

Summary
Islet β-cells from newborn mammals exhibit high basal insulin secretion and poor glucose-stimulated-insulin-secretion (GSIS). Here we show that β cells of newborns secrete more insulin than adults in response to similar intracellular Ca2+ concentrations, suggesting differences in the Ca2+ sensitivity of insulin secretion. Synaptotagmin-4 (Syt4), a non-Ca2+ binding paralogue of the β-cell Ca2+ sensor Syt7, increased by ~8-fold during β-cell maturation. Syt4 ablation increased basal insulin secretion and compromised GSIS. Precocious Syt4 expression repressed basal insulin secretion but also impaired islet morphogenesis and GSIS. Syt4 was localized on insulin granules and Syt4 levels inversely related to the number of readily releasable vesicles. Thus, transcriptional regulation of Syt4 impacts insulin secretion; Syt4 expression is regulated in part by Myt transcription factors, which repress Syt4 transcription. Finally, human SYT4 regulated GSIS in EndoC-βH1 cells, a human β-cell line. These findings reveal the role that altered Ca2+ sensing plays in regulating β-cell maturation.
eTOC
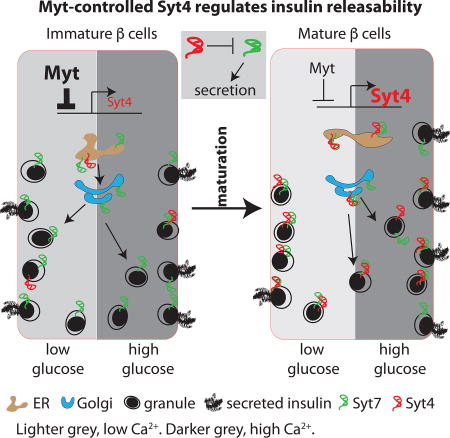
In immature pancreatic beta cells, high glucose does not prompt an increase in insulin secretion. Huang et al. show that this poor response is due to greater Ca2+ sensitivity in immature cells, producing a higher basal secretion rate. Furthermore, Ca2+ sensitivity is regulated by Synaptotagmin-4, whose levels increase during maturation.
Introduction
Whole body euglycemia is mediated in large part by insulin secreted from islet β cells. However, the precise mechanisms that govern insulin secretion, particularly in neonates, have not been completely characterized. In contrast to adult islet β cells, fetal and neonatal cells secrete more insulin in response to low basal glucose levels and have only modest GSIS (Grasso et al., 1968; Pildes et al., 1969). There are many potential stages at which GSIS can be regulated in β cells, including gap junctional or paracrine communication amongst islet cells, intracellular glucose metabolism, glucose-stimulated Ca2+ entry, as well as insulin vesicle formation, fusion, and release [(Liu and Hebrok, 2017) and references therein]. Understanding the postnatal maturation of the β-cell secretory response will provide important insight for producing functional and therapeutically relevant β cells from human ES/iPS cells in vitro, as this process represents a major limiting step in their generation (Kieffer, 2016). Such knowledge will presumably also provide an understanding of islet β-cell dysfunction in diabetes.
Several pathways and transcription factors (TFs) have been suggested to direct postnatal β-cell development. For example, Ca2+/calcineurin signaling facilitates β-cell maturation by promoting insulin vesicle biogenesis (Goodyer et al., 2012); thyroid hormones activate expression of the MafA transcription factor (Aguayo-Mazzucato et al., 2013), which enhances the transcription of insulin and GSIS-related genes (Hang and Stein, 2011); secretory molecules, including Ucn3 from β cells [via somatostatin in δ cells (Blum et al., 2012; van der Meulen et al., 2015)] and neurotransmitters /hormones, can modulate β-cell secretion by regulating β-cell signal transduction or vesicular properties (Scarlett and Schwartz, 2015). Inactivation of several other TFs, including NeuroD (Gu et al., 2010) and Myt1 (Wang et al., 2007) reduce GSIS, suggesting their roles in postnatal β-cell maturation. In part, NeuroD1 functions through repression of glycolytic enzyme expression (Gu et al., 2010), although how Myt1 regulates β-cell function remains unclear, largely due to the redundancy with two paralogues, Myt1L (Nzf1) and St18 (Nzf3) (Wang et al., 2007). How these pathways and signals integrate to control the changes in β-cell GSIS that occur during maturation remains unknown.
Changes in glucose metabolism and ATP-regulated channel activity play an important part in improving GSIS during β-cell maturation (Rorsman et al., 1989). This entails reduced expression of various enzymes favoring glycolysis (e.g. hexokinases and lactate dehydrogenase, or disallowed factors) and increasing transcript abundance of those facilitating mitochondrial oxidative phosphorylation (Lemaire et al., 2016). The molecular mechanisms regulating the expression of these metabolic enzymes involve epigenetic modifications (Dhawan et al., 2015), miRNAs (Jacovetti et al., 2015), and nuclear receptors (Yoshihara et al., 2016).
Although changes in metabolism lead to changes in ion channel activity, these pathways are not sufficient to induce the alterations in GSIS that occurs during β-cell maturation. Notably, influx of Ca2+, a key mediator of insulin secretion, is similar in P2 (two days after birth) and adult β cells (Rozzo et al., 2009) even though physiological GSIS is not observed until postnatal day 9 (P9) or later (Blum et al., 2012; Nishimura et al., 2006). The number of releasable insulin vesicles does not limit the P2 GSIS response since these β cells possess high basal and KCl-stimulated insulin secretion properties (Blum et al., 2012). These observations suggest that under-developed Ca2+-secretion coupling of immature β cells could contribute to their impaired glucose responses. To this end, the availability of vesicles for release and/or Ca2+-sensitivity of vesicle fusion with the plasma membrane could contribute to this immaturity (Kalwat and Cobb, 2017). Indeed, many components of the SNARE (Soluble N-ethylmaleimide-sensitive-factor Attachment Protein Receptor) vesicle fusion complex are Ca2+ sensitive, including syntaxin 1A (Stx1A), synaptosomal-associated protein 25 (Snap25), and Synaptotagmins (Syts). The Syts are particularly interesting because they are known to regulate Ca2+-secretion coupling in nerve cells (Craxton, 2004; Sudhof, 2012). While Syt7 is reported to promote insulin secretion (Dolai et al., 2016; Gustavsson et al., 2008; Wu et al., 2015), the broader influence of the Syt family of proteins in β-cell maturation and GSIS is unknown.
There are 17 distinct Syt-encoding genes in mammals. Their ability to stimulate secretion depends on Ca2+ binding (Berton et al., 2000; Dean et al., 2009; Fukuda et al., 2003). Those that have a high affinity for Ca2+ (Syt1, 2, 3, 5, 6, 7, 9, and 10) can potentiate microsome fusion (Bhalla et al., 2008), while those who lack significant Ca2+ affinity repress SNARE-mediated membrane fusion (Bhalla et al., 2008; Littleton et al., 1999; Thomas et al., 1999). For example, Syt4 can inhibit vesicle secretion in cochlear inner ear hair cell synapses or PC12 cells (Johnson et al., 2010; Machado et al., 2004; Moore-Dotson et al., 2010), which involves direct binding of Syt4 to Ca2+ sensitive Syt1 (Littleton et al., 1999).
Here we demonstrate that insulin vesicles in immature β cells have higher sensitivity to cytoplasmic Ca2+ levels, which is regulated by Syt4. Moreover, we provide evidence that elevated Syt4 expression, partially regulated by Myt transcription factors, during β-cell maturation plays a key role in regulating the GSIS response.
Results
The improvement in GSIS is not regulated by Ca2+ entry into maturing islet β-cells
Depolarization-induced Ca2+ entry is the trigger for insulin secretion in β cells (Rorsman and Renstrom, 2003). To examine if there was a difference in depolarization-stimulated insulin secretion (DSIS) between immature (P1 or P4) and mature islet β cells (P12 or adults), extracellular potassium was elevated to depolarize the cell membrane potential and activate voltage-gated Ca2+ channels (VGCCs). Glucose-independent DSIS occurred in each of these β cell populations (Figure 1A, left columns). Immature β-cells actually showed greater (~4-fold) DSIS following a 10 or 45 minutes KCl stimulation (Figure 1A), a temporal response similar to the first and second phase of GSIS (Rorsman and Renstrom, 2003). Addition of 2.8 mM glucose during DSIS produced findings comparable to KCl alone, with immature β cells secreting ~4-fold more insulin than mature β cells (Figure 1A, right columns). The higher levels of insulin secretion in immature β cells could not be explained by the amount of insulin within each β cell, because mature cells contain 1.6 fold more insulin than immature β cells (Figure 1B). Nor is it a result of differing quantities of insulin in each vesicle, as the diameter of the dense insulin protein core was similar between immature and mature β cells (Figure S1A–E).
Figure 1. Equivalent Ca2+ influx results in more insulin secretion in immature β cells.

Handpicked islets were used for secretion and Ca2+ assays. (A) The percentage of insulin secretion induced by 25 mM KCl in islets at 0 (left columns) or 2.8 mM (right columns) glucose. Data from three time points, before KCl stimulation (“0”), 10 minutes or 45 minutes (m) with KCl stimulation (“10” or “45”) were presented. The data at “0” minute represent insulin secretion within a 45 minutes time window without KCl stimulation. The indicated statistical analysis (top brackets) was calculated between P1 and P12 islets. (B) Insulin content per β cell, assayed with purified β cells of RipmCherry mice. (C) Average islet cytoplasmic Ca2+ responses to 25 mM KCl-induced depolarization in 2.8 mM basal glucose; these responses were assayed with Fura2AM. (D) Quantification of the KCl-induced islet Ca2+ response defined with area-under-curve after KCl addition (from 2–8 minute). (E) The greatest total Ca2+ levels reached in islets of different ages stimulated by 25mM KCl. (F) Insulin secretion induced by ionomycin (Iono) in P1 and adult islets. Diazoxide (Dia) was included in these assays. (G) Islet DSIS with or without glucose-induced degranulation, achieved by incubating islets in 5.6 mM glucose for one hour prior to DSIS. (*: P< 0.05. T-test).
These findings suggest that immature β cells undergo a greater number of secretion events following a depolarizing stimulus than mature cells, which might be explained by differences in cytoplasmic Ca2+ handling. While KCl treatment induced β-cell cytoplasmic Ca2+ influx in islets of all stages (Figure 1C), the total amount of KCl-induced Ca2+ entry was significantly higher in mature β cells compared to immature β cells (Figure 1D, E). However, immature β cells (P1, P4) had a higher concentration of basal cytoplasmic Ca2+ compared to mature β cells [P12, adult, (Blum et al., 2012)](Figure 1C, ~120%), consistent with their higher glycolysis rate (Jermendy et al., 2011). These data suggest that the greater DSIS in immature β cells is not a result of elevated Ca2+ influx.
It is also possible that changes in VGCC isoforms or subcellular localization influence Ca2+ entry and insulin secretion during maturation. To test this, we monitored insulin secretion from islets treated with a K+ channel activator (i.e. diazoxide) and an ionophore (ionomycin) to induce equivalent Ca2+ entry across the entire β-cell membrane without VGCC activation (Do et al., 2014). Under these conditions, the immature islets again showed ~3-fold higher insulin secretion compared to mature islets (Figure 1F), suggesting that the hyper insulin secretion in immature β cells does not depend on particular sites of Ca2+ entry.
Since immature β cells secrete a greater percentage of total cellular insulin content, it was predicted that prolonged stimulation would deplete the readily releasable insulin vesicle. This was tested by pre-incubating islets with 5.6 mM glucose for one hour (degranulation), which will only stimulate high insulin secretion from the immature β cells. As expected, there was significant reduction of DSIS in immature β cells following pre-incubation in 5.6 mM glucose (degranulation) compared to untreated controls, but no reduction in adult islets (Figure 1G).
Increasing cAMP levels stimulate GSIS in a Ca2+-independent fashion (Ammala et al., 1993). Yet, elevating the intracellular cAMP level with 3-isobutyl-1-methylxanthine (IBMX), an inhibitor of the phosphodiesterase that degrades cAMP, potentiated DSIS by ~5-fold in adult islets and only ~2-fold in P1 (Figure S1F). This finding suggests that the cAMP-mediated pathway is less active in immature β cells and unlikely to account for their hypersensitive Ca2+-secretion coupling.
Collectively, our results imply that the greater DSIS of immature β cells is not a result of elevated depolarization-induced Ca2+ influx or cAMP generation. Hence, we found that the insulin vesicles in immature β cells undergo greater Ca2+-induced fusion with the plasma membrane than adult islet cells, conditions producing less cAMP. In the following experiments, we will show that Syt4 regulates vesicle sensitivity to Ca2+ and GSIS during β cell maturation.
Increased Syt4 expression correlates with β-cell maturation
RNA-Sequencing (RNA-seq) was performed on β cells purified from P1, P12, and P60 MipeGFP mouse islets to identify gene networks that change over the β-cell maturation process. P1 represented immature β cells, P12 represented β cells that have proper GSIS with no exposure to the post-weaning high carbohydrate diet (Blum et al., 2012), and P60 represented adult functional β cells (Stolovich-Rain et al., 2015). As expected, most of the genes known to promote β-cell maturation, including MafA, NeuroD1, and Ucn3 (Table S1), displayed significantly increased expression from immature to mature state.
We focused here on vesicular genes that could regulate Ca2+-secretion coupling. Eight of the 17 Synaptotagmin genes (Syt) were expressed in these β cell populations (i.e., Syt3, 4, 5, 7, 9, 11, 13, and 14, Table S1). The expression change in each of these key SNARE gene products was verified by real time RT-PCR in FACS purified P1, P12, and adult β cells from a second mouse line, RipmCherry mice. No significant difference was found in Syt5, Syt7, Syt9, and Syt13 levels, whereas the mRNA levels of Syt3, Syt4, Syt11, and Syt14 increased during islet maturation (Figure 3A. repeated measure ANOVA, P≤0.019). Notably, only Syt4 and Syt14 displayed significant expression changes (~>5 folds) between the P1 and P12 islets (T-test. P≤0.04). Subsequent analysis focused on Syt4 because its transcript levels were ~8-fold higher than Syt14 (Figure 3A). Most importantly, this resulted in increased Syt4 production during the process of β-cell maturation, Figure 2A).
Figure 3. Syt4OE promotes β-cell maturation but impairs islet morphogenesis and GSIS.

Control (“con”, including TetOSyt4 and RiprTTA mice) and Syt4OE littermates were used for all assays. Images were captured with identical parameters. (A) The time frame used for Dox administration and islet/mouse characterization. After intercrossing, Dox feeding started at E16.5 in plugged females and last to P10, or to the point of tissue collection before P10. (B) Real-time RT PCR showing Syt4 overexpression in isolated P2 islets of three independent mouse lines. (C) GSIS in P4 and P7 Syt4OE islets. Shown are the % of insulin secretion within a 45 minutes time window. (D) Random feeding blood glucose in control (con) and Syt4OE (OE) mice. (E–G) Immunoassays in P10 islets with/without Syt4OE, highlighting islet morphology (E), Glut2 (F), and MafB production (G). (H, I) β-cell proliferation assays and quantification with Ki67 labeling in P10 islets. (J) β-cell mass in P10 pancreata. (K–M) Gene expression in P35 islets in mice with/without transient Syt4OE between E16.5 to P10. Panels L1 and L2 were split, with a single Glut2 channel to highlight the change in Glut2 levels. (N, O) Islet GSIS and IPGTT assays in P35 control and Syt4OE mice. Scale bars=20 µm. *: P<0.05, T-tests.
Figure 2. Increased Syt4 expression is required for β-cell maturation.

(A) LSM confocal images of Ins/Syt4 immunofluorescence in P1 to adult β cells. Single optical slices of merged images were shown, taken under identical confocal parameters. Note that isolated islets were partially dissociated and quickly attached onto slides with Cytospin methods, followed by fixation and staining. Under this condition, the islet vesicles appeared disrupted so that insulin immunofluorescence appeared as diffusive signals. (B) SIM images of Ins/Syt4 staining in a representative adult β cell. Shown are projections of 8 optical slices taken 125 nm apart, with insulin (B1), merged (B2), and Syt4 channel (B3) shown. Arrows and arrowheads in B2 showed examples of vesicles with or without co-localizing Syt4 signals. Green arrows and broken circles in B3 highlighted Syt4 patches devoid of Ins signals. (C) An enlarged single optical slice (~100 nm thickness) to highlight the co-localization of Ins and Syt4 in several vesicles (arrows). Arrowheads pointed to several vesicles that are close but not contacting the Syt4 signals. Scale bars=1 µm. (D) IPGTT in 8-week old control (WT) and Syt4 null mice. P value is calculated with Repeated Measure ANOVA. Glc, Glucose. (E) Insulin sensitivity assay in 8-week old mice after 4-hour fasting. Presented are the ratios of blood glucose levels over that before insulin injection. (F) Serum insulin levels detected during IPGTT test (8-week old mice). (G) GSIS of 8-week old Syt4−/− islets. (H) GSIS and DSIS of P14 islets. (I, J) P14 islet cytoplasmic Ca2+ induced by 20 mM glucose (I) or 25 mM KCl (J). *: P<0.05, T-test.
Syt4 localizes to both vesicles and non-vesicular β-cell compartments
Super-resolution Structured-Illumination-Microscopy (SIM) was used to determine the subcellular localization of Syt4 in β cells. SIM clearly resolved the structure of individual vesicles (Figure 2B1). Importantly, we observed that a majority of insulin vesicles co-localized with Syt4 signals (Figure 2B2), which also displayed granular non-uniform distribution (Figure 2B3). The co-localization of Syt4-insulin signals was seen under 3-D projection (Figure 2B) as well as single optical slice images (Figure 2C). A small portion of bright insulin vesicles (~10% in 50 β cells examined with strong insulin immunofluorescence signals) did not co-localize with Syt4 (Figure 2B2, arrowheads). Why these vesicles are different from the rest of the pool is not clear, but they may have no or only low levels of Syt4, or they may be vesicles in the process of being degraded by autophagy. Interestingly, some strong Syt4 signals did not co-localize with insulin vesicles (Figure 2A–C and Figure S2B), but in the Golgi (Figure S2C, D) and the ER (Figure S2E, F). While the role of Syt4 in ER and Golgi biology will be addressed elsewhere, we focused on the potential role of Syt4 in regulating Ca2+-induced insulin secretion.
Syt4 is required for β-cell maturation
Syt4 knockout (Syt4−/−) mice were used to determine the functional involvement of this protein in islet β cells. Eight-week old Syt4−/− mice displayed a modest, but significant, defect in glucose clearance (Figure 2D), despite their normal insulin sensitivity (Figure 2E). In addition, Syt4−/− serum insulin levels were lower during glucose tolerance tests (Figure 2F). GSIS in 8-week old Syt4−/− islets was also blunted compared to controls (Figure 2G). At P10 and adult stages, Syt4−/− islet and vesicular morphology was unchanged as were the levels of many key endocrine genes associated with cell activity, including the endocrine hormones, the Glut2 glucose transporter, and key islet transcription factors [MafA, MafB, Pdx1, and Nkx6.1 (Figure S3) and data not shown]. These observations indicated that the Syt4 effect on insulin secretion in vivo was not mediated by changes in islet cell identity or vesicular biosynthesis, but the response of vesicles to glucose stimulation.
Interestingly, a significantly higher basal GSIS was observed in pre-weaned, two-week old Syt4−/− islets (Figure 2H), implying that Syt4 increases the threshold of Ca2+ needed to trigger insulin secretion. However, the response of these islets to high glucose levels remained largely normal at this stage (Figure 2H), indicating an overall leftward shift of Ca2+ sensitivity. This possibility was examined using DSIS by pre-incubating mutant and control islets in Krebs-Ringer Bicarbonate Buffer (KRB) without glucose for one hour in order to prevent glucose-induced degranulation. DSIS from non-degranulated Syt4−/− islets was significantly higher than control islets (Figure 2H), supporting the hypothesis that Syt4 reduces Ca2+ sensitive insulin vesicle fusion with the plasma membrane. Notably, preventing degranulation before the DSIS normalized insulin secretion at basal conditions (physiological KCl) in control and Syt4−/− islets, because of the increased basal secretion in control cells (Figure 2H, showing the basal insulin secretion levels in control islets with GSIS versus DSIS). These changes in DSIS in Syt4−/− islets were not due to Ca2+ handling, since P14 control and Syt4−/− islets had similar Ca2+ influx properties during both glucose-stimulation and KCl-induced depolarization (Figure2I, J).
Syt4 over-expression (Syt4OE) represses basal insulin secretion and impairs islet morphogenesis
We next examined whether precocious Syt4OE would expedite β-cell maturation. Three TetO-inducible transgenic mouse lines (termed #1, 2, and 3) were derived that enabled inducible Syt4 production in TetOSyt4; RiprTTA (Syt4OE) β cells. Syt4OE embryos/mice were exposed to doxycycline (Dox) from E16.5 via drinking water from the plugged mice (Figure 3A). This procedure induced detectable Syt4OE only after E17.5, which reached ~6 fold in P2 islets in all three lines (Figure 3B). Lines #1 and #2 were used to test the physiological effects of Syt4OE in β cells, which resulted in significantly repressed basal insulin secretion in perinatal islets at P4 and P7 but without significant effect on GSIS (Figure 3C). Corresponding to the reduced basal insulin secretion, two-weeks old Syt4OE mice displayed a trend of higher random blood glucose levels (i.e. ~20%. Figure 3D).
We examined the islet morphologies and gene expression patterns in P4 pancreatic sections of Syt4OE pancreas to determine if the observed differences in insulin secretion resulted from abnormal β-cell development. No differences were observed in the expression levels/patterns of endocrine hormones (indicative of islet morphogenesis) and key β-cell markers including MafA, MafB, Pdx1, and Glut2 (Figure S4A, B). These findings, together with the P4 insulin secretion profiles, suggest that Syt4OE reduced basal insulin secretion without compromising β-cell development or islet morphogenesis.
Interestingly, at P10, Syt4OE islets showed noticeable abnormalities in islet morphology, gene expression, and β-cell proliferation. At this stage, >83% of islets had the typical β-cell-enriched core organization (Figure 3E1). Only 51.4±3.4% of Syt4OE islets had this architecture, with nearly half displayed non-β cells in the core of islets (Figure 3E2). Moreover, Syt4OE β cells had reduced Glut2 and increased MafB levels in β cells (Figure 3F,G). In contrast, expression of several other transcription factors was unaffected, including MafA, Myt1, Nkx6.1, and Pdx1 (Figure S4C, D). Furthermore, islet β-cell proliferation was significantly compromised at P10 as assayed with Ki67 positivity (Figure 3H, I), which resulted in reduced β-cell mass (Figure 3J).
The abnormal islet phenotype of Syt4OE mice persisted for nearly four weeks after cessation of Syt4OE upon Dox withdrawal (P35), with non-β cells consistently observed in the center of islets (Figure 3K) and reduced Glut2 levels on the β-cell membrane (Figure 3L). In contrast, MafB expression was reduced to undetectable levels in β cells (Figure 3M). GSIS was also compromised in Syt4OE islets (Figure 3N), and there was a trend toward defective glucose clearance in post-weaning mice (Figure 3O, P=0.06 with repeated measure ANOVA. P<0.05 for time points 30 and 60 minutes, T-test). These data suggest that transient Syt4 over-expression at neonatal stages, through repressing basal secretion, has long lasting detrimental effects on islet morphogenesis and function.
Syt4 partially co-localizes and interacts with Syt7 in islet β cells
We next explored if interactions between Syt4 and other family members could influence insulin secretion, a mechanism used to regulate Ca2+-induced secretion in neurons. For example, Syt4 binding to Syt1 reduces the efficiency of vesicle plasma membrane fusion induced by Ca2+ (Bhalla et al., 2008; Littleton et al., 1999). Notably, the Syt1 functional paralog, Syt7, is highly expressed in β cells and has been found to act as a Ca2+ sensor to promote insulin secretion (Dolai et al., 2016; Gustavsson et al., 2008; Wu et al., 2015).
Syt7 was detected in the majority of insulin vesicles and non-vesicular compartments when assayed using SIM, in both maxi-projection (Figure 4A) and single optical slice images (Figure 4B). Furthermore, Syt7 partially co-localized with Syt4 in dispersed islet β cells (Figure 4C). Proximity ligation analysis (PLA) showed that Syt4 and Syt7 localized very close to each other (<40 nanometer, Figure 4D, E), suggesting their direct interaction. Indeed, interaction between these proteins was also observed in co-immunoprecipitation assays performed in 293 cells with Syt mutant fragments lacking their transmembrane domains to minimize nonspecific binding (Figure 4F). Moreover, insulin secretion was severely attenuated in immature Syt7−/− islets (Figure 4G). These data are consistent with Syt4:Syt7 interactions reducing Ca2+-induced insulin secretion in maturing β cells.
Figure 4. Syt4 co-localizes with Syt7 in β cells.

(A, B) SIM images showing the subcellular localization of Syt7 in a representative adult β cell. Panels A are projections of 8 optical slices taken 125 nm apart. Panel B are single SIM slices for better resolution of Ins/Syt7 localization. Note the large portions of vesicles with overlapping/close localization of Ins/Syt7 (arrows in A1, B1). Also note the vesicles without obvious contact with Syt7 (arrowheads). (C) Co-localization of Syt4 and Syt7 in β cells shown with SIM. Arrows, spots of Syt4-Syt7 co-localization. Arrowheads, spots of single Syt4 or Syt7 signal. The cell identity is confirmed by insulin expression, not included here. Bars=0.5 µm. (D) PLA assays verifying the close localization of Syt4 and Syt7, accepted as indication of direct association. Hand picked islets attached onto slides with cytospin method were used. Images were captured with LSM and Z-stack images were presented to show signals in the entire cell. Note that merged images (D1, D3) between DAPI and PLA signal and single PLA signal images (D2, D4) were shown. Syt4−/− islet cells (D3, D4) were included as negative controls. Arrows and arrowheads in D2 and D4 are the positions of line-scan to compare the relative PLA signal intensity presented in panel (E), in horizontal and vertical directions. Scale bar=20 µm. (F) Immunoprecipitation showing Syt4-Syt7 interactions. Lysates from HEK293 cells transfected with constructs that expressing HA-tagged Syt4 and Syt7 cytoplasmic domains were used. Proteins were immunoprecipitated by anti-HA, then immuno-blotted with anti-Syt7 antibodies. (G) GSIS of P4 Syt7−/− and control immature islets. *: P=0.02, T-tests.
Syt4 regulates insulin vesicle localization to the plasma membrane
Syt7 has been found to promote insulin vesicle transport to the cell membrane (Dolai et al., 2016; Osterberg et al., 2015). Consequently, TEM was used to determine if Syt4, presumably through interaction with Syt7, could also impact insulin vesicle density on/near the plasma membrane.
While Syt4OE did not significantly change the size of mature insulin vesicles (recognized by their dense core surrounded by a ring of electron light materials, arrows in Figure 5A, B and Figure S5), it caused a ~20% increase in the number of mature vesicles in β cells at P4 and P7 (Figure 5E). Additionally, Syt4OE reduced the number of insulin vesicles docked within 20 nm of the plasma membrane (Figure 5F). In contrast, neither insulin vesicle size nor density was changed in Syt4−/− β cells (Figure S5B), but docked vesicles density was increased in P14 β cells, when the endogenous Syt4 levels were high (Figure 5C–F). Interestingly, Syt4−/− β cells have a reduction in the number of docked vesicles just after weaning at P24, which is consistent with the reduced GSIS in post-weaning Syt4−/− islets (Figure 2G). These combined findings suggest that Syt4 regulates the localization of insulin vesicles to the cell membrane.
Figure 5. Syt4 regulates insulin vesicle docking to the plasma membrane.

(A–D) Representative TEM images of β cells with Syt4 overexpression or inactivation. (A, B) P1 control and Syt4OE β cells. (C, D) P24 control and Syt4−/− β cells. Green arrows point to several examples of docked mature vesicles. Blue arrowheads point to several immature vesicles, recognized by the low electron density of the insulin core. The cell-cell junctions were outlined with thin broken red lines. (E) Density of vesicles at different stage in islets of control, Syt4OE, or Syt4−/− null mouse β cells. (F) Quantification of docked vesicles in β cells. See Figure S5 for more images used for quantification. *: P<0.05
Myelin transcription factors (Myt) regulate Syt4 expression
We searched for the transcription factors that regulate the expression of Syt4. Inactivation of MafA did not change the expression of Syt4 (Artner et al., 2010). Strikingly, we found that total loss the Myt1, Myt1L, and St18 production in pancreatic progenitors yielded a phenotype similar to Syt4OE islets. Our previous work had demonstrated that expression of the Myt1 paralogs, Myt1L and St18, were induced in the embryonic Myt1−/− pancreas, a mutant mouse with deficiencies in GSIS in adult islets (Wang et al., 2007). Furthermore, we found that all three factors were produced in postnatal islet cells (Figure 6A). To address the overall significance of the Myt family in the pancreas, all three Myt genes were inactivated in pancreatic progenitors using Pdx1Cre and newly derived Myt1LF and St18F floxed alleles (Figure S6). This allowed for efficient inactivation of all three genes in the endocrine islets of Myt1F/F; Myt1LF/F; St18F/F; Pdx1Cre mice (termed 6F; Cre, Figure 6B).
Figure 6. Myt factors repress Syt4 transcription.

(A, B) Myt protein detection in P35 control (con, 6F in this case) and 6F; Cre mutant islets. Note that A4 and B4 showed examples of co-staining of St18 and Pdx1 of panels A3 and B3 to locate the islets, respectively. (C) Fasting glucose in 6F; Cre mutant mice. (D) Islet GSIS and DSIS of weaned mice (P24). (E) Immunofluorescence to show the relative insulin level in P21 control and 6F; Cre β cells. (F) TEM images of insulin secretory vesicles in P21 control and 6F; Cre β cells. Arrows, examples of mature insulin vesicles. Arrowheads, examples of immature vesicles. (G) RNA-seq data of several genes in P1 control and 6F; Cre β cells (n=3). The P-values (t-test) of several genes were marked on the top. (H) Real-time RT-PCR assays of Syt4 in hand-picked P14 islets in control and 6F; Cre islets (n=3). (I) Expression levels of Myt genes from purified β cells (via MipeGFP expression) at different postnatal stages via RNA-seq (n=3). (J–M) Immunofluorescence assays of Myt1 protein levels in β cells of different ages. Wild type (CD1) mice were used. For quantification in (M), ~200 cells were used from each stage.
The body weight and blood glucose levels of neonatal 6F; Cre mice were normal until about one week after birth compared with wild type, Myt1F/F; Myt1LF/F; St18F/F (6F), and Pdx1Cre control mice. Yet the 6F; Cre mice developed elevated fasting blood glucose several days after weaning (Figure 6C), concomitant with reduced GSIS and DSIS, like Syt4OE islets (compare Figure 6D to Figure 3N). The 6F; Cre islets lacked visible defects in their insulin levels, assayed by insulin immunofluorescence (Figure 6E), and vesicular morphology, determined with TEM (Figure 6F). These results suggest the involvement of Myt factors with β -cell function, likely involved the maturation steps.
RNA-seq was performed to analyze Syt4 levels in purified β cells by virtue of MipeGFP expression from newly born (P1) 6F; Cre; MipeGFP and control MipeGFP islets. There was a significant 2.4-fold up-regulation of Syt4 in 6F; Cre β cells (Figure 6G). Interestingly, we also observed significant up-regulation of NeuroD1 (1.6-fold, P=0.002) and trends of up-regulation of Ucn3 (P=0.14) and MafA (P=0.16). The expression of Nkx6.1 and Pdx1 was unchanged and Glut2 expression was significantly decreased in the 6F; Cre β cells (Figure 6G). Increased Syt4 expression was also observed in P14 islets (Figure 6H), although the expression of the other genes except Glut2 was normalized (data not shown).
The above findings suggest that the loss of Myt factors have de-repressed Syt4 transcription and a normal function of Myt proteins is to repress Syt4. This conclusion predicts that Myt expression levels could decrease during β-cell maturation to allow for Syt4 upregulation. Indeed, the Myt1 expression level showed a significant reduction from P1 immature to P12/adult mature β cells, at both mRNA (Figure 6I) and protein levels (Figure 6J–M), whereas the expression levels of Myt1L and St18 stayed relatively unchanged (Figure 6I).
SYT4 regulates insulin secretion in a human β cell line
SYT4 is expressed in human β cells, and like in the mouse it is produced at higher levels in adult when compared to fetal cells [i.e. ~3-fold (Blodgett et al., 2015)]. With the objective of investigating whether SYT4 regulates Ca2+-secretion coupling in human β cells, we tested the effect of SYT4 overexpression and knockdown in human EndoC-βH1 cells. While lentiviral vector-mediated overexpression of SYT4 (~7-fold) had little impact on many genes linked β-cell function (e.g. GCK, GLUT1, INSULIN, MAFA, MAFB, NEUROD1, NKX6.1, PDX1, and SLC30A8 (Figure 7A)), there was a significant reduction in basal insulin secretion (P=0.04) (Figure 7B). Moreover, reducing SYT4 levels by ~80% also had no detectable effect on the expression of most these key β-cell genes, except a ~20% reduction in INS transcripts, in EndoC-βH1 cells (Figure 7C). Yet this knockdown significantly reduced GSIS (20 mM, P=0.002. Figure 7D). Interestingly, this reduction in Syt4 did not increase basal insulin secretion (Figure 7D), like in Syt4 mutant mouse islets. However, when we examined the ratio of insulin secretion at 20mM over 2.8 mM glucose, we observed a strong trend of reduced high glucose response (P=0.08) in SYT4 knockdown cells (Figure 7E). The implication is that at lowered levels of SYT4, the secretable insulin vesicles had a high probability of being released at low glucose to deplete the releasable insulin vesicles, a similar conclusion as drawn in the mouse β cells. We also overexpressed SYT4 in human embryonic stem cell-derived β cells. Lentivirus-based transduction can overexpress SYT4 in ~30% of pancreatic progenitor cells derived from human ES cells (Figure S7A). While not statistically significant, we observed a clear trend of SYT4 toward reduced basal insulin secretion (Figure S7B). These results suggest that the manner by which SYT4 controls insulin secretion is conserved between rodent and humans.
Figure 7. SYT4 regulates human β-cell GSIS.

Lentivirus was used to introduce human SYT4 overexpression, while siRNA were used for SYT4 knockdown. (A) Gene expression in EndoC-βH1 cells with SYT4OE (OE), normalized against empty virus-infected control (con) cells. (B) GSIS in EndoC-βH1 cells with SYT4OE. (C) Gene expression in EndoC-βH1 cells with SYT4KD (KD), normalized against cells treated with scrabbled siRNA (con). (D, E) Insulin secretion in EndoC-βH1 cells with SYT4KD, presented as the % of insulin secretion at different level of glucose (D), or as the ratio of insulin release at 20 mM over 2.8 mM glucose (E). *: P<0.05. (F) A model with two pathways that converge to regulate β-cell maturation: one aspect includes glucose metabolism/subsequent oxidative phosphorylation and membrane excitability, which ensure efficient glucose metabolism, ATP production, ionic activity, and Ca2+ entry. Another is the modulation of Ca2+ sensitivity of the vesicles. In this pathway, immature β cells have lower levels and mature β cells have higher levels of non-Ca2+ binding Syts. The Ca2+-binding Syts remain unchanged during maturation. The increased ratio between non-Ca2+ to Ca2+ binding Syts can desensitize the vesicles so that high secretion only occurs at high Ca2+ in the mature β cells. This desensitization can be achieved by 1) using Syt4 localized on vesicles to inhibit Syt7-SNARE interactions, 2) reducing Syt7 levels on vesicle surface, or both.
Discussion
Calcium is the primary signal that drives β-cell GSIS. Both the influx of Ca2+ and secretion-inducing capabilities of Ca2+ are optimized during β-cell maturation to allow for this physiological response. However, the mechanism(s) involved in adapting to the changes in Ca2+ influx and Ca2+ signaling during β-cell maturation have not been well-defined. A clearer understanding of how Ca2+ signaling changes during maturation has the potential to uncover postnatal β-cell mechanisms that control GSIS and thus glucose homeostasis (Jacobson and Tzanakakis, 2017). Our results strongly suggest that Syt4 is involved in controlling the Ca2+ sensitivity of insulin vesicle secretion during β-cell maturation and Syt4 control is conserved between rodent and human β-cells.
We showed that immature β cells have high insulin secretion despite similar levels of Ca2+ influx with mature β cells. This observation suggests that the insulin granule fusion machinery in immature β cells is more sensitive to Ca2+. This sensitivity was reduced during β-cell maturation. Increased production of Syt4 contributes to the regulation of this sensitivity (Figure 7F). Consistent with this model, Syt4 in neurons inhibits the ability of Syt1 to induce Ca2+-dependent membrane fusion. The mechanism involves Syt4 competing with Syt1 for Syt-SNARE interactions, resulting in non-Ca2+ responsive Syt4-SNARE complexes due to the inability of Syt4 to bind Ca2+ (Bhalla et al., 2008). The primary Ca2+ sensor in β cells is Syt7 (Dolai et al., 2016; Gustavsson et al., 2008; Wu et al., 2015). Interestingly, we found that Syt4 and Syt7 are both localized to the insulin vesicle and non-vesicular compartments and they physically interact. Thus, one possibility is that Syt4 interacts with and modulates the ability of Syt7 to bind with SNARE components at the plasma membrane, with the levels of Syt4 inversely regulating insulin secretion (Figure 7F, top). Alternatively, Syt4 molecules localized in the ER or Golgi could interact with Syt7, which reduces vesicular levels of Syt7 and their probability of release (Figure 7F, bottom). Future sensitive assays on Syt7 levels on mature vesicles could help to resolve these possibilities.
Syt4 control of insulin secretion is further tuned by the size of the RRP of insulin vesicles in β cells. Mature β cells have a limited number of these primed insulin vesicles (Rorsman and Renstrom, 2003). Our data extend this concept to immature β cells, suggesting that a limited number of releasable vesicles and higher basal insulin secretion depletes these vesicles and results in poor GSIS. Consequently, when Syt4 is low and insulin granules are being secreted at low Ca2+ concentration, the readily releasable insulin granules pool is predicted to become depleted, accounting for the poor GSIS in immature β cells. This concept is supported by the accumulation of the RRP of vesicles following treatment of newly matured islets with glucose-free media, which promotes DSIS. Moreover, loss of Syt4 results in an increase in the readily releasable insulin granules, suggesting that Syt4 control of insulin granule Ca2+ sensitivity modifies the quantity of insulin vesicles in RRP.
Interestingly, the insulin RRP decreases in Syt4 deficient post-weaning mice, when islet GSIS demand increases with high carbohydrate diet (Stolovich-Rain et al., 2015). Along a similar line, dysfunctional islets from type II diabetic patients expressed lower levels of SYT4 (Andersson et al., 2012). These findings further imply that Syt4, in conjunction with Syt7, may have specific postnatal function in regulating vesicle biosynthesis/transport/docking during β-cell maturation. Indeed, neuronal loss of Syt4 reduces synaptic vesicle transport to the plasma membrane (Arthur et al., 2010). It will be important to determine the unique postnatal function(s) of Syt4 in trafficking and the release of the insulin granule pool with temporally-controlled β-cell specific Syt4 loss of function models.
It is noteworthy that besides Syt4, several other non-Ca2+ binding Syts (e.g., Syt11, 13, and 14) have trends or statistically significant up-regulation during mouse β-cell maturation (Figure S2). Interestingly, these other Syts are also upregulated during human β-cell maturation (Blodgett et al., 2015). Thus, it is likely that these other Syts products can work together with Syt4 to regulate the Ca2+ sensitivity of insulin vesicles, in a redundant fashion. Future studies to inactivate each and/or all of these Syts would be interesting to test their specificity and redundancy in modulating Ca2+-secretion coupling in β cells.
While increasing Syt4 limited insulin secretion in neonatal islets, high levels of Syt4 also caused compromised postnatal islet development. This may indicate that factors secreted from immature β cells control normal islet morphogenesis, islet gene expression, and proliferation. Indeed, factors packaged inside insulin vesicles such as insulin, C-peptide, IGF2, IAPP, granins, and small molecules such as GABA and ATP (Suckale and Solimena, 2010) may have autocrine effects on postnatal islet development. Particularly relevant to this possibility is the finding that mTOR signaling can impact islet β-cell maturation and islet morphogenesis (Sinagoga et al., 2017). Interfering β-cell secretion with Syt4 overexpression could interrupt these signaling processes, including mTORC2, which impairs the islet morphogenesis (Sinagoga et al., 2017). Similar examples have been proposed in other cell types such as peptidergic nerve terminals, where changes in Syt4 levels tune neuropeptide secretion, which has been proposed to influence neuroendocrine transitions (Zhang et al., 2009). Another possibility is that intracellular Syt4 serves in regulatory capacities beyond secretion. For example, Syt4 is localized to the Golgi in many cells such as hippocampal neurons, where it plays an important role in regulating Golgi morphology in addition to synaptic vesicle formation (Arthur et al., 2010). As a substantial amount of β-cell Syt4 localizes to the ER, future studies are required to examine these Syt4 effects on islet development and function, for example, by regulating the general Ca2+ signaling in the ER of β cells.
The regulation of Syt4 expression remains largely unknown. Our findings suggest that Myt proteins repress Syt4 transcription in newborn β cells, which likely prevents the detrimental effects of precocious Syt4 activation and β-cell maturation. It is not clear, however, whether Myt factors directly repress Syt4. To this end, Myt1L was reported to bind the promoters of several Syt genes (Syt1, 2, 3, 7, and 12) and to mediate their repression in fibroblasts (Mall et al., 2017). It is likely that Myts in β cells use analogous mechanisms to repress Syt4 expression. Examining occupancy of the Syt4 promoter by Myts in β cells would answer this question. Neither do we know all the factors that up-regulate Syt4 in neonatal stages. In this regard, inactivation of MafA does not alter Syt4 expression (Artner et al., 2010). Neither does the weaning process (data not shown). Inactivation of nuclear receptor ERRγ reduces, but does not eliminate, Syt4 expression (Yoshihara et al., 2016). Interestingly, prolactin receptor-mediated signaling, required for perinatal β-cell development (Auffret et al., 2013), positively regulates Syt4 expression (Cunha et al., 2006). Moreover, different cell types appeared to use different signals to activate Syt4 expression. For example, diabetic conditions in human correlated with reduced Syt4 expression in islets (Andersson et al., 2012) but increased in adipose tissues (Rahimi et al., 2015). Thus, the further identification of unknown factors and mechanisms of Syt4 expression control is needed in order to fully understand stimulus-secretion coupling in β cells.
Although both mouse and human β-cell GSIS is modulated by SYT4, there are significant species-related differences in how β cells respond to Syt4 manipulation. Inactivating Syt4 in mouse β cells elevates basal insulin secretion; however, SYT4 knockdown in human cells causes a trend toward lower basal insulin secretion, although the GSIS in these SYT4 knockdown cells was even lower. It is possible that Syt4 functions differently in mouse and human cells due to the different expression profiles of different Syt paralogues in mouse and human cells. In this regard, several other SYTs with no/low Ca2+ affinity (SYT11, SYT13, SYT14, and SYT16) also displayed increased expression in maturing human β cells (Blodgett et al., 2015). They could cooperate with SYT4 to regulate Ca2+-secretion coupling or other aspects of insulin vesicle biogenesis, including insulin production, packaging, and transport. It is also possible that the fetal nature of EndoC-βH1 cells impinges on SYT4 impact on basal insulin secretion. This possibility is consistent with the high expression in EndoC-βH1 cells of many markers not found in mature β-cells such as HK1, HK2, and LDHA (Dhawan et. al., 2015). Finally, the species-related differences in GSIS could also be due to an in vivo knockout occurring before birth versus an acute knockdown in a cell line. Resolving this issue requires future studies to examine the roles of different Syts in fully-functional β cells from both species.
In summary, our studies reveal a previously unrecognized Syt4-mediated mechanism that regulates β-cell maturation in both rodents and humans. Manipulating the Ca2+ sensitivity of insulin vesicles through control of Syt4 expression, together with altering the metabolic profiles, will likely help the production of mature human β cells for transplantation-based diabetes therapy.
STAR Methods
Contact for Reagents and Resources Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the lead author, Dr. Guoqiang Gu (Guoqiang.gu@vanderbilt.edu).
Experimental models
Mice
Mouse usage conforms to protocols approved by the Vanderbilt University IACUC for Dr. Gu, in compliance with regulations of AAALAC. All mice were housed in a level 6 facility, in a particular room that has no detectable pathogens. Mice are housed in 51-squre inch ages, with have autoclaved hard-wood derived bedding and ad lib access to water and chew. For new born mice, one litter will be housed in each cage. For post weaning mice (genotyped around day 14–20), a maximum of 5 mice of same sex were housed. For husbandry, one male mouse was housed together with two females of breeding age until cross. Plugs were checked daily to identify plugged females, which were transferred to new cages for birth.
Wild-type CD1 (ICR) mice were from Charles River Laboratories. Flipe, RiprtTA and Syt4+/− mice were from the Jackson Laboratories. RipmCherry mice were derived in Vanderbilt Transgenic Mouse/ES Cell Shared Resource and reported in (Zhu et al., 2015). The TetOSyt4, Myt1LF, and St18F mice were derived in Vanderbilt as well.
Cell line
HEK293T. These cells were isolated from a female human embryonic kidney (HEK), transformed with large T antigen.
Method details
Mouse Derivation and phenotyping
TetOSyt4 mice were derived by pronuclear injection with a DNA construct made by ligating a TetO-CMV promoter (PTRE2, Clontech), the Syt4 coding sequence, and a SV40 polyA signal. The DNA construct was verified by sequencing. Three independent lines were derived, all without recognizable defects. For Syt4 over expression, doxycycline (2 mg/ml) was added to the drinking water of pregnant mice from E16.5 until P10 or to the time of tissue collection (if tissue collection was before P10). Both male and female mice were included for all studies, with similar numbers of both sexes.
Myt1LF and St18F derivation used routine targeting (Figure S6). Briefly, BAC clones RP22-191C21 and RP22-214C11 were used to isolate Myt1L and St18 genomic fragments via BAC recombineering (Warming et al., 2005). Targeting vectors were made so that the exons 12+13 in Myt1L (coding amino acid residues 491–566) and exons 9+10 in St18 (coding amino acid resides 355–429) were flanked with LoxP sites (Figure S6). Such designs introduced frame shifts in Myt1L and St18 mRNAs after Cre-mediated recombination. The homologous recombination arms were 7.2/2.6 kb for Myt1L and 3.1/8.2 kb for St18. Properly targeted TL1 (male) ES cell clones were verified by southern blots on both 5’ and 3’ probes. After chimera production and selection cassette removal with Flipe mice, PCR were used for genotyping (with DNA oligos listed in Table S2).
The functionality of the Myt1LF and St18F alleles were authenticated by producing Myt1LF/F, St18F/F, Myt1LF/F; Pdx1Cre, and St18F/F; Pdx1Cre mice. None of these mice have detectable phenotypes. For unknown reasons that might be related with nonsense-mediated degradation or protein instability, the Myt1LF/F; Pdx1Cre, and St18F/F; Pdx1Cre islets did not produce detectable N-terminal Myt1L or St18 protein fragments, which were expected to be translated from mRNAs transcribed from the Myt1L− and St18− alleles after Cre-mediated recombination. The newly derived Myt1L and St18 antibodies used in this study were raised using antigens localized to the N-terminal side of the deleted protein region (see information about Myt1L and St18 antibody derivation and Figure 6B).
Glucose and insulin tolerance test
For IPGTT, and ITT, mice were fasted overnight or 4 hours, respectively. Glucose or insulin was injected at 2mg/kg or 1 unit/kg via intraperitoneal injection, respectively. Blood glucose was then measured via tail snip at the time points indicated in the manuscript. For serum insulin assays, whole blood was collected via retro-orbital bleeding into EDTA-coated tubes. Plasma were then prepared by centrifugation and used for down stream assays.
Pancreatic islet isolation
Pancreata were directly digested (for P1 and P12 pancreata) (Huang and Gu, 2017) or perfused (for pancreata older than 2 weeks) (Zhao et al., 2010) with 0.5 mg/ml Type IV collagenase dissolved in Hanks Balanced Salt Solution (HBSS) with Ca2+/Mg2+. After digestion at 37 °C, lysates were washed in RPMI 1640 with 5.6 mM glucose and 10% fetal bovine serum (i.e. RPMIFBS) for 4 times. Islets were handpicked for down stream usage.
Cell purification, RNA-seq-, and Real-time PCR-based gene expression assays
Handpicked MipeGFP or Ripmcherry islets were washed in HBSS without Ca2+/Mg2+ for 5 minutes and dissociated into single cells with 0.25% trypsin at 37 °C as in (Gu et al., 2004). The dissociation usually lasted for less than 10 minutes. Cells were pipetted once every 2 minutes to aide dissociation. When ~half of the cells appeared as singles, the digestion was stopped by washing with RPMIFBS 3 times in 1.5 ml tubes. Cells were then filtered, stained with propidium iodide, and sorted using a BD FACSAria III cell sorter. Live/single/fluorescence+ cells were collected and verified under microscopes. MipeGFP mice express human growth hormone (hGH), which can impact the expression of some genes (Brouwers et al., 2014), while Ripmcherry mice have no hGH sequence.
For RNA-seq, RNA was prepared with TRIzol (Life Technologies) and a DNA free RNA™ kit following manufacturer’s protocols (Zymo Research). 200 ng total RNAs with RINs above 8 (assessed using Agilent Bioanalyzer 2100) were sequenced, with 3 biological replicas for each stage/genotype, following Illumina protocols on HiSeq-2000. Raw reads were processed and analyzed with TopHat and Cufflinks to determine the relative abundance of gene expression at each stages, reported as FPKM (Fragments Per Kilobase of transcript per Million mapped reads) (Trapnell et al., 2012). Real time RT-PCR utilized SYBR green master mix of the Bio-Rad system, with oligos listed in Table S2.
Ca2+ clamp and GSIS/DSIS assays in islets
Secretion was assayed with the % of total insulin secreted within a 45-minute time window, unless noted. Hand-picked islets were allowed to recover in RPMIFBS for ~2 hours or overnight. Islets were washed twice with pre-warmed KRB solution (2.8 mM glucose, 102 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 2.7 mM CaCl2, 20 mM HEPES, 5 mM NaHCO3, and 10 mg/ml BSA, pH 7.4). Islets were then incubated in KRB (37 °C) for one hour, washed with pre-warmed KRB once again. ~10 islet were then transferred into each wells of 12-well plates with 1 ml pre-warmed KRB to start the secretion assays. For ionomycin-mediated Ca2+ clamp, hand-picked islets were pre-incubated with 0.25 mM diazoxide for one hour. 50 µM ionomycin was then added to induce insulin secretion. For all assays, at least six mice of each genotype were used for islet isolation, with islets from two or more mice examined in each independent experiment (as three-four technical replicas).
Insulin was assayed with Elisa kit from Alpco following manufacturer protocol. For assaying insulin per cell, specific numbers of sorted cells were directly extracted with ethanol-HCl for total insulin quantification (Zhao et al., 2010).
Immunolabeling, β-cell mass assays, and electron microscopy
Immunoflorescence staining followed published procedures (Wang et al., 2010; Wang et al., 2007) with antibodies in the Key Resource Table. Briefly, tissue sections or cells attached on slides were washed in 1XPhosphate Buffered Saline (PBS) 3 time, 5 minutes each. The frozen sections were permeabilized in basal staining buffer (1XPBS+ 0.1% triton X100 + 0.1% Tween-20) for 30 minutes at room temperature (slides from paraffin embedded tissues did need permeabilization). Samples were blocked with blocking buffer (1XPBS+ 0.1% triton X100 + 0.1% Tween-20 + 0.1% BSA + 0.05% donkey serum, 100 micro-litter per slide) for another 30 minutes at room temperature. Primary antibodies (100 micro-litter per slide) diluted in blocking buffer were then added (see below for dilution) and incubate at 4 °C overnight. After three time wash in basal staining buffer, secondary antibodies diluted in blocking buffer were overlaid on slides (100 micro litter each slide), incubate for 1 hour at room temperature, washed three times with basal buffer, and mounted for observation. All primary and secondary antibodies except that against Syt4 (1:200 dilution) and Syt7 (1:500 dilution) used 1:1000 dilutions.
For hormones and transcription factor staining, frozen or paraffin tissue sections were both used. Tissue section preparations followed routine procedures: pancreata were dissected; large pancreata (older than P14) were cut into 5–10 small pieces and fixed in 4% paraformaldehyde overnight at 4 °C; fixed tissues were washed with PBS 3 times (10 minutes each) and prepared for frozen or paraffin blocks (following dehydration through PBS, 40%, 70%, 85%, 100% ethanol, and HistoClear; 15 and 6 µm thick sections were prepared for frozen and paraffin sections, respectively. For antibodies against Syt4, Syt7, GM130, and PDI, islet cells attached to glass coverslips were used. For Syt4 quantification, post-fixation (after freshly isolated islets were partially dissociated and attached onto slides with cytospin) was used, which appeared to produce better dynamic range of immunofluorescence signals. For subcellular localization, islets were isolated and partially dissociated into small cell clusters (~5–20 cells per cluster) with trypsin. Cells were then cultured on glass coverslips coated with human fibronectin in RPMIFBS. After 24 hours, samples were fixed in 4% paraformaldehyde for 15 minutes and processed for immunolabeling. Paraffin sections were de-waxed through HistoClear, 100%, 85%, 70%, 40% ethanol, and PBS solutions for antibody staining.
For co-labeling with antibodies raised in the same host (amongst PDI, Syt4, and Syt7), one round of labeling would be completed first with proper fluorophore-conjugated secondary antibodies. The samples were blocked with anti-IgG of the host, followed by routine labeling for the second antibody with another fluorophore.
Guinea-pig anti Myt1L and rat anti-St18 were raised against the amino acids 267–493 and 60–297 of each protein, respectively. Recombinant proteins were produced (as GST-fusion proteins) and purified and sent to Strategic Bio-Solutions (Newark, De) for antibody production in guinea pig and rat, respectively. Final bleeds were obtained 45 days after immunization.
For imaging, laser scanning microscopy (LSM) and structured illumination microscopy (SIM) were used. When expression levels of a protein between samples were compared, slides/cells were processed side-by-side, and images were captured utilizing identical optical/electronic settings. For protein level quantification in nuclei, LSM images taken under identical parameters were selected for Image J-based particle quantification.
Transmission electron microscopy (TEM) followed established procedure (Zhao et al., 2010). Briefly, handpicked islets were fixed in 2.5% glutaraldehyde in 0.1M cacodylate buffer (pH 7.4) overnight at room temperature. Islets were then washed and treated in 1% osmium tetroxide in 0.1M cacodylate buffer for 1 hour. Islets were then washed, embedded, thin-sectioned for imaging.
Proximity ligation assays (PLA), co-immunoprecipitation, and western blotting
For PLA, islets from adult control and Syt4−/−mice were isolated and dissociated into small clusters with trypsin (~5–20 cells/cluster). Cytospin were used to attach cells onto slides, followed by fixation with 4% paraformaldehyde for 15 minutes at room temperature. PLA strictly followed manufacturer’s instruction. For immunoprecipitation, HEK293T cells were co-transfected with plasmids expressing the cytoplasmic C2 domains of Syt4 (tagged by HA) and Syt7. 48 hours after transfection, cells were lysed by co-IP Buffer (150 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 1% NP-40, 0.5% sodium deoxycholate, protease inhibitor cocktails). Cell lysates were mixed with anti-HA magnetic beads (at 4°C for 4 h. The beads were washed with TBST (20 mM Tris-HCl pH7.6, 150 mM NaCl, 0.1% Tween-20) five times, and bound proteins were analyzed by Western blotting.
Free cytoplasmic Ca2+ recording
Ca2+ recording and analysis followed published methods (Jacobson et al., 2010). Briefly, islets were cultured on ploy-lysine coated glass for 48 hours in RPMI medium containing 10% FBS and 11 mM glucose. Islets were loaded with 2 mM FURA2AM (Invitrogen) for 25 minutes in culture medium followed by 20 min starvation in REC buffer (119 mM NaCl, 2.5 mM CaCl2, 4.7 mM KCl, 10 mM Hepes, 1.2 mM MgSO4 and 1.2 mM KH2PO4) with 2mM glucose. Islets were then transferred to REC buffer with 2.8 mM glucose for three minutes for recording before stimulation. Buffer changes were achieved by perfusion at 2 ml/min. Stimulating glucose and KCl were 20 and 25 mM, respectively. Imaging was performed using a Nikon Eclipse TE2000-U microscope. The ratios of emitted fluorescence intensities at excitation wavelengths of 340 and 380 nm (F340/F380) were determined every 5 second. The total Ca2+ was calculated using a fura-2 calibration kit (Grynkiewicz et al., 1985).
Gene expression and GSIS assays in human EndoC-βH1 cells
Human EndoC-βH1 cells were grown in DMEM containing 5.6 mM glucose, 2% BSA, 50 µM 2-mercaptoethanol, 10 mM nicotinamide, 5.5 µg/mL transferrin, 6.7 ng/mL selenite, 100 units/mL penicillin, and 100 units/mL streptomycin (Ravassard et al., 2011). For gene overexpression, cells were infected with lentiviral particles (carrying a human SYT4 full coding region and eGFP reporter) one day after plating. For siRNA transfection (with a mix of three individual siRNAs), dissociated cells were incubated with siRNA (at 100 nana-molar concentration, with RNAiMax) for 5 minutes before plating. Gene expression assays were performed three days after infection or transfection. For GSIS, cells were incubated with 1mM glucose overnight, briefly washed three times using KRB with 2.8 mM glucose, before insulin secretion assays as described above. The cells were equilibrated in KRB with 2.8 mM glucose for 1 hour before GSIS assays.
Quantification and Statistical analysis
All experiments contained at least two biological replicas and two technical replicas. For quantifications of β cell mass and replication index throughout the pancreas, the frozen pancreatic block was sectioned at 20 µm intervals. One third of all sections were labeled and scanned using Aperio Scanscope. β-cell mass was then calculated based on the total pancreas weight and the percentage of tissues area that labeled for insulin. For vesicle quantification, images from different sections were used. 30–50 images from different β cells were counted, under double-blind settings (image capture and vesicle counting were done by different personnel).
Statistical analyses utilized standard student T test for pairwise comparisons or One-way ANOVA for comparing multiple groups of data points. A P-value of 0.05 or lower was considered significant. Quantification data were presented as (mean ± SEM). For all assays the “n” noted in all figures and manuscript referred to the number of independent experiments, including both biological replicas and technical replicas.
Data availability
The original RNA-seq data from P1, P12, and P60 β cells were deposited in ArrayExpress under ID codes E-MTAB-2266 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2266/) and E-MTAB-6615 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6615/).
Supplementary Material
Highlights.
Immature β-cells are more sensitive to Ca2+ stimulation during GSIS
Increased Syt4 production promotes β-cell maturation by reducing Ca2+ sensitivity
Myt transcription factors repress the expression of Syt4 in β cells
Syt4, interacting with Syt7, regulates GSIS in mouse and human β cells
Acknowledgments
This study is supported by grants from NIDDK (DK065949 for GG, DK089523 for MM, DK097392 for DAJ, DK105831 and DK108666 for MH), JDRF (1-2009-371 for GG). Confocal and TEM imaging were performed with VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
G.G., M.G., D.A.J., M.A.M. and C.S.Jr. conceptualized the work and designed the experiments. C.H., G.G, R.H., Y. X. and W.Z. did gene expression and secretion assays in mice. Y.X. and S.W. derived the conditional Myt gene alleles and examined mutant phenotypes. D.K.P. recorded Ca2+ responses in islets. J.L., G.G.N., and M.H. performed and discussed Syt4 function in human ES-cell derived β-cells. E.W. and R.S. did analysis in human β-cell lines. J.W. did TEM imaging. J.M. analyzed the TEM images and helped with islet isolation. T.S. did bioinformatics data processing and deposition. W.H. and T.H.Y.A. provided Syt7−/− pancreata. All authors participated the manuscript preparation and editing.
Declaration of interest
The authors declare no competing interests.
References
- Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, Weir GC, Sharma A, Larsen PR, Bonner-Weir S. Thyroid hormone promotes postnatal rat pancreatic beta-cell development and glucose-responsive insulin secretion through MAFA. Diabetes. 2013;62:1569–1580. doi: 10.2337/db12-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammala C, Ashcroft FM, Rorsman P. Calcium-independent potentiation of insulin release by cyclic AMP in single beta-cells. Nature. 1993;363:356–358. doi: 10.1038/363356a0. [DOI] [PubMed] [Google Scholar]
- Andersson SA, Olsson AH, Esguerra JL, Heimann E, Ladenvall C, Edlund A, Salehi A, Taneera J, Degerman E, Groop L, et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Molecular and cellular endocrinology. 2012;364:36–45. doi: 10.1016/j.mce.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Arthur CP, Dean C, Pagratis M, Chapman ER, Stowell MH. Loss of synaptotagmin IV results in a reduction in synaptic vesicles and a distortion of the Golgi structure in cultured hippocampal neurons. Neuroscience. 2010;167:135–142. doi: 10.1016/j.neuroscience.2010.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59:2530–2539. doi: 10.2337/db10-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffret J, Freemark M, Carre N, Mathieu Y, Tourrel-Cuzin C, Lombes M, Movassat J, Binart N. Defective prolactin signaling impairs pancreatic beta-cell development during the perinatal period. Am J Physiol Endocrinol Metab. 2013;305:E1309–1318. doi: 10.1152/ajpendo.00636.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton F, Cornet V, Iborra C, Garrido J, Dargent B, Fukuda M, Seagar M, Marqueze B. Synaptotagmin I and IV define distinct populations of neuronal transport vesicles. The European journal of neuroscience. 2000;12:1294–1302. doi: 10.1046/j.1460-9568.2000.00013.x. [DOI] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, Chapman ER. Analysis of the synaptotagmin family during reconstituted membrane fusion. Uncovering a class of inhibitory isoforms. The Journal of biological chemistry. 2008;283:21799–21807. doi: 10.1074/jbc.M709628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, Kim S, Kucukural A, Davis RJ, Kent SC, et al. Novel Observations From Next-Generation RNA Sequencing of Highly Purified Human Adult and Fetal Islet Cell Subsets. Diabetes. 2015;64:3172–3181. doi: 10.2337/db15-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nature biotechnology. 2012;30:261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers B, de Faudeur G, Osipovich AB, Goyvaerts L, Lemaire K, Boesmans L, Cauwelier EJ, Granvik M, Pruniau VP, Van Lommel L, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 2014;20:979–990. doi: 10.1016/j.cmet.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton M. Synaptotagmin gene content of the sequenced genomes. BMC Genomics. 2004;5:43. doi: 10.1186/1471-2164-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha DA, Amaral ME, Carvalho CP, Collares-Buzato CB, Carneiro EM, Boschero AC. Increased expression of SNARE proteins and synaptotagmin IV in islets from pregnant rats and in vitro prolactin-treated neonatal islets. Biological research. 2006;39:555–566. doi: 10.4067/s0716-97602006000300016. [DOI] [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nature neuroscience. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. DNA methylation directs functional maturation of pancreatic beta cells. J Clin Invest. 2015;125:2851–2860. doi: 10.1172/JCI79956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do OH, Low JT, Gaisano HY, Thorn P. The secretory deficit in islets from db/db mice is mainly due to a loss of responding beta cells. Diabetologia. 2014;57:1400–1409. doi: 10.1007/s00125-014-3226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolai S, Xie L, Zhu D, Liang T, Qin T, Xie H, Kang Y, Chapman ER, Gaisano HY. Synaptotagmin-7 Functions to Replenish Insulin Granules for Exocytosis in Human Islet beta-Cells. Diabetes. 2016;65:1962–1976. doi: 10.2337/db15-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Ogata Y, Saegusa C, Kim T, Loh YP, Yamamoto A. Nerve growth factor-dependent sorting of synaptotagmin IV protein to mature dense-core vesicles that undergo calcium-dependent exocytosis in PC12 cells. The Journal of biological chemistry. 2003;278:3220–3226. doi: 10.1074/jbc.M208323200. [DOI] [PubMed] [Google Scholar]
- Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Developmental cell. 2012;23:21–34. doi: 10.1016/j.devcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso S, Messina A, Saporito N, Reitano G. Serum-insulin response to glucose and aminoacids in the premature infant. Lancet. 1968;2:755–756. doi: 10.1016/s0140-6736(68)90954-9. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell metabolism. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B, Melton DA. Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development. 2004;131:165–179. doi: 10.1242/dev.00921. [DOI] [PubMed] [Google Scholar]
- Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ, Li C, Radda GK, Sudhof TC, Han W. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3992–3997. doi: 10.1073/pnas.0711700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang Y, Stein R. MafA and MafB activity in pancreatic beta cells. Trends Endocrinol Metab. 2011;22:364–373. doi: 10.1016/j.tem.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Gu G. Effective Isolation of Functional Islets from Neonatal Mouse Pancreas. J Vis Exp. 2017 doi: 10.3791/55160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. The Journal of physiology. 2010;588:3525–3537. doi: 10.1113/jphysiol.2010.190207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson EF, Tzanakakis ES. Human pluripotent stem cell differentiation to functional pancreatic cells for diabetes therapies: Innovations, challenges and future directions. J Biol Eng. 2017;11:21. doi: 10.1186/s13036-017-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, Regazzi R. Postnatal beta-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat Commun. 2015;6:8084. doi: 10.1038/ncomms9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, Weir GC, Sgroi D, Bonner-Weir S. Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia. 2011;54:594–604. doi: 10.1007/s00125-010-2036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Franz C, Kuhn S, Furness DN, Ruttiger L, Munkner S, Rivolta MN, Seward EP, Herschman HR, Engel J, et al. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nature neuroscience. 2010;13:45–52. doi: 10.1038/nn.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalwat MA, Cobb MH. Mechanisms of the amplifying pathway of insulin secretion in the beta cell. Pharmacol Ther. 2017;179:17–30. doi: 10.1016/j.pharmthera.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer TJ. Closing in on Mass Production of Mature Human Beta Cells. Cell Stem Cell. 2016;18:699–702. doi: 10.1016/j.stem.2016.05.014. [DOI] [PubMed] [Google Scholar]
- Lemaire K, Thorrez L, Schuit F. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu Rev Nutr. 2016;36:45–71. doi: 10.1146/annurev-nutr-071715-050808. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- Liu JS, Hebrok M. All mixed up: defining roles for beta-cell subtypes in mature islets. Genes Dev. 2017;31:228–240. doi: 10.1101/gad.294389.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado HB, Liu W, Vician LJ, Herschman HR. Synaptotagmin IV overexpression inhibits depolarization-induced exocytosis in PC12 cells. Journal of neuroscience research. 2004;76:334–341. doi: 10.1002/jnr.20072. [DOI] [PubMed] [Google Scholar]
- Mall M, Kareta MS, Chanda S, Ahlenius H, Perotti N, Zhou B, Grieder SD, Ge X, Drake S, Euong Ang C, et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature. 2017;544:245–249. doi: 10.1038/nature21722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore-Dotson JM, Papke JB, Harkins AB. Upregulation of synaptotagmin IV inhibits transmitter release in PC12 cells with targeted synaptotagmin I knockdown. BMC neuroscience. 2010;11:104. doi: 10.1186/1471-2202-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, Sharma A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Developmental biology. 2006;293:526–539. doi: 10.1016/j.ydbio.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterberg JR, Chon NL, Boo A, Maynard FA, Lin H, Knight JD. Membrane Docking of the Synaptotagmin 7 C2A Domain: Electron Paramagnetic Resonance Measurements Show Contributions from Two Membrane Binding Loops. Biochemistry. 2015;54:5684–5695. doi: 10.1021/acs.biochem.5b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pildes RS, Hart RJ, Warrner R, Cornblath M. Plasma insulin response during oral glucose tolerance tests in newborns of normal and gestational diabetic mothers. Pediatrics. 1969;44:76–83. [PubMed] [Google Scholar]
- Rahimi M, Vinciguerra M, Daghighi M, Ozcan B, Akbarkhanzadeh V, Sheedfar F, Amini M, Mazza T, Pazienza V, Motazacker MM, et al. Age-related obesity and type 2 diabetes dysregulate neuronal associated genes and proteins in humans. Oncotarget. 2015;6:29818–29832. doi: 10.18632/oncotarget.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, Scharfmann R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Arkhammar P, Bokvist K, Hellerstrom C, Nilsson T, Welsh M, Welsh N, Berggren PO. Failure of glucose to elicit a normal secretory response in fetal pancreatic beta cells results from glucose insensitivity of the ATP-regulated K+ channels. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:4505–4509. doi: 10.1073/pnas.86.12.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang SB, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Annals of the New York Academy of Sciences. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- Scarlett JM, Schwartz MW. Gut-brain mechanisms controlling glucose homeostasis. F1000Prime Rep. 2015;7:12. doi: 10.12703/P7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinagoga KL, Stone WJ, Schiesser JV, Schweitzer JI, Sampson L, Zheng Y, Wells JM. Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development. 2017;144:2402–2414. doi: 10.1242/dev.146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, Dor Y. Weaning triggers a maturation step of pancreatic beta cells. Dev Cell. 2015;32:535–545. doi: 10.1016/j.devcel.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Suckale J, Solimena M. The insulin secretory granule as a signaling hub. Trends in endocrinology and metabolism: TEM. 2010;21:599–609. doi: 10.1016/j.tem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Ferguson GD, Herschman HR, Elferink LA. Functional and biochemical analysis of the C2 domains of synaptotagmin IV. Mol Biol Cell. 1999;10:2285–2295. doi: 10.1091/mbc.10.7.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Donaldson CJ, Caceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, Huising MO. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat Med. 2015;21:769–776. doi: 10.1038/nm.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Yan J, Anderson DA, Xu Y, Kanal MC, Cao Z, Wright CV, Gu G. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Developmental biology. 2010;339:26–37. doi: 10.1016/j.ydbio.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang J, Zhao A, Hipkens S, Magnuson MA, Gu G. Loss of Myt1 function partially compromises endocrine islet cell differentiation and pancreatic physiological function in the mouse. Mechanisms of development. 2007;124:898–910. doi: 10.1016/j.mod.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Wei S, Petersen N, Ali Y, Wang X, Bacaj T, Rorsman P, Hong W, Sudhof TC, Han W. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from beta-cells. Proc Natl Acad Sci U S A. 2015;112:9996–10001. doi: 10.1073/pnas.1513004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, Tseng T, Dai Y, Yu RT, Liddle C, et al. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell Metab. 2016;23:622–634. doi: 10.1016/j.cmet.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Bhalla A, Dean C, Chapman ER, Jackson MB. Synaptotagmin IV: a multifunctional regulator of peptidergic nerve terminals. Nature neuroscience. 2009;12:163–171. doi: 10.1038/nn.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A, Ohara-Imaizumi M, Brissova M, Benninger RK, Xu Y, Hao Y, Abramowitz J, Boulay G, Powers AC, Piston D, et al. Galphao represses insulin secretion by reducing vesicular docking in pancreatic beta-cells. Diabetes. 2010;59:2522–2529. doi: 10.2337/db09-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hu R, Brissova M, Stein RW, Powers AC, Gu G, Kaverina I. Microtubules Negatively Regulate Insulin Secretion in Pancreatic beta Cells. Dev Cell. 2015;34:656–668. doi: 10.1016/j.devcel.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original RNA-seq data from P1, P12, and P60 β cells were deposited in ArrayExpress under ID codes E-MTAB-2266 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2266/) and E-MTAB-6615 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6615/).