

Abstract
New therapeutic options are needed for treatment of human African trypanosomiasis (HAT) caused by protozoan parasite Trypanosoma brucei. S-Adenosylmethionine decarboxylase (AdoMetDC) is an essential enzyme in the polyamine pathway of T. brucei. Previous attempts to target this enzyme were thwarted by the lack of brain penetration of the most advanced series. Herein, we describe a T. brucei AdoMetDC inhibitor series based on a pyrimidineamine pharmacophore that we identified by target-based high-throughput screening. The pyrimidineamines showed selectivity for T. brucei AdoMetDC over the human enzyme, inhibited parasite growth in whole-cell assay, and had good predicted blood–brain barrier penetration. The medicinal chemistry program elucidated structure–activity relationships within the series. Features of the series that were required for binding were revealed by determining the X-ray crystal structure of TbAdoMetDC bound to one analog. The pyrimidineamine series provides a novel starting point for an anti-HAT lead optimization.
Keywords: human African trypanosomiasis, Trypanosoma brucei, AdoMetDC, pyrimidineamine, co-crystallization
TOC image
Introduction
Human African trypanosomiasis (HAT), also known as sleeping sickness, is caused by the single celled protozoal parasite Trypanosoma brucei and is classified by the WHO as a Neglected Tropical Disease (NTD).1 HAT is transmitted by the tsetse fly to humans and animals where it replicates extracellularly in the blood and lymph causing fever and influenza-like symptoms. Later in infection, the parasite crosses the blood–brain barrier (BBB) and invades the central nervous system (CNS) leading to a variety of neurological symptoms, including disruption of the sleep–wake cycle, coma, and eventual death in most patients.2 The number of reported cases has declined steadily since peaking in 1998 at nearly 40,000 cases a year.3, 4 Control and surveillance programs, which include both vector control and identification and treatment of infected individuals, have been credited with this decline.3 The WHO has targeted HAT for sustainable elimination by 2030;5 however, the recent discovery that some infected individuals are asymptomatic carriers threatens the elimination program.6 Additionally, it is now recognized that skin and fat serves as a significant reservoir for the parasites and thus screening of blood underestimates the magnitude of the parasite burden in the endemic communities.7–9
Currently approved HAT treatments have significant liabilities hindering efforts to control the disease.3 Particularly problematic are treatments for the CNS stage of the disease. Nifurtimox–eflornithine combination therapy (NECT) has replaced the poorly tolerated melarsoprol as a front-line therapy against Gambian form of the infection.10 However, both the high cost of this therapy 10 and its limited efficacy against Rhodesian form of HAT has led to continued use of melarsoprol.11 Thus, it is recognized that a safer and less expensive therapy is needed that would cure both early- and CNS-stage infections caused by either T. brucei subspecies, eliminating the need to stage the disease. There are currently only two drug candidates in clinical development: oxaborole SCYX-715812 and nitroheterocyclic fexinidazole.13 Both have the potential to meet the outlined treatment goals but it is too early to know if either will make it to registration.
Of the approved drugs only eflornithine has a well understood mechanism of action. It targets ornithine decarboxylase, a key enzyme in polyamine biosynthesis, suggesting that other enzymes in the pathway would also provide potential targets for drug discovery.14, 15 A second key enzyme in the pathway, S-adenosylmethionine decarboxylase (AdoMetDC), has been shown to be essential by genetic studies,16 and small-molecule inhibitors of AdoMetDC have been reported that show in vivo activity against T. brucei in mouse models.14, 17–19 However, T. brucei AdoMetDC inhibitors, such as CGP 40215, mechanism-based MDL 73811, and its derivative Genz-644131 (1), were deemed unsuitable for anti-HAT development as their physicochemical properties were not consistent with good blood–brain barrier (BBB) penetration and thus they were not effective for CNS stage of the disease.14, 17, 18 AdoMetDCs from trypanosomatids have a novel subunit configuration that differentiates them from the human enzyme: they are allosterically activated by heterodimerization with an inactive paralogous pseudoenzyme, we termed prozyme.20 Structural analysis by X-ray crystallography demonstrated that heterodimerization leads to displacement of an autoinhibitory sequence and to a coupled structural reorganization that stabilizes the active conformation through insertion of the N-terminus into the heterodimer interface.21 These structural differences suggested that selective inhibition of the parasite enzyme over the host enzyme was plausible.
We recently described mass spectrometry-based high-throughput screening (HTS) campaign that identified 13 classes of novel small-molecule inhibitors of T. brucei AdoMetDC that also inhibited parasite growth.22 Several of the identified series demonstrated high propensity to cross the BBB and were at least partially on-target in the parasite cells.22 Herein, we characterize a pyrimidineamine chemotype that was identified as a hit in the HTS. Initially, the hit failed to demonstrate substantial AdoMetDC inhibition in validation studies and thus was not previously reported. We later discovered that the potency of the initial hit compound was pH-dependent, triggering re-prioritization of the pyrimidineamines for a medicinal chemistry effort to establish the structure–activity relationships (SAR) within the series. A number of pyrimidineamine analogs with low-micromolar activity on both the enzyme and the parasites were identified. Compounds in the series also showed good selectivity versus the human enzyme and were predicted to cross the blood–brain barrier. Finally, a crystal structure of TbAdoMetDC bound to one analog was determined, providing insight into the structural basis for the pH dependence of inhibitor binding to T. brucei AdoMetDC, and paving the way for future lead development. This work has led to the validation of the pyrimidineamines as a hit series that could be taken forward into a hit-to-lead optimization program for the treatment of HAT.
Results
Synthesis
Compound 7 was purchased from Maybridge (part of Thermo Fisher Scientific), and compounds 56-59, 74-88, and 90 were purchased from ChemBridge (San Diego, CA, USA). The synthesis of all other analogs is shown in Scheme 1.
Scheme 1. Synthesis of pyrimidineaminesa.
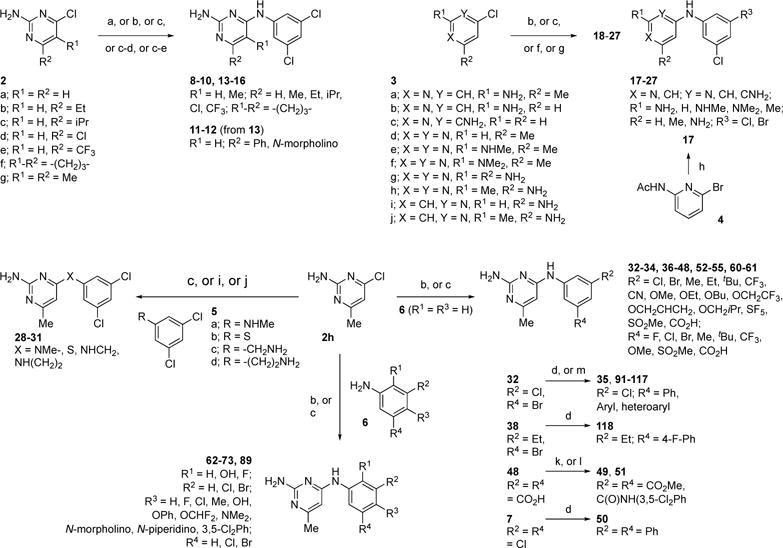
a Reagents and conditions: (a) 2a, 3,5-Cl2PhNH2, DMSO, 100 °C, 18 h (8); (b) 2a-c,f-h or 3g, aniline (3,5-Cl2PhNH2 or 3-Br-5-ClPhNH2 or 6), conc. HCl, EtOH, microwave (140 °C), 1.5 h (9, 10, 15, 16, 26, 32-34, 36, 37, 44, 45, 53, 54, 61-68, 72, 73, 89); (c) 2a,d,e,h or 3d-f,h, aniline (3,5-Cl2PhNH2 or 3-Br-5-ClPhNH2 or 5a or 6) conc. HCl, EtOH, reflux, 18 h (13, 14, 23-25, 27, 28, 38-43, 46-48, 52, 55, 60, 69-71); (d) 7 or 13 or 32 or 38, ArylB(OH)2, Pd(OAc)2, PPh3, Na2CO3, H2O/DME, 90 °C, 18 h (11, 35, 50, 91-109, 116-118); (e) 13, DIPEA, morpholine, CH3CN, microwave (140 °C), 2 h (12); (f) 3,5-Cl2PhNH2, 3a,c,i,j, conc. HCl, BuOH, microwave (185 °C), 4 h (18-20, 22); (g) 3,5-Cl2PhNH2, 3b, conc. HCl, MeOH/H2O (1/1), sealed tube, 100 °C, 18 h (21); (h) 4, 3,5-Cl2PhNH2, cat. Pd2(dba)3, BINAP, NaOtBu, toluene, 70 °C, 18 h; then 3N aq. NaOH, EtOH, reflux, 18 h (17); (i) 2h, 5b, Et3N, DMF, rt, 24 h (29); (j) 2a, 5c,d, DIPEA, CH3CN, microwave (140 °C), 2 h (30, 31); (k) 48, conc. H2SO4, MeOH, 4Å mol. sieves, reflux, 18 h (49); (l) 48, 3,5-Cl2PhNH2, EDI, py/DCM/DMF (0.4/1/1), rt, 18 h (51); (m) 32, heteroaryl boronic acid MIDA-ester, cat. Pd(OAc)2, PPh3, sodium triphosphate, dioxane/H2O) (4/1), 90 °C, 18 h (110-115).
Compounds 9-10, 13-16, 18-28, 32-34, 36-48, 52-55, 60-73, and 89 were readily available via reaction of the appropriate chloropyrimidine (2a-h, 3d-h) or chloropyridine (3a-c, 3i-j) with a variety of substituted anilines in the presence of acidic ethanol or butanol under conventional23 or microwave heating.24 Compound 8 was obtained similarly but under neutral conditions via heating of chloropyrimidine 2a with 3,5-Cl2PhNH2 in DMSO at 100 °C, whereas amine-linker analogs 30-31 resulted from microwave heating of chloropyrimidine 2h with amines 5c-d in acetonitrile in the presence of iPr2NEt.25 Stirring thiol 5b with chloropyrimidine 2h and triethylamine in DMF at room temperature yielded thioether-linked analog 29.26 Analog 12 was obtained from chloride-displacement in 13 with morpholine in the presence of iPr2NEt under microwave heating in acetonitrile.27 The bis-ester and amide analogs 49 and 50 resulted from Fisher esterification and EDI-mediated amide coupling of 48, respectively. Pyridine analog 17 was obtained via palladium-catalyzed amination of pyridinylbromide 4 with 3,5-Cl2PhNH2 followed by base-mediated hydrolysis of the acetamide.28 Finally, all biaryl-containing compounds (11, 35, 50, 91-118) were prepared via Suzuki cross-coupling of the corresponding arylchloride (7, 13) or arylbromide (32, 38) analogs with arylboronic acids29 or the corresponding MIDA boronates.30
Discovery of pyrimidineamines as T. brucei AdoMetDC inhibitors
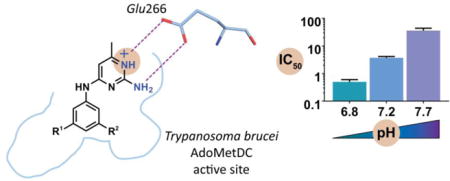
The pyrimidineamine 7 was identified as a singleton in our previously reported HTS campaign to identify inhibitors of TbAdoMetDC (Figure 1A).22 Compound 7 did not comprise substructural motifs characteristic of pan-assay interference compounds (PAINS),31 according to our analysis performed as described in ref 22. The compound was repurchased and tested for inhibition of T. brucei cell growth in vitro demonstrating a half-maximal effective growth-inhibitory concentration (EC50) of 5.6 μM after 48 h treatment. However, retesting of the TbAdoMetDC enzyme activity yielded a half-maximal inhibitory concentration (IC50) of 30 μM (assay pH 7.7). Thus, the compound was not presented in the original HTS publication, where the IC50 threshold for an active hit was set at 25 μM.
Figure 1.
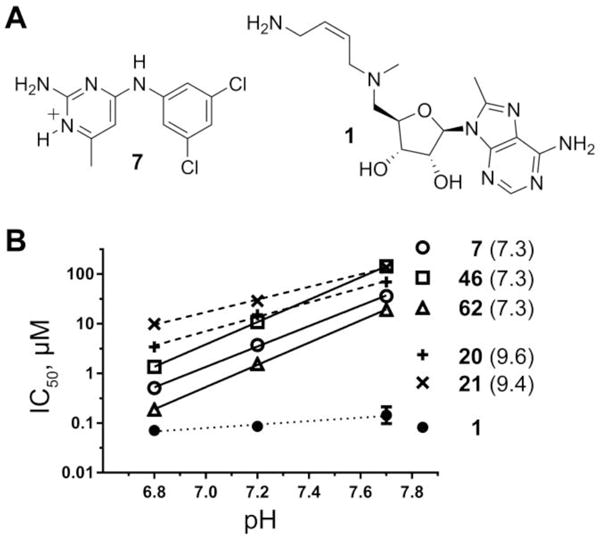
Pyrimidineamines are pH-dependent TbAdoMetDC inhibitors. (A) Structure of the protonated form of pyrimidineamine 7 (N1 is predicted to have pKa of 7.3), and structure of mechanism-based inhibitor Genz-644131 (1). (B) IC50 of pyrimidineamines plotted as a function of the assay buffer pH and fitted with linear regression analysis in Prism (GraphPad). Predicted pKa(N1) values are shown in parentheses. The slopes for pyrimidineamines with pKa(N1) = 7.3 (solid lines) are compared with those for analogs with pKa(N1) > 9 (dashed lines), and with that for a non-pyrimidineamine control (dotted line).
Later, characterization of the pH dependence of the TbAdoMetDC inhibition profile showed that there was a 60-fold increase in inhibitory activity of 7 at pH 6.8 compared with pH 7.7 (Figure 1B; IC50 = 30, 3.7, and 0.50 μM at pH 7.7, 7.2, and 6.8, respectively). The pKa value of the conjugated acid of the aromatic nitrogen in position 1 (further denoted as pKa(N1)) was estimated using MarvinSketch (version 16.3.28.0, ChemAxon, Budapest, Hungary) to be 7.3 (Figure 1A). Thus, we hypothesized that protonation of N1 in 7 was important for binding to the T. brucei AdoMetDC. Pyrimidineamine analogs (described below) with predicted pKa(N1) of 7.3 had similar slopes of IC50–pH dependence, while analogs with pKa(N1)) > 9 had much shallower slopes reflecting more comparable percentages of protonated species among the three pH points (Figure 2B). IC50 values for the structurally unrelated 1 tested as a control were not appreciably pH-dependent, as only a 2-fold difference was observed between potencies at pH 6.8 and pH 7.7 (Figure 2B).
Figure 2.
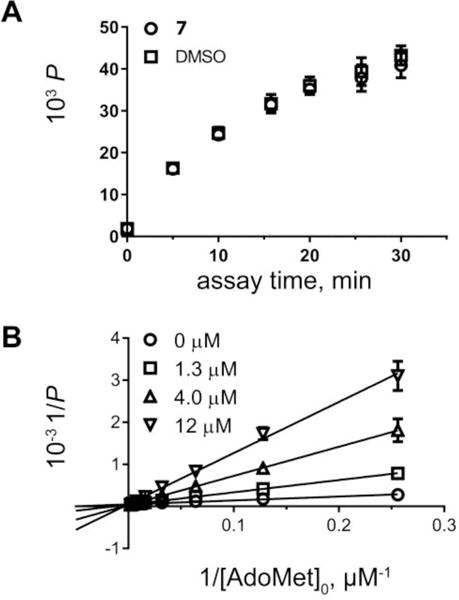
Mechanism of inhibition studies for 7. (A) Evaluation of reversibility. Recovery of enzyme activity after a 30 min incubation with inhibitor followed by a rapid 100-fold dilution of the inhibitor. The progression of the reaction was measured as product formation using RapidFire–MS analysis. (B) Determination of the inhibition modality. Double-reciprocal Lineweaver-Burk plot of substrate titrations in the presence of four inhibitor concentrations shows competitive inhibition.
Encouragingly, compound 7 was predicted to have a high likelihood for the ability to cross the BBB as assessed using an MDCKII-hMDR1 monolayer cell assay in the presence or absence of P-glycoprotein 1 (Pgp) inhibitor N-(4-(2-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)phenyl)-5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxamide (GF120918). The assay was performed as previously described.22 The apparent permeability of 7 was 135 nm/s ± inhibitor, suggesting that 7 is not a substrate of Pgp-mediated efflux. This permeability rate is close to an ideal threshold value of 150 nm/s cited as an indicator of a good likelihood that the compound will cross into the CNS.32
Mechanism of inhibition studies
Reversibility of inhibition by 7 was tested in a rapid dilution assay.33 TbAdoMetDC was equilibrated with 7 (36 μM ~ IC90 at pH 7.2) for 30 min and then diluted 100-fold to a concentration that was no longer inhibitory (<IC10) into reaction buffer containing ~Km concertation (50 μM) of substrate, S-adenosyl-L-methionine (AdoMet), to initiate the reaction. Aliquots were taken out at 5 min intervals, enzyme was quenched, and produced decarboxylated AdoMet (dcAdoMet) was quantified using RapidFire–mass spectrometry (MS) (Figure 2A). The data were compared with those obtained for the vehicle control. Enzyme activity was fully recovered after diluting out the inhibitor following the 30 min treatment, which is consistent with rapidly reversible inhibition.
To determine the modality of the reversible inhibition, we followed dcAdoMet produced at varied concentrations of AdoMet and 7. The double-reciprocal Lineweaver–Burk plot of the substrate titrations at four inhibitor concentrations is consistent with the competitive inhibition (Ki = 2.6 (2.2–3.0) μM; 95% confidence interval in parentheses) (Figure 2B).
Hit expansion and development of structure–activity relationships (SAR)
On the basis of a tractable chemotype and anticipated BBB permeability, 7 was prioritized for a hit-to-lead expansion. The atomic structure of TbAdoMetDC was not available at the start of this project. Thus, the medicinal chemistry effort to elucidate the SAR relied on iterative structure alterations informed by enzyme and T. brucei cell-based assays. AdoMetDC inhibitory activity was determined at three pH values (6.8, 7.2, and 7.7) to assess the importance of N1 protonation for binding; only pH 7.2 data are reported in the main text figures and tables. Data collected at the alternative pH values are provided in Supplemental Information (Table S1). Additionally, the inhibition of HsAdoMetDC at pH 7.2 was measured to assess parasite–host enzyme selectivity.
SAR studies: pyrimidine ring substituents
We began the SAR studies by modifying the pyrimidine ring (Table 1). Replacement of the methyl-substituent at C6 with ethyl (9) did not disrupt activity in the enzyme or cell-based assays, while increasing bulk at C6 by addition of isopropyl (10) led to a ~3-fold increase in the IC50 for AdoMetDC inhibition, as did C5–C6 cyclization (15) or removal of the C6-Me (8). Introduction of aromatic (11), heterocyclic (12), or electronegative (13, 14) functionality at C6 caused a sharp reduction in inhibitor potency. It is unclear whether this drop is due to steric hindrance, adverse effect on the N1 protonation at the assay pH, or a combination of both. Notably, there was a lack of a corresponding drop in the cell growth assay, suggesting off-target activity. C5 could be methylated (16) without loss of enzyme inhibitory activity.
Table 1.
SAR of pyrimidine C5 and C6 positions
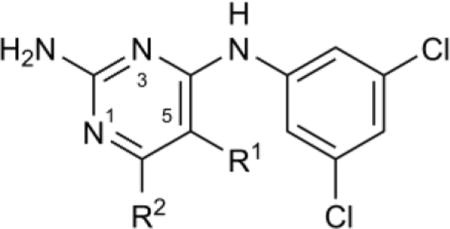
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | pKa(N1)a | Tb IC50,b μM | Hs IC50,c μM | Tb427 EC50,d μM |
| 7 | H | Me | 7.3 | 3.7 (3.3–4.1) | 27% at 180 | 5.6 (4.5–7.1) |
| 8 | H | H | 6.6 | 13 (11–15) | >180 | 9.9 (7.7–13) |
| 9 | H | Et | 7.1 | 3.0 (2.7–3.3) | 36% at 180 | 4.7 (4.1–5.4) |
| 10 | H | iPr | 7.0 | 12 (10–14) | >180 | 4.7 (3.9–5.8) |
| 11 | H | Ph | 6.1 | 24% at 21 | >21 | 7.2 (6–8.5) |
| 12 | H | N-morpholino | 6.0 | >180 | >180 | 13 (10–17) |
| 13 | H | Cl | –2.2 | >180 | >180 | 8.9 (7.3–11) |
| 14 | H | CF3 | –3.3 | >180 | >180 | 10 (7.5–14) |
| 15 | −(CH2)3− | 6.8 | 12 (11–14) | >21 | 19 (15–24) | |
| 16 | Me | Me | 7.0 | 4.8 (4.5–5.2) | 25% at 180 | 17 (14–20) |
The pKa value of the conjugated acid of the pyrimidine nitrogen N1 estimated using MarvinSketch.
Mean IC50 for T. brucei AdoMetDC/prozyme at pH 7.2 determined using the RapidFire–MS-based enzyme activity assay, in triplicate, with 95% confidence interval (CI) shown in parentheses.
Mean IC50 for human AdoMetDC at pH 7.2 determined in the RapidFire–MS-based enzyme activity assay, in triplicate, with 95% CI shown in parentheses.
Mean EC50 as measured in the ATP–bioluminescence-based bloodstream form T. brucei Lister 427 cell viability assay, in triplicate, with 95% CI shown in parentheses. Percent inhibition at the maximal tested concentration (in μM) is shown when maximum mean inhibition was <50%.
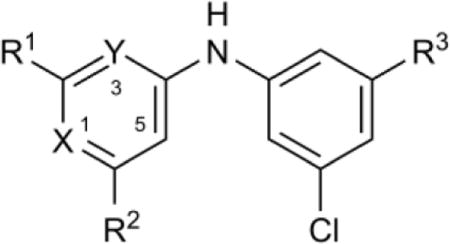
SAR studies: pyrimidine ring nitrogens and amino group
Next, compounds were prepared to probe the function of the pyrimidine ring nitrogen atoms and C2-amino group (Table 2). Substitution of N1 with CH (17 vs. 8) led to complete loss of enzyme inhibitory activity consistent with the hypothesized importance of protonated NH+ base for binding. This activity loss of 1-deaza analogs could not be rescued by moving the C2-amino group to the C6 position (18 and 19). Conversely, replacement of N3 with CH led to only a 2–4-fold decrease in inhibitory activity (20 and 21 compared with the respective parent compounds 7 and 8). Interestingly, the pKa(N1) for compounds with CH in place of N3 was predicted to increase by 2–3 units compared with the parent compounds. Thus the concentration of the N1-protonated species was expected to be >99% at pH 7.2, compared with only 55% or 20%, respectively, for the parent compounds, which was predicted to led to an increase in inhibitory activity. These data suggest that the positive gains of increased N1 protonation were offset by loss of other interactions with N3.
Table 2.
SAR of pyrimidine ring nitrogens
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | X | Y | R1 | R2 | R3 | pKa(N1)a | TbIC50,b μM | Hs IC50,c μM | Tb427 EC50,d μM |
| 7 | N | N | NH2 | Me | Cl | 7.3 | 3.7 (3.3–4.1) | 27% at 180 | 5.6 (4.5–7.1) |
| 8 | N | N | NH2 | H | Cl | 6.6 | 13 (11–15) | >180 | 9.9 (7.7–13) |
| 17 | CH | N | NH2 | H | Cl | NA | >180 | >180 | 96 (66–140) |
| 18 | CH | N | H | NH2 | Cl | NA | >180 | >180 | 38 (31–47) |
| 19 | CH | N | Me | NH2 | Cl | NA | 24% at 180 | >180 | 13 (8.2–22) |
| 20 | N | CH | NH2 | Me | Cl | 9.6 | 15 (13–18) | >180 | 2.2 (1.7–2.9) |
| 21 | N | CH | NH2 | H | Cl | 9.4 | 29 (27–31) | >180 | 11 (9.5–12) |
| 22 | N | CNH2 | H | H | Cl | 8.4 | 21% at 180 | >180 | 18 (14–24) |
| 23 | N | N | H | Me | Cl | 5.2 | >180 | >180 | 27 (22–33) |
| 24 | N | N | NHMe | Me | Cl | 7.1 | >63 | >63 | 2.7 (2.4–3.1) |
| 25 | N | N | NMe2 | Me | Cl | 7.0 | >180 | >180 | 14 (8.1–24) |
| 26 | N | N | NH2 | NH2 | Cl | 6.4 | 48 (43–54) | >180 | 14 (12–17) |
| 27 | N | N | Me | NH2 | Br | 6.4 | 24% at 180 | >180 | 20 (17–24) |
See footnotes for Table 1; NA, not applicable.
Deletion (23) or methylation (24 and 25) of the C2-amino group led to the loss of the enzyme inhibition. While the former decreased the pKa(N1) to 5.2, the latter had little effect on the predicted pKa for N1 protonation but introduced steric interactions preventing optimal engagement of Glu266 (vide infra, Fig. 3B). Additionally, moving the C2-amino group to C3 in the background of a Me → H substitution at C6 led to further loss of inhibitory activity (22 vs. 21). Taken together, these data suggest the importance of the primary C2-amine and N1 for binding interactions, which was later supported by crystallographic data (vide infra) showing H-bonding and salt-bridge of a protonated pyrimidineamine to Glu266 (Fig. 3B).
Figure 3.
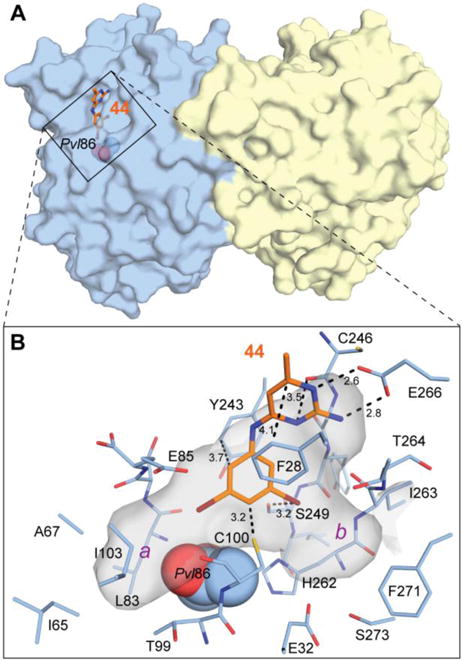
Crystal structure of TbAdoMetDC/prozyme in complex with 44. (A) Surface representation of TbAdoMetDC/prozyme (blue/yellow) with 44 (orange sticks) occupying the catalytic site. Pyruvoyl residue (Pvl) is shown as spheres. (B) Surface representation of the catalytic site. Residues forming the catalytic-site cavity are shown as sticks. Select interatomic distances are shown as dashed lines with values in Å. The catalytic-site cavity is shown as a semi-transparent gray surface. Two pockets that are partially occupied by bromines are labeled a and b in purple.
Substituting an amino group for methyl at C6 (26) or swapping the C2-amino and C6-methyl groups (27; compare with the parent compound 44 in Table 4) resulted in lower pKa(N1) and a marked drop in the enzymatic inhibition. This is reflected in a better activity for 26 at pH 6.8 (5.8 μM against Tb AdoMetDC, see Table S1). Poorer activity of 27 can be rationalized by the loss of the C2-amino group essential for interaction with Glu266 (vide infra, Fig. 3B).
Table 4.
SAR of meta aniline ring substitutions
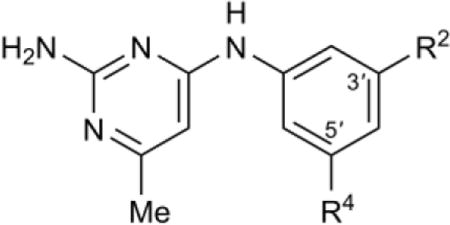
| |||||
|---|---|---|---|---|---|
| Compound | R2 | R4 | Tb IC50,b μM | Hs IC50,c μM | Tb427 EC50,d μM |
| 7 | Cl | Cl | 3.7 (3.3–4.1) | 27% at 180 | 5.6 (4.5–7.1) |
| 32 | Br | Cl | 2.2 (2.0–2.4) | 33% at 180 | 4.7 (3.9–5.8) |
| 33 | CN | Cl | 3.9 (3.2–4.8) | 29% at 180 | 12 (10–14) |
| 34 | CF3 | Cl | 3.9 (3.4–4.4) | 39% at 180 | 13 (11–15) |
| 35 | Ph | Cl | 3.3 (2.8–3.9) | 30% at 63 | 4.5 (2.1–9.4) |
| 36 | SF5 | Cl | 16 (14–18) | >180 | 13 (11–15) |
| 37 | SF5 | Br | 8.7 (7.4–10) | >180 | 0.45 (0.32–0.62) |
| 38 | Et | Br | 7.6 (5.7–10) | >180 | 5.5 (4.6–6.6) |
| 39 | OEt | Br | 6.0 (5.2–6.9) | 36% at 180 | 4.4 (4.2–4.7) |
| 40 | OCH2CF3 | Br | 1.6 (1.1–2.4) | 44 | 4.2 (3.6–4.8) |
| 41 | OCH2CHCH2 | Br | 4.5 (4.1–4.9) | 28% at 180 | 4.4 (3.7–5.3) |
| 42 | OBu | Br | 4.5 (4.0–5.1) | >180 | 1.9 (1.7–2.1) |
| 43 | OCH2iPr | Br | 11 (9.5–12) | >180 | 4.4 (3.5–5.5) |
| 44 | Br | Br | 1.9 (1.7–2.1) | 31% at 180 | 4.8 (4.1–5.6) |
| 45 | SO2Me | SO2Me | 1.5 (1.3–1.7) | 39% at 180 | >100 |
| 46 | CF3 | CF3 | 11 (9.8–12) | >180 | 14 (11–17) |
| 47 | OMe | OMe | 17 (14–20) | 21% at 180 | 41 (33–51) |
| 48 | CO2H | CO2H | >180 | >180 | >100 |
| 49 | CO2Me | CO2Me | 40% at 180 | >180 | 42 (22–78) |
| 50 | Ph | Ph | >180 | >180 | 1.5 (1.3–1.6) |
| 51 | N-(3,5-Cl2Ph)−acetamide | N-(3,5-Cl2Ph)−acetamide | >180 | >180 | 2.0 (1.2–3.7) |
| 52 | Me | Me | 141 (119–167) | >180 | 19 (17–22) |
| 53 | tBu | tBu | >63 | >63 | 4.1 (4.1–4.5) |
| 54 | SF5 | H | 23% at 180 | >180 | 14 (11–17) |
| 55 | Cl | H | 35% at 180 | >180 | 42 (34–52) |
| 56 | Br | H | 41% at 180 | >180 | 22 (19–26) |
| 57 | F | H | >180 | >180 | >25 |
| 58 | SMe | H | 149 (122–181) | 23% at 180 | >25 |
| 59 | H | H | >180 | >180 | >25 |
| 60 | F | F | 180 (160–201) | >180 | 70 (61–80) |
| 61 | F | Cl | 32 (13–82) | >180 | 17 (15–20) |
All compounds have pKa(N1)a of 7.3.
See footnotes for Table 1.
Notably, most compounds that showed poor enzyme inhibitory activity, with the exception of 17, retained anti-trypanosomal activity, suggesting the presence of at least a partial off-target component in the mechanism of the cell growth inhibition.
SAR studies: linker variation
Having established the pyrimidine ring traits required for activity, we shifted our focus to understanding the role of the linker (Table 3).
Table 3.
SAR of the linker
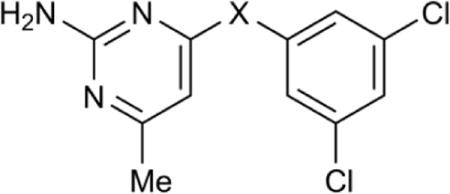
| |||||
|---|---|---|---|---|---|
| Compound | −X− | pKa(N1)a | Tb IC50,b μM | Hs IC50,c μM | Tb427 EC50,d μM |
| 7 | −NH− | 7.3 | 3.7 (3.3–4.1) | 27% at 180 | 5.6 (4.5–7.1) |
| 28 | −NMe− | 7.2 | 81 (74–89) | >180 | 38 (34–43) |
| 29 | −S− | 4.3 | >180 | >180 | >25 |
| 30 | −NH(CH2)− | 8.1 | 139 (130–150) | >180 | 11 (9.9–13) |
| 31 | −NH(CH2)2− | 8.2 | 28 (25–32) | 29% at 180 | 5.7 (1.6–21) |
See footnotes for Table 1.
Methylation of the linking amine (28) resulted in a sharp decrease in inhibitory activity in both enzyme and cell growth assays. The replacement of the amine with a thiol linker (29) eliminated enzyme inhibitory activity completely, presumably due to the effect on N1 acidity (pKa(N1) = 4.3). Extension of the linker decreased potency to varying degrees. Addition of a single methylene (30) led to a 40-fold decrease in enzyme inhibition, while extension by two methylene units (31) decreased enzyme inhibition only by 8-fold.
SAR studies: aniline ring substitutions
We next focused our efforts on elucidating the role of the substituted aniline ring. Both the size and electronic nature of the substituents in the meta positions were probed first (Table 4). Replacement of one of the two chlorines in 7 with moderately sized or large functional groups (Br, CN, CF3, Ph in 32–35) was well tolerated and these analogs had TbAdoMetDC IC50 values similar to the parent dichloro compound 7. In contrast, pentafluorosulfanyl substitution led to 4-fold increase in the IC50 for TbAdoMetDC inhibition (36 and 37). Since Br was a good substituent for Cl, we used 3-bromo analogs to probe the second meta positions of the aniline ring with alkyl and O-alkyl functionalities of varying length, including branched and unsaturated chains (39–43). All of these analogs showed T. brucei AdoMetDC inhibitory activity. The trifluoroethoxy of 40 conferred the highest potency of the tested substitutions, showing 3.8-fold lower IC50 than the similarly sized ethoxy of 39. This underlines the importance of electronics in the terminal position of the substituent. Linear allyloxy and butoxy of 41 and 42, respectively, performed 2.4-fold better than isobutoxy of 43 suggesting a preference for linear over branched substituents. Also, ethyl in 38 conferred activity similar to that of ethoxy in 39, suggesting that the phenolic ether was dispensable.
When both chlorines were simultaneously replaced with either bromines (44) or methylsulfonyls (45), inhibitor potency on the enzyme was improved by 2-fold (although 45 had no antitrypanosomal activity, presumably due to impaired cell uptake). Conversely, other moderately sized nonpolar and polar (Me, t-Bu, CF3, OMe, CO2H, CO2Me) or larger aryl (Ph and N-(3,5-(di-Cl)-Ph)-acetamide) substituents in both meta positions led to reduced (46 and 47) or nearly absent (48–53) enzyme inhibitory activity. Removal of one or both meta-substituents (54-59) or replacement with a smaller fluorine-substituent (60, 61) substantially reduced (54-56, 58, 60, 61) or eliminated (57, 59) enzyme inhibitory activity.
The correlation between enzyme and cell growth inhibition was dependent on the aniline-ring substitution pattern. Both enzyme and cell-based activity correlated reasonably for analogs 32-44, 46-49, 52, 57-61, whereas enzyme-inactive compounds 50, 51, 53 retained substantial antitrypanosomal activity indicating off-target activity. The very weak AdoMeDC inhibitors 54-56 did retain some, albeit reduced cellular activity indicating a partial off-target activity or increased cellular uptake/accumulation.
Having established the effects of meta-substitution, we looked at SAR related to substitution patterns that include ortho and para positions (Table 5).
Table 5.
SAR of ortho- and para-aniline ring substitutions
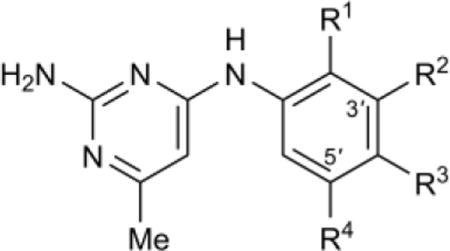
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Tb IC50,b μM | Hs IC50,c μM | Tb427 EC50,d μM |
| 7 | H | Cl | H | Cl | 3.7 (3.3–4.1) | 27% at 180 | 5.6 (4.5–7.1) |
| 62 | H | Cl | F | Cl | 1.5 (1.4–1.8) | 160 (135–191) | 4.8 (3.3–7) |
| 63 | H | Br | F | Br | 1.6 (1.3–1.8) | 43% at 180 | 4.6 (3.6–5.9) |
| 64 | H | Cl | Cl | Cl | 11 (8.8–13) | >63 | 4.5 (3.4–5.9) |
| 65 | H | Cl | Me | Cl | 20 (17–24) | >63 | 4.4 (4.2–4.8) |
| 66 | H | Cl | OPh | Cl | 38% at 180 | >180 | 1.1 (0.97–1.3) |
| 67 | H | Cl | OH | Cl | 33% at 180 | >180 | 13 (10–16) |
| 68 | H | Cl | OCHF2 | Cl | >63 | >63 | 4.5 (3.7–5.4) |
| 69 | H | Cl | NMe2 | Cl | >180 | >180 | 5.8 (4.2–7.9) |
| 70 | H | Cl | N-morpholino | Cl | >180 | >180 | 18 (14–24) |
| 71 | H | Cl | N-piperidino | Cl | >180 | >180 | 2.1 (1.3–3.2) |
| 72 | OH | Cl | H | Cl | 37 (29–46) | >180 | 26% at 100 |
| 73 | F | Cl | F | Cl | 2.2 (2.0–2.6) | >63 | 10 (8.5–12) |
| 74 | Cl | H | H | Cl | 115 (89–150) | >180 | 23% at 25 |
| 75 | Cl | Cl | H | H | 137 (95–198) | >180 | 27% at 25 |
| 76 | Cl | H | Cl | H | >180 | >180 | 28% at 25 |
| 77 | Me | H | H | H | >180 | >180 | >25 |
| 78 | SMe | H | H | H | >180 | >180 | >25 |
| 79 | Br | H | H | H | >180 | >180 | >25 |
| 80 | Me | Cl | H | H | >180 | >180 | 29% at 25 |
| 81 | Me | H | H | Cl | >180 | >180 | 28% at 25 |
| 82 | Me | H | Cl | H | >180 | >180 | >25 |
| 83 | F | H | Br | H | >180 | >180 | 25% at 25 |
| 84 | Cl | H | H | CF3 | 199 (164–240) | >180 | 28% at 25 |
| 85 | H | H | Cl | H | >180 | >180 | 28% at 25 |
| 86 | H | H | Et | H | >180 | >180 | 19 (17–22) |
| 87 | H | H | iPr | H | >180 | >180 | 11 (8.5–15) |
| 88 | H | H | CF3 | H | >180 | >180 | 15 (13–17) |
| 89 | H | H | 3,5-Cl2Ph | H | >180 | >180 | 1.0 (0.88–1.2) |
| 90 | H | F | F | H | 33% at 180 | >180 | 25% at 25 |
All compounds have pKa(N1)a of 7.3 except 67 (pKa(N1)= 7.0); 72 (pKa(N1)= 7.1); 73, 74, and 83 (pKa(N1)= 7.2).
See footnotes for Table 1.
Introduction of a fluorine into the para position led to up to 2-fold enhancement in potency towards TbAdoMetDC (62, 63 vs. 7, 44), whereas chlorine and methyl-substitution (64, 65) reduced activity 3 to 6-fold vs. comparator 7. All other para-substituents that were explored, irrespective of size or electronic nature, led to a complete loss of inhibitory activity towards the enzyme. Retrospectively, this SAR is in agreement with the co-crystal structure data, which indicates a size-restriction in the AdoMetDC binding pocket along the para-position vector (vide infra, Fig. 3B). The ortho position tolerated addition of a fluoro (73), but substitution with OH (72) resulted in ten-fold reduced inhibitory activity vs. 7. Addition of a Cl, Br, Me, or SMe substituent in the ortho-position of the inactive unsubstituted phenyl analog 59 or very weakly active mono meta-chloro or bromo analogs 55 or 56 was ineffective at introducing any activity for compounds 74-84 (some additional inactive compounds are shown in Table S2).
In this series, there was a reasonable correlation between the activity on the enzyme and cellular activity for compounds without para-substituents or with para-halogens (62-64, 72-85, 90). However, this correlation breaks down for every other para-substituted compounds tested (65-71, 86-89). Notably, enzyme inactive para-substituted analogs 66-71 and 86-89 retained substantial antitrypanosomal activity, with EC50 values between 1 and 20 μM.
The data so far suggested that meta-positioning of substituents on the aniline ring was crucial for activity and minimizing off-target antitrypanosomal activity. Minimally, one of the two meta-positions requires substitution with a chlorine or bromine, whereas the other meta-position needs to be substituted but has more flexibility such that halogens, cyano, CF3, Et, alkylethers, and Ph can be accommodated. However, while these modifications were tolerated, none resulted in a compound with substantially increased potency over the parent compound. Since we discovered that phenyl in one of the two meta-positions yielded an active compound (35, see Table 4), we prepared a number of substituted phenyl, heteroaromatic, and fused-ring analogs, aiming to further tune the inhibitory activity and off-target antitrypanosomal activity (Table 6).
Table 6.
SAR of meta-aryl aniline ring substitutions
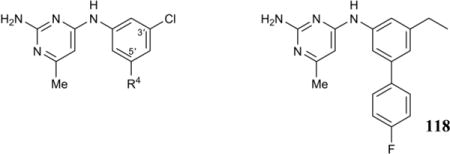
| ||||
|---|---|---|---|---|
| Compound | R4 | Tb IC50,b μM | Hs IC50,c μM | Tb427 EC50,d μM |
| 35 | Ph | 3.3 (2.8–3.9) | 30% at 63 | 4.5 (2.1–9.4) |
| 91 | 4-FPh | 1.1 (0.95–1.2) | >21 | 1.6 (0.94–2.8) |
| 92 | 4-ClPh | 1.7 (1.6–2.0) | >63 | 2.2 (1.8–2.7) |
| 93 | 4-MeOPh | 2.3 (2.1–2.6) | >180 | 4.5 (3.1–6.6) |
| 94 | 4-MePh | 4.6 (4.0–5.3) | >63 | 4.3 (3.3–5.7) |
| 95 | 4-CF3Ph | 4.7 (3.9–5.7) | >21 | 2.0 (1.4–3) |
| 96 | 4-CNPh | 7.6 (3.2–18) | >7 | 1.0 (0.96–1.1) |
| 97 | 4-AcPh | 28% at 21 | >21 | 6.7 (4.1–11) |
| 98 | 4-HOPh | 143 (115–178) | >180 | 9.7 (7.5–13) |
| 99 | 4-PhPh | 167 (150–190) | >180 | 0.91 (0.82–1) |
| 100 | 4-NMe2Ph | 34% at 180 | >180 | 5.0 (4.2–6) |
| 101 | 2-ClPh | 26 (22–33) | >180 | 8.5 (7.6–9.5) |
| 102 | 3-ClPh | 81 (57–114) | >180 | 1.9 (1.6–2.3) |
| 103 | 3,4-(Cl)2Ph | 102 (82–125) | >180 | 2.9 (1.3–3.6) |
| 104 | 3-Cl-4-FPh | 157 (95–260) | >180 | 3.2 (2.8–3.7) |
| 105 | 3-CF3-4-ClPh | 53 (46–61) | >180 | 1.5 (1–2.2) |
| 106 | 3,5-(CF3)2Ph | 28 (25–31) | >180 | 0.53 (0.47–0.59) |
| 107 | 2-naphthyl | 57 (51–64) | >180 | 1.7 (1.4–2) |
| 108 | 9-phenanthrenyl | >180 | >180 | 1.8 (1.5–2.1) |
| 109 | 5-benzo-1,3-dioxolyl | 6.2 (4.6–8.3) | >21 | 2.3 (1.6–3.2) |
| 110 | 5-benzofurazanyl | 27 (21–35) | >180 | 0.57 (0.13–2.5) |
| 111 | 3-furanyl | 7.4 (6.2–8.7) | 30% at 180 | 7.1 (5.7–8.8) |
| 112 | 2-benzofuranyl | 23% at 63 | >63 | 1.4 (1.4–1.5) |
| 113 | 3-thiopheneyl | 26 (22–32) | 30% at 180 | 29 (23–35) |
| 114 | 2-benzothiopheneyl | 41 (36–46) | >180 | 1.6 (1.5–1.8) |
| 115 | 5-thiazolyl | 42% at 21 | >21 | 3.6 (3.1–4.1) |
| 116 | N-Me-4-pyrazolyl | 164 (132–203) | >180 | 6.7 (4.7–9.6) |
| 117 | 5-pyrimidinyl | >180 | >180 | 58 (33–100) |
| 118 | 2.7 (2.2–3.4) | >180 | 1.3 (1.1–1.5) | |
All compounds have pKa(N1)a of 7.3.
See footnotes for Table 1.
Compared to the parent meta-phenyl substituted biaryl analog 35, 4-FPh and 4-ClPh substituted biaryl analogs 91 and 92 were about 2 to 3-fold more potent in the enzyme and cell-based assays, whereas 4-OMe, 4-Me, and 4-CF3 substitution delivered roughly equipotent compounds in both assays (93–95). A 2-fold loss of enzyme inhibition was observed for the 4-CNPh analog (96), but this was accompanied by an increase in cellular activity (~5-fold). Moving to 4-Ac (97), 4-OH (98), and 4-NMe2 (100) substituents, enzyme inhibition dropped off markedly but cell-based inhibition remained at single-digit micromolar EC50 levels, indicating off-target activity. Similarly, moving chlorine to the 2- or 3-position (101, 102), adding an extra Cl or CF3 to the 3-position (103–105), or 3,5-(CF3)2-substitution (106) also abolished enzyme inhibition while retaining the off-target cell-based activity.
Biaryls (4-PhPh, 99), polycyclic aryls (naphthyl 107, phenanthrenyl 108), benzofused heteroaryls (110, 112, 114), or 5-membered heteroaromatics (5-thiazolyl 115, N-Me-4-pyrazolyl 116) were largely incompatible with enzyme inhibition but retained potent off-target cell activity. Enzyme and cell activity tracked better for benzodioxolyl (109), furanyl (111), thiophenyl (113), and pyrimidinyl (117) substituents, with the former two (109, 111) active in the low micromolar range, and the latter two (113, 117) largely devoid of activity in both assays. Finally, we prepared and tested compound 118 that combined 4-fluorophenyl and ethyl meta-substitutions. Compared to the corresponding 3-(4-FPh)-5-Cl analog 91, the similar activity of 118 indicates that meta-Cl can be exchanged for meta-Et without affecting enzyme or cell-based potency.
Crystal structure of TbAdoMetDC/prozyme in complex with a pyrimidineamine analog reveals binding interactions
To assist in rational design of pyrimidineamine analogs with improved binding properties, we solved the crystal structure of the TbAdoMetDC/prozyme complex co-crystallized with 44. Compound 44 was selected for co-crystallization on the basis of its potency and solubility in aqueous solutions. Additionally, the presence of two bromines facilitated location of the bound compound in the complex, and the anomalous diffraction data collected at the Br K-edge were instrumental in determining the correct orientation of this quasi-symmetrical molecule in the binding pocket (See Experimental methods). The structure was solved by molecular replacement using our previously determined TbAdoMetDC/prozyme heterodimeric structure (Protein Data Bank (PDB) identifier 5TVM)21 as a search model, and refined to 3.0 Å resolution with Rwork/Rfree = 20.0/25.3%. Overall, the structure was similar to the unliganded active heterodimer (RMSD of 0.28 Å between current structure and 5TVM as determined by TM-align34 for 636 equivalent Cα atoms).
A single molecule of 44 was found in the catalytic site of the TbAdoMetDC in the heterodimer of TbAdoMetDC/prozyme, positioned 3.9 Å from the catalytic pyruvate (Figure 3A). This binding mode is consistent with biochemically demonstrated competitive mode of inhibition (see Figure 2B). The binding interactions observed between 44 and the enzyme also explain much of the above described SAR for the series. The requirement for N1 to be protonated and for C2 to have a primary amine is explained by the finding that both groups are within hydrogen-bonding distance of the side-chain carboxyl oxygens of Glu266 in the binding pocket. It is thus reasonable to assume that a protonated pyrimidineamine participates in favorable bidentate H-bonding and salt-bridge interactions (Figure 3B).
Additional binding energy is also likely contributed by π– π stacking interactions between the pyrimidine ring and Phe28, and between aniline ring and Tyr243. The proximity of the catalytic Cys100 to the aniline ring of 44 (3.2 Å between SH and C4′ of the ring) is suggestive of the aromatic–thiol interaction (Figure 3B), though the angle of the interaction is not ideal as determined by previously described interactions of this type.35, 36 Finally, an H-bonding interaction37 between the partially negative lateral end of one of the aniline ring meta-bromines and Ser249 hydroxyl group is also potentially present, and may contribute to the binding energy (Figure 3B).
Importantly, the structure of TbAdoMetDC co-crystallized with 44 revealed that the 3′- and 5′-substituents on the aniline ring projected into two distinct pockets designated a and b (Figure 3B). Building on the structural data and the observations reported in Tables 4–6, we hypothesize that of the two pockets, only pocket a could accommodate a phenyl-sized substitution at the meta-position of the aniline ring. Conversely, the shape and depth of pocket b suggested that it would be accessible to linear, flexible substituents (for example, alkyls and alkyl ethers 38-43 in Table 4). The fact that 118, functionalized with p-fluorophenyl at one meta-position and ethyl at the other, inhibited the enzyme with good potency (IC50 = 2.7 μM, see Table 6) suggests that both pockets can be accessed simultaneously in the context of a single compound. Additionally, our structure suggested that para-substituents larger than F, Cl, or Me would be inaccessible as confirmed by the enzyme inhibition data in Table 5 (66-71).
Structural basis for parasite–host selectivity
HsAdoMetDC (PDB identifier 3DZ7)38 was aligned to the TbAdoMetDC bound to 44 to provide insight into the structural basis for selectivity of this and other pyrimidineamines for T. brucei AdoMetDC compared with the human enzyme (Figure 4). The catalytic sites are mostly conserved between the two species, and their geometries are similar. One residue at the lid of the pocket, however, differs between them. This residue (His5 in human AdoMetDC and Arg26 in T. brucei AdoMetDC) is positioned differently in the two structures. His5 in human AdoMetDC is within 3.2 Å of 44 in the overlaid enzyme, and may function to restrict the ligand-binding site compared with the T. brucei structure (Figure 4). The analogous position in T. brucei AdoMetDC (Arg26) was not well resolved in the electron density maps of the structure reported herein; however, it was observed in previous structures (PDB depositions 5TVF and 5TVM) where it was positioned away from the opening of the binding pocket (Figure 4).
Figure 4.
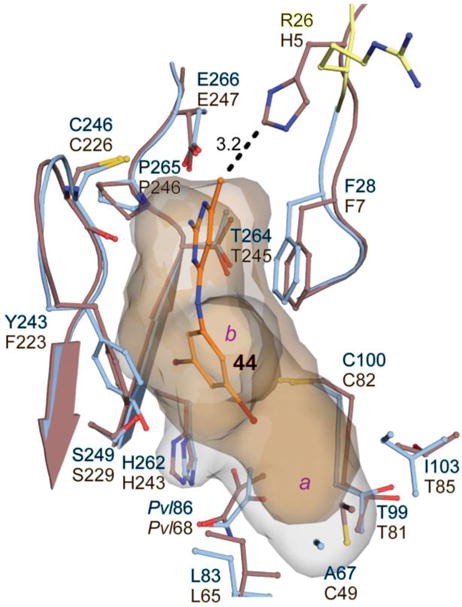
Comparison of human and T. brucei AdoMetDC active sites. Human AdoMetDC (PDB 3DZ7, brown) was aligned with TbAdoMetDC structure (blue) co-crystallized with 44 (orange). The analogous residues are labeled in the corresponding colors (T. brucei residues on top). Alignment was done in PyMol, and the side chains within 4 Å of an inhibitor (with the exception of TbGlu86/HsGlu68) are shown here. A dashed line represents a distance, in Å, between His5 of the human protein and 44 in the aligned T. brucei structure. Residues Ser25 and Arg26, which were not modeled in 44-liganded structure, are from the previously reported unliganded TbAdoMetDC structure (PDB 5TVM, yellow). The active-site cavities for human (brown) and 44-bound T. brucei (blue) AdoMetDCs are shown as semi-transparent surfaces (pocket b is pointing away from the viewer).
Additionally, the substitution of Cys49 in the human structure for Ala67 in T. brucei AdoMetDC restricts the size of the human pocket a relative to T. brucei (Figure 4). This size restriction would be expected to provide additional selectivity for analogs that have larger substituents in pocket a, such as the p-fluorophenyl of 91 and 118.
On-target mechanism-of-action studies
As was noted above, the correlation between the enzyme and cell-growth inhibition was weak. This suggested that trypanocidal effect was at least in part due to off-target mechanism of action. Thus, we sought to identify whether the pyrimidineamines were inhibiting AdoMetDC in T. brucei cells. We previously reported that the levels of prozyme protein markedly increased when AdoMetDC activity in T. brucei cells was reduced by either genetic knockdown of AdoMetDC or by chemical inhibition.16, 39 We exploited this feedback loop to evaluate whether hit compounds identified in the HTS were affecting the parasite cell growth through intracellular AdoMetDC inhibition.22 Here, we compared prozyme protein levels in cells treated for 24 h with representative pyrimidineamine analogs that 1) inhibited both T. brucei cell growth and AdoMetDC (7 and 8), 2) inhibited only T. brucei growth (23 and 13), or 3) were inactive in both assays (29). Only compounds that were active on both the enzyme and cells induced prozyme above the vehicle-only control levels (Figure 5). These results show that the pyrimidineamines with TbAdoMetDC inhibitory activity on the purified enzyme also block AdoMetDC activity in T. brucei cells. However, these results also confirm that there was a substantial off-target component to pyrimidineamine-induced cell growth inhibition, as 23 and 13, which did not inhibit TbAdoMetDC — either in the purified-enzyme assay or in the cell (as evident from vehicle-only levels of the prozyme protein) — inhibited cell growth with low-micromolar potencies (Figure 5).
Figure 5.
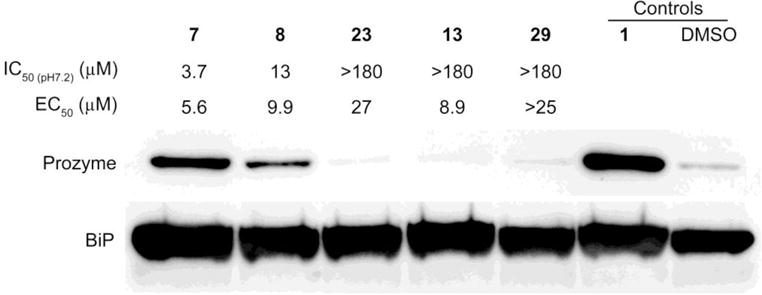
Mechanism of action studies in T. brucei bloodstream form parasites. Western blot analysis of prozyme levels in T. brucei cells treated with active or inactive pyrimidineamine analogs for 24 h. Three types of compounds were compared: 1) compounds that demonstrated low-micromolar inhibition of both TbAdoMetDC at pH 7.2 (IC50 (pH7.2)) and of T. brucei cells in culture (EC50) (7 and 8); 2) compounds that did not inhibit TbAdoMetDC but did inhibit T. brucei cell growth (23 and 13); and 3) a compound that did not inhibit either TbAdoMetDC or the cells (29). Cells were treated with 1.5 × EC50 concentrations of inhibitors (or 30 μM 29). T. brucei binding protein (BiP) was used as a loading control.
Discussion and Conclusions
The number of cases of HAT has decreased 10-fold since the beginning of the 21st century owing to proactive surveillance and control measures set in place in endemic countries.3 Yet the disease has a history of resurgence following years of a downward trend.3 Sustainability of HAT elimination relies in part on the availability of patient-friendly treatment options.3 Neither melarsoprol nor eflornithine, alone or as a part of NECT, are ideal due to high toxicity of the former and high price and requirement for multiple intravenous infusions of the latter.2, 10 The potential for eflornithine resistance to emerge is another negative factor.40 Thus, new safe anti-trypanosomal drugs with CNS-stage efficacy are needed.
We identified and characterized a pyrimidineamine series as a new class of reversible, competitive inhibitors of T. brucei AdoMetDC. This series has a number of positive attributes, including species-selective inhibition of the T. brucei AdoMetDC compared with the human enzyme and antitrypanosomal activity against T. brucei in whole-cell assays. Importantly, the series is predicted to have good brain penetration on the basis of the monolayer permeability assay performed on the parent compound. Thus, the pyrimidineamines have the potential to be used for treatment of both early-stage and late-stage HAT. Herein, we have established the preliminary SAR within the series and have solved the crystal structure of one analog bound to T. brucei AdoMetDC, providing insight for future lead optimization of the series.
While the pyrimidineamines look to be a promising series for further optimization, three key issues were identified that would need to be addressed in future chemistry programs. Firstly, while the pyrimidineamines did show activity against both the enzyme and the parasite, the correlation between these activities was poor. On-target activity was present as evident from the finding that prozyme was upregulated in cells treated with compounds that inhibited AdoMetDC. In addition, good correlation between enzyme inhibition and anti-parasite activity was observed for most active analogs with two meta-substituents in the aniline ring, with at least one substituent being Cl or Br. If the second meta-position is a phenyl-ring substituted in the 4-position with F, Cl, OMe, Me, or CF3, good activity in both assays was observed. However, there was also clear evidence for an off-target activity, as a substantial fraction of pyrimidineamines that had lost TbAdoMetDC inhibitory activity elicited cell growth inhibition in the low-micromolar range. Off-target activity was especially noticeable for para-substituted aniline rings (Table 5) and bis-meta-substituted aniline rings with on meta-position occupied by 2-, 3-, 2,3- or other specifically substituted aryls, biaryls, polycyclic aryls and heteroaryls (Table 6). Secondly, despite synthesis of a number of analogs of the original parent compound, we were unable to substantially improve compound potency below the low-micromolar level. This issue may also relate to the off-target activity, and thus identification of more potent analogs may lead to reduced off-target activity as the on-target component improves. As the analogs described herein had been synthesized prior to obtaining the crystal structure with an inhibitor bound, there remains room for a structure-based lead optimization program to facilitate the identification of more potent, on-target compounds. Lastly, 7 showed poor metabolic stability; however, this liability was not the focus of the current chemistry program and remains to be addressed during further optimization of the series.
A notable feature of pyrimidineamines is the finding that inhibitor binding to TbAdoMetDC is pH-dependent in the relevant physiological range (pH 6.8–7.7). This dependence has its basis in the pKa of N1 in the pyrimidine ring, which is predicted to be 7.3 in the parent compound 7. Within the series, modifying the ring substituents to lower the pKa(N1) (<6) led to reduced inhibitory activity at physiological pH, while optimizing the ring electronics to yield a higher pKa(N1) (>9) reduced the pH dependence of binding. The crystal structure of TbAdoMetDC bound to 44 revealed that N1 forms a salt bridge with Glu266, explaining the requirement for protonation of N1 for inhibitor binding. While we were able to identify compounds with higher pKa, this was achieved through removal of N3 (20 and 21), which led to a loss of the possible interaction between N3 and main-chain carbonyl of Cys246 (3.5 Å away, see Figure 3B). Thus we did not accomplish the goal of improved binding affinity by this strategy. One potential direction for future optimization of this series will be to identify compounds that retain full charge at N1 at physiological pH, without compromising other binding interactions.
In addition to the protonation state of N1, we identified a number of other features of the pharmacophore that were required for TbAdoMetDC inhibition. A primary amine is required at C2, and it is involved in an H-bond with the Glu266 carboxyl group. The amine linker could not be modified as there was a loss of potency when the linker was methylated (28), extended by one or two methylene links (30 and 31), or replaced with sulfur (29). The amine linker does not make any clear interactions with the protein, so its importance may relate to geometry and steric considerations. For sulfur-linked analog 29, the decreased pKa at N1 (4.3) could explain the loss of enzyme inhibition. Finally, both pyrimidine and aniline rings are required, and these form stabilizing π–π stacking interactions with the two aromatic residues, Phe28 and Tyr243, on opposite sides of the catalytic site.
The aniline ring is the most buried part of the ligand. SAR and the crystal structure indicate that substituents in both of the meta-positions are required for good binding. We identified both steric and electronic requirements to the meta-substitutions. At least one of the meta-positions can include an alkyl, alkyloxy, halide, or 4-substituted phenyl ring; whereas the second position is best occupied by Cl, or Br. Notably, both an alkyl (ethyl) and a 4-substituted phenyl could be accommodated at the same time as indicated by anti-TbAdoMetDC activity of 118. A hydrogen bond between Ser249 γ-hydroxyl and a partially negative acceptor in a meta position, such as halide or alkoxy may contribute to the binding energy and explain the finding.37 The next steps, informed by the co-crystal structure data, will involve a much more in depth combinatorial exploration of both meta-positions in such a manner as to ideally occupy both a and b pockets as well as maximize π–π stacking interactions.
All active analogs were strongly selective for T. brucei AdoMetDC over the human enzyme. The selectivity ratio (HsIC50/TbIC50) was at least 27 (40) and in many cases exceeded 100 (e.g., 44 and 62). Yet, the structural basis for species selectivity was not immediately obvious given very similar geometries and residue composition of the catalytic sites in the two species. Specifically, the human site has Phe7 and Phe223 in the positions analogous to T. brucei Phe28 and Tyr243, which could provide π–π stacking interactions with a compound (see Figure 4). Also, Glu247 in the human enzyme closely aligns with T. brucei Glu266, and could potentially also form H-bonding and salt-bridge interactions with pyrimidineamine part of an inhibitor. One possible hypothesis suggested by our co-crystal structure is that His5 in the human AdoMetDC sterically limits access to the binding site, excluding certain classes of inhibitors and thus may be responsible for the observed species selectivity of the pyrimidineamines. In T. brucei, the equivalent position is occupied by an Arg residue with a more labile side chain (see Figure 4). Notably, all 13 classes of validated active hits from our HTS campaign were also selective inhibitors of the T. brucei enzyme,22 suggesting this may be a general principle of parasite–host selectivity. In contrast, a number of compounds that have greater binding affinity for the human enzyme have also been described, such as methylglyoxal bis(guanylhydrazone), or MGBG,41 and CGP 40215,42 so evidently this mechanism does not limit general access to the active site. An additional possible origin for species selectivity can be inferred from the allosteric mechanism of T. brucei AdoMetDC activation: its N-terminal residues form an autoinhibitory lid blocking the active site in the monomer that is displaced from the active site upon binding to prozyme.21 Conversely, the N-terminus of the human enzyme is 21 residues shorter than that of the T. brucei enzyme, and it has not been observed to bind in the active site in any of the published X-ray crystal structures.38, 43–45 However, these data do not rule out the possibility that these residues undergo dynamic transitions during catalysis or ligand binding. Thus, potentially selectivity could arise if the N-terminal residues of the human enzyme compete for ligand binding. In this scenario, T. brucei selective inhibitors would be unable to effectively compete with the N-terminal residue lid, whereas human selective inhibitors and substrate would be effective competitors. Finally, we note that pocket a in human AdoMetDC is smaller than in the T. brucei enzyme, and this difference will enhance selectivity for compounds that fully occupy this pocket (e.g., 91), though this difference is unlikely to explain the selectivity of compounds with smaller substitutions in pocket a (e.g. 44).
The work described herein has led to the discovery of pyrimidineamines with low-micromolar activity against both T. brucei AdoMetDC and the parasite. Through a combination of medicinal chemistry and protein crystallography, we have defined the key components of the pharmacophore that are required for TbAdoMetDC inhibitory activity. If this series is to advance to lead optimization, future work will need to focus on identifying more potent compounds. Two pockets were discovered in the crystal structure that were not fully exploited in the current work. Pocket a is larger of the two and is lined with both polar (Glu85 and Pvl86) and nonpolar (Ile65, Ala67, Leu83, and Ile103) side chains (see Figure 3B). The p-substituted phenyl analogs such as 91 provided for good size complementarity with the available pocket but the crystal structure and activity data suggest that more polar substituents that could engage in H-bonding interactions with Glu85 and backbone carbonyls in the pocket might provide better binding energy (see Table 6 and Figure 3B). Alternatively, a nucleophilic group (e.g, aminooxy) might allow formation of a Schiff base with the prosthetic pyruvoyl group, transforming pyrimidineamines into mechanism-based inhibitors, as described for substrate analogs.46 Pocket b also has substantial polar character with several opportunities for H-bonding interactions presented by side chains of Glu32, Ser273, Ser249, and His262 (see Figure 3B). Current ring substituents did not fully exploit these opportunities and thus further series development should focus on optimizing these potential polar interactions at the meta-positions of the aniline ring. The search of the suitable substituents can be aided by computational protein–ligand docking through virtual screening of meta-position analogs. Iterative co-crystallization with new analogs will aid development.
Experimental Section
Chemistry
General Procedures
Unless otherwise specified, all commercially available reagents were used as received. All reactions using dried solvents were carried out under an atmosphere of argon in flame-dried glassware with magnetic stirring. Dry solvent was dispensed from a solvent purification system that passes solvent through two columns of dry neutral alumina. Silica gel chromatographic purifications were performed by flash chromatography with silica gel (Sigma, grade 62, 60-200 mesh) packed in glass columns; the eluting solvent was determined by thin layer chromatography (TLC). Analytical TLC was performed on glass plates coated with 0.25 mm silica gel using UV for visualization. Melting points are uncorrected. Routine 1H and proton-decoupled 13C NMR spectra were obtained on a Bruker 400 MHz NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) from low to high field relative to residual solvent. Multiplicities are given as: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), m (multiplet). All synthetic compounds exhibited >95% purity as determined by LC-MS analysis performed on an Agilent 1100 HPLC system using an Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm; Agilent) that was coupled to an Agilent G1956A (or 6120) ESI mass spectrometer run in the positive mode with a scan range of 100 to 800 (or 1000) m/z. Liquid chromatography was carried out at a flow rate of 0.5 mL/min at 30 °C with a 5 μL injection volume, using the gradient elution with aqueous acetonitrile containing 0.1% formic acid: 20–50% over 10 min followed by 50–90% for the next 10 min. All tested purchased compounds were verified to be >95% pure using the method described above with the following modifications: Agilent Infinity II HPLC system coupled to an Agilent 6120 ESI mass spectrometer was used; the range was from 100 to 1000 m/z; flow rate was 0.8 mL/min; and gradient elution was done with aqueous acetonitrile containing 0.1% trifluoroacetic acid: 10-100% over 7 min followed by 100% for the next 8 min.
General Procedure A (for the synthesis of compounds 13, 14, 23-25, 27, 28, 38-43, 46-48, 52, 55, 60, 69-71)
To a stirred solution of chloropyrimidine 2a,d,e,h or 3d-f,h (1 equiv.) and substituted aniline (1 equiv.) in EtOH (4 mL/mmol) was added conc. HCl (0.1 mL/mmol). The solution was then heated to reflux for 18 h. After cooling to rt, the volatiles were concentrated in vacuo. The residual oil was partitioned between EtOAc (4 mL/mmol) and 10% aq. NaOH (4 mL/mmol). The layers were separated and the aqueous layer was extracted with EtOAc (3 ×; 4 mL/mmol). The combined organic layers were dried (Na2SO4) and concentrated. The resulting residue was purified by flash chromatography.
General Procedure B (for the synthesis of compounds 18-20, 22)
The appropriate chloropyridine 3a,c,i,j (1 equiv.) was dissolved in n-BuOH (1.23 mL/mmol chloropyridine) in a microwave vial before 3,5-dichloroaniline (1.5 equiv.) was added followed by concentrated HCl (0.154 mL/mmol chloropyridine). The vial was sealed and the solution was heated in the microwave at 185 °C for 4 h before being cooled to rt and diluted with 30 mL of EtOAc (4.6 mL/mmol chloropyridine). 10% Aq. NaOH (4.6 mL/mmol chloropyridine) was added, the layers were separated and the aqueous layer was extracted with EtOAc (3 ×; 2.3 mL/mmol chloropyridine). The organic layers were combined, dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by flash chromatography.
General Procedure C (for the synthesis of compounds 9, 10, 15, 16, 26, 32-34, 36, 37, 44, 45, 53, 54, 61-68, 72, 73, 89)
The appropriate aniline (1 equiv.) and chloropyrimidine 2a-c,f-h or 3g (1 equiv.) were dissolved in EtOH (1.03 mL/mmol) containing concentrated HCl (0.129 mL/mmol) in a microwave vial. The vial was sealed and the solution was heated in the microwave at 140 °C for 1.5 hours before being cooled to rt and diluted with 10 mL of EtOAc (5.15 mL/mmol). 10% Aq. NaOH (5.15 mL/mmol) was added, the layers were separated and the aqueous layer was extracted with EtOAc (3 ×; 5.15 mL/mmol). The organic layers were combined, dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by flash chromatography.
General Procedure D (for the synthesis of compounds 11, 50, 91-109, 116-118)
To a solution of sodium carbonate (3 equiv.) in water (1 mL/mmol carbonate) and DME (3.33 mL/mmol carbonate) was added arylchloride 7 or 13 or arylbromide 32 or 38 (1 equiv.), phenylboronic acid (1.1 equiv.) (note: 2.2 equiv. are used for bis-arylation) and PPh3 (0.244 equiv.). The solution was evacuated and backfilled with Ar 5× before Pd(OAc)2 (0.122 equiv.) was added. The solution was again evacuated and backfilled with Ar 5× before being heated to 90 °C for 18 h. The solution was cooled to rt and filtered through Celite. The filtrate was diluted with EtOAc (29 mL/mmol arylhalide) and the solution was washed with 10% aq. NaOH followed by brine. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo to afford a residue that was purified by flash chromatography.
General Procedure E (for the synthesis of compounds 110-115)
To a solution of sodium triphosphate (7.48 equiv.) in water:dioxane (1:4; 13 mL/mmol 32) was added bromopyrimidine 32 (1 equiv.), and PPh3 (0.3 equiv.). The solution was evacuated and backfilled with Ar 5× before the appropriate heteroaryl boronic acid MIDA ester (1.19 equiv.) was added. The solution was again evacuated and backfilled with Ar 5× and Pd(OAc)2 (0.145 equiv.) was added. The solution was again evacuated and backfilled with Ar 5× before being heated to 90 °C for 18 h. The solution was cooled to rt and filtered through Celite. The filtrate was diluted with EtOAc (48 mL/mmol 32) and the solution was washed with 10% aq. NaOH followed by brine. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo to afford a residue that was purified by flash chromatography.
N2-(3,5-Dichlorophenyl)pyridine-2,4-diamine (18)
According to General Procedure B, 2-chloropyridin-4-amine (1.0 g, 7.76 mmol) and 3,5-dichloroaniline (2.50 g, 15.5 mmol) yielded compound 18 as an off-white solid (0.75 g, 39%); mp >250 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.95 (s, 1H), 7.73 (d, J = 5.5 Hz, 1H), 7.06 (d, J = 5.5 Hz, 1H), 6.97 (d, J = 1.8 Hz, 2H), 6.93 (t, J = 1.8 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 144.6, 138.6, 136.3, 135.5, 135.2, 121.6, 120.3, 116.0, 111.9. LC/MS (ESI) calcd for C11H10Cl2N3 (M + H)+ 254.0, found 254.0.
N2-(3,5-Dichlorophenyl)-6-methylpyridine-2,4-diamine (19)
According to General Procedure B, 2-chloro-6-methylpyridin-4-amine (0.18 g, 1.28 mmol) and 3,5-dichloroaniline (0.41 g, 2.56 mmol) yielded compound 19 as a purple oil (0.07 g, 20%). 1H NMR (400 MHz, Methanol-d4) δ 7.26 (t, J = 1.8 Hz, 1H), 7.22 (d, J = 1.8 Hz, 2H), 6.21 (d, J = 2.1, Hz, 1H), 6.16 (d, J = 2.1 Hz, 1H), 2.33 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 156.9, 156.1, 155.1, 144.9, 134.6, 118.8, 115.3, 103.1, 92.0, 22.4. LC/MS (ESI) calcd for C12H11Cl2N3 (M + H)+ 268.0, found 268.0.
N4-(3,5-Dichlorophenyl)-6-methylpyridine-2,4-diamine (20)
According to General Procedure B, 4-chloro-6-methylpyridin-2-amine (0.25 g, 1.75 mmol) and 3,5-dichloroaniline (0.426 g, 2.62 mmol) yielded compound 20 as an off-white foam (0.09 g, 19%). 1H NMR (400 MHz, Methanol-d4) δ 7.26 (t, J = 1.8 Hz, 1H), 7.22 (d, J = 1.8 Hz, 2H), 6.21 (d, J = 2.7 Hz, 1H), 6.16 (d, J = 2.7 Hz, 1H), 2.33 (s, 3H). 13C NMR (101 MHz, CD3CN) δ 154.9, 148.3, 141.1, 136.2, 135.3, 124.3, 120.6, 102.1, 88.2, 18.6. LC/MS (ESI) calcd for C11H12Cl2N3 (M + H)+ 268.1, found 268.1.
N4-(3,5-Dichlorophenyl)pyridine-2,4-diamine (21)
4-Chloropyridin-2-amine (1.00 g, 7.76 mmol) and 3,5-dichloroaniline (1.23 g, 7.76 mmol) were dissolved in 22 mL of a 50:50 mixture of MeOH/H2O in a sealed tube. To this solution was added 1 mL of conc. HCl and the tube was sealed and heated to 100 °C for 18 h. After cooling to rt, the volatiles were concentrated in vacuo. The residual oil was partitioned between 30 mL of EtOAc and 30 mL of 10 % NaOH. The layers were separated and the aqueous layer was extracted with 3 × 30 mL of EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. The resulting oil was purified by flash chromatography (0–10% MeOH/DCM) to give amine 21 as an off-white foam (0.41 g, 21%). 1H NMR (400 MHz, Methanol-d4) δ7.66 (d, J = 5.9 Hz, 1H), 7.10 (s, 2H), 7.00 (s, 1H), 6.30–6.24 (m, 2H). 13C NMR (101 MHz, Methanol-d4) δ160.3, 151.2, 146.7, 143.7, 135.2, 121.1, 117.3, 102.6, 92.4. LC/MS (ESI) calcd for C11H10Cl2N3 (M+H)+ 254.0, found 254.0.
N4-(3,5-Dichlorophenyl)pyridine-3,4-diamine (22)
According to General Procedure B, 4-chloropyridin-3-amine (0.835 g, 6.50 mmol) and 3,5-dichloroaniline (1.58 g, 9.73 mmol) yielded compound 22 as a yellow solid (0.53 g, 32%). 1H NMR (400 MHz, CD3OD) δ 7.95 (s, 1H), 7.73 (d, J = 5.5 Hz, 1H), 7.06 (d, J = 5.5 Hz, 1H), 6.97 (d, J = 1.8 Hz, 2H), 6.93 (t, J = 1.8 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 144.6, 138.6, 136.3, 135.5, 135.2, 121.6, 120.3, 116.0, 111.9.
N-(3,5-Dichlorophenyl)-6-methylpyrimidin-4-amine (23)
According to General Procedure A, 4-chloro-6-methylpyrimidine (0.69 g, 5.37 mmol) and 3,5-dichloroaniline (0.87 g, 5.37 mmol) yielded compound 23 (0.86 g, 63 %) as a white foam.1H NMR (400 MHz, Methanol-d4) δ 8.55 (s, 1H), 7.74 (s, 2H), 7.03 (s, 1H), 6.62 (s, 1H), 2.37 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ164.7, 160.6, 157.0, 142.0, 134.6, 121.5, 117.5, 105.9, 21.9. LC/MS (ESI) calcd for C11H10Cl2N3 (M + H)+ 254.0, found 254.0.
N4-(3,5-Dichlorophenyl)-N2,6-dimethylpyrimidine-2,4-diamine (24)
According to General Procedure A, 4-chloro-N,6-dimethylpyrimidin-2-amine (0.53 g, 3.37 mmol) and 3,5-dichloroaniline (0.55 g, 3.37 mmol) yielded compound 24 (0.56 g, 67%) as an off-white solid; mp 200–201 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.81 (s, 2H), 6.95 (s, 1H), 5.87 (s, 1H), 2.94 (s, 3H), 2.18 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 164.8, 162.1, 161.4, 142.7, 134.4, 120.6, 117.1, 95.1, 27.2, 21.7. LC/MS (ESI) calcd for C12H13Cl2N4 (M+H)+ 283.1, found 283.1.
N4-(3,5-Dichlorophenyl)-N2,N2,6-trimethylpyrimidine-2,4-diamine (25)
According to General Procedure A, 4-chloro-N,N,6-trimethylpyrimidin-2-amine (0.86 g, 4.98 mmol) and 3,5-dichloroaniline (0.81 g, 4.98 mmol) yielded compound 25 (0.63 g, 48%) as an off-white foam. 1H NMR (400 MHz, Methanol-d4) δ 7.72 (s, 2H), 6.85 (s, 1H), 5.77 (s, 1H), 3.10 (s, 6H), 2.17 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.4, 161.8, 160.6, 142.9, 134.3, 120.1, 116.7, 94.6, 36.4, 22.4. LC/MS (ESI) calcd for C13H15Cl2N4 (M+H)+ 297.1, found 297.1.
N4-(3,5-Dichlorophenyl)pyrimidine-2,4,6-triamine (26)
According to General Procedure C, 6-chloropyrimidine-2,4-diamine (1.00 g, 6.91 mmol) and 3,5-dichloroaniline (1.46 g, 8.99 mmol) yielded compound 26 as an off-white solid (0.95 g, 51%); mp 119–120 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.97 (s, 1H), 7.75 (d, J = 1.8 Hz, 2H), 6.95 (t, J = 1.8 Hz, 1H), 6.04 (s, 2H), 5.92 (s, 2H), 5.21 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 165.0, 163.1, 161.2, 144.7, 134.2, 119.3, 116.6, 78.1. LC/MS (ESI) calcd for C10H10Cl2N5 (M + H)+ 270.0, found 270.0.
N4-(3-Bromo-5-chlorophenyl)-2-methylpyrimidine-4,6-diamine (27)
According to General Procedure A, 6-chloro-2-methylpyrimidin-4-amine (0.50 g, 3.48 mmol) and 3-chloro-5-bromoaniline (0.94 g, 4.52 mmol) yielded compound 27 as a white solid (0.82 g, 75%); mp 178–179 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.69 (t, J = 1.8 Hz, 1H), 7.61 (t, J = 1.8 Hz, 1H), 7.10 (t, J = 1.8 Hz, 1H), 5.72 (s, 1H), 2.34 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 166.1, 163.6, 160.5, 143.0, 134.7, 123.6, 122.1, 120.3, 117.8, 82.8, 23.7. LC/MS (ESI) calcd for C11H10BrClN4 (M + H)+ 313.0, found 313.0.
N4-(3,5-Dichlorophenyl)-N4,6-dimethylpyrimidine-2,4-diamine (28)
According to General Procedure A, pyrimidineamine (0.56 g, 2.83 mmol) and 3,5-dichloro-N-methylaniline (0.41 g, 2.83 mmol) yielded compound 28 (0.47 g, 59%) as an off-white solid; mp 156-157 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.39 (t, J = 1.9 Hz, 1H), 7.30 (d, J = 1.9 Hz, 2H), 5.73 (s, 1H), 3.38 (s, 3H), 2.13 (s, 3H). 13C NMR (101 MHz Methanol-d4) δ 164.3, 163.5, 161.9, 147.0, 135.4, 126.3, 125.7, 93.7, 37.0, 21.6. LC/MS (ESI) calcd for C12H13Cl2N4 (M+H)+ 283.1, found 283.1.
4-((3,5-Dichlorophenyl)thio)-6-methylpyrimidin-2-amine (29)
To a stirred solution of 4-chloro-6-methylpyrimidin-2-amine (0.45 g, 3.11 mmol) and 3,5-dichlorothiophenol (0.56 g, 3.11 mmol) in 6.2 mL of DMF was added Et3N (0.87 mL, 6.22 mmol). The solution was stirred at rt for 24 h The mixture was partitioned between 20 mL of EtOAc and 20 mL of H2O. The layers were separated and the aqueous layer was extracted with 3 × 20 mL of EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. The resulting dark oil was purified by flash chromatography (0–50% EtOAc/Hexanes) to afford thioether 29 as a white foam (0.58 g, 65%). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 1.9 Hz, 2H), 7.39 (t, J = 1.9 Hz, 1H), 6.04 (s, 1H), 5.59 (br s, 2H), 2.18 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.2, 167.63 162.2, 135.5, 133.2, 131.9, 129.7, 106.9, 23.8. LC/MS (ESI) calcd for C11H10Cl2N3S (M+H)+ 286.0, found 286.0.
N4-(3,5-Dichlorobenzyl)-6-methylpyrimidine-2,4-diamine (30)
4-Chloro-6-methylpyrimidin-2-amine (0.68 g, 4.73 mmol) and (3,5-dichlorophenyl)methanamine (1.00 g, 5.67 mmol) were charged into a microwave vial followed by DIPEA (1.65 mL, 9.46 mmol) and 8 mL of CH3CN. The vial was capped and heated to 140 °C for 2 h in the microwave. After cooling to rt, the volatiles were concentrated in vacuo. The residual oil was partitioned between 20 mL of EtOAc and 20 mL of 10% NaOH. The layers were separated and the aqueous layer was extracted with 3 × 20 mL of EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. The resulting dark oil was purified by flash chromatography (0–10% MeOH/DCM) to give amine 30 as a white solid (0.66 g, 49%); mp 129–130 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.17 (app s, 3H), 5.69 (s, 1H), 4.44 (s, 2H), 2.06 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 164.3, 163.8, 162.7, 143.8, 134.6, 126.4, 125.4, 94.2, 47.3, 42.7, 21.0. LC/MS (ESI) calcd for C12H13Cl2N4 (M + H)+ 283.0, found 283.0.
N4-(3,5-Dichlorophenethyl)-6-methylpyrimidine-2,4-diamine (31)
4-Chloro-6-methylpyrimidin-2-amine (0.22 g, 1.53 mmol) and 2-(3,5-dichlorophenyl)ethan-1-amine (0.35 g, 1.83 mmol) were charged into a microwave vial followed by DIPEA (0.5 mL, 3.06 mmol) and 4 mL of CH3CN. The vial was capped and heated to 140 °C for 2 h in the microwave. After cooling to rt, the volatiles were concentrated in vacuo. The residual oil was partitioned between 20 mL of EtOAc and 20 mL of 10% NaOH. The layers were separated and the aqueous layer was extracted with 3 × 20 mL of EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. The resulting dark oil was purified by flash chromatography (0–10% MeOH/DCM) to give amine 31 as a clear oil (0.29 g, 64%). 1H NMR (400 MHz, CD3CN) δ 7.30 (d, J = 1.9 Hz, 1H), 7.22 (t, J = 1.9 Hz, 2H), 6.09 (s, 1H), 5.71 (s, 1H), 4.08 (br s, 2H), 3.52 (q, J = 6.7 Hz, 2H), 2.84 (t, J = 6.7 Hz, 2H), 2.10 (s, 3H). 13C NMR (101 MHz, CD3CN) δ 163.9, 143.8, 134.3, 134.3, 127.6, 126.0, 125.9, 93.5, 41.2, 34.6, 34.4. LC/MS (ESI) calcd for C13H15Cl2N4 (M + H)+ 297.1, found 297.1.
N4-(3-Bromo-5-chlorophenyl)-6-methylpyrimidine-2,4-diamine (32)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (1.32 g, 8.07 mmol) and 3-bromo-5-chloroaniline (2.00 g, 9.68 mmol) yielded compound 32 as a white solid (2.20 g, 87%); mp 155–156 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 7.89 (t, J = 1.9 Hz, 1H), 7.84 (t, J = 1.9 Hz, 1H), 7.13 (t, J = 1.9 Hz, 1H), 6.32 (s, 2H), 5.83 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.3, 163.2, 161.2, 144.0, 134.5 123.0, 122.4, 119.9, 117.5, 95.9, 23.9. LC/MS (ESI) calcd for C11H11BrClN4 (M + H)+ 313.0, found 313.0.
3-((2-Amino-6-methylpyrimidin-4-yl)amino)-5-chlorobenzonitrile (33)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.47 g, 2.73 mmol) and 3-chloro-5-cyanoaniline (0.50 g, 3.27 mmol) yielded compound 33 as an off-white solid (0.52 g, 73%); mp270–271 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 8.21 (t, J = 1.8 Hz, 1H), 8.01 (t, J = 1.8 Hz, 1H), 7.45 (t, J = 1.8 Hz, 1H), 6.41 (s, 2H), 5.85 (s, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.6, 163.2, 161.1, 143.6, 134.4, 123.7, 122.7, 120.4, 118.3, 113.5, 95.9, 24.0. LC/MS (ESI) calcd for C11H11ClN5 (M + H)+ 260.1, found 260.1.
N4-(3-Chloro-5-(trifluoromethyl)phenyl)-6-methylpyrimidine-2,4-diamine (34)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.38 g, 2.64 mmol) and 3-chloro-5-trifluromethylaniline (0.62 g, 3.17 mmol) yielded compound 34 as a white solid (0.62 g, 81%); mp 159–160 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 8.30 (t, J = 1.9 Hz, 1H), 7.84 (t, J = 1.9 Hz, 1H), 7.25 (t, J = 1.9 Hz, 1H), 6.34 (br s, 2H), 5.85 (s, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO- d6) δ 166.5, 163.2, 161.2, 143.6, 134.6, 131.3 (q, JCF = 32.0 Hz), 125.2, 121.7, 117.0, 113.9, 95.9, 23.9. LC/MS (ESI) calcd for C12H11ClF3N4 (M + H)+ 303.1, found 303.1.
N4-(5-Chloro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (35)
According to General Procedure D, bromo-pyrimidineamine 32 (0.50 g, 1.59 mmol) and phenylboronic acid (0.21 g, 1.75 mmol) yielded compound 35 as an off-white foam (0.33 g, 67%). 1H NMR (400 MHz, CDCl3) δ 7.53–7.50 (m, 2H), 7.45–7.40 (m, 3H), 7.39–7.34 (m, 2H), 7.28 (m, 1H), 6.86 (s, 1H), 5.99 (s, 1H), 4.98 (s, 2H), 2.22 (s, 3H). (101 MHz, CDCl3) 13C NMR (100 MHz, CDCl3) δ 167.6, 162.7, 161.6, 143.7, 140.4, 139.4, 135.0, 128.9, 128.18, 127.0, 122.6, 120.1, 118.3, 95.0, 24.0. LC/MS (ESI) calcd for C17H16ClN4 (M + H)+ 311.1, found 311.1.
N4-(3-Chloro-5-(pentafluoro-λ6-sulfaneyl)phenyl)-6-methylpyrimidine-2,4-diamine (36)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.14 g, 0.97 mmol) and 3-chloro-5-(pentafluorosulfanyl)aniline (0.30 g, 1.18 mmol) yielded compound 36 as a white solid (0.22 g, 63%); mp194–195 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H), 8.43 (s, 1H), 7.95 (s, 1H), 7.39 (s, 1H), 6.40 (s, 2H), 5.86 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.6, 163.1, 161.2, 153.7 (sext, JCF = 17.1 Hz), 143.3, 134.1, 121.6, 117.4 114.5, 96.0, 23.8. LC/MS (ESI) calcd for C11H11ClF5N4S (M + H)+ 361.1, found 361.1.
N4-(3-Bromo-5-(pentafluoro- λ6-sulfaneyl)phenyl)-6-methylpyrimidine-2,4-diamine (37)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.09 g, 0.58 mmol) and 3-bromo-5-(pentafluorosulfanyl)aniline (0.21 g, 0.70 mmol) yielded compound 37 as a white solid (0.21 g, 77%); mp 175–176 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 8.48 (t, J = 1.8 Hz, 1H), 8.05 (t, J = 1.8 Hz, 1H), 7.54 (t, J = 1.8 Hz, 1H), 6.36 (s, 2H), 5.86 (s, 1H), 2.11 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.6, 163.1, 161.2, 143.4, 124.5, 122.1, 120.1, 120.0, 114.9, 96.0, 23.9. LC/MS (ESI) calcd for C11H11BrF5N4S (M + H)+ 405.0, found 405.0.
N4-(3-Bromo-5-ethylphenyl)-6-methylpyrimidine-2,4-diamine (38)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.28 g, 1.92 mmol) and 3-bromo-5-ethylaniline (0.50 g, 2.50 mmol) yielded compound 38 as an off-white foam (0.48 g, 81%). 1H NMR (400 MHz, Methanol-d4) δ 7.75 (t, J = 1.8 Hz, 1H), 7.30 (t, J = 1.8 Hz,1H), 6.96 (t, J = 1.8 Hz, 1H), 5.90 (s, 1H), 2.57 (q, J = 7.6 Hz, 2H), 1.19 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 164.7, 162.5, 161.9, 146.7, 141.3, 124.4, 121.7, 120.0, 118.0, 95.6, 28.2, 21.8, 14.5. LC/MS (ESI) calcd for C13H16BrN4 (M + H)+ 307.1, found 307.1.
N4-(3-Bromo-5-ethoxyphenyl)-6-methylpyrimidine-2,4-diamine (39)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.15 g, 1.07 mmol) and 3-bromo-5-ethoxyaniline (0.35 g, 1.29 mmol) yielded compound 39 as a orange oil (0.25 g, 71%). 1H NMR (400 MHz, Methanol-d4) δ 7.35 (t, J = 1.9 Hz, 1H), 7.27 (t, J = 1.9 Hz, 1H), 6.66 (t, J = 1.9 Hz, 1H), 5.90 (s, 1H), 3.98 (q, J = 7.0 Hz, 2H), 2.15 (s, 3H), 1.35 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 164.9, 162.6, 161.9, 160.0, 142.2, 122.0, 114.7, 111.0, 104.9, 95.1, 63.4, 21.8, 13.6. LC/MS (ESI) calcd for C13H16BrN4O (M + H)+ 323.0, found 323.0.
N4-(3-Bromo-5-(2,2,2-trifluoroethoxy)phenyl)-6-methylpyrimidine-2,4-diamine (40)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.12 g, 0.86 mmol) and 3-bromo-5-(2,2,2-trifluoroethoxy)aniline (0.35 g, 1.29 mmol) yielded compound 40 as a white solid (0.21 g, 65%); mp 94–95 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.53–7.44 (m, 2H), 6.87 (s, 1H), 5.96 (s, 1H), 4.54 (q, JHF = 8.6 Hz, 2H), 2.22 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 162.1, 159.9, 158.3, 141.6, 125.0, 122.2, 116.7, 112.1, 105.8, 96.4, 65.2 (q, JCF = 35.4 Hz), 19.7. LC/MS (ESI) calcd for C13H13BrF3N4O (M + H)+ 377.0, found 377.0.
N4-(3-(Allyloxy)-5-bromophenyl)-6-methylpyrimidine-2,4-diamine (41)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.15 g, 1.01 mmol) and 3-(allyloxy)-5-bromoaniline (0.30 g, 1.32 mmol) yielded compound 41 as an off-white foam (0.25 g, 73%). 1H NMR (400 MHz, Acetone-d6) δ 8.38 (s, 1H), 7.57–7.43 (m, 2H), 6.70 (s, 1H), 6.01 (m, 1H), 5.93-5.86 (m 3H), 5.40 (dd, J = 17.4, 1.8 Hz, 1H), 5.24 (dd, J = 10.6, 1.8 Hz, 1H), 4.57 (d, J = 5.5 Hz, 2H), 2.10 (s, 3H). 13C NMR (101 MHz, Acetone-d6) δ 166.2, 163.2, 161.6, 159.8, 143.2, 133.4, 121.9, 116.8, 114.5, 110.9, 104.8, 95.5, 68.6, 22.9. LC/MS (ESI) calcd for C14H16BrN4O (M + H)+ 335.0, found 335.0.
N4-(3-Bromo-5-butoxyphenyl)-6-methylpyrimidine-2,4-diamine (42)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.14 g, 0.94 mmol) and 3-bromo-5-butoxyaniline (0.30 g, 1.23 mmol) yielded compound 42 as a tan oil (0.2 g, 61%). 1H NMR (400 MHz, Acetone-d6) δ 8.35 (s, 1H), 7.53 (s, 1H), 7.37 (s, 1H), 6.68 (s, 1H), 5.94 (s, 1H), 5.85 (s, 2H), 3.99 (t, J = 5.6 Hz, 2H), 2.11 (s, 3H), 1.81–1.63 (m, 2H), 1.56–1.41 (m, 2H), 0.95 (t, J = 5.8 Hz, 3H). 13C NMR (101 MHz, Acetone-d6) δ 166.2, 163.2, 161.7, 160.3, 143.2, 122.0, 114.3, 110.7, 104.5, 95.5, 67.7, 31.1, 22.9, 18.9, 13.2. LC/MS (ESI) calcd for C15H20BrN4O (M + H)+ 351.1, found 351.1.
N4-(3-Bromo-5-isobutoxyphenyl)-6-methylpyrimidine-2,4-diamine (43)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.14 g, 0.99 mmol) and 3-bromo-5-isobutoxyaniline (0.30 g, 1.22 mmol) yielded compound 43 as a tan oil (0.22 g, 65%). 1H NMR (400 MHz, Acetone-d6) δ 8.38 (s, 1H), 7.56 (s, 1H), 7.33 (s, 1H), 6.69 (s, 1H), 6.09–5.84 (m, 3H), 3.74 (d, J = 6.8 Hz, 2H), 2.11 (s, 3H), 2.04 (m, 1H), 0.99 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, Acetone-d6) δ 166.2, 163.3, 161.7, 160.4, 143.1, 122.0, 114.4, 110.8, 104.6, 95.5, 74.3, 28.0, 22.9, 18.5. LC/MS (ESI) calcd for C15H20BrN4O (M + H)+ 351.1, found 351.1.
N4-(3,5-Dibromophenyl)-6-methylpyrimidine-2,4-diamine (44)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (1.08 g, 6.64 mmol) and 3,5-dibromoaniline (2.00 g, 7.97 mmol) yielded compound 44 as an off-white solid (1.92 g, 81%); mp 179–180 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.25 (s, 1H), 7.95 (d, J = 1.8 Hz, 2H), 7.21 (t, J = 1.8 Hz, 1H), 6.39 (s, 2H), 5.85 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.3, 163.2, 161.3, 144.1, 125.6, 122.6, 120.3, 96.0, 23.8. LC/MS (ESI) calcd for C11H11Br2N4 (M + H)+ 357.0, found 357.0.
N4-(3,5-Bis(methylsulfonyl)phenyl)-6-methylpyrimidine-2,4-diamine (45)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.24 g, 1.67 mmol) and 3,5-bis(methylsulfonyl)aniline (0.50 g, 2.00 mmol) yielded compound 45 as a white solid (0.35 g, 59%); mp >290 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, 1H), 8.50 (t, J = 1.5 Hz, 2H), 7.86 (d, J = 1.5 Hz, 1H), 6.34 (s, 2H), 5.91 (s, 1H), 3.33 (s, 6H), 2.12 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.8, 163.1, 161.1, 143.2, 142.8, 121.3, 117.0, 96.0, 43.7, 24.0. LC/MS (ESI) calcd for C13H17N4O4S2 (M + H)+ 357.1, found 357.1.
N4-(3,5-Bis(trifluoromethyl)phenyl)-6-methylpyrimidine-2,4-diamine (46)
According to General Procedure A, pyrimidineamine (1.22 g, 8.52 mmol) and 3,5-bis-trifluoromethylaniline (1.32 g, 8.52 mmol) yielded compound 46 (1.57 g, 55%) as white solid; mp 119–120 °C. 1H NMR (400 MHz, Methanol-d4) δ 8.31 (s, 2H), 7.56 (s, 1H), 6.05 (s, 1H), 2.27 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 162.1, 160.2, 159.7, 141.2, 132.21, 131.9; 131.7 (q, J = 33.2 Hz); 131.6, 131.2, 127.4, 124.7, 121.9, 119.5, 115.3, 96.7, 20.6. LC/MS (ESI) calcd for C13H11F6N4 (M+H)+ 337.1, found 337.1.
N4-(3,5-Dimethoxyphenyl)-6-methylpyrimidine-2,4-diamine (47)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.72 g, 5.00 mmol) and 3,5-dimethoxyaniline (0.76 g, 5.00 mmol) yielded amine 47 as a white solid (1.08 g, 83%); mp 184–185 °C. 1H NMR (400 MHz, CD3OD) δ 6.81 (d, J = 2.3 Hz, 2H), 6.17 (t, J = 2.3 Hz, 1H), 5.94 (s, 1H), 3.75 (s, 6H), 2.15 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 164.6, 162.6, 162.2, 161.0, 141.4, 98.6, 95.4, 94.5, 54.3, 21.8. LC/MS (ESI) calcd for C11H10Cl2N3 (M + H)+ 254.0, found 254.0.
5-((2-Amino-6-methylpyrimidin-4-yl)amino)isophthalic acid (48)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (1.00 g, 6.69 mmol) and 5-aminoisophthalic acid (1.21 g, 6.69 mmol) yielded compound 48 as a white solid (1.25 g, 65%); mp >300 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.13 (br s, 2H), 11.18 (br s, 1H), 8.38 (s, 2H), 8.21–8.18 (m, 3H), 6.27 (s, 1H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO- d6) δ 167.0, 162.3, 156.2, 153.5, 138.9, 132.4, 126.8, 126.2, 97.9, 18.7. LC/MS (ESI) calcd for C13H13N4O4 (M + H)+ 289.1, found 289.1.
Dimethyl 5-((2-amino-6-methylpyrimidin-4-yl)amino)isophthalate (49)
To a stirred solution of 5-((2-amino-6-methylpyrimidin-4-yl)amino)isophthalic acid 48 (0.05 g, 0.17 mmol) in 25 mL of MeOH was added 0.8 mL of conc. H2SO4. 4Å molecular sieves were added and the solution was heated to reflux for 18 h. After cooling to rt, the volatiles were concentrated in vacuo. The residual oil was partitioned between 20 mL of EtOAc and 20 mL of 10% NaOH. The layers were separated and the aqueous layer was extracted with 3 × 20 mL of EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. The resulting dark oil was purified by flash chromatography (0–10% MeOH/DCM) to give amine 49 as a white foam (0.05 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 9.43 (s, 1H), 8.51 (d, J = 1.6 Hz, 2H), 8.00 (t, J = 1.6 Hz, 1H), 6.13 (s, 2H), 5.86 (s, 1H), 3.86 (s, 6H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.7, 166.3, 166.0, 163.2, 142.2, 131.0, 123.9, 122.3, 95.8, 52.9, 23.9. LC/MS (ESI) calcd for C15H17N4O4 (M + H)+ 317.1, found 317.1.
N4-([1,1′:3′,1″-Terphenyl]-5′-yl)-6-methylpyrimidine-2,4-diamine (50)
According to General Procedure D, dibromo-pyrimidineamine 44 (0.5 g, 1.40 mmol) and phenylboronic acid (0.41 g, 3.35 mmol) yielded compound 50 as a yellow foam (0.36 g, 72%). 1H NMR (500 MHz, CDCl3) δ 7.65–7.62 (m, 4H), 7.61–7.58 (m, 2H), 7.52 (d, J = 1.6 Hz, 2H), 7.50–7.45 (m, 4H), 7.43–7.38 (m, 2H), 6.10 (s, 1H), 5.29 (s, 2H), 2.24 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 167.4, 162.9, 162.4, 142.9, 140.6, 139.6, 128.9, 127.7, 127.2, 122.29, 120.2, 94.4, 24.0. LC/MS (ESI) calcd for C23H21N4 (M + H)+ 353.2, found 353.2.
5-((2-Amino-6-methylpyrimidin-4-yl)amino)-N1,N3-bis(3,5-dichlorophenyl)isophthalamide (51)
To a solution of diacid 48 (0.32 g, 1.09 mmol) and 3,5-dichloroaniline (0.45 g, 2.74 mmol) in a solution of 3 mL pyridine, 7 mL DCM and 7 mL of DMF, was added EDI (0.63 g, 3.27 mmol). The solution was stirred at rt for 18 h before being diluted with 50 mL of water. The solution was then extracted with 3 × 50 mL of EtOAc. The combined organic layers were then washed with water followed by brine, dried (Na2SO4) and concentrated in vacuo to afford a brown oil. The oil was purified by flash chromatography (0–20% MeOH/DCM gradient) to afford diamide 51 as a white solid (0.43 g, 69%); mp 235–236 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.71 (s, 2H), 9.48 (s, 1H), 8.41 (d, J = 1.4 Hz, 2H), 8.04 (t, J = 1.4 Hz, 1H), 7.93 (d, J = 1.9 Hz, 4H), 7.37 (t, J = 1.9 Hz, 2H), 6.23 (s, 2H), 5.96 (s, 1H), 2.14 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.3, 166.2, 163.2, 161.5, 141.9, 141.9, 135.7, 134.4, 123.4, 121.9, 119.7, 118.7, 95.6, 24.0. LC/MS (ESI) calcd for C25H19Cl4N6O2 (M + H)+ 575.0, found 575.0.
N4-(3,5-Dimethylphenyl)-6-methylpyrimidine-2,4-diamine (52)
According to General Procedure A, pyrimidineamine (0.76 g, 6.10 mmol) and 3,5-dimethylaniline (0.88 g, 6.10 mmol) yielded compound 52 (0.85 g, 61%) as white solid; mp 163–164 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.10 (s, 2H), 6.67 (s, 1H), 5.89 (s, 1H), 2.25 (s, 6H), 2.12 (s, 3H). 13C NMR (101 MHz Methanol-d4) δ 164.7, 162.7, 162.4, 139.3, 138.1, 124.5, 118.7, 94.8, 21.8, 20.1. LC/MS (ESI) calcd for C13H17N4 (M+H)+ 229.1, found 229.1.
N4-(3,5-Di-tert-butylphenyl)-6-methylpyrimidine-2,4-diamine (53)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.32 g, 2.24 mmol) and 3,5-di-tert-butylaniline (0.55 g, 2.68 mmol) yielded compound 53 as a white solid (0.56 g, 80%); mp 194–195 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 7.39 (d, J = 1.8 Hz, 2H), 7.06 (t, J = 1.8 Hz, 1H), 6.71 (s, 2H), 5.97 (s, 1H), 3.13 (s, 2H), 2.13 (m, 2H), 1.25 (s, 18H). 13C NMR (101 MHz, DMSO-d6) δ 162.1, 160.0, 159.6, 151.1, 139.0, 117.3, 115.9, 95.9, 35.0, 31.7. LC/MS (ESI) calcd for C19H29N4 (M + H)+ 313.2, found 313.2.
6-Methyl-N4-(3-(pentafluoro- λ6-sulfaneyl)phenyl)pyrimidine-2,4-diamine (54)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.27 g, 1.90 mmol) and 3-pentafluorosulfanylaniline (0.50 g, 2.28 mmol) yielded compound 54 as a white solid (0.24 g, 87%); mp 175–176 °C. 1H NMR (400 MHz, Methanol-d4) δ 8.05 (t, J = 2.0 Hz, 1H), 7.96 (m, 1H), 7.40–7.28 (m, 2H), 5.90 (s, 1H), 2.13 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.3, 162.6, 161.8, 153.7 (sext, JCF = 16.6 Hz), 140.8, 128.6, 122.6, 118.6, 116.7, 95.8, 21.9. LC/MS (ESI) calcd for C11H12F5N4S (M + H)+ 327.1, found 327.1.
N4-(3-Chlorophenyl)-6-methylpyrimidine-2,4-diamine (55)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.73 g, 5.06 mmol) and 3-chloroaniline (0.534 g, 5.06 mmol) yielded compound 55 (0.57 g, 48%) as white solid; mp 142–143 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.75 (s, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.16 (t, J = 8.2 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 5.88 (s, 1H), 2.13 (s, 3H). 13C NMR (101 MHz Methanol-d4) δ 165.1, 162.7, 161.9, 141.4, 133.9, 129.5 121.8, 119.6, 118.0, 95.7, 22.0. LC/MS (ESI) calcd for C11H12ClN4 (M+H)+ 235.1, found 235.1.
N4-(3,5-Difluorophenyl)-6-methylpyrimidine-2,4-diamine (60)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (1.09 g, 7.61 mmol) and 3,5-fluoroaniline (0.98 g, 7.61 mmol) yielded compound 60 (0.97 g, 54%) as a white solid; mp 179–180 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.38–7.33 (m, 2H), 6.43 (tt, J = 9.2, 2.4 Hz, 1H), 5.88 (s, 1H), 2.15 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.2, 163.1 (dd, JCF = 242.8, 15.2 Hz), 162.6, 161.7, 142.90 (t, JCF = 13.8 Hz), 101.96 – 101.47 (m), 96.0, 95.9 (dd, JCF = 52.7, 3.4 Hz); 21.9. LC/MS (ESI) calcd for C11H11F2N4 (M+H)+ 237.1, found 237.1.
N4-(3-Chloro-5-fluorophenyl)-6-methylpyrimidine-2,4-diamine (61)
According to General Procedure C, 3-chloro-5-fluoroaniline (0.28 g, 1.94 mmol) and 4-chloro-6-methylpyrimidin-2-amine (0.28 g, 1.94 mmol) yielded compound 61 as a white solid (0.378 g, 77%); mp 143–144 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.63 (br s, 1H), 7.80 (dt, J = 10.2, 2.1 Hz, 1H), 7.53 (q, J = 2.1 Hz, 1H), 6.74 (dt, J = 10.2, 2.1 Hz, 1H), 6.15 (br s, 2H), 5.94 (s, 1H), 2.12 (s, 3H). 13C NMR (101 MHz, Acetone-d6) δ 166.6, 162.9 (d, JCF = 243.6 Hz), 161.4, 163.3, 143.7 (d, JCF = 13.1 Hz), 134.2 (d, JCF = 13.3 Hz), 114.5 (d, JCF = 2.9 Hz), 108.0 (d, JCF = 25.8 Hz), 104.5 (d, JCF = 27.0 Hz) 95.7, 22.90. LC/MS (ESI) calcd for C10H12ClFN4 (M + H)+ 253.1, found 225.1.
N4-(3,5-Dichloro-4-fluorophenyl)-6-methylpyrimidine-2,4-diamine (62)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (1.00 g, 6.69 mmol) and 3,5-dichloro-4-fluoroaniline (1.24 g, 6.69 mmol) yielded compound 62 as a white solid (1.55 g, 81%); mp 204–205 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.48 (br s, 1H), 7.93 (d, JHF = 6.1 Hz, 2H), 5.95 (br s, 2H), 5.91 (s, 1H), 2.11 (s, 3H). 3C NMR (101 MHz, Acetone-d6) δ 166.7, 163.1, 161.3, 148.4 (d, J = 242.2 Hz), 138.2 (d, J = 3.8 Hz), 121.1 (d, J = 17.9 Hz), 119.1, 95.5, 23.0. LC/MS (ESI) calcd for C11H9Cl2FN4 (M + H)+ 287.0, found 287.0.
N4-(3,5-Dibromo-4-fluorophenyl)-6-methylpyrimidine-2,4-diamine (63)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.26 g, 1.84 mmol) and 3,5-dibromo-4-fluoroaniline (0.50 g, 2.39 mmol) yielded compound 63 as a white solid (0.59 g, 85%); mp 205–206 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.02 (d, JHF = 5.7 Hz, 2H), 6.33 (s, 2H), 5.80 (s, 1H), 2.07 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.2, 163.1, 161.2, 149.7 (d, J = 237.2 Hz), 139.6 (d, J = 3.3 Hz), 122.4, 108.8 (d, J = 22.5 Hz), 95.6, 23.9. LC/MS (ESI) calcd for C11H10Br2FN4 (M + H)+ 374.9, found 374.9.
6-Methyl-N4-(3,4,5-trichlorophenyl)pyrimidine-2,4-diamine (64)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.54 g, 3.79 mmol) and 3,4,5-trichloroaniline (0.80 g, 3.79 mmol) yielded compound 64 as a white solid (0.94 g, 82%); mp 219–220 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.00 (s, 2H), 6.37 (s, 2H), 5.83 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 163.1, 161.1, 141.6, 133.0, 120.8, 118.9, 95.9, 23.9. LC/MS (ESI) calcd for C11H10Cl3N4 (M + H)+ 303.0, found 303.3.
N4-(3,5-Dichloro-4-methylphenyl)-6-methylpyrimidine-2,4-diamine (65)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.31 g, 2.18 mmol) and 3,5-dichloro-4-methylaniline (0.50 g, 2.84 mmol) yielded compound 65 as a white solid (0.51 g, 83%); mp 185–186 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 7.80 (s, 2H), 6.30 (s, 2H), 5.81 (s, 1H), 2.28 (s, 3H), 2.07 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.1, 163.2, 161.3, 140.7, 134.5, 125.3, 118.1, 95.7, 23.9, 16.8. LC/MS (ESI) calcd for C12H13Cl2N4 (M + H)+ 283.1, found 283.1.
N4-(3,5-Dichloro-4-phenoxyphenyl)-6-methylpyrimidine-2,4-diamine (66)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.09 g, 0.67 mmol) and 3,5-dichloro-4-phenoxyaniline (0.50 g, 0.78 mmol) yielded compound 66 as an off-white solid (0.20 g, 71%); mp 100–101 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 7.96 (s, 2H), 7.48–7.23 (m, 2H), 7.07 (m, 1H), 6.78 (m, 2H), 6.35 (s, 2H), 5.85 (s, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.1, 163.1, 161.2, 157.2, 140.1, 139.6, 130.3, 128.7, 122.9, 119.1, 114.9, 95.8, 23.9. LC/MS (ESI) calcd for C17H15Cl2N4O (M + H)+ 361.1, found 361.1.
4-((2-Amino-6-methylpyrimidin-4-yl)amino)-2,6-dichlorophenol (67)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.62 g, 4.32 mmol) and 4-amino-2,6-dichlorophenol (1.00 g, 5.62 mmol) yielded compound 67 as an off-white solid (1.00 g, 82%); mp 250 °C (decomposes). 1H NMR (500 MHz, DMSO-d6) δ 8.99 (s, 1H), 7.72 (s, 2H), 6.25 (s, 2H), 5.80 (s, 1H), 2.09 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.6, 163.2, 161.5, 143.9, 134.5, 122.8, 119.6, 95.2, 23.9. LC/MS (ESI) calcd for C11H11Cl2N4O (M + H)+ 285.0, found 285.0.
N4-(3,5-Dichloro-4-(difluoromethoxy)phenyl)-6-methylpyrimidine-2,4-diamine (68)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.13 g, 0.87 mmol) and 3,5-dichloro-4-difluoromethoxy aniline (0.24 g, 1.05 mmol) yielded compound 68 as an off-white solid (0.22 g, 62%); mp 169–170 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 7.93 (s, 2H), 7.03 (t, JHF = 72.8 Hz, 1H), 6.36 (s, 2H), 5.83 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.4, 163.1, 161.1, 140.9, 136.8, 128.7, 118.8, 118.0 (t, J = 261.8 Hz), 95.82, 23.91. LC/MS (ESI) calcd for C12H11Cl2F2N4O (M + H)+ 335.0, found 335.0.
N4-(3,5-Dichloro-4-(dimethylamino)phenyl)-6-methylpyrimidine-2,4-diamine (69)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.36 g, 2.50 mmol) and 2,6-dichloro-N1,N1-dimethylbenzene-1,4-diamine (0.62 g, 3.00 mmol) yielded compound 69 as a clear oil (0.56 g, 72%). 1H NMR (400 MHz, Methanol-d4) δ 7.60 (s, 2H), 5.85 (s, 1H), 2.77 (s, 6H), 2.12 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.1, 162.6, 161.6, 139.9, 138.3, 135.3 119.7, 95.8, 41.2, 22.0. LC/MS (ESI) calcd for C13H16Cl2N5 (M + H)+ 312.1, found 312.1.
N4-(3,5-Dichloro-4-morpholinophenyl)-6-methylpyrimidine-2,4-diamine (70)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.11 g, 0.75 mmol) and 3,5-dichloro-4-morpholinoaniline (0.22 g, 0.89 mmol) yielded compound 70 as a white solid (0.21 g, 81%); mp 114–115 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 7.76 (s, 2H), 6.29 (s, 2H), 5.81 (s, 1H), 3.71–3.54 (m, 4H), 3.11–2.98 (m, 4H), 2.07 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.1, 161.2, 140.0, 137.3, 134.9, 119.1, 95.7, 67.5, 49.9, 23.8. LC/MS (ESI) calcd for C15H18Cl2N5O (M + H)+ 354.1, found 354.1.
N4-(3,5-Dichloro-4-(piperidin-1-yl)phenyl)-6-methylpyrimidine-2,4-diamine (71)
According to General Procedure A, 4-chloro-6-methylpyrimidin-2-amine (0.37 g, 2.54 mmol) and 3,5-dichloro-4-(piperidin-1-yl)aniline (0.75 g, 3.06 mmol) yielded compound 71 as a yellow foam (0.70 g, 79%). 1H NMR (400 MHz, DMSO-d6) δ 9.14 (s, 1H), 7.73 (s, 2H), 6.24 (s, 2H), 5.80 (s, 1H), 3.05 – 2.95 (m, 4H), 2.05 (s, 3H), 1.62-1.44 (m, 6H). 13C NMR (101 MHz DMSO-d6) δ 166.1, 163.2, 161.3, 139.4, 139.0, 134.7, 119.1, 95.6, 51.1, 26.8, 24.3, 23.9. LC/MS (ESI) calcd for C16H20Cl2N5(M + H)+ 352.1, found 352.1.
2-((2-Amino-6-methylpyrimidin-4-yl)amino)-4,6-dichlorophenol (72)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (1.00 g, 6.96 mmol) and 2-amino-4,6-dichlorophenol (1.48 g, 8.35 mmol) yielded compound 72 as a tan solid (1.40 g, 71%); mp 216–217 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.53 (d, J = 2.6 Hz, 1H), 7.13 (d, J = 2.6 Hz, 1H), 6.02 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.7, 161.9, 161.1, 144.2, 131.8, 123.5, 123.4, 122.9, 120.6, 95.6, 23.7. LC/MS (ESI) calcd for C11H11Cl2N4O (M + H)+ 285.0, found 285.0.
N4-(3,5-Dichloro-2,4-difluorophenyl)-6-methylpyrimidine-2,4-diamine (73)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.30 g, 2.10 mmol) and 3,5-dichloro-2,4-difluoroaniline (0.50 g, 2.52 mmol) yielded compound 73 as a white solid (0.63 g, 81%); mp 203–204 °C. 1H NMR (400 MHz, Methanol-d4) δ 8.38 (t, JHF = 7.9 Hz, 1H), 6.03 (s, 1H), 2.18 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.8, 162.6, 161.9, 122.2, 95.6, 21.9 (halogenated carbons not visible). LC/MS (ESI) calcd for C11H8Cl2F2N4 (M + H)+ 305.0, found 305.0.
N4-(3′,5′-Dichloro-[1,1′-biphenyl]-4-yl)-6-methylpyrimidine-2,4-diamine (89)
According to General Procedure C, 4-chloro-6-methylpyrimidin-2-amine (0.38 g, 2.62 mmol) and 3′,5′-dichloro-[1,1′-biphenyl]-4-amine (0.75 g, 3.15 mmol) yielded compound 89 as a white solid (0.69 g, 76%); mp 237–238 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.65 (s, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.46 (s, 1H), 6.20 (br s, 2H), 5.90 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 163.3, 161.5, 144.0, 142.1, 135.0, 129.9, 127.6, 126.3, 125.0, 119.7, 95.6, 23.9. LC/MS (ESI) calcd for C17H15Cl2N4 (M + H)+ 345.1, found 345.1.
N4-(5-Chloro-4′-fluoro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (91)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 91 as an off-white solid (0.20 g, 65%); mp 105–106 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.81 (t, J = 1.9 Hz, 1H), 7.62 (t, J = 1.9 Hz, 1H), 7.60–7.55 (m, 2H), 7.17–7.11 (m, 3H), 5.92 (s, 1H), 2.16 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.1, 164.0, 162.8 (d, JCF = 245.9 Hz), 161.9, 141.9 (d, JCF = 16.2 Hz), 135.99, 135.95, 134.4, 128.5 (d, JCF = 8.2 Hz), 120.0, 118.1, 116.2, 115.2 (d, JCF = 21.9 Hz), 95.7, 21.8. LC/MS (ESI) calcd for C17H15ClFN4 (M + H)+ 329.1, found 329.1.
N4-(4′,5-Dichloro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (92)
According to General Procedure D, bromo-pyrimidineamine 32 (0.50 g, 1.59 mmol) yielded compound 92 as a yellow foam (0.39 g, 72%). 1H NMR (400 MHz, DMSO-d6) δ 9.26 (s, 1H), 7.93 (t, J = 1.8 Hz, 1H), 7.83 (t, J = 1.8 Hz, 1H), 7.66 (d, J = 8.5 Hz, 2H), 7.47 (d, J = 8.5 Hz, 2H), 7.20 (t, J = 1.8 Hz, 1H), 6.32 (s, 2H), 5.88 (s, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.2, 161.6, 143.2, 141.3, 138.2, 134.3, 133.3, 129.4, 129.0, 119.2, 117.9, 116.0, 95.8, 23.9. LC/MS (ESI) calcd for C17H15Cl2N4 (M + H)+ 345.1, found 345.1.
N4-(5-Chloro-4′-methoxy-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (93)
According to General Procedure D, bromo-pyrimidineamine 32 (0.46 g, 1.45 mmol) yielded compound 93 as a yellow foam (0.30 g, 61%). 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 7.90 (t, J = 1.8 Hz, 1H), 7.71 (t, J = 1.8 Hz, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.15 (t, J = 1.8Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 6.25 (s, 2H), 5.87 (s, 1H), 3.77 (s, 3H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.2, 161.6, 159.7, 143.1, 142.3, 134.1, 131.6, 128.3, 118.8, 117.0, 115.5, 114.8, 95.7, 55.6, 23.9. LC/MS (ESI) calcd for C18H18ClN4O (M + H)+ 341.1, found 341.1.
N4-(5-Chloro-4′-methyl-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (94)
According to General Procedure D, bromo-pyrimidineamine 32 (0.50 g, 1.59 mmol) yielded compound 94 as a yellow oil (0.40 g, 81%). 1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 7.98 (t, J = 1.8 Hz, 1H), 7.73 (t, J = 1.8 Hz, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 7.16 (t, J = 1.8 Hz, 1H), 6.30 (s, 2H), 5.89 (s, 1H), 2.30 (s, 3H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.3, 161.6, 143.1, 142.6, 137.8, 136.5, 134.1, 130.0, 127.0, 119.1, 117.4, 115.8, 95.8, 23.9, 21.1. LC/MS (ESI) calcd for C18H18ClN4 (M + H)+ 325.1, found 325.1.
N4-(5-Chloro-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (95)
According to General Procedure D, bromo-pyrimidineamine 32 (0.5 g, 1.59 mmol) yielded compound 95 as a yellow foam (0.41 g, 67%). 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 7.96 (m, 2H), 7.85 (d, J = 8.3 Hz, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.25 (t, J = 1.7 Hz, 1H), 6.38 (s, 2H), 5.91 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.3, 161.6, 143.4, 141.0, 134.3, 128.0, 128.8 (q, JCF = 31.9 Hz), 126.2 (q, JCF = 3.7 Hz), 126.0, 123.3, 119.5, 118.4, 116.3, 95.9, 23.8. LC/MS (ESI) calcd for C18H15ClF3N4 (M + H)+ 379.1, found 379.1.
3′-((2-Amino-6-methylpyrimidin-4-yl)amino)-5′-chloro-[1,1′-biphenyl]-4-carbonitrile (96)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 96 as a tan solid (0.28 g, 88%); mp 197–198 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 7.95 (m, 1H), 7.92 (t, J = 1.8 Hz, 1H), 7.90–7.82 (m, 4H), 7.27 (t, J = 1.8 Hz, 1H), 6.32 (s, 2H), 5.87 (s, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.1, 163.2, 161.4, 143.9, 143.3 140.7, 134.5, 133.3, 128.2, 119.5, 119.2, 118.5, 116.2, 111.0, 95.8, 23.9. LC/MS (ESI) calcd for C18H15ClN5 (M + H)+ 336.1, found 336.1.
1-(3′-((2-Amino-6-methylpyrimidin-4-yl)amino)-5′-chloro-[1,1′-biphenyl]-4-yl)ethan-1-one (97)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 97 as a white solid (0.24 g, 72%); mp 225 °C (chars). 1H NMR (400 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.02 (d, J = 8.3 Hz, 2H), 7.97 (s, 1H), 7.91 (s, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.29 (s, 1H), 6.28 (s, 2H), 5.87 (s, 1H), 2.59 (s, 3H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 197.9, 166.1, 163.2, 161.5, 143.6, 143.3, 141.3, 136.5, 134.3, 129.4, 127.45, 119.4, 118.3, 116.2, 95.8, 27.2, 23.9. LC/MS (ESI) calcd for C19H18ClN4O (M + H)+ 353.1, found 353.1.
3′-((2-Amino-6-methylpyrimidin-4-yl)amino)-5′-chloro-[1,1′-biphenyl]-4-ol (98)
According to General Procedure D, bromo-pyrimidineamine 32 (0.31 g, 0.99 mmol) yielded compound 98 as a white solid (0.22 g, 69%); mp 142–143 °C. 1H NMR (400 MHz DMSO-d6) δ 9.18 (s, 1H), 7.86 (s, 1H), 7.66 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.11 (s, 1H), 6.82 (d, J = 8.0 Hz, 2H), 6.24 (s, 2H), 5.86 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.9, 163.2, 161.6 158.0, 143.0, 142.7, 134.0, 130.0, 128.3, 118.6, 116.7, 116. 2, 115.3, 95.7, 23.9. LC/MS (ESI) calcd for C17H16ClN4O (M + H)+ 327.1, found 327.1.
N4-(5-Chloro-[1,1′:4′,1″-terphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (99)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 99 as a tan solid (0.25 g, 69%); mp 104–105 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.25 (s, 1H), 7.96 (t, J = 1.7 Hz, 1H), 7.84 (t, J = 1.7 Hz, 1H), 7.75 (s, 4H), 7.70 (d, J = 7.6 Hz, 2H), 7.46 (d, J = 7.6 Hz, 2H), 7.35 (m, 1H), 7.27 (t, J = 1.7 Hz, 1H), 6.27 (s, 2H), 5.88 (s, 1H), 2.09 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.3, 161.6 143.2, 142.0, 140.1, 139.9, 138.3, 134.2, 129. 5, 128.1, 127.7, 127.7, 127.0, 119.2, 117.7, 115.9, 95.8, 24.0. LC/MS (ESI) calcd for C23H20ClN4 (M + H)+ 387.2, found 387.2.
N4-(5-Chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (100)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 100 as a yellow solid (0.28 g, 82%); mp 104–105 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.70 (t, J = 1.8 Hz, 1H), 7.51 (t, J = 1.8 Hz, 1H), 7.40 (d, J = 8.8 Hz, 2H), 7.12 (t, J = 1.8 Hz, 1H), 6.75 (d, J = 8.8 Hz, 2H), 5.91 (s, 1H), 2.90 (s, 6H), 2.14 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.0, 162.7, 161.9, 150.5, 143.2, 141.5, 134.2, 127.3, 127.0, 119.4, 117.0, 115.4, 112.6, 95.7, 39.3, 21.9. LC/MS (ESI) calcd for C19H21ClN5 (M + H)+ 354.2, found 354.2.
N4-(2′,5-Dichloro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (101)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 101 as an off-white solid (0.22 g, 66%); mp 83–84 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.91 (m, 1H), 7.48 (m, 1H), 7.44 (t, J = 1.7 Hz, 1H), 7.37 – 7.30 (m, 3H), 6.99 (t, J = 1.7 Hz, 1H), 5.95 (s, 1H), 2.16 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.1, 162.7, 161.9, 141.2, 141.2, 139.2, 133.5, 131.9, 130.9, 129.6, 128.9, 126.9, 122.5, 118.8, 118.5, 95.7, 21.8. LC/MS (ESI) calcd for C17H15Cl2N4 (M + H)+ 345.1, found 345.1.
N4-(3′,5-Dichloro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (102)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 102 as an off-white solid (0.23 g, 71%); mp 169–170 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.91 (m, 1H), 7.57 (t, J = 1.9 Hz, 1H), 7.54 (t, J = 1.9 Hz, 1H), 7.47 (m, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.33 (m, 1H), 7.15 (t, J = 1.9 Hz, 1H), 5.92 (s, 1H), 2.16 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.1, 162.7, 161.8, 141.9, 141.7, 141.5, 134.5, 134.4, 130.0, 127.5, 126.5, 125.0, 120.1, 118.6, 116.2, 95.8, 21.9. LC/MS (ESI) calcd for C17H15Cl2N4 (M + H)+ 345.1, found 345.1.
6-Methyl-N4-(3′,4′,5-trichloro-[1,1′-biphenyl]-3-yl)pyrimidine-2,4-diamine (103)
According to General Procedure D, bromo-pyrimidineamine 32 (0.29 g, 0.93 mmol) yielded compound 103 as a yellow foam (0.27 g, 76%). 1H NMR (400 MHz, CDCl3) δ 7.55 (s, 1H), 7.49–7.42 (m, 2H), 7.34–7.27 (m, 2H), 7.20–7.09 (m, 2H), 5.94 (s, 1H), 5.18 (s, 2H), 2.21 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 162.7, 161.4, 141.0, 140.7, 139.4, 135.3, 133.0, 132.3, 130.8, 128.8, 126.2, 122.0, 120.5, 117.8, 95.2, 23.9. LC/MS (ESI) calcd for C17H14Cl3N4 (M + H)+ 379.0, found 379.0.
(3′,5-Dichloro-4′-fluoro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (104)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 104 as an off-white solid (0.22 g, 64%); mp 129–130 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.90 (t, J = 1.8 Hz, 1H), 7.70 (m, 1H), 7.64 (t, J = 1.8 Hz, 1H), 7.56 (m, 1H), 7.31 (m, 1H), 7.19 (t, J = 1.8 Hz, 1H), 5.95 (s, 1H), 2.17 (s, 3H). LC/MS (ESI) calcd for C17H14Cl2FN4 (M + H)+ 363.1, found 363.1.
N4-(4′,5-Dichloro-3′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (105)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 105 as an off-white foam (0.22 g, 67%). 1H NMR (500 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.07 (m, 1H), 8.01 (m, 2H), 7.91–7.78 (m, 2H), 7.39 (s, 1H), 6.32 (s, 2H), 5.91 (s, 1H), 2.13 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 166.1, 163.2, 161.5, 143.36, 139.9, 138.9, 134.4, 134.3 132.9, 132.7, 126.4, 119.5, 118.4, 117.0, 116.2, 95.8, 24.0 (CF3 carbon not visible). LC/MS (ESI) calcd for C18H14Cl2F3N4 (M + H)+ 413.1, found 413.1.
N4-(5-Chloro-3′,5′-bis(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (106)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 106 as a tan solid (0.26 g, 61%); mp 159–160 °C. 1H NMR (400 MHz, Methanol-d4) δ 8.13 (s, 2H), 8.08 (t, J = 1.8 Hz, 1H), 7.96 (s, 1H), 7.67 (t, J = 1.8 Hz, 1H), 7.29 (t, J = 1.8 Hz, 1H), 5.94 (s, 1H), 2.17 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 165.3, 162.7, 161.8, 142.3, 142.3, 139.7, 135.0, 131.9 (q, JCF = 33.2 Hz), 127.1, 122.0, 121.0, 120.1, 119.4, 116.3, 95.9, 21.8. LC/MS (ESI) calcd for C19H14ClF6N4 (M + H)+ 447.1, found 447.1.
N4-(3-Chloro-5-(naphthalen-2-yl)phenyl)-6-methylpyrimidine-2,4-diamine (107)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 107 as tan solid (0.26 g, 75%); mp 189–190 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.20 (m, 1H), 8.04 (t, J = 1.8 Hz, 1H), 8.02–7.96 (m, 2H), 7.94–7.88 (m, 2H), 7.80 (m, 1H), 7.56–7.47 (m, 2H), 7.37 (t, J = 1.8 Hz, 1H), 6.31 (s, 2H), 5.90 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.3, 161.6, 143.3, 142.5, 136.7, 134.3, 133.7, 132.9, 129.0, 128.8, 127.9, 126.9, 126.8, 126.0, 125.4, 119.6, 117.8, 116.3, 95.8, 23.9. C/MS (ESI) calcd for C21H18ClN4 (M + H)+ 361.1, found 361.1.
N4-(3-Chloro-5-(phenanthren-9-yl)phenyl)-6-methylpyrimidine-2,4-diamine (108)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 108 as a white solid (0.28 g, 71%); mp 110–111 °C. 1H NMR (400 MHz, Methanol-d4) δ 8.75 (d, J = 8.4 Hz, 1H), 8.69 (d, J = 8.4 Hz, 1H), 7.94 (s, 1H), 7.90–7.78 (m, 2H), 7.64–7.46 (m, 6H), 7.05 (s, 1H), 5.89 (s, 1H), 2.12 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 164.98, 162.6, 161.9, 142.6, 141.4, 137.2, 133.9, 131.3, 130.6, 130.4, 129.9, 128.4, 127.1, 126.6, 126.6, 126.3, 126.3, 126.1, 123.0, 122.7, 122.1, 119.2, 118.3, 95.8, 21.8. LC/MS (ESI) calcd for C25H20ClN4 (M + H)+ 411.1, found 411.1.
N4-(3-(Benzo[d][1,3]dioxol-5-yl)-5-chlorophenyl)-6-methylpyrimidine-2,4-diamine (109)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 109 a tan solid (0.27 g, 82%); mp 225–226 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 7.93 (t, J = 2.0 Hz, 1H), 7.64 (t, J = 2.0 Hz, 1H), 7.20 (t, J = 2.0 Hz, 1H), 7.16–7.08 (m, 2H), 6.96 (m, 1H), 6.26 (s, 2H), 6.04 (s, 2H), 5.87 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.3, 161.6, 148.4, 147.7, 143.0, 142.4, 134.0, 133.6, 121.0, 119.2, 117.3, 115.9, 109.1, 107.6, 101.7, 95.7, 23.9. LC/MS (ESI) calcd for C18H16ClN4O2 (M + H)+ 355.1, found 355.1.
N4-(3-(Benzo[c][1,2,5]oxadiazol-5-yl)-5-chlorophenyl)-6-methylpyrimidine-2,4-diamine (110)
According to General Procedure E, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 110 as a yellow solid (0.24 g, 71%); mp 234–235 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.29 (d, J = 1.6 Hz, 1H), 8.12 (m, 1H), 8.05–7.96 (m, 2H), 7.93 (m, 1H), 7.42 (d, J = 1.6 Hz, 1H), 6.34 (s, 2H), 5.89 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.2, 163.3, 161.5, 149.9, 148.9, 143.3, 143.1, 140.3, 134.4, 133.8, 119.9, 119.0, 117.15, 116.7, 113.3, 95.8, 24.0. LC/MS (ESI) calcd for C17H14ClN6O (M + H)+ 353.1, found 353.1.
N4-(3-Chloro-5-(furan-3-yl)phenyl)-6-methylpyrimidine-2,4-diamine (111)
According to General Procedure E, bromo-pyrimidineamine 32 (0.30 g, 0.95 mmol) yielded compound 111 as a white foam (0.21 g, 72%). 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.24 (s, 1H), 7.91 (m, 1H), 7.72 (t, J = 1.8 Hz, 1H), 7.66 (t, J = 1.8 Hz, 1H), 7.21 (t, J = 1.8 Hz, 1H), 6.95 (m, 1H), 6.32 (s, 2H), 5.86 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.9, 163.3, 161.6, 144.8, 143.2, 140.6, 134.5, 134.0, 125.4, 118.2, 117.1, 114.5, 109.2, 95.3, 23.9. LC/MS (ESI) calcd for C15H14ClN4O (M + H)+ 301.1, found 301.1.
N4-(3-(Benzofuran-2-yl)-5-chlorophenyl)-6-methylpyrimidine-2,4-diamine (112)
According to General Procedure E, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 112 as an off-white foam (0.23 g, 69%). 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.09 (t, J = 1.8 Hz, 1H), 8.01 (t, J = 1.8 Hz, 1H), 7.72–7.58 (m, 2H), 7.52 (s, 1H), 7.48 (t, J = 1.8 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 7.25 (t, J = 7.5 Hz, 1H), 6.34 (s, 2H), 5.89 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.1, 163.2, 161.5, 154.7, 154.2, 143.4, 134.4, 132.0, 129.1, 125.4, 123.8, 121.8, 118.6, 117.0, 113.4, 111.7, 103.7, 95.9, 23.4. LC/MS (ESI) calcd for C19H15ClN4O (M + H)+ 351.1, found 351.1.
N4-(3-Chloro-5-(thiophen-3-yl)phenyl)-6-methylpyrimidine-2,4-diamine (113)
According to General Procedure E, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 113 as a yellow foam (0.22 g, 74%). 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 7.97 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 2.2 Hz, 1H), 7.60 (m, 1H), 7.53 (s, 1H), 7.30 (d, J = 2.2 Hz, 1H), 6.34 (s, 2H), 5.89 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 163.3, 161.6, 143.2, 140.6, 137.5, 134.1, 127.7 126.6, 122.6, 118.76, 117.3, 115.2, 95.8, 23.9. LC/MS (ESI) calcd for C15H14ClN4S (M + H)+ 317.1, found 317.1.
N4-(3-(Benzo[b]thiophen-2-yl)-5-chlorophenyl)-6-methylpyrimidine-2,4-diamine (114)
According to General Procedure E, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 114 as an off-white foam (0.21 g, 64%). 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.01–7.90 (m, 4H), 7.84 (m, 1H), 7.38–7.34 (m, 3H), 6.31 (s, 2H), 5.89 (s, 1H), 2.10 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.2, 163.2, 161.5, 143.4, 142.2, 140.7, 139.1, 135.8, 134.4, 125.4, 125.3, 124.5, 122.9, 121.6, 118.3, 118.2, 115.1, 95.9, 23.9. LC/MS (ESI) calcd for C19H16ClN4S (M + H)+ 367.1, found 367.1.
N4-(3-Chloro-5-(thiazol-5-yl)phenyl)-6-methylpyrimidine-2,4-diamine (115)
According to General Procedure E bromo-pyrimidineamine 32 (0.30 g, 0.96 mmol) yielded compound 115 as a yellow solid (0.25 g, 81%); mp 220–221 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.29 (s, 1H), 9.09 (s, 1H), 8.36 (s, 1H), 7.95 (t, J = 1.8 Hz, 1H), 7.81 (t, J = 1.8 Hz, 1H), 7.30 (t, J = 1.8 Hz, 1H), 6.30 (s, 2H), 5.86 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.1, 163.2, 161.4, 154.6, 143.4, 140.7, 137.8, 134.4, 133.0, 118.6, 118.2, 115.6, 95.8, 23.9. LC/MS (ESI) calcd for C14H13ClN5S (M + H)+ 318.1, found 318.1.
N4-(3-Chloro-5-(1-methyl-1H-pyrazol-4-yl)phenyl)-6-methylpyrimidine-2,4-diamine (116)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 116 as a white solid (0.29 g, 88%); mp 204–205 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.13 (s, 1H), 8.15 (s, 1H), 7.98–7.79 (m, 2H), 7.54 (s, 1H), 7.15 (s, 1H), 6.29 (s, 2H), 5.85 (s, 1H), 3.84 (s, 3H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.9, 163.23, 161.6, 143.2, 136.7, 135.1, 133.9, 128.8, 121.3, 117. 6, 116.1, 113.9, 95.7, 39.1, 23.9. LC/MS (ESI) calcd for C15H16ClN6 (M + H)+ 315.1, found 315.1.
N4-(3-Chloro-5-(pyrimidin-5-yl)phenyl)-6-methylpyrimidine-2,4-diamine (117)
According to General Procedure D, bromo-pyrimidineamine 32 (0.3 g, 0.95 mmol) yielded compound 117 as a white solid (0.25 g, 84%); mp 184–185 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.32 (s, 1H), 9.19 (s, 1H), 9.10 (s, 2H), 7.99 (s, 1H), 7.90 (s, 1H), 7.39 (s, 1H), 6.33 (s, 2H), 5.87 (s, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 161.7, 157.5, 155.0, 147.0, 146.0, 145.8, 142.9, 136.3, 119.8, 119.1, 116.1, 95.9, 22.5. LC/MS (ESI) calcd for C15H14ClN6 (M + H)+ 313.1, found 313.1.
N4-(5-Ethyl-4′-fluoro-[1,1′-biphenyl]-3-yl)-6-methylpyrimidine-2,4-diamine (118)
According to General Procedure D, bromo-pyrimidineamine 38 (0.21 g, 0.70 mmol) yielded compound 118 a tan oil (0.19 g, 83%). 1H NMR (500 MHz, Methanol-d4) δ 7.65–7.59 (m, 3H), 7.36 (t, J = 1.7 Hz, 1H), 7.19–7.13 (m, 2H), 7.10 (t, J = 1.7 Hz, 1H), 5.97 (s, 1H), 2.70 (q, J = 7.6 Hz, 2H), 2.18 (s, 3H), 1.29 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, Methanol-d4) δ 164.8, 163.4, 162.3, 162.1 (d, J CF = 162.1 Hz), 145.5, 140.7, 140.2, 137.5 (d, J CF = 3.3 Hz), 128.4 (d, JCF = 8.0 Hz), 120.9, 119.0, 116.7, 115.0 (d, JCF = 21.5 Hz), 95.2, 28.6, 21.9, 14.8. LC/MS (ESI) calcd for C19H20FN4 (M + H)+ 323.2, found 323.2.
Biological Assays
Materials
Reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA) unless specified. S-Adenosyl-L-methionine (AdoMet) sulfate p-toluenesulfonate salt was purchased from Affymetrix (Santa Clara, CA, USA), Gibco Iscove’s modified Dulbecco’s medium (IMDM) was from Thermo Fisher Scientific (Waltham, MA, USA). Compound 1 was kindly provided by Genzyme (now Sanofi-Genzyme, Waltham, MA, USA). Pyrimidineamine compound stocks were made in dimethyl sulfoxide (DMSO), with an initial stock concentration of 25 or 50 mM (or the highest achievable concentration for compounds with lower solubility). Stocks were stored at –20 °C and thawed once to generate fresh solutions in DMSO for enzyme and cell-based assays. DMSO based dilution series for the enzyme assays were generated using Echo 555 acoustic liquid dispenser (Labcyte, Sunnyvale, CA, USA); final DMSO concentrations in the assays were 2%. Cell growth inhibition was determined for a 3-fold serial dilution series generated in DMSO so that final DMSO concentrations in the assay were 0.1%.
Protein Expression and Purification
Heterologous expression and purification of recombinant TbAdoMetDC/prozyme heterodimer and HsAdoMetDC used in the enzyme activity assays and for structure determination were carried out as previously described.21, 47 The purified TbAdoMetDC/prozyme complex was concentrated to 73 mg/mL and stored at –80 °C.
Mass Spectrometry-Based AdoMetDC Activity Assay
The steady-state activity of TbAdoMetDC/prozyme and HsAdoMetDC were measured using RapidFire–MS-based assay as previously described.22 Inhibition data were acquired in a concentration–response format based on a 3-fold dilution series over the range from 0.01 μM to 180 μM final of inhibitor. Data were normalized to a fully inhibited control (0.5 μM 1) and fitted to the normalized response versus log(inhibitor concentration) equation in Prism constraining bottom and Hill slope to 0 and –1, respectively, to determine the IC50.
Parasite Culture, Cell Viability Assay, and Western Blot Analysis
T. brucei brucei Lister 427 bloodstream-form parasites were cultured in HMI-19 medium supplemented with fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA, USA) in 96-well plates for the cell viability assay or in culture flasks for the prozyme Western blot analysis, as previously described.22 Cell densities in the cell viability assay were analyzed using ATP–bioluminescence assay with CellTiter-Glo reagent (Promega, Madison, WI, USA) after 48 h culturing in the presence of serial dilutions of compound (ranging from 0.0038 to 25 μM or from 0.015 to 100 μM final in the assay) and at a fixed concentration of vehicle (0.1% DMSO) as previously described.22 The data were fitted to the normalized response versus log(inhibitor concentration) equation in Prism constraining bottom to 0. Hill slope was constrained to –8 (based on the best-fit value of a four-parameter fit in Prism) when the steepness of the curve resulted in a fit with an ambiguous EC50 value (see Table S1). The Western blot analysis of prozyme levels in the treated cell lysates was performed as previously described.22
In Vitro Metabolism and Distribution Analysis
Compound metabolic stability was assessed using male CD-1 mouse and mixed-gender human hepatic microsomes as previously described.22 BBB permeability assay was done using MDCKII-hMDR1 monolayer cells as previously described.22
X-ray Crystallography
Protein Crystallization and Data Collection
TbAdoMetDC/prozyme was diluted to final concentration of 4 mg/mL into a crystallization buffer (50 mM bis-tris propane, pH 7.2, 50 mM NaCl, 4 mM tris(2-carboxyethyl)phosphine (TCEP), 2 mM putrescine) containing 0.5 mM 44 added as 50 mM DMSO stock yielding a final DMSO concentration of 1%. Protein and the inhibitor were allowed to equilibrate at room temperature for 3 h, after which the sample was filtered at 23 °C using Ultrafree-MC 0.22 μm PVDF centrifugal filter units (Merck Millipore, Tullagreen, Ireland). Crystals were obtained using hanging-drop vapor diffusion method by combining 1 μL of the protein in crystallization buffer, 1 μL of reservoir solution, and 0.5 μL of unliganded protein microseeds obtained using Seed–Bead method (Hampton Research, Aliso Viejo, CA, USA) and diluted 50- fold as previously described.21 Reservoir solution contained 17% PEG 6000 and 100 mM bis-tris propane, pH 7.0. Crystals formed on day 5 and were prepared for data collection by cryoprotection in a combination of the crystallization buffer and reservoir solution supplemented with 18% ethylene glycol. Crystals were then mounted and cryo-chilled in liquid nitrogen. Diffraction data were collected at the Br K-edge at the Structure Biology Center at the Advance Photon Source, Beamline 19-ID at 100 K(see Table S2). Data reduction was carried out by HKL package.48
Structure Determination and Model Refinement
Previously solved atomic coordinates of TbAdoMetDC/prozyme (PDB identifier 5TVM)21 were used as a search model for molecular replacement in Phaser49 as implemented in Phenix (v1.10.1-2155).50 The coordinates and geometry restraints for 44 were generated from a chemical formula in Elbow.51 The ligand was placed after initial positional refinement in Phenix44 on the basis of mFo−DFc positive density in the active site and the anomalous diffraction map (Figure S1). The structure was further improved through alternating manual model building in Coot52 and positional, individual B-factor, occupancy, and anomalous groups refinement in Phenix. Visualization of the atomic coordinates and electron densities was done using PyMOL molecular graphics system (Version 1.8, Schrödinger).
Supplementary Material
Table S1 with structures and biological assay data for all tested compounds.
Table S2 with X-ray crystallography data collection and refinement statistics.
Figure S1 with electron density maps supporting ligand placement in the X-ray crystal structure.
Acknowledgments
This work was supported by National Institutes of Health Grants 2R37AI034432 (to M.A.P.) and R01AI090599 (to M.A.P. and J.K.D.B.). M.A.P. and J.K.D.B. acknowledge the support of the Welch Foundation (Grants I-1257 and I-1422). M.A.P. holds the Sam G. Winstead and F. Andrew Bell Distinguished Chair in Biochemistry, and J.K.D.B. the Julie and Louis Beecherl, Jr. Chair in Medical Science. The authors thank Sara Schock for help with in vitro permeability studies, and Thomas E. Richardson, both of Scynexis, Inc. (now Avista Pharma Solutions) for helpful medicinal chemistry discussions; and from UT Southwestern, Diana R. Tomchick for advise on crystallization studies, Karen MacMillan for her help in preparing the manuscript, Melissa McCoy and Bruce A. Posner for the RapidFire–mass spectrometry analysis of the enzyme assay, Lin You and Chuo Chen for LC–MS analysis of compound purity, and Shihua Zhong for performing parasite viability assays. Structural data shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Abbreviations
- HAT
human African trypanosomiasis
- AdoMet
S-adenosyl-L-methionine
- AdoMetDC
S-adenosylmethionine decarboxylase
- dcAdoMet
decarboxylated S-adenosyl-L-methionine
- Tb
T. brucei
- Hs
Homo sapiens
- BBB
blood−brain barrier
- CNS
central nervous system
- NECT
nifurtimox–eflornithine combination therapy
- HTS
high-throughput screening
- SAR
structure−activity relationship
- MS
mass spectrometry
- Pgp
P-glycoprotein 1
- DMSO
dimethyl sulfoxide
- Pvl
pyruvoyl prosthetic group
- PDB
Protein Data Bank
Footnotes
Copies of H- and 13C-NMR spectra of synthesized analogs.
Major datasets
|
| |||
| Authors | Dataset title | Database information | Dataset URL |
|
| |||
| Volkov, O.A., Chen, Z., Phillips, M.A. | Crystal structure of Trypanosoma brucei AdoMetDC/prozyme heterodimer in complex with pyrimidineamine inhibitor 44 | Publicly available at the RCSB Protein Data Bank (accession number 6BM7) | http://www.rcsb.org/pdb/explore/explore.do?structureId=6BM7 |
|
| |||
Authors will release the atomic coordinates and experimental data upon article publication.
References
- 1.WHO. Neglected tropical diseases. http://www.who.int/neglected_diseases/diseases/en/ (accessed Jun 12,2017)
- 2.Kennedy PGE. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness) Lancet Neurol. 2013;12:186–194. doi: 10.1016/S1474-4422(12)70296-X. [DOI] [PubMed] [Google Scholar]
- 3.Franco JR, Simarro PP, Diarra A, Jannin JG. Epidemiology of human African trypanosomiasis. Clin Epidemiol. 2014;6:257–275. doi: 10.2147/CLEP.S39728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO. Trypanosomiasis, human African (sleeping sickness) http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed Jun 12, 2017)
- 5.Franco JR, Simarro PP, Diarra A, Ruiz-Postigo JA, Jannin JG. The journey towards elimination of gambiense human African trypanosomiasis: not far, nor easy. Parasitology. 2014;141:748–760. doi: 10.1017/S0031182013002102. [DOI] [PubMed] [Google Scholar]
- 6.Berthier D, Breniere SF, Bras-Goncalves R, Lemesre JL, Jamonneau V, Solano P, Lejon V, Thevenon S, Bucheton B. Tolerance to trypanosomatids: a threat, or a key for disease elimination? Trends Parasitol. 2016;32:157–168. doi: 10.1016/j.pt.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Capewell P, Cren-Travaille C, Marchesi F, Johnston P, Clucas C, Benson RA, Gorman TA, Calvo-Alvarez E, Crouzols A, Jouvion G, Jamonneau V, Weir W, Stevenson ML, O’neill K, Cooper A, Swar NK, Bucheton B, Ngoyi DM, Garside P, Rotureau B, Macleod A. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi: 10.7554/eLife.17716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trindade S, Rijo-Ferreira F, Carvalho T, Pinto-Neves D, Guegan F, Aresta-Branco F, Bento F, Young SA, Pinto A, Van Den Abbeele J, Ribeiro RM, Dias S, Smith TK, Figueiredo LM. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe. 2016;19:837–848. doi: 10.1016/j.chom.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanowitz HB, Scherer PE, Mota MM, Figueiredo LM. Adipose tissue: a safe haven for parasites? Trends Parasitol. 2017;33:276–284. doi: 10.1016/j.pt.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simarro PP, Franco J, Diarra A, Postigo JaR, Jannin J. Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology. 2012;139:842–846. doi: 10.1017/S0031182012000169. [DOI] [PubMed] [Google Scholar]
- 11.Iten M, Mett H, Evans A, Enyaru JC, Brun R, Kaminsky R. Alterations in ornithine decarboxylase characteristics account for tolerance of Trypanosoma brucei rhodesiense to D,L-alpha-difluoromethylornithine. Antimicrob Agents Chemother. 1997;41:1922–1925. doi: 10.1128/aac.41.9.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drugs for Neglected Diseases initiative: SCYX-7158 Oxaborole. doi: 10.4314/ajhs.v12i1.30793. https://www.dndi.org/diseases-projects/portfolio/scyx-7158/ (accessed Jun 13, 2017) [DOI] [PubMed]
- 13.Drugs for Neglected Diseases initiative: Fexinidazole (HAT) https://www.dndi.org/diseases-projects/portfolio/fexinidazole/ (accessed Jun 13, 2017)
- 14.Jacobs RT, Nare B, Phillips MA. State of the art in African trypanosome drug discovery. Curr Top Med Chem. 2011;11:1255–1274. doi: 10.2174/156802611795429167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willert E, Phillips MA. Regulation and function of polyamines in African trypanosomes. Trends Parasitol. 2012;28:66–72. doi: 10.1016/j.pt.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Willert EK, Phillips MA. Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 2008;4:e1000183. doi: 10.1371/journal.ppat.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacchi CJ, Brun R, Croft SL, Alicea K, Buhler Y. In vivo trypanocidal activities of new S-adenosylmethionine decarboxylase inhibitors. Antimicrob Agents Chemother. 1996;40:1448–1453. doi: 10.1128/aac.40.6.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacchi CJ, Barker RH, Jr, Rodriguez A, Hirth B, Rattendi D, Yarlett N, Hendrick CL, Sybertz E. Trypanocidal activity of 8-methyl-5′-{[(Z)-4-aminobut-2-enyl]-(methylamino)}adenosine (Genz-644131), an adenosylmethionine decarboxylase inhibitor. Antimicrob Agents Chemother. 2009;53:3269–3272. doi: 10.1128/AAC.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bitonti AJ, Byers TL, Bush TL, Casara PJ, Bacchi CJ, Clarkson AB, Mccann PP, Sjoerdsma A. Cure of Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense infections in mice with an irreversible inhibitor of S-adenosylmethionine decarboxylase. Antimicrob Agents Chemother. 1990;34:1485–1490. doi: 10.1128/aac.34.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willert EK, Fitzpatrick R, Phillips MA. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc Natl Acad Sci USA. 2007;104:8275–8280. doi: 10.1073/pnas.0701111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volkov OA, Kinch L, Ariagno C, Deng X, Zhong S, Grishin N, Tomchick DR, Chen Z, Phillips MA. Relief of autoinhibition by conformational switch explains enzyme activation by a catalytically dead paralog. Elife. 2016;5:e20198. doi: 10.7554/eLife.20198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volkov OA, Cosner CC, Brockway AJ, Kramer M, Booker M, Zhong S, Ketcherside A, Wei S, Longgood J, Mccoy M, Richardson TE, Wring SA, Peel M, Klinger JD, Posner BA, De Brabander JK, Phillips MA. Identification of Trypanosoma brucei AdoMetDC inhibitors using a high-throughput mass spectrometry-based assay. ACS Infect Dis. 2017;3:512–526. doi: 10.1021/acsinfecdis.7b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valente S, Liu Y, Schnekenburger M, Zwergel C, Cosconati S, Gros C, Tardugno M, Labella D, Florean C, Minden S, Hashimoto H, Chang Y, Zhang X, Kirsch G, Novellino E, Arimondo PB, Miele E, Ferretti E, Gulino A, Diederich M, Cheng X, Mai A. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J Med Chem. 2014;57:701–713. doi: 10.1021/jm4012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borman RA, Coleman RA, Clark KL, Oxford AW, Hynd G, Archer JA, Aley A, Harris NV. 5 HT2B Receptor Antagonists. 2005/012263. Patent WO. 2005;A1
- 25.Sander K, Kottke T, Tanrikulu Y, Proschak E, Weizel L, Schneider EH, Seifert R, Schneider G, Stark H. 2,4-Diaminopyrimidines as histamine H4 receptor ligands–Scaffold optimization and pharmacological characterization. Bioorg Med Chem. 2009;17:7186–7196. doi: 10.1016/j.bmc.2009.08.059. [DOI] [PubMed] [Google Scholar]
- 26.Sekiya K, Takashima H, Ueda N, Kamiya N, Yuasa S, Fujimura Y, Ubasawa M. 2-Amino-6-arylthio-9-[2-(phosphonomethoxy)ethyl]purine bis(2,2,2-trifluoroethyl) esters as novel HBV-specific antiviral reagents. J Med Chem. 2002;45:3138–3142. doi: 10.1021/jm020036x. [DOI] [PubMed] [Google Scholar]
- 27.Medina JR, Becker CJ, Blackledge CW, Duquenne C, Feng Y, Grant SW, Heerding D, Li WH, Miller WH, Romeril SP, Scherzer D, Shu A, Bobko MA, Chadderton AR, Dumble M, Gardiner CM, Gilbert S, Liu Q, Rabindran SK, Sudakin V, Xiang H, Brady PG, Campobasso N, Ward P, Axten JM. Structure-based design of potent and selective 3-phosphoinositide-dependent kinase-1 (PDK1) inhibitors. J Med Chem. 2011;54:1871–1895. doi: 10.1021/jm101527u. [DOI] [PubMed] [Google Scholar]
- 28.Yi B, Long S, Gonzalez-Cestari TF, Henderson BJ, Pavlovicz RE, Werbovetz K, Li C, Mckay DB. Discovery of benzamide analogs as negative allosteric modulators of human neuronal nicotinic receptors: pharmacophore modeling and structure-activity relationship studies. Bioorg Med Chem. 2013;21:4730–4743. doi: 10.1016/j.bmc.2013.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wijtmans M, Scholten DJ, Roumen L, Canals M, Custers H, Glas M, Vreeker MC, De Kanter FJ, De Graaf C, Smit MJ, De Esch IJ, Leurs R. Chemical subtleties in small-molecule modulation of peptide receptor function: the case of CXCR3 biaryl-type ligands. J Med Chem. 2012;55:10572–10583. doi: 10.1021/jm301240t. [DOI] [PubMed] [Google Scholar]
- 30.Knapp DM, Gillis EP, Burke MD. A general solution for unstable boronic acids: slow-release cross-coupling from air-stable MIDA boronates. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 32.Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 33.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery : a Guide for Medicinal Chemists and Pharmacologists. Wiley-Interscience; Hoboken, N.J: 2005. [PubMed] [Google Scholar]
- 34.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duan G, Smith VH, Weaver DF. Characterization of aromatic-thiol π-type hydrogen bonding and phenylalanine-cysteine side chain interactions throughab initiocalculations and protein database analyses. Mol Phys. 2001;99:1689–1699. [Google Scholar]
- 36.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed Engl. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 37.Cavallo G, Metrangolo P, Milani R, Pilati T, Priimagi A, Resnati G, Terraneo G. The halogen bond. Chem Rev. 2016;116:2478–2601. doi: 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mccloskey DE, Bale S, Secrist JA, 3rd, Tiwari A, Moss TH, 3rd, Valiyaveettil J, Brooks WH, Guida WC, Pegg AE, Ealick SE. New insights into the design of inhibitors of human S-adenosylmethionine decarboxylase: studies of adenine C8 substitution in structural analogues of S-adenosylmethionine. J Med Chem. 2009;52:1388–1407. doi: 10.1021/jm801126a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao Y, Nguyen S, Kim SH, Volkov OA, Tu BP, Phillips MA. Product feedback regulation implicated in translational control of the Trypanosoma brucei S-adenosylmethionine decarboxylase regulatory subunit prozyme. Mol Microbiol. 2013;88:846–861. doi: 10.1111/mmi.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincent IM, Creek D, Watson DG, Kamleh MA, Woods DJ, Wong PE, Burchmore RJ, Barrett MP. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 2010;6:e1001204. doi: 10.1371/journal.ppat.1001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bitonti AJ, Dumont JA, Mccann PP. Characterization of Trypanosoma brucei brucei S-adenosyl-L-methionine decarboxylase and its inhibition by Berenil, pentamidine and methylglyoxal bis(guanylhydrazone) Biochem J. 1986;237:685–689. doi: 10.1042/bj2370685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brun R, Buhler Y, Sandmeier U, Kaminsky R, Bacchi CJ, Rattendi D, Lane S, Croft SL, Snowdon D, Yardley V, Caravatti G, Frei J, Stanek J, Mett H. In vitro trypanocidal activities of new S-adenosylmethionine decarboxylase inhibitors. Antimicrob Agents Chemother. 1996;40:1442–1447. doi: 10.1128/aac.40.6.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tolbert WD, Zhang Y, Cottet SE, Bennett EM, Ekstrom JL, Pegg AE, Ealick SE. Mechanism of human S-adenosylmethionine decarboxylase proenzyme processing as revealed by the structure of the S68A mutant. Biochemistry. 2003;42:2386–2395. doi: 10.1021/bi0268854. [DOI] [PubMed] [Google Scholar]
- 44.Bale S, Brooks W, Hanes JW, Mahesan AM, Guida WC, Ealick SE. Role of the sulfonium center in determining the ligand specificity of human S-adenosylmethionine decarboxylase. Biochemistry. 2009;48:6423–6430. doi: 10.1021/bi900590m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bale S, Lopez MM, Makhatadze GI, Fang Q, Pegg AE, Ealick SE. Structural basis for putrescine activation of human S-adenosylmethionine decarboxylase. Biochemistry. 2008;47:13404–13417. doi: 10.1021/bi801732m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shantz LM, Stanley BA, Secrist JA, Pegg AE. Purification of human S-adenosylmethionine decarboxylase expressed in Escherichia coli and use of this protein to investigate the mechanism of inhibition by the irreversible inhibitors, 5′-deoxy-5′-[(3-hydrazinopropyl)methylamino]adenosine and 5′-{[(Z)-4-amino-2-butenyl]methylamino}-5′-deoxyadenosine. Biochemistry. 1992;31:6848–6855. doi: 10.1021/bi00144a027. [DOI] [PubMed] [Google Scholar]
- 47.Beswick TC, Willert EK, Phillips MA. Mechanisms of allosteric regulation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry. 2006;45:7797–7807. doi: 10.1021/bi0603975. [DOI] [PubMed] [Google Scholar]
- 48.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 49.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, Mccoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moriarty NW, Grosse-Kunstleve RW, Adams PD. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 with structures and biological assay data for all tested compounds.
Table S2 with X-ray crystallography data collection and refinement statistics.
Figure S1 with electron density maps supporting ligand placement in the X-ray crystal structure.