, 1H), 2.07 (m, 2H), 2.34 (m, 1H), 2.38 (s, 3H), 2.46 (m, 2H), 2.59 (m, 1H), 2.67 (m, 1H), 2.84 (s, 4H), 3.01 (m, 1H), 3.75 (m, 1H), 3.79 (m, 1H), 3.97 (m, 1H), 4.00 (s, 1H), 4.09 (m, 1H), 4.13 (m, 2H), 4.17 (d, J = 8.5 Hz, 1H), 4.32 (d, J = 8.5 Hz, 1H), 4.87 (m, 1H), 4.89 (s, 3H), 5.10 (m, 1H), 5.59 (m, 1H), 5.69 (d, J = 6.9 Hz, 1H), 6.15 (t, J = 8.5 Hz, 1H), 6.30 (s, 1H), 6.46 (s, 1H), 6.54 (d, J = 8.5 Hz, 1H), 6.75 (d, J = 8.5 Hz, 1H), 6.87 (m, 2H), 6.98 (m, 1H), 7.26 (m, 2H), 7.31 (m, 2H), 7.47 (t, J = 7.6 Hz, 2H), 7.60 (t, J = 7.6 Hz, 1H), 7.67 (t, J = 7.5 Hz, 1H), 7.72 (t, J = 7.5 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 8.11 (d, J = 7.6 Hz, 2H), 8.27 (d, J = 7.5 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 8.7, 8.8, 10.8, 12.8, 14.2, 14.5, 18.5, 18.8, 18.9, 20.3, 20.4, 21.0, 21.4, 22.3, 23.7, 25.6, 25.7, 26.2, 27.5, 28.2, 29.7, 30.2, 30.3, 31.6, 33.3, 35.4, 38.7, 38.8, 43.2, 45.5, 46.8, 56.0, 60.4, 68.0, 71.5, 71.7, 71.8, 74.6, 74.8, 75.0, 76.3, 78.7, 79.8, 80.6, 83.9, 100.7, 105.7, 113.9, 114.7, 117.4, 119.9, 127.9, 128.0, 128.4, 128.6, 128.9, 129.2, 129.6, 129.8, 130.1, 130.3, 130.5, 130.6, 130.8, 130.9, 131.0, 131.3, 132.1, 132.5, 133.4, 133.6, 134.2, 137.1, 137.8, 141.9, 150.5, 154.3, 154.9, 159.0, 163.6, 165.5, 166.8, 168.1, 169.1, 169.5, 170.0, 171.1, 171.7, 172.8, 185.6, 202.5. HRMS (TOF) [M + H]+ calcd for C90H101N2O26S2+ 1689.6078, found 1689.6060 (Δ = −1 ppm).
7-FOB-Taxoid-Linker-SS-Tether-PEG3-(CH2)2-N324
To a stirred solution of 22 (73 mg, 0.043 mmol) in DCM (4 mL, 0.01 M) was slowly added a solution of 11-azido-3,6,9-trioxaundecan-1-amine (18)72 (9.4 mg, 0.043 mmol) in DCM (2 mL) at rt. The progress of the reaction was monitored by TLC. Upon completion, the solvent was evaporated in vacuo and the resulting crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 24 (57 mg, 70% yield) as an orange solid: 1H NMR (500 MHz, CD3OD) δ 0.69 (d, J = 6.7 Hz, 3H), 0.70 (d, J = 6.7 Hz, 3H), 0.95 (m, 2H), 1.02 (m, 2H), 1.17 (s, 3H), 1.20 (s, 3H), 1.30 (m, 3H), 1.44 (s, 9H), 1.59 (m, 1H), 1.69 (m, 1H), 1.77 (s, 3H), 1.79 (s, 3H), 1.80 (m, 2H), 1.92 (m, 3H), 1.93 (m, 1H), 2.07 (m, 2H), 2.27 (m, 2H), 2.34 (m, 1H), 2.43 (s, 3H), 2.47 (m, 2H), 2.55 (m, 2H), 2.91 (m, 1H), 3.37 (m, 3H), 3.52 (m, 2H), 3.60 (m, 2H), 3.66 (m, 8H), 3.75 (m, 1H), 3.78 (m, 1H), 3.96 (m, 1H), 4.02 (m, 1H), 4.12 (m, 1H), 4.21 (m, 2H), 4.25 (m, 2H), 4.97 (s, 2H), 5.01 (m, 1H), 5.32 (m, 1H), 5.62 (t, J = 8.9 Hz, 1H), 5.69 (d, J = 6.9 Hz, 1H), 6.17 (m, 1H), 6.33 (s, 1H), 6.52 (s, 1H), 6.60 (d, J = 9.7 Hz, 1H), 6.96 (d, J = 8.9 Hz, 1H), 7.06 (m, 2H), 7.27 (m, 2H), 7.34 (m, 2H), 7.44 (d, J = 7.3 Hz, 1H), 7.52 (t, J = 7.6 Hz, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.80 (t, J = 7.5 Hz, 2H), 7.85 (t, J = 7.4 Hz, 1H), 8.14 (d, J = 7.6 Hz, 2H), 8.31 (d, J = 7.5 Hz, 1H). 13C NMR (125 MHz, CD3OD) δ 7.7, 7.8, 10.0, 12.3, 13.7, 13.8, 17.2, 17.8, 17.9, 19.5, 19.6, 20.7, 21.8, 23.4, 24.7, 25.4, 27.4, 27.5, 29.8, 31.3, 32.8, 32.9, 35.2, 38.1, 38.2, 39.0, 43.2, 45.9, 46.0, 46.8, 49.3, 50.3, 55.8, 68.1, 69.1, 69.7, 69.9, 70.1, 70.2, 70.3, 71.4, 71.5, 71.7, 74.5, 75.0, 75.1, 75.8, 77.5, 79.1, 80.5, 83.7, 100.6, 100.7, 104.1, 114.8, 116.7, 119.8, 127.6, 127.7, 128.0, 128.3, 129.4, 129.7, 129.9, 130.2, 130.4, 130.5, 130.9, 131.0, 131.2, 132.6, 132.8, 133.2, 133.6, 137.2, 137.3, 137.4, 141.3, 154.7, 155.0, 156.1, 160.1, 164.9, 165.0, 165.5, 166.1, 168.8, 170.1, 170.8, 172.1, 173,1, 173.7, 185.7, 202.5. HRMS (TOF) [M + H]+ calcd for C94H114N5O26S2+ 1792.7188, found 1792.7177 (Δ = −0.6 ppm).
Taxoid-Linker-SS-Tether-PEG3-(CH2)2-N32575
To a solution of SB-T-1214-linker activated ester 23 (114 mg, 0.1 mmol) in DCM (10 mL) was added 1 equivalent of 18 (21.5 mg, 0.1 mmol) in DCM (2 mL) dropwise under room temperature for 23 h. The progress of the reaction was monitored by TLC. Upon completion, the solvent was evaporated in vacuo and the crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to give 25 (118 mg, 0.082 mmol, 85% yield). 1H NMR (500 MHz, MeOD) δ 1.12 (s, 3 H), 1.22 (s, 3 H), 1.25 (d, 3 H), 1.32 (s, 9 H), 1.63 (s, 3 H), 1.68 (d, 6 H), 1.72 (m, 1H), 1.88 (s, 6 H), 2.16–2.20 (m, 2H), 2.33 (s, 3 H), 2.46–2.53 (m, 1H), 2.68 (broad, 1H), 2.86–2.91 (m, 1H), 3.34–3.37 (m, 4H), 3.48–3.50 (q, J = 3 Hz, 2H), 3.59 (d, J = 3.75 Hz, 3H), 3.63–3.65 (m, 10H), 3.77 (d, J =7 Hz, 3H), 3.92–3.96 (dd, 1H), 4.06–4.09 (d, 1H), 4.14 (d, J = 8.4 Hz, 1H), 4.37 (q, J = 6.85 Hz, 1H), 4.92 (m, 4H), 5.08 (m, 1H), 5.63 (d, J = 7.1 Hz, 1H), 6.03 (m, 1H), 6.14 (t, J = 8.9 Hz, 1H), 6.26 (s, 1H), 7.21–7.32 (m, 2H), 7.44 (t, J = 7.75 Hz, 2H), 7.57 (t, J = 7.45 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 8.07 (d, J = 7.75 Hz, 2H). 13C NMR (125 MHz, MeOD) δ 9.1, 9.3, 9.5, 13.0, 14.8, 18.5, 20.6, 20.7, 22.2, 22.4, 25.7, 26.6, 28.2, 31.2, 33.4, 33.5, 35.4, 35.5, 38.7, 38.8, 39.2, 43.1, 45.6, 46.2, 46.3, 49.0, 50.6, 58.4, 69.7, 70.0, 70.2, 70.5, 70.6, 70.6, 70.7, 71.7, 72.1, 75.0, 75.2, 75.4, 76.3, 79.2, 79.7, 80.9, 84.4, 119.9, 127.8, 128.3, 128.6, 129.3, 130.1, 130.4, 130.5, 130.9, 131.0, 132.4, 133.3, 133.6, 137.4, 137.5, 137.7, 143.3, 155.0, 166.9, 168.1, 169.6, 170.3, 172.1, 175.0, 204.1. HRMS (TOF) [M + H]+ calcd for C66H90N5O20S2+ 1336.5615, found 1336.5620 (Δ = 0.4 ppm). These data are consistent with the literature values.75
Biotin-PEG3-Tether-Fluorescein 28
A solution of N-(2-(2-aminoethoxy)ethyl)-5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamide (biotin-PEG3-amine)63 (25 mg, 0.058 mmol) and FOB-OSu 20 (33 mg, 0.058 mmol) in DCM was stirred for 16 h at rt, and the progress of the reaction was monitored by TLC. Upon completion, the solvent was removed and the crude product was purified by column chromatography on silica gel (DCM/MeOH with 99/1 to 9/1 gradient) to afford fluorescent biotin probe 28 (43 mg, 85% yield) as an orange solid: 1H NMR (700 MHz, CD3OD) δ 0.70 (d, J = 6.7 Hz, 3H), 0.71 (d, J = 6.7 Hz, 3H), 1.30 (m, 1H), 1.42 (m, 2H), 1.59 (m, 2H), 1.65 (m, 2H), 1.71 (m, 1H), 2.14 (quint, J = 6.7 Hz, 2H), 2.20 (t, J = 7.3 Hz, 2H), 2.43 (t, J = 7.3 Hz, 2H), 2.69 (d, J = 12.3 Hz, 1H), 2.90 (dd, Ja = 12.3 Hz, Jb = 5.3 Hz, 1H), 3.19 (m, 1H), 3.35 (q, J = 5.3 Hz, 2H), 3.38 (q, J = 5.3 Hz, 2H), 3.53 (t, J = 5.4 Hz, 2H), 3.54 (t, J = 5.4 Hz, 2H), 3.61 (m, 10H), 3.73 (m, 1H), 3.78 (m, 1H), 4.21 (t, J = 5.4 Hz, 2H), 4.29 (m, 1H), 4.48 (m, 1H), 6.51 (s, 1H), 6.59 (d, J = 9.6 Hz, 1H), 6.96 (d, J = 9.6 Hz, 1H), 7.06 (d, J = 7.6 Hz, 2H), 7.25 (s, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.85 (t, J = 7.6 Hz, 1H), 8.00 (m, 1H), 8.09 (m, 1H), 8.31 (d, J = 7.6 Hz, 1H). 13C NMR (176 MHz, CD3OD) δ 17.8, 17.9, 24.8, 25.4, 25.5, 27.4, 28.1, 28.3, 31.8, 31.9, 35.3, 35.4, 38.9, 39.0, 39.1, 39.6, 55.6, 60.2, 61.9, 68.1, 69.2, 69.8, 70.1, 71.4, 100.6, 104.1, 114.6, 114.8, 116.7, 128.0, 129.5, 129.9, 130.3, 130.5, 130.9, 131.2, 132.6, 133.6, 154.7, 154.9, 160.0, 164.8, 165.4, 173.8, 174.7, 185.7. HRMS (TOF) [M + H]+ calcd for C46H59N4O11S+ 875.3896, found 875.3909 (Δ = 1.5 ppm).
ABTD-TTC-1 1
A mixture of click-ready asymmetric bow-tie dendrimer 6 (10 mg, 0.78 μmol), taxoid-linker-SS-PEG3-azide 25(75) (4.3 mg, 3.1 μmol), copper sulfate pentahydrate (0.75 mg, 3.1 μmol), and sodium ascorbate (0.5 mg, 3.1 μmol) in MeOH/distilled water (4/1, 5.0 mL) was stirred for 24 h at rt, and the progress of the reaction was monitored by FIA-MS. Upon completion, the reaction mixture was subjected to dialysis using an MWCO 6000–8000 tubing membrane for 48 h against MeOH. The resulting solution was filtrated to remove insoluble solid and then lyophilized to give ABTD-TTC-1 (1) (10.3 mg, 72% yield) as a light yellow solid: 1H NMR (700 MHz, CD3OD) δ 0.86 (m, 8H), 0.97 (m, 8H), 1.01 (m, 4H), 1.07 (m, 4H), 1.17 (s, 12H), 1.27 (dd, Ja = 2.7 Hz, Jb = 6.8 Hz, 12H), 1.40 (s, 36H), 1.60 (m, 12H), 1.65 (s, 16H), 1.73 (s, 12H), 1.75 (s, 12H), 1.91 (s, 12H), 2.14 (m, 4H), 2.22 (m, 8H), 2.24 (m, 12H), 2.37 (s, 12H), 2.45 (m, 16H), 2.56 (m, 16H), 2.71 (m, 4H), 2.86 (m, 8H), 2.93 (m, 4H), 2.98 (m, 8H), 3.12 (m, 12H), 3.19 (m, 12H), 3.33 (s, 12H), 3.35 (t, 16H), 3.39 (m, 4H), 3.49 (m, 12H), 3.53 (m, 12H), 3.58 (m, 16H), 3.61 (m, 16H), 3.64 (m, 40H), 3.72 (m, 12H), 3.77 (m, 4H), 3.84 (d, J = 7.1 Hz, 4H), 3.99 (dd, Ja = 4.6 Hz, Jb = 16.8 Hz, 4H), 4.10 (d, J = 16.8 Hz, 4H), 4.18 (q, J = 8.3 Hz, 8H), 4.30 (q, J = 7.1 Hz, 8H), 4.50 (m, 8H), 4.92 (s, 4H), 4.99 (d, J = 9.7 Hz, 4H), 5.27 (broad, 4H), 5.66 (d, J = 7.1 Hz, 4H), 6.13 (m, 4H), 6.45 (s, 4H), 6.82 (m, 4H), 7.26 (m, 4H), 7.31 (m, 8H), 7.49 (t, J = 7.6 Hz, 8H), 7.61 (t, J = 7.6 Hz, 4H), 7.79 (m, 4H), 8.11 (d, J = 7.6 Hz, 8H). 13C NMR (176 MHz, CD3OD) δ 7.7, 7.8, 9.0, 12.3, 13.5, 13.6, 17.2, 19.4, 19.5, 20.9, 21.8, 24.6, 25.5, 27.3, 28.1, 28.4, 31.3, 31.4, 32.7, 32.9, 35.4, 36.1, 38.1, 38.9, 39.0, 43.1, 45.8, 45.9, 46.6, 48.4, 50.3, 55.7, 57.8, 60.2, 61.9, 66.9, 69.1, 69.2, 69.7, 69.8, 70.1, 70.2, 70.9, 71.5, 74.9, 75.3, 77.7, 80.9, 84.4, 100.0, 127.5, 127.9, 128.2, 129.7, 130.0, 130.1, 130.2, 131.0, 133.1, 133.4, 137.2, 137.3, 141.2, 156.0, 164.6, 166.2, 168.8, 170.0, 170.9, 172.7, 173.6, 173.7, 174.6, 203.7. GPC analysis (see General Methods for conditions): tR = 22.0 min; >99% purity (see Figure S12).
ABTD-TTC-2 2
A mixture of 6 (7.5 mg, 0.58 μmol), FOB-taxoid-linker-SS-tether-PEG3-azide 24 (4.2 mg, 2.3 μmol), copper sulfate pentylhydrate (0.6 mg, 2.3 μmol), and sodium ascorbic acid (0.4 mg, 2.3 μmol) in MeOH/distilled water (4/1, 2.5 mL) was stirred for 24 h at rt, and the progress of the reaction was monitored by FIA-MS. Upon completion, the reaction mixture was subjected to dialysis using an MWCO 6000–8000 tubing membrane for 48 h against MeOH. The resulting solution was filtrated to remove insoluble solid and then lyophilized to give ABTD-TTC-2 (2) (8.0 mg, 69% yield) as a yellow solid: 1H NMR (700 MHz, CD3OD) δ 0.65 (t, J = 6.7 Hz, 12H), 0.67 (t, J = 6.7 Hz, 12H), 0.86 (m, 32H), 0.92 (m, 16H), 0.99 (m, 8H), 1.13 (s, 16H), 1.17 (m, 16H), 1.27 (m, 56H), 1.35 (m, 16H), 1.40 (s, 36H), 1.43 (m, 16H), 1.58 (m, 36H), 1.66 (m, 24H), 1.73 (m, 24H), 1.76 (24H), 1.86 (m, 8H), 1.89 (m, 16H), 2.03 (m, 8H), 2.11 (m, 8H), 2.16 (m, 8H), 2.22 (m, 16H), 2.24 (m, 8H), 2.31 (m, 12H), 2.39 (s, 16H), 2.46 (m, 48H), 2.53 (m, 24H), 2.64 (s, 4H), 2.66 (m, 4H), 2.70 (m, 12H), 2.87 (m, 16H), 2.92 (m, 18H), 2.97 (m, 24H), 3.19 (m, 24H), 3.33 (m, 36H), 3.39 (m, 8H), 3.47 (m, 16H), 3.50 (m, 8H), 3.53 (m, 32H), 3.62 (m, 108H), 3.72 (m, 32H), 3.75 (m, 8H), 3.83 (m, 4H), 3.86 (m, 4H), 3.93 (m, 8H), 4.17 (m, 8H), 4.20 (m, 12H), 4.26 (m, 4H), 4.31 (m, 16H), 4.49 (m, 16H), 4.52 (m, 8H), 4.68 (m, 4H), 4.93 (m, 8H), 4.99 (m, 8H), 5.27 (m, 8H), 5.59 (m, 8H), 5.65 (d, J = 6.9 Hz, 8H), 6.13 (broad, 4H), 6.29 (s, 4H), 6.50 (s, 4H), 6.57 (d, J = 6.9 Hz, 4H), 6.95 (d, J = 9.2 Hz, 4H), 7.04 (m, 8H), 7.26 (m, 8H), 7.31 (m, 8H), 7.41 (d, J = 7.5 Hz, 4H), 7.49 (t, J = 7.6 Hz, 8H), 7.61 (m, 4H), 7.63 (m, 4H), 7.71 (m, 4H), 7.78 (m, 8H), 7.82 (t, J = 7.5 Hz, 4H), 8.11 (d, J = 7.6 Hz, 8H), 8.28 (d, J = 7.5 Hz, 4H). 13C NMR (176 MHz, CD3OD) δ 7.7, 10.0, 12.2, 13.0, 13.7, 16.9, 17.2, 17.8, 19.4, 19.5, 20.7, 21.2, 21.7, 22.3, 23.4, 24.6, 25.0, 25.1, 25.4, 25.5, 25.7, 27.3, 27.4, 28.1, 28.4, 28.8, 29.0, 29.2, 29.3, 29.8, 31.6, 32.9, 34.9, 35.1, 35.2, 35.9, 36.3, 38.6, 28.7, 38.9, 39.7, 43.2, 45.8, 45.9, 49.7, 50.3, 55.6, 55.8, 56.9, 60.2, 61.9, 66.9, 67.4, 68.1, 68.5, 69.0, 69.1, 69.2, 69.7, 69.9, 70.1, 70.2, 70.5, 71.4, 71.7, 74.5, 75.0, 75.8, 77.5, 79.0, 80.5, 83.7, 100.7, 104.1, 114.8, 116.7, 119.8, 127.6, 127.9, 128.2, 128.4, 129.4, 129.7, 130.2, 130.3, 130.5, 130.9, 131.0, 131.2, 132.6, 133.2, 133.6, 137.2, 137.3, 154.8, 155.0, 160.1, 161.4, 161.6, 161.8, 164.6, 165.0, 166.1, 167.8, 168.8, 170.1, 172.6, 173.1, 173.7, 174.6, 185.8, 202.5. GPC analysis (see General Methods for conditions): tR = 11.5 min; >98% purity (see Figure S13).
ABTD-TTC-3 3
A mixture of 6 (11 mg, 0.85 μmol), fluorescein-tether-PEG3-azide 19 (2.3 mg, 3.4 μmol), copper sulfate pentylhydrate (1.7 mg, 6.8 μmol), and sodium ascorbic acid (1.2 mg, 6.8 μmol) in MeOH/distilled water (4/1, 2.5 mL) was stirred for 24 h at rt, and the progress of the reaction was monitored by FIA-MS. Upon completion, the reaction mixture was subjected to dialysis using an MWCO 6000–8000 tubing membrane for 48 h against MeOH. The resulting solution was filtrated to remove insoluble solid and then lyophilized to give ABTD-TTC-3 (3) (10 mg, 76% yield) as a yellow solid: 1H NMR (700 MHz, CD3OD) δ 0.70 (m, 8H), 0.89 (m, 32H), 1.19 (m, 16H), 1.30 (m, 32H), 1.39 (m, 12H), 1.46 (m, 48H), 1.62 (m, 48H), 1.67 (m, 32H), 1.76 (m, 28H), 1.94 (m, 8H), 2.19 (m, 24H), 2.19 (m, 32H), 2.31 (m, 8H), 2.42 (m, 24H), 2.48 (m, 120H), 2.74 (m, 24H), 2.95 (m, 36H), 3.19 (m, 16H), 3.23 (m, 32H), 3.26 (m, 16H), 3.36 (s, 16H), 3.38 (m, 32H), 3.42 (m, 8H), 3.55 (m, 68H), 3.65 (m, 188H), 3.75 (m, 40H), 4.34 (m, 16H), 4.53 (m, 16H), 4.62 (m, 8H), 4.93 (s, 16H), 4.99 (m, 8H), 6.66 (m, 4H), 6.94 (m, 4H), 7.30 (m, 4H), 7.49 (m, 4H), 7.62 (m, 4H), 7.74 (m, 4H), 8.15 (m, 4H), 8.21 (m, 4H). 13C NMR (176 MHz, CD3OD) δ 7.5, 9.9, 11.2, 11.3, 16.7, 17.8, 19.2, 21.3, 21.7, 22.3, 24.8, 25.0, 25.1, 25.2, 25.9, 27.2, 27.4, 28.1, 28.4, 28.8, 29.2, 29.3, 29.5,. 35.4, 35.8, 38.9, 39.7, 43.2. 50.3, 55.6, 55.8, 56.9, 60.2, 61.9, 66.9, 67.5, 68.4, 68.5, 69.0, 69.2, 69.7, 69.9, 70.1, 70.2, 100.7, 128.4, 129.4, 129.7, 130.2, 130.3, 130.4, 130.9, 132.0, 131.7, 132.6, 163.6, 164.9, 166.1, 166.8, 170.1, 172.6, 174.7, 174.6. GPC analysis (see General Methods for conditions): tR = 22.1 min; >99% purity (see Figure S14).
Biological Evaluations
Cell Culture
ID-8 (ovary), MX-1 (breast), and WI-38 (human lung fibroblast) cell lines were cultured as monolayers on 100 mm tissue culture dishes in the RPMI-1640 cell culture medium (Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) as well as 1% (v/v) penicillin and streptomycin (P/S) at 37 °C in a humidified atmosphere with 5% CO2. The cells were harvested and collected by centrifugation at 950 rpm for 5 min and finally suspended in fresh cell culture medium containing different cell densities for subsequent biological experiments and analysis.
Incubation of Cells with Fluorescent Probes
The cell suspension at 5 × 105 cells/well was split into each well of a six-well plate with 3 mL of RPMI-1640 cell culture medium and allowed to grow for 24 h. The fluorescent probe 28 or 3 in dimethyl sulfoxide (DMSO) was diluted in RPMI-1640 cell culture medium (3 mL) at a final concentration of 10 or 20 μM for multibinding effect study and internalization through RME study, respectively, and was used to replace the medium in each well followed by incubation at 37 °C for different periods (16, 6, 4, 2, and 1 h for multibinding effect study, and 3 and 1 h for internalization through RME study). One well with cells was incubated without any fluorescent probes as control for different periods (16 h for multibinding effect study, and 3 h for internalization through RME study). After incubation, the medium was aspirated and the cells were washed twice with PBS, collected by centrifugation, and resuspended in 300 μL of PBS for flow cytometry fluorescein measurement and confocal microscopy imaging.
Flow Cytometry Analysis of Treated Cells
Flow cytometry analysis of the treated cells was performed with a flow cytometer, FACSCalibur, operating at a 488 nm excitation wavelength and detecting emission wavelengths with a 530/30 nm bandpass filter. At least 10 000 cells were counted for each experiment using CellQuest 3.3 software (Becton Dickinson), and the distribution of fluorescence was analyzed using WinMDI 2.8 freeware (Joseph Trotter, Scripps Research Institute). Propidium iodide staining was used in all experiments to rule out dead cell count in the analysis.
Confocal Microscopy Analysis of Treated Cells
Cells treated as described above were dropped onto an uncoated glass dish (MatTek Corp.). Confocal fluorescence microscopy (CFM) experiments were performed using a Zeiss LSM 510 META NLO two-photon laser scanning confocal microscope system, operating at a 488 nm excitation wavelength and at 527 nm (23 nm detecting emission wavelength using a 505–550 nm bandpass filter). Images were captured using a C-Apochromat 63×/1.2 water (corr.) objective or a Plan-Apochromat 100×/1.45 oil objective. Acquired data were analyzed using LSM 510 META software.
Cytotoxicity Assay
Cytotoxicities of paclitaxel, SB-T-1214 (4), SB-T-1214-fluorescein (5), biotin-PEG3-linker-SB-T-1214 (BLT-S) (29), ABTD-TCC-1 (1), ABTD-TTC-2 (2), and ABTD-TTC-3 (3) were evaluated in vitro against ID-8 (ovary, BR++) and MX-1 (breast, BR+) cancer cell lines, wherein biotin receptor (BR) is overexpressed, as well as WI-38 (human lung fibroblast cell line, normal), by means of a quantitative colorimetric assay using a tetrazolium salt-based analysis (“MTT assay”; MTT = 3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma Chemical Co.). The inhibitory activity of each compound is represented by the IC50 value, which is defined as the concentration required for inhibiting 50% of the cell growth. The cells were harvested, collected, and resuspended in 200 μL of medium at a concentration of ∼0.5 × 106 cells (ID-8) or ∼1.5 × 106 (MX-1 and WI-38) per well over a 96-well plate. The cells were allowed to descend to the bottom of the plates overnight, and fresh medium was added to each well upon removal of the old medium. A drug or drug conjugate in DMSO stock solution was diluted to a series of concentrations in the cell culture medium to prepare test solutions. These test solutions at different concentrations ranging from 500 pM to 5 μM (100 μL each) were added to the wells in the 96-well plate, and the cells were subsequently cultured for 72 h. After removing the old medium by aspiration, 50 μL of DPBS buffer containing MTT (0.5 mg/mL) was added to each well and incubated at 37 °C for 3 h. The resulting DPBS buffer was then removed and the as-produced insoluble violet formazan crystals were dissolved in 50 μL of 0.1 N HCl in isopropanol to give a violet solution. The plate was allowed to shake for 8 min to fully dissolve the violet formazan crystal and then the spectrophotometric absorbance measurement of each well in the 96-well plate was run at 568 nm. The IC50 values and their standard errors were calculated from the viability–concentration curve using the Four Parameter Logistic Model of Sigmaplot. The concentration of DMSO per well was ≤1% in all cases.
Acknowledgments
This research was supported by a grant from the National Institutes of Health (CA103314 to I.O.). The authors are grateful to Dr. Anne Savitt and Rebecca Rowehl for their valuable help with cell culture preparations at the Cell Culture and Hybridoma Facility of Stony Brook University, Dr. Todd Hunt at the Flow Cytometry Center for FACS measurements, and Dr. Guo-Wei Tian at the Central Microscopy Imaging Center of Stony Brook University for his technical assistance with confocal fluorescence microscopy. They also thank Dr. Béla Ruzsicska for his helpful discussions on mass spectrometry analyses. The authors thank Dr. Galina Botchkina, Dr. Jonathan Rudick, and Dr. Miriam Rafailovich for their helpful advice.
Glossary
Abbreviations
- ABC
ATP-binding cassette
- ABTD
asymmetric bow-tie dendrimer
- BF-S
biotin-PEG3-(CH2)2-fluorescein
- BLT-S
biotin-PEG3-(CH2)2-(SS-Linker)-SB-T-1214
- CFM
confocal fluorescence microscopy
- CuAAC
copper-catalyzed azide–alkene cycloaddition
- DCM
dichloromethane
- DDS
drug-delivery system
- DHB
2,5-dihydroxybenzoic acid
- DIAD
diisopropyl azodicarboxylate
- DIC
N,N′-diisopropylcarbodiimide
- DMAP
4-N,N-dimethylaminopyridine
- DMF
dimethylformamide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- EPR
enhanced permeability and retention
- FITC
fluorescein isothiocyanate
- GSH
glutathione
- GSH-OEt
glutathione ethyl ester
- HPLC
high-performance liquid chromatography
- ID-8
murine ovarian cancer cell line
- mAb
monoclonal antibody
- MALDI-TOF
matrix-assisted laser desorption/ionization time of flight
- MDR
multidrug resistance
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MWCO
molecular weight cut-off
- MX-1
human breast carcinoma cell line
- NHS
N-hydroxysuccinimide
- NMM
N-methylmorpholine
- PAMAM
poly(amidoamine)
- PEG
poly(ethylene)glycol
- P-gp
P-glycoprotein
- PUFA
polyunsaturated fatty acids
- RME
receptor-mediated endocytosis
- SAR
structure–activity relationship
- SMDCs
small-molecule drug conjugates
- SWNT
single-walled carbon nanotubes
- TBDMSCl
tert-butyldimethylsilyl chloride
- TCEP
tris(2-carboxyethyl)phosphine
- TFA
trifluoroacetic acid
- TLC
thin-layer chromatography
- TTC
tumor-targeted conjugate
- TTDDS
tumor-targeted drug-delivery system
- TTM
tumor-targeting moiety
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00409.
1H and 13C NMR spectra of all new compounds, MALDI-TOF and LC-UV-TOF analyses of key intermediates, GPC analysis for ABTD-TTC conjugates 1–3, confocal fluorescence microscopy, and flow cytometry analyses (PDF)
Author Present Address
§ Kashiv Pharma, LLC, 995 US HWY 202/206, Bridgewater, New Jersey 08807, United States. E-mail: taow@kashivpharma.com.
The authors declare no competing financial interest.
Supplementary Material
References
- Jemal A.; Bray F.; Center M. M.; Ferlay J.; Ward E.; Forman D. Global cancer statistics. Ca-Cancer J. Clin. 2011, 61, 69–90. 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Siegel R.; Ma J.; Zou Z.; Jemal A. Cancer statistics, 2014. Ca-Cancer J. Clin. 2014, 64, 9–29. 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- Jaracz S.; Chen J.; Kuznetsova L. V.; Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- Ducry L.; Stump B. Antibody–Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjugate Chem. 2010, 21, 5–13. 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- Chen J.; Jaracz S.; Zhao X.; Chen S.; Ojima I. Antibody–cytotoxic agent conjugates for cancer therapy. Expert Opin. Drug Delivery 2005, 2, 873–890. 10.1517/17425247.2.5.873. [DOI] [PubMed] [Google Scholar]
- Wu X.; Ojima I. Tumor Specific Novel Taxoid-Monoclonal Antibody Conjugates. Curr. Med. Chem. 2004, 11, 429–438. 10.2174/0929867043455963. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Geng X.; Wu X.; Qu C.; Borella C. P.; Xie H.; Wilhelm S. D.; Leece B. A.; Bartle L. M.; Goldmacher V. S.; Chari R. V. J. Tumor-Specific Novel Taxoid–Monoclonal Antibody Conjugates. J. Med. Chem. 2002, 45, 5620–5623. 10.1021/jm025540g. [DOI] [PubMed] [Google Scholar]
- Hamann P. R.; Hinman L. M.; Hollander I.; Beyer C. F.; Lindh D.; Holcomb R.; Hallett W.; Tsou H.-R.; Upeslacis J.; Shochat D.; Mountain A.; Flowers D. A.; Bernstein I. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody–Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate Chem. 2002, 13, 47–58. 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- Chari R. V. J. Targeted delivery of chemotherapeutics: tumor-activated prodrug therapy. Adv. Drug Delivery Rev. 1998, 31, 89–104. 10.1016/S0169-409X(97)00095-1. [DOI] [PubMed] [Google Scholar]
- Bradley M. O.; Webb N. L.; Anthony F. H.; Devanesan P.; Witman P. A.; Hemamalini S.; Chander M. C.; Baker S. D.; He L.; Horwitz S. B.; Swindell C. S. Tumor Targeting by Covalent Conjugation of a Natural Fatty Acid to Paclitaxel. Clin. Cancer Res. 2001, 7, 3229–3238. [PubMed] [Google Scholar]
- Kuznetsova L.; Chen J.; Sun L.; Wu X.; Pepe A.; Veith J. M.; Pera P.; Bernacki R. J.; Ojima I. Syntheses and evaluation of novel fatty acid-second-generation taxoid conjugates as promising anticancer agents. Bioorg. Med. Chem. Lett. 2006, 16, 974–977. 10.1016/j.bmcl.2005.10.089. [DOI] [PubMed] [Google Scholar]
- Leamon C. P.; Reddy J. A. Folate-targeted chemotherapy. Adv. Drug Delivery Rev. 2004, 56, 1127–1141. 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Low P. S. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Delivery Rev. 2002, 54, 675–693. 10.1016/S0169-409X(02)00042-X. [DOI] [PubMed] [Google Scholar]
- Wagner E.; Curiel D.; Cotten M. Delivery of drugs, proteins and genes into cells using transferrin as a ligand for receptor-mediated endocytosis. Adv. Drug Delivery Rev. 1994, 14, 113–135. 10.1016/0169-409X(94)90008-6. [DOI] [Google Scholar]
- Nagy A.; Schally A. V.; Halmos G.; Armatis P.; Cai R.-Z.; Csernus V.; Kovács M.; Koppán M.; Szepesházi K.; Kahán Z. Synthesis and biological evaluation of cytotoxic analogs of somatostatin containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1998, 95, 1794–1799. 10.1073/pnas.95.4.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T. C.; Marks J. W.; Lavery L. A.; Faulkner S.; Rosenblum M. G.; Ellington A. D.; Levy M. Aptamer:Toxin Conjugates that Specifically Target Prostate Tumor Cells. Cancer Res. 2006, 66, 5989–5992. 10.1158/0008-5472.CAN-05-4583. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Bernshaw N.; Lu Z.-R.; Kopecek J.; Prestwich G. Targeted Delivery of Doxorubicin by HPMA Copolymer-Hyaluronan Bioconjugates. Pharm. Res. 2002, 19, 396–402. 10.1023/A:1015170907274. [DOI] [PubMed] [Google Scholar]
- Russell-Jones G.; McTavish K.; McEwan J.; Rice J.; Nowotnik D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J. Inorg. Biochem. 2004, 98, 1625–1633. 10.1016/j.jinorgbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Rowinsky E. K. The Development and Clinical Utility of the Taxane Class of Antimicrotubule Chemotherapy Agents. Annu. Rev. Med. 1997, 48, 353–374. 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- Vredenburg M. R.; Ojima I.; Veith J.; Pera P.; Kee K.; Cabral F.; Sharma A.; Kanter P.; Greco W. R.; Bernacki R. J. Effects of Orally Active Taxanes on P-Glycoprotein Modulation and Colon and Breast Carcinoma Drug Resistance. J. Natl. Cancer Inst. 2001, 93, 1234–1245. 10.1093/jnci/93.16.1234. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Slater J. C.; Michaud E.; Kuduk S. D.; Bounaud P.-Y.; Vrignaud P.; Bissery M.-C.; Veith J. M.; Pera P.; Bernacki R. J. Syntheses and Structure–Activity Relationships of the Second-Generation Antitumor Taxoids: Exceptional Activity against Drug-Resistant Cancer Cells. J. Med. Chem. 1996, 39, 3889–3896. 10.1021/jm9604080. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Slater J. C.; Kuduk S. D.; Takeuchi C. S.; Gimi R. H.; Sun C.-M.; Park Y. H.; Pera P.; Veith J. M.; Bernacki R. J. Syntheses and Structure–Activity Relationships of Taxoids Derived from 14β-Hydroxy-10-deacetylbaccatin III. J. Med. Chem. 1997, 40, 267–278. 10.1021/jm960563e. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Lin S.; Wang T. Recent advances in the medicinal chemistry of taxoids with novel beta-amino acid side chains. Curr. Med. Chem. 1999, 6, 927–954. [PubMed] [Google Scholar]
- Ojima I.; Wang T.; Miller M. L.; Lin S.; Borella C. P.; Geng X.; Pera P.; Bernacki R. J. Synthesis and structure-activity relationships of new second-generation taxoids. Bioorg. Med. Chem. Lett. 1999, 9, 3423–3428. 10.1016/S0960-894X(99)00629-0. [DOI] [PubMed] [Google Scholar]
- Ferlini C.; Distefano M.; Pignatelli F.; Lin S.; Riva A.; Bombardelli E.; Mancuso S.; Ojima I.; Scambia G. Antitumour activity of novel taxanes that act at the same time as cytotoxic agents and P-glycoprotein inhibitors. Br. J. Cancer 2000, 83, 1762–1768. 10.1054/bjoc.2000.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geney R.; Chen J.; Ojima I. Recent advances in the new generation taxane anticancer agents. Med. Chem. 2005, 1, 125–139. 10.2174/1573406053175292. [DOI] [PubMed] [Google Scholar]
- Ehrlichova M.; Vaclavikova R.; Ojima I.; Pepe A.; Kuznetsova L.; Chen J.; Truksa J.; Kovar J.; Gut I. Transport and cytotoxicity of paclitaxel, docetaxel, and novel taxanes in human breast cancer cells. Naunyn Schmiedebergs Arch. Pharmacol. 2005, 372, 95–105. 10.1007/s00210-005-1080-4. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Chen J.; Sun L.; Borella C. P.; Wang T.; Miller M. L.; Lin S.; Geng X.; Kuznetsova L.; Qu C.; Gallager D.; Zhao X.; Zanardi I.; Xia S.; Horwitz S. B.; Mallen-St. Clair J.; Guerriero J. L.; Bar-Sagi D.; Veith J. M.; Pera P.; Bernacki R. J. Design, Synthesis, and Biological Evaluation of New-Generation Taxoids. J. Med. Chem. 2008, 51, 3203–3221. 10.1021/jm800086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovář J.; EhrlichovÁ M.; ŠMejkalovÁ B.; Zanardi I.; Ojima I.; Gut I. Comparison of Cell Death-inducing Effect of Novel Taxane SB-T-1216 and Paclitaxel in Breast Cancer Cells. Anticancer Res. 2009, 29, 2951–2960. [PMC free article] [PubMed] [Google Scholar]
- Ojima I.; Zuniga E. S.; Berger W. T.; Seitz J. D. Tumor-targeting drug delivery of new-generation taxoids. Future Med. Chem. 2012, 4, 33–50. 10.4155/fmc.11.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Ojima I. Guided Molecular Missiles for Tumor-Targeting Chemotherapy-Case Studies Using the Second-Generation Taxoid as Warheads. Acc. Chem. Res. 2008, 41, 108–119. 10.1021/ar700093f. [DOI] [PubMed] [Google Scholar]
- Vlahov I. R.; Leamon C. P. Engineering Folate–Drug Conjugates to Target Cancer: From Chemistry to Clinic. Bioconjugate Chem. 2012, 23, 1357–1369. 10.1021/bc2005522. [DOI] [PubMed] [Google Scholar]
- Srinivasarao M.; Galliford C. V.; Low P. S. Principles in the design of ligandtargeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discovery 2015, 203–219. 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- Noguchi Y.; Wu J.; Duncan R.; Strohalm J.; Ulbrich K.; Akaike T.; Maeda H. Early Phase Tumor Accumulation of Macromolecules: A Great Difference in Clearance Rate between Tumor and Normal Tissues. Jpn. J. Cancer Res. 1998, 89, 307–314. 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R. M.; Kannan S.; Romero R.; Navath R. S.; Dai H.; Menjoge A. R.. Dendrimer Based Nanodevices for Therapeutic and Imaging Purposes. US20120003155 A1, 2012.
- Trivedi V.; Patel U.; Bhimani B.; Daslaniya D.; Patel G.; Vyas B. Dendrimer-Polymer of 21st Century. IJPRBS J. 2012, 1, 1–21. [Google Scholar]
- Esfand R.; Tomalia D. A. Poly(amidoamine) (PAMAM) dendrimers: From biomimicry to drug delivery and biomedical applications. Drug Discovery Today 2001, 6, 427–436. 10.1016/S1359-6446(01)01757-3. [DOI] [PubMed] [Google Scholar]
- El Kazzouli S.; Mignani S.; Bousmina M.; Majoral J.-P. Dendrimer therapeutics: covalent and ionic attachments. New J. Chem. 2012, 36, 227–240. 10.1039/C1NJ20459A. [DOI] [Google Scholar]
- Gillies E. R.; Fréchet J. M. J. Dendrimers and dendritic polymers in drug delivery. Drug Discovery Today 2005, 10, 35–43. 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- Hsu H.-J.; Bugno J.; Lee S.-r.; Hong S. Dendrimer-based nanocarriers: a versatile platform for drug delivery. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2017, 9, e1409 10.1002/wnan.1409. [DOI] [PubMed] [Google Scholar]
- Medina S. H.; El-Sayed M. E. H. Dendrimers as Carriers for Delivery of Chemotherapeutic Agents. Chem. Rev. 2009, 109, 3141–3157. 10.1021/cr900174j. [DOI] [PubMed] [Google Scholar]
- Mignani S.; El Kazzouli S.; Bousmina M.; Majoral J.-P. Expand classical drug administration ways by emerging routes using dendrimer drug delivery systems: A concise overview. Adv. Drug Delivery Rev. 2013, 65, 1316–1330. 10.1016/j.addr.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Mignani S.; Rodrigues J.; Tomas H.; Zablocka M.; Shi X.; Caminade A.-M.; Majoral J.-P. Dendrimers in combination with natural products and analogues as anti-cancer agents. Chem. Soc. Rev. 2018, 47, 514–532. 10.1039/C7CS00550D. [DOI] [PubMed] [Google Scholar]
- Sharma A. K.; Gothwal A.; Kesharwani P.; Alsaab H.; Iyer A. K.; Gupta U. Dendrimer nanoarchitectures for cancer diagnosis and anticancer drug delivery. Drug Discovery Today 2017, 22, 314–326. 10.1016/j.drudis.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Svenson S. The dendrimer paradox - high medical expectations but poor clinical translation. Chem. Soc. Rev. 2015, 44, 4131–4144. 10.1039/C5CS00288E. [DOI] [PubMed] [Google Scholar]
- Wolinsky J. B.; Grinstaff M. W. Therapeutic and diagnostic applications of dendrimers for cancer treatment. Adv. Drug Delivery Rev. 2008, 60, 1037–1055. 10.1016/j.addr.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Majoros I. J.; Myc A.; Thomas T.; Mehta C. B.; Baker J. R. PAMAM Dendrimer-Based Multifunctional Conjugate for Cancer Therapy: Synthesis, Characterization, and Functionality. Biomacromolecules 2006, 7, 572–579. 10.1021/bm0506142. [DOI] [PubMed] [Google Scholar]
- Zong H.; Thomas T. P.; Lee K.-H.; Desai A. M.; Li M.-h.; Kotlyar A.; Zhang Y.; Leroueil P. R.; Gam J. J.; Banaszak Holl M. M.; Baker J. R. Bifunctional PAMAM Dendrimer Conjugates of Folic Acid and Methotrexate with Defined Ratio. Biomacromolecules 2012, 13, 982–991. 10.1021/bm201639c. [DOI] [PubMed] [Google Scholar]
- Thomas T. P.; Choi S. K.; Li M.-H.; Kotlyar A.; Baker J. R. Jr. Design of riboflavin-presenting PAMAM dendrimers as a new nanoplatform for cancer-targeted delivery. Bioorg. Med. Chem. Lett. 2010, 20, 5191–5194. 10.1016/j.bmcl.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen D. G.; Desai A. M.; Waddell J. N.; Cheng X.-m.; Kelly C. V.; McNerny D. Q.; Majoros I. J.; Baker J. R.; Sander L. M.; Orr B. G.; Banaszak Holl M. M. The Implications of Stochastic Synthesis for the Conjugation of Functional Groups to Nanoparticles. Bioconjugate Chem. 2008, 19, 1748–1752. 10.1021/bc8002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Kukowska-Latallo J. F.; Tang S.; Zong H.; Johnson K. B.; Desai A.; Gordon C. L.; Leroueil P. R.; Baker J. R. Jr. The facile synthesis of multifunctional PAMAM dendrimer conjugates through copper-free click chemistry. Bioorg. Med. Chem. Lett. 2012, 22, 3152–3156. 10.1016/j.bmcl.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Teng G. Y.-H.; Ojima I.. Synthesis of a novel asymmetrical bow-tie PAMAM dendrimer-based drug conjugate bearing biotin and a second-generation taxoid, Abstract 244th American Chemical Society National Meeting, MEDI 69, Philadelphia, PA, August 19–23, 2012.
- Ojima I.; Wang T.; Teng Y.-H.. Synthesis of Asymmetric Bow-Tie PAMAM Dendrimer-Based Conjugates for Tumor-Targeting Drug Delivery. WO2015038493A1, 2015.
- Gaertner H. F.; Cerini F.; Kamath A.; Rochat A.-F. o; Siegrist C.-A.; Menin L.; Hartley O. Efficient Orthogonal Bioconjugation of Dendrimers for Synthesis of Bioactive Nanoparticles. Bioconjugate Chem. 2011, 22, 1103–1114. 10.1021/bc1005653. [DOI] [PubMed] [Google Scholar]
- Caminade A.-M.; Laurent R.; Delavaux-Nicot B.; Majoral J.-P. “Janusis” dendrimers: syntheses and properties. New J. Chem. 2012, 36, 217–226. 10.1039/C1NJ20458K. [DOI] [Google Scholar]
- Gillies E. R.; Dy E.; Fréchet J. M. J.; Szoka F. C. Biological Evaluation of Polyester Dendrimer: Poly(ethylene oxide) “Bow-Tie” Hybrids with Tunable Molecular Weight and Architecture. Mol. Pharm. 2005, 2, 129–138. 10.1021/mp049886u. [DOI] [PubMed] [Google Scholar]
- Gillies E. R.; Fréchet J. M. J. Designing Macromolecules for Therapeutic Applications: Polyester DendrimerPoly(ethylene oxide) “Bow-Tie” Hybrids with Tunable Molecular Weight and Architecture. J. Am. Chem. Soc. 2002, 124, 14137–14146. 10.1021/ja028100n. [DOI] [PubMed] [Google Scholar]
- Gillies E. R.; Fréchet J. M. J. Synthesis and Self-Assembly of Supramolecular Dendritic “Bow-Ties”: Effect of Peripheral Functionality on Association Constants. J. Org. Chem. 2004, 69, 46–53. 10.1021/jo035329s. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Gillies E. R.; Fox M. E.; Guillaudeu S. J.; Fréchet J. M. J.; Dy E. E.; Szoka F. C. A single dose of doxorubicin-functionalized bow-tie dendrimer cures mice bearing C-26 colon carcinomas. Proc Natl. Acad. Sci. U.S.A. 2006, 103, 16649. 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Zhao X.; Chen J.; Chen J.; Kuznetsova L.; Wong S. S.; Ojima I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjugate Chem. 2010, 21, 979–987. 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vineberg J. G.; Zuniga E. S.; Kamath A.; Chen Y.-J.; Seitz J. D.; Ojima I. Design, Synthesis, and Biological Evaluations of Tumor-Targeting Dual-Warhead Conjugates for a Taxoid–Camptothecin Combination Chemotherapy. J. Med. Chem. 2014, 57, 5777–5791. 10.1021/jm500631u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vineberg J. G.; Wang T.; Zuniga E. S.; Ojima I. Design, Synthesis, and Biological Evaluation of Theranostic Vitamin–Linker–Taxoid Conjugates. J. Med. Chem. 2015, 58, 2406–2416. 10.1021/jm5019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz J. D.; Vineberg J. G.; Herlihy E.; Park B.; Melief E.; Ojima I. Design, Synthesis and Biological Evaluation of a Highly-Potent and Cancer Cell Selective Folate-Taxoid Conjugate. Bioorg. Med. Chem. 2015, 23, 2187–2194. 10.1016/j.bmc.2015.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Chen S.; Zhao X.; Kuznetsova L. V.; Wong S. S.; Ojima I. Functionalized Single-walled Carbon Nanotubes as Rationally Designed Vehicles for Tumor-Targeted Drug Delivery. J. Am. Chem. Soc. 2008, 130, 16778–16785. 10.1021/ja805570f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima I. Tumor-Targeting Drug Delivery of Chemotherapeutic Agents. Pure Appl. Chem. 2011, 83, 1685–1698. 10.1351/PAC-CON-11-02-10. [DOI] [Google Scholar]
- Carter R. L.; Johnson B. F.; Sood A.; Rishel M. J.; Valliant J. F.; Stephenson K. A.; Wu T.; Yang Y.. Preparation of Biotin Stannanes for HPLC-Free Radioiodination. US20130261311A1, 2013.
- Brauch S.; Henze M.; Osswald B.; Naumann K.; Wessjohann L. A.; van Berkel S. S.; Westermann B. Fast and efficient MCR-based synthesis of clickable rhodamine tags for protein profiling. Org. Biomol. Chem. 2012, 10, 958–965. 10.1039/C1OB06581E. [DOI] [PubMed] [Google Scholar]
- Adamczyk M.; Grote J. Synthesis of novel spirolactams by reaction of fluorescein methyl ester with amines. Tetrahedron Lett. 2000, 41, 807–809. 10.1016/S0040-4039(99)02197-8. [DOI] [Google Scholar]
- Adamczyk M.; Grote J. Efficient Fluorescein Spirolactam and Bis-spirolactam Synthesis. Synth. Commun. 2001, 31, 2681–2690. 10.1081/SCC-100105396. [DOI] [Google Scholar]
- Goswami L. N.; Houston Z. H.; Sarma S. J.; Jalisatgi S. S.; Hawthorne M. F. Efficient synthesis of diverse heterobifunctionalized clickable oligo(ethylene glycol) linkers: potential applications in bioconjugation and targeted drug delivery. Org. Biomol. Chem. 2013, 11, 1116–1126. 10.1039/c2ob26968f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabacher A. W.; Lane J. W.; Schiesher M. W.; Leigh K. M.; Johnson C. W. Desymmetrization Reactions: Efficient Preparation of Unsymmetrically Substituted Linker Molecules. J. Org. Chem. 1998, 63, 1727–1729. 10.1021/jo971802o. [DOI] [Google Scholar]
- Seitz J. D.; Vineberg J.; Wei L.; Khan J. F.; Lichtenthal B.; Lin C.-F.; Ojima I. Design, Synthesis and Application of Fluorine-Labeled Taxoids as 19F NMR Probes for the Metabolic Stability Assessment of Tumor-Targeted Drug Delivery Systems. J. Fluorine Chem. 2015, 171, 148–161. 10.1016/j.jfluchem.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P. S.; Zuniga E. S.; Ojima I.; Carrico I. S. Targeted and armed oncolytic adenovirus via chemoselective modification. Bioorg. Med. Chem. Lett. 2011, 21, 4985–4988. 10.1016/j.bmcl.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernaro I.; Hartmann S.; Sommer L.; Hausmann H.; Rohner C.; Ruehl M.; Hoffmann-Roeder A.; Schlecht S. Synthesis of tumor-associated MUC1-glycopeptides and their multivalent presentation by functionalized gold colloids. Org. Biomol. Chem. 2015, 13, 81–97. 10.1039/C4OB01339E. [DOI] [PubMed] [Google Scholar]
- Keil S.; Claus C.; Dippold W.; Kunz H. Zur Entwicklung von Antitumor-Impfstoffen: ein synthetisches Konjugat aus tumorassoziiertem MUC1-Glycopeptidantigen und dem Tetanustoxin-Epitop. Angew. Chem. 2001, 113, 379–382. . [DOI] [Google Scholar]
- McReynolds K. D.; Hadd M. J.; Gervay-Hague J. Synthesis of Biotinylated Glycoconjugates and Their Use in a Novel ELISA for Direct Comparison of HIV-1 Gp120 Recognition of GalCer and Related Carbohydrate Analogues. Bioconjugate Chem. 1999, 10, 1021–1031. 10.1021/bc990050x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.