

Here, Rawal et al. analyzed Gcn4-induced genes in mutants lacking the catalytic subunits of SWI/SNF or RSC and uncovered cooperation between these remodelers in evicting nucleosomes from different locations in the promoter and repositioning the +1 nucleosome downstream to produce wider NDRs—highly depleted of nucleosomes—during transcriptional activation. Their findings show that SWI/SNF acts comparably with RSC in forming wide nucleosome-free NDRs to achieve high-level transcription but only at the most highly expressed genes exhibiting the greatest SWI/SNF occupancies.
Keywords: nucleosome, SWI/SNF, RSC, transcription, yeast
Abstract
The nucleosome remodeling complex RSC functions throughout the yeast genome to set the positions of −1 and +1 nucleosomes and thereby determines the widths of nucleosome-depleted regions (NDRs). The related complex SWI/SNF participates in nucleosome remodeling/eviction and promoter activation at certain yeast genes, including those activated by transcription factor Gcn4, but did not appear to function broadly in establishing NDRs. By analyzing the large cohort of Gcn4-induced genes in mutants lacking the catalytic subunits of SWI/SNF or RSC, we uncovered cooperation between these remodelers in evicting nucleosomes from different locations in the promoter and repositioning the +1 nucleosome downstream to produce wider NDRs—highly depleted of nucleosomes—during transcriptional activation. SWI/SNF also functions on a par with RSC at the most highly transcribed constitutively expressed genes, suggesting general cooperation by these remodelers for maximal transcription. SWI/SNF and RSC occupancies are greatest at the most highly expressed genes, consistent with their cooperative functions in nucleosome remodeling and transcriptional activation. Thus, SWI/SNF acts comparably with RSC in forming wide nucleosome-free NDRs to achieve high-level transcription but only at the most highly expressed genes exhibiting the greatest SWI/SNF occupancies.
In the yeast Saccharomyces cerevisiae, nucleosomes are organized in a stereotypical pattern relative to the promoter regions of most genes transcribed by RNA polymerase II (Pol II). A nucleosome-depleted region (NDR) of average width ∼120 base pairs (bp) situated upstream of gene coding sequences (CDSs) is flanked by highly positioned “−1” and “+1” nucleosomes, with the transcription start site (TSS) generally located within the +1 nucleosome (+1_Nuc) downstream from the NDR. Promoter elements and upstream activator-binding sites (UAS elements) reside within the NDR and sometimes extend upstream into the −1_Nuc (Jiang and Pugh 2009; Wang et al. 2011; Cui et al. 2012; Rando and Winston 2012). The location of the TSS within the +1_Nuc dictates that the latter is frequently evicted (Boeger et al. 2003; Reinke and Horz 2003) or shifted in the 3′ direction (Reja et al. 2015; Nocetti and Whitehouse 2016) at least transiently during assembly of the Pol II transcription preinitiation complex (PIC). Certain highly regulated genes exhibit greater nucleosome occupancies in NDRs—occluding promoter elements and transcription factor-binding sites—in their repressed states (Cairns 2009; Ganguli et al. 2014; Parnell et al. 2015). Consequently, the yeast ATP-dependent chromatin remodeling complexes SWI/SNF and RSC, capable of sliding or evicting nucleosomes, are required for robust transcriptional activation of many genes (Rando and Winston 2012). Consistent with this, both RSC and SWI/SNF are enriched at locations both 5′ and 3′ of NDRs and at promoter-proximal nucleosomes in CDSs throughout the yeast genome (Yen et al. 2012).
RSC has been shown to function at the majority of yeast genes both in vivo (Badis et al. 2008; Parnell et al. 2008; Hartley and Madhani 2009) and in a reconstituted in vitro system (Krietenstein et al. 2016) to maintain the native NDR width by opposing encroachment of the −1_Nuc or +1_Nuc into the NDR. Shifts in the +1_Nuc toward the NDR on RSC inactivation are generally accompanied by movement in the same direction of the downstream array of phased nucleosomes (+2 to +5) with little change in nucleosome spacing (Ganguli et al. 2014; Parnell et al. 2015), suggesting that RSC acts primarily to determine the locations of the +1_Nuc and −1_Nuc. This activity could be instrumental in gene activation by preventing the +1_Nuc from sliding upstream and occluding promoter elements (e.g., the TATA box) or the TSS within the nucleosome core (Zhang and Reese 2007; Reja et al. 2015; Nocetti and Whitehouse 2016). Indeed, a widespread reduction in expression of many genes has been observed on RSC inactivation (Parnell et al. 2008). However, the most highly affected group of RSC-dependent genes, which also displayed the highest RSC promoter occupancies, did not exhibit a well-defined NDR or display decreased NDR width on RSC inactivation, suggesting a more dynamic mobilization of promoter nucleosomes by RSC in activating these promoters (Parnell et al. 2015). Other studies have revealed higher RSC occupancies in the coding regions versus promoters of many genes, which is directly correlated with transcription rate (Ganguli et al. 2014; Spain et al. 2014), and such genes frequently exhibit reduced transcription on RSC inactivation, especially those weakly expressed in wild-type cells, all of which suggests a role for RSC in promoting transcription elongation (Spain et al. 2014).
Whereas RSC acts primarily to exclude the −1_Nuc and +1_Nuc from the NDR, the chromatin remodelers ISW1 and CHD1 have overlapping functions in establishing the regular spacing and phasing of nucleosomes in the CDSs at most genes, and ISW1 is particularly important for the proper linker length separating these nucleosomes in vivo (Gkikopoulos et al. 2011; van Bakel et al. 2013; Ocampo et al. 2016). Consistent with this, one or more ISWI complexes were required in addition to RSC to partially reconstitute in vitro phased nucleosome arrays downstream from the NDR in conjunction with general regulatory factors Abf1 or Reb1 (Krietenstein et al. 2016).
In contrast to RSC, ISWI, and CHD1 chromatin remodelers, the SWI/SNF complex does not appear to function broadly in determining the width or nucleosome occupancies of NDRs or in the phasing or spacing of downstream nucleosomes. Thus, only a small fraction of genes with wide NDRs and poor phasing was found to exhibit increased nucleosome occupancies in their NDRs in snf2Δ cells, suggesting a specialized role for SWI/SNF (Ganguli et al. 2014). Consistent with this, we showed previously that SWI/SNF acts in cooperation with the histone acetyltransferase (HAT) Gcn5 and Hsp70 cochaperone Ydj1 in the eviction of promoter nucleosomes at a subset of genes strongly induced by transcriptional activator Gcn4 in amino acid-deprived cells (Qiu et al. 2016).
Gcn4 synthesis is up-regulated at the translational level by amino acid starvation and coordinately induces transcription of hundreds of genes, including nearly all amino acid biosynthetic genes (Hinnebusch 2005). We found previously that approximately one-third of the genes most highly induced during starvation for isoleucine and valine (Ile/Val) exhibit substantial eviction of promoter nucleosomes and that a pronounced defect in nucleosome eviction and transcriptional activation of these genes occurs on elimination or depletion of Snf2, the SWI/SNF catalytic subunit. In many cases, however, marked defects occurred only in mutants also lacking Ydj1 or Gcn5. Similar functional cooperation among these cofactors was observed for the subset of constitutively transcribed genes displaying the highest Pol II occupancies, implicating SWI/SNF at the most highly transcribed genes in yeast (Qiu et al. 2016). These findings agree with previous results implicating SWI/SNF in robust nucleosome remodeling and transcriptional activation of certain highly induced genes, including PHO8 (Gregory et al. 1999) and PHO5 (Adkins et al. 2007; Barbaric et al. 2007)—where it has overlapping functions with other remodeling complexes in mobilizing promoter nucleosomes (Musladin et al. 2014)—as well as SUC2 (Hirschhorn et al. 1992; Schwabish and Struhl 2007), GAL1 (Bryant et al. 2008), HO (Gkikopoulos et al. 2009), RNR1 (Sharma et al. 2003), CHA1 (Ansari et al. 2014), and heat-shock genes (Shivaswamy and Iyer 2008).
Having observed previously that nucleosome eviction in the induced Gcn4 transcriptome was only partially impaired in snf2Δ cells, we surmised that SWI/SNF cooperates with one or more other remodeling factors in evicting promoter nucleosomes. Considering that RSC and SWI/SNF belong to the same family of remodeling complexes (Clapier et al. 2017) and that each has been implicated in both nucleosome eviction and sliding (Lorch et al. 1999; Whitehouse et al. 2007), we asked whether SWI/SNF and RSC cooperate in nucleosome eviction at genes induced by Gcn4 and also at genes expressed constitutively at high levels where SWI/SNF cooperates with Gcn5 and Ydj1 (Qiu et al. 2016). We also explored whether SWI/SNF resembles RSC in determining the positions of −1_Nuc and +1_Nuc and hence NDR width at such highly expressed genes. To determine nucleosome positions, we used micrococcal nuclease (MNase) digestion to produce nucleosome-protected DNA fragments for immunoprecipitation with H3 antibodies (MNase-ChIP-seq [chromatin immunoprecipitation combined with high-throughput sequencing). To measure eviction of promoter nucleosomes, we relied primarily on ChIP-seq analysis of sonicated chromatin (ChIP-seq) using antibodies against H3 or H2B and quantified histone occupancies in the promoter intervals spanning the −1_Nuc, NDR, and +1_Nuc (dubbed [−1,NDR,+1]) that were defined by MNase-ChIP-seq. While ChIP-seq lacks the high positional resolution of MNase-ChIP-seq, it avoids complications of differential susceptibility of different nucleosomes to MNase digestion (Chereji et al. 2016, 2017; Mieczkowski et al. 2016).
Our findings reveal a pronounced widening of NDRs in addition to eviction of the −1_Nuc and +1_Nuc upon induction of Gcn4 target genes in wild-type cells and demonstrate that SWI/SNF and RSC have distinct and critical roles in achieving wide nucleosome-free NDRs for robust transcription at these induced genes. We also uncovered strong cooperation between SWI/SNF and RSC in nucleosome positioning and eviction at the most highly transcribed subset of constitutively expressed genes, suggesting their general cooperation in achieving high transcription throughout the genome. The occupancies of both remodelers were found to be greatest at the highly expressed or induced genes that depend on their functions for nucleosome eviction and transcriptional activation, consistent with their direct roles in sliding or evicting promoter nucleosomes.
Results
Both nucleosome eviction and sliding occur at sulfometuron methyl (SM)-induced genes to widen NDRs and expose promoter sequences
Depriving yeast cells of an amino acid, including starvation for Ile/Val achieved with the inhibitor SM, evokes substantial reprogramming of transcription, which is mainly dependent on the transcriptional activator Gcn4 (Jia et al. 2000; Natarajan et al. 2001; Saint et al. 2014). Previously, we conducted ChIP-seq analysis of cross-linked chromatin using antibodies against Pol II subunit Rpb3 to measure transcriptional activation and identified a group of 204 genes exhibiting twofold or greater induction of Rpb3 occupancies averaged across the CDSs on treatment with SM. ChIP-seq analysis with antibodies against histone H3 was used to quantify nucleosome occupancy and revealed that 70 of these SM-induced genes exhibit a marked reduction in nucleosome occupancy in the interval spanning the consensus positions of the −1_Nuc and +1_Nuc and intervening NDRs at those genes. Having observed that substantial eviction of promoter nucleosomes at most of the 70 SM-induced genes remained intact in snf2Δ mutant cells (Qiu et al. 2016), we asked whether RSC contributes to the residual nucleosome eviction occurring in the absence of SWI/SNF function.
We began by extending our analysis of nucleosome remodeling of SM-induced genes by determining the changes in histone H2B occupancies that accompany gene activation in wild-type cells. Sonicated cross-linked chromatin isolated from cells growing exponentially in defined medium with or without a 25-min treatment with SM was immunoprecipitated with antibodies against H2B followed by paired-end sequencing of the immunoprecipitated DNA (H2B ChIP-seq). ChIP-seq analysis with H3 antibodies was conducted in parallel on the same samples. The results revealed similar marked eviction of H2B and H3 from the promoter regions of four canonical Gcn4 target genes: ARG1, ARG4, HIS4, and CPA2 [Supplemental Fig. S1A, H2B (S-C), H3 (S-C)]. Averaging the results for the group of 70 SM-induced genes indicated similar reductions in H2B and H3 occupancies across the promoters of these highly remodeled genes upon SM treatment (Fig. 1A). Calculating the average occupancy per nucleotide across the [−1,NDR,+1] interval for all 70 genes revealed no significant differences between the median occupancies of H3 versus H2B in untreated cells or between the reduced median occupancies of H3 versus H2B in SM-treated cells (Fig. 1B). Similar trends were observed for the remaining 134 SM-induced genes among the previously identified group of 204 induced genes (Fig. 1C), which showed less pronounced H3 eviction compared with the group of 70 highly remodeled genes (Qiu et al. 2016). The H3 and H2B occupancies in the [−1,NDR,+1] intervals at these 134 genes are somewhat higher to begin with in untreated cells and also exhibit smaller reductions on SM treatment compared with the group of 70 genes (Fig. 1, C vs. B). The similar reductions of H3 and H2B occupancies at both sets of genes are consistent with the idea that SM-induced transcriptional activation evokes primarily elimination of complete +1_Nucs and −1_Nucs.
Figure 1.

Promoter nucleosomes are disassembled and repositioned on SM-induced transcriptional activation. (A) Plots of H3 and H2B occupancies at each base pair, normalized to the average occupancy on the respective chromosome for each gene, calculated from ChIP-seq data of sonicated chromatin, averaged over the 70 SM-induced genes, and aligned to the TSS. (Blue and yellow) WT_U; (red and purple) WT_I. (B,C) Notched box plots of H3 and H2B occupancies per nucleotide in the [−1,NDR,+1] region calculated from ChIP-seq data of sonicated chromatin from at least three replicates of WT_U and WT_I cultures for 70 SM-induced genes (B) and 134 SM-induced genes (C). If the notches of two plots do not overlap, there is 95% confidence that the true medians of the two distributions differ. Each box depicts the interquartile range containing 50% of the data, intersected by the median; the notch indicates a 95% confidence interval (CI) around the median. (D) Heat map depictions of changes in H3 or Rpb3 occupancies upon SM induction of wild-type cells (WTI vs. WTU) calculated from MNase (H3) or (Rpb3) ChIP-seq data for the 204 SM-induced genes, divided between 70 (top) and 134 (bottom) SM-induced genes, and sorted by induced Rpb3 levels for each group. (Panel i) H3 occupancy differences relative to the +1_Nuc dyad. (Panel ii) Rpb3 occupancies averaged over the CDSs in WTU and WTI cells. (Panel iii) Differences in Rpb3. (Panel iv) Rpb3 induction ratios between WTI and WTU cells for the same gene order, color-coded as shown at the right of each panel. (E) Average dyad density from MNase-ChIP-seq data aligned to the +1_Nuc for 70 SM-induced genes. Midpoints (dyads) of nucleosomal size sequences between 120 and 160 bp were determined with respect to the +1_Nuc and summed for 70 genes. Average profiles were smoothed using a moving average filter with a span of 31 bp. The data were normalized internally to the average value for each data set. (F) Box plots depicting shift in +1_Nuc and −1_Nuc positions and change in NDR width for 70 SM-induced genes, calculated from H3 MNase-ChIP-seq data by calculating change in dyad peak position in WTI versus WTU cells for the +1_Nuc and −1_Nuc, respectively. (G). Box plots depicting nucleosome spacing for 70 SM-induced genes in SM-induced or uninduced wild-type cells for the array of +1_Nuc to +5_Nuc, calculated from H3 MNase-ChIP-seq data.
To examine changes in locations of promoter nucleosomes, we conducted H3 ChIP-seq analysis of fragments generated from cross-linked chromatin by digestion with MNase (H3 MNase-ChIP-seq), which yields primarily a mixture of mononucleosome-sized fragments of ∼150 bp and various subnucleosome-sized fragments (Supplemental Fig. S2A). (Differences in fragment length distributions among replicates did not produce any obvious differences in dyad peak positions or H3 occupancies for SM-induced or constitutively expressed genes [Supplemental Fig. S2B,C].) The results confirmed the occurrence of strong promoter nucleosome eviction upon SM induction for the four canonical Gcn4 target genes noted earlier [Supplemental Fig. S1A, H3 (MN-C)] and also for the two groups of 70 and 134 SM-induced genes, with the expected greater reductions in average H3 occupancy on induction of the 70 versus 134 genes (Supplemental Fig. S1B,C). Results for individual genes are displayed in a heat map quantifying the differences in H3 occupancies surrounding the dyads of +1_Nucs in SM-induced versus uninduced cells, with genes sorted vertically on induced Rpb3 occupancies (Fig. 1D, panel i) and with the Rpb3 occupancies depicted separately in adjacent heat maps for uninduced and induced wild-type cells (Fig. 1D, panels ii–iv). For the group of 70 genes, H3 occupancies of both the −1_Nucs and +1_Nucs were markedly reduced by SM treatment for essentially the entire cohort (Fig. 1D, panel i, top sector, dark-blue shades). The decreases in H3 occupancy were smaller overall for the 134 induced genes (Fig. 1D, panel i, bottom sector); however, considerable H3 eviction at the +1_Nuc is evident for all but the most weakly expressed genes in this cohort near the bottom of the lower sector of the map (Fig. 1D, panel i, 134 genes). In contrast, the −1_Nuc is relatively more stable for the group of 134 genes compared with the group of 70 genes (Fig. 1D, panel i, cf. 70 and 134 genes). Among both groups of genes, however, those with the highest induced Rpb3 occupancies (Fig. 1D, panel i, at the top of each sector) also display substantial nucleosome eviction from the CDSs, extending to the +6_Nuc for the group of 70 genes but mostly confined to the +2_Nuc to +4_Nuc for the less remodeled 134 induced genes (Fig. 1D, panel i). These findings confirm the direct relationship between the magnitude of induced transcription and the extent of nucleosome eviction in both promoters and CDSs among SM-induced genes (Qiu et al. 2016).
Because the TSS generally resides within the upstream boundary of the +1_Nuc (Ocampo et al. 2016), the latter may impose a barrier to PIC assembly that can be overcome by its repositioning downstream (Zhang et al. 2011; Reja et al. 2015; Nocetti and Whitehouse 2016). In fact, examination of the H3 MNase-ChIP-seq occupancy plots for the groups of 70 and 134 induced genes suggests a downstream shift away from the NDR for the entire array of +1_Nucs to +5_Nucs on SM induction (Supplemental Fig. S1B,C). To quantify this shift, we plotted the midpoints of the immunoprecipitated nucleosome-sized fragments, which approximate the positions of the nucleosome dyads (Fig. 1E; Supplemental Fig. S1D). For the 70 SM-induced genes, we observed a downstream shift in the +1_Nuc and an upstream shift of the −1_Nuc by median values of +11 and −6 bp, respectively, increasing the median NDR width by ∼19 bp upon SM induction (Fig. 1E,F). In contrast, there is no significant change in nucleosome spacing (Fig. 1G), indicating that the entire array of phased +1_Nucs to +6_Nucs shifts downstream coordinately upon SM induction of wild-type cells. Relatively smaller upstream and downstream shifts of +6 and −3 bp, respectively, occurred for the −1_Nucs and +1_Nucs upon SM induction of the group of 134 genes for a less pronounced median NDR widening of ∼10 bp, again without a significant change in nucleosome spacing (Supplemental Fig. S1D–F). As the 134 genes exhibit a lower Rpb3 induction ratio overall compared with the group of 70 genes (Fig. 1D, panel iv), the degree of NDR widening is correlated with the extent of transcriptional induction by SM, consistent with the notion that decreased nucleosome occlusion of the promoter and TSS stimulates PIC assembly or transcription initiation. These findings agree with previous results indicating that wider NDRs are characteristic of highly expressed genes in yeast (Chereji and Morozov 2015). In summary, the results indicate that both nucleosome displacement and eviction occur during activation of SM-induced genes to widen the NDR and expose the promoter and TSS sequences.
SWI/SNF and RSC cooperate in promoter nucleosome disassembly and repositioning at SM-induced genes
To determine the roles of SWI/SNF and RSC in nucleosome sliding and eviction at SM-induced genes, we measured the effects of inactivating these remodelers on histone occupancies by both ChIP-seq and MNase-ChIP-seq. Whereas SWI/SNF could be inactivated by deleting SNF2, cells lacking the gene encoding the catalytic subunit of RSC (STH1) are inviable. Accordingly, we replaced the STH1 promoter with a doxycycline-repressible promoter (PTET-STH1) to conditionally deplete Sth1 protein by adding doxycycline to the growth medium (Supplemental Fig. S3A). We observed that culturing PTET-STH1 cells with doxycycline for 8 h dramatically depletes Sth1, as judged by Western blot analysis, without substantially impairing induction of Gcn4 protein by SM (Supplemental Fig. S3B, cf. lanes 2 and 3). Moreover, cell growth continued at a low rate during further incubation with doxycycline from 8 to 16 h, probably indicating incomplete elimination of Sth1 (Supplemental Fig. S3C).
Analysis of histone H3 and H2B occupancies by ChIP-seq revealed that deleting SNF2 or depleting Sth1 confers comparable defects in eviction of promoter nucleosomes at the 70 SM-induced genes, since the averaged occupancies of both H3 and H2B upstream of and downstream from the TSS showed similar increases in the two SM-treated mutants compared with the relatively lower levels observed in SM-induced wild-type (WTI) cells (Supplemental Fig. S3D, left and right, yellow and purple vs. red). Quantification of H3 and H2B occupancies in the [−1,NDR,+1] intervals led to the same conclusions, since the median average H3 and H2B occupancies were substantially elevated in each single mutant versus wild type upon SM treatment (Fig. 2A,B, induced). Considerably smaller increases were observed in uninduced snf2Δ or PTET-STH1 cells, which were statistically significant only for the latter (Fig. 2A,B, uninduced). Interestingly, a greater eviction defect was observed for both H3 and H2B in the SM-treated snf2ΔPTET-STH1 double mutant (Supplemental Fig. S3D, left and right, green vs. yellow and purple), with histone occupancies in the [−1,NDR,+1] intervals increasing to the elevated levels observed in uninduced wild-type (WTU) cells (Fig. 2A,B, snf2ΔPTET-STH1 induced vs. wild-type uninduced). Highly similar findings were made for the group of 134 genes (Supplemental Fig. S3E,F). Thus, both SWI/SNF and RSC are required for wild-type levels of promoter nucleosome disassembly during transcriptional activation of SM-induced genes, and RSC makes a small but significant contribution to nucleosome eviction at these genes in noninducing conditions.
Figure 2.

SWI/SNF and RSC remodelers cooperate in promoter nucleosome disassembly and repositioning at SM-induced genes. (A,B) Notched box plots of average H3 (A) or H2B (B) occupancies per nucleotide in the [−1,NDR,+1] region, calculated from ChIP-seq data from at least three replicates of the indicated yeast strains for the 70 SM-induced genes. (C) Heat map depictions of changes in H3 occupancies from MNase-ChIP-seq data (left large panels) as well as differences (middle) or fold change (right) in Rpb3 occupancies averaged over CDSs between wild-type and the indicated mutant cultures under inducing conditions for 204 SM-induced genes, sorted and color-coded as in Figure 1D. (D) Average dyad densities calculated from H3 MNase-ChIP-seq data, aligned to the +1_Nuc for 70 SM-induced genes (depicted as in Fig. 1E) between wild type and the indicated mutants under inducing conditions. Average profiles were smoothed using a moving average filter with a span of 31 bp. (E,F). Box plots depicting changes in +1_Nuc and −1_Nuc dyad positions (E) or nucleosome spacing (F)—calculated from H3 MNase-ChIP-seq data for the 70 SM-induced genes—between wild type and the indicated mutants under inducing conditions, determined as in Figure 1, F and G.
Functional cooperation between SWI/SNF and RSC was also revealed by H3 MNase-ChIP-seq analysis, and the higher resolution of this technique unveiled distinct contributions of these two remodelers to nucleosome eviction. The H3 occupancy difference heat map shown for the group of 70 genes in the top sector of Figure 2C, panel i, reveals that eliminating Snf2 evoked a large increase in H3 occupancies at the −1_Nucs, somewhat smaller increases at the +1_Nucs, and moderate increases in NDRs compared with wild-type cells upon SM induction. In contrast, depleting Sth1 conferred a relatively greater increase in H3 occupancies in NDRs, along with appreciable increases at the −1_Nucs but little change at the +1_Nucs of these 70 genes (Fig. 2C, panel ii, top sector). The snf2Δ PTET-STH1 double mutant exhibits a composite defect in H3 eviction that spans the entire [−1,NDR,+1] interval, with relatively larger increases in all locations than observed in either single mutant (Fig. 2C, panel iii, top sector). The visual impressions from these heat maps are confirmed in the gene-averaged H3 occupancy plots for the 70 induced genes (Supplemental Fig. S4A, yellow, purple, and green vs. red). Qualitatively similar but less pronounced defects in nucleosome eviction were observed for the group of 134 less remodeled genes (Fig. 2C, panels i–iii, bottom sectors; Supplemental Fig. S4B), with RSC making a greater and more widespread contribution than SWI/SNF in clearing or evicting nucleosomes from NDRs, and SWI/SNF playing a more prominent role than RSC in removing the −1_Nucs and +1_Nucs.
Interestingly, dyad peaks of the +1 to +5 array of nucleosomes were shifted upstream toward the NDR at the 70 SM-induced genes in all three remodeler mutants versus wild type under SM-inducing conditions, with the smallest upstream shift occurring in PTET-STH1 cells, a larger shift seen in snf2Δ cells, and the greatest shift observed in the double mutant (Fig. 2D, panels i–iii, red vs. blue traces). Consistent with this, the +1_Nuc and −1_Nuc were repositioned toward the NDR by median values of −6 and +4 bp in snf2Δ cells and −12.5 and +7 bp in snf2ΔPTET-STH1 cells compared with wild-type cells under inducing conditions but shifted by negligible amounts in the PTET-STH1 single mutant (Fig. 2E, panels i–iii). Despite the latter, dyad shifts toward the NDR by ≥5 bp were observed for the +1_Nuc or −1_Nuc in PTET-STH1I versus WTI cells at 23 and 28 genes, respectively. Moreover, the number of genes showing ≥5-bp shifts of the +1_Nuc or −1_Nuc is greater in the induced snf2ΔPTET-STH1 double mutant versus the snf2ΔI single mutant (57 out of 47 genes for +1_Nuc/−1_Nuc shifts versus 41/34 +1_Nuc/−1_Nuc shifts in snf2ΔPTET-STH1 vs. snf2Δ, respectively). Together, these findings indicate that RSC plays an appreciable role in shifting the +1_Nuc and −1_Nuc away from the NDR upon SM induction, albeit less than that of SWI/SNF. No significant changes in median spacings between nucleosomes were observed for the 70 genes in any of the mutants versus wild type under inducing conditions (Fig. 2F).
Analysis of the less remodeled 134 induced genes revealed shifts in the −1_Nuc and +1_Nuc dyads back toward the NDR in all three mutants that were comparable in magnitude in the induced snf2Δ and PTET-STH1 single mutants but substantially greater in the double mutant compared with wild-type cells under inducing conditions (Supplemental Fig. S5A,B, panels i–iii). Thus, the contribution of RSC is relatively larger and on par with that of SWI/SNF in shifting +1_Nuc/−1_Nuc away from the NDR for this gene cohort compared with the 70 induced genes (cf. Fig. 2E and Supplemental Fig. S5B for snf2Δ and PTET-STH1). Again, none of the mutants displayed significant changes in median nucleosome spacing compared with wild-type cells for the group of 134 induced genes (Supplemental Fig. S5C).
Aligning the dyad peaks of the 70 induced genes relative to the TSS indicates that the nucleosome sliding evoked by SM in wild-type cells moves the +1_Nuc downstream from the promoter and TSS in a manner largely reversed in snf2Δ cells and completely eliminated in the snf2ΔPTET- STH1 double mutant (Supplemental Fig. S5D). Thus, during SM induction of these highly remodeled genes, SWI/SNF acts more prominently than RSC in sliding the +1_Nuc away from the TSS. For the set of 134 induced genes, the downstream shift of the +1_Nuc from the TSS evoked by SM in wild-type cells is again reversed in the double mutant, but RSC appears to play a larger role than SWI/SNF at these genes, as the shift is largely prevented in the PTET-STH1 single mutant but not in the snf2Δ strain (Supplemental Fig. S5E).
As discussed further below, our findings that RSC plays a greater role than SWI/SNF in reducing H3 occupancies within NDRs but a relatively smaller or comparable role in repositioning the −1_Nucs and +1_Nucs at SM-induced genes implies that RSC can evict nucleosomes from NDRs in addition to sliding the −1_Nucs/+1_Nucs to maintain proper NDR widths.
Reductions in SWI/SNF and RSC function confer additive defects in transcription of SM-induced and highly expressed constitutive genes
The additive defects in repositioning and eviction of promoter nucleosomes conferred by the snf2Δ and PTET- STH1 mutations described above are associated with cumulative reductions in transcriptional activation of SM-induced genes. Thus, the CDS occupancies of Rpb3 and also the extent of Rpb3 induction are reduced in both snf2Δ and PTET-STH1 cells compared with wild-type cells for the sets of 70 and 134 SM-induced genes, as indicated by the pervasive blue shading in the heat maps of Figure 2C (panels i,ii, two right panels), but relatively larger reductions in Rpb3 occupancies and induction ratios (Fig. 2C, panels i,ii, darker-blue shades) occur in the snf2ΔPTET-STH1 double mutant (Fig. 2C, panel iii). Consistent with this, the median Rpb3 occupancies for both sets of SM-induced genes are reduced more extensively in the snf2ΔPTET-STH1 double mutant than in either single mutant compared with WTI cells (Fig. 3A,B). These findings are consistent with the idea that both eviction of the +1_Nuc and −1_Nuc by SWI/SNF and clearing of nucleosomes from the NDR by RSC are critical for strong transcriptional induction of SM-induced genes and that a compound defect in the double mutant confers a cumulative reduction in SM-induced transcription.
Figure 3.

Both SWI/SNF and RSC are required for robust transcription of highly expressed SM-induced and constitutive genes. (A,B) Notched box plots of log2 values of Rpb3 occupancies averaged over CDSs for the groups of 70 (A) or 134 (B) SM-induced genes or deciles of 3619 constitutive genes sorted in descending order of Rpb3 occupancies in induced cells (C), calculated from at least three biological replicates of the indicated wild-type and mutant yeast strains under inducing (I) or uninducing (U) conditions.
Interestingly, the defects in transcriptional activation of SM-induced genes conferred by impairing SWI/SNF and RSC function are not observed genome-wide for noninduced genes. We identified a group of 3619 genes exhibiting little or no transcriptional activation by SM in wild-type cells (Rpb3_I/Rpb3_U < 1.2; dubbed constitutive genes), divided them into 10 equal deciles according to CDS occupancies of Rpb3 in WTI cells, and examined the effects of the remodeler mutants for each decile. Only the most highly expressed genes in decile 1 exhibit reductions in median Rpb3 occupancies in snf2Δ and PTET-STH1 single mutants plus a relatively greater reduction in the double mutant compared with WTI cells (Fig. 3C, decile 1, green, blue, and purple vs. tan), just as noted above for the SM-induced genes (Fig. 3A,B). In fact, the most weakly expressed genes in deciles 9 and 10 displayed increased Rpb3 occupancies in the mutants compared with WTI cells, with the double mutant showing larger increases than either single mutant (Fig. 3C, constitutive deciles 9 and 10). These last findings might be attributable to a redistribution of the transcriptional machinery in the remodeler mutants to favor weakly expressed genes able to compete more effectively for limiting factors when transcription of the most highly expressed genes is reduced (Qiu et al. 2016). Because the absolute transcription level of all genes is expected to be reduced in the PTET-STH1 mutants (Parnell et al. 2008), the weakly expressed genes might merely exhibit smaller reductions than highly expressed genes. Nevertheless, the results indicate that chromatin remodeling by SWI/SNF and RSC is more critical for wild-type transcription at highly expressed versus weakly expressed genes.
SWI/SNF cooperates with RSC in NDR formation and promoter nucleosome eviction at only the most highly expressed yeast genes
Having found that impairing SWI/SNF and RSC reduces the relative transcription levels of only the most highly transcribed constitutive genes, we sought to understand the genome-wide contributions of these remodelers to promoter nucleosome organization. To this end, we constructed heat maps of H3 occupancy differences in MNase-ChIP-seq data for all 3619 constitutive genes, centered on the +1_Nuc dyad and sorted by the Rpb3 occupancies in CDSs of WTI cells. As expected, these maps revealed relatively few differences in H3 occupancies of the [−1,NDR,+1] regions between WTI and WTU cells (Fig. 4A, panel i, left). The snf2Δ mutation conferred small increases in H3 occupancies at the −1_Nuc and a shift in the +1_Nuc toward the NDR for the majority of the constitutively expressed genes (Fig. 4A, panel ii, yellow hues). In contrast, depleting Sth1 evoked marked differences in H3 occupancies for the majority of constitutive genes (Fig. 4A, panel iii, left, bottom sector), substantially reducing occupancies at the +1_Nuc (Fig. 4A, panel iii, blue hues), with a concomitant increase in H3 occupancies in NDRs coupled with a shift in the +1_Nuc toward the NDRs (Fig. 4A, panel iii, orange to red hues). Interestingly, both changes were muted for the 200 most highly expressed genes at the top of the map, of which approximately two-thirds are ribosomal protein genes. Except for these “top 200 genes,” the H3 occupancy changes observed in the PTET-STH1 snf2Δ double mutant are similar to those observed in the PTET-STH1 single mutant (Fig. 4A, panel iv vs. panel iii, left, bottom sector). In contrast, the top 200 genes exhibit greater increases in H3 occupancy at the +1_Nucs and the CDS nucleosomes in the double mutant versus either single mutant (Fig. 4A, panel iv vs. panels ii,iii, top sector), suggesting that SWI/SNF contributes to promoter nucleosome remodeling selectively at these most highly expressed constitutive genes.
Figure 4.

SWI/SNF and RSC have comparable complementary roles in NDR formation and promoter nucleosome eviction at highly expressed constitutive genes. (A) Difference maps of H3 occupancies calculated from MNase-ChIP-seq data (left panels) or Rpb3 occupancies averaged over CDSs (right strips) for the indicated culture conditions or strains for the group of 3619 constitutive genes, as in Figure 2C. The most highly expressed 200 genes are above the white line. (B) Notched box plots of average H3 occupancies per nucleotide in the [−1,NDR,+1] region for the top 200 constitutive genes and indicated deciles of 3619 constitutive genes, computed from sonication ChIP-seq data for the indicated yeast strains. (C) Box plots depicting changes in +1_Nuc and −1_Nuc dyad positions calculated from H3 MNase-ChIP-seq data for all 3619 constitutive genes (gray) or the top 200 expressed constitutive genes (red).
The increased H3 occupancies in the double mutant are associated with appreciable reductions in Rpb3 occupancy only for the most highly expressed subset of constitutive genes at the top of the heat map (Fig. 4A, panel iv, right). This was to be expected, as these genes belong to decile 1 of Rpb3 occupancies, for which a relatively greater reduction in Rpb3 levels occurs in the PTET-STH1 snf2Δ double mutant than in either single remodeler mutant (Fig. 3C, decile 1).
A selective role for SWI/SNF in promoter nucleosome depletion at the most highly expressed constitutive genes was confirmed by examining plots of average H3 occupancies in the [+1,NDR,−1] regions for genes of different expression levels (Fig. 4B). Thus, median H3 occupancies were increased comparably in the snf2Δ and PTET-STH1 single mutants and more substantially in the double mutant for both the top 200 genes and the genes in Rpb3 occupancy decile 1, whereas for deciles 2 and 10, the PTET- STH1 strain and double mutant exhibit comparable occupancy increases exceeding those in the snf2Δ single mutant (Fig. 4B).
In addition to its broader role in determining promoter nucleosome occupancies, RSC is also more important than SWI/SNF in determining NDR widths genome-wide. Analysis of median dyad positions of the −1_Nuc and +1_Nuc determined from the H3 MNase-ChIP-seq data revealed that in the PTET-STH1 and PTET-STH1 snf2Δ mutants, the median position of the +1_Nuc shifted upstream by ∼8–10 bp, whereas the median position of the −1_Nuc shifted downstream by 6–7 bp (Fig. 4C, panels ii,iii, gray boxes), thus narrowing the NDR considerably by 14–17 bp. Relatively smaller shifts for both the −1_Nuc and +1_Nuc were observed in snf2Δ versus wild-type cells (Fig. 4C, panel i, gray boxes). Nucleosome spacing was only slightly reduced in the snf2Δ and PTET-STH1 snf2Δ mutants and was unaffected in the PTET-STH1 single mutant (Supplemental Fig. S6A, gray boxes). Overall, these results support previous findings that RSC functions genome-wide to prevent encroachment of the −1_Nuc and +1_Nuc into the NDR to maintain proper NDR widths (Ganguli et al. 2014; Parnell et al. 2015).
For the top 200 most highly expressed constitutive genes, however, SWI/SNF functions on par with RSC in determining NDR widths, and the two remodelers cooperate extensively at these genes. Thus, the upstream shift in median dyad position of the +1_Nuc was somewhat greater in induced snf2Δ versus induced PTET-STH1 cells (∼6 bp vs. ∼2 bp), and both the upstream and downstream shifts in median dyad positions of the +1_Nuc and −1_Nuc, respectively, were larger in the double mutant (∼9 bp for each shift) than in either single mutant (Fig. 4C, panels i–iii, red boxes).
The selective role of SWI/SNF in NDR formation at highly expressed constitutive genes was confirmed by comparing the effects of the remodeler mutations on median NDR widths. For genes with the low Rpb3I levels in deciles 2 and 10, the median NDR width determined from H3 MNase-ChIP-seq data was reduced comparably in the PTET-STH1 and PTET-STH1 snf2Δ mutants by 16–19 bp but by only 6–7 bp in the snf2Δ single mutant (Fig. 5A). For decile 1, in contrast, the median NDR width was reduced moderately in each of the single mutants, and the double mutation was required to produce the greater reductions in NDR width conferred by depleting Sth1 alone for the other nine deciles (Fig. 5A). The top 200 genes resemble decile 1 but with an even greater effect of the snf2Δ mutation. Essentially the same conclusions emerged from analyzing the H3 occupancies within NDRs from the MNase-ChIP-seq data (Supplemental Fig. S6B).
Figure 5.

SWI/SNF and RSC cooperate in NDR widening and blocking nucleosome formation in NDRs of highly expressed genes. (A) Notched box plots of changes in NDR widths calculated from H3 MNase-ChIP-seq data for the indicated strains, the top 200 constitutive genes, and indicated deciles of 3619 constitutive genes as in Figure 4B. (B) Integrated Genomics Viewer (IGV) tracks depicting H3 occupancies from MNase-ChIP-seq data in genomic regions surrounding YMR016C (panel i), YBR092C (panel ii), and YLR042C (panel iii) genes in the indicated yeast strains/conditions. Vertical dashed lines demarcate NDRs discussed in the text. (C) Plots comparing normalized and averaged H3 occupancies from MNase-ChIP-seq reads aligned to the NDR center for constitutive genes selected for exhibiting nucleosome peaks within NDRs in snf2Δ PTET-STH1 cells, plotted separately for subsets of genes displaying multiple (panel i) or single (panel ii) nucleosome peaks in NDRs or NDRs filling without nucleosome peaks (panel iii).
In summary, whereas RSC plays a substantially larger role than SWI/SNF in widening and clearing nucleosomes from NDRs at most constitutive genes, these remodelers act comparably and function additively in NDR formation at the most highly expressed genes (decile 1 and top 200), which also tend to have the widest NDRs among all yeast genes (Chereji and Morozov 2015).
The different contributions of SWI/SNF and RSC in NDR formation are also well illustrated by examining the H3 MNase-ChIP-seq results for specific genes with different NDR widths. Within the very wide (∼850-bp) NDR upstream of YMR016C, H3 occupancies are only slightly increased in the PTET-STH1 single mutant compared with wild-type, whereas additional nucleosomes appear in snf2Δ cells and become more prominent in the double mutant (Fig. 5B, panel i, dotted region). In the moderately wide (∼300-bp) NDR at YBR092C, a single nucleosome peak is much more evident in either snf2Δ or double-mutant cells versus the PTET-STH1 single mutant (Fig. 5B, panel ii). In contrast, for the relatively narrow (∼150-bp) NDR at YLR042C, depleting Sth1 increases NDR H3 occupancies without the appearance of an obvious nucleosome peak, whereas eliminating Snf2 has no effect in the presence or absence of Sth1 (Fig. 5B, panel iii).
Using an algorithm to identify genes in which nucleosomes appear within NDRs in the double mutant that are not present in wild-type cells (described in the Materials and Methods), we identified 24 constitutive genes with particularly wide wild-type NDRs in which multiple dyad peaks appear within the NDR in the double mutant versus wild-type cells (as for YMR016C in Fig. 5B, panel i). We further identified 44 genes with moderately wide NDRs in which a single new dyad peak is observed (as for YBR029C in Fig. 5B, panel ii) and 36 genes with relatively narrow NDRs that exhibit primarily NDR filling (as exemplified by YLR042C in Fig. 5B, panel iii) in the double mutant versus wild-type cells (genes listed in sheet 2 of Supplemental File 1). The fact that nucleosome peaks appear in the double mutant only in NDRs wide enough to accommodate one or more conventional nucleosomes suggests that the new peaks correspond to nucleosomes that are ordinarily evicted from the NDRs but spared in the remodeler mutants. Comparing the averaged MNase-ChIP-seq H3 occupancies in the single versus double mutants for these three sets of genes indicates that SWI/SNF plays the predominant role in clearing nucleosomes from the widest NDRs (Fig. 5C, panel i) and that SWI/SNF and RSC function comparably in removing nucleosomes from NDRs of moderate width (Fig. 5C, panel ii), while RSC is largely responsible for preventing NDR filling by the −1_Nucs and +1_Nucs (Fig. 5C, panel iii).
We confirmed that the appearance of new nucleosome peaks in the NDRs of constitutive genes in the snf2Δ PTET- STH1 double mutant is not restricted to SM-inducing conditions by examining MNase-ChIP-seq data from uninduced cells. Heat maps of H3 occupancies aligned relative to NDR centers and sorted by increasing wild-type NDR width revealed that in genes with narrower NDRs, shown in the top halves of the maps, H3 occupancies increase within NDRs only in Sth1-depleted cells (Supplemental Fig. S7, top half, green to yellow hues in PTET-STH1 and snf2Δ PTET-STH1 vs. blue hues in wild type). In contrast, in genes with wider NDRs, in the bottom portions of the maps, increases in NDR H3 occupancies are evident only in the snf2Δ PTET-STH1 double mutant and, to a lesser extent, snf2Δ cells (Supplemental Fig. S7 bottom half, intermittent green hues in snf2Δ PTET-STH1 and snf2Δ).
SWI/SNF and RSC occupancies increase on activation of SM-induced genes and correlate with Pol II occupancies at constitutively expressed genes
To provide evidence that SWI/SNF and RSC act directly in nucleosome remodeling of SM-induced genes, we conducted ChIP–chip analysis of genome-wide occupancies of Myc13 epitope-tagged versions of Snf2 and Sth1. Gene-averaged occupancy plots reveal an appreciable increase in occupancies of both factors in the promoters and across the CDSs of the 70 highly remodeled SM-induced genes and lesser but significant increases at the less remodeled 134 SM-induced genes (Fig. 6A,B). We also observed higher average occupancies of both factors at the top 200 highly expressed constitutive genes compared with all 3619 constitutive genes and comparable with those seen for the 70 SM-induced genes (Fig. 6C,D). As shown in Figure 6E, the median H2B and H3 occupancies in WTI cells in the [−1,NDR,+1] intervals for the group of 200 constitutive genes and the 70 and 134 SM-induced genes are well below the median values for the group of 3619 constitutively expressed genes, and the median histone occupancies for the 70 genes are below those for the 134 induced genes. Thus, the occupancies of SWI/SNF and RSC are inversely related to the promoter H3/H2B occupancies for these sets of genes. On the other hand, the occupancies of the remodelers are directly correlated with the median Rpb3 occupancies for these groups of genes (Fig. 6F). Together, these results establish a strong correlation between the degree of histone depletion in the promoter, the transcription level of the gene, and the occupancies of both SWI/SNF and RSC across the gene for both SM-induced and constitutively expressed genes, all consistent with a direct role for both remodelers in evicting and repositioning promoter nucleosomes for robust transcription.
Figure 6.

Chromatin remodelers SWI/SNF and RSC are enriched at highly expressed genes. Metagene profiles of Snf2-myc (A,C) or Sth1-myc (B,D) occupancies from ChIP–chip analysis in wild-type cells treated or untreated with SM for the groups of 70 or 134 SM-induced genes (A,B) or for either all 3619 constitutive genes or the top 200 expressed constitutive genes (C,D). The log2 (immunoprecipitation/input) values are aligned to the TSS or the transcription termination site (TTS) and shown for 500 bp surrounding the TSS/TTS. (E,F) Notched box plots of average H3 and H2B occupancies per nucleotide in [−1,NDR,+1] regions (E) and log2 values of Rpb3 occupancies averaged over CDSs (F) for the indicated gene sets from ChIP-seq data from at least three replicates of WTI cells are shown.
Discussion
Previous studies have demonstrated a broad role for RSC in establishing the proper widths of NDRs in the yeast genome, which is achieved by impeding invasion of the −1_Nuc or +1_Nuc into the NDR (Badis et al. 2008; Parnell et al. 2008, 2015; Hartley and Madhani 2009; Ganguli et al. 2014) without affecting the spacing between phased nucleosomes in the CDS (Ganguli et al. 2014; Parnell et al. 2015). In contrast, analysis of a snf2Δ mutant indicated that SWI/SNF was not required for wild-type NDR widths at the vast majority of genes, at least in cells containing RSC and all other remodeling complexes, but functioned at a small fraction of genes containing wide NDRs and poorly phased nucleosomes (Ganguli et al. 2014). Previously, we showed that SWI/SNF acts together with the HAT Gcn5 and Hsp70 cochaperone Ydj1 in evicting promoter nucleosomes throughout the genome, being particularly important at genes highly induced by Gcn4 in SM-treated cells (Qiu et al. 2016). However, it was unknown whether SWI/SNF and RSC functionally cooperate in evicting promoter nucleosomes and maintaining proper NDR widths for the Gcn4 transcriptome and at other genes exhibiting high-level constitutive expression.
Using ChIP-seq to measure changes in H3 occupancy in mutants functionally impaired for SWI/SNF, RSC, or both remodelers, we found that RSC and SWI/SNF are equally important in removing nucleosomes from the promoter regions extending from the −1_Nuc to +1_Nuc of SM-induced genes, as increases in nucleosome occupancy throughout these intervals are greater in the double mutant defective for both remodelers than in either single mutant. Interestingly, the higher-resolution data afforded by MNase-ChIP-seq revealed that they function in complementary rather than fully redundant ways, with SWI/SNF generally playing the predominant role in evicting the −1 and +1 promoter nucleosomes, and RSC being relatively more focused on preventing these (or other) nucleosomes from populating NDRs, although SWI/SNF also contributes to the latter. Eliminating Snf2 confers a greater defect in evicting the −1_Nuc versus the +1_Nuc of the SM-induced genes (Fig. 2C, panel i; Supplemental Fig. S4A,B), and we note that the defect in +1_Nuc eviction in snf2Δ cells could at least partly reflect reduced Pol II occupancy and diminished competition between Pol II and the +1_Nuc.
Our MNase-ChIP-seq data also revealed that transcriptional activation of SM-induced genes in wild-type cells is accompanied by repositioning of the +1_Nuc downstream from and the −1_Nuc upstream of the NDR by median values of ∼11 and ∼6 bp, respectively, for 70 highly SM-induced genes, substantially widening their NDRs. Eliminating Snf2 diminished these repositioning events, and eliminating/depleting both remodelers simultaneously completely reversed the repositioning at most of the 70 induced genes and actually evoked a narrowing of their NDRs compared with the normal widths in uninduced cells (Fig. 2E). RSC was relatively more important at the less remodeled 134 SM-induced genes, where it functioned comparably with SWI/SNF in repositioning of −1_Nuc/+1_Nuc (Supplemental Fig. S5B), but, again, the double mutant showed stronger positioning defects than either single mutant.
The MNase-ChIP-seq data revealed a greater increase in H3 NDR occupanices for both the 70 and 134 groups of SM-induced genes in the PTET-STH1 versus snf2Δ mutant (Supplemental Fig. S4A,B). Increased H3 occupancy in the NDR could result from either a failure to prevent nucleosomes from forming in the NDR or from encroachment of the −1_Nuc/+1_Nuc into the NDR. Because we observed a larger (in the group of 70 genes) or comparable (group of 134 genes) repositioning of the −1_Nuc/+1_Nuc toward the NDR in the snf2Δ versus PTET-STH1 mutant, it appears that a substantial component of the increased H3 occupancy observed in PTET-STH1 cells involves defective nucleosome eviction rather than sliding, particularly for the group of 70 genes.
The defects in nucleosome remodeling at both groups of SM-induced genes in the remodeler mutants are generally accompanied by reduced transcription, which is more pronounced in the double mutant compared with the single mutants (Fig. 3A), which supports the conclusion that eviction or sliding of promoter nucleosomes increases access of the transcriptional machinery to the promoter/TSS to stimulate transcription initiation at these induced genes (model in Fig. 7). However, we note that not all of the 70 SM-induced genes exhibit appreciable reductions in transcription even in the double mutant (Fig. 2C, panel iii, right panels, 70 genes), which might indicate that residual nucleosome remodeling occurs at some of these genes carried out by another remodeling factor that is sufficient for robust transcription or that some other aspect of PIC assembly or transcription initiation is more rate-limiting at these genes.
Figure 7.
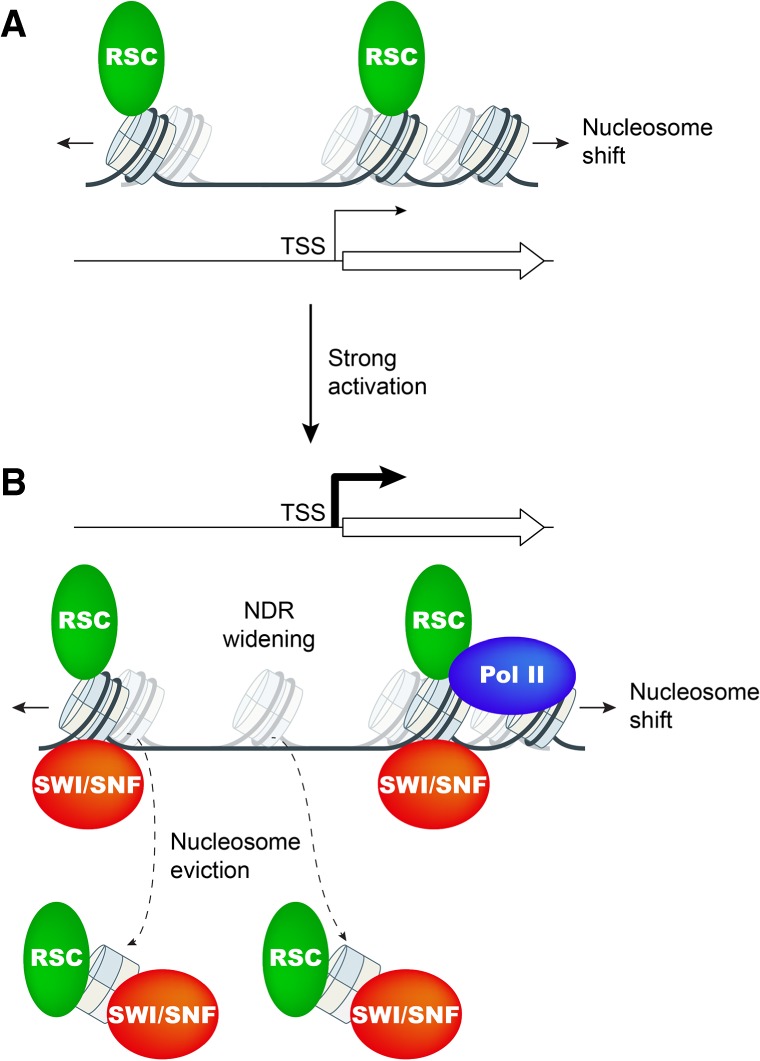
Model for cooperation between chromatin remodelers SWI/SNF and RSC in promoter nucleosome disassembly and repositioning at highly expressed genes. (A) At weakly expressed genes, relatively low levels of RSC (green oval) suffice to maintain wild-type NDR formation primarily by keeping nucleosomes (sectored cylinders) from crossing NDR boundaries. Cylinders with faint shades indicate nucleosome positions in cells depleted of functional RSC (Sth1). (B) Upon gene activation by Gcn4 or at highly expressed constitutive genes, SWI/SNF (red oval) and RSC are actively recruited and cooperate in evicting and repositioning the −1_Nuc and +1_Nuc to both widen the NDR and keep nucleosomes from assembling there. Cylinders with faint shades indicate nucleosome positions in cells lacking SWI/SNF or depleted of RSC. Pol II may also contribute to eviction of +1_Nucs.
Consistent with previous findings (Ganguli et al. 2014), we observed that the majority of 3619 constitutively expressed genes is considerably more dependent on RSC than SWI/SNF for NDR formation. Depletion of Sth1 leads to a repositioning of the −1_Nuc and +1_Nuc that narrows the NDR by a median value of ∼16 bp at the majority of these genes (Fig. 4C, panel ii, gray). Because the increased H3 occupancy in the NDR is accompanied by decreased occupancy of the +1_Nuc (Fig. 4A, panel iii), invasion of NDRs by +1_Nucs via nucleosome sliding upstream is a prominent defect evoked by loss of RSC function. At most of these constitutively expressed genes, SWI/SNF plays a relatively minor role (Fig. 4A, panel ii), although it does appear to augment RSC function in opposing upstream sliding by the +1 to +5 nucleosome array (Fig. 4C, panels i,iii, gray).
A strikingly different interplay between SWI/SNF and RSC occurs, however, at the 200 most highly transcribed constitutive genes, where eliminating SWI/SNF has an effect equal to depleting Sth1 on nucleosome occupancies across the promoter, and both remodelers are required for wild-type nucleosome eviction (Fig. 4B). SWI/SNF and RSC also function comparably in determining NDR widths and occupancies, with clear additive effects on both parameters when both remodelers are impaired in the double mutant (Fig. 5A; Supplemental Fig. S6B). Thus, as we observed for the SM-induced genes, SWI/SNF plays a critical role and cooperates substantially with RSC in nucleosome remodeling at the promoters of the most highly expressed constitutive genes. It should be noted that at the highly expressed constitutive or SM-induced genes, where the PTET-STH1 strain displays an impairment comparable with or lower than that of the snf2Δ strain in nucleosome loss or NDR widening, we cannot exclude the possibility that residual RSC resulting from incomplete depletion of Sth1 is acting on these promoters and partially masking RSC's contribution to remodeling.
The complementary functions of RSC and SWI/SNF in maintaining wide NDRs free of nucleosomes were clearly on display for the subset of genes in which NDR occupancies increase the most in the PTET-STH1 snf2Δ double mutant versus wild type. For such genes with NDRs too narrow to accommodate a nucleosome, it appeared that RSC was largely responsible for preventing NDR filling by encroachment of the −1_Nuc/+1_Nuc. For genes with an NDR large enough to accommodate a single nucleosome, RSC and SWI/SNF functioned comparably to prevent a nucleosome from assembling. For genes with even larger NDRs, SWI/SNF seemed to play a greater role than RSC in blocking multiple nucleosomes from forming inside the NDRs, although the greater H3 occupancies in these NDRs observed in the double mutant versus the snf2Δ strain also indicate nucleosome eviction by RSC at these NDRs as well (Fig. 5C). These findings, coupled with our conclusion above that RSC evicts nucleosomes from the NDRs of many SM-induced genes, provide strong evidence that both RSC and SWI/SNF can promote NDR formation by evicting nucleosomes and not merely by preventing encroachment into the NDR by the −1_Nuc and +1_Nuc and that they cooperate in both functions at the most highly expressed genes in yeast.
Materials and methods
Plasmid and yeast strain construction
The yeast strains used are listed in Supplemental Table S1 and were either purchased from Research Genetics (Winzeler et al. 1999) or constructed as described in the Supplemental Material.
ChIP-seq and MNase-ChIP-seq analysis of H3, H2B, and Rpb3 genome-wide occupancies
Wild-type and mutant strains were cultured in synthetic complete medium lacking isoleucine and valine (SC − Ilv) to A600 of 0.6–0.8, and SM was added at 1 µg/mL for 25 min to induce Gcn4 synthesis prior to treating cells with formaldehyde as described previously (Qiu et al. 2016). ChIP-seq was conducted and DNA libraries for Illumina paired-end sequencing were prepared as described previously (Cole et al. 2014) with modifications described in Qiu et al. (2016) using polyclonal antibodies against H3 (Abcam, ab1791), H2B (Abcam, ab1790), and Rpb3 (NeoClone, W0012).
For MNase-ChIP-seq, yeast cells were grown, treated with SM, and cross-linked with formaldehyde as described for CHIP-seq (Qiu et al. 2016) except cells were grown in 500-mL cultures and harvested as five equal aliquots. Cells were lysed with glass beads in FA lysis buffer, and the chromatin-rich fraction was recovered and digested with MNase as described (Wal and Pugh 2012), with the modifications described in the Supplemental Material. For each yeast strain, MNase-digested samples with 70%–80% mononucleosomes were immunoprecipitated as follows. Aliquots containing 5.0 µg of DNA were adjusted to 500 µL in 1× FA lysis buffer and mixed with 2 µg of anti-H3 antibodies (Abcam) conjugated with anti-rabbit Dynabeads and immunoprecipitated overnight. Immune complexes were washed and treated to reverse cross-linking, DNA was precipitated, and paired-end libraries were prepared and sequenced as described previously (Qiu et al. 2016) for ChIP-seq of sonicated samples.
Paired-end sequencing (50 nucleotides from each end) was conducted by the DNA Sequencing and Genomics Core Facility of the National Heart, Lung, and Blood Institute of the National Institutes of Health. Sequence data were aligned to the SacCer3 version of the genome sequence using Bowtie2 (Langmead and Salzberg 2012) with parameters -X 1000 --very-sensitive to map sequences up to 1 kb with maximum accuracy. PCR duplicates from ChIP-seq data were removed using the SAMtools rmdup package. Numbers of aligned paired reads from each ChIP-seq or MNase-ChIP-seq experiment are summarized in Supplemental Table S2. Raw genome-wide occupancy profiles for H3, H2B, and Rpb3 were computed in Matlab and R by counting the number of DNA fragments that overlapped with every base pair using the bioinformatics toolbox from Matlab and the coverage methods for GRanges objects from the GenomicRanges package in R. Distributions of DNA fragment centers were constructed by stacking only the nucleotides corresponding to the fragment centers. To allow the comparison between different samples, each profile was normalized to an average of 1 for each chromosome. To assess the quality of the MNase-ChIP-seq data, two-dimensional occupancy heat maps were generated in R using the plot2DO package (https://github.com/rchereji/plot2DO). Heat maps showing alignments of multiple loci were generated in Matlab using the bioinformatics toolbox to import data and the “heatmap” plotting function (http://www.mathworks.com/matlabcentral/fileexchange/24253-customizable-heat maps) or in R using custom scripts (https://github.com/rchereji/bamR). To visualize specific loci, igvtools was used to create tracks (.tdf files) that were loaded in the Integrative Genomics Viewer (IGV) (Robinson et al. 2011). Transcript end coordinates (TSS and transcription termination site) were obtained from Pelechano et al. (2013).
Procedures to determine the median positions of +1_Nucs and −1_Nucs and the internuclesomal positions for each gene and an iterative algorithm used to identify nucleosomes located within NDRs of the of the PTET-STH1 snf2Δ strain but absent in wild type are described in the Supplemental Material.
ChIP–chip analysis of Snf2 and Sth1 occupancies
SNF2-myc (HQY367) and STH1-myc (HQY459) yeast strains (Swanson et al. 2003) were grown, SM-treated, cross-linked, lysed with glass beads, sonicated with a Bioruptor, and immunoprecipitated as described for ChIP-seq except that chromatin was immunoprecipitated with anti-myc antibody (Roche). The ChIP and corresponding input DNA samples were amplified using GenomePlex whole-genome amplification kit (catalog no. WGA2, lot no. SLBQ6074V) as per the manufacturer's instruction. The amplified DNA samples were purified on the Qiagen PCR purification columns, and DNA was quantified using Nanodrop. Agilent Sure Tag DNA-labeling kit was used to label ChIP and input DNA with Cy5 and Cy3, respectively, and the labled DNA after purification on columns (provided with the kit) were competitively hybridized on Agilent 4x180K high-resolution custom arrays (G4821A). The data were extracted using the Feature Extraction software (Agilent).
Data normalization was performed using the limma pakage from Bioconductor as described previously (Venkatesh et al. 2012). The occupancy profiles for Snf2 and Sth1 were generated using the VAP program (Brunelle et al. 2015). The genes were split in half such that the probes corresponding to the first half were aligned to the TSSs, and those corresponding to the latter half were aligned to the transcription end sites.
Data access
Raw and analyzed data have been deposited in the NCBI Gene Expression Omnibus database under the accession number GSE110379.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institutes of Health (NIH) and used the computational resources of the NIH High-Performance Computing Biowulf Cluster (http://hpc.nih.gov) and the National Heart, Lung, and Blood Institute DNA Sequencing and Genomics Core Facility. C.K.G. is supported by grants from the NIH (GM095514) and Center for Biomedical Research (Oakland University).
Author contributions: Y.R., R.V.C., H.Q., D.J.C., and A.G.H. conceived the study. Y.R., R.V.C., C.K.G., D.J.C., and A.G.H. performed the formal analyses. Y.R., R.C.V., H.Q., and S.A. performed the investigations. Y.R., R.V.C., C.K.G., D.J.C., and A.G.H wrote the manuscript. All of the authors reviewed and edited the manuscript. A.G.H., D.J.C., and C.K.G. supervised the study and acquired the funding.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.312850.118.
References
- Adkins MW, Williams SK, Linger J, Tyler JK. 2007. Chromatin disassembly from the PHO5 promoter is essential for the recruitment of the general transcription machinery and coactivators. Mol Cell Biol 27: 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari SA, Paul E, Sommer S, Lieleg C, He Q, Daly AZ, Rode KA, Barber WT, Ellis LC, LaPorta E, et al. 2014. Mediator, TATA-binding protein, and RNA polymerase II contribute to low histone occupancy at active gene promoters in yeast. J Biol Chem 289: 14981–14995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32: 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaric S, Luckenbach T, Schmid A, Blaschke D, Horz W, Korber P. 2007. Redundancy of chromatin remodeling pathways for the induction of the yeast PHO5 promoter in vivo. J Biol Chem 282: 27610–27621. [DOI] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. 2003. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell 11: 1587–1598. [DOI] [PubMed] [Google Scholar]
- Brunelle M, Coulombe C, Poitras C, Robert MA, Markovits AN, Robert F, Jacques PE. 2015. Aggregate and heatmap representations of genome-wide localization data using VAP, a versatile aggregate profiler. Methods Mol Biol 1334: 273–298. [DOI] [PubMed] [Google Scholar]
- Bryant GO, Prabhu V, Floer M, Wang X, Spagna D, Schreiber D, Ptashne M. 2008. Activator control of nucleosome occupancy in activation and repression of transcription. PLoS Biol 6: 2928–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR. 2009. The logic of chromatin architecture and remodelling at promoters. Nature 461: 193–198. [DOI] [PubMed] [Google Scholar]
- Chereji RV, Morozov AV. 2015. Functional roles of nucleosome stability and dynamics. Brief Funct Genomics 14: 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Kan TW, Grudniewska MK, Romashchenko AV, Berezikov E, Zhimulev IF, Guryev V, Morozov AV, Moshkin YM. 2016. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res 44: 1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Ocampo J, Clark DJ. 2017. MNase-sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol Cell 65: 565–577 e563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Iwasa J, Cairns BR, Peterson CL. 2017. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol 18: 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole HA, Ocampo J, Iben JR, Chereji RV, Clark DJ. 2014. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res 42: 12512–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui F, Cole HA, Clark DJ, Zhurkin VB. 2012. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res 40: 10753–10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguli D, Chereji RV, Iben JR, Cole HA, Clark DJ. 2014. RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res 24: 1637–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikopoulos T, Havas KM, Dewar H, Owen-Hughes T. 2009. SWI/SNF and Asf1p cooperate to displace histones during induction of the Saccharomyces cerevisiae HO promoter. Mol Cell Biol 29: 4057–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen-Hughes T. 2011. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 333: 1758–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PD, Schmid A, Zavari M, Munsterkotter M, Horz W. 1999. Chromatin remodelling at the PHO8 promoter requires SWI–SNF and SAGA at a step subsequent to activator binding. EMBO J 18: 6407–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59: 407–450. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Brown SA, Clark CD, Winston F. 1992. Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev 6: 2288–2298. [DOI] [PubMed] [Google Scholar]
- Jia MH, Larossa RA, Lee JM, Rafalski A, Derose E, Gonye G, Xue Z. 2000. Global expression profiling of yeast treated with an inhibitor of amino acid biosynthesis, sulfometuron methyl. Physiol Genomics 3: 83–92. [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietenstein N, Wal M, Watanabe S, Park B, Peterson CL, Pugh BF, Korber P. 2016. Genomic nucleosome organization reconstituted with pure proteins. Cell 167: 709–721.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch Y, Zhang M, Kornberg RD. 1999. Histone octamer transfer by a chromatin-remodeling complex. Cell 96: 389–392. [DOI] [PubMed] [Google Scholar]
- Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, et al. 2016. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 7: 11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musladin S, Krietenstein N, Korber P, Barbaric S. 2014. The RSC chromatin remodeling complex has a crucial role in the complete remodeler set for yeast PHO5 promoter opening. Nucleic Acids Res 42: 4270–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. 2001. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol 21: 4347–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocetti N, Whitehouse I. 2016. Nucleosome repositioning underlies dynamic gene expression. Genes Dev 30: 660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo J, Chereji RV, Eriksson PR, Clark DJ. 2016. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res 44: 4625–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell TJ, Huff JT, Cairns BR. 2008. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J 27: 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell TJ, Schlichter A, Wilson BG, Cairns BR. 2015. The chromatin remodelers RSC and ISW1 display functional and chromatin-based promoter antagonism. Elife 4: e06073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. 2013. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497: 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Chereji RV, Hu C, Cole HA, Rawal Y, Clark DJ, Hinnebusch AG. 2016. Genome-wide cooperation by HAT Gcn5, remodeler SWI/SNF, and chaperone Ydj1 in promoter nucleosome eviction and transcriptional activation. Genome Res 26: 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Winston F. 2012. Chromatin and transcription in yeast. Genetics 190: 351–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke H, Horz W. 2003. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell 11: 1599–1607. [DOI] [PubMed] [Google Scholar]
- Reja R, Vinayachandran V, Ghosh S, Pugh BF. 2015. Molecular mechanisms of ribosomal protein gene coregulation. Genes Dev 29: 1942–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint M, Sawhney S, Sinha I, Singh RP, Dahiya R, Thakur A, Siddharthan R, Natarajan K. 2014. The TAF9 C-terminal conserved region domain is required for SAGA and TFIID promoter occupancy to promote transcriptional activation. Mol Cell Biol 34: 1547–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabish MA, Struhl K. 2007. The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol Cell Biol 27: 6987–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VM, Li B, Reese JC. 2003. SWI/SNF-dependent chromatin remodeling of RNR3 requires TAF(II)s and the general transcription machinery. Genes Dev 17: 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaswamy S, Iyer VR. 2008. Stress-dependent dynamics of global chromatin remodeling in yeast: dual role for SWI/SNF in the heat shock stress response. Mol Cell Biol 28: 2221–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain MM, Ansari SA, Pathak R, Palumbo MJ, Morse RH, Govind CK. 2014. The RSC complex localizes to coding sequences to regulate Pol II and histone occupancy. Mol Cell 56: 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MJ, Qiu H, Sumibcay L, Krueger A, Kim S-J, Natarajan K, Yoon S, Hinnebusch AG. 2003. A Multiplicity of coactivators is required by Gcn4p at individual promoters in vivo. Mol Cell Biol 23: 2800–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C. 2013. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9: e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. 2012. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489: 452–455. [DOI] [PubMed] [Google Scholar]
- Wal M, Pugh BF. 2012. Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol 513: 233–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bai L, Bryant GO, Ptashne M. 2011. Nucleosomes and the accessibility problem. Trends Genet 27: 487–492. [DOI] [PubMed] [Google Scholar]
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. 2007. Chromatin remodelling at promoters suppresses antisense transcription. Nature 450: 1031–1035. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Reese JC. 2007. Exposing the core promoter is sufficient to activate transcription and alter coactivator requirement at RNR3. Proc Natl Acad Sci 104: 8833–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ma H, Pugh BF. 2011. Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res 21: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.