

Abstract
Azolopyrimidines are efficiently prepared by direct imidoyl C–H bond activation. Annulations of N-azolo imines with sulfoxonium ylides and diazoketones under redox-neutral conditions, and alkynes under oxidizing conditions, provide products with various arrangements of nitrogen atoms and carbon substituents. We have also probed the mechanism of this first example of Rh(III)-catalyzed direct imidoyl C–H activation by structural characterization of a catalytically competent rhodacycle obtained after C–H activation and by kinetic isotope effects.
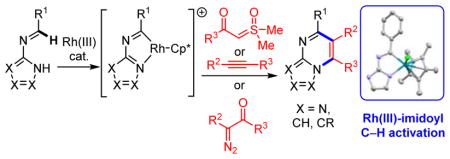
Graphical Abstract
Bridgehead N-fused [5,6]-bicyclic heterocycles are privileged pharmacophores increasingly found in drug leads, clinical candidates and approved drugs. We recently reported the synthesis of azolopyridines by annulations of C-alkenyl azoles with alkynes or diazoketones as mediated by alkenyl C(sp2)–H functionalization (Scheme 1a).1 Other researchers had also previously reported on annulations to give tricyclic N-fused heterocycles by C(sp2)–H functionalization of C-aryl azoles with alkynes.2,3
Scheme 1.

Catalytic C(sp2)–H functionalization for the preparation of bridgehead N-fused [5,6]-bicyclic heterocycles.
Azolopyrimidines are another class of bridgehead N-fused [5,6]-bicyclic heterocycles found in many drugs and drug candidates.4 We envisioned that annulations of N-azolo imines might provide efficient entry to these compounds (Scheme 1b). An attractive aspect of such a strategy would be that many commercial aminoazoles and, in particular, aldehyde inputs would allow for rapid and convenient access to a wide range of N-azolo imine substrates. However, while imidoyl C–H activation has been reported,5 to the best of our knowledge, it has not been applied to annulations to give nitrogen heterocycles. Moreover, while Rh(III) and Co(III) are particularly effective catalysts for nitrogen heterocycle annulations,3 neither has been reported for imidoyl C–H activation. Herein we disclose the first heterocycle formation by catalytic, direct imidoyl C–H activation. Specifically, Rh(III)-catalyzed annulation of N-azolo imines with sulfoxonium ylides, diazoketones and alkynes generate azolopyrimidines with a wide variety of substitution patterns. We have obtained a crystal structure of a catalytically competent rhodacycle C–H activation intermediate, which together with kinetics and deuterium isotope studies, allow us to posit a catalytic mechanism.
Initially, we focused on Rh(III)-catalyzed annulations of N-azolo imines with sulfoxonium ylides. These reagents are typically well-behaved crystalline solids that demonstrate high stability and serve as carbene precursors that do not generate gasses as by-products, which can cause safety issues in large scale processes.6 Recently the groups of Li, Aïssa, and Cheng have shown that these reagents are very effective for Rh(III)-catalyzed acyl methylations7 and annulations8 of arenes. We envisioned that azolopyrimidines could be obtained by imidoyl C–H acyl methylation followed by intramolecular condensation.
After identification of optimal reaction conditions for the desired coupling (see Table S1 in the Supporting Information), the reactivities of several N-azolo imines with sulfoxonium ylide 2a were assessed (Scheme 2). Imines derived from 2-aminoimidazole and various benzaldehydes provided imidazopyrimidines in generally high yields (3a–3f). These substrates included electron-rich (3b) and electron-deficient (3c and 3d) benzaldimines. Bromide (3e) and chloride (3f) substituents are also compatible with the reaction conditions, affording products capable of subsequent cross-coupling. The chloro derivative (3f) additionally establishes that ortho-substituted benzaldimines undergo reaction with only a modest reduction in yield. Reaction of a furfural-derived imine also provided heterocycle 3g. Moreover, imines derived from aminopyrazole starting materials proceeded to pyrazolopyrimidines 3h–3j in good to excellent yields. Preparation of N-azolo imines from enolizable aldehydes proved difficult, and therefore, annulation of this class of substrates was not studied. Finally, preparation of 3a was achieved outside the glovebox on 1 mmol scale in comparable (82%) isolated yield.
Scheme 2. Rh(III)-Catalyzed C–H Functionalization of Azoloaldimines 1 with Sulfoxonium Ylide 2aa.

aReactions performed on 0.3 mmol scale. bReaction performed with tetrahydrofuran as solvent. cReaction performed with 4.0 equiv of PivOH at 120 °C. dReaction performed on 1 mmol scale on the benchtop under an inert atmosphere.
We next investigated the scope of this new transformation with respect to other sulfoxonium ylides (Scheme 3). Although an electron-rich aryl ylide coupled efficiently (3k), the reaction of an electron-deficient ylide proceeded less favorably (3l). Sulfoxonium ylides with bromophenyl (3m) and thiophene (3n) substituents also successfully participated in these couplings. In addition to aromatic ylides, aliphatic ylides can undergo couplings (3o–3q), with carbamate 3q primed for further functionalization following Boc-deprotection. Finally, N-pyrazolo imines afforded products bearing electron-rich aryl (3r), electron-deficient aryl (3s) and alkyl (3t) substituents in excellent yields.
Scheme 3. Rh(III)-Catalyzed C–H Functionalization of Azolobenzaldimines 1 with Sulfoxonium Ylides 2a.

aReactions performed on 0.3 mmol scale. bReaction performed with tetrahydrofuran as solvent.
Next, we expanded this reaction protocol to include the stabilized diazoketone 4. Like, sulfoxonium ylides, diazoketones are redox-neutral carbene equivalents, and they have seen extensive use in Rh(III)-catalyzed C–H functionalization chemistry.9 An initial screen revealed that the conditions used for sulfoxonium ylides were also optimal for diazoketone 4 (see Table S2 in the Supporting Information). Thus, we observed coupling of diazoketone 4 with several imines (Scheme 4). In addition to the parent imine (5a), reactions proceeded well with an electron-rich (5b) and electron-deficient (5c and 5d) imines. Imines derived from 3-bromobenzaldehyde (5e), furfural (5f), and cinnamaldehyde (5g) also participated in moderate to high yields. N-Pyrazolo imines also demonstrated good reactivity with this coupling partner (5h–5j).
Scheme 4. Rh(III)-Catalyzed C–H Functionalization of Azoloaldimines 1 with Diazoketone 4a.

aReactions performed on 0.3 mmol scale. bReaction performed with 4.0 equiv of PivOH at 120 °C.
We also evaluated N-azolo imines 1 in oxidative annulation processes, and for this purpose, studied oxidative couplings with alkynes 6 (Scheme 5). Preliminary experiments (see Table S3 in the Supporting Information) revealed 2,2,2-trifluoroethanol to be the optimal solvent with copper(II) acetate as the terminal oxidant. Under these conditions, 3-hexyne reacted with the standard suite of electronically and sterically differentiated N-imidazo imines (7a–7f). Coupling also proceeded in 93% yield starting from an imine prepared from 2-aminobenzimid-azole (7g). Additionally, furfural- (7h) and cinnamaldehyde-derived (7i) imines engaged in effective couplings with 3-hexyne. Use of diphenylacetylene as the alkyne resulted in isolation of imidazopyrimidine 7j in 75% yield, and use of 1-phenyl-1-butyne provided a separable 2.8:1 mixture of regioisomers 7k. As in the redox-neutral reactions, pyrazolopyrimidines were also obtained, with 7l and 7m isolated in 46% and 83% yields, respectively. However, for these pyrazolopyrimidines AgOAc needed to be used as the stoichiometric oxidant, with Cu(OAc)2 resulting in considerable decomposition and < 5% yield of the desired products.
Scheme 5. Rhodium(III)-Catalyzed C–H Functionalization of Azoloaldimines 1 with Alkynes 6a.

aReactions performed on 0.3 mmol scale. bReaction conditions: [Cp*RhCl2]2 (5 mol %), AgOAc (2.2 equiv), PivOH (4.0 equiv), THF, 100 °C, 18 h.
We next turned our attention to the mechanism. To assess the viability of the proposed rhodacyclic intermediate (i.e. Scheme 1b), we set out to isolate and characterize this key C–H activated species by X-ray crystallography. Treatment of imine 1a with stoichiometric (based on metal) [Cp*RhCl2]2 and NaOAc cleanly afforded rhodacycle 8 (Scheme 6a) in nearly quantitative yield. Single crystals of 8 were characterized by X-ray analysis and provided the first structure of a complex obtained by Rh(III)-imidoyl activation.10
Scheme 6.

Formation and Reaction of Rhodacycle 8
We confirmed the catalytic activity of this proposed intermediate in both a redox-neutral and an oxidative coupling. Reaction of imine 1a with ylide 2a in the presence of 10 mol % of rhodacycle 8 and AgSbF6 provided a 58% isolated yield of heterocycle 3a (Scheme 6b).11 Likewise, the oxidative coupling of imine 1a with 3-hexyne 6a afforded a 78% isolated yield of imidazopyrimidine 7a (Scheme 6c). These experiments substantiate the likelihood of the cationic variant of rhodacycle 8 as an intermediate along the catalytic pathway.
To better understand the fundamental C–H activation step, we submitted deuteroimine 1a-D and ylide 2a to the standard reaction conditions, then halted the coupling at 33% yield of product 3a (Scheme 7a). Remaining imine was analyzed for hydrogen incorporation, revealing less than 4% conversion of deuteroimine 1a-D to protioimine 1a. This low level of scrambling is indicative of a slightly reversible C–H activation step. Given the limited reversibility of this step, we investigated the initial rates of reactions of 1a and deuterated 1a-D with 2a to reveal a primary KIE of 1.5 ± 0.09 (Scheme 7b). This result is consistent with rate-limiting C–H activation and rules out alternative nucleophilic addition to the imine, as this would result in rehybridization and a secondary KIE.
Scheme 7.

Deuterium Isotope Studies with 1a-D
We propose the catalytic mechanism depicted in Scheme 8. Based on the findings in Scheme 7, we suggest concerted metalation/deprotonation of imine 1a to form rhodacycle 8a is rate determining. According to the results in Scheme 6, we believe this intermediate lies on the active catalytic pathway, and undergoes carbene insertion to rhodacycle 9 with sulfoxonium ylide 2, resulting in the extrusion of DMSO.7a Protonolysis releases ketone 10 and regenerates free Rh(III) catalyst. Finally, cyclodehydration of imidazole 10 results in the formation of heterocycle 3. The related catalytic cycle for couplings with diazoketone 4 and the catalytic cycle for oxidative couplings with alkynes 6 are provided in the Supporting Information (Schemes S2 and S3).
Scheme 8.

Proposed Mechanism of Rhodium(III)-Catalyzed C–H Functionalizations of Iminyl Azoles
The modular and regiospecific nature of annulation by imidoyl C-H activation is worth noting. For example, condensation of an aminoazole with a 1,3-diketone also proceeds by cyclodehydration of an intermediate analogous to 10. However, differently substituted 1,3-diketones must first be obtained and generally condense with poor regiocontrol.12
In conclusion, Rh(III)-catalyzed direct imidoyl C–H activation and coupling with sulfoxonium ylides, diazoketones and alkynes provides varied azolopyrimidines. We have probed the unique imine C–H activation pathway by isolating and characterizing by X-ray crystallography a catalytically active rhodacycle intermediate and by obtaining a primary kinetic isotope effect consistent with rate limiting C–H activation. The reported methodology should find utility in the pharmaceutical sector, where azolopyrimidines are frequently employed.4
Supplementary Material
Acknowledgments
This work was supported by the NIH (R35GM122473 to J.A.E. and F32GM114880 to M.R.W.) and the Villum Foundation (VKR023371 to K.S.H.).
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Experimental details, characterization data, and NMR spectra (PDF)
Crystallographic data for 3c, 7a, and 8 (CIF)
References
- 1.Halskov KS, Roth HS, Ellman JA. Angew Chem Int Ed. 2017;56:9183. doi: 10.1002/anie.201703967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For select examples, see: Morimoto K, Hirano K, Satoh T, Miura M. Org Lett. 2010;12:2068. doi: 10.1021/ol100560k.Li X, Zhao M. J Org Chem. 2011;76:8530. doi: 10.1021/jo201530r.Ma W, Graczyk K, Ackermann L. Org Lett. 2012;14:6318. doi: 10.1021/ol303083n.Kavitha N, Sukumar G, Kumar VP, Mainkar PS, Chandrasekhar S. Tetrahedron Lett. 2013;54:4198.Algarra AG, Cross WB, Davies DL, Khamker Q, Macgregor SA, McMullin CL, Singh K. J Org Chem. 2014;79:1954. doi: 10.1021/jo402592z.Zheng L, Hua R. J Org Chem. 2014;79:3930. doi: 10.1021/jo500401n.
- 3.For recent reviews on heterocycle synthesis by C-H functionalization, see: Gulías M, Mascareñas JL. Angew Chem Int Ed. 2016;55:11000. doi: 10.1002/anie.201511567.Yoshino T, Matsunaga S. Adv Synth Catal. 2017;359:1245.
- 4.For information on selected azolopyrimidine drugs and phase II and III clinical candidates: anagliptin, zaleplon, divaplon, verucerfont, lorediplon, dicoglurant, filibuvir, dinaciclib and DSM-265, search the compound name in PubChem.
- 5.Jun C-H, Lee H, Hong J-B. J Org Chem. 1997;62:1200.Park YJ, Park JW, Jun CH. Acc Chem Res. 2008;41:222. doi: 10.1021/ar700133y.. Low valent Co: Yang J, Seto YW, Yoshikai N. ACS Catal. 2015;5:3054.. Ru(II): Park YJ, Jo E-A, Jun C-H. Chem Commun. 2005:1185. doi: 10.1039/b415810e.. Pd(0): Zhao J, Yue D, Campo MA, LaRock RCJ. Am Chem Soc. 2007;129:5288. doi: 10.1021/ja070657l.Fukutani T, Umeda N, Hirano K, Satoh T, Miura M. Chem Commun. 2009:5141. doi: 10.1039/b910198e.Xu P, Wang G, Wu Z, Ii S, Zhu C. Chem Sci. 2017;8:1303. doi: 10.1039/c6sc03888c.
- 6.Oost R, Neuhaus JD, Merad J, Maulide N. Structure and Bonding. Springer; Berlin, Heidelberg: 2017. Sulfur Ylides in Organic Synthesis and Transition Metal Catalysis; pp. 1–43. [Google Scholar]
- 7.(a) Xu Y, Zhou X, Zheng G, Li X. Org Lett. 2017;19:5256. doi: 10.1021/acs.orglett.7b02531. [DOI] [PubMed] [Google Scholar]; (b) Barday M, Janot C, Halcovitch NR, Muir J, Aïssa C. Angew Chem Int Ed. 2017;56:13117. doi: 10.1002/anie.201706804. [DOI] [PubMed] [Google Scholar]; (c) Wu X, Xiong H, Sun S, Cheng J. Org Lett. 2018;20:1396. doi: 10.1021/acs.orglett.8b00119. [DOI] [PubMed] [Google Scholar]
- 8.(a) Zheng G, Tian M, Xu Y, Chen X, Li X. Org Chem Front. 2018;5:998. [Google Scholar]; (b) Xu Y, Zheng G, Yang X, Li X. Chem Commun. 2018;54:670. doi: 10.1039/c7cc07753j. [DOI] [PubMed] [Google Scholar]
- 9.Xia Y, Qiu D, Wang J. Chem Rev. 2017;117:13810. doi: 10.1021/acs.chemrev.7b00382. [DOI] [PubMed] [Google Scholar]
- 10.Rhodacycles obtained by Rh(I) imidoyl C–H oxidative addition have been characterized: Albinati A, Arz C, Pregosin PS. J Organomet Chem. 1987;335:379.. Imidoyl rhodacycles obtained by insertion of isocyanides: Werner H, Heinemann A, Windmüller B, Steinert P. Chem Ber. 1996;129:903.Vicente J, Chicote MT, Vicente-Hernández I, Bautista D. Inorg Chem. 2007;46:8939. doi: 10.1021/ic700963h.
- 11.[Cp*RhCl2]2 (5 mol %) and AgSbF6 (20 mol %) as the catalytic system resulted in 66% yield (see Table S1 in the Supporting Information).
- 12.Maquestiau A, Taghret H, Vanden Eynde J-J. Bull Soc Chim Belg. 1992;101:131. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.