

Abstract
Mice with deficiency in tocopherol (alpha) transfer protein gene develop peripheral tocopherol deficiency and sensory neurodegeneration. Ttpa−/− mice maintained on diets with deficient α-tocopherol (α-TOH) had proprioceptive deficits by six months of age, axonal degeneration and neuronal chromatolysis within the dorsal column of the spinal cord and its projections into the medulla. Transmission electron microscopy revealed degeneration of dorsal column axons. We addressed the potential pathomechanism of α-TOH deficient neurodegeneration by global transcriptome sequencing within the spinal cord and cerebellum. RNA-sequencing of the spinal cord in Ttpa−/− mice revealed upregulation of genes associated with the innate immune response, indicating a molecular signature of microglial activation as a result of tocopherol deficiency. For the first time, low level Ttpa expression was identified in the murine spinal cord. Further, the transcription factor liver X receptor (LXR) was strongly activated by α-TOH deficiency, triggering dysregulation of cholesterol biosynthesis. The aberrant activation of transcription factor LXR suppressed the normal induction of the transcription factor retinoic-related orphan receptor-α (RORA), which is required for neural homeostasis. Thus we find that α-TOH deficiency induces LXR, which may lead to a molecular signature of microglial activation and contribute to sensory neurodegeneration.
Keywords: RNA-sequencing, transcription, vitamin E
Graphical abstract
Introduction
Free radical-mediated oxidation products highly correlate with the pathogenesis and progression of many diseases including Alzheimer’s disease, Down syndrome, Parkinson’s disease, and stroke [1]. Vitamin E (vitE) is a major antioxidant that scavenges free radicals generated by lipid peroxidation. Presently, 90% of the population does not consume the recommended dietary allowance (RDA) of 15 mg of vitE per day but averages closer to 7 mg/day [2]. Additionally, vitE is not routinely tested in standard clinicopathologic evaluations.
The most well-documented biologic phenotype of vitE deficiency in humans is its role in neuronal function [3]. This pivotal link between vitE and neurologic health was promulgated by the discovery of a form of neurodegeneration in humans, termed ataxia with vitamin E deficiency (AVED), which is due to mutations in the α-tocopherol transfer protein gene (TTPA) [4, 5]. The encoded protein, TTP, is a hepatic cytosolic transport protein responsible for maintaining plasma α-tocopherol (α-TOH) concentrations by facilitating α-TOH export from the liver. Various genetic mutations in TTPA result in severe vitE deficiency that can manifest a variety of clinical signs, but most strikingly causes neurodegeneration that is surprisingly early in onset compared with many neurodegenerative diseases. Age of onset for AVED is broad, between 2-52 y, with most patients demonstrating symptoms at <20 y [5, 6]. This age of symptom onset depends on the type of mutation in the TTPA [6]. Symptoms of AVED include cerebellar ataxia, dysarthria, reduced or absent deep tendon reflexes, and vibratory-sense disturbances [6].
A transgenic mouse model of AVED was produced by deleting the translational start codon of Ttpa (Ttpa−/−)[7]. Transcription of Ttpa therefore occurs in these mice; however, the protein is not produced. Mice demonstrate an ataxia and retinal degeneration after one year of age and histologic lesions, including reduction of myelinated fibers in the gracile fasciculus and gliosis of the nucleus gracilis, by 20 months (mo) of age [4]. Using high-density oligonucleotide arrays on a variety of organs from affected transgenic mice maintained on diets containing normal concentrations of vitE (35-150 mg dl-alpha-tocopheryl acetate/kg diet), gene expression studies revealed dysregulated cerebral cortical transcripts involved in synaptogenesis and myelination [8, 9]. In particular, a 13-fold decrease in the expression of a nuclear receptor, the retinoic-related orphan receptor-α (RORA) mRNA was identified [8] and validated in a second study [9]. Nuclear receptor activation by the fat-soluble vitamins A and D have been well-documented [10] but this was the first report demonstrating an effect of vitE on nuclear receptor expression. When incorporating the effect of age (birth versus 9-12 mo), markers of synaptic degeneration and demyelination were most apparent in the newborn mice [9]. While seemingly counterintuitive, it was hypothesized that Ttpa−/− mice lose their synapses at a faster rate and reach a plateau at a young age. However, the main neuroanatomic tracts affected with α-TOH deficiency, located within the spinal cord, medulla oblongata, and cerebellum [4, 11], were not profiled in these studies.
Within the cerebellum, cellular atrophy and diminished dendritic branching of Purkinje neurons has been demonstrated in Ttpa−/− mice fed diets deficient in α-TOH (6.6 mg/kg dl-alpha-tocopheryl acetate/kg diet) at 17 mo of age [11]. Additionally, a threefold increase in cerebellar protein-bound 3-nitrotyrosine (3-NT) was identified [11], complementing previous studies demonstrating increased oxidative stress markers in these mice including thiobarbituric acid reactive substances (TBARS) [4], F2-isoprostanes [7], and total hydroxyoctadecadienoic acids (t-HODEs) [12]. Cerebellar pathology and associated neurologic deficits were prevented by supplementation of Ttpa−/− mice with a diet containing 600 mg dl-alpha-tocopheryl acetate/kg [11]. These studies demonstrate that the interaction of genotype, dietary α-TOH concentration, and time point in postnatal development is crucial to the development of the neurodegenerative phenotype.
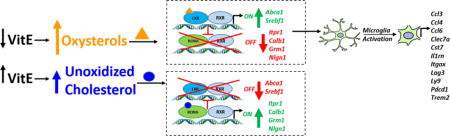
Recently, we have documented upregulation of liver X receptor (LXR) target genes in a naturally occurring α-TOH deficiency in the horse [13]. Mutual suppression between RORA and LXR has been previously reported, with loss of RORA increasing expression of LXR target genes and, reciprocally, loss of LXR resulting in activation of select RORA target genes [14]. Other fat-soluble vitamins, including vitamins A and D, are known ligands for transcription factors and play a role in cell signaling. Therefore, we postulate that vitE may play either a direct or indirect role in cell signaling through activation of nuclear receptors.
In order to build on these previous studies [8, 9, 13], the primary goal of this study was to identify differentially expressed genes and pathways by global transcriptome analysis of the spinal cord and cerebellum of mice varying in genotype, age, and diet. Our hypothesis was that the dysregulated synaptic and myelinating pathways in Ttpa−/− mice were associated with transcriptome alterations of altered neuronal and myelin homeostasis. Additionally, we hypothesized that nuclear receptor activation may be altered in Ttpa−/− α-TOH deficient mice during postnatal development.
Materials and Methods
Mice
Animals were housed and cared for in accordance with the University of California Davis standing committee on animal use and care (IACUC) as well as the Guide for the Care and Use of Laboratory animals (8th edition, 2011). All procedures performed were also approved by the University IACUC. A rederived colony [7] of mixed (50% C57BL6 and 50% 129/SvJae) mice heterozygous for the deletion (Ttpa+/−) were crossed with C57BL6/J (Ttpa−/−) mice to establish a colony of Ttpa−/− mice. Offspring were genotyped using specific primers for Ttpa, as previously described [7]. Of the 686 offspring produced, 125 were Ttpa−/−, significantly less than the expected value of 172 mice (X2=9.22, P<0.01) and suggestive of fetal mortality in Ttpa−/− mice. Genotypes were confirmed for each animal by western blot analysis of hepatic TTP using an anti-TTP antibody as previously described [7].
At weaning, Ttpa+/− and Ttpa−/− mice were fed a normal diet (35 mg of dl-alpha-tocopheryl acetate/kg, vitE+), α-TOH-deficient diet (<10 mg of dl-alpha-tocopheryl acetate/kg, vitE−), or α-TOH-supplemented diet (600 mg of dl-alpha-tocopheryl acetate/kg, vitE+++) diet. Custom Teklad non-irradiated vacuum-packaged diets were ordered through Harlan Laboratories (Madison, WI) and were identical to those previously used in this mouse model [11]. To prevent oxidation, diets were maintained at −20°C and used within six months. Mouse diets were replaced once per week. High-performance liquid chromatography with fluorescence detection to confirm dietary α-TOH concentrations was performed as previously described [15].
Sex has been shown to influence tissue α-TOH concentrations [16]. Therefore, experimental groups were assigned by sex to minimize data variability in RNA-sequencing. At 6 or 12 mo of age, male and female mice (n=3-4 per group; supplemented mice only evaluated at 6 mo of age) underwent intracardiac perfusion for histology. Briefly, mice were anesthetized with pentobarbital (60 mg/kg intraperitoneally) and perfused sequentially with 10 mL each of sodium nitrite, saline, and fixative. For routine histology, mice were perfused with 3.7% paraformaldehyde, the skull and spinal column were dissected and fixed in 3.7% formaldehyde followed by 70% EtOH. Samples were then subjected to a 24-hour combined fixation/decalcification in Formical (StatLab Medical Products) after which the tissue was trimmed and processed for paraffin embedding. For electron microscopy, mice were perfused with a transmission electron microscopic (TEM) Karnovsky aldehyde fixative at 14-16 mo of age (n=3 per group). At either weaning or 6 mo of age, female mice (n=6 per group) were sacrificed with pentobarbital (>100 mg/kg IP) and tissues were flash-frozen for RNA sequencing. Validation of differentially expressed transcripts was performed with qPCR on 7-8 mice per group, consisting of both males and females.
Neurobehavioral Testing
Examination of balance and coordination were performed on mice at one and six mo of age. Mice were housed under conditions of constant temperature (20°C), light (6:00 am to 6:00 pm), and with access to food and water ad libitum. Testing apparatus was cleaned with 70% ethanol to remove animal odors before and after each test. Previous power analyses calculated for the TreadScan system revealed that 6-9 animals per group were required to separate sham animals from animals with mild spinal cord contusion [17]. Experiments were therefore performed with 8-18 mice per group, consisting of ~50% male and 50% female. Each test was performed at one time point, 1-2 days prior to sacrifice.
Foot misplacement testing for motor coordination
Foot misplacement test was performed as previously described [18]. Symmetrically removed rungs or “traps” were introduced at the 15th, 30th, 45th, and 60th positions. A total of four trials were run per mouse (two trials with no “traps”; two trials with “traps”).
Gait Analysis via TreadScan®
Mice were not trained to the treadmill as adverse acclimatization effects have been demonstrated in mice in response to training on the TreadScan [17]. Two trials were performed for each mouse. The first was at 8 meters per minute (mpm) and the second at 12mpm, with each TreadScan trial generating data for 208 gait metrics.
Von Frey Assay
The Von Frey assay was performed as previously described [19]. A total of six recordings were performed per mouse (initial reflex, followed by five “up-down” stimuli).
Statistical Analysis
All neurobehavioral data was evaluated for normality using a D’Agostino-Pearson test. For weaning time points, the effect of genotype on each dependent variable was analyzed with an unpaired T test. At 6-mo time points, a one-way ANOVA compared the 6 genotype/diet groups followed by a Tukey post-hoc test. Significance was set at P<0.05.
Histologic and Transmission Electron Microscopic Evaluation
Paraffin-embedded sections of brain and spinal cord were cut at 5μm, processed according to standard laboratory procedure, and stained with hematoxylin and eosin (HE). Tissue for TEM were aldehyde fixed, post-fixed with osmium tetroxide, in-block stained with uranyl acetate, dehydrated, infiltrated with epoxy resin, and polymerized into blocks. The blocks are cut on an ultra-microtome-thick (survey) sections at 500nm and thin sections (for the TEM) at 60-80nm. TEM sections were post-stained with uranyl acetate and lead citrate and viewed on the TEM. Pathologic evaluation was performed by a board-certified veterinary anatomic pathologist blinded to the genotype and diet of each mouse (ADM).
Alpha-tocopherol concentrations
Cerebral cortex samples were selected to assess central nervous system α-TOH concentrations as spinal cord and cerebellar samples were used for RNA-sequencing and cerebral cortex α-TOH concentrations have been previously demonstrated to adequately represent spinal cord and cerebellar α-TOH concentrations in Ttpa−/− mice [20]. Samples from n=9-10 mice from each experimental group (weaning Ttpa+/+, weaning Ttpa−/−, 6 mo Ttpa+/+ vitE+, 6 mo Ttpa+/+ vitE−, 6 mo Ttpa+/+ vitE+++, 6 mo Ttpa−/− vitE−, 6 mo Ttpa−/− vitE+, and 6 mo Ttpa−/− vitE++6+) were collected immediately upon euthanasia and frozen in liquid nitrogen. Samples were stored at −80°C until analysis, performed within 12 mo of collection.
Alpha-tocopherol extraction
Fifty mg of mouse brain were spiked with 10 μL of 5 μg/mL internal standard (TD6, (alpha-tocopherol) (5-methyl-D3,7-methyl-D3, Cambridge Isotope Laboratories, Tewksbury, MA) in methanol and 10 μL of a 25 mg/mL butylated hydroxy- toluene (BHT, MP Biomedicals, Solon, OH) in methanol before addition of 200 μL of DI water and 730 μL of acetonitrile. Brains were then homogenized with 2 stainless steel, 4-mm diameter grinding balls using a GenoGrinder (SPEX SamplePrep, Metuchen, NJ) pre-cooled to −20°C, for 2×30 sec at the 1500 rpm setting. Homogenates were shaken at room temperature for 15 minutes (Innova 2000 Platform Shaker, New Brunswick Scientific, Edison, NJ), then centrifuged for 3 min at 9900 rcf (Thermo IEC Micromax RF refrigerated microcentrifuge, Thermo Scientific, Waltham, MA) at 10 °C. A 400 μL aliquot of supernatant was added to each well of a Phenomenex Phree phospholipid removal 96-well plate (Phenomenex, Torrance, CA). After the flow-through was discarded, the plate was eluted twice with 600μl of acetonitrile/isopropanol (v/v 90:10) and the combined eluate taken to dryness under a stream of nitrogen (GlasCol well evaporator, Terre Haute, IN) at 20 LPM flow setting, while heating to 40°C using a block heating unit (Fisher Scientific, Pittsburg, PA). The dry residues were reconstituted in 200 μL of methanol containing 2 ng/mL CUDA (12-[[(cyclohexylamino)carbonyl]amino]-dodecanoic acid, Cayman Chemical, Ann Arbor, MI) as the internal standard. The plate was briefly vortexed, then sonicated for 2 min (Branson Model 1510 Ultrasonic water bath) and centrifuged for 3 min at 2100 rcf and 10°C (Eppendorf Centrifuge 5804R, Eppendorf, Hauppauge, NY).
LC-MS Analysis
Of the supernatants, 120 μL was transferred to pre-labeled LC vials with 150 μL inserts for LC-MS analysis. LC-MS analysis was carried out using a Bruker EVOQ LC-TQ Mass Spectrometer coupled with a Bruker Advance HPLC system (Bruker Corp, Freemont, CA, USA). The analytical column was a Waters Acquity UPLC BEH C18 column, 1.7 μm, 2.1 mm × 100 mm with matching Waters Vanguard precolumn (Waters, Milford, MA), kept at 50 °C. Gradient elution was achieved with mobile phase A, water with 0.1% formic acid and B, methanol with 0.1% formic acid at a flow rate of 250 μl/min, starting for 0.5 min at 80% B followed by a linear gradient from 80% to 90% B from minute 0.5 to 2, going to 95% B by minute 10 and 98% by minute 15.5, then back to 80% B within 0.5 min and re-equilibration at 80% B from 16 to 20 min, for a total run time of 20 minutes. Mass spectral data for α-TOH were acquired in the positive ion electrospray ionization (ESI) mode with the following retention times (RT) and scan events: α-TOH - RT 12.1 min, MRM 431.2>165.0 (CE 25.0V) and 5-methyl-D3,7-methyl-D3 (TD6) - RT 12.0 min, MRM 437.4>171.0 (CE 27.0V); and CUDA - RT 1.9 min, MRM 341.0>216.0 (CE 14.0V). ESI parameters were as follows: positive ionization mode with spray voltage 4500 V, cone temperature 300°C, heated probe temperature 350°C, cone gas flow 15, probe gas flow 35 L/min, nebulizer gas flow 40 L/min. The injection volume for all standards and sample extracts was 2 μl. Reagent blanks, controls, and fortified samples were analyzed along with the samples. Quantification was carried out using an 11-point calibration curve covering the range from 1 ng/mL to 2500 ng/mL and linear regression. Each level of calibration standard contained the internal standard TD6 at 100 ng/mL and CUDA at 2 ng/mL, matching their final concentrations in the analysis samples. Analysis of negative control reagent blanks for α-TOH by the LC-MRM technique described above showed clean chromatograms with no background contribution from contaminations.
Data Analysis
Data was analyzed with a one-way ANOVA with experimental group as a factor and post-hoc Tukey test with P values adjusted for multiple comparisons.
RNA-Sequencing
RNA Isolation and Quality Control
For all spinal cord and cerebellar samples, total RNA was extracted using TRIzol reagent (Thermofisher, Wilmington, DE, USA) as previously described [13]. The resulting amount of RNA and integrity scores are included in Table A1. For RT-qPCR, RNA was washed and eluted on columns (Direct-zol™ RNA MiniPrep Plus, Zymo Research, Irvine, CA) and treated with DNase I (Zymo Research, Irvine, CA, USA) according to manufacturer’s instructions. Quantification and quality of RNA, along with degree of rRNA contamination, was assessed using the Pico chip on the Agilent Bioanalyzer 2100 (Santa Clara, CA, USA), with a RNA integrity number (RIN) ≥ 6.5.
Sequencing
Samples of total RNA (1 μg) with RNA integrity numbers (RIN) of ≥ 6.5 were used for construction of strand-specific, poly-A enriched libraries. Following KAPA Stranded mRNA-Seq Kit (Illumina® platform, Cape Town, South Africa) manufacturer’s protocol, poly-A enrichment followed by cDNA synthesis and A tailing adapter ligation was performed. The distribution of the library fragments was assessed with Agilent 2100 Bioanalyzer. Spinal cord libraries were combined into 1 pool and sequenced across 5 lanes with specification of 50-bp paired-end (50PE) on Illumina HiSeq 2500.
Sequence reads quality assessment, trimming, and alignment
Raw reads were processed with expHTS [21] to trim low quality sequences, adapter contamination, and to remove PCR duplicates. Trimmed reads for each sample were aligned to the mm10 murine genome with GRCm38 annotation (Ensembl), using STAR v. 2.5.1a aligner [22], which also generated raw counts per gene that were the input to the statistical analysis.
Analysis of differential gene expression
Count data were normalized with TMM normalization in the Bioconductor package edgeR, version 3.14.049. For each tissue (spinal cord and cerebellum), differential expression analyses were conducted using the limma-voom pipeline (version 3.28.17) [23] using a statistical model that factors genotype (Ttpa+/+, Ttpa−/−), diet (vitE−, vitE+, vitE+++), age (weaning, 6 mo) all two-and three-way interactions among these, and RIN. Standard errors were adjusted for within-litter correlations. Contrasts were then subsequently evaluated (Tables A2 and A3). Statistical significance was assigned to genes associated with Benjamini-Hochberg false discovery rate adjusted p value of <0.05.
GO enrichment analysis
GO enrichment analysis was conducted with Bioconductor package topGO, version 2.24.0 (topGO: Enrichment Analysis for Gene Ontology. R package version 2.24.0). The data is presented in raw p-value for Kolmogorov-Simonov test for enrichment of GO terms.
Quantitative real-time PCR Validation
Prioritized genes of interest and Ttpa were selected for validation in an independent biologic replicate set of mice. The replicate set consisted of 6-mo old mice (male and female, n=6 per group) on either the vitE− or vitE+ diets. For ten prioritized genes in the spinal cord (Itgb2, Itgax, Lilrb4a, Pdcd1, Ccl3, Tyrobp, Mmp12, Cd68, Plek, and Rgs1), one in the cerebellum (Necab1), and reference gene (Actb), primers were designed to cover exons that were included in all the reported transcript isoforms. Primers were designed in Primer3plus [24] and RT-qPCR performed as previously reported [13]. Primers are listed in Table A4. Efficiencies for each gene were calculated at 90-110%. Relative quantitation of gene expression was calculated by comparative threshold cycle method (2−ΔΔCt) using the Ct of the housekeeping gene. Data were analyzed using the non-parametric Mann-Whitney test due to the small sample size with P<0.05 set as significant.
Immunohistochemistry
Five μm sections were cut from each block. The slides were deparaffinized in xylene and hydrated with graded alcohols. Peroxide diluted to 3% in phosphate buffered saline was used to block endogenous peroxidase activity. Antigen retrieval for anti-Iba1 antibody (catalog #019-19741 Wako Chemicals, [Richmond, VA, USA]) was done by microwaving the slides for twenty minutes in citrate buffer (Vector Laboratories [Burlingame, CA, USA]). Proteinase K treatment (Dako [Carpinteria, CA, USA]) for 3 minutes was used as antigen retrieval for anti-glial fibrillary acidic protein (GFAP) antibody (catalog # Z0334 Dako). Avidin/biotin block (Invitrogen [Carlsbad, CA, USA]) was used to block any biotin present in the tissue and serum-free protein block (Dako) was used to block non-specific binding. Anti-Iba1 antibody was diluted 1:500 in antibody diluent (Dako) and incubated for 30 minutes at room temperature. Anti-Gf antibody was diluted 1:2500 and incubated for 1 hour at room temperature. The slides were incubated for 30 minutes with biotinylated goat anti-rabbit (Vector Laboratories), diluted at 1:200 in antibody diluent (Dako). The slides were incubated with Vectastain ABC Elite reagent (Vector Laboratories) for 30 minutes at room temperature. Signal was developed with DAB chromogen (Dako) and then counter stained with Mayer’s hematoxylin (Dako). Positive controls consisted of mouse brain tissue and negative controls consisted of isotype matched antibody used in lieu of the primary antibody.
Fluorescent Immunohistochemistry
Five μm sections were cut from each block. The slides were deparaffinized in xylene and hydrated with graded alcohols. An immunofluorescence triple-labeling assay was performed in spinal cord sections from 1-year old Ttpa+/+ vitE+ and Ttpa−/− vitE− mice. Tissues were labeled with primary antibodies, which included 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) blue-fluorescent stain, produced by Thermo Fisher Scientific (Waltham, MA), and rabbit anti-beta III tubulin (B-III Tubulin), Alexa Fluor® 488 conjugate antibody, purchased from Millipore Sigma (St. Louis MO). Terminal deoxynucleotidyl transferase (TdT) nick end labeling, or TUNEL (Roche Industries), was used to enzymatically label double strand breaks with TMR red. Slides underwent a permeabilization treatment of 0.1% Triton X-100 and 0.1% Sodium Citrate for 8 minutes. After 2 washes with PBS, the slides were treated with TUNEL as per manufacturer’s instruction. Slides were then rinsed twice with PBS and incubated with 5% BSA for 30 minutes at 22°C. B-III Tubulin was diluted 1:200 and incubated for 1 hour at 22°C. After 2 washes with PBS, DAPI was diluted 1:1000 and incubated for 5 minutes. After a final wash in PBS, the slides were mounted with ProLong Gold Antifade Mountant, purchased from Thermo Fisher Scientific. DRG were visualized under a Leica TCS SP8 STED 3X fluorescence microscope from Leica Camera (Weltzlar, Germany). The images were digitally recorded as Z-stacks and processed with LAS X software platform by Leica Camera.
Spinal Cord Cholesterol and Oxysterol Concentrations
Spinal cord cholesterol, 7-dehydrocholesterol, 8-dehydrocholesterol, desmosterol, lanosterol and six oxysterol concentrations (24-epoxycholesterol, 24-ketocholesterol, 24-hydroxycholesterol, 4β-hydroxycholesterol, 7-ketocholesterol and 7-hydroxycholesterol) were determined in five of the experimental groups (Ttpa+/+ weaning, Ttpa−/− weaning, 6 mo Ttpa+/+ vitE+, 6 mo Ttpa−/− vitE−, and 6 mo Ttpa−/− vitE+++, n=5-6 per group).
Materials
Optima LC/MS grade solvents (methylene chloride, chloroform, methanol, water, and formic acid) were purchased from Thermo Fisher Scientific. d7-7-Ketocholesterol was prepared as reported previously [25]. 200 proof ethanol (USP Specs) from Decon Laboratories, Inc. was used. Potassium hydroxide (ACS certified) and sodium chloride (ACS certified) were purchased from Thermo Fisher Scientific.
Lipid extraction from tissues and fluids
Prior to lipid extraction, the internal standard, d7-7-ketocholesterol (500 ng), d6-24-epoxycholesterol (500 ng), and d7-cholesterol (2.5 μg) was added to each sample. For spinal cord, the tissues were homogenized in Folch solution (3mL, chloroform:methanol = 2:1) by a blade homogenizer. For serum (100 μL), 3 mL of Folch solution was added directly to each sample. NaCl aqueous solution (0.9%, 1 mL) was then added and the resulting mixture was briefly vortexed and centrifuged for 5 minutes. The lower organic phase was recovered and dried at room temperature using the speed vacuum (Thermo Fisher Savant), and then re-dissolved in methylene chloride (1 mL for tissue samples and 500 μL for CSF samples). For the serum samples, the extracts were dried under SpeedVac and the dried samples were then reconstituted in 2 mL of 2.5% KOH in 90% EtOH and incubated in a water bath at 55°C for 45 minutes. Another lipid extraction was performed on the solution from the base hydrolysis and the extracts were dried again using the SpeedVac. Each sample was re-constituted in methylene chloride (500 μL).
HPLC-APCI-MS/MS analyses of sterols and oxysterols
Analysis of cholesterol, sterols, and oxysterols were performed by UHPLC-MS/MS using a triple quadrupole mass spectrometer (Sciex 4000) equipped with atmospheric pressure chemical ionization (APCI). For analysis, an appropriate amount of sample was transferred to an LC vial, dried under a stream of argon, and reconstituted in 90% methanol with 0.1% formic acid (for spinal cord, 60 μL was reconstituted in 30 μL; for serum 100 μL to 50 μL; for CSF, 100 μL to 50 μL). Reverse-phase chromatography was performed with the following conditions: C18 column (1.7um, 100 mm × 2.1 mm, Phenomenex Kinetex); flow rate, 0.4 mL/min; elution solvent, 90% methanol with 0.1% formic acid. MS conditions: spray voltage, 5000 V; curtain gas, 10 psi ion source gas, 20 psi; collision gas, high; entrance potential, 10 V; collision energy, 25 V; declustering potential, 80 V; temperature, 300°C. For MS analysis, selective reaction monitoring (SRM) was employed to monitor the dehydration process of the ion [M+H]+ or [M+H-H2O]+ as described previously [13]. The internal standard, d7-7-ketocholesterol, was used to quantify the analytes 7-ketocholesterol, 4β-hydroxycholesterol, and 7-hydroxycholesterol, d6-24-epoxycholesterol was used to quantify 24-epoxycholesterol, 24-ketocholesterol, and 24-hydroxycholesterol, and d7-cholesterol was used to quantify cholesterol. SRM mass transitions are: d7-7-ketocholesterol m/z 408.3 ➔ 390.3; d6-24-epoxycholesterol m/z 389.3 ➔ 371.3; 7-ketocholesterol, m/z 401.3 ➔ 383.3; 7-hydroxycholesterol, 4β-hydroxycholesterol, and 24-hydroxycholesterol, m/z 385.3 ➔ 367.3; 24-epoxycholesterol and 24-ketocholesterol, m/z 383.3 ➔ 365.3; d7-cholesterol, m/z 376.3 ➔ 376.3; cholesterol, m/z 369.3 ➔ 369.3. Ratios of oxysterol to cholesterol concentrations were used for quantification.
Data Analysis
Cholesterol and oxysterol concentrations were evaluated with a two-way ANOVA with experimental group and age as factors and accounting for all 2-way interactions. A post-hoc Sidak’s test was performed with P values adjusted for multiple comparisons at the 95% confidence interval.
Data Access
The RNA-sequencing data from this study has been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sta/) under accession number SUB3127283.
Results
Spinal cord and medulla oblongata are the initial site of pathology with α-TOH deficiency
The initial pathology in 6 mo Ttpa−/− mice on vitE deficient diets was confined to the spinal cord and caudal medulla oblongata in the nucleus gracilis (Fig. 1). Evaluation of the dorsal spinal columns by TEM revealed retraction of large diameter axons with loosely organized myelin that had multifocally lost its electron density (Fig. 1D). The myelin around these axons was also less tightly concentric and lamellar than the control section. Cerebellar pathology was not evident in any sample at this age.
Figure 1.
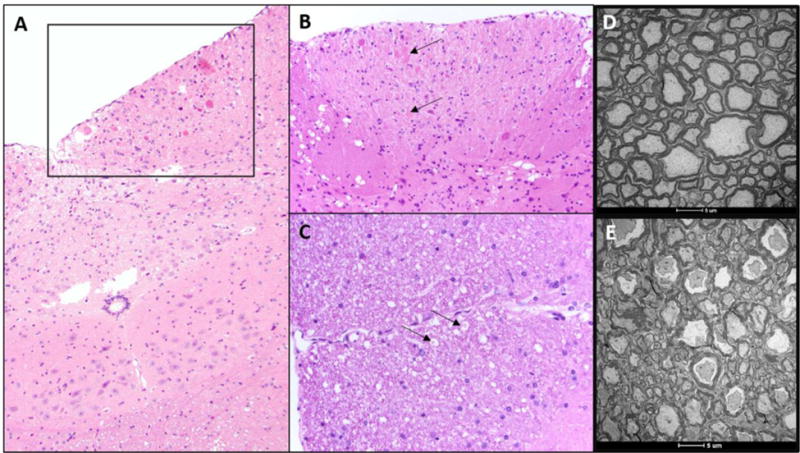
(A,B [inset of A]): Chromatolytic neurons (arrows) within the nucleus gracilis of the medulla oblongata and (C) Digestion chambers (arrows), consistent with myelin degeneration of the dorsal column of the spinal cord of Ttpa−/− mice on vitE deficient diets, n=3-5. (D) Dorsal spinal column from 1-year old Ttpa+/+ mice on vitE+ diet demonstrating healthy myelin. (E) Loosely organized myelin with loss of electron density in the dorsal spinal column of 1-year old Ttpa−/− mice on vitE deficient diet. n=2-3
While no differences in neurobehavioral testing were observed at weaning, the dorsal column pathology at 6 mo of age was associated with a longer time taken to navigate the horizontal ladder using foot misplacement testing in Ttpa−/− mice on vitE deficient diets (Fig. 2). TreadScan data did not reveal any significant disturbances in gait and no evidence of a peripheral neuropathy was observed between groups (data not shown). Foot misplacement neurologic deficits and histologic lesions were not rescued in Ttpa−/− mice when fed a vitE adjusted diet (35 IU/kg feed) at weaning, but instead required a vitE supplemented diet (600 IU/kg feed) to fully rescue the neuropathologic phenotype (Fig. 2).
Figure 2.
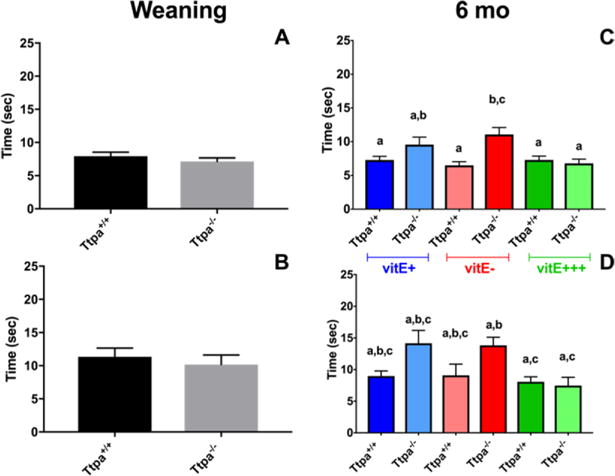
Foot misplacement testing in diet/Ttpa genotype groups at weaning (A,B) and 6 mo of age (C,D). Time to navigate horizontal ladder with no traps (A,C) and with traps (B, D) Number of forelimb errors. (mean ± SEM, n=8-15; t-test or 1-way ANOVA, different superscripts denote significant differences between experimental groups, P<0.05).
Brain α-TOH concentrations significantly differed in experimental groups
Concentrations of α-TOH in the cerebral cortex were below the limit of detection (0.3 ng α-TOH/100 mg mouse brain tissue) in Ttpa−/− mice on vitE− diets (Fig. 3). Only on the vitE+++ diet did Ttpa−/− mice achieve brain α-TOH concentrations comparable to Ttpa+/+ vitE− mice.
Figure 3.
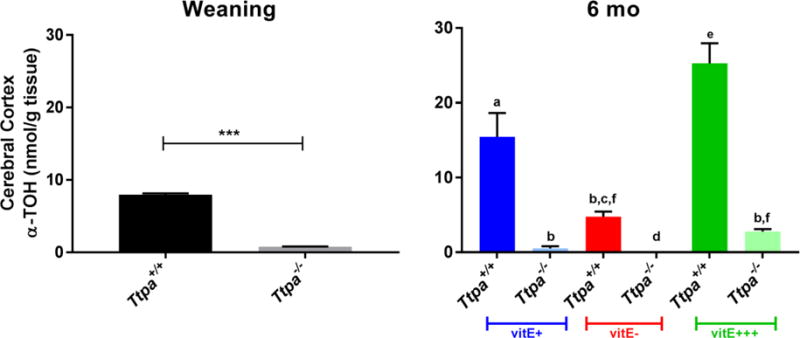
Alpha-tocopherol concentrations within the cerebral cortex at weaning and 6 mo (mean ± SEM, n=9-10; t-test or 1-way ANOVA, ***P<0.0001, different superscripts denote significant differences between experimental groups, P<0.05).
Ttpa is expressed in spinal cord and downregulated in Ttpa−/− mice regardless of diet
Ttpa was lowly expressed and significantly downregulated (log2fold change= −1.5; PFDR<0.05) in the spinal cord of all Ttpa−/− mice irrespective of dietary vitE group (Table 1). An additional transcribed region in Ttpa was identified between exons 2 and 3 (chr4:20,014,785-20,019,569; Fig. A.1) in ~80% of cerebella reads and ~75% of spinal cord reads. This transcript variant has not been previously identified for Ttpa and may be an artifact of this particular mouse strain.
Table 1.
Number of differentially expressed transcripts (DETs) for each contrast in RNA-sequencing from spinal cord. DET=differentially expressed transcript, FDR=false discovery rate, #=distinct sequencing batch and analysis, red=upregulated, green=downregulated, GO=gene ontology pathway ID
| Contrast | # DETs at FDR<0.05
|
Dysregulated Pathways (FDR<0.05) | ||
|---|---|---|---|---|
| Down | Up | |||
| Baseline | Weaning Ttpa−/− vs Ttpa+/+ |
1 (Ttpa) | 0 | None |
| 6 mo genotype/diet | 6 mo vitE+ Ttpa−/− vs Ttpa+/+ |
1 (Ttpa) | 16 | Translation (GO:0006412) Innate immune response (GO:0045087) Inflammatory response (GO:0006954) Defense response to bacterium (GO:0042742) |
| 6 mo vitE− Ttpa−/− vs Ttpa+/+ |
5 (Ttpa, 4930412C18Rik, Lipg, Eml1, Gm27040) | 152 | Innate immune response (GO:0045087) Inflammatory response (GO:0006954) Positive regulation of angiogenesis (GO:0045766) G-protein coupled receptor signaling pathway (GO: 0007186) Defense response to bacterium (GO:0042742) Defense response to virus (GO:0051607) Negative regulation of immune response (GO:0050728) Defense response to protozoa (GO:0042832) Chemokine mediated signaling pathway (GO:0070098) Regulation of cell proliferation (GO:0042127) Immune response (GO:0006955) Positive regulator of tumor necrosis factor production (GO:0032760) Receptor mediated endocytosis (GO:0006898) Cellular response to interferon gamma (GO:0071346) Chemotaxis (GO:0006935) Positive regulator of interleukin 10 production (GO:0032733) Positive regulation of ERK1 and ERK2 cascade (GO:0070374) Intrinsic apoptotic signaling pathway to DNA damage (GO: 0008630) Positive regulation of macrophage differentiation (GO: 0045651) Biologic process (GO:0008150) Positive regulation of interferon-gamma production (GO:0032729) |
|
| 6 mo Ttpa−/− vitE− vs Ttpa+/+ vitE+ |
20 (including Ttpa) |
129 | Inflammatory response (GO:0006954) Antigen processing and presentation via MHC Class I (GO: 0002479) Positive regulator of tumor necrosis factor production (GO:0032760) Positive regulation of angiogenesis (GO:0045766) |
|
| 6 mo vitE+++ Ttpa−/− vs Ttpa+/+# |
1 (Ttpa) | 0 | None | |
| Effect of age on genotype/diet |
Ttpa+/+ vitE+ Wean vs 6 mo |
2258 | 2114 | Cholesterol biosynthetic process (GO:0006695) |
|
Ttpa+/+ vitE− Wean vs 6 mo |
1648 | 1256 | Cholesterol biosynthetic process (GO:0006695) Myelination (GO:0042552) |
|
|
Ttpa−/− vitE+ Wean vs 6 mo |
1335 | 855 | Cholesterol biosynthetic process (GO:0006695) Myelination (GO:0042552) |
|
|
Ttpa−/− vitE− Wean vs 6 mo |
973 | 798 | Cholesterol biosynthetic process (GO:0006695) Myelination (GO:0042552) |
|
Innate immune transcripts were upregulated in the spinal cord of Ttpa−/− mice at 6 mo
Spinal cord Ttpa expression did not change with age between weaning and 6 mo of age. At 6 mo of age, 17 transcripts were dysregulated if Ttpa−/− mice were maintained on the vitE+ diet whereas 157 transcripts (Table 1 and Table A2), primarily representing genes involved in the innate immune response and inflammation, were differentially expressed when on the vitE− diet (Fig. 4). These same dysregulated transcripts were identified when comparing Ttpa−/− vitE− mice to Ttpa+/+ vitE+ mice at 6 mo. Pathway analysis revealed dysregulation of four pathways in 6 mo Ttpa−/− mice maintained on the vitE+ diet, whereas on the vitE− diet, dysregulation of an additional 17 pathways, most related to immune and inflammatory responses, was identified (Table 1). The effect of age could not be assessed in the vitE+++ groups due to the batch effect of sequencing.
Figure 4.
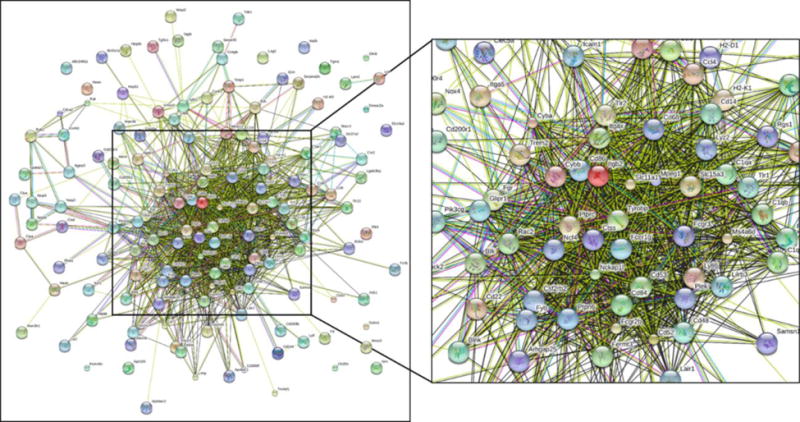
String diagram of the interconnections between the upregulated transcripts from the 6 mo vitE deficient KO vs. WT contrast. Genes within this cluster were associated with GO:IDs innate immune response, inflammatory response, apoptosis, and G-protein coupled receptor signaling.
Myelination was significantly downregulated in the spinal cord during aging in vitE deficient mice
From weaning to 6 mo of age, vitE deficiency led to fewer DETs in the spinal cord. Cholesterol biosynthesis was downregulated with age in all experimental groups; however, myelination was only significantly downregulated with age in the three vitE− groups (Ttpa+/+ vitE−, Ttpa−/− vitE+, Ttpa−/− vitE−) (Table 1).
Minimal differential expression was identified between genotypes in the cerebellum at 6 mo
In all contrasted groups at 6 mo, very few transcripts were differentially expressed in the cerebella of Ttpa−/− mice (Table 2 and Table A3). N-terminal EF-hand calcium binding protein 1 (Necab1), a calcium ion binding transcript, was downregulated in the Ttpa−/− for all genotype contrasts (average log2FC= −1.98, PFDR=0.002). Kelch-like family member 32 (Klhl32), encoding for a protein involved in protein ubiquitination, was downregulated between Ttpa−/− vitE− and vitE+ mice when compared to Ttpa+/+ vitE+ mice. Gamma-glutamyl hydrolase (Ggh), involved in glutamine metabolism, was downregulated between genotypes at weaning and between 6 mo. Ttpa−/− vitE− and Ttpa+/+ vitE+ mice. There were no dysregulated pathways identified in any contrast at weaning or 6 mo of age.
Table 2.
Number of differentially expressed transcripts (DETs) for each contrast in RNA-sequencing from cerebellum DET=differentially expressed transcript, FDR=false discovery rate, red=upregulated, green=downregulated
| Contrast | # of DETs at FDR<0.05
|
Dysregulated Pathways (FDR<0.05) | ||
|---|---|---|---|---|
| Down | Up | |||
| Baseline | Weaning Ttpa−/− vs Ttpa+/+ |
3 (Ttpa, Necab1, Gdh) | 1 (Opn3) | None |
| 6 mo genotype/diet | 6 mo vitE+ Ttpa−/− vs Ttpa+/+ |
2 (Necab1, Klhl32) | 0 | None |
| 6 mo vitE− Ttpa−/− vs Ttpa+/+ |
1 (Necab1) | 0 | None | |
| 6 mo vitE+++ Ttpa−/− vs Ttpa+/+ |
2 (Ttpa, Necab1) | 0 | None | |
| 6 mo Ttpa−/− vitE− vs Ttpa+/+ vitE+ |
3 (Necab1, Klhl32, Gdh) | 0 | None | |
| Effect of aging on genotype/diet |
Ttpa+/+ vitE+++ Wean vs 6 mo |
1372 (Ttpa PFDR=0.05) | 1170 | None |
|
Ttpa+/+ vitE+ Wean vs 6 mo |
904 (including Ttpa) | 783 | None | |
|
Ttpa+/+ vitE− Wean vs 6 mo |
817 (Ttpa PFDR=0.06) | 587 | None | |
|
Ttpa−/− vitE +++ Wean vs 6 mo |
887 (including Ttpa) | 862 | Myelination (GO:0042552) | |
|
Ttpa−/− vitE+ Wean vs 6 mo |
968 | 815 | None | |
|
Ttpa−/− vitE− Wean vs 6 mo |
1037 | 813 | Myelination (GO:0042552) | |
Cerebellar Ttpa expression significantly downregulated during aging only with sufficient vitE
In the cerebellum, Ttpa was the only transcript that downregulated significantly during aging with sufficient vitE (Ttpa+/+ vitE+ and vitE+++) while expression approached significance in the Ttpa+/+ vitE− and Ttpa−/− vitE+++ groups (Table 2). In the two most deficient groups (Ttpa−/− vitE+ and vitE−), Ttpa was not downregulated. Necab1 was not differentially expressed with age in any experimental group. Myelination was significantly dysregulated in Ttpa−/− mice on vitE−and vitE+++ diets.
Cerebellar myelination downregulation with age correlated to vitE status
The central transcripts associated with myelination, including proteolipid protein (Plp), myelin basic protein (Mbp), and myelin-associated oligodendrocyte basic protein (Mobp) were not dysregulated between experimental groups at 6 mo of age. However, from weaning to 6 mo of age, all three transcripts were significantly downregulated in all experimental groups. The degree of downregulation was positively correlated to the vitE status of the group in the spinal cord and, to a lesser degree, in the cerebellum (Fig. 5). Additional statistical analysis was precluded as log2FC is calculated based on the overall change among biologic replicates.
Figure 5.
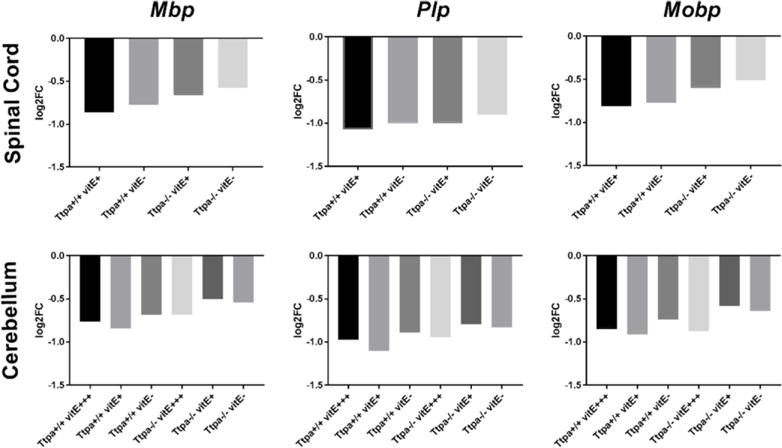
Downregulation of central myelin transcripts, Mbp, Plp and Mobp between weaning and 6 mo of age in spinal cord and cerebellum of mice of various experimental groups. The degree of downregulation was positively correlated with the vitE status of each group, which was more pronounced for spinal cord than cerebellum.
Nuclear orphan receptors
Based on previous gene expression studies in vitE deficient animals [8, 9, 13], gene targets for both RORA and LXR were evaluated in all datasets.
RORA
In Ttpa+/+ mice, RORA was upregulated in the spinal cord, but not cerebellum, as mice aged from weaning to 6 mo on both diets examined (vitE− and vitE+) (Table 3). In Ttpa−/− mice, RORA was not upregulated with either the vitE− or vitE+ diets between weaning and 6 mo of age.
Table 3.
Nuclear receptor activation in the spinal cord between weaning and 6 mo. of age from mice of various genotype and diet combinations. Each transcript is up- or downregulated in relation to weaning with (log2FC, PFDR values) provided.
| Ttpa+/+ vitE+ | Ttpa+/+ vitE− | Ttpa−/− vitE+ | Ttpa−/− vitE− | |
|---|---|---|---|---|
| Retinoic orphan receptor alpha | ||||
| RORA | Up (0.50, 0.03) | Up (0.46, 0.05) | ||
| RORA-targeted transcripts | ||||
| Circadian Rhythm | ||||
| Per3 | Down (−0.53, 0.0) | Down (−0.39, 0.08)# | ||
| Dbp | Down (−0.62, 0.04) | |||
| Per1 | Down (−0.87, 0.001) | |||
| Per2 | ||||
| Clock | Up (0.43, 0.006) | Up (0.39, 0.02) | ||
| Cry2 | Up (0.27, 0.07)# | Up (0.35, 0.03) | Up (0.40, 0.01) | Up (0.41, 0.02) |
| Arntl | Up (0.34, 0.06)# | |||
| Neuronal [26] [27] | ||||
| Itpr1 | ||||
| Calb1 Grm1 |
Up (0.57, 1.00 × 10−4) | Up (0.68, 1.25 × 10−5) | Up (0.41, 0.005) | Up (0.42, 0.007) |
| Shh | Up (0.45, 0.006) | |||
| Rbfox1 | Up (0.51, 0.02) | Up (0.52, 0.02) | ||
| Nlgn1 | Up (0.42, 0.08)# | Up (0.47, 0.06)# | ||
| Ntrk2 | Up (0.47, 0.03) | |||
| Up (0.57, 0.03) | Up (0.30, 0.04) | |||
| Up (0.23, 0.09)# | ||||
| Liver X Receptors | ||||
| Nrlh3 | Up (0.44, 0.03) | Up (0.44, 0.05)# | ||
| Nrlh2 | Up (0.16, 0.08) | Up (0.20, 0.04) | ||
| LXR-targeted transcripts | ||||
| Lipid Homestasis; activate | ||||
| Abca1 | Up (0.46, 0.03) | Up (0.46, 0.04) | Up (0.72, 0.001) | Up (0.8, 6.2 × 10−4) |
| Pltp | Down (−0.72, 0.02) | |||
| Srebf1 | Up (0.28, 0.08) | Up (0.42, 0.009) | Up (0.44, 0.008) | |
| Apoe | ||||
| Acaca | Down (−0.35, 0.005) | Down (−0.22, 0.009) | Down (−0.29, 0.03) | |
| Immunity | ||||
| Eng (up) | Down (−0.96, 0.008) | Down (−0.70, 0.07)# | Down (−0.68, 0.08)# | |
| Nos2 (down) | Down (−1.35, 0.04) | |||
| Il6 (up) | ||||
| Oxidized cholesterol processing Cathepsins | ||||
| Ctsb | Up (0.28, 0.005) | Up (0.27, 0.01) | ||
| Ctsz | Up (0.95, 6.43 × 10−5) | Up (0.97, 1.01 × 10−4) | ||
| Oxidized lipoprotein receptor | ||||
| Olr1 | Up (1.63, 0.04) | Up (1.77, 0.04) |
=PFDR <0.1 but not <0.05)
RORA targeted genes for upregulation (Clock, Cry2, Itpr1, Calb1, Grm1, Rbfox1, Nlgn1, Ntrk2) were upregulated in Ttpa+/+ mice on vitE + diets and, for some transcripts, even on vitE− diets; however, this upregulation with age did not occur for these target genes in the Ttpa−/− mice on either diet. Transcripts targeted for downregulation with RORA, including Per3, Dbp, and Per1 were downregulated in Ttpa+/+ mice, but either unchanged or upregulated in Ttpa−/− mice on vitE deficient diets. Taken together, these results suggest that RORA is transcriptionally activated in the spinal cord during normal aging in mice with sufficient vitE and this activation does not occur in Ttpa−/− mice on diets with nominal (+) or deficient (−) vitE.
LXR
In the spinal cord, transcription of LXR alpha (Nr1h3) and beta (Nr1h2) were both upregulated with aging in Ttpa−/− mice on vitE− diets. Nr1h3 was also upregulated in Ttpa−/− vitE+ mice (Table 3). Major targets for LXR activation, Abca1 and Srebf1, were increasingly upregulated with aging and positively correlated with cerebral cortex vitE concentrations. Another LXR target, Pltp, was downregulated in the control group with age (Ttpa+/+ mice on vitE+) and did not down-regulate in any experimental vitE deficient groups. Another LXR target, Acaca was also downregulated with age in Ttpa+/+ mice regardless of diet and Ttpa−/− mice on vitE+ diets but did not similarly downregulate with age in the Ttpa−/− mice on vitE− diets. Transcripts associated with processing oxidized cholesterol, the major ligand for LXR, including cathepsins and the oxidized lipoprotein receptor 1 (Olr1) were upregulated with age in the spinal cord of Ttpa−/− mice regardless of diet. Taken together, these results provide evidence of LXR activation in the spinal cord during aging in mice that are deficient in vitE.
RT-qPCR validates differential expression of prioritized genes
Of the 11 transcripts of interest that were dysregulated in the spinal cord RNA-seq experiment, eight were differentially expressed in the spinal cord in the independent biologic replicate group (Table 4). Additionally, Necab1 was validated as significantly downregulated in the cerebellum of Ttpa−/− vitE− mice in an independent group of mice (Table 4). Upregulation of Nr1h3 in Ttpa−/− vitE− mice (1.56 fold change, P=0.015) but not Ttpa+/+ vitE− mice (1.02 fold change, P=0.51) with aging was confirmed in an independent group of mice (n=7-8). Upregulation of RORA in Ttpa+/+ vitE+ mice was not validated in this small subset of mice (0.85 fold change, P>0.05).
Table 4.
RT-qPCR validation of prioritized genes in 6 mo. Ttpa+/+ vitE+ mice vs. Ttpa−/− vitE− mice. n=6-8, Mann-Whitney U test
| Transcript | Fold Change | qPCR Validation P value (confidence interval) | |
|---|---|---|---|
| Spinal Cord | Itgb2 | 2.43 | 0.02 (0.36 to 3.87) |
| Itgax | 11.6 | 0.0002 (0.36 to 5.00) | |
| Lilr4ba | 4.40 | 0.03 (0.18 to 6.25) | |
| Pdcd1 | 6.54 | 0.0003 (0.9 to 4.53) | |
| Ccl3 | 11.0 | 0.0015 (1.64 to 5.82) | |
| Tyrobp | 2.63 | 0.13 (−1.36 to 5.37) | |
| Mmp12 | 4.69 | 0.04 (0.63 to 5.18) | |
| Cd68 | 1.29 | 0.48 (−3.52 to 3.75) | |
| Plek | 1.27 | 0.82 (−2.45 to 1.81) | |
| Rgs1 | 5.60 | 0.0007 (1.00 to 3.95) | |
| Trem2 | 3.91 | 0.015 (0.46 to 3.86) | |
| Cerebellum | Necab1 | 0.20 | 0.006 (−3.31 to −1.63) |
Activation of immune pathways without neuroinflammation
Based on the RNA-seq results, immunohistochemical (IHC) staining was performed to determine if there were noticeable changes in microglia (brain macrophages; Iba1) or astrocytes (GFAP) as a response to the altered innate immunity profile. There were no notable increases in microglia in the nuclei or spinal cord that were affected in the Ttpa−/− mice on vitE− diets (Fig. A.2). Similarly, GFAP did not have any staining differences between the groups.
Evidence for apoptosis with long-term vitE deficiency in DRG neurons
To understand the mechanism of long-term alterations that could explain neurodegeneration, an immunofluorescence triple-labeling assay in DRG was performed. At 1-year of age, Ttpa−/− vitE− mice demonstrated an increase in TUNEL staining of both neurons and microglia, supporting an underlying apoptotic mechanism of neurodegeneration (Fig. 6).
Figure 6.
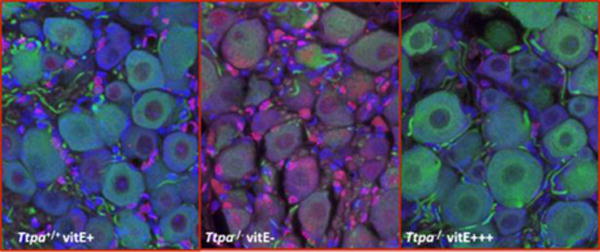
(Red: TUNEL; Green: beta-tubulin III (neuronal cells), Blue: DAPI nuclei) Fluorescent Immunohistochemistry of DRG from Ttpa+/+ and Ttpa−/− (12-month) old mice. Ttpa+/+ vitE+ mice (left panel). Triple-labeling identified evidence of primarily glial cell apoptosis. Under similar conditions, DRG neurons from Ttpa−/− vitE− mice (right panel) demonstrated increased neuronal TUNEL immunostaining.
Alternate routes of cholesterol metabolism in vitE deficient mice
A significant (P<0.0001) decrease with age was noted in cholesterol, cholesterol precursors (7-dehydrocholesterol, 8-dehydrocholesterol, desmosterol, lanosterol), 24-epoxycholesterol, 24-ketocholesterol, and 7-ketocholesterol, with no difference between groups. Alternatively, for enzymatically derived oxysterols (24-hydroxycholesterol and 4β-hydroxycholesterol), significant increases were observed with age (P<0.0001) in all groups (Fig 7). For these two oxysterols, Ttpa+/+ vitE+ mice had significantly higher concentrations within the spinal cord at 6 mo (Fig 7). When normalized to tissue weight, 7-hydroxycholesterol, a marker of lipid peroxidation, decreased most significantly with age (P<0.0001) in the Ttpa+/+ vitE+ mice and least significantly in the Ttpa−/− vitE− mice. This was likely due to slightly higher cholesterol concentrations in the Ttpa−/− vitE− mice as the effect was not significant when normalized to cholesterol.
Fig. 7.
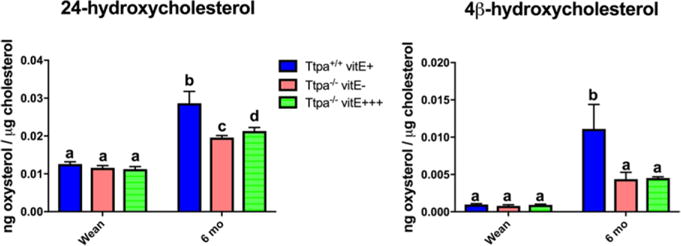
24-hydroxycholesterol and 4β-hydroxycholesterol concentrations within the spinal cord at weaning and 6 mo. In order to analyze the effect of aging on the 3 groups, data for mice at weaning is replicated for Ttpa−/− vitE− mice as Ttpa−/− vitE+++ mice as the experimental diets were not initiated until after weaning. (mean ± SEM, n=5-6; 2-way ANOVA, different superscripts denote significant differences between experimental groups, P<0.05).
Discussion
Spinal cord is the site of initial gene dysregulation with α-TOH deficiency
To date, most gene expression studies using the Ttpa−/− mouse model have focused on the entire brain or the cerebellum as the primary site of dysfunction [8, 9, 11]. While the cerebellum is likely targeted at the later stages of the disease (i.e. >12 mo of age in the Ttpa−/− mouse [4, 11]), we have demonstrated that most of the initial pathology is confined to the spinal cord and caudal medulla oblongata in the Ttpa−/− mouse and evident by 6 mo. Pathology is primarily restricted to the dorsal columns and associated with mild proprioceptive deficits. Previous studies of long-term vitE deficiency in rats have identified a similar increase in spheroids within the dorsal column [28]. Complete rescue of the ataxic phenotype, gene dysregulation, and resulting pathology in Ttpa−/− mice was not attained by providing a basal vitE diet (35 mg/kg feed), but instead required a diet supplemented with ~17× the amount of vitE (600 mg/kg feed) at weaning. Cerebral cortex α-TOH concentrations in our wild-type and Ttpa−/− mice on diets containing similar concentrations of α-TOH were comparable to previous studies in 3-4 mo mice [20]. Consistent with other studies [29], high dose supplementation did not completely restore cerebral cortex α-TOH concentrations to those of wild-type mice (Fig. 3).
Ttpa is lowly expressed in murine spinal cord and significantly downregulates with age in the cerebellum of mice with sufficient vitE
For the first time, low levels of expression of Ttpa were identified in murine spinal cord. At 6 mo, Ttpa was downregulated in Ttpa−/− mice regardless of diet. As mice aged, however, there was no change in spinal cord Ttpa gene expression of any experimental group. In contrast, while demonstrating no difference at 6 mo of age, Ttpa was downregulated in the cerebellum of Ttpa+/+ mice on all three diets, and in Ttpa−/− vitE+++ mice, from weaning until 6 mo of age. In the Ttpa−/− vitE+ and vitE− mice, the α-TOH deficiency may have prevented the physiologic downregulation of cerebellar Ttpa. Previous studies using radioactive α-TOH have identified the cerebellar gray matter having the highest uptake of radioactive α-TOH from blood [30]. It was suggested that the cerebellum is therefore particularly active in the metabolism of vitE and thus may be more susceptible to vitE deficiency than other areas [30]. VitE has also been demonstrated to be required for Purkinje neuron integrity [11]. Therefore, while early gene dysregulation, defined by innate immune and inflammatory responses, occurs within the spinal cord of vitE− mice, evidence exists for dysregulation of Ttpa with aging in the cerebellum, even though morphologic changes were not detected histologically.
Downregulation of cholesterol biosynthesis and myelination with age
During myelinogenesis, astrocytes and neurons produce cholesterol prolifically but, as the brain matures, neurons downregulate the expression of genes involved in cholesterol biosynthesis [31]. In the spinal cord of both Ttpa+/+ and Ttpa−/− mice, the cholesterol biosynthesis pathway downregulated between weaning and 6 mo of age regardless of diet. This was further supported by decreasing sterol levels with aging. Downregulation of cholesterol biosynthesis has previously been demonstrated in the spinal cord in rats during aging from ~15 to 30 months [32].
In the spinal cord, the overall myelination pathway significantly decreased with age in all vitE deficient mice (Table 1) and was decreased in the cerebellum in two of the Ttpa−/− groups (vitE+++ and vitE−) (Table 2). In addition, analysis of specific genes involved in myelination, including MOBP, PLP, and MBP were downregulated from weaning to 6 mo in all experimental groups in both the spinal cord and cerebellum, with the degree of downregulation correlating to the vitE status of each group (Fig. 5). Genes involved in myelination, including Mobp and Plp, have been demonstrated to be downregulated in healthy rats during aging from ~3-30 months [32] and in Ttpa-null mice at 1-year of age [9]. Our results indicate that vitE status can affect the degree of downregulation of myelin-associated transcripts in the spinal cord and cerebellum during postnatal development.
Cerebellar Necab1 is downregulated in Ttpa−/− mice regardless of diet
In the cerebellum, differential expression of Necab1 reliably distinguished between Ttpa genotypes regardless of diet. Necab1 is the target protein of synaptotagmin I C2A-domain, with expression localized to layer 4 cortical pyramidal neurons, inhibitory interneurons, and hippocampal CA2 pyramidal cells in mouse brain [33, 34]. Expression of Necab1 has recently been documented in mouse dorsal root ganglia and the dorsal horn of the spinal cord and most Necab1/2 neurons are glutaminergic [35]. As previous gene expression studies of cerebral cortex from Ttpa−/− mice demonstrated dysregulation of transcripts involved in synaptogenesis, the downregulation of Necab1 during vitE deficiency suggests a specific alteration in synaptotagmin I binding warranting further investigation.
Spinal cord RORA activation in vitE sufficient states versus LXR activation during vitE deficiency
Similar to other gene expression studies in heart [36]and lungs [37] of Ttpa−/− mice, there was no significant change in the expression of classical antioxidant genes (catalase, superoxide dismutase, glutathione peroxidase) in Ttpa−/− mice. We have previously demonstrated similar results in the spinal cord of horses affected with an inherited vitE deficiency [13]. In these horses, activation of LXR targeted transcripts in the spinal cord was instead identified [13]. In our Ttpa−/− mice, alternate nuclear receptor targets were identified with aging. In Ttpa+/+ mice with sufficient vitE, upregulation of RORA mRNA and associated RORA targeted transcripts occurred. In contrast, activation of LXR mRNA (Nr1h3, Nr1h2) and associated LXR targeted transcript dysregulation was identified in Ttpa−/− vitE+ and vitE− mice. Mutual suppression between RORA and LXR has been previously reported. Loss of RORA increases the expression of LXR target genes and, reciprocally, mice deficient in LXR α and β isoforms show activation of select RORA target genes [14]. Therefore, we propose that activation of LXR during vitE deficiency results in suppression of RORA. This theory is supported by previous gene expression studies in the cerebral cortex of Ttpa−/− mice at 3-4 mo of age, where a 13-fold decrease in the expression of RORA mRNA was identified [8] and later validated [9].
RORA activation is essential for synaptic maintenance [38, 39]. RORA-null mice, termed staggerer mice, demonstrate Purkinje cell defects that worsen with postnatal age [40, 41]. The neuroprotective effect of RORA is mediated by glutathione peroxidase 1 and peroxiredoxin 6, protecting neurons against oxidative stress-induced apoptosis [42]. The suppression of RORA with vitE deficiency during a critical window in postnatal development may result in increased susceptibility of neurons to oxidative stress.
Instead of RORA activation, vitE deficient mice demonstrated activation of LXR during postnatal development. LXRα/β double knockout (LXR dKO) mice have both motor coordination and spatial learning alterations linked to cerebellar deficits. These mice have thinner myelin sheaths and decreased myelin gene expression (Plp and Mbp) [43]. Additional studies demonstrate that LXRα and LXRβ are required for correct myelination of peripheral nerve axons [44, 45]. Activation of LXR targeted genes within the spinal cord of Ttpa−/− vitE+ and vitE− mice should therefore promote myelination from weaning to 6 mo of age [43]. However, activation of LXR results in a decrease in cholesterol biosynthesis, which may result in decreased myelination during a critical period of postnatal development [46]. As differential expression of target genes controlled by these two nuclear receptors was evident with postnatal aging, distinct nuclear receptor activation may play a role in the α-TOH deficient phenotype.
Decreased enzymatic metabolism of spinal cord cholesterol in vitE− mice with age
Between 1-6 months of age, a significant decrease in cholesterol and cholesterol precursors, including 7-dehydrocholesterol, 8-dehydrocholesterol, desmosterol, and lanosterol was observed across all experimental groups. A profound decrease in cholesterol biosynthesis has been reported between 4-26 weeks of age in the spinal cord of healthy mice, although minimal changes in overall cholesterol content were noted [47]. This effect continues with aging through 30 months [32]. Aging of the spinal cord is also associated with upregulation of cholesterol transport and metabolism genes, including the LXR targets, Apoe and Abca1 [32].
The enzymatically produced oxysterol 24-hydroxycholesterol increased with age most notably in vitE+ mice. The efflux of 24-hydroxycholesterol is the main mechanism by which the brain facilities removal of cholesterol [48]. An alternate mechanism, through Cyp3a4 conversion to 4β-cholesterol, was also differentially increased with age in the vitE+ vs. vitE−mice. Therefore, the enzymatic removal of cholesterol in the spinal cord may be altered both in Ttpa−/− mice and eNAD/EDM affected horses during postnatal development [13]. As various cholesterol products differ in lipophilicity and are therefore differentially eliminated across the blood brain barrier, this may contribute to the pathophysiology of α-TOH deficient neurodegeneration.
The increased production of 24-hydroxycholesterol in the Ttpa+/+ vitE+ mice during postnatal development would presumably lead to activation of the LXR, as this oxysterol is one of the main ligands for the LXR [49]. However, the levels of 24-hydroxycholesterol were significantly lower in Ttpa−/− vitE− mice than in Ttpa+/+ vitE+ mice. We postulate that the discrepancy between high amounts of LXR ligand without activation of LXR in the Ttpa+/+ vitE+ mice is due to inactivation of LXR by RORA [14].
7-hydroxycholesterol was increased in Ttpa−/− vitE− at 6 months of age but this appeared to be due to higher cholesterol concentrations. 7α-hydroxycholesterol is produced by both enzymatic (Cyp7a1) and free radical oxidation and 7β-hydroxycholesterol is formed via free radical oxidation. As Cyp7a1 is not expressed in murine spinal cord, the majority of 7-hydroxycholesterol is most likely due to free radical oxidation, which may be higher in Ttpa−/− vitE− mice.
Activation of spinal cord innate immune and inflammatory transcripts with vitE deficiency
Of the 157 dysregulated transcripts in 6 mo Ttpa−/− vitE− mice, 54 were associated with the innate immune response [50] (Table A2), including upregulation of complement components (C1qa, C1qb, C1qc), complement receptors (C3ar1, Cd11c, Itgax, Itgb2), lymphocyte antigens (Ly86, Ly9), signaling lymphocyte-activating molecule family members (Slamf6, Slamf9), and toll-like receptors (Tlr1, Tlr13, Tlr2). A similar innate immune response has been reported with complex I deficiency in retinas of Ndufs4-null mice [51], with associated activation of microglia and astrocytes. Innate immune activation in the nervous system is a delicate balance of pro- and anti-inflammatory mediators and leads to astrocyte and microglial responses. Within the Ttpa−/− vitE− mice, the most upregulated transcripts were associated with microglial and astrocyte activation including chemokines (Ccl3, Ccl4), costimulatory agents expressed on antigen-presenting cells necessary for immune activation (Cd68, Cd86), matrix metalloproteinase 12 (Mmp12), and the cell signaling molecule pleckstrin (Plek). Mmp12, Ccl3, and Ccl4 were also upregulated in Ttpa−/− mice on vitE+ diets, demonstrating that basal concentrations of vitE are not sufficient to completely prevent the innate immune and inflammatory response in the spinal cord.
An increased inflammatory response within the lung and liver was previously reported in Ttpa−/− mice injected with lipopolysaccharide [52]. Genome-wide screening in heart tissues of Ttpa−/− mice identified activation of genes related to lipid metabolism and inflammation and repression of immune system genes [36]. Our results extend the hypothesis that suboptimal α-TOH concentrations derange the inflammatory-immune responses in the central nervous system. While there is a molecular signal of some degree of glial activation, IHC did not support infiltration of microglia or astrocytes to chromatolytic neurons. Glial fibrillary acidic protein (GFAP) and allograft inflammatory factor 1 (Aif1), encoding Iba1, were not dysregulated in our study, supporting the IHC results. While multiple innate immunity genes are upregulated in the current study, there are only a select few immunohistochemical reagents that perform reliably in the mouse and both Iba1 and GFAP did not reveal any differences.
Various states of microglial activation were recently reported using myeloid expression profiling. Categories included proliferation, interferon-related, LPS-related and neurodegeneration-related and each category was associated with specific insults to the central nervous system (viral, neoplasic, LPS or neurodegenerative) [53]. In our Ttpa−/− vitE− mice, 17/85 dysregulated immune and inflammatory transcripts could be classified in the neurodegeneration-related microglial activation state, with none identified in the proliferation or interferon-related states and only 4/85 in the LPS-related classification. Additionally, three of most dysregulated neurodegeneration-related transcripts (Clec7, Cst7, Itgax) [53] were also among the top dysregulated transcripts in the Ttpa−/− vitE− mice. Surveilling microglia can be activated by LXR [54].We postulate that molecular activation of microglia may occur secondary to the cholesterol dysregulation incurred by LXR activation in Ttpa−/− vitE− mice.
The overall transcriptomic profile within the spinal cord of Ttpa−/− vitE− mice suggests altered cholesterol homeostasis and activation of innate immune and inflammatory pathways. An increase in inflammatory and immune response transcripts has been previously reported in aging spinal cord of healthy rats, from 3-30 months of age [32]. This was associated with perturbations in cholesterol homeostasis and it was hypothesized that this altered status within the spinal cord white matter could result in impaired function with aging [32]. Of note, the most severely perturbed neuroanatomic tracts in this study were within the white matter of the dorsal funiculus. As the neurologic phenotype in Ttpa−/− vitE− mice localizes to this anatomic region and appears to be associated with altered cholesterol homeostasis, we postulate that a lack of vitE is accelerating cholesterol dysregulation and associated inflammation within the spinal cord. Nuclear receptors most likely play a role in modulating cholesterol homeostasis in the spinal cord during postnatal development and activating glia into specific molecular states.
Upregulation of three apoptosis inhibitory proteins (Naip2, Naip5, Naip6; log2fold change ~1.00) and programmed cell death (Pdcd1, log2fold change 2.32) support an underlying apoptotic mechanism for neuronal death with vitE deficiency. This was further corroborated in the DRG by fluorescent IHC (Fig. 6). It has been previously demonstrated in vitE−deficient rats that vitE deficiency induced a reversible proapoptotic response in lung cells, sensitizing them for additional insults [55]. These observations, combined with the upregulation of the intrinsic apoptotic signaling pathway to DNA damage in Ttpa−/− vitE− mice, support an apoptotic response of neurons in states of vitE deficiency.
Conclusion
In conclusion, activation of specific nuclear receptors and upregulation of innate immunity define temporal neuroaxonal degeneration with α-TOH deficiency. These pathways provide novel insight into the cellular mechanism of the vitE deficient phenotype during the critical window of postnatal development.
Supplementary Material
Table A.1: Quality of RNA generated from each sample used for RNA-sequencing.
{kind=link}
Table A.2: Transcripts that were uniquely expressed in spinal cord tissue from all murine analyses.
{kind=link}
Table A.3: Transcripts that were uniquely expressed in cerebellum tissue from all murine analyses.
Table A.4: Primers used for qPCR validation of select RNA-sequencing results.
Figure A.1: Integrated Genomics Viewer (http://software.broadinstitute.org/software/igv/) of spinal cord RNA-seq data from four experimental mice at 6 months of age (Ttpa+/+ vitE+, Ttpa+/+ vitE−, Ttpa−/− vitE+ and Ttpa−/− vitE−) aligned to the Ensembl annotation of Ttpa. The red arrow denotes a non-annotated exon with reads identified in all four experimental mouse groups.
Figure A.2: Nucleus gracilis of a Ttpa+/+ mouse on a vitE+ diet and a (B) Ttpa−/− mouse on a vitE− diet at 6 months of age. Arrows denote chromatolytic neurons that are associated with only a slight increase in Iba1+ microglia and an equivocal decrease in processes.
Highlights.
Neurologic deficits can be observed as early as 6 months of age in Ttpa−/− mice
RNA-sequencing of spinal cord in Ttpa−/− mice revealed upregulation of genes associated with the innate immune response
Ttpa expression was identified in the murine spinal cord
The transcription factor liver X receptor was strongly activated in Ttpa−/− mice
Acknowledgments
Support for this work was provided by the National Institutes of Health (NIH) to C.J.F. (1K01OD015134 and L40 TR001136) and to L.X (R00 HD073270 and R01HD092659). Additional postdoctoral fellowship support was provided by the Morris Animal Foundation (D14EQ-021) to C.J.F. and support for sequencing provided by the University of Minnesota Equine Center.
Abbreviations
- 3-NT
3-nitrotyrosine
- α-TOH
Alpha-tocopherol
- AVED
Ataxia with vitamin E deficiency
- CNS
Central nervous system
- DET
Differentially expressed transcript
- GFAP
Glial fibrillary acidic protein
- HE
Hematoxylin and eosin
- IHC
Immunohistochemistry
- LXR
Liver X receptor
- MPM
Meters per minute
- RORA
Retinoic-related orphan receptor-α
- RXR
Retinoid X receptor
- TBARS
Thiobarbituric acid
- TEM
Transmission electron microscopy
- t-HODE
Total hydroxyoctadecadienoic acids
- TTP
Tocopherol (alpha) transfer protein
- VitE
Vitamin E
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shichiri M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr. 2014;54:151–160. doi: 10.3164/jcbn.14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fulgoni VL, 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011;141:1847–1854. doi: 10.3945/jn.111.142257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller DP. Vitamin E and neurological function. Mol Nutr Food Res. 2010;54:710–718. doi: 10.1002/mnfr.200900460. [DOI] [PubMed] [Google Scholar]
- 4.Yokota T, Igarashi K, Uchihara T, Jishage K, Tomita H, Inaba A, Li Y, Arita M, Suzuki H, Mizusawa H, Arai H. Delayed-onset ataxia in mice lacking alpha -tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proc Natl Acad Sci U S A. 2001;98:15185–15190. doi: 10.1073/pnas.261456098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gotoda T, Arita M, Arai H, Inoue K, Yokota T, Fukuo Y, Yazaki Y, Yamada N. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N Engl J Med. 1995;333:1313–1318. doi: 10.1056/NEJM199511163332003. [DOI] [PubMed] [Google Scholar]
- 6.Cavalier L, Ouahchi K, Kayden HJ, Di Donato S, Reutenauer L, Mandel JL, Koenig M. Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet. 1998;62:301–310. doi: 10.1086/301699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terasawa Y, Ladha Z, Leonard SW, Morrow JD, Newland D, Sanan D, Packer L, Traber MG, Farese RV., Jr Increased atherosclerosis in hyperlipidemic mice deficient in alpha -tocopherol transfer protein and vitamin E. Proc Natl Acad Sci U S A. 2000;97:13830–13834. doi: 10.1073/pnas.240462697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gohil K, Schock BC, Chakraborty AA, Terasawa Y, Raber J, Farese RV, Jr, Packer L, Cross CE, Traber MG. Gene expression profile of oxidant stress and neurodegeneration in transgenic mice deficient in alpha-tocopherol transfer protein. Free Radic Biol Med. 2003;35:1343–1354. doi: 10.1016/s0891-5849(03)00509-4. [DOI] [PubMed] [Google Scholar]
- 9.Gohil K, Godzdanker R, O'Roark E, Schock BC, Kaini RR, Packer L, Cross CE, Traber MG. Alpha-tocopherol transfer protein deficiency in mice causes multi-organ deregulation of gene networks and behavioral deficits with age. Ann N Y Acad Sci. 2004;1031:109–126. doi: 10.1196/annals.1331.012. [DOI] [PubMed] [Google Scholar]
- 10.Kato S, Fujiki R. Transcriptional controls by nuclear fat-soluble vitamin receptors through chromatin reorganization. Biosci Biotechnol Biochem. 2011;75:410–413. doi: 10.1271/bbb.100807. [DOI] [PubMed] [Google Scholar]
- 11.Ulatowski L, Parker R, Warrier G, Sultana R, Butterfield DA, Manor D. Vitamin E is essential for Purkinje neuron integrity. Neuroscience. 2014;260:120–129. doi: 10.1016/j.neuroscience.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida Y, Itoh N, Hayakawa M, Habuchi Y, Saito Y, Tsukamoto Y, Cynshi O, Jishage K, Arai H, Niki E. The role of alpha-tocopherol in motor hypofunction with aging in alpha-tocopherol transfer protein knockout mice as assessed by oxidative stress biomarkers. J Nutr Biochem. 2010;21:66–76. doi: 10.1016/j.jnutbio.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Finno CJ, Bordbari MH, Valberg SJ, Lee D, Herron J, Hines K, Monsour T, Scott E, Bannasch DL, Mickelson J, Xu L. Transcriptome profiling of equine vitamin E deficient neuroaxonal dystrophy identifies upregulation of liver X receptor target genes. Free Radic Biol Med. 2016;101:261–271. doi: 10.1016/j.freeradbiomed.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao L, Xie X, Zhai Y. Functional crosstalk of CAR-LXR and ROR-LXR in drug metabolism and lipid metabolism. Adv Drug Deliv Rev. 2010;62:1316–1321. doi: 10.1016/j.addr.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Higgins JK, Puschner B, Kass PH, Pusterla N. Assessment of vitamin E concentrations in serum and cerebrospinal fluid of horses following oral administration of vitamin E. Am J Vet Res. 2008;69:785–790. doi: 10.2460/ajvr.69.6.785. [DOI] [PubMed] [Google Scholar]
- 16.Gohil K, Oommen S, Quach HT, Vasu VT, Aung HH, Schock B, Cross CE, Vatassery GT. Mice lacking alpha-tocopherol transfer protein gene have severe alpha-tocopherol deficiency in multiple regions of the central nervous system. Brain Res. 2008;1201:167–176. doi: 10.1016/j.brainres.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beare JE, Morehouse JR, DeVries WH, Enzmann GU, Burke DA, Magnuson DS, Whittemore SR. Gait analysis in normal and spinal contused mice using the TreadScan system. J Neurotrauma. 2009;26:2045–2056. doi: 10.1089/neu.2009.0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cummings BJ, Engesser-Cesar C, Cadena G, Anderson AJ. Adaptation of a ladder beam walking task to assess locomotor recovery in mice following spinal cord injury. Behav Brain Res. 2007;177:232–241. doi: 10.1016/j.bbr.2006.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinov T, Mack M, Sykes A, Chatterjea D. Measuring changes in tactile sensitivity in the hind paw of mice using an electronic von Frey apparatus. J Vis Exp. 2013:e51212. doi: 10.3791/51212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Traber MG, Siddens LK, Leonard SW, Schock B, Gohil K, Krueger SK, Cross CE, Williams DE. Alpha-tocopherol modulates Cyp3a expression, increases gamma-CEHC production, and limits tissue gamma-tocopherol accumulation in mice fed high gamma-tocopherol diets. Free Radic Biol Med. 2005;38:773–785. doi: 10.1016/j.freeradbiomed.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 21.Streett D, Petersen K, Gerritsen A, Hunter S, Settles M. expHTS: analysis of high throughput sequence data in an experimental framework. 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics; September 9-12, 2015; Atlanta, GA, USA. 2015. [Google Scholar]
- 22.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu L, Korade Z, Rosado DA, Jr, Liu W, Lamberson CR, Porter NA. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J Lipid Res. 2011;52:1222–1233. doi: 10.1194/jlr.M014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gold DA, Baek SH, Schork NJ, Rose DW, Larsen DD, Sachs BD, Rosenfeld MG, Hamilton BA. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40:1119–1131. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarachana T, Hu VW. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism. 2013;4:14. doi: 10.1186/2040-2392-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pillai SR, Traber MG, Kayden HJ, Cox NR, Toivio-Kinnucan M, Wright JC, Braund KG, Whitley RD, Gilger BC, Steiss JE. Concomitant brainstem axonal dystrophy and necrotizing myopathy in vitamin E-deficient rats. J Neurol Sci. 1994;123:64–73. doi: 10.1016/0022-510x(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 29.Lim Y, Schock BC, Gohil K, Leonard SW, Packer L, Cross CE, Traber MG. Gene-nutrient interactions exemplified by the alpha-tocopherol content of tissues from alpha-tocopherol transfer protein-null mice fed different dietary vitamin E concentrations. Ann N Y Acad Sci. 2004;1031:328–329. doi: 10.1196/annals.1331.033. [DOI] [PubMed] [Google Scholar]
- 30.Vatassery GT, Angerhofer CK, Knox CA, Deshmukh DS. Concentrations of vitamin E in various neuroanatomical regions and subcellular fractions, and the uptake of vitamin E by specific areas, of rat brain. Biochim Biophys Acta. 1984;792:118–122. doi: 10.1016/0005-2760(84)90211-x. [DOI] [PubMed] [Google Scholar]
- 31.Vitali C, Wellington CL, Calabresi L. HDL and cholesterol handling in the brain. Cardiovasc Res. 2014;103:405–413. doi: 10.1093/cvr/cvu148. [DOI] [PubMed] [Google Scholar]
- 32.Parkinson GM, Dayas CV, Smith DW. Perturbed cholesterol homeostasis in aging spinal cord. Neurobiol Aging. 2016;45:123–135. doi: 10.1016/j.neurobiolaging.2016.05.017. [DOI] [PubMed] [Google Scholar]
- 33.Sugita S, Sudhof TC. Specificity of Ca2+-dependent protein interactions mediated by the C2A domains of synaptotagmins. Biochemistry. 2000;39:2940–2949. doi: 10.1021/bi9920984. [DOI] [PubMed] [Google Scholar]
- 34.Sugita S, Ho A, Sudhof TC. NECABs: a family of neuronal Ca(2+)-binding proteins with an unusual domain structure and a restricted expression pattern. Neuroscience. 2002;112:51–63. doi: 10.1016/s0306-4522(02)00063-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhang MD, Tortoriello G, Hsueh B, Tomer R, Ye L, Mitsios N, Borgius L, Grant G, Kiehn O, Watanabe M, et al. Neuronal calcium-binding proteins 1/2 localize to dorsal root ganglia and excitatory spinal neurons and are regulated by nerve injury. Proc Natl Acad Sci U S A. 2014;111:E1149–1158. doi: 10.1073/pnas.1402318111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasu VT, Hobson B, Gohil K, Cross CE. Genome-wide screening of alpha-tocopherol sensitive genes in heart tissue from alpha-tocopherol transfer protein null mice (ATTP(−/−)) FEBS Lett. 2007;581:1572–1578. doi: 10.1016/j.febslet.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gohil K, Oommen S, Vasu VT, Aung HH, Cross CE. Tocopherol transfer protein deficiency modifies nuclear receptor transcriptional networks in lungs: modulation by cigarette smoke in vivo. Mol Aspects Med. 2007;28:453–480. doi: 10.1016/j.mam.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Landis DM, Sidman RL. Electron microscopic analysis of postnatal histogenesis in the cerebellar cortex of staggerer mutant mice. J Comp Neurol. 1978;179:831–863. doi: 10.1002/cne.901790408. [DOI] [PubMed] [Google Scholar]
- 39.Sotelo C, Changeux JP. Transsynaptic degeneration 'en cascade' in the cerebellar cortex of staggerer mutant mice. Brain Res. 1974;67:519–526. doi: 10.1016/0006-8993(74)90499-5. [DOI] [PubMed] [Google Scholar]
- 40.Yoon CH. Developmental mechanism for changes in cerebellum of “staggerer” mouse, a neurological mutant of genetic origin. Neurology. 1972;22:743–754. doi: 10.1212/wnl.22.7.743. [DOI] [PubMed] [Google Scholar]
- 41.Vogel MW, Sinclair M, Qiu D, Fan H. Purkinje cell fate in staggerer mutants: agenesis versus cell death. J Neurobiol. 2000;42:323–337. doi: 10.1002/(sici)1097-4695(20000215)42:3<323::aid-neu4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Boukhtouche F, Vodjdani G, Jarvis CI, Bakouche J, Staels B, Mallet J, Mariani J, Lemaigre-Dubreuil Y, Brugg B. Human retinoic acid receptor-related orphan receptor alpha1 overexpression protects neurones against oxidative stress-induced apoptosis. J Neurochem. 2006;96:1778–1789. doi: 10.1111/j.1471-4159.2006.03708.x. [DOI] [PubMed] [Google Scholar]
- 43.Meffre D, Shackleford G, Hichor M, Gorgievski V, Tzavara ET, Trousson A, Ghoumari AM, Deboux C, Nait Oumesmar B, Liere P, et al. Liver X receptors alpha and beta promote myelination and remyelination in the cerebellum. Proc Natl Acad Sci U S A. 2015;112:7587–7592. doi: 10.1073/pnas.1424951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makoukji J, Shackleford G, Meffre D, Grenier J, Liere P, Lobaccaro JM, Schumacher M, Massaad C. Interplay between LXR and Wnt/beta-catenin signaling in the negative regulation of peripheral myelin genes by oxysterols. J Neurosci. 2011;31:9620–9629. doi: 10.1523/JNEUROSCI.0761-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shackleford GG, Grenier J, Abi Habib W, Massaad C, Meffre D. Liver X Receptors differentially modulate central myelin gene mRNA levels in a region-, age- and isoform-specific manner. J Steroid Biochem Mol Biol. 2017;169:61–68. doi: 10.1016/j.jsbmb.2016.02.032. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Rogers PM, Su C, Varga G, Stayrook KR, Burris TP. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem. 2008;283:26332–26339. doi: 10.1074/jbc.M804808200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quan G, Xie C, Dietschy JM, Turley SD. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res Dev Brain Res. 2003;146:87–98. doi: 10.1016/j.devbrainres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 48.Ohyama Y, Meaney S, Heverin M, Ekstrom L, Brafman A, Shafir M, Andersson U, Olin M, Eggertsen G, Diczfalusy U, et al. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J Biol Chem. 2006;281:3810–3820. doi: 10.1074/jbc.M505179200. [DOI] [PubMed] [Google Scholar]
- 49.Olkkonen VM, Beaslas O, Nissila E. Oxysterols and their cellular effectors. Biomolecules. 2012;2:76–103. doi: 10.3390/biom2010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–1233. doi: 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu AK, Song L, Murray KD, van der List D, Sun C, Shen Y, Xia Z, Cortopassi GA. Mitochondrial complex I deficiency leads to inflammation and retinal ganglion cell death in the Ndufs4 mouse. Hum Mol Genet. 2015;24:2848–2860. doi: 10.1093/hmg/ddv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schock BC, Van der Vliet A, Corbacho AM, Leonard SW, Finkelstein E, Valacchi G, Obermueller-Jevic U, Cross CE, Traber MG. Enhanced inflammatory responses in alpha-tocopherol transfer protein null mice. Arch Biochem Biophys. 2004;423:162–169. doi: 10.1016/j.abb.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer's Disease Not Evident in Mouse Models. Cell Rep. 2018;22:832–847. doi: 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
- 54.Holtman IR, Skola D, Glass CK. Transcriptional control of microglia phenotypes in health and disease. J Clin Invest. 2017;127:3220–3229. doi: 10.1172/JCI90604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sinha P, Kolleck I, Volk HD, Schlame M, Rustow B. Vitamin E deficiency sensitizes alveolar type II cells for apoptosis. Biochim Biophys Acta. 2002;1583:91–98. doi: 10.1016/s1388-1981(02)00189-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table A.1: Quality of RNA generated from each sample used for RNA-sequencing.
Table A.2: Transcripts that were uniquely expressed in spinal cord tissue from all murine analyses.
Table A.3: Transcripts that were uniquely expressed in cerebellum tissue from all murine analyses.
Table A.4: Primers used for qPCR validation of select RNA-sequencing results.
Figure A.1: Integrated Genomics Viewer (http://software.broadinstitute.org/software/igv/) of spinal cord RNA-seq data from four experimental mice at 6 months of age (Ttpa+/+ vitE+, Ttpa+/+ vitE−, Ttpa−/− vitE+ and Ttpa−/− vitE−) aligned to the Ensembl annotation of Ttpa. The red arrow denotes a non-annotated exon with reads identified in all four experimental mouse groups.
Figure A.2: Nucleus gracilis of a Ttpa+/+ mouse on a vitE+ diet and a (B) Ttpa−/− mouse on a vitE− diet at 6 months of age. Arrows denote chromatolytic neurons that are associated with only a slight increase in Iba1+ microglia and an equivocal decrease in processes.