

Abstract
Background
Gastric cancer is currently the fourth leading cause of cancer-related death worldwide. Gastric cancer is often diagnosed at advanced stages and the outcome of the treatment is often poor. Therefore, identifying new therapeutic targets for this cancer is urgently needed. Integrin alpha 2 (ITGA2) subunit and the beta 1 subunit form a heterodimer for a transmembrane receptor for extracellular matrix, is an important molecule involved in tumor cell proliferation, survival and migration. Integrin α2β1 is over-expressed on a variety of cancer cells, but is low or absent in most normal organs and resting endothelial cells.
Results
In this report, we assessed the ITGA2 as the potential therapeutic target with the bioinformatics tools from the TCGA dataset in which composed of 375 gastric cancer tissues and 32 gastric normal tissues. According to the information from the Cancer Cell Line Encyclopedia (CCLE) database, the AGS cell line with ITGA2 high expression and the SUN-1 cell line with low expression were chosen for the further investigation. Interestingly, the anti-ITGA2 antibody (at 3 μg/ml) inhibited approximately 50% survival of the AGS cells (over-expressed ITGA2), but had no effect in SNU-1 cells (ITGA2 negative). The extents of antibody-mediated cancer inhibition positively correlated with the expression levels of the ITGA2. We further showed that the anti-ITGA2 antibody induced apoptosis by up-regulating the RhoA-p38 MAPK signaling to promote the expressions of Bim, Apaf-1 and Caspase-9, whereas the expressions of Ras and Bax/Bcl-2 were not affected. Moreover, blocking ITGA2 by the specific antibody at lower doses also inhibited cell migration of gastric cancer cells. Blockade of ITGA2 by a specific antibody down-regulated the expression of N-WASP, PAK and LIMK to impede actin organization and cell migration of gastric cancer cells.
Conclusions
Here, we showed that the mRNA expression levels of ITGA2 comparing to normal tissues significantly increased. In addition, the results revealed that targeting integrin alpha 2 subunit by antibodies did not only inhibit cell migration, but also induce apoptosis effect on gastric cancer cells. Interestingly, higher expression level of ITGA2 led to significant effects on apoptosis progression during anti-ITGA2 antibody treatment, which indicated that ITGA2 expression levels directly correlate with their functionality. Our findings suggest that ITGA2 is a potential therapeutic target for gastric cancer.
Electronic supplementary material
The online version of this article (10.1186/s12575-018-0073-x) contains supplementary material, which is available to authorized users.
Keywords: Integrin alpha-2, ITGA2, Gastric cancer, Cell migration, Proliferation, Apoptosis, Therapeutic target
Background
Gastric cancer is the fourth most common leading cause of cancer-related death in the world, with approximately 952,000 new cases and 723,000 cancer-related deaths per year, according to the GLOBOCAN 2012 report [1]. Although surgery has been the main treatment modality for non-metastatic gastric cancer, the majority of patients diagnosed at advanced stages often unfit for surgery. The prognoses for the patients are therefore less favorable, with a 5-year survival rate lower than 30% [2–4].
Currently, monoclonal antibody has become a new venue of cancer treatments [5, 6]. Some of the antibodies such as trastuzumab (an anti-human epidermal growth factor receptor 2 (HER2) antibody) and bevacizumab (an anti-vascular endothelial growth factor (VEGF) antibody) have been used for advanced gastric cancer [7–9]. However, these therapeutic antibodies do not treat all gastric cancer patients since less than 20% of gastric cancers showed HER2 gene amplification and approximately 25% of gastric cancers showed HER2 protein overexpression [10]. Furthermore, multiple clinical trials on the efficacies of trastuzumab and bevacizumab indicate that these antibodies may not improve the patient survival over chemotherapy alone [11–13]. Therefore, identifying new markers and develop new effective and safe therapies for advanced gastric cancer is urgently needed.
ITGA2 encodes the alpha 2 integrin that may form a heterodimer with beta 1 subunit of the α2β1 integrin as a transmembrane receptor for cell adhesions to extracellular matrix (ECM) [14, 15]; alpha 2 beta1 integrin as the major collagen receptor also can bind laminin, fibronectin and E-cadherin proteins [16–19]. Moreover, several evidences have shown that α2β1 integrin is primarily expressed in newborn vessels, activated endothelial cells and immune cells in vivo [20–22]. It is also over-expressed on a variety of cancer cells, but is absent or low in most normal organs and resting endothelial cells [20, 23]. Functional studies indicate that α2β1 promotes cancer cells migration and invasion as well as angiogenesis [24–26]. In addition, numerous studies have investigated that expression profiles ofα2β1 integrin on cancer cells positively correlate the aggressive behaviors during cancer progression [27–30]. Therefore, α2 integrin may represent a promising marker for developing targeted therapeutic agents of cancers.
In this study, we found that blockade of ITGA2 by a specific antibody inhibited gastric cancer cells by two mechanisms: reduced cell viability (increased apoptosis) and reduced cell migration. The extents of inhibition positively correlated to the expression of ITGA2 as the anti-cancer effects were more pronounced in ITGA2-overexpressing cells. We further identified the mitochondrial apoptotic pathway and the actin assembly pathway as the major molecular mechanisms induced or inhibited, respectively, by the anti-ITGA2 antibody. These data suggest that ITGA2 may be a potential therapeutic target for gastric cancer.
Methods
Dataset for ITGA2 mRNA Expression Levels in Gastric Cancer
The mRNA data of gastric normal and tumor tissues were downloaded from The Cancer Genome Atlas (TCGA) Research Network portal (cancergenome.nih.gov). The dataset contained 32 normal gastric tissues and 375 gastric cancer tissues, which involved 27 pairs of gastric cancer tissues and their matched non-cancerous tissue samples. The total RNA-seq sample data were integrated into a matrix with log2 transformed for the downstream analysis, and listed in Additional file 1: Table S2.
Genetic characterization of 38 human gastric cancer cell lines were accessed from the Cancer Cell Line Encyclopedia (CCLE) database (portals.broadinstitute.org/ccle).
Cell Lines and Cell Culture
The human gastric adenocarcinoma cell line AGS (ATCCno.CRL-1739) and SNU-1 cells (ATCC no. CRL-5971) were purchased from the Bioresource Collection and Research Center (Hsinchu, Taiwan). Cells were maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 2.2 g/L of sodium bicarbonate (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Invitrogen) at 37 °C in a humidified atmosphere with 5% CO2.
RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
The total RNA was extracted from the cells by TRIZOL reagent (Invitrogen), according to the manufacturer’s instructions. Reverse transcriptions of 5 μg total cellular RNA were performed using the SuperScript™ III One-Step RT-PCR System with Platinum™ Taq DNA Polymerase Kit (Invitrogen). Each PCR was run for 35 cycles by repeating denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min. The last cycle was followed by an additional incubation at 72 °C for 10 min. The sequences of the specific primers selected for the RT-PCR reactions are listed in Additional file 2: Table S1. The PCR products were size fractioned by 2% agarose gel electrophoresis and then imaged by a CCD camera. Gene expressions were presented as the relative intensities of the target RT-PCR products, which were normalized by the RT-PCR product of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene.
Immunoblotting Analysis
The cells were lysed in lysis buffer (5% glycerol, 1 mM sodium EDTA, 1 mM dithiothreitol, 40 μg/ml leupeptin, 40 μg/ml aprotinin, 20 μg/ml pepstatin, 1 mM PMSF, 0.5% Triton X-100 in PBS) and then incubated on ice for 30 min. After centrifugation at 10,000 xg for 30 min at 4 °C, supernatants were collected and the protein concentrations were determined by Bradford protein assay (Bio-Rad, Hercules, CA, USA). Fifty μg of the protein lysates were resolved by 10% SDS-PAGE. The resolved proteins were transferred onto 0.2 μm PVDF membranes (PALL). After blocking the membranes with 5% skim milk in TBST buffer (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20), a mouse anti-human ITGA2 monoclonal antibody (1:1000 dilution; Santa Cruz Biotechnology, Paso Robles, CA, USA) was added to the membranes in TBST supplemented with 5% skim milk at 4 °C overnight or at room temperature for 1 h. After extensive washing in TBST, the membranes were then probed with a goat anti-mouse peroxidase-conjugated antibody (1:10,000 dilution; Sigma–Aldrich) at room temperature for 1 h and then washed in TBST. Finally, the bound proteins were revealed by the WesternBright ECL Western blotting detection kit (Advansta, Menlo Park, CA, USA) and imaged by a CCD camera.
Flow Cytometry for Analysis of ITGA2 Expression Levels on the Cell Surface
AGS and SNU-1 cells (2 × 105) were stained by the mouse anti-human ITGA2 monoclonal antibody (1:100 dilution; Santa Cruz Biotechnology) at 4 °C for 1 h, and washed with cold 1X PBS. The cells were then incubated with a FITC-labeled goat anti-mouse IgG (H + L) antibody (1:500 dilution; Jackson Immuno Research) at 4 °C for 30 min. The antibody-treated cells were washed and resuspended in 500 μl PBS containing 1 μg/ml propidium iodide (Thermo Fisher Scientific) to distinguish live and dead cells. Cells were then analyzed by a FACS SCAN flow cytometer (BD Biosciences, San Jose, CA, USA).
Cell Proliferation Assay
AGS and SNU-1 cells were seeded at a density of 1 × 104 per well in 96-well plates overnight. The cells were treated with graded concentrations of mouse anti-human ITGA2 monoclonal antibody or isotype control antibody for another 48 h. Cell viability was measured by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Sigma–Aldrich, St. Louis, MO, USA).
Analysis of Cell Apoptosis
AGS cells were seeded at a density of 1 × 105 per well in 24-well plates and treated with 0.3 μg of mouse anti-human ITGA2 monoclonal antibody or isotype control for 48 h. The cells were detached by EDTA and washed cells with cold PBS. The cells were then stained by the PE Active Caspase-3 Apoptosis Kit (BD Biosciences), according to the manufacturer’s instructions. The apoptosis responses (activated caspase-3) of the treated cells were then analyzed by a FACS SCAN flow cytometer.
Cell Migration Assay
Migration assay was performed in triplicate using a 24-well Transwell chamber system (Corning Inc., NY, USA) equipped with a filter membrane with 8 μm pores. AGS cells were seeded in the upper chamber at 1 × 105 cells/well and treated with 0.1 μg of mouse anti-human ITGA2 monoclonal antibody or isotype control antibody in 0.3 ml of serum-free DMEM media. Media supplemented with 10% fetal bovine serum was placed in the bottom well in the volume of 0.5 ml. After 18 h incubation, the filter membranes were fixed with methanol and stained with 50 μg/ml propidium iodide (Thermo Fisher Scientific) for 30 min. The amount of cells that had migrated to the lower surface of the filter membrane was counted in ten random fields under an inverted fluorescence microscope (Olympus IX71, Stanford, CA, USA) at 200 × .
Cell Morphology Analysis
AGS cells were seeded at 1 × 105 on coverslips overnight (Marienfeld-Superior, Lauda-Königshofen, Germany), than the attachment of cells were treated with 0.3 μg of mouse anti-human ITGA2 monoclonal antibody or isotype control antibody for 12 h. The cells were fixed with 4% paraformaldehyde (Sigma–Aldrich) for 30 min, and permeabilized cells with 0.25% Triton X-100 (AMRESCO, Solon, OH, USA) for 15 min. After blocking with 5% BSA for 1 h, the cells were stained by FITC-conjugated phalloidin (Thermo Fisher Scientific) for 1 h. The cells were washed three times with PBS and incubated with 10 mg/ml of Hoechst 33,342 (Thermo Fisher Scientific) dye for 20 min. After another set of washes, cells were mounted onto slides with 20 μl of Dako Cytomation fluorescent mounting medium (DAKO, Glostrup, Denmark). Observation was performed in a 63× objective lens on a multiphoton and confocal microscope (Leica, Mannheim, Germany).
Statistical Analysis
Data were analyzed using the SAS statistical software package (SAS Institute Inc., Cary, USA).The data were expressed as the means ± SD. Student’s t-test was used when comparing two independent samples and ANOVA was used when comparing multiple samples. Differences with p < 0.05 were considered statistically significant.
Results
Expressions of ITGA2 in Gastric Cancer Cell Lines
To determine whether ITGA2 was over-expressed in gastric cancers, mRNA expression of ITGA2 in 32 normal gastric tissues and 375 gastric cancer tissues from the Caner Genome Atlas project (TCGA) were investigated. As shown in Fig. 1a, the mRNA expressions of ITGA2 were significantly higher in gastric cancers than in the normal gastric tissues (cancer expression (mean ± SD): 13.1 ± 10 vs. normal expression: 4.6 ± 3.98, p < 0.0001). To compare the expression of ITGA2 in gastric cancer and normal tissue in a same patient, 27 pairs of gastric cancer tissues and their matched non-cancerous tissue samples were compared. ITGA2 was found to be over-expressed in 74% (20/27) of the patients (Fig. 1b). Statistical analysis using the paired t-test indicated a significant difference in the mRNA levels of ITGA2 between the gastric cancer and non-cancerous tissues (cancer expression: 11.1 ± 9.39 vs. normal expression: 4.9 ± 4.24, p < 0.01). These data indicate that ITGA2 may play important role(s) in gastric cancer development.
Fig. 1.
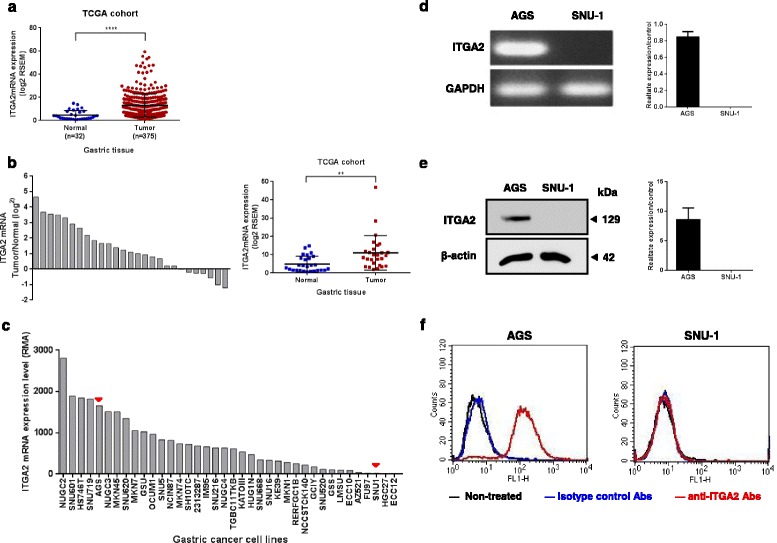
Analysis of ITGA2 expression levels in gastric cancers. a Data of ITGA2 mRNA levels were extracted from 32 normal gastric tissue and 375 gastric cancer tissue in the TCGA datasets. The result was expressed as mean ± standard deviation (SD), and statistical comparisons were made by Wilcoxon signed rank test. **** p < 0.0001. b The mRNA levels of ITGA2 in 27 available paired gastric normal and tumor tissues were compared, and a positive log2 (tumor/normal) value indicates increased expression, while a negative log2 value indicates decreased expression, of ITGA2 in the gastric tumor tissues. ITGA2 mRNA was significantly overexpressed in gastric tumor tissues as compared with normal tissues (p < 0.01, paired t-test). Result was expressed as mean ± standard deviation (SD). c mRNA expression profiles for ITGA2 across gastric cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE) database. Red arrowheads mark the expression levels of AGS and SUN-1 cells. d The expression of ITGA2 mRNA was measured by quantitative real-time PCR in AGS and SNU-1 cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. e The ITGA2 protein expression was semi-quantified by western blot analyses. d and e The densitometric measurements of ITGA2 in AGS and SNU-1 cells were normalized to the internal control (β-actin). f The surface expressions of ITGA2 were determined on PI-negative cells, using flow cytometry. Data are representative of three independent experiments
Additionally, the ITGA2 mRNA expression levels of 38 human gastric cancer cell lines were analyzed from the Cancer Cell Line Encyclopedia (CCLE) database. A spectrum of ITGA2 expression was noted across the gastric cancer cell lines studied. Seven human gastric cancer cell lines expressed high (defined as RMA > mean + 1 SD) and 5 human gastric cancer cell lines expressed low (defined as robust multi-array average (RMA) < mean-1 SD) ITGA2 mRNA levels (Fig. 1c), among which the AGS cells expressed high level of ITGA2 (RMA 1656.7) and the SUN-1 cells express low level of ITGA2 (RMA 18.0). We chose AGS and SUN-1 for further verifications by RT-PCR (normalized AGS expression: 0.9 ± 0.06 vs. normalized SUN-1 expression: non-detected, p < 0.0001, Fig. 1d) and western blot (normalized AGS expression: 8.6 ± 1.94 vs. SUN-1 expression: non-detected, p < 0.01, Fig. 1e). From these results, it is clear that AGS expressed high levels of mRNA and proteins of ITGA2, whereas ITGA2 expression in SUN-1 was low or undetectable. Furthermore, ITGA2 was expressed on the plasma membrane in AGS cells, but no expression on SNU-1 cells was noted (AGS mean fluorescence intensity (MFI): 178.7 vs. SUN-1 MFI: 4.2, p < 0.0001, Fig. 1f). Thus, we confirmed that the AGS and SNU-1 cells display contrasting ITGA2 expressions and we used these two cell lines to further investigate the potential role of ITGA2 in gastric cancer.
Anti-ITGA2 Antibody Induced Apoptosis in ITGA2-Overexpressed Gastric Cancer Cells
To investigate whether blocking ITGA2 could affect survival of gastric cancer cells, AGS and SNU-1 cells were treated with graded concentrations of an anti-ITGA2 antibody or an isotype control antibody and examined for cell numbers by an MTT assay. The anti-ITGA2 antibody dose-dependently inhibited the cell proliferation in AGS cells (Fig. 2a). The anti-ITGA2 antibody could inhibit AGS cells, even at low doses (0.01 μg/ml), albeit effect was minimal. Higher doses of the anti-ITGA2 antibody more efficiently inhibited the survival (~ 50% inhibition at 3 μg/ml) of the AGS cells. On the contrary, the anti-ITGA2 antibody did not cause any inhibition in SNU-1 cells. The anti-ITGA2 antibodies caused membrane shrinkage and apoptotic body-like structures in anti-ITG2 antibody-treated AGS cells (Additional file 3: Figure S1). Thus, we investigated whether anti-ITGA-2 antibody induce apoptosis in AGS cells. The levels of active caspase-3 were significantly increased in AGS cells treated with anti-ITGA2 antibodies compared to isotype control (anti-ITGA2 antibody MFI: 19.3 vs. isotype control MFI: 7.4, p < 0.001, Fig. 2b). These results suggested that blocking ITGA2 by a specific antibody induces apoptosis in ITGA2-overexpressing cells.
Fig. 2.
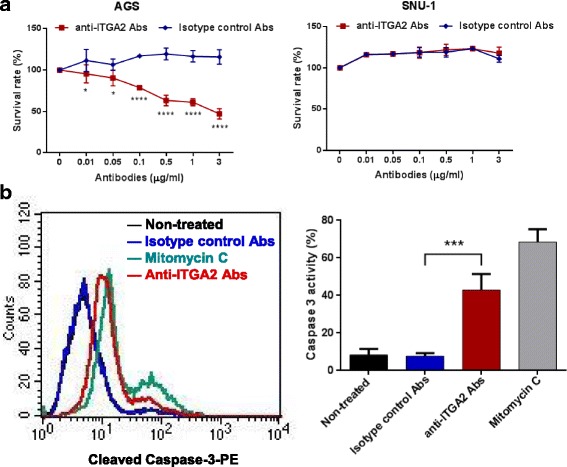
The anti-ITGA2 antibody induced apoptosis in AGS cells but not SUN-1 cells. a Human gastric cancer cell lines AGS and SUN-1 were treated with different concentrations of an anti-ITGA2 antibody for 48 h, and cell survivals were measured by an MTT assay. An isotype control antibody was used as a negative control. b AGS cells were treated with mitomycin C as a positive control, or incubated with 0.3 μg of anti-ITGA2 antibodies or isotype control (negative control) for 48 h. Cells were stained with PE-conjugated anti-active caspase-3 antibodies, and analyzed by flow cytometry. The percentages of caspase-3 positive-staining cells summarized in the right panel, showing mean ± standard deviation (S.D). Statistical comparisons were made by one-way ANOVA or two-way ANOVA with Bonferroni comparisons. * p < 0.05 and **** p < 0.0001. Data are representative of three independent experiments
ITGA2 Blockade Up-Regulated Bim-Mediated Mitochondrial Apoptotic Pathway
To characterize the molecular mechanism by which blockade of ITGA2 induced apoptosis in gastric cancer cells, we analyzed the mRNA expression of genes that are involved in the apoptotic pathways. RhoA and p38 MAPK mRNAs were strongly up-regulated in AGS cells after treatment by the anti-ITGA2 antibody, consistent with previous findings that activated RhoA-p38 pathway may induce apoptosis [31, 32]. Expression of the pro-apoptosis factor Bim was highly up-regulated in AGS cells after prolonged exposure to anti-ITGA2 antibody (anti-ITGA2 antibody: 7.1 ± 0.11 vs. isotype control: 1.0 ± 0.01, p < 0.0001), whereas expression of the anti-apoptotic factor Bcl-2 did not change (Fig. 3a and b). Interestingly, expression of the other pro-apoptotic factor Bax and anti-apoptotic factor Bcl-2 were unaltered by the anti-ITGA2 antibody treatment. Bim up-regulation can trigger cytochrome c release from mitochondria and induce the formation of the apoptosome (Apaf1 and cytochrome c) that led to activation of Caspase-9 and apoptosis. Thus, we analyzed the expression of Apaf-1 and Caspase-9 expression. ITGA2 blockade strongly induced Apaf-1 (anti-ITGA2 antibody: 10.3 ± 0.45 vs. isotype control: 0.8 ± 0.01, p < 0.0001) and Caspase-9 (anti-ITGA2 antibody: 87.2 ± 9.44 vs. isotype control: 1.1 ± 0.07, p < 0.0001) expression (Fig. 3a and b). These results indicate that ITGA2 blockade induces apoptosis through Bim, cytochrome c/Apaf-1 and Caspase-9, but not through Bcl-2 or Bax.
Fig. 3.
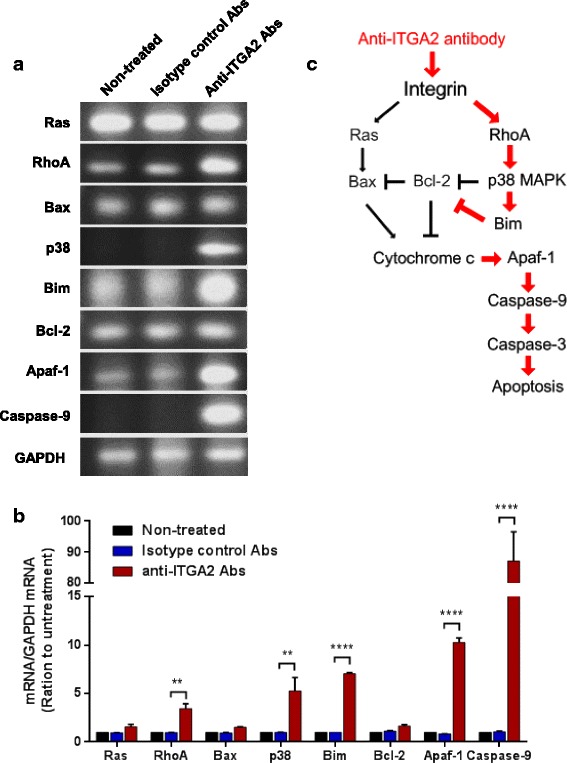
The anti-ITGA2 antibody induced a RhoA-p38 MAPK-mediated apoptotic pathway in AGS cells. a mRNA expression of Ras, RhoA, Bax, p38, Bim, Bcl-2, Apaf-1 and Caspase-9 in AGS cells treated with 0.3 μg of anti-ITGA2 antibodies or isotype control antibodies (negative control) for 48 h were analyzed by RT-PCR. Each analysis was derived from the same experiment and gels were processed in parallel. Cropped gels are displayed to compare gene expressions in different treatment groups. b Densitometric measurements on the intensities of each RT-PCR product was normalized to the mRNA level of GAPDH, and displayed as mean ± standard deviation (S.D). Statistical comparisons were made by two-way ANOVA with Bonferroni comparisons. ** p < 0.01, **** p < 0.0001. Data are representative of three independent experiments. c Summary of the anti-ITGA2 antibody-mediated apoptosis signaling pathway in AGS gastric cancer cells
ITGA2 Blockade Reduced Migration of ITGA2-Overexpressing Gastric Cancer Cells
Integrins are well known for regulating cell mobility through actin rearrangement. Thus we performed transwell cell migration assay to determine whether ITGA2 blockade could inhibit migration of gastric cancer cells. Since high dosage and prolonged incubation of the anti-ITGA2 antibody induced apoptosis in ITGA2-overexpressing AGS cells, we used a non-cytotoxic dosage of the anti-ITGA2 antibody (0.1 μg/ml, Additional file 4: Figure S2) and shorter exposure time (18 h) to study the effect of ITGA2 blockade on cell migration. As expected, low dose of anti-ITGA2 antibody treatments reduced the cells that migrated through the membrane by ~ 6.6 fold, as compared to the isotype control treatments (Fig. 4a). Decreased cell migration of AGS cells was accompanied by morphological alterations in the anti-ITGA2 antibody treated cells (Fig. 4b). Lamellipodia and filopodia that correlated with cell migration were markedly reduced and cells rounded up upon treatment with anti-ITGA2 antibodies. The confocal microscopy and image analysis revealed a ring-like actin structure, indicating disruption of F-actin formation, induced by low dose anti-ITGA2 antibodies. These findings suggested that ITGA2 blockade inhibit cell migration through destabilizing F-actin.
Fig. 4.
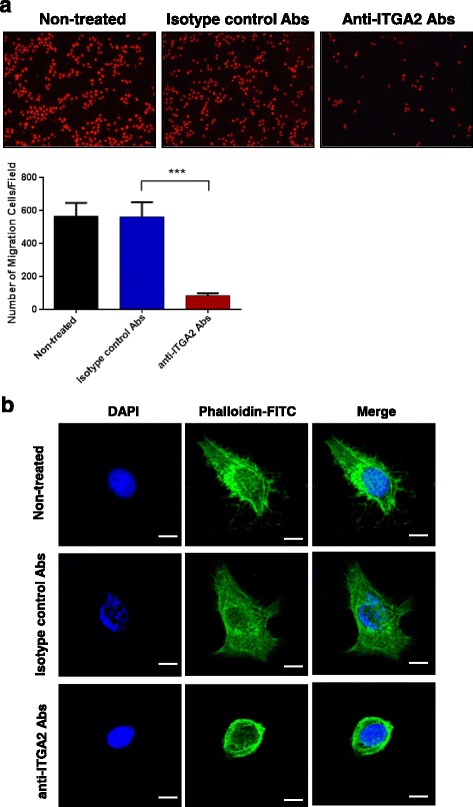
Blockade of ITGA2 reduced migration of AGS cells. a AGS cells were treated with 0.1 μg anti-ITGA2 antibodies or isotype control antibodies (negative control) for 18 h. Cells in the lower face of transwell membranes were stained by PI and imaged (upper panel) and data summarized as mean ± standard deviation (S.D) (lower panel). Statistical comparisons were made by two-way ANOVA with Bonferroni comparisons. *** p < 0.001. b AGS cells were treated with 0.3 μg of the anti-ITGA2 antibody for 12 h. F-actin (green) was stained with FITC-conjugated phalloidin, and nuclei stained by DAPI. Scale bar = 10 μm. Data are representative of three independent experiments
ITGA2 Blockade Reduced Actin Polymerization through the Rac1/CDC42 Signaling Pathway
To investigate how ITGA2 blockade may affect F-actin turnover, we examined the expression of RhoA, Rac1, CDC42, PAK, LIMK and N-WASP in anti-ITGA2 antibody treated AGS cells. mRNA expression of RhoA (anti-ITGA2 antibody: 0.9 ± 0.22 vs. isotype control: 0.9 ± 0.26), Rac1 (anti-ITGA2 antibody: 0.4 ± 0.06 vs. isotype control: 1.0 ± 0.03, p < 0.0001), CDC42 (anti-ITGA2 antibody: 0.6 ± 0.07 vs. isotype control: 1.0 ± 0.01, p < 0.0001), PAK (anti-ITGA2 antibody: 0.7 ± 0.45 vs. isotype control: 0.9 ± 0.08, p < 0.01), LIMK (anti-ITGA2 antibody: 0.4 ± 0.11 vs. isotype control: 1.0 ± 0.04, p < 0.0001) and N-WASP (anti-ITGA2 antibody: 0.4 ± 0.45 vs. isotype control: 0.9 ± 0.11, p < 0.0001) were markedly reduced in anti-ITGA2 antibodies treated cells (Fig. 5a and b). These results suggested that ITGA2 blockade down-regulated Rac 1 and CDC42 to inhibit actin filament assembly and impeded filopodia formation and lamellipodial protrusion (Fig. 5c). Accordingly, ITGA2 signaling inhibition may prevent metastasis of gastric cancer through down-regulating Rac 1/CDC42 signaling pathway.
Fig. 5.
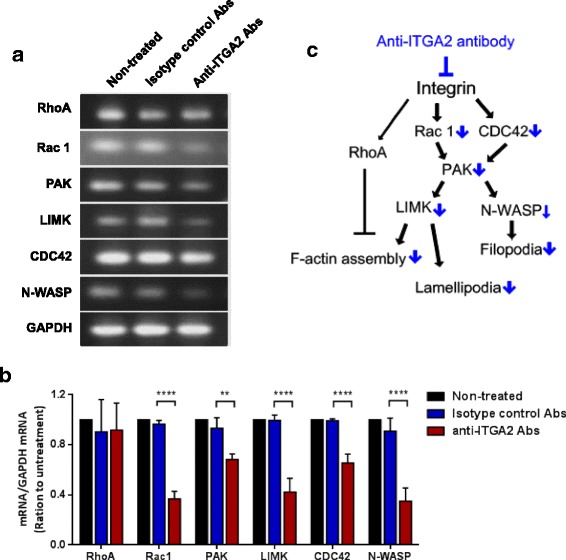
Anti-ITGA2 antibody reduced cell migration of AGS cells through down-regulation of Rac1 and CDC42 signaling pathway. a Gene expression of LIMK, Rac1, PAK, CDC42 and N-WASP in AGS after treatment with 0.1 μg anti-ITGA2 antibodies or isotype control antibodies (negative control) for 18 h. Each analysis was derived from the same experiment and gels were processed in parallel. Cropped gels are displayed for comparison between gene expressions in different groups. b Densitometric measurements on the intensities of each RT-PCR product was normalized to the mRNA level of GAPDH, and displayed as mean ± standard deviation (S.D). Statistical comparisons were made by two-way ANOVA with Bonferroni comparisons. ** p < 0.01 and **** p < 0.0001. c Summary of the anti-ITGA2 antibody-mediated migration signaling pathway in AGS gastric cancer cells
Discussion
Gastric cancer has few or no symptoms in the early stages thus ~ 65% patients present as advanced cancers (T3, T4) [33, 34]. Treatment outcomes for gastric cancer (by cisplatin/fluoropyrimidine or by trastuzumab in combination with chemotherapy for HER2-positive advanced gastric cancer) are unsatisfying; the 5-year survival rate of patients with advanced gastric cancer is below 20% and has not been improved in recent 20 years [35–37]. Although several chemical drugs (such as taxane and irinotecan [38]) or monoclonal antibodies (such as pembrolizumab and ramucirumab [39, 40]) are currently tested in phase II and III clinical trials, increasing reports indicates that these drugs do not significantly improve the therapeutic outcome over conventional chemotherapy [41–44] and the prognosis remains poor. To address this important issue, our lab is dedicated to finding new molecule candidates for the developments of targeted therapies. Our data indicated that ITGA2 may represent a promising candidate because a majority (74%) of gastric cancers over-expressed ITGA2. Moreover, blockade of ITGA2 by a specific antibody dually suppresses gastric cancer by inhibiting cell migration and inducing apoptosis. Thus the development of targeted therapies against ITGA2 may improve survival of advanced gastric cancer patients.
Recent studies showed that the ITGA2 gene over-expression is associated with increased risks of prostate cancer, breast cancer and gastric cancer [45–47]. Up-regulation of ITGA2 seems to be one of the important factors accelerating tumor progression and metastasis in various types of cancers, including gastric cancer [22, 48, 49]. In line with this notion, blocking ITGA2 was shown to inhibit cell migration and metastasis of gastric cancer [50, 51]. Surprisingly, we found that prolonged blockade of ITGA2 by a specific antibody at a relatively low dose (3 μg/ml) also suppresses gastric cancer cell survival, which is previously unknown. Blockade of ITGA2 by the antibody may induce apoptosis through RhoA-p38 MAPK signaling pathway in gastric cancer cells, consistent with previous reports that the RhoA and p38 MAPK are rapidly activated, upon cellular cholesterol depletion, to induce apoptosis [31]. In addition, RhoA and p38 MAPK activation is responsible for simvastatin-induced apoptosis in osteosarcoma cells [32]. However, other reports suggest RhoA activation may lead to cell death via activation of JNK-dependent signaling pathways and activation of pro-apoptotic protein Bim [52–54]. We did not address whether JNK pathways have involved the gastric cancer cell death treated with the anti-ITGA2 antibody at the moment. Nevertheless, further studies are needed to better delineate the mechanism for developing a targeted therapy for gastric cancer.
In this study, we found that caspase-3 expression was increased in AGS gastric cancer cells after treatment with the anti-ITGA2 antibody. Furthermore, we identified that RhoA, p38 MAPK and Bim were remarkably up-regulated by anti-ITGA2 antibody. This pathway against the activity of Bcl-2, the anti-apoptotic element, will facilitate apoptosis. Moreover, the increased Apaf-1, Caspase-9 and 3 will further activate the mitochondrial apoptotic death pathway (Fig. 3c). The molecular mechanisms underlying its apoptosis effects are incompletely understood, however, the intracellular (mitochondrial) apoptotic pathway is most likely involved. Bim, Apaf-1 and Caspase-9 were markedly up-regulated by anti-ITGA2 antibody, whereas Bcl-2 and Bax were unaltered. It is well known that Bim activates apoptosis by binding to Bcl-2 and disrupt Bcl-2-Bax complexes to release Bax. Free Bax then forms homodimer, or forms heterodimer with Bak, to subsequently promote cytochrome c release from the mitochondria [55–59]. Cytosolic cytochrome c form complex with Apaf-1 to activate caspase-9 which in turn activates caspase-3. Thus, although the expression of Bax and Bcl-2 were unaltered by the ITGA2 blockade, increased Bim expression is expected to promote Bax homo- or heterodimer formations to promote induction of apoptosis. On the other hand, although p38 MAPK was shown to mediate apoptosis by down-regulating anti-apoptotic Bcl-2 family proteins (e.g. Bcl-2, Bcl-XL) [60–62] and up-regulating the pro-apoptotic Bcl-2 family proteins (e.g. Bax, Bid and Bim) [54, 63, 64], RhoA-p38 MAPK pathway do not seem to regulate Bcl-2 and Bax expression in the case of ITGA2 blockade.
It is well established that F-actin assembly is necessary for protrusion dynamics and shaping cell morphology as well as cell migration [65–68]. In addition, epithelial-mesenchymal transition (EMT), a process where epithelial cells transformed into mesenchymal phenotypes, depends on the reorganization of cytoskeletal structures, which result in cell shape changes, cell elongation and membrane protrusions promote gastric cancer aggressiveness [69–71]. Integrins can either promote or suppress actin cytoskeleton with mediated cellular signaling pathway involved in cell movement and function [72–74]. We show that block ITGA2 has a dramatic impact on cell morphology in AGS cells. Furthermore, we found that a non-cytotoxic dose of the anti-ITGA2 antibody in shorter exposure reduced significantly migration of AGS cells, but had no effect on total cell number. In addition, our results indicated that blocking ITGA2 significantly decreased Rac 1 and CDC42 expressions, which further reduced the expressions of PAK. Reduced PAK signaling might also decrease the expressions of both N-WASP and LIMK, which impaired the filopodia formation, F-actin filament assembly and lamellipodial protrusion (Fig. 5c). These results are consistent with previous studies that CDC42 and Rac 1 are involved in intracellular signaling pathways downstream of integrins and affected actin filament organization [75–78]. For instance, modulation of CDC42 interaction with N-WASP has been shown to be implicated in filopodia formation [79, 80] and Rac1 recognized as a key regulator in the formation of lamellipodia [81, 82]. Thus, blocking ITGA2 is expected to eliminate both filopodia and lamellipodia formation by down-regulate Rac1 and Cdc42. It is also interesting to speculate whether blockade of ITGA2 can inhibit EMT since the small Rho GTPases, Rho, Rac and CDC42, have been shown to promote the EMT transition via down-regulated expression of E-cadherin and rearrange actin cytoskeleton that drive cell polarity and invasiveness [76, 83, 84]. Collectively, these data also support that blocking ITGA2 may be a promising method to inhibit migration of gastric cancers.
The integrin α2 is expressed in epithelial cells, platelets, megakaryocytes and fibroblasts [20–22]. Direct application of anti-ITGA2 antibodies to treat gastric cancer may induce considerable cytotoxicity to these cells. Several strategies may be used to reduce the risks of this complication and improve the safety of the antibodies, including designing pro-antibodies by masking the binding sites of antibodies which may cleaved by proteases that highly expressed at tumor sites [85–87]. This strategy may direct the therapeutic activity of the anti-ITGA2 antibodies to tumor lesions, but not the normal tissues. This development may alleviate the cytotoxicity associated with the antibody to achieve a better therapeutic index for gastric cancers.
Conclusions
In this study, our results demonstrated that ITGA2 is over-expressed in a majority of gastric cancer. Blockade of ITGA2 dually inhibited gastric cancer cells by inducing apoptosis and inhibiting cell migration, depending on the dose and duration of exposure. Notably, we showed that the RhoA-p38 MAPK signaling pathway promoted up-regulation of Bim to elicit the cytochrome c release, and following Apaf-1, Caspase-9 and 3 activated to induce the mitochondrial apoptotic death pathway, whereas the expression of Ras and Bax/Bcl-2 was not affected. We also discussed the down-regulation of N-WASP, PAK and LIMK, downstream of Cdc42 and Rac1, which altered actin organization and was involved in targeting ITGA2 inhibited cell migration of gastric cancer cells. Thus, targeting ITGA2 may be a promising therapy to improve the survival of gastric cancer patients.
Additional files
Table S2. RNA-seq sample annotation for each of the 32 normal gastric tissues and 375 gastric cancer tissues from TCGA. (DOCX 16 kb)
Table S1. Primer list for reverse transcription-polymerase chain reactions (RT-PCR). (DOCX 15 kb)
Figure S1. Effect of anti-ITGA2 antibody on cell morphology. The AGS cells were treated with a 3 μg of the anti-ITGA2 antibodies or isotype control antibodies (negative control) for 48 h, and cell morphology was observed at 200X magnification. Data are representative of three independent experiments. (PPTX 1463 kb)
Figure S2. Low dose of anti-ITGA2 antibodies did not induce cell death in AGS cells. Photography and quantitative analyses on cell number of the AGS cells treated with 0.1 μg anti-ITGA2 antibodies or isotype control antibodies (negative control) for 18 h. Data are expressed as mean ± standard deviation (S.D). Statistical comparisons were made by one-way ANOVA with Bonferroni comparisons. Data are representative of three independent experiments. (PPTX 784 kb)
Acknowledgements
We thank the core facility of the multiphoton and confocal microscope system of the National Chiao Tung University (NCTU).
Funding
This study was supported by the Aiming for the Top University Program of the Mackay Memorial Hospital Research Foundation (MMH-106-112 and MNH-104-2311-B-034-001), and in part by the Ministry of Science and Technology (MOST105–2314-B-182A-133 and MOST104–2311-B-034-001) and Chang-Gung Memorial Hospital Research Foundation (CMRPG2F0141~ 2 and CMRPG2D0091~ 3).
Abbreviations
- ECM
Extracellular matrix
- HER2
Human epidermal growth factor receptor 2
- ITGA2
Integrin alpha 2
- RT-PCR
Reverse transcription polymerase chain reaction
- VEGF
Vascular endothelial growth factor
Authors’ Contributions
YCC, HYW and CHC performed experiments, data analysis and approved the manuscript. YLL and SCT contributed to experimental procedures, and helped draft the manuscript. TYJ, PRC, YCC, CHL, THH and CCW participated in the design of the experiments. YLC and KWL conceived of the study, designed the study, coordinated the study and draft the manuscript. All authors gave final approval for publication.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12575-018-0073-x) contains supplementary material, which is available to authorized users.
Contributor Information
Yi-Lin Chan, Phone: +886-2-28610511, Email: phd.elainechan@gmail.com.
Kuang-Wen Liao, Phone: +886-3-5712121, Email: liaonms@mail.nctu.edu.tw.
References
- 1.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin D, Forman D, Bray F. GLOBOCAN 2012 v1. 0, cancer incidence and mortality worldwide: IARC CancerBase no. 11 [internet]. 2013. International Agency for Research on Cancer.globocan iarc fr/Default aspx: Lyon, France; 2014. [Google Scholar]
- 2.Bria E, De Manzoni G, Beghelli S, Tomezzoli A, Barbi S, Di Gregorio C, Scardoni M, Amato E, Frizziero M, Sperduti I, et al. A clinical-biological risk stratification model for resected gastric cancer: prognostic impact of Her2, Fhit, and APC expression status. Ann Oncol. 2013;24:693–701. doi: 10.1093/annonc/mds506. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto H, Watanabe Y, Maehata T, Morita R, Yoshida Y, Oikawa R, Ishigooka S, Ozawa S, Matsuo Y, Hosoya K, et al. An updated review of gastric cancer in the next-generation sequencing era: insights from bench to bedside and vice versa. World J Gastroenterol. 2014;20:3927–3937. doi: 10.3748/wjg.v20.i14.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akhondi-Meybodi M, Ghane M, Akhondi-Meybodi S, Dashti G. Five-year survival rate for gastric cancer in Yazd Province, Central Iran, from 2001 to 2008. Middle East J Dig Dis. 2017;9:39–48. doi: 10.15171/mejdd.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orditura M, Galizia G, Sforza V, Gambardella V, Fabozzi A, Laterza MM, Andreozzi F, Ventriglia J, Savastano B, Mabilia A, et al. Treatment of gastric cancer. World J Gastroenterol. 2014;20:1635–1649. doi: 10.3748/wjg.v20.i7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Digklia A, Wagner AD. Advanced gastric cancer: current treatment landscape and future perspectives. World J Gastroenterol. 2016;22:2403–2414. doi: 10.3748/wjg.v22.i8.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smyth EC, Cunningham D. Targeted therapy for gastric cancer. Curr Treat Options in Oncol. 2012;13:377–389. doi: 10.1007/s11864-012-0192-6. [DOI] [PubMed] [Google Scholar]
- 8.Kanat O, O'Neil B, Shahda S. Targeted therapy for advanced gastric cancer: a review of current status and future prospects. World J Gastrointest Oncol. 2015;7:401–410. doi: 10.4251/wjgo.v7.i12.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523–1529. doi: 10.1093/annonc/mdn169. [DOI] [PubMed] [Google Scholar]
- 10.Phan DAT, Nguyen VT, Hua TNH, Ngo QD, Doan TPT, Nguyen ST, Thai AT, Nguyen VT. HER2 status and its heterogeneity in gastric carcinoma of Vietnamese patient. J Pathol Transl Med. 2017;51:396–402. doi: 10.4132/jptm.2017.04.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtsu A, Shah MA, Cutsem EV, Rha SY, Sawaki A, Park SR, Lim HY, Yamada Y, Wu J, Langer B, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol. 2011;29:3968–3976. doi: 10.1200/JCO.2011.36.2236. [DOI] [PubMed] [Google Scholar]
- 12.Shen L, Li J, Xu J, Pan H, Dai G, Qin S, Wang L, Wang J, Yang Z, Shu Y, et al. Bevacizumab plus capecitabine and cisplatin in Chinese patients with inoperable locally advanced or metastatic gastric or gastroesophageal junction cancer: randomized, double-blind, phase III study (AVATAR study) Gastric Cancer. 2015;18:168–176. doi: 10.1007/s10120-014-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabernero J, Hoff PM, Shen L, Ohtsu A, Yu R, Eng-Wong J, Kang Y-K. Pertuzumab (P) with trastuzumab (T) and chemotherapy (CTX) in patients (pts) with HER2-positive metastatic gastric or gastroesophageal junction (GEJ) cancer: an international phase III study (JACOB). J Clin Oncol. 2013;31:TPS4150–0.
- 14.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 15.Tuckwell D, Calderwood DA, Green LJ, Humphries MJ. Integrin alpha 2 I-domain is a binding site for collagens. J Cell Sci. 1995;108(Pt 4):1629–1637. doi: 10.1242/jcs.108.4.1629. [DOI] [PubMed] [Google Scholar]
- 16.Whittard JD, Craig SE, Mould AP, Koch A, Pertz O, Engel J, Humphries MJ. E-cadherin is a ligand for integrin α2β1. Matrix Biol. 2002;21:525–532. doi: 10.1016/S0945-053X(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 17.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 18.Stipp CS. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert Rev Mol Med. 2010;12:e3. doi: 10.1017/S1462399409001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gout SP, Jacquier-Sarlin MR, Rouard-Talbot L, Rousselle P, Block MR. RhoA-dependent switch between alpha2beta1 and alpha3beta1 integrins is induced by laminin-5 during early stage of HT-29 cell differentiation. Mol Biol Cell. 2001;12:3268–3281. doi: 10.1091/mbc.12.10.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madamanchi A, Santoro SA, Zutter MM. alpha2beta1 Integrin. Adv Exp Med Biol. 2014;819:41–60. doi: 10.1007/978-94-017-9153-3_3. [DOI] [PubMed] [Google Scholar]
- 21.Silva R, D'Amico G, Hodivala-Dilke KM, Reynolds LE. Integrins: the keys to unlocking angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1703–1713. doi: 10.1161/ATVBAHA.108.172015. [DOI] [PubMed] [Google Scholar]
- 22.Zutter MM, Santoro SA. Widespread histologic distribution of the alpha 2 beta 1 integrin cell-surface collagen receptor. Am J Pathol. 1990;137:113–120. [PMC free article] [PubMed] [Google Scholar]
- 23.McCall-Culbreath KD, Zutter MM. Collagen receptor integrins: rising to the challenge. Curr Drug Targets. 2008;9:139–149. doi: 10.2174/138945008783502494. [DOI] [PubMed] [Google Scholar]
- 24.Yang C, Zeisberg M, Lively JC, Nyberg P, Afdhal N, Kalluri R. Integrin alpha1beta1 and alpha2beta1 are the key regulators of hepatocarcinoma cell invasion across the fibrotic matrix microenvironment. Cancer Res. 2003;63:8312–8317. [PubMed] [Google Scholar]
- 25.San Antonio JD, Zoeller JJ, Habursky K, Turner K, Pimtong W, Burrows M, Choi S, Basra S, Bennett JS, DeGrado WF, Iozzo RV. A key role for the integrin alpha2beta1 in experimental and developmental angiogenesis. Am J Pathol. 2009;175:1338–1347. doi: 10.2353/ajpath.2009.090234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall CL, Dubyk CW, Riesenberger TA, Shein D, Keller ET, van Golen KL. Type I collagen receptor (alpha2beta1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia. 2008;10:797–803. doi: 10.1593/neo.08380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med. 1999;222:124–138. doi: 10.1046/j.1525-1373.1999.d01-122.x. [DOI] [PubMed] [Google Scholar]
- 28.Danen EH. Integrins: regulators of tissue function and cancer progression. Curr Pharm Des. 2005;11:881–891. doi: 10.2174/1381612053381756. [DOI] [PubMed] [Google Scholar]
- 29.Danen EH, Sonnenberg A. Integrins in regulation of tissue development and function. J Pathol. 2003;201:632–641. doi: 10.1002/path.1472. [DOI] [PubMed] [Google Scholar]
- 30.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 31.Calleros L, Lasa M, Rodriguez-Alvarez FJ, Toro MJ, Chiloeches A. RhoA and p38 MAPK mediate apoptosis induced by cellular cholesterol depletion. Apoptosis. 2006;11:1161–1173. doi: 10.1007/s10495-006-6980-3. [DOI] [PubMed] [Google Scholar]
- 32.Kamel WA, Sugihara E, Nobusue H, Yamaguchi-Iwai S, Onishi N, Maki K, Fukuchi Y, Matsuo K, Muto A, Saya H, Shimizu T. Simvastatin-induced apoptosis in osteosarcoma cells: a key role of RhoA-AMPK/p38 MAPK signaling in antitumor activity. Mol Cancer Ther. 2017;16:182–192. doi: 10.1158/1535-7163.MCT-16-0499. [DOI] [PubMed] [Google Scholar]
- 33.Leslie H, Sobin MKG. Christian Wittekind: TNM classification of malignant Tumours. 7. Oxford: Wiley-Blackwell; 2011. [Google Scholar]
- 34.Pal N, Axisa B, Yusof S, Newcombe RG, Wemyss-Holden S, Rhodes M, Lewis MP. Volume and outcome for major upper GI surgery in England. J Gastrointest Surg. 2008;12:353–357. doi: 10.1007/s11605-007-0288-7. [DOI] [PubMed] [Google Scholar]
- 35.Kang B, Guo RF, Tan XH, Zhao M, Tang ZB, Lu YY. Expression status of ataxia-telangiectasia-mutated gene correlated with prognosis in advanced gastric cancer. Mutat Res. 2008;638:17–25. doi: 10.1016/j.mrfmmm.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Weledji EP. How surgery can improve multimodal treatment of gastric cancer?Annals of Laparoscopic and Endoscopic. Surgery. 2016;1
- 37.Kurokawa T, Masuda T. A statistics about the causes of delay in the diagnosis of stomach cancer. Tohoku J Exp Med. 1951;54:294. doi: 10.1620/tjem.54.294. [DOI] [PubMed] [Google Scholar]
- 38.Kanagavel D, Fedyanin M, Tryakin A, Tjulandin S. Second-line treatment of metastatic gastric cancer: current options and future directions. World J Gastroenterol. 2015;21:11621–11635. doi: 10.3748/wjg.v21.i41.11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran PN, Sarkissian S, Chao J, Klempner SJ. PD-1 and PD-L1 as emerging therapeutic targets in gastric cancer: current evidence. Gastrointestinal cancer : targets and therapy. 2017;7:1–11. doi: 10.2147/GICTT.S113525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Catenacci DVT, Tebbutt NC, Davidenko I, Murad AM, Al-Batran SE, Ilson DH, Tjulandin S, Gotovkin E, Karaszewska B, Bondarenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1467–1482. doi: 10.1016/S1470-2045(17)30566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SY, Oh SC. Changing strategies for target therapy in gastric cancer. World J Gastroenterol. 2016;22:1179–1189. doi: 10.3748/wjg.v22.i3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marin JJ, Al-Abdulla R, Lozano E, Briz O, Bujanda L, Banales JM, Macias RI. Mechanisms of resistance to chemotherapy in gastric cancer. Anti Cancer Agents Med Chem. 2016;16:318–334. doi: 10.2174/1871520615666150803125121. [DOI] [PubMed] [Google Scholar]
- 43.Shi WJ, Gao JB. Molecular mechanisms of chemoresistance in gastric cancer. World J Gastrointest Oncol. 2016;8:673–681. doi: 10.4251/wjgo.v8.i9.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimoyama S. Unraveling trastuzumab and lapatinib inefficiency in gastric cancer: future steps (review) Mol Clin Oncol. 2014;2:175–181. doi: 10.3892/mco.2013.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Liu NN, Li JQ, Yang L, Zeng Y, Zhao XM, Xu LL, Luo X, Wang B, Wang XR. Association between ITGA2 C807T polymorphism and gastric cancer risk. World J Gastroenterol. 2011;17:2860–2866. doi: 10.3748/wjg.v17.i23.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langsenlehner U, Renner W, Yazdani-Biuki B, Eder T, Wascher TC, Paulweber B, Clar H, Hofmann G, Samonigg H, Krippl P. Integrin alpha-2 and beta-3 gene polymorphisms and breast cancer risk. Breast Cancer Res Treat. 2006;97:67–72. doi: 10.1007/s10549-005-9089-4. [DOI] [PubMed] [Google Scholar]
- 47.Chin SP, Marthick JR, West AC, Short AK, Chuckowree J, Polanowski AM, Thomson RJ, Holloway AF, Dickinson JL. Regulation of the ITGA2 gene by epigenetic mechanisms in prostate cancer. Prostate. 2015;75:723–734. doi: 10.1002/pros.22954. [DOI] [PubMed] [Google Scholar]
- 48.Chan BM, Matsuura N, Takada Y, Zetter BR, Hemler ME. In vitro and in vivo consequences of VLA-2 expression on rhabdomyosarcoma cells. Science. 1991;251:1600–1602. doi: 10.1126/science.2011740. [DOI] [PubMed] [Google Scholar]
- 49.Ura H, Denno R, Hirata K, Yamaguchi K, Yasoshima T. Separate functions of alpha2beta1 and alpha3beta1 integrins in the metastatic process of human gastric carcinoma. Surg Today. 1998;28:1001–1006. doi: 10.1007/BF02483952. [DOI] [PubMed] [Google Scholar]
- 50.Wu YJ, Pagel MA, Muldoon LL, Fu R, Neuwelt EA. High alphav integrin level of cancer cells is associated with development of brain metastasis in Athymic rats. Anticancer Res. 2017;37:4029–4040. doi: 10.21873/anticanres.11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai QQ, Dong YW, Wang R, Qi B, Guo JX, Pan J, Liu YY, Zhang CY, Wu XZ. MiR-124 inhibits the migration and invasion of human hepatocellular carcinoma cells by suppressing integrin alphaV expression. Sci Rep. 2017;7:40733. doi: 10.1038/srep40733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neisch AL, Speck O, Stronach B, Fehon RG. Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J Cell Biol. 2010;189:311–323. doi: 10.1083/jcb.200912010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai NP, Wei LN. RhoA/ROCK1 signaling regulates stress granule formation and apoptosis. Cell Signal. 2010;22:668–675. doi: 10.1016/j.cellsig.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stankiewicz TR, Linseman DA. Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci. 2014;8 [DOI] [PMC free article] [PubMed]
- 55.Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gogvadze V, Orrenius S, Zhivotovsky B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim Biophys Acta. 2006;1757:639–647. doi: 10.1016/j.bbabio.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 57.Nhan TQ, Liles WC, Schwartz SM. Physiological functions of caspases beyond cell death. Am J Pathol. 2006;169:729–737. doi: 10.2353/ajpath.2006.060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci. 2010;123:3209–3214. doi: 10.1242/jcs.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang M, Zheng J, Nussinov R, Ma B. Release of cytochrome C from Bax pores at the mitochondrial membrane. Sci Rep. 2017;7:2635. doi: 10.1038/s41598-017-02825-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, et al. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 61.He W, Zhang MF, Ye J, Jiang TT, Fang X, Song Y. Cordycepin induces apoptosis by enhancing JNK and p38 kinase activity and increasing the protein expression of Bcl-2 pro-apoptotic molecules. J Zhejiang Univ Sci B. 2010;11:654–660. doi: 10.1631/jzus.B1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bachelor MA, Bowden GT. Ultraviolet A-induced modulation of Bcl-XL by p38 MAPK in human keratinocytes: post-transcriptional regulation through the 3′-untranslated region. J Biol Chem. 2004;279:42658–42668. doi: 10.1074/jbc.M406626200. [DOI] [PubMed] [Google Scholar]
- 63.Cai B, Xia Z. p38 MAP kinase mediates arsenite-induced apoptosis through FOXO3a activation and induction of Bim transcription. Apoptosis. 2008;13:803–810. doi: 10.1007/s10495-008-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porras A, Zuluaga S, Black E, Valladares A, Alvarez AM, Ambrosino C, Benito M, Nebreda AR. p38α Mitogen-activated Protein Kinase Sensitizes Cells to Apoptosis Induced by Different Stimuli. Mol Biol Cell. 2004;15:922–933. doi: 10.1091/mbc.E03-08-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kirfel G, Rigort A, Borm B, Herzog V. Cell migration: mechanisms of rear detachment and the formation of migration tracks. Eur J Cell Biol. 2004;83:717–724. doi: 10.1078/0171-9335-00421. [DOI] [PubMed] [Google Scholar]
- 66.Bugyi B, Carlier M-F. Control of actin filament Treadmilling in cell motility. Annu Rev Biophys. 2010;39:449–470. doi: 10.1146/annurev-biophys-051309-103849. [DOI] [PubMed] [Google Scholar]
- 67.Knecht DA, LaFleur RA, Kahsai AW, Argueta CE, Beshir AB, Fenteany G. Cucurbitacin I inhibits cell motility by indirectly interfering with actin dynamics. PLoS One. 2010;5:e14039. doi: 10.1371/journal.pone.0014039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le Clainche C, Carlier M-F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- 69.Huang L, Wu RL, Xu AM. Epithelial-mesenchymal transition in gastric cancer. Am J Transl Res. 2015;7:2141–2158. [PMC free article] [PubMed] [Google Scholar]
- 70.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng Z, Wang CX, Fang EH, Wang GB, Tong Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J Gastroenterol. 2014;20:5403–5410. doi: 10.3748/wjg.v20.i18.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wolfenson H, Lavelin I, Geiger B. Dynamic regulation of the structure and functions of integrin adhesions. Dev Cell. 2013;24:447–458. doi: 10.1016/j.devcel.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khyrul WA, LaLonde DP, Brown MC, Levinson H, Turner CE. The integrin-linked kinase regulates cell morphology and motility in a rho-associated kinase-dependent manner. J Biol Chem. 2004;279:54131–54139. doi: 10.1074/jbc.M410051200. [DOI] [PubMed] [Google Scholar]
- 74.Delon I, Brown NH. Integrins and the actin cytoskeleton. Curr Opin Cell Biol. 2007;19:43–50. doi: 10.1016/j.ceb.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 75.Price LS, Leng J, Schwartz MA, Bokoch GM. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 77.Deroanne C, Vouret-Craviari V, Wang B, Pouyssegur J. EphrinA1 inactivates integrin-mediated vascular smooth muscle cell spreading via the Rac/PAK pathway. J Cell Sci. 2003;116:1367–1376. doi: 10.1242/jcs.00308. [DOI] [PubMed] [Google Scholar]
- 78.Arthur WT, Noren NK, Burridge K. Regulation of rho family GTPases by cell-cell and cell-matrix adhesion. Biol Res. 2002;35:239–246. doi: 10.4067/S0716-97602002000200016. [DOI] [PubMed] [Google Scholar]
- 79.Rohatgi R, Ho HY, Kirschner MW. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J Cell Biol. 2000;150:1299–1310. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martinez-Quiles N, Rohatgi R, Anton IM, Medina M, Saville SP, Miki H, Yamaguchi H, Takenawa T, Hartwig JH, Geha RS, Ramesh N. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat Cell Biol. 2001;3:484–491. doi: 10.1038/35074551. [DOI] [PubMed] [Google Scholar]
- 81.Kato T, Kawai K, Egami Y, Kakehi Y, Araki N. Rac1-dependent lamellipodial motility in prostate cancer PC-3 cells revealed by optogenetic control of Rac1 activity. PLoS One. 2014;9:e97749. doi: 10.1371/journal.pone.0097749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–759. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401. doi: 10.1242/jcs.100115. [DOI] [PubMed] [Google Scholar]
- 84.Patel S, Takagi KI, Suzuki J, Imaizumi A, Kimura T, Mason RM, Kamimura T, Zhang Z. RhoGTPase activation is a key step in renal epithelial mesenchymal transdifferentiation. J Am Soc Nephrol. 2005;16:1977–1984. doi: 10.1681/ASN.2004110943. [DOI] [PubMed] [Google Scholar]
- 85.Chen IJ, Chuang CH, Hsieh YC, Lu YC, Lin WW, Huang CC, Cheng TC, Cheng YA, Cheng KW, Wang YT, et al. Selective antibody activation through protease-activated pro-antibodies that mask binding sites with inhibitory domains. Sci Rep. 2017;7:11587. doi: 10.1038/s41598-017-11886-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, Hall SS, Ruoslahti E, Daugherty PS. Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J Control Release. 2012;161:804–812. doi: 10.1016/j.jconrel.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. RNA-seq sample annotation for each of the 32 normal gastric tissues and 375 gastric cancer tissues from TCGA. (DOCX 16 kb)
Table S1. Primer list for reverse transcription-polymerase chain reactions (RT-PCR). (DOCX 15 kb)
Figure S1. Effect of anti-ITGA2 antibody on cell morphology. The AGS cells were treated with a 3 μg of the anti-ITGA2 antibodies or isotype control antibodies (negative control) for 48 h, and cell morphology was observed at 200X magnification. Data are representative of three independent experiments. (PPTX 1463 kb)
Figure S2. Low dose of anti-ITGA2 antibodies did not induce cell death in AGS cells. Photography and quantitative analyses on cell number of the AGS cells treated with 0.1 μg anti-ITGA2 antibodies or isotype control antibodies (negative control) for 18 h. Data are expressed as mean ± standard deviation (S.D). Statistical comparisons were made by one-way ANOVA with Bonferroni comparisons. Data are representative of three independent experiments. (PPTX 784 kb)