

Kinetic and structural comparison of the cotranslational and refolding pathways of HaloTag reveals how translation reroutes the folding pathway to avoid aggregation.
Abstract
Precise protein folding is essential for the survival of all cells, and protein misfolding causes a number of diseases that lack effective therapies, yet the general principles governing protein folding in the cell remain poorly understood. In vivo, folding can begin cotranslationally and protein quality control at the ribosome is essential for cellular proteostasis. We directly characterize and compare the refolding and cotranslational folding trajectories of the protein HaloTag. We introduce new techniques for both measuring folding kinetics and detecting the conformations of partially folded intermediates during translation in real time. We find that, although translation does not affect the rate-limiting step of HaloTag folding, a key aggregation-prone intermediate observed during in vitro refolding experiments is no longer detectable. This rerouting of the folding pathway increases HaloTag’s folding efficiency and may serve as a general chaperone-independent mechanism of quality control by the ribosome.
INTRODUCTION
Biophysical characterization of protein energy landscapes has provided key insights into the mechanisms of protein folding and misfolding, design, and structure prediction. These in vitro studies, however, often fail to recapitulate the folding process in vivo (1, 2). In the cell, the ribosome synthesizes proteins one amino acid at a time, and the translational machinery is a major hub for protein quality control (3, 4). During translation, the nascent chain has the opportunity to explore regions of the energy landscape in the absence of the protein’s entire sequence. Therefore, cotranslational folding is fundamentally different from typical refolding experiments, where the full-length protein is denatured and then allowed to refold (5). Cotranslational folding has thus become a highly active area of research (6–10) and has revealed insights into the mechanisms of protein-misfolding diseases (11).
Recent studies on stalled ribosome-nascent chain complexes (RNCs) have illuminated some of the features that guide cotranslational protein folding, that is, effects due to the tethering and proximity of the ribosome (12–15). These elegant studies, however, fail to recapitulate the dynamic process of translation; isolated, stalled RNCs are not sufficient for understanding the interplay between translation and protein folding. The importance of cotranslational folding is highlighted by biochemical studies, which have demonstrated that concurrent translation can increase the fidelity of protein folding and quaternary structure formation (16–21). Small changes in protein folding efficiency (the fraction of produced protein that folds to its native state) can have marked effects—they can overwhelm the cellular proteostasis machinery and lead to protein-misfolding diseases (4, 22). Thus, proper cotranslational folding is essential for maintaining cellular and organismal proteostasis. The structural details for the folding process, however, have only been characterized during in vitro refolding where the protein is refolded via dilution from a chemically or thermally denatured state. To understand how translation modulates protein folding, a direct comparison of the cotranslational and refolding trajectories is essential. However, we lack the high-resolution tools needed to monitor cotranslational folding.
Here, we determine the structural basis by which cotranslational folding increases the folding efficiency of the protein HaloTag and present new techniques to directly compare the structural and energetic differences between a protein’s cotranslational and standard refolding trajectories. We find that translation inhibits formation of a folding intermediate without changing the observed rate of overall folding, providing a general, chaperone-independent mechanism for increasing folding efficiency in vivo.
RESULTS
HaloTag refolding can be monitored by fluorescence polarization
HaloTag is a modified haloalkane dehalogenase commonly used as a tool for in vivo imaging (Fig. 1, A and B) that covalently binds a ligand in its native conformation [k = 2.7 × 106 M−1 s−1, ~27.0 s−1 at 10 μM tetramethylrhodamine (TMR)–ligand (the concentration used in this study)] (23). If folding is much slower than 27.0 s−1, then we reasoned that the amount of protein-bound ligand in a folding experiment would be a direct measurement of folded HaloTag at any specific time.
Fig. 1. Cotranslational folding of HaloTag monitored in real-time with FP.
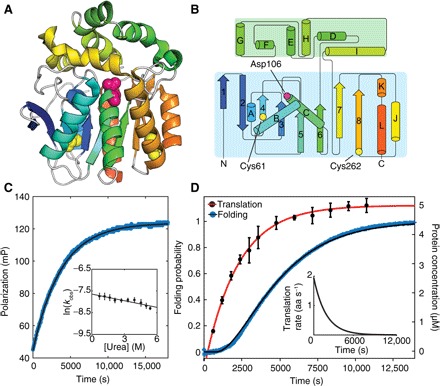
(A) Cartoon representation of the crystal structure of HaloTag from PDB 5UY1 (41). Cys61 and Cys262, as well as the active-site Asp (D106), are represented as spheres. (B) Secondary structure map of HaloTag (42). (C) Refolding of HaloTag as a function of time monitored by FP. HaloTag refolding as a function of denaturant concentration (inset). (D) HaloTag cotranslational folding (blue; left axis) measured by FP and HaloTag synthesis (black circles with red fit; right axis) measured by gel (see fig. S1). Elongation rate as a function of time is shown (inset). aa, amino acid.
We monitored refolding of HaloTag in the presence of a free TMR-functionalized ligand using fluorescence polarization (FP). FP reports on the relative tumbling time of the fluorophore and thus is related to its apparent molecular weight. Rapid dilution of unfolded HaloTag into folding conditions (for example, 8 to 0.8 M urea), results in single exponential kinetics (kobs = 4.7 ± 0.9 × 10−4 s−1 at a final urea concentration of 0.8 M urea; Fig. 1). Linear extrapolation of the natural log of the folding rate, ln(kobs), as a function of the final urea concentration (24) yields a folding rate in the absence of a denaturant (kf,H2O,FP = 4.8 ± 0.6 × 10−4 s−1) that is >10,000 times slower than ligand binding (Table 1) and similar to the folding rate determined by circular dichroism (CD; see below). Thus, changes in FP are measures of HaloTag folding.
Table 1. Summary of kinetic and thermodynamic data of HaloTag and its mutants (error bars are SDs of at least three separate measurements).
| Data from equilibrium experiments | ||
| ΔGunf (kcal⋅mol−1) (CD) | 6.03 ± 0.39 | |
| m value (kcal⋅mol−1 M−1) | 1.57 ± 0.11 | |
| Data from kinetic experiments | ||
| ΔGunf (kcal⋅mol−1) | 5.24 ± 2.0 | |
| m value (kcal⋅mol−1 M−1) | 1.41 ± 0.58 | |
| kf,H2O (s−1) | 0.04 ± 0.02 | |
| mf (kcal⋅mol−1 M−1) | 1.46 ± 0.71 | |
| kconstant, H2O (s−1) | 6.6 ± 0.71 × 10−4 | |
| mconstant (kcal⋅mol−1 M−1) | 0.02 ± 0.1 | |
| kf, H2O,FP (s−1) | 4.8 ± 0.6 × 10−4 | |
| mFP (kcal⋅mol−1 M−1) | −0.1 ± 0.04 | |
| kNI, H2O (s−1) | 8.47 ± 20 ± 10−6 | |
| mNI (kcal⋅mol−1 M−1) | −0.44 ± 0.3 | |
| kIU, H2O (s−1) | 3.3 ± 9.9 × 10−4 | |
| mIU (kcal⋅mol−1 M−1) | 0.70 ± 0.15 | |
| Data from cysteine accessibility experiments | ||
| kWT,refolding, slow (s−1) | 7.8 ± 0.6 ± 10−4 | |
| kWT,refolding, fast (s−1) | 0.03 ± 0.02 | |
| kWT,IVT (s−1) | 4.7 ± 0.3 × 10−4 | |
| kM129C,refolding (s−1) | >0.01 | |
| kM129C,IVT (s−1) | 3.2 ± 0.2 × 10−4 | |
| kI126C,refolding (s−1) | >0.01 | |
| kI126C,IVT (s−1) | 2.2 ± 0.2 × 10−4 | |
| kE121C,refolding (s−1) | >0.01 | |
| kE121C,IVT (s−1) | >0.01 | |
Cotranslational folding can be monitored by FP
To monitor cotranslational folding in real time, we harnessed the same methodology, following FP, during in vitro translation. We initiated the IVT (in vitro transcription and translation) reaction directly in the fluorimeter by adding DNA encoding HaloTag to the coupled IVT system, PURExpress (New England Biolabs). Figure 1D reveals biphasic kinetics: a lag phase and an exponential phase. The observed kinetics are independent of the TMR-ligand concentration and are specific to the HaloTag gene (fig. S1 and table S1). To confirm that the changes in TMR polarization monitor cotranslational folding and not protein synthesis, we independently determined the time dependence of protein production using a gel-based assay (Fig. 1D) (25). The observed kinetics of protein synthesis are also biphasic but with a lag phase significantly shorter than that observed by FP. In addition, we observed an exponential increase in FP signal even after the addition of the translation inhibitor neomycin, confirming that the change in FP reports on HaloTag folding and is not translation-limited (see fig. S1).
Analysis of translation and folding kinetics
These data were analyzed with a kinetic model to account for the asynchronous nature of both protein synthesis and protein folding (see Materials and Methods). The protein synthesis data were analyzed to determine the translation lag phase (251 ± 35 s), which represents the time to synthesize detectable protein levels, and the time-dependent translation rate. The average translation rate, ~1 aa s−1, is similar to translation rates determined for other in vitro systems (26, 27). To determine the cotranslational folding rate, we augmented this model to include a protein-folding component. The lag time observed for the change in polarization is fourfold larger than that observed for protein synthesis, 811 ± 9 s versus 251 ± 35 s, respectively. The resulting cotranslational folding rate is thus 4.42 ± 0.02 × 10−4 s−1, similar to that obtained in the absence of translation (Table 1).
Refolding studies of HaloTag
We then characterized the stability and refolding of HaloTag using recombinant, purified protein (Fig. 2 and Table 1). Folding kinetics, determined by CD, fit to two exponential phases: a fast-folding phase and a urea-independent slow phase (Fig. 2, A to C). The slow phase, as measured by CD, corresponds to the refolding rate determined by FP (Table 1). Often, urea-independent folding is attributed to a cis-trans proline isomerization. However, both refolding and cotranslational folding in the presence of the proline isomerase cyclophilin A (CypA) revealed no effect, which suggests that this may not be due to proline isomerization (fig. S2, Tables 1, and table S3). Surprisingly, refolding to below 1.0 M urea resulted in visible precipitation and protein aggregation (Fig. 2 and fig. S3), although no aggregation was observed in the above cotranslational folding experiments that take place at 0 M urea. Aggregation occurred after an initial decrease in CD signal with a rate similar to the fast refolding phase observed in nonaggregating conditions. Using centrifugation, we determined the fraction of soluble protein to be 0.70 ± 0.06 under these conditions (Fig. 3A).
Fig. 2. Characterization of HaloTag folding kinetics and stability.
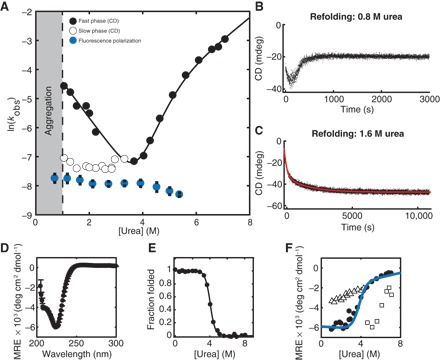
(A) Chevron plot of HaloTag folding and unfolding rates as a function of urea concentration. Fast phase (black circles) and slow phase (white circles, black outline). Refolding as measured by FP is shown in blue. Refolding traces of HaloTag at (B) 0.8 M urea, where there is visible protein aggregation, and (C) 1.6 M urea, where no precipitation is observed. (D) CD spectrum of HaloTag at 0 M urea. (E) Equilibrium denaturant melt of HaloTag. (F) Burst-phase amplitudes for refolding (white triangles with black outline) and unfolding (white squares with black outline). Kinetic final amplitudes (black circles) overlay well with the fit of equilibrium data (blue line). Error bars represent the SD of three separate experiments.
Fig. 3. HaloTag folding is more efficient during in vitro translation than after refolding.
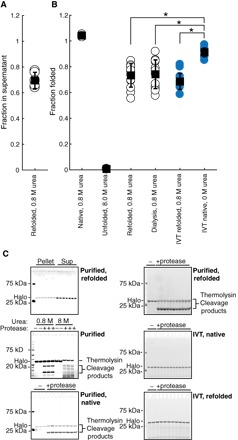
(A) Fraction of total protein remaining in supernatant after centrifugation following refolding of HaloTag to 0.8 M urea. (B) Fraction folded as measured by pulse proteolysis in conditions as indicated—either after refolding, after in vitro translation, or both. Blue circles are in vitro–translated protein. (C) Representative gels for (A) and (B). All error bars are the SDs of at least 15 separate experiments except for HaloTag in 0.8 and 8.0 M urea, which are the SDs of three experiments. *P < 0.01, Student’s unpaired t test.
HaloTag cotranslational folding is more efficient than refolding
To compare the efficiencies of refolding and cotranslational folding, that is, the fraction of protein that reaches the native state, we used pulse proteolysis (28), a gel-based method for measuring the amount of folded protein (Fig. 3). Purified HaloTag incubated in 0.8 M urea is completely folded when evaluated by pulse proteolysis, but when refolded by dilution from 8.0 to 0.8 M urea, the efficiency is only 0.73 ± 0.03, consistent with that determined by centrifugation above. By contrast, cotranslational folding is significantly more efficient than refolding: 0.91 ± 0.03 versus 0.73 ± 0.03 (P < 0.01, Student’s unpaired t test; n > 12; Fig. 3 and table S2). Note that IVT reactions are carried out at a higher protein concentration than the less-efficient refolding studies (>5 and 3 μM, respectively; see fig. S1 and Materials and Methods). To rule out any possible chemical differences between in vitro–translated protein and recombinant protein, we measured the refolding efficiency of IVT protein and determined it to be similar to that of purified protein: 0.69 ± 0.06 versus 0.70 ± 0.06, respectively (Fig. 3). Why is cotranslational folding significantly more efficient than refolding? How does translation alter the folding pathway of HaloTag?
Structural characterization of the in vitro refolding pathway using HX-MS
To compare the refolding and cotranslational folding pathways of HaloTag, we first used pulse-labeling hydrogen-deuterium exchange coupled with proteolysis and mass spectrometry (HX-MS) to obtain structural information about the conformations formed during HaloTag refolding (29, 30). We applied pulses of hydrogen exchange at various refolding times, and monitored the mass of individual peptides as a function of refolding time. Changes in mass are a measure of backbone amide accessibility at that particular refolding time. Figure 4A shows the fraction deuterated for each peptide after 10 s of refolding and reveals two populations: those that are at least 25% deuterated by 10 s (“fast,” red) and those that are not (“slow,” blue). Plotting the mean-normalized fraction deuterated for both slow and fast peptides further highlights that these two groups of peptides have distinct behaviors throughout the folding trajectory (Fig. 4B). On average, fast peptides are more deuterated at all time points analyzed than slow peptides. Data for all peptides used in this analysis are available in table S4. These data, together with the biphasic CD kinetics (Fig. 2), suggest that the early protection is a result of the formation of a fast-folding intermediate. The early phase corresponds to protection of peptides comprising the Rossman-fold core of the protein, while the entire lid domain and β-strand 8 are protected more slowly (Fig. 4C). It is possible that the formation of this intermediate is directly responsible for HaloTag’s aggregation. For instance, helix B and β-strand 4 remain unprotected, despite the fact they both make critical contacts with the rest of the Rossman fold (β-strands 1 and 2 and helix C; Fig. 4C). This likely results in a large, exposed hydrophobic surface. Thus, during the early steps of folding, not only is the lid domain completely unprotected but there is also a large exposed hydrophobic surface. Moreover, because this intermediate is comprised of residues distant in sequence space, it is possible that cotranslational folding does not involve formation of this intermediate.
Fig. 4. The HaloTag folding trajectory changes during cotranslational folding.
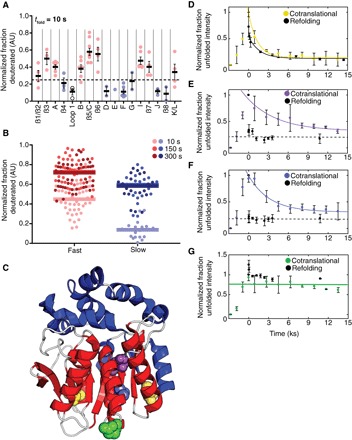
(A) Peptides derived from HX-MS experiments after 10 s of refolding were plotted according to their corresponding secondary structural element. Helices are lettered, whereas β sheets are numbered. Secondary structural elements were then divided into fast-folding (red circles) or slow-folding (blue circles) regions based on the average fraction deuterated (solid line) for peptides within those secondary structures at the 10-s time point (above or below dashed line). Error bars represent SEM. (B) Normalized fraction deuterated for all peptides (filled circles) plotted with the mean fraction deuterated for each group of peptides (solid lines) is shown for three time points. A full list of peptides is available in table S4. (C) Crystal structure of HaloTag with slow (blue) and fast (red) secondary structural elements colored. Loops are colored in white. Cysteines probed in (D) to (G) are represented as spheres (yellow, Cys61 and Cys262; purple, M129C; blue, I126C; green, E121C). (D to G) Cysteine accessibility experiments during in vitro translation (colored lines and circles) and refolding (dotted lines and black dots). (D) WT HaloTag. (E) Halo* M129C. (F) Halo* I126C. (G) Halo* E121C. Error bars represent the SD of three separate experiments except for (A) where error bars are the SEM. Gels are shown in fig. S6. AU, arbitrary units.
Comparison of the HaloTag refolding and cotranslational folding trajectories using pulsed cysteine labeling
Unfortunately, the above HX-MS studies are currently not feasible for investigating cotranslational folding. Therefore, to determine whether HaloTag populates the same folding intermediate during cotranslational folding and refolding, we designed specific thiol probes based on the above HX-MS data. Labeling of reactive cysteines has been used successfully in the past on stalled RNCs (7). We monitored thiol accessibility during both refolding and cotranslational folding using a fluorescein-conjugated maleimide (FSM), detected by in-gel fluorescence.
Both of HaloTag’s native cysteines, positioned at the base of two β-strands, β4 and β8, are completely protected in the folded state and accessible in the unfolded state (modified within 30 s; see fig. S4). One of these, Cys262, is in the region we anticipate to be structured in the intermediate, and the other, Cys61, is not. Pulsed thiol labeling during refolding of the wild-type (WT) protein showed two phases with similar rates to those obtained by CD. By contrast, pulsed thiol labeling during cotranslational folding resulted in only a single exponential indistinguishable from HaloTag’s slow folding rate during refolding (Fig. 4D and Table 1).
We then created three site-specific cysteine variants to probe the very early stages of folding (E121C, I126C, and M129C) in an otherwise cysteine-free background (Halo*). Residues 126 and 129 are both buried side chains on β6, and during refolding, both are protected within the burst phase of the experiment (Fig. 4, E and F). E121C is on the surface of HaloTag and remains unprotected throughout the folding reaction (Fig. 4G). All three variants bind TMR and display similar folding kinetics as WT HaloTag (fig. S5). In contrast to the previous refolding experiments, during in vitro translation, sites 126 and 129 are not protected early but rather show slow protection corresponding to the overall folding rate of the protein (Fig. 4D). Thus, the folding pathway of HaloTag is altered during translation.
DISCUSSION
Together, our results suggest that the HaloTag refolding intermediate, which is likely the precursor for aggregation, is not populated during translation-coupled folding. This change in the folding trajectory is likely responsible for HaloTag’s increased cotranslational folding efficiency. Moreover, this model also provides an explanation for the recent report that the mutation K73T, located within the structured region of the refolding intermediate, leads to increased HaloTag aggregation (31). The specific cysteines characterized here, however, do not yield further insight into other potential intermediates that may form during translation. The overall rate of folding is not changed during cotranslational folding, and thus, the rate-limiting step for folding does not appear to require the formation of this specific intermediate.
Intermediates in protein folding can play both positive and negative roles. Intermediates are often beneficial to the folding process by narrowing conformational space, while access to transient intermediates is also a major determinant for the formation of toxic aggregates associated with disease (32, 33). Previous studies have suggested that formation of translation-specific intermediates may help to guide the folding process; our data support the hypothesis that destabilization of potentially toxic or off-pathway intermediates that form during translation are also advantageous (12, 34, 35). Thus, we have determined an additional mechanism by which translation helps to avoid aggregation of the emerging protein.
Our findings highlight the interplay between the rates of translation and folding (36–38). For instance, the relatively slow rate of translation in our IVT setup may aid in increasing HaloTag folding efficiency. Using the methods described here, it will now be possible to measure how folding efficiency and folding trajectories are modulated by the rate of translation.
HaloTag is ideally suited for these kinds of studies. HaloTag folding can be monitored by FP, thus folding experiments can be performed with high throughput and in the presence of many other biologically active molecules including during IVT. This is a powerful system to systematically investigate how the translational and quality control machinery modulates protein folding. These types of unbiased approaches will lead to the discovery of general and quantitative rules that govern not only protein folding during translation but also protein folding in other high-complexity environments.
MATERIALS AND METHODS
Protein expression and purification
Protein expression
BL21(DE3) cells were transformed with expression vectors containing the WT or mutant HaloTag cDNA. Single colonies were used to seed starter cultures grown overnight to saturation. Large-scale cultures were inoculated with 5 ml of overnight culture, grown at 37°C to an optical density at 600 nm of 0.6 to 0.8 and induced with 1 mM isopropyl-β-D-thiogalactopyranoside for 2 to 3 hours at 37°C. After induction, cultures were pelleted at 5000g for 10 min at 4°C, flash-frozen, and stored at −80°C.
Purification
Cell pellets were resuspended in 10 mM tris/H2SO4 (pH 7.5) and 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP; lysis buffer) and lysed by sonication on ice. Lysates were cleared by centrifugation for 30 min at 20,000g at 4°C and subsequently filtered through 0.2-μm filters. After clearing, the lysate was dialyzed into at least a 10-fold volume excess of lysis buffer, loaded onto a HiPrep Q XL 16/10 column equilibrated with lysis buffer, and eluted with a gradient of lysis buffer plus 0 to 600 mM NaCl. Fractions containing the HaloTag protein were dialyzed into at least a 10-fold volume excess of 20 mM sodium acetate (pH 5.0; Q buffer) loaded onto a HiPrep Q XL16/10 column equilibrated with Q buffer and eluted with a gradient of Q buffer plus 0 to 800 mM NaCl. Fractions containing HaloTag protein were then concentrated and purified on a HiLoad 16/600 Superdex 75 pg column equilibrated with 50 mM ammonium bicarbonate or 25 mM Hepes KOH (pH 7.5), 15 mM MgOAc, 150 mM KCl, and 0.1 mM TCEP (HKMT), and the fractions with the retention volume corresponding to the size of monomeric HaloTag were either lyophilized (ammonium bicarbonate runs) and subsequently stored at −80°C or concentrated and immediately used for experiments (HKMT runs). All lyophilized protein was resuspended in HKMT and spun-filtered at 4°C before use in experiments.
Fluorescence polarization
Data collection
All experiments were performed at 37°C unless otherwise noted. FP was performed on a BioTek Synergy Neo2 plate reader in 384-well, black flat-bottom plates for IVT reactions (Corning) or 96-well, clear flat-bottom plates (refolding experiments). Acquisitions were collected using polarizers and 530-nm/590-nm filters with a side gain set at 45 and a top gain set at 40. Read height was 7.5 mm, and 10 measurements were made per data point. After a 15-min incubation at 37°C, readings were initialized by the addition of DNA (IVT reactions) or unfolded protein (refolding experiments). Measurements were taken every 20 s for 5 hours after 30 s of mixing and a 90-s delay for temperature equilibration.
IVT reactions
IVT reactions using a PURExpress system were set up on ice per the manufacturer’s protocols for a 30-μl reaction with the addition of 1 μl of ribonuclease (RNase) inhibitior, murine, and 1 μl of 300 μM TMR [in 100% anhydrous dimethyl sulfoxide (DMSO), for a final TMR concentration of 10 μM] and pipetted into wells. Plates were covered with clear titer tops to prevent evaporation and equilibrated at 37°C for at least 15 min. Reactions were initiated with 2 μl of plasmid DNA (125 ng/μl).
Refolding experiments
Refolding experiments were performed in HKMT buffer plus appropriate concentrations of urea and TMR (to a final concentration of 5 μM TMR and 3.33% DMSO). Plates were sealed and incubated at 37°C for 15 min until reactions were initiated by adding 10 μl of 20 μM HaloTag in 8 M urea that had been incubated at 37°C for at least 12 hours. Refolding traces were fit to the following equation in Matlab, using bi-square fitting and “k” bounded at zero
Urea concentrations were measured using a refractometer as previously described (28).
Circular dichroism
Kinetic and equilibrium experiments were performed using a 0.5-cm cuvette at 37°C with constant stirring at 3 μM (0.1 mg/ml) in HKMT buffer. Equilibrium and kinetic experiments were performed as previously described (39) but at a wavelength of 225 nm instead of 222 nm to increase the signal-to-noise ratio. Analysis was performed as described (39). Wavelength experiments were performed in a 0.1-cm cuvette at 37°C with 15 μM protein (~0.5 mg/ml) in HKMT buffer.
Determination of folding efficiency
All reactions were performed at 37°C at a final concentration of 3 μM protein in HKMT buffer unless otherwise noted.
Centrifugation assay
Proteins were refolded by the dilution of protein in 8 M urea to the proper urea concentration and allowed to reach equilibrium for at least 12 hours. Samples were then centrifuged at 21,130g for 30 min, and the supernatant was carefully removed. The pellet was resuspended in an equal volume of 8 M urea. Both the supernatant and pellet were then mixed with a 6× SDS–polyacrylamide gel electrophoresis (PAGE) loading dye and run on a 4 to 12% bis-tris gel in an MES run buffer and subsequently stained with SYPRO Red for 30 min in 10% acetic acid. After destaining in 10% acetic acid for an hour, gels were imaged using a Typhoon Trio (GE Healthcare) and analyzed with ImageJ.
Pulse proteolysis
IVT reactions were performed as per the manufacturer’s instructions but with the addition of 1 μl of RNase inhibitor, murine, and 1.25 μl of FluoroTect GreenLys (Promega) per 30 μl of IVT reaction (25). IVT reactions were quenched after 1 hour to a final concentration of 2 mM chloramphenicol and RNase A (0.1 mg/ml). Refolding experiments were performed as described above. IVT reactions and refolding reactions were allowed to reach equilibrium for at least 12 hours. Subsequently, reactions were aliquoted to 10 μl, and 1 μl of thermolysin (1 mg/ml; Sigma) was added to each reaction for 1 min and quenched with EDTA to a final concentration of 83 mM. SDS-PAGE loading dye was then added to each reaction, and each reaction was run on a 4 to 12% bis-tris gel in an MES run buffer. Imaging and analysis was performed as described previously (28).
Refolding of IVT-translated protein
IVT reactions were performed and quenched as described above. A 10-fold volume excess of 8 M urea in HKMT buffer was then added and mixed with the IVT translation reaction and allowed to equilibrate at 37°C overnight. Reactions were then concentrated in a 0.5-ml 10-kDa cutoff spin concentrator (Amicon) and diluted to 0.8 M urea. After equilibration at 37°C overnight, pulse proteolysis was performed and analyzed as described above.
Translation rate measurement
IVT reactions were performed as per the manufacturer’s instructions but with the addition of 1 μl of RNase inhibitor, murine, and 1.25 μl of FluoroTect GreenLys (Promega) per 30 μl of IVT reaction and initiated with 250 ng of DNA. At each time point, 1.5 μl of IVT reaction was quenched into a final concentration of 2 mM chloramphenicol and RNase A (0.1 mg/ml) and then SDS-PAGE loading dye. Reactions were then run on a 4 to 12% bis-tris gel in an MES run buffer and imaged using a Typhoon Trio. Analysis was performed using ImageJ.
Cysteine protection assays
Purified protein
Refolding reactions were initiated as described above. At each time point, a 50-fold molar excess of FSM was added for 30 s and quenched into an equal volume of SDS-PAGE loading dye containing β-mercaptoethanol (BME) to a final BME concentration of 2.15 M. Reactions were then run on 4 to 12% bis-tris gel in an MES run buffer and imaged using a Typhoon Trio. Analysis was performed using ImageJ. Traces were fit to the following equation in Matlab, using bi-square fitting and “k” bounded at zero (for those data which displayed exponential kinetics)
Because folded Halo**E121C cysteine reactivity is time-dependent over the labeling time of the reaction, intensities after refolding was initiated were normalized to the reactivity at that labeling time as determined in fig. S7.
IVT reactions
IVT reactions were initiated as described above. At each time point, an equal volume of 2 mM FSM was mixed with IVT reaction for 30 s and quenched into SDS-PAGE loading dye as above. At 45 min, reactions were halted by the addition of chloramphenicol to a final concentration of 2 mM. Reactions were then run on a 4 to 12% bis-tris gel in an MES run buffer and imaged using a Typhoon Trio. A sample of purified, TMR-bound HaloTag was run to determine the size of the Halo-FSM band marked with an “*” in Fig. 6 (A to D). Analysis was performed using ImageJ. Intensities were normalized to a major protein product running at ~65 kDa to control for effects of evaporation, fluorescein bleaching, and gel loading. Traces were fit to the following equation in Matlab after exclusion of points before 45 min, using bi-square fitting and “k” bounded at zero
Pulsed labeling HX-MS
Pulse-labeling scheme
The HX-MS pulsed labeling experiments were based on previously described approaches (29, 30). Deuterated protein was prepared by lyophilizing unfolded HaloTag in 8 M urea followed by resuspension in D2O, repeated four times. Refolding experiments were carried out using a BioLogic QFM-4 apparatus in interrupted flow mode. To initiate refolding, we diluted deuterated protein in 8 M urea [deuterated HKMT buffer (pDread 7.9), 10°C] with 10 volumes of deuterated HKMT to a final urea concentration of 1.6 M. After a variable delay time (refolding time), D-to-H exchange at still-exposed sites was induced by a high-pH pulse of protonated buffer (200 mM glycine; 10 ms; 5 volumes; final pH, 10.00). Because of the large volume changes, the final solution is only 31% protonated. The pulse was quenched by dilution with a low-pH buffer (1 M glycine; 5 volumes; final pH, 2.00) to slow any further exchange. Protein samples were then collected and injected into a custom liquid chromatography–mass spectrometry (LC/MS) system. A folded control sample was prepared by subjecting deuterated, native protein to the same pulse/quench sequence, and an unfolded control was measured by performing the pulse/quench using fully deuterated, unfolded protein.
LC/MS system
A custom high-performance liquid chromatography system was used for in-line protease digestion, desalting, and separation of peptides. Peptides were eluted from the trap column and separated on an analytical C8 column using an acetonitrile gradient (5 to 90% acetonitrile) at 17 μl/min. The output of this system was directly injected into a Thermo Scientific LTQ Orbitrap Discovery using electrospray ionization.
Data analysis of HX-MS pulsed labeling
Peptides were identified using a SEQUEST search using a Proteome Discoverer 2.0 software. Peptide mass envelopes were fit using HDExaminer (Sierra Analytics) followed by a manual confirmation of each peptide. Deuterium content was assessed by examination of the centroid of each fitted peptide mass envelope. Only peptides with a high signal-to-noise ratio at each time point were used for further analysis. For each peptide at each time point, the fraction deuterated was determined by comparison to the folded and unfolded control samples.
Kinetic modeling of translation and folding
Model for protein production in an IVT experiment
To derive a chemical kinetic expression for the amount of protein produced as a function of time, we note that the rate of protein synthesis is the rate at which L amino acids link together covalently to create a protein P. Thus, the simplest reaction scheme for this process is
| (1) |
This reaction occurs with some rate k, which is a function of many processes including translation-initiation, translation-elongation, and translation-termination. We consider L number of amino acids as one “bundle” (B) of monomer subunits; one bundle is required to synthesize one protein molecule. If NAA is the total number of free amino acids in solution, then the total number of bundles NB is NAA/L. Thus, to express these reactants in concentration form, we write the reaction scheme as
| (2) |
that is, state B consists of L free amino acids that are converted into P. The time evolution of the protein concentration, [P(t)], is governed by the master equation
| (3) |
Solving Eq. 3 under boundary conditions, [P(t)] = 0 at t = 0, yields
| (4) |
The maximum protein concentration [Pmax] at time t → ∞ is equal to [B(0)]. Therefore
| (5) |
There must be a time lag between the time at which transcription starts and the time at which the first protein molecule is fully synthesized. If this time lag is t0, then Eq. 5 becomes
| (6) |
We fit the experimentally measured protein production curve using Eq. 6 and extracted the overall rate of protein synthesis k and time lag t0.
Translation speed
We estimated the average translation speed based on the method described in the study of Capece et al. (40). We calculated the rate of protein production J(t) by taking the derivative of Eq. 6
| (7) |
Next, we normalized this quantity with the ribosome concentration (that is, 0.45 μM), which we denote as J(t). Here, J(t) is the rate of protein production from a single ribosome. On average, then, 1/J(t) is the time required to synthesize a protein molecule, and a ribosome goes through a round of initiation, elongation, termination, and recycling during this time. Thus, 1/J(t) is an upper bound on the gene translation time, and if elongation is the rate-limiting step, then J(t)L is an estimate of the average codon translation rate.
Analytical derivation for the fraction of folded protein
To derive an expression for the probability of protein folding in an IVT experiment, we assume that the protein is released in the unfolded state. Unfolded proteins fold posttranslationally with rate kF, and the backward transition occurs with rate kU. In this situation, the following chemical kinetic equations govern the time evolution of the concentration of proteins in the unfolded and folded state
| (8) |
| (9) |
and
| (10) |
[F(t)] and [U(t)] in Eqs. 8 to 10 are the concentration of proteins in the folded and unfolded state, respectively, whereas [P(t)] (Eq. 6) is the total protein concentration at time t. Solving Eqs. 8 to 10 yields
| (11) |
and
where is the concentration of folded protein at t → ∞. We used Eq. 11 to fit the experimentally measured folding probability and extract the numerical value of kF, assuming kU = 0.
Supplementary Material
Acknowledgments
We thank the entire Marqusee Lab for advice on experiments, paper writing, and support; S. A. Lim for help with kinetic fitting; and M. K. Jensen for help with the manuscript. We also thank A. Martin and his laboratory for equipment and reagents, D. Costanzo for advice relating to FSM labeling, J. S. Fraser for advice on the manuscript, and J. Chen for technical support. Funding: This work was supported by NIH grant GM050945 (to S.M.), NSF grant MCB-1553291 (to A.K.S.), and NIH grant 1R35GM124818-01 (to E.P.O.). Author contributions: A.J.S., E.B., and S.M. designed experiments and wrote the paper. A.J.S. and S.M.C. performed experiments other than HX-MS. E.B. performed HX-MS experiments. A.J.S. and E.B. carried out the HX-MS data analysis. A.K.S. and E.P.O. derived Eqs. 6 and 11 and analyzed the protein synthesis and FP data. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaas9098/DC1
fig. S1. Cotranslational folding of HaloTag can be measured using FP.
fig. S2. Addition of the peptidyl-proline isomerase CypA does not affect HaloTag refolding or cotranslational folding rates.
fig. S3. Aggregation of HaloTag.
fig. S4. Cysteine accessibility of WT HaloTag.
fig. S5. Characterization of Halo* cysteine mutants.
fig. S6. Gels for data shown in Fig. 4.
fig. S7. Characterization of Halo* E121C cysteine accessibility.
table S1. Kinetic data obtained for HaloTag folding using FP.
table S2. Determination of HaloTag folding efficiency under different conditions.
table S3. Folding rates of HaloTag and variants measured by FP.
table S4. Normalized HX-MS data.
REFERENCES AND NOTES
- 1.Braselmann E., Chaney J. L., Clark P. L., Folding the proteome. Trends Biochem. Sci. 38, 337–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartl F. U., Hayer-Hartl M., Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Pechmann S., Willmund F., Frydman J., The ribosome as a hub for protein quality control. Mol. Cell 49, 411–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balchin D., Hayer-Hartl M., Hartl F. U., In vivo aspects of protein folding and quality control. Science 353, aac4354 (2016). [DOI] [PubMed] [Google Scholar]
- 5.O’Brien E. P., Christodoulou J., Vendruscolo M., Dobson C. M., New scenarios of protein folding can occur on the ribosome. J. Am. Chem. Soc. 133, 513–526 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Cabrita L. D., Dobson C. M., Christodoulou J., Protein folding on the ribosome. Curr. Opin. Struct. Biol. 20, 33–45 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Lu J., Deutsch C., Folding zones inside the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 12, 1123–1129 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Kramer G., Boehringer D., Ban N., Bukau B., The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 16, 589–597 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Nilsson O. B., Nickson A. A., Hollins J. J., Wickles S., Steward A., Beckmann R., von Heijne G., Clarke J., Cotranslational folding of spectrin domains via partially structured states. Nat. Struct. Mol. Biol. 24, 221–225 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Nilsson O. B., Hedman R., Marino J., Wickles S., Bischoff L., Johansson M., Müller-Lucks A., Trovato F., Puglisi J. D., O’Brien E. P., Beckmann R., von Heijne G., Cotranslational protein folding inside the ribosome exit tunnel. Cell Rep. 12, 1533–1540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim S. J., Yoon J. S., Shishido H., Yang Z., Rooney L. A., Barral J. M., Skach W. R., Translational tuning optimizes nascent protein folding in cells. Science 348, 444–448 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Samelson A. J., Jensen M. K., Soto R. A., Cate J. H. D., Marqusee S., Quantitative determination of ribosome nascent chain stability. Proc. Natl. Acad. Sci. U.S.A. 113, 13402–13407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight A. M., Culviner P. H., Kurt-Yilmaz N., Zou T., Ozkan S. B., Cavagnero S., Electrostatic effect of the ribosomal surface on nascent polypeptide dynamics. ACS Chem. Biol. 8, 1195–1204 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kaiser C. M., Goldman D. H., Chodera J. D., Tinoco I. Jr., Bustamante C., The ribosome modulates nascent protein folding. Science 334, 1723–1727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabrita L. D., Cassaignau A. M. E., Launay H. M. M., Waudby C. A., Wlodarski T., Camilloni C., Karyadi M.-E., Robertson A. L., Wang X., Wentink A. S., Goodsell L. S., Woolhead C. A., Vendruscolo M., Dobson C. M., Christodoulou J., A structural ensemble of a ribosome–nascent chain complex during cotranslational protein folding. Nat. Struct. Mol. Biol. 23, 278–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labbadia J., Morimoto R. I., The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G., Hubalewska M., Ignatova Z., Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 16, 274–280 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Ugrinov K. G., Clark P. L., Cotranslational folding increases GFP folding yield. Biophys. J. 98, 1312–1320 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frydman J., Erdjument-Bromage H., Tempst P., Hartl F. U., Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat. Struct. Biol. 6, 697–705 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Fedorov A. N., Baldwin T. O., Process of biosynthetic protein folding determines the rapid formation of native structure. J. Mol. Biol. 294, 579–586 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Shieh Y.-W., Minguez P., Bork P., Auburger J. J., Guilbride D. L., Kramer G., Bukau B., Operon structure and cotranslational subunit association direct protein assembly in bacteria. Science 350, 678–680 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Gloge F., Becker A. H., Kramer G., Bukau B., Co-translational mechanisms of protein maturation. Curr. Opin. Struct. Biol. 24, 24–33 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., Simpson D., Mendez J., Zimmerman K., Otto P., Vidugiris G., Zhu J., Darzins A., Klaubert D. H., Bulleit R. F., Wood K. V., HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Matouschek A., Kellis J. T. Jr., Serrano L., Fersht A. R., Mapping the transition state and pathway of protein folding by protein engineering. Nature 340, 122–126 (1989). [DOI] [PubMed] [Google Scholar]
- 25.Mallam A. L., Jackson S. E., Knot formation in newly translated proteins is spontaneous and accelerated by chaperonins. Nat. Chem. Biol. 8, 147–153 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Guet C. C., Bruneaux L., Min T. L., Siegal-Gaskins D., Figueroa I., Emonet T., Cluzel P., Minimally invasive determination of mRNA concentration in single living bacteria. Nucleic Acids Res. 36, e73 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zubay G., In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7, 267–287 (1973). [DOI] [PubMed] [Google Scholar]
- 28.Park C., Marqusee S., Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2, 207–212 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Hu W., Walters B. T., Kan Z.-Y., Mayne L., Rosen L. E., Marqusee S., Englander S. W., Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 110, 7684–7689 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayne L., Kan Z.-Y., Chetty P. S., Ricciuti A., Walters B. T., Englander S. W., Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass Spectrom. 22, 1898–1905 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y., Fares M., Dunham N. P., Gao Z., Miao K., Jiang X., Bollinger S. S., Boal A. K., Zhang X., AgHalo: A facile fluorogenic sensor to detect drug-induced proteome stress. Angew. Chem. Int. Ed. Engl. 56, 8672–8676 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Jahn T. R., Radford S. E., Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 469, 100–117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim K. H., Dasari A. K. R., Hung I., Gan Z., Kelly J. W., Wemmer D. E., Structural changes associated with transthyretin misfolding and amyloid formation revealed by solution and solid-state NMR. Biochemistry 55, 1941–1944 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hulleman J. D., Balch W. E., Kelly J. W., Translational attenuation differentially alters the fate of disease-associated fibulin proteins. FASEB J. 26, 4548–4560 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trovato F., O’Brien E. P., Fast protein translation can promote co- and posttranslational folding of misfolding-prone proteins. Biophys. J. 112, 1807–1819 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang G., Ignatova Z., Folding at the birth of the nascent chain: Coordinating translation with co-translational folding. Curr. Opin. Struct. Biol. 21, 25–31 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Pechmann S., Frydman J., Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 20, 237–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans M. S., Sander I. M., Clark P. L., Cotranslational folding promotes β-helix formation and avoids aggregation in vivo. J. Mol. Biol. 383, 683–692 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connell K. B., Miller E. J., Marqusee S., The folding trajectory of RNase H is dominated by its topology and not local stability: A protein engineering study of variants that fold via two-state and three-state mechanisms. J. Mol. Biol. 391, 450–460 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capece M. C., Kornberg G. L., Petrov A., Puglisi J. D., A simple real-time assay for in vitro translation. RNA 21, 296–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y., Miao K., Dunham N. P., Liu H., Fares M., Boal A. K., Li X., Zhang X., The cation–π interaction enables a halo-tag fluorogenic probe for fast no-wash live cell imaging and gel-free protein quantification. Biochemistry 56, 1585–1595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stivala A., Wybrow M., Wirth A., Whisstock J. C., Stuckey P. J., Automatic generation of protein structure cartoons with pro-origami. Bioinformatics 27, 3315–3316 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaas9098/DC1
fig. S1. Cotranslational folding of HaloTag can be measured using FP.
fig. S2. Addition of the peptidyl-proline isomerase CypA does not affect HaloTag refolding or cotranslational folding rates.
fig. S3. Aggregation of HaloTag.
fig. S4. Cysteine accessibility of WT HaloTag.
fig. S5. Characterization of Halo* cysteine mutants.
fig. S6. Gels for data shown in Fig. 4.
fig. S7. Characterization of Halo* E121C cysteine accessibility.
table S1. Kinetic data obtained for HaloTag folding using FP.
table S2. Determination of HaloTag folding efficiency under different conditions.
table S3. Folding rates of HaloTag and variants measured by FP.
table S4. Normalized HX-MS data.