

Abstract
In diabetes mellitus, the polyol pathway is highly active and consumes approximately 30% glucose in the body. This pathway contains 2 reactions catalyzed by aldose reductase (AR) and sorbitol dehydrogenase, respectively. AR reduces glucose to sorbitol at the expense of NADPH, while sorbitol dehydrogenase converts sorbitol to fructose at the expense of NAD +, leading to NADH production. Consumption of NADPH, accumulation of sorbitol, and generation of fructose and NADH have all been implicated in the pathogenesis of diabetes and its complications. In this review, the roles of this pathway in NADH/NAD + redox imbalance stress and oxidative stress in diabetes are highlighted. A potential intervention using nicotinamide riboside to restore redox balance as an approach to fighting diabetes is also discussed.
Keywords: diabetes mellitus, fructose, NADH/NAD+, oxidative stress, polyol pathway, redox imbalance stress
1. INTRODUCTION
Diabetes mellitus is a debilitating disease. It impairs the biological function of many organs in the body. The underlying mechanism of diabetic pathogenesis is hyperglycemia‐induced chronic glucotoxicity,1, 2, 3, 4, 5, 6 which impairs numerous pathways in the biological metabolome. During development and progression of diabetes, many pathways are upregulated in an attempt to handle the overflow of glucose in the body. These pathways include the polyol pathway,7, 8, 9, 10, 11, 12 the glycation pathway,13, 14, 15 the protein kinase c pathway,16, 17, 18, 19 the hexosamine pathway,20, 21, 22 and the enediol/alpha‐ketoaldehyde pathway.23, 24, 25 It is now believed that all the pathways converge on elevation of reactive oxygen species (ROS) by a variety of ROS generation systems.25, 26, 27, 28
Under normoglycemic conditions, the major purpose of glucose combustion is to produce energy in the form of ATP, and to produce NADPH and ribose via the pentose phosphate pathway (Figure 1A). Excess glucose can be further stored in the body as either glycogen or fatty acids (Figure 1A).29 As glucose metabolism involves electron extraction, storage, and transportation, nearly all the biochemical reactions in glucose metabolism are actually redox reactions. For example, splitting of glucose to 2 molecules of pyruvate during glycolysis stores the extracted electrons in NADH, as does the pyruvate dehydrogenase complex pathway whereby pyruvate is decarboxylated to form acetyl‐CoA. After entry of acetyl‐CoA into the Krebs cycle, electrons are stored in both NADH and FADH2. These electron donors then donate their electrons to complex I (NADH) or complex II (FADH2) in the mitochondrial electron transport chain. Oxygen is only used at the last step whereby complex IV transports electrons from cytochrome c to oxygen.
Figure 1.
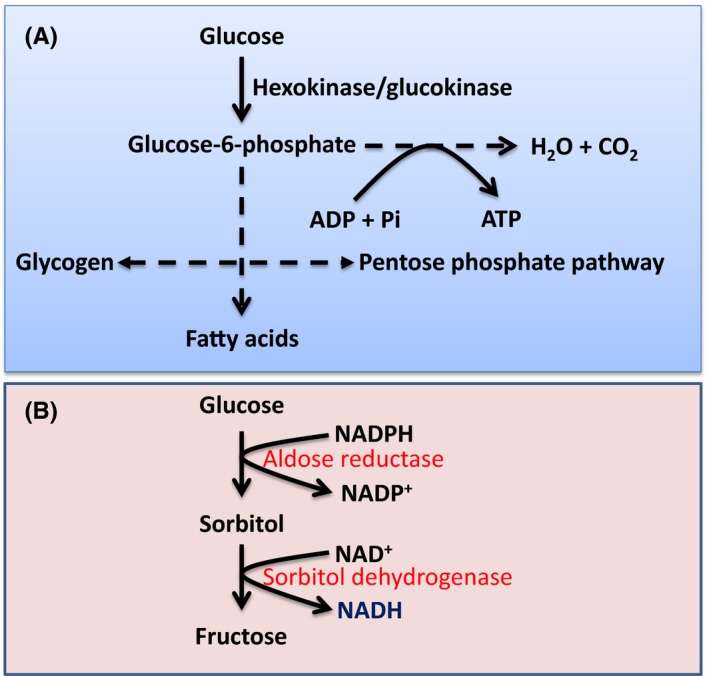
Glucose metabolic pathways under euglycemic and hyperglycemic conditions. A, Under normal physiological conditions, glucose is used for energy (ATP) production via glycolysis and the Krebs cycle pathways. Glucose can also be fluxed to the pentose phosphate pathway that makes NADPH and ribose. Excess glucose can be stored as glycogen or fatty acids. B, Under diabetic conditions, approximately 30% of glucose can be fluxed to the polyol pathway, whereby glucose is converted to fructose via 2 consecutive reactions that also transform NADPH to NADH
As glucose provides electrons that are mainly stored in NADH, the higher the blood glucose levels, the higher the NADH contents. This can tilt the redox balance between NADH and NAD+ toward the side of NADH, resulting in redox imbalance.6, 30 This is indeed what occurs in diabetes31, 32 and the polyol pathway is known to play a major role in breaking the redox balance between NADH and NAD+.33, 34, 35, 36
2. THE POLYOL PATHWAY
The polyol pathway consists of 2 reactions catalyzed by 2 respective enzymes.7, 10, 35 As shown in Figure 1B, the first reaction is reduction of glucose to sorbitol, which is catalyzed by aldose reductase (AR). This reaction is the rate‐limiting reaction37 in this pathway and also converts NADPH to NADP+. The second reaction converts sorbitol to fructose and is catalyzed by sorbitol dehydrogenase, which makes NADH from NAD+. So the overall products of the polyol pathway are sorbitol, fructose, and NADH. NADH production results from the consumption of NADPH. Because nearly 30% of blood glucose can flux through the polyol pathway in diabetes,38, 39 this pathway has been thought to be the major pathway contributing to NADH/NAD+ redox imbalance in diabetes.7, 8, 26, 34 I will now dissect each of the pathway's components (Figure 2) and their role in redox imbalance stress and diabetes mellitus.
Figure 2.
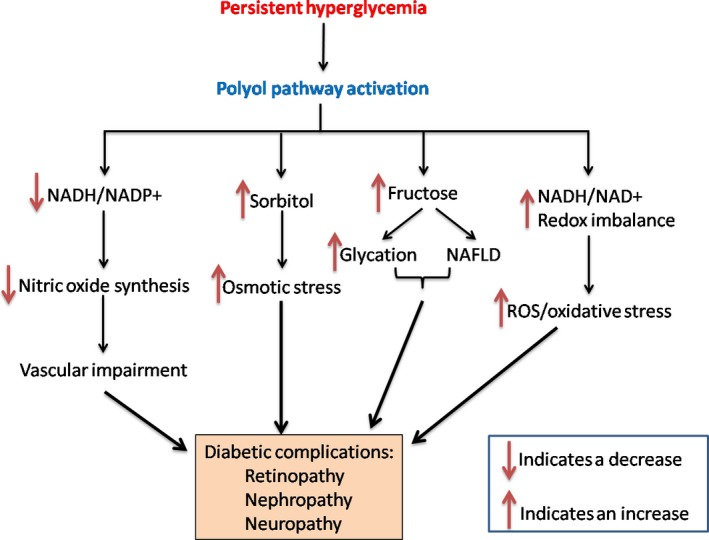
Pathophysiological effects of the polyol pathway activated by persistent hyperglycemia. Activation of the polyol pathway can (1) decrease the NADPH/NADP + ratio and nitric oxide production; (2) induce sorbitol accumulation and osmotic stress; (3) increase fructose content, leading to increased protein glycation and development of non‐alcoholic fatty liver disease (NFALD); (4) increase NADH/NAD + ratio leading to ROS production and oxidative stress. The consequences of these events are diabetic complications including retinopathy, nephropathy, and neuropathy
2.1. Aldose reductase
The physiological function of this enzyme still remains murky, but it is usually thought that the enzyme, under normal physiological conditions, can degrade toxic aldehyde byproducts formed by lipid peroxidation such as 4‐hydroxy‐nonenal (HNE) and its glutathione conjugates (GSH‐HNE).40, 41 However, its ability to catalyze glucose reduction is nearly negligible under physiological conditions due to the high K m of its reaction with glucose.42 In contrast, when the glucose level is high, this enzyme and the polyol pathway becomes a major pathway in disposing of glucose.7, 35, 36, 37, 43
The role of AR in diabetes has been well elucidated by using its inhibitors and by AR knockout animal models. It has been found that AR inhibitors can ameliorate diabetes mellitus.44, 45 In fact, numerous AR inhibitors have been tested and evaluated.46, 47, 48, 49, 50 For example, the AR inhibitor zopolrestato can lower acetate utilization in the diabetic heart,45 indicating increased glucose combustion via the glycolytic pathway and the Krebs cycle, that otherwise is inhibited in diabetes. Another example is the use of AR inhibitor sorbinil,51 which has been clinically used to stabilize diabetic corneal epithelial disorders.52 One caveat of inhibiting the polyol pathway is that it could be over‐inhibited, leading to increased protein glycation by glucose.53
With respect to AR deletion or knockout studies, it has been demonstrated that AR deletion from mice could inhibit diabetes‐induced retinal capillary degeneration mediated by superoxide production.54 It has also been demonstrated that the AR knockout mouse is resistant to the development of diabetic nephropathy.55
2.2. Consumption of NADPH and redox imbalance of NADPH and NADP+
As glucose flux through the polyol pathway consumes NADPH, it has been suggested that the level of NADPH could be significantly decreased.56 Indeed, we have found that this is the case in diabetic lung and pancreas,31, 57 whereby NADPH content is lower than that in controls. It has been established that there is about a 15% decrease in NADPH in the diabetic lens.35 The NADPH decrease could further impair the GSH/GSSG redox balance, as GSSG reduction by glutathione reductase requires NADPH as a cofactor.58, 59 NADPH is also involved in the biosynthesis of biological molecules such as fatty acids and nitric oxide, so its decrease or depletion should have deleterious effects on many anabolic pathways.60 Additionally, from a chemical point of view, the polyol pathway can also compete with glutathione reductase for NADPH,61, 62 leading to further impairment in glucose metabolism.
2.3. Accumulation of sorbitol
In certain tissues such as retina, sorbitol dehydrogenase content is low,63 so sorbitol formed from glucose reduction can accumulate.35 This accumulation can change cellular membrane osmotic pressure and triggers osmotic stress.35 This osmotic stress has been thought to be the main underlying mechanism for diabetic retinopathy64, 65 and has also been implicated in diabetic kidney dysfunction or nephropathy.9 It should be noted that even in the same organ, different cell populations may have different levels of sorbitol dehydrogenase;66 hence, the effect of sorbitol on diabetic tissue is differential.
2.4. NADH overproduction and NAD+ depletion
The second reaction of the polyol pathway involves NADH production from NAD+. This pathway has therefore been regarded as the major source of NADH/NAD+ redox imbalance.5, 6, 26, 32 On one hand, NADH is overproduced, which could lead to reductive stress followed by oxidative stress.26, 28 This is because elevated levels of NADH could overwhelm mitochondrial complex I, leading to more ROS production from the mitochondrial electron transport chain.26 Additionally, excess NADH can also inhibit the glycolytic pathway, the pyruvate dehydrogenase complex, and the Krebs cycle,12, 67 leading to more flux of glucose through the polyol pathway. On the other hand, an NAD+ decrease also imposes deleterious effects on a variety of metabolic pathways.6 A major one is the sirtuin pathway,6 which is responsible for protein deacetylation.68 A decrease in NAD+ would inactivate sirtuins, leading to over‐acetylation of proteins and less efficient glucose metabolism.69, 70, 71
In the case of NADH/NAD+ redox imbalance, it has been demonstrated that restoring the redox balance by supplementing with an NAD+ precursor or analogue is a valuable approach.72 In this regard, the recently identified precursor nicotinamide riboside is very promising as this chemical is more tolerant and has fewer side‐effects than niacin.73 For example, it has been reported that nicotinamide riboside can ameliorate diabetes and diabetic neuropathy in mice, and can enhance metabolism and prevent development of obesity induced by a high fat diet.74, 75
2.5. Fructose
As the polyol pathway consumes approximately 30% of blood glucose in diabetes,39 fructose is overproduced in the body. Overproduction of fructose can lead to severe metabolic consequences. On one hand, fructose can chemically glycate proteins,76 leading to protein dysfunction. It is known that fructose can be further metabolized to produce 3‐deoxyglucose and fructose‐3‐phosphate, both of which are very potent nonenzymatic glycation agents.76 On the other hand, as fructose metabolism by fructokinase, with the consumption of ATP, can bypass the regulation of the glycolytic pathway,77, 78 acetyl‐CoA could be overproduced78 and ATP could be depleted.79 Acetyl‐CoA overproduction could cause non‐alcoholic fatty liver disease (NAFLD), as acetyl‐CoA is the precursor of fatty acid,77, 80, 81, 82 while ATP depletion could cause cell death. Additionally, overproduction of acetyl‐CoA can result in more protein acetylation, leading to protein functional impairment.83, 84, 85 Protein acetylation can worsen when sirtuin proteins are inactive due to lack of NAD+ in diabetes.6, 86 Therefore, fructose accumulation due to activation of the polyol pathway by hyperglycemia can accentuate diabetes and its complications.
2.6. Effect of redox imbalance on sirtuins
Sirtuins are protein deacetylases that use NAD+ as their substrate.87 So when NAD+ levels decrease during diabetes, sirtuin activities will be decreased,69, 88 and this can also be modulated by decreased expression of sirtuin proteins. Indeed, numerous studies including ours, have demonstrated attenuated expression of sirtuin proteins in diabetes.31, 57, 88, 89 As a consequence, protein acetylation is increased (Figure 3A), leading to functional changes of numerous proteins.83, 90 Accordingly, studies have demonstrated that supplementing with NAD+ precursors or analogous can serve as an approach for enhancing sirtuin activity, thereby augmenting protein deacetylation, which can lead to amelioration of diabetes.70, 71, 91
Figure 3.
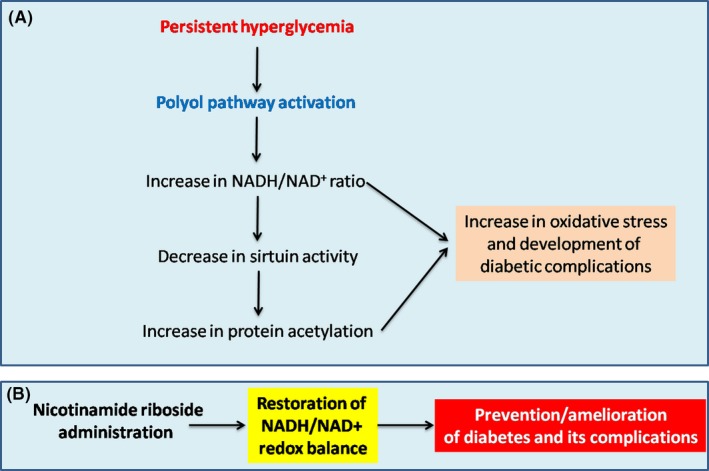
A, NADH/NAD + redox imbalance induced by activation of the polyol pathway can attenuate sirtuin activity and increase protein acetylation which eventually leads to oxidative stress and development of diabetic complications. B, Restoration or normalization of NADH/NAD + redox balance by nicotinamide riboside, which can serve as a promising approach to preventing or ameliorating diabetes and its complications
2.7. Effect of redox imbalance on poly‐ADP‐ribosylase function
In diabetes, it is usually thought that DNA damage occurs first, which triggers the upregulation of poly‐ADP‐ribosylase (PARP) activity.32, 92, 93 This upregulation can deplete NAD+, as PARP also uses NAD+ as its substrate during repair of damaged DNA.94, 95, 96 Indeed, PARP knockout mouse has been found to be resistant to diabetes development97, 98 and inhibition of PARP can also retard the development of diabetes.99, 100, 101, 102 On the other hand, it is also possible that decreased levels of NAD+ caused by activation of the polyol pathway could impair PRAP activity, leading to accentuation of diabetes, as it is likely that damaged DNA would not get repaired promptly. Nonetheless, the crosstalk between the polyol pathway and the PARP pathway will need to be further investigated. This author tends to believe that the 2 pathways may form a vicious cycle that will worsen the situation during progression of diabetes.
2.8. Redox imbalance and oxidative stress
One of the major consequences of NADH/NAD+ redox imbalance is oversupply of electron donors to the mitochondrial electron transport chain.26 Oversupply of NADH would overwhelm complex I, which relays electrons from NADH to CoQ.32 One feature of complex I electron transport is that the more electrons it transports, the more superoxide it will produce.103, 104, 105, 106 This is because more electrons could leak and partially reduce oxygen, leading to overproduction of superoxide which is the precursor of all the ROS.107, 108, 109, 110 Hence, oversupply of NADH in diabetes driven by constant hyperglycemia can devastate cells with enhanced oxidative stress, impaired mitochondrial function, and increased cell death, as has been demonstrated by numerous investigators.17, 27, 28, 111, 112, 113, 114, 115, 116, 117, 118
2.9. Targeting redox imbalance as an approach for diabetes therapy
It is reasonable to say that diabetes is a redox imbalance disease.32 Hence restoration of NADH/NAD+ redox balance may serve to combat diabetes. One approach, as mentioned above, is supplementing with NAD+ precursors or analogues (Figure 3B). In particular, the utilization of nicotinamide riboside in a variety of experimental settings has demonstrated the beneficial effects of this compound.10, 74, 75, 119 Additionally, plant extracts or compounds that are antioxidants in nature have also been evaluated for their effects in mitigating oxidative stress and promoting cell survival.120, 121, 122, 123 As these compounds can counteract the deleterious effects of the activated polyol pathway that is responsible for redox imbalance in diabetes, an understanding of how they work in alleviating diabetes and its complications should provide insights into the design of novel strategies for fighting this epidemic and devastating disease.
3. CONCLUDING REMARKS
The active polyol pathway in diabetes mellitus is a major contributor to NADH/NAD+ redox imbalance due to its ability to convert NADPH to NADH. Not only can excess NADH induce oxidative stress via generation of ROS through the mitochondrial electron transport chain and other pathways, but lowered NADPH content can also induce oxidative stress by impairing glutathione metabolism. Approaches to restoration of redox balance by targeting the polyol pathway have been explored and should remain a research focus in order to provide novel strategies for fighting diabetes and its complications.
CONFLICT OF INTEREST
None.
ACKNOWLEDGEMENTS
The writing of this review article was supported in part by UNTHSC seed grants RI10015 and RI10039 and by the National Institutes of Health (grant no. R01NS079792).
Yan LJ. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim Models Exp Med. 2018;1:7–13. 10.1002/ame2.12001
REFERENCES
- 1. Del Prato S. Role of glucotoxicity and lipotoxicity in the pathophysiology of type 2 diabetes mellitus and emerging treatment strategies. Diabet Med. 2009;26:1185–1192. [DOI] [PubMed] [Google Scholar]
- 2. Brahma MK, Pepin ME, Wende AR. My sweetheart is broken: role of glucose in diabetic cardiomyopathy. Diabetes Metab J. 2017;41:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta‐cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab. 2003;16:5–22. [DOI] [PubMed] [Google Scholar]
- 4. Brunner Y, Schvartz D, Priego‐Capote F, Coute Y, Sanchez JC. Glucotoxicity and pancreatic proteomics. J Proteomics. 2009;71:576–591. [DOI] [PubMed] [Google Scholar]
- 5. Zheng H, Wu J, Jin Z, Yan LJ. Protein Modifications as manifestations of hyperglycemic glucotoxicity in diabetes and its complications. Biochem Insights. 2016;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo X, Wu J, Jing S, Yan LJ. Hyperglycemic stress and carbon stress in diabetic glucotoxicity. Aging Dis. 2016;7:90–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes‐induced oxidative stress. J Am Soc Nephrol. 2003;14(8 Suppl. 3):S233–S236. [DOI] [PubMed] [Google Scholar]
- 8. Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S3–S12. [DOI] [PubMed] [Google Scholar]
- 9. Hotta N. New concepts and insights on pathogenesis and treatment of diabetic complications: polyol pathway and its inhibition. Nagoya J Med Sci. 1997;60:89–100. [PubMed] [Google Scholar]
- 10. Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999;13:23–30. [DOI] [PubMed] [Google Scholar]
- 11. Li Q, Hwang YC, Ananthakrishnan R, Oates PJ, Guberski D, Ramasamy R. Polyol pathway and modulation of ischemia‐reperfusion injury in type 2 diabetic BBZ rat hearts. Cardiovasc Diabetol. 2008;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang WH, Wu S, Wong TM, Chung SK, Chung SS. Polyol pathway mediates iron‐induced oxidative injury in ischemic‐reperfused rat heart. Free Radic Biol Med. 2008;45:602–610. [DOI] [PubMed] [Google Scholar]
- 13. Lyons TJ, Jenkins AJ. Glycation, oxidation, and lipoxidation in the development of the complications of diabetes: a carbonyl stress hypothesis. Diabetes Rev (Alex). 1997;5:365–391. [PMC free article] [PubMed] [Google Scholar]
- 14. Vlassara H, Striker GE. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol Metab Clin North Am. 2013;42:697–719. [DOI] [PubMed] [Google Scholar]
- 15. Wolff SP, Jiang ZY, Hunt JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med. 1991;10:339–352. [DOI] [PubMed] [Google Scholar]
- 16. Feng B, Ruiz MA, Chakrabarti S. Oxidative‐stress‐induced epigenetic changes in chronic diabetic complications. Can J Physiol Pharmacol. 2013;91:213–220. [DOI] [PubMed] [Google Scholar]
- 17. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xia L, Wang H, Munk S, et al. Reactive oxygen species, PKC‐beta1, and PKC‐zeta mediate high‐glucose‐induced vascular endothelial growth factor expression in mesangial cells. Am J Physiol Endocrinol Metab. 2007;293:E1280–E1288. [DOI] [PubMed] [Google Scholar]
- 19. Xia L, Wang H, Munk S, et al. High glucose activates PKC‐zeta and NADPH oxidase through autocrine TGF‐beta1 signaling in mesangial cells. Am J Physiol Renal Physiol. 2008;295:F1705–F1714. [DOI] [PubMed] [Google Scholar]
- 20. Beyer AM, Weihrauch D. Hexosamine pathway activation and O‐linked‐N‐acetylglucosamine: novel mediators of endothelial dysfunction in hyperglycemia and diabetes. Vascul Pharmacol. 2012;56:113–114. [DOI] [PubMed] [Google Scholar]
- 21. Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S13–S18. [DOI] [PubMed] [Google Scholar]
- 22. Yanagida K, Maejima Y, Santoso P, et al. Hexosamine pathway but not interstitial changes mediates glucotoxicity in pancreatic beta‐cells as assessed by cytosolic Ca2+ response to glucose. Aging (Albany NY). 2014;6:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mira ML, Martinho F, Azevedo MS, Manso CF. Oxidative inhibition of red blood cell ATPases by glyceraldehyde. Biochim Biophys Acta. 1991;1060:257–261. [DOI] [PubMed] [Google Scholar]
- 24. Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem J. 1987;245:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–42354. [DOI] [PubMed] [Google Scholar]
- 26. Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res. 2014;2014:137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abdul‐Ghani MA, DeFronzo RA. Oxidative stress in type 2 diabetes In: Miwa S, Beckman KB, Muller FL, eds. Oxidative Stress in Aging Humana Press; 2008:191–212. [Google Scholar]
- 28. Araki E, Nishikawa T. Oxidative stress: a cause and therapeutic target of diabetic complications. J Diabetes Investig. 2010;1:90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lieberman M, Marks AD. Marks’ Basic Medical Biochemistry: A Clinical Approach (4th ed.). Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 30. Luo X, Li R, Yan LJ. Roles of pyruvate, NADH, and mitochondrial complex I in redox balance and imbalance in β cell function and dysfunction. J Diabetes Res. 2015;2015:12 10.1155/2015/512618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu J, Jin Z, Yan LJ. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox Biol. 2017;11:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu J, Jin Z, Zheng H, Yan LJ. Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes. 2016;9:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ido Y, Williamson JR. Hyperglycemic cytosolic reductive stress ‘pseudohypoxia’: implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:1467–1470. [PubMed] [Google Scholar]
- 34. Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813. [DOI] [PubMed] [Google Scholar]
- 35. Kador PF, Kinoshita JH. Role of aldose reductase in the development of diabetes‐associated complications. Am J Med. 1985;79:8–12. [DOI] [PubMed] [Google Scholar]
- 36. Chung SS, Chung SK. Aldose reductase in diabetic microvascular complications. Curr Drug Targets. 2005;6:475–486. [DOI] [PubMed] [Google Scholar]
- 37. Yabe‐Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev. 1998;50:21–33. [PubMed] [Google Scholar]
- 38. Gonzalez RG, Barnett P, Aguayo J, Cheng HM, Chylack LT Jr. Direct measurement of polyol pathway activity in the ocular lens. Diabetes. 1984;33:196–199. [DOI] [PubMed] [Google Scholar]
- 39. Fantus IG. The pathogenesis of the chronic complications of the diabetes mellitus. Endocrinol Rounds. 2002;2:1–8. [Google Scholar]
- 40. Srivastava SK, Yadav UC, Reddy AB, et al. Aldose reductase inhibition suppresses oxidative stress‐induced inflammatory disorders. Chem Biol Interact. 2011;191:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maccari R, Ottana R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. J Med Chem. 2015;58:2047–2067. [DOI] [PubMed] [Google Scholar]
- 42. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 43. Kador PF. The role of aldose reductase in the development of diabetic complications. Med Res Rev. 1988;8:325–352. [DOI] [PubMed] [Google Scholar]
- 44. Demir Y, Isik M, Gulcin I, Beydemir S. Phenolic compounds inhibit the aldose reductase enzyme from the sheep kidney. J Biochem Mol Toxicol. 2017;31 (9). [DOI] [PubMed] [Google Scholar]
- 45. Trueblood N, Ramasamy R. Aldose reductase inhibition improves altered glucose metabolism of isolated diabetic rat hearts. Am J Physiol. 1998;275:H75–H83. [DOI] [PubMed] [Google Scholar]
- 46. Hao X, Han Z, Li Y, et al. Synthesis and structure‐activity relationship studies of phenolic hydroxyl derivatives based on quinoxalinone as aldose reductase inhibitors with antioxidant activity. Bioorg Med Chem Lett. 2017;27:887–892. [DOI] [PubMed] [Google Scholar]
- 47. Han Z, Hao X, Ma B, Zhu C. A series of pyrido[2,3‐b]pyrazin‐3(4H)‐one derivatives as aldose reductase inhibitors with antioxidant activity. Eur J Med Chem. 2016;121:308–317. [DOI] [PubMed] [Google Scholar]
- 48. Zhu S, Hao X, Zhang S, Qin X, Chen X, Zhu C. Synthesis of benzothiadiazine derivatives exhibiting dual activity as aldose reductase inhibitors and antioxidant agents. Bioorg Med Chem Lett. 2016;26:2880–2885. [DOI] [PubMed] [Google Scholar]
- 49. Zhu S, Zhang S, Hao X, et al. Pyridothiadiazine derivatives as aldose reductase inhibitors having antioxidant activity. J Enzyme Inhib Med Chem. 2016;31:126–130. [DOI] [PubMed] [Google Scholar]
- 50. Zou Y, Qin X, Hao X, et al. Phenolic 4‐hydroxy and 3,5‐dihydroxy derivatives of 3‐phenoxyquinoxalin‐2(1H)‐one as potent aldose reductase inhibitors with antioxidant activity. Bioorg Med Chem Lett. 2015;25:3924–3927. [DOI] [PubMed] [Google Scholar]
- 51. Datiles MB, Kador PF, Kashima K, Kinoshita JH, Sinha A. The effects of sorbinil, an aldose reductase inhibitor, on the corneal endothelium in galactosemic dogs. Invest Ophthalmol Vis Sci. 1990;31:2201–2204. [PubMed] [Google Scholar]
- 52. Narayanan S. Aldose reductase and its inhibition in the control of diabetic complications. Ann Clin Lab Sci. 1993;23:148–158. [PubMed] [Google Scholar]
- 53. Jagdale AD, Bavkar LN, More TA, Joglekar MM, Arvindekar AU. Strong inhibition of the polyol pathway diverts glucose flux to protein glycation leading to rapid establishment of secondary complications in diabetes mellitus. J Diabetes Complications. 2016;30:398–405. [DOI] [PubMed] [Google Scholar]
- 54. Tang J, Du Y, Petrash JM, Sheibani N, Kern TS. Deletion of aldose reductase from mice inhibits diabetes‐induced retinal capillary degeneration and superoxide generation. PLoS ONE. 2013;8:e62081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu H, Luo Y, Zhang T, et al. Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia. 2011;54:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Diaz‐Flores M, Ibanez‐Hernandez MA, Galvan RE, et al. Glucose‐6‐phosphate dehydrogenase activity and NADPH/NADP+ ratio in liver and pancreas are dependent on the severity of hyperglycemia in rat. Life Sci. 2006;78:2601–2607. [DOI] [PubMed] [Google Scholar]
- 57. Wu J, Luo X, Thangthaeng N, et al. Pancreatic mitochondrial complex I exhibits aberrant hyperactivity in diabetes. Biochem Biophys Rep. 2017;11:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bravi MC, Pietrangeli P, Laurenti O, et al. Polyol pathway activation and glutathione redox status in non‐insulin‐dependent diabetic patients. Metabolism. 1997;46:1194–1198. [DOI] [PubMed] [Google Scholar]
- 59. Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21:5164–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mapanga RF, Essop MF. Damaging effects of hyperglycemia on cardiovascular function: spotlight on glucose metabolic pathways. Am J Physiol Heart Circ Physiol. 2016;310:H153–H173. [DOI] [PubMed] [Google Scholar]
- 61. Cumbie BC, Hermayer KL. Current concepts in targeted therapies for the pathophysiology of diabetic microvascular complications. Vasc Health Risk Manag. 2007;3:823–832. [PMC free article] [PubMed] [Google Scholar]
- 62. Ravindranath TM, Mong PY, Ananthakrishnan R, et al. Novel role for aldose reductase in mediating acute inflammatory responses in the lung. J Immunol. 2009;183:8128–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aldebasi Y, El‐Gendy SM, Kamel A, Mohieldein A. Aldo‐keto reductase and sorbitol dehydrogenase enzymes in Egyptian diabetic patients with and without proliferative diabetic retinopathy. Clin Exp Optom. 2013;96:303–309. [DOI] [PubMed] [Google Scholar]
- 64. Kinoshita JH. Cataracts in galactosemia. The Jonas S. Friedenwald Memorial Lecture. Invest Ophthalmol. 1965;4:786–799. [PubMed] [Google Scholar]
- 65. Robison WG Jr, Kador PF, Kinoshita JH. Retinal capillaries: basement membrane thickening by galactosemia prevented with aldose reductase inhibitor. Science. 1983;221:1177–1179. [DOI] [PubMed] [Google Scholar]
- 66. Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol. 2002;50:325–392. [DOI] [PubMed] [Google Scholar]
- 67. Hwang YC, Bakr S, Ellery CA, Oates PJ, Ramasamy R. Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. FASEB J. 2003;17:2331–2333. [DOI] [PubMed] [Google Scholar]
- 68. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133–171. [DOI] [PubMed] [Google Scholar]
- 69. Yang T, Sauve AA. NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 2006;8:E632–E643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Turkmen K, Karagoz A, Kucuk A. Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J Diabetes. 2014;5:894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kitada M, Kume S, Kanasaki K, Takeda‐Watanabe A, Koya D. Sirtuins as possible drug targets in type 2 diabetes. Curr Drug Targets. 2013;14:622–636. [DOI] [PubMed] [Google Scholar]
- 72. Lee CF, Chavez JD, Garcia‐Menendez L, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134:883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. [DOI] [PubMed] [Google Scholar]
- 74. Canto C, Houtkooper RH, Pirinen E, et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high‐fat diet‐induced obesity. Cell Metab. 2012;15:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Trammell SA, Weidemann BJ, Chadda A, et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016;6:26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gugliucci A. Formation of fructose‐mediated advanced glycation end products and their roles in metabolic and inflammatory diseases. Adv Nutr. 2017;8:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jegatheesan P, De Bandt JP. Fructose and NAFLD: the multifaceted aspects of fructose metabolism. Nutrients. 2017;9 (3). [Google Scholar]
- 78. Diggle CP, Shires M, Leitch D, et al. Ketohexokinase: expression and localization of the principal fructose‐metabolizing enzyme. J Histochem Cytochem. 2009;57:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Johnson RJ, Rodriguez‐Iturbe B, Roncal‐Jimenez C, et al. Hyperosmolarity drives hypertension and CKD–water and salt revisited. Nat Rev Nephrol. 2014;10:415–420. [DOI] [PubMed] [Google Scholar]
- 80. Lanaspa MA, Ishimoto T, Li N, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013;4:2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Choi Y, Abdelmegeed MA, Song BJ. Diet high in fructose promotes liver steatosis and hepatocyte apoptosis in C57BL/6J female mice: role of disturbed lipid homeostasis and increased oxidative stress. Food Chem Toxicol. 2017;103:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ishimoto T, Lanaspa MA, Rivard CJ, et al. High‐fat and high‐sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. 2013;58:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 2014;54:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Baeza J, Smallegan MJ, Denu JM. Site‐specific reactivity of nonenzymatic lysine acetylation. ACS Chem Biol. 2015;10:122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Paik WK, Pearson D, Lee HW, Kim S. Nonenzymatic acetylation of histones with acetyl‐CoA. Biochim Biophys Acta. 1970;213:513–522. [DOI] [PubMed] [Google Scholar]
- 86. Madsen AS, Andersen C, Daoud M, et al. Investigating the Sensitivity of NAD+‐dependent Sirtuin Deacylation Activities to NADH. J Biol Chem. 2016;291:7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family's role in aging and age‐associated pathologies. J Clin Invest. 2013;123:973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. de Kreutzenberg SV, Ceolotto G, Papparella I, et al. Downregulation of the longevity‐associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes. 2010;59:1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Caton PW, Richardson SJ, Kieswich J, et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia. 2013;56:1068–1077. [DOI] [PubMed] [Google Scholar]
- 90. Hirschey MD, Shimazu T, Jing E, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Huynh FK, Hershberger KA, Hirschey MD. Targeting sirtuins for the treatment of diabetes. Diabetes Manag (Lond). 2013;3:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP‐ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mueller‐Dieckmann C, Kernstock S, Lisurek M, et al. The structure of human ADP‐ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP‐ribosylation. Proc Natl Acad Sci U S A. 2006;103:15026–15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dolle C, Rack JG, Ziegler M. NAD and ADP‐ribose metabolism in mitochondria. FEBS J. 2013;280:3530–3541. [DOI] [PubMed] [Google Scholar]
- 95. Du X, Matsumura T, Edelstein D, et al. Inhibition of GAPDH activity by poly(ADP‐ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Horvath EM, Magenheim R, Kugler E, et al. Nitrative stress and poly(ADP‐ribose) polymerase activation in healthy and gestational diabetic pregnancies. Diabetologia. 2009;52:1935–1943. [DOI] [PubMed] [Google Scholar]
- 97. Masutani M, Suzuki H, Kamada N, et al. Poly(ADP‐ribose) polymerase gene disruption conferred mice resistant to streptozotocin‐induced diabetes. Proc Natl Acad Sci U S A. 1999;96:2301–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pieper AA, Brat DJ, Krug DK, et al. Poly(ADP‐ribose) polymerase‐deficient mice are protected from streptozotocin‐induced diabetes. Proc Natl Acad Sci U S A. 1999;96:3059–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Obrosova IG, Minchenko AG, Frank RN, et al. Poly(ADP‐ribose) polymerase inhibitors counteract diabetes‐ and hypoxia‐induced retinal vascular endothelial growth factor overexpression. Int J Mol Med. 2004;14:55–64. [PubMed] [Google Scholar]
- 100. Sarras MP Jr, Mason S, McAllister G, Intine RV. Inhibition of poly‐ADP ribose polymerase enzyme activity prevents hyperglycemia‐induced impairment of angiogenesis during wound healing. Wound Repair Regen. 2014;22:666–670. [DOI] [PubMed] [Google Scholar]
- 101. Long CA, Boulom V, Albadawi H, et al. Poly‐ADP‐ribose‐polymerase inhibition ameliorates hind limb ischemia reperfusion injury in a murine model of type 2 diabetes. Ann Surg. 2013;258:1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Szkudelski T. Streptozotocin‐nicotinamide‐induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood). 2012;237:481–490. [DOI] [PubMed] [Google Scholar]
- 103. Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH‐ubiquinone oxidoreductase (complex I). J Biol Chem. 2011;286:27103–27110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cooper JM, Mann VM, Krige D, Schapira AH. Human mitochondrial complex I dysfunction. Biochim Biophys Acta. 1992;1101:198–203. [DOI] [PubMed] [Google Scholar]
- 106. Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36:976–980. [DOI] [PubMed] [Google Scholar]
- 107. Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. [DOI] [PubMed] [Google Scholar]
- 108. Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. [DOI] [PubMed] [Google Scholar]
- 109. Laustsen C, Nielsen PM, Norlinger TS, et al. Antioxidant treatment attenuates lactate production in diabetic nephropathy. Am J Physiol Renal Physiol. 2017;312:F192–F199. [DOI] [PubMed] [Google Scholar]
- 110. Yan LJ. Positive oxidative stress in aging and aging‐related disease tolerance. Redox Biol. 2014;2:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bhatt NM, Aon MA, Tocchetti CG, et al. Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose. Am J Physiol Heart Circ Physiol. 2015;308:H291–H302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tocchetti CG, Stanley BA, Sivakumaran V, et al. Impaired mitochondrial energy supply coupled to increased H2O2 emission under energy/redox stress leads to myocardial dysfunction during Type I diabetes. Clin Sci (Lond). 2015;129:561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. [DOI] [PubMed] [Google Scholar]
- 114. de M Bandeira S, da Fonseca LJ, da S Guedes G, Rabelo LA, Goulart MO, Vasconcelos SM. Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. Int J Mol Sci. 2013;14:3265–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Haldar SR, Chakrabarty A, Chowdhury S, Haldar A, Sengupta S, Bhattacharyya M. Oxidative stress‐related genes in type 2 diabetes: association analysis and their clinical impact. Biochem Genet. 2015;53:93–119. [DOI] [PubMed] [Google Scholar]
- 116. Henriksen EJ, Diamond‐Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2011;51:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lenzen S. Oxidative stress: the vulnerable beta‐cell. Biochem Soc Trans. 2008;36:343–347. [DOI] [PubMed] [Google Scholar]
- 118. Shah S, Iqbal M, Karam J, Salifu M, McFarlane SI. Oxidative stress, glucose metabolism, and the prevention of type 2 diabetes: pathophysiological insights. Antioxid Redox Signal. 2007;9:911–929. [DOI] [PubMed] [Google Scholar]
- 119. Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel‐induced peripheral neuropathy in female rats. Pain. 2017;158:962–972. [DOI] [PubMed] [Google Scholar]
- 120. Parveen K, Ishrat T, Malik S, Kausar MA, Siddiqui WA. Modulatory effects of Pycnogenol in a rat model of insulin‐dependent diabetes mellitus: biochemical, histological, and immunohistochemical evidences. Protoplasma. 2013;250:347–360. [DOI] [PubMed] [Google Scholar]
- 121. Ku CR, Lee HJ, Kim SK, Lee EY, Lee MK, Lee EJ. Resveratrol prevents streptozotocin‐induced diabetes by inhibiting the apoptosis of pancreatic beta‐cell and the cleavage of poly (ADP‐ribose) polymerase. Endocr J. 2012;59:103–109. [DOI] [PubMed] [Google Scholar]
- 122. Chanpoo M, Petchpiboonthai H, Panyarachun B, Anupunpisit V. Effect of curcumin in the amelioration of pancreatic islets in streptozotocin‐induced diabetic mice. J Med Assoc Thai. 2010;93(Suppl. 6):S152–S159. [PubMed] [Google Scholar]
- 123. Ding Y, Zhang Z, Dai X, et al. Grape seed proanthocyanidins ameliorate pancreatic beta‐cell dysfunction and death in low‐dose streptozotocin‐ and high‐carbohydrate/high‐fat diet‐induced diabetic rats partially by regulating endoplasmic reticulum stress. Nutr Metab (Lond). 2013;10:51. [DOI] [PMC free article] [PubMed] [Google Scholar]