

Abstract
TP53 mutations frequently occur in head and neck squamous cell carcinoma (HNSCC) patients without human papillomavirus infection. The recurrence rate for these patients is distinctly high. It has been actively explored to identify agents that target TP53 mutations and restore wild-type (WT) TP53 activities in HNSCC. PRIMA-1 (p53-Reactivation and Induction of Massive Apoptosis-1) and its methylated analogue PRIMA-1Met (also called APR-246) were found to be able to reestablish the DNA-binding activity of p53 mutants and reinstate the functions of WT p53. Herein we report that piperlongumine (PL), an alkaloid isolated from Piper longum L., synergizes with APR-246 to selectively induce apoptosis and autophagic cell death in HNSCC cells, whereas primary and immortalized mouse embryonic fibroblasts (MEFs) and spontaneously immortalized non-tumorigenic human skin keratinocytes (HaCat) are spared from the damage by the cotreatment. Interestingly, PL-sensitized HNSCC cells to APR-246 are TP53 mutation-independent. Instead, we demonstrated that glutathione S-transferase pi 1 (GSTP1), a GST family member that catalyzes the conjugation of GSH with electrophilic compounds to fulfill its detoxification function, is highly expressed in HNSCC tissues. Administration of PL and APR-246 significantly suppresses GSTP1 activity, resulting in the accumulation of ROS, depletion of GSH, elevation of GSSG, and DNA damage. Ectopic expression of GSTP1 or pretreatment with antioxidant N-acetyl-L-cysteine (NAC) abrogates the ROS elevation and decreases DNA damage, apoptosis, and autophagic cell death prompted by PL/APR-246. In addition, administration of PL and APR-246 impedes UMSCC10A xenograft tumor growth in SCID mice. Taken together, our data suggest that HNSCC cells are selectively sensitive to the combination of PL and APR-246 due to a remarkably synergistic effect of the cotreatment in the induction of ROS by suppression of GSTP1.
Keywords: head and neck squamous cell carcinoma (HNSCC), piperlongumine (PL), p53-reactivator, glutathione S-transferase pi 1 (GSTP1), cell death
Introduction
Normal cells physiologically retain redox homeostasis by balancing the generation and elimination of reactive oxygen species (ROS). The redox dynamics fluctuate within a tolerable range. Due to the increased proliferation and disturbed metabolism, cancer cells, however, possess a high level of ROS. When the increase of ROS reaches a critical level (the threshold), it may potentially overwhelm the cellular antioxidant capacity and trigger the cell-death.1 This common effect suggests that cancer cells may be more vulnerable to oxidant stress. Indeed, applications of small molecules that increase ROS accumulation have been proposed for cancer therapy.2-10
Piperlongumine (PL) is an alkaloid originally identified in Piper longum L. PL bears multiple functions on anti-platelet aggregation, anti-inflammatory, and anti-cancer.11-25 Raj et al. screened several thousand biologically active compounds and found that PL is the strongest activator to promote p53-mediated transcription.15 The antitumor effect of PL was much more dramatic than that of cisplatin in a melanoma allografted mouse model.14 In a spontaneous mouse mammary tumorigenesis model, PL outperformed paclitaxel.15 lGutathione-S-transferase-P1 (GSTP1) and carbonyl reductase 1 (CBR1), two enzymes participating in cellular response to oxidative stress, were among the most enriched proteins after PL exposure, indicating the possible involvement of ROS in the process. Indeed, the authors demonstrated that treatment of PL augmented the levels of ROS in cancer cells but not in non-transformed cells.15,16
PRIMA-1 (p53-Reactivation and Induction of Massive Apoptosis-1) and PRIMA-1Met (methylated analogue of PRIMA-1, also called APR-246) are quinuclidine compounds, which can restore the DNA-binding activity of p53 mutants and reinstate the functions of wild-type (WT) p53.26-28 PRIMA-1 and APR-246 are converted to methylene quinuclidinone (MQ), a Michael acceptor in the cell. MQ binds covalently to cysteines in mutant p53 and unfold WT p53, hence reestablishing the action of p53.28-30 Consequently, p53 target genes, such as PUMA, NOXA, and BAX, are up-regulated and caspases -2, -3 and -9 are activated by PRIMA-1 or APR-246.28 Recent studies showed that MQ may also possess p53-independent functions in the induction of cell death in a variety of tumor types.31 MQ modifies and converts antioxidant thioredoxin reductase 1 (TrxR1) into pro-oxidant NADPH oxidase, promoting the generation of ROS.32-34 In addition, APR-246 also decreases intracellular content of glutathione (GSH), hence enhancing the accumulation of ROS.35
In the current study, we investigated the synergistic effect of PL and APR-246 in the induction of cell death in HNSCC, in which mutations of TP53 are frequently identified and relevant to tumor progression and recurrence.36-38 The combination is based on the strong induction of ROS by both agents, which may create a reciprocal sensitization. In addition, reactivation of p53 by APR-246 may provide a mechanistic basis for the action of PL.15 Our results support the premise and suggest that co-treatment of PL and APR-246 synergistically induces apoptosis and autophagical cell death in HNSCC cells.
Results
PL enhances APR-246-mediated cell death in HNSCC cells
To determine whether PL possesses a synergistic effect with TP53 reactivator, we exposed UMSCC10A and FaDu, two HNSCC cell lines with TP53 mutation to PL and/or APR-246. As shown in Figures 1a-d, both PL and APR-246 individually suppressed cell survival and bore a modest effect on the killing of HNSCC cells. Co-treatment with PL and APR-246 led to a marked reduction in cell viability and an increase in cell death (Figures 1a-d). Similar results were acquired for UMSCC1 and UMSCC17A cell lines (See below Figure 3).
Figure 1. PL and APR-246 synergistically induce HNSCC cell death.
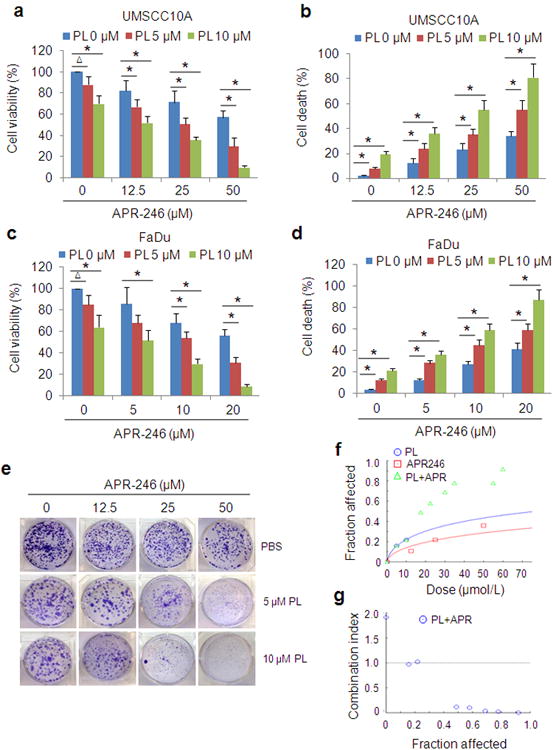
(a-d) UMSCC10A and FaDu cells were treated with 0-10 μM PL in the presence or absence of APR-246 (0-50 μM) for 72 h. Cell viability was determined by the MTT assay. Cell death was measured with the trypan blue exclusion assay. The assays were performed in triplicate samples, and the results are representative of three independent experiments (ΔP < 0.05; * P < 0.01). (e-g) UMSCC10A cells were treated with various concentrations of PL and/or APR-246 for 16 h. Clonogenic survival assays were performed. (e) Representative images of the clonogenic survival assays are presented. (f, g) Assessment of the synergy between PL and APR-246 in the cells using the CompuSyn software (ComboSyn, Inc. Paramus, NJ 2007). Drug interactions are expressed as fraction affected (FA) curves (f) and combination index (CI) plots (g). The CI values were automatically generated by the software over a range of FA levels from growth inhibition percentages. A CI < 1.0 was considered as synergy. Synergy was remarkable at relevant FA values being 50% or greater (g).
Figure 3. PL- and APR-246-induced cell death is independent of TP53.
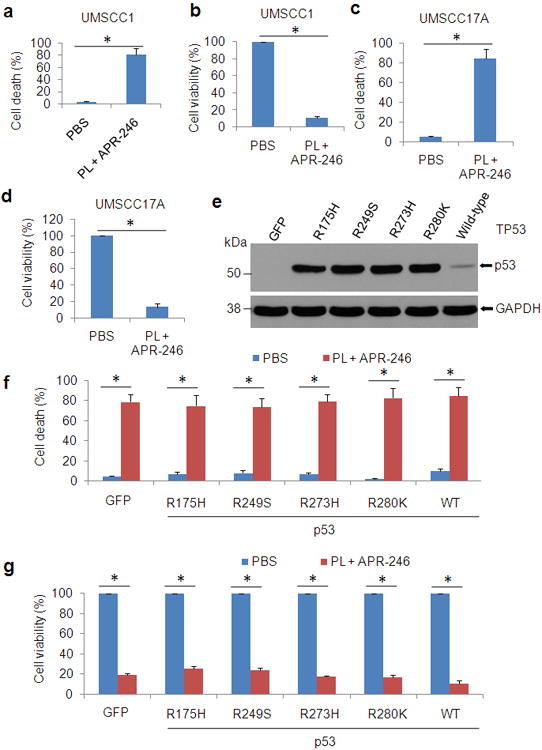
(a-d) UMSCC1 (null TP53) cells (a, b) and UMSCC17A (wild-type TP53) cells (c, d) were exposed to 10 μM PL and 25 μM APR-246 for 72 h. After the treatment, cell death and cell viability were determined as described in Figure 1. (e-g) UMSCC1 cells were infected with lentiviruses expressing TP53 mutants R175H, R249S, R273H, R280K, and wild-type TP53, or GFP (control). Cell transduction efficiency was about 60% with the fluorescence microscopy analysis at 48 h after the infection. Immunoblot was used for the detection of p53 expression in the transduced cells (e). Cell death (f) and cell viability (g) in the cells treated with 10 μM PL and 25μM APR-246 for additional 72 h were determined as described in Figure 1. The data represent the mean ± S.D. * P < 0.01, n = 3.
To further evaluate the synergistic effect of the co-treatment, we exposed UMSCC10A cells to PL and/or APR-246 and assessed cell growth using the clonogenic survival assay. As shown in Figure 1e, colony formation was suppressed with the treatment of PL or APR-246 and the effect was strikingly enhanced with the co-treatment. Fraction affected (FA) levels from growth inhibition percentages were markedly higher in co-treatment groups as compared with those treated with PL or APR-246 alone (Figure 1f). Combination index (CI), a quantitative representation for the degree of drug interaction and an indicator for drug synergy, was calculated over a range of FA levels from growth inhibition percentages.39 A CI < 1.0 can be considered as synergy.40 As shown in Figure 1g, the CI plots in the co-treated cells revealed a strongly synergistic effect when FA values were 50% or greater (Figure 1g, Supplementary Figure 1). These results suggest that PL and APR-246 possess a synergistic effect against HNSCC cells.
PL and/or APR-246 have a marginal impact on the survival of non-transformed cells
To determine whether the combined effect of PL and APR-246 was uniquely to cancer cells (HNSCC), we exposed the primary and immortalized mouse embryonic fibroblasts (MEFs) to the co-treatment. It showed that there was only marginal reduction in cell viability (Supplementary Figures 2a and b). Consistently, we detected only a minimal decrease in cell viability in spontaneously immortalized non-tumorigenic human skin keratinocytes (HaCat) (Supplementary Figure 2c). These results indicate that PL and APR-246 only have a limited impact on the survival of non-transformed cells.
PL and APR-246 prompt both apoptosis and autophagic cell death
To characterize the cell death enhanced by PL in APR-246-treated cells, we assessed apoptosis and autophagy in UMSCC10A cells with the co-treatment. As shown in Figure 2a and b, administration of PL or APR-246 induced the activation of parp-1, caspase-7, and caspase-9, which was markedly enhanced by the combination of the two agents. DNA fragments released from the nuclei in the treated cells were remarkably increased, supporting the existence of apoptosis (Figures 2a and b). Interestingly, immunoblot detection also revealed that under the treatment of PL- and/or APR-246, a significant part of light chain 3 (LC-3, also known as autophagy-related gene 8, ATG8) were converted from isoform I (LC3-I) to isoform II (LC3-II), indicating the induction of autophagy (Figure 2a). To visualize autophagy, we established UMSCC10A/green fluorescent protein (GFP)-LC3 cells, a stable cell line that expresses GFP-tagged LC3.13 GFP-LC3 formed cytoplasmic puncta in cells treated with PL and/or APR-246 but not in control cells (Figures 2c), supporting the notion that cotreatment of PL and APR-246 leads to both apoptosis and autophagy.
Figure 2. PL enhances APR-246-induced apoptosis and autophagy in HNSCC cells.
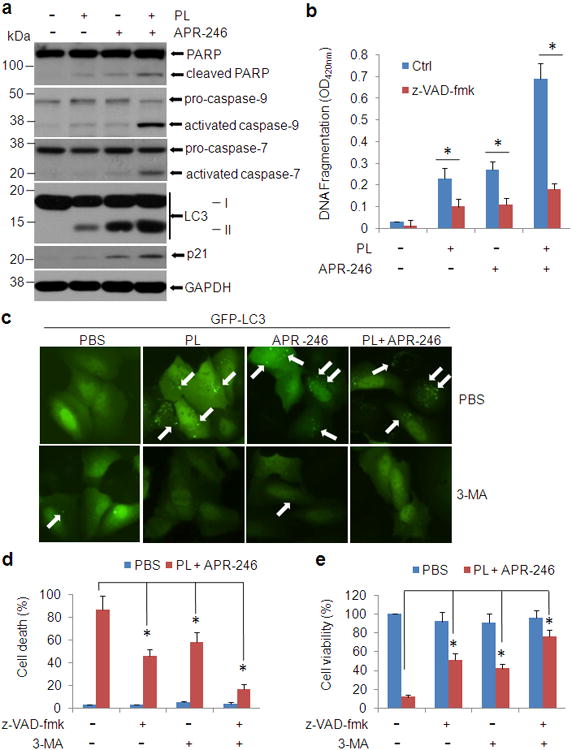
(a) UMSCC10A cells were treated with 10 μM PL and/or 25 μM APR-246 for 24 h. After the treatments, whole cell extracts were collected for the western blot analysis. Thirty μg proteins were loaded in each lane. GAPDH serves as a loading control. (b) UMSCC10A cells were treated with 10 μM PL and/or 25μM APR-246 in the presence or absence of 20 μM z-VAD-fmk for 72 h. After the treatment, cell apoptosis was quantified using a cell death ELISA kit (Roche Diagnostics) showing enrichment of nucleosomes in the cytoplasmic fraction of the cells. Values represent the mean ± S.D. * P < 0.01, n = 3. (c) UMSCC10A cells were transfected with pcDNA3/GFP-LC3 for 24 h. The cells were then treated with 10 μM PL and/or 25μM APR-246 for additional 24 h in the presence or absence of 1 mM 3-MA. Autophagy (GFP-LC3 punctuates) wasmonitored under an Olympus IX51 fluorescence microscope. White arrows show the autophagosomes/autolysomes. (d, e) UMSCC10A cells were co-treated with 10 μM PL and 25 μM APR-246 in the presence or absence of 20 μM z-VAD-fmk or 1 mM 3-MA for 72 h. After the treatment, cell death (d) and cell viability (e) were determined as described in Figure 1. Values represent the mean ± S.D. * P < 0.01 as compared with respective treatment without z-VAD and 3-MA, n = 3.
To validate the contribution of apoptosis in PL and APR-246-induced cell death, we pre-treated UMSCC10A cells with pan-apoptotic inhibitor benzyloxycarbonylvalyl-alanyl-aspartic acid (O-methyl)-fluoro-methylketone (zVAD-fmk) before PL and/or APR-246 were added. As expected, PL and APR-246-induced DNA fragment was markedly reduced (Figure 2b). Consistently, cell death was decreased and the cell viability was improved by the pre-treatment with zVAD-fmk (Figure 2d and e). However, cells pre-treated with zVAD-fmk still underwent a certain level of cell death and the cell viability was not completely recovered when PL/APR-246 were added (Figures 2d and e). In addition, although PL or APR-246 alone induced a notable cell death (Figure 1), PARP cleavage and activation of caspases were relatively low (Figure 2a). Collectively, these results support the notion that additional cell death (autophagic cell death) may also be involved.
To determine the impact of autophagy on PL and APR-246-induced cell death, we pre-treated UMSCC10A cells with an autophagy inhibitor 3-methyladenine (3-MA)13, which blocks class III PI3Ks that are critical during the late stage of vesicle expansion, and analyzed autophagy formation after the addition of PL and APR-246. As shown in Figure 2c, addition of 3-MA blocked the autophagy formation in PL/APR-246-treated cells. More importantly, pre-treatment with 3-MA reduced cell death and improved cell viability resulted from the co-treatment of PL and APR-246 although both were not completely reversed (Figures 2d and e), indicating the participation of autophagy in the cell death induced by the PL/APR-246. Consistent with these observations, pre-treatment with both z-VAD-fmk and 3-MA strikingly reduced the cell death and further improved cellular viability in the PL/APR-246-co-treated cells (Figures 2d and e), supporting our notion that PL and APR-246 induce apoptosis and autophagy, both of which contribute to the cell death in the co-treatment.
PL-induced sensitization of HNSCC cells to APR-246 is independent of TP53
PRIMA-1 and APR-246 were originally developed as mutant p53 re-activators.20,25 However, recent studies revealed that a range of functions that the family compounds have may be independent of p53 mutation.28-30 To define whether the cell death resulted from the cotreatment of PL and APR-246 is relevant to p53 status, we detected cell viability and cell death in UMSCC17A (WT p53) and UMSCC1 (p53 deficient) cells with the cotreatment. Similar to the responses of p53 mutated UMSCC10A and FaDu cells in Figures 1a-d, bothUMSCC1 and UMSCC17A cells reacted to the cotreatment and shared a similar pattern that cellular viability was reduced and cell death was elevated by the co-treatment (Figures 3a-d). To further assess the involvement of p53 in PL- and APR-246-induced cell death, we transfected WT and mutant p53 vectors into UMSCC1 cells (Figure 3e). Consistently, a similar response to the co-treatment was observed in cells with WT and mutant p53 (Figures 3f and g). Collectively, our data suggest that PL-enhanced sensitivity of HNSCC cells to APR-246 is independent of TP53.
Co-treatment of PL and APR-246 leads to the accumulation of ROS in HNSCC cells
We and other groups previously reported that PL treatment leads to an increase in ROS levels and induces cell death in a variety of cells.13-16 Interestingly, APR-246 is also able to induce ROS by disturbing the cellular redox balance.32-35 Thus, application of PL and APR-246 may synergistically elevate intracellular ROS, rendering cancer cells a severe oxidant stress. To test this hypothesis, we detected intracellular ROS levels in PL- and APR-246-treated UMSCC10A cells. We found that application of PL and/or APR-246 individually or together markedly increased intracellular ROS contents (Figure 4a). However, PL and/or APR-246 only marginally increased ROS levels in non-transformed MEFs (Figure 4b). Consistent with increased ROS in the cells, treatment with PL and/or APR-246 decreased the GSH and increased the oxidized glutathione (GSSG) levels in UMSCC10A cells (Figures4c and d). Moreover, pre-treatment with the reducing agent 5 mM NAC,), which quenches ROS, prevented the cotreatment-induced ROS accumulation, GSH depletion, and GSSG increase in UMSCC10A cells (Figures 4a, c, and d). Taken together, our results suggest that PL exposure promotes the accumulation of ROS in APR-246-treated cells.
Figure 4. PL and APR-246 promote the accumulation of ROS, the depletion of GSH, and the elevation of GSSG in HNSCC cells, which are reversed by the administration of antioxidant NAC.

UMSCC10A cells (a, c, d) and MEFs (b) were treated with 10 μM PL and/or 25μM APR-246 in the presence or absence of 5 mM NAC for 24 h. (a, b) ROS in the cells. One hour prior to the termination of the treatment, 100 ng/ml dihydroethidium was added to the medium. The cells were collected, washed and analyzed by flow cytometry with the red laser channel (FL-3) using a FACscan analyzer. (c) GSH contents in the cells. GSH was analyzed using a commercial kit from Abcam. The values were then normalized to mg protein. The number in PBS-treated cells was set as “100%”. (d) GSSG levels in the cells. GSSG was analyzed using a commercial kit from Abcam. The values were then normalized to mg protein. The number in PBS-treated cells was set as “1”. Means ± SD for three independent experiments were presented. *P < 0.01;Δ P < 0.05; NS = Not Significant.
Treatment with PL and APR-246 induces DNA damage
A direct outcome of elevated intracellular ROS is DNA damage, which may eventually result in genomic instability and cell death. The DNA adduct, 8-oxo-7, 8-dihydro-20 -deoxyguanine (8-oxo-dG) is formed by the reaction of OH radical with the DNA guanine base, leading to oxidative DNA damage.41 To determine whether PL and APR-246-prompted ROS accumulation is able to cause oxidative DNA damage, we exposed UMSCC10A cells to PL and/or APR-246 and measured 8-oxo-dG. As shown in Figure 5a, both PL and APR-246 exposures led to a high level of 8-oxo-dG, in particular under the treatment of combination. To further identify the DNA damage in PL and APR-246-treated cells, we exposed UMSCC10A cells to both agents and detected the formation and repair of DNA damage by the comet assay.41 The tail moment that reflects the frequency of DNA breaks was measured to quantify DNA damage. In both PL and APR-246 treated cells, levels of DNA damage were noticeably higher than those in control (PBS)-treated cells (Figures 5b and c). When both agents were used for the co-treatment, damaged DNAs in the tail moment in cells were strikingly elevated (Figures 5b and c). Phosphorylation of histone H2AX (γ-H2AX) occurs at DNA breakage sites and is another DNA damage marker.41 We found thatγ-H2AX was increased in PL or APR-246 treated cells and elevated by the cotreatment both in the immunofluorescence assay and in the immunoblot analysis (Figures 5d and e). Taken together, our results suggest that treatment with PL and APR-246 not only elevates the level of intracellular ROS but also induces DNA damage in HNSCC cells.
Figure 5. PL/APR-246 treatment promotes DNA damage.
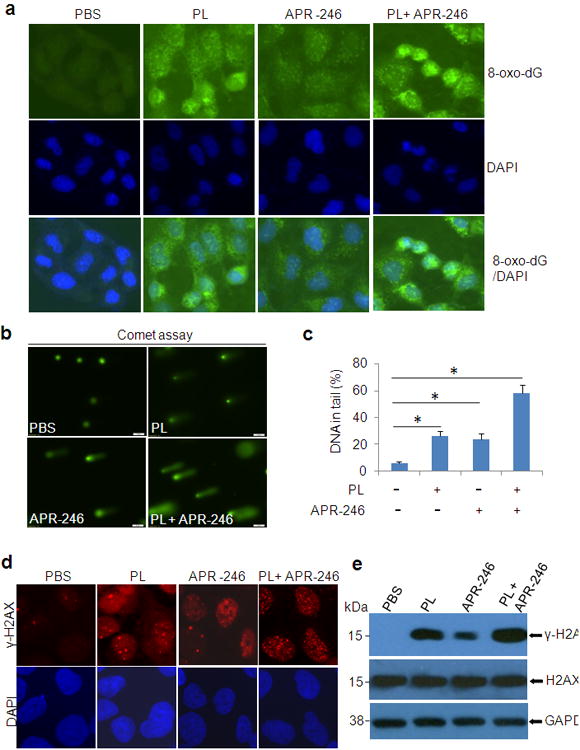
(a) 8-oxo-dG in UMSCC10A cells treated with PL and/or APR-246. UMSCC10A cells growing on fibronectin-coated coverslips were treated with 10 μM PL and/or 25 μM APR-246 in serum-free and phenol-red-free medium for 16 h at 37°C, fixed in absolute methanol (- 20°C, 20 min) and permeabilized with 0.1% Triton X-100 at room temperature for 15 min. After blocking, the cells were stained with FITC-conjugated avidin for 1 h at 37°C. Fluorescence is captured with an Olympus IX51 fluorescence microscope. (b, c)Comet assay showing elevated DNA damage in cells treated with PL and APR-246. UMSCC10A cells were treated with 10 μM PL and/or 25μM APR-246 for 16 h. The cells were then trypsinized and washed with PBS. Two thousand cells were mixed with 100 μl low melting agarose for alkaline comet assay. Cells in the gel were stained and visualized with epifluorescence microscopy (b). (c) Percentage of DNAs in the tail (damaged DNA) was calculated. *P < 0.01, n = 3. (d, e) Expression of γ-H2AX in PL/APR-246-treated cells. UMSCC10A cells were treated with 10 μM PL and/or 25 μM APR-246 for 16 h. After the treatment, the cells were collected for the immunofluorescent analysis of γ-H2AX foci formation (d) or immunoblot analysis of γ-H2AX and H2AX expression (e).
GSTP1 mediates the activity of PL and APR-246 in HNSCC cells
PL treatment selectively increased ROS levels (Figures 4a and b) and induced cell death in cancer cells relative to normal cells (Figures 1a-d and 3a-d, Supplementary Figure 2). The variant responses of cancer cells and normal cells to the treatment with PL are partially due to the interaction with and suppression of increased GSTP1, CBR1, and TrxR1 activity in cancer cells, in particular GSTP1 that was the highest-confidence hit in a recent screening assay to identify targets and their associated complexes that bind to PL.15,41 To determine whether GSTP1 also plays a role in APR-246-induced ROS accumulation, in particular in PL- and APR-246 cotreatment-induced oxidative stress and cell death, we detected the expression of GSTP1 with immunoblot in UMSCC10A cells with the cotreatment. Surprisingly, neither PL nor APR-246 affected the protein level of GSTP1 (Figure 6a). We next measured the activity of GSTP1 in PL- and/or APR-246-treated cells. It showed that PL and/or APR-246 inhibited the activity of GSTP1, in particular under the co-treatment (Figure 6b, Supplementary Figure 3). Application of antioxidant NAC partially recovered the activity of GSTP1 (Supplementary Figure 4). Further, stably overexpressing GSTP1 markedly reduced PL- and APR-246-induced ROS elevation, autophagy induction, and cell death (Figures 6c-f). In a complementary study, knockdown of GSTP1 marginally increased the levels of ROS in PL- and APR-246-treated cells and almost bore no influence on autophagy induction and cell death occurred in PL/APR-246 treatment (Figures 6f-i), suggesting that other relevant antioxidant targets may also be regulated by the agents or that there might have partially overlapping functions between GSTP1 knockdown and the co-treatment in the cell. Taken together, these data support the notion that PL- and/or APR-246 induce cell death by interfering with redox and ROS homeostatic regulators such as GSTP1.
Figure 6. Treatment of PL/APR-246 inactivates GSTP1, manipulations of which affect the function of PL/APR-246.
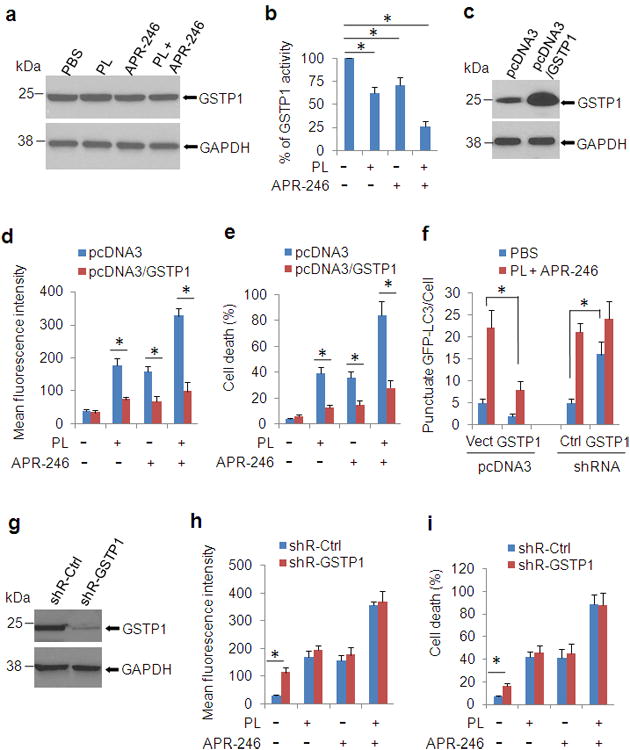
(a, b) PL/APR-246 treatment reduces the activity of GSTP1. UMSCC10A cells were treated with 10 μM PL and/or 25 μM APR-246 for 24 h. After the treatment, the cells were collected for the immunoblot analysis of GSTP1 expression (a) or the measurement of GSTP1 activity (b). Measurements of the GSTP1 activity were performed using a commercial kit from Abcam following the manufacturer's instructions. The values were then normalized to mg protein. The number in PBS-treated cells was set as “100%”. The data represent means ± SD from three independent experiments (*P < 0.01). (c-e) Overexpression of GSTP1 reduces PL- and APR-246- induced ROS escalation and cell death. UMSCC10A/pcDNA3-GSTP1 cells were exposed to 10 μM PL and/or 25 μM APR-246 for 48 h. The cells were collected for the determination of GSTP1 expression by western blot (c), ROS determination (d), or cell death measurement (e). (f) Manipulations of GSTP1 affect autophagy induction by the cotreatment of PL and APR-246. UMSCC10A cells with GSTP1 overexpression or knockdown were transfected with pcDNA3/GFP-LC3 for 24 h. The cells were then treated with 10 μM PL and 25 μM APR-246 for additional 24 h. Autolysosomes/autophagosomes (GFP-LC3 punctuates) were monitored under an Olympus IX51 fluorescence microscope. The average punctate GFP-LC3 fluorescence dots were calculated in 200 cells. Data are shown as mean ± S.D. (g - i) Knockdown of GSTP1 increases intracellular ROS levels, but only marginally escalates ROS in PL- and APR-246-treated cells. Scramble shRNA and shR-GSTP1 were packaged with lentiviral system and transduced into UMSCC10A cells. After selection with puromycin for 2 weeks, the cells were verified for the knock-down by western blot (f). The cells were then exposed to 10 μM PL and/or 25μM APR-246 for 72 h for intracellular ROS determination (h) or cell death detection (i). ROS were measured as described in Figure 4. Cell death was determined as described in Figure 1b and d. Means ± SD for three independent experiments were presented. *P < 0.01.
Application of PL and APR-246 reduces xenograft tumor growth of HNSCC cells
To validate the role of PL and APR-246 in the suppression of HNSCC cells, we subcutaneously inoculated UMSCC10A cells into SCID mice. Application of PL or APR-246 alone reduced tumor volumes (p < 0.01), whereas co-treatment with the two agents substantially increased the suppressive effect and led to marked reductions in tumor volumes (Figure 7a). Consistent with the observation that the expression of GSTP1 was not affected by PL/APR-246 (Figure 6a), treatments with these agents bore no impact on the level of GSTP1 by the immunohistochemistry staining (Figure 7b).
Figure 7. GSTP1 is highly expressed in HNSCC tissues. Application of PL and APR-246 reduces xenograft tumor growth of HNSCC cells.
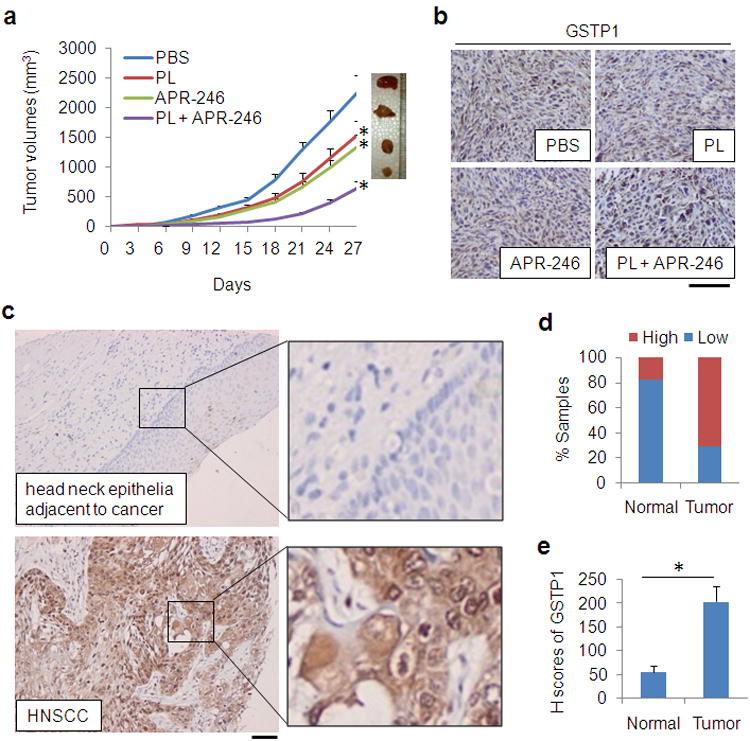
(a, b) 5 × 106 UMSCC10A cells were inoculated subcutaneously under the flank of SCID mice. Three days later, the mice were randomized into4 groups (n = 10 / group) receiving PBS (control), PL (4 mg/kg/day), APR-246 (100 mg/kg/day), or PL (4 mg/kg/day) + APR-246 (100 mg/kg/day) by i. p. Treatment was performed daily for 24 days. (a) Tumor volumes were measured every 3 days. *P < 0.01 as compared with control treatment group. (b) The tumors were removed from euthanized mice. IHC was used to detect GSTP1. Scale bar = 100 μm. (c - e) HNSCC tissues from healthy (n = 28) and HNSCC (n = 194) subjects were assessed for the expression of GSTP1 by IHC. (c) Representative IHC staining of GSTP1 in a normal head and neck epithelial tissue and in an HNSCC tissue. Scale bar = 100 μm. (d) Quantification of GSTP1 expression in human head and neck tissues. Low: overall negative or weak staining; High: overall moderate or strong staining. The Pearson's chi-square test was used to analyze the distribution difference of GSTP1 between healthy and HNSCC tissues (P < 0.01). (e) H-scores of GSTP1 in head and neck tissues (*P< 0.01).
GSTP1 is highly expressed in HNSCC tissues
To investigate the pathological significance of GSTP1 in HNSCC, we assessed its expression in human HNSCC tissues using IHC. Tissues from normal (n = 28) and HNSCC (n = 194) were analyzed. Healthy head and neck epithelial tissues or normal tissues adjacent to cancer generally displayed weak GSTP1 signals (Figure 7c). In contrast, some 70% HNSCC cases were positive for GSTP1 (Figures 7c and d). The H score42 also demonstrated an intense signal of GSTP1 in cancerous tissues (Figure 7e). Taken together, these data are consistent with our in vitro observation that GSTP1 levels are elevated in HNSCC cells and it may be worthy exploring it as a potential target for precision therapy of HNSCC as we demonstrated in this study.
Discussion
In this study, we found that combination of PL and APR-246 resulted in a marked increase of cell death in various HNSCC cell lines, including FaDu, UMSCC1, UMSCC10A, and UMSCC17A. Further, we showed that the cytotoxicity of PL and APR-246 was selective to malignant cells, but not to non-transformed cells. The different responses of malignant cells and non-transformed cells to the combination of PL and APR-246 may provide a therapeutic window for effectively targeting cancer cells with limited off-target effects.
It sounds rationale to postulate that the combination might work specifically on TP53 mutated cells since APR-246 was originally developed for targeting TP53 mutation and restored the activity of p53 in the cells.20,25 To our surprise, UMSCC1 (TP53 deficient), UMSCC17A (wild-type TP53), and FaDu and UMSCC10A (TP53 mutation) cells were responsive to PL and APR-246 similarly (Figures 1a-d and 3a-d). More importantly, we transfected various mutant and wild-type TP53 constructs into TP53-null UMSCC1 cells, and the transduction did not improve or reduce the response of the cells to the combined treatment of APR-246 and PL, further suggesting the independence of TP53 for the function we observed in the cotreated cells. These results are consistent with recently reports showing that APR-246 and its analogue PRIMA-1 possess TP53 independent effect on the killing of tumor cells.28-30
We and other groups previously reported that PL treatment increases intracellular ROS and hence induces cell death.13-16 Interestingly, APR-246 is also able to induce ROS by disturbing the cellular redox balance.28-30 Thus, application of PL and APR-246 may synergistically elevate intracellular ROS, rendering cancer cells a severe oxidant stress. Our results supported this hypothesis and demonstrated that combination of PL and APR-246 led to a strikingly elevation of ROS in cancer cells, but not in non-transformed MEFs (Figure 4), providing a concrete evidence for the selectivity of the co-treatment against cancer cells. Unlike normal cells that keep redox homeostasis by balancing the production and removal of ROS and the redox dynamics fluctuates in a tolerable range, cancer cells possess high levels of ROS due to active cell metabolism and the hypoxia.43,44 Therefore, agents that increase ROS generation or decrease ROS clearance may push the level of ROS in the tumor over the threshold of life and death to prompt cell death.1
Previous reports demonstrated that PL interacts with and suppresses the activity of GSTP1, CBR1, and TrxR1 in cancer cells. GSTP1 was the highest-confidence hit in a recent screening assay to identify targets and their associated complexes that bind to PL.15,45-47 In keeping with these findings, we found that around 70% HNSCC tissues bore a high level of GSTP1 expression whereas only about 14% normal tissues possessed elevated GSTP1 (Figure 7c-e). Overexpression of GSTP1 mitigated ROS accumulation and prevented PL/APR-246-induced cell death to a certain level, indicating that GSTP1 may play a critical role in the action of the co-treatment.48,49 On the other hand, how, depletion of GSTP1 with shRNA increased the ROS, but could not reach that in PL-/APR-246 treated cells (Figures 6c-i), suggesting that additional ROS-relevant enzymes, such as TrxR1 and CBR1, may also participate in the oxidant stress in the co-treatment. It is interesting to clarify the contribution of these enzymes to the ROS in PL/APR-246 co-treatment in the future.
Taken together, we identified that combination of PL and APR-246 selectively induces HNSCC cell death by inactivating GSTP1 and hence elevating intracellular ROS levels. Complementary to the in vitro findings, our in vivo investigation provides further evidence demonstrating that cotreatment with PL and APR-246 could be a valuable means for controlling HNACC tumor growth. Due to the selectivity and high efficiency, combination of the two compounds for the treatment of HNSCC may possess a great feasibility and bear a high potential.
Materials and Methods
Cell lines, primary MEFs, and drug treatment
FaDu and HaCat were from ATCC (Manassas, VA). UMSCC1, UMSCC10A, and UMSCC17A were kindly provided by Dr. Thomas Carey's labin the University of Michigan.50 MEFs were isolated from C57BL6 mice and immortalized by culturing and overcoming the crisis stage at passage 11-16.41 Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS (Atlanta Biologicals, Atlanta, GA). PL, APR-246, NAC, and 3-MA were ordered from Sigma-Aldrich (St. Louis, MO). zVAD-fmk was from Biomol International (Plymouth Meeting, PA).
Plasmids and transfection
WT and mutation p53 plasmids (R175H, R249S, R273H, and R280K) were reported previously51,52 and recloned into pEF1a lentiviral vector and packaged with 293T cells. Lentiviruses expressing WT and mutant p53 were used for the infection of UMSCC1 cells, followed by selection with puromycin. shRNA-control and shRNA-GSTP1 were purchased from Thermo Scientific (Lafayette, CO), packaged with lentiviral expression system, and applied for the transduction.
Assays for cell survival and death
Cell viability was determined using the MTT assay.41 Cell death was detected with trypan blue exclusion assay. Apoptosis in cells exposed to different treatments was measured with a kit from Roche Diagnostics quantifying nucleosome enrichment in cytoplasm.
Clonogenic survival assay and analysis of combined drug effects
Three thousand UMSCC10A cells each well were planted in a 6-well plate and treated concurrently with different dosages of PL and APR-246. Sixteen hours later, the reagents were removed by washing the cells with 1 × PBS for three times. The cells were cultured for 14 days before fixation for clonogenic determination. Colonies with 50 or more cells were calculated. Survival rate of cells in each treatment was the ratio of value to control group. FA was calculated by “1 - survival rate”. CI was calculated with the CompuSyn software (ComboSyn, Inc. Paramus, NJ 2007).39 A CI < 1.0 was considered as synergy.40
Antibody and immunoblot
Whole cell extracts in RIPA buffer with proteasome inhibitors were isolated for SDS-PAGE and used for the western blot analysis. Anti- p53, caspase-7, and caspase-9 antibodies were ordered from BD Pharmingen (San Diego, CA). Antibodies against total and phosphorylated H2AX, p21, and GAPDH were purchased from Abcam Inc. (Cambridge, MA). Antibodies against PARP and GSTP1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-LC3 polyclonal antibody was ordered from Novus Biological Inc. (Littleton, CO).
Analysis with fluorescent microscope
UMSCC10A cells were transduced with pcDNA3/GFP-LC3 for 24 h and exposed to PL and/or APR-246 in the presence or absence of 3-MA.13,53 Autophagy was determined by calculating GFP-LC3 dots (autophagosomes/autolysosomes) with the Olympus IX51 fluorescence microscope (Center Valley, PA, USA).
Measurement of ROS
One hundred ng/ml dihydroethidium (DHE) was added to cell culture medium at one hour before the cells were trypsinized and collected. Cells were then washed with 1 × PBS for three times and analyzed by flow cytometry.41
Measurement of GSH, GSSG, and GSTP1
Clarified supernatants from lysates of 2 × 105 cells were isolated for the measurement of GST enzymatic activity, GSH, and GSSG. GST enzymatic activity was determined with a GST Activity Assay Kit from Abcam (ab65326). GSH and GSSG were measured with a GSH/GSSG Ratio Detection Assay Kit (ab138881).
Comet assay
Alkaline comet assay was performed as described previously.41 After the treatment, cells were collected and run in alkaline buffer for 20 min and analyzed with the Olympus IX51 fluorescence microscope.
Detection of 8-oxo-dG
Cells were cultured in serum-free and phenol-red-free medium in the presence or absence of PL and/or APR-246 for 2 h. Cells were fixed with absolute methanol for 20 minutes in - 20°C, After permeabilization, cells were washed with 1 × PBS for three times and incubated with FITC-conjugated avidin for 1 h. Fluorescence was captured with the Olympus IX51 fluorescence microscope.41
Xenograft tumor model
SCID mice were housed in Huanhu Hospital (Tianjin, China) animal barrier facility. The animal protocol was approved by the IACUC committee of the Hospital and Jilin University, China. Early-passage UMSCC10A cells were harvested and mixed 2:1 in matrigel (BD Biosciences). Five million cells were injected subcutaneously into the flank of SCID mice. Both male and female mice were used (n= 20 for each sex). Three days later, the mice were randomized into treatments (PL, APR-246, or PL + APR-246) or control PBS groups (10 mice / group). Four mg/kg/day of PL46 and/or 100 mg/kg/day of APR-246 54 dissolved in 200 μl PBS were applied by i.p. on day 4 to day 27. Xenograft tumors were measured every three days with calipers. Tumor volumes were calculated based on the formula: a2 × b × 0.4. In the formula, “a” is the smallest diameter of the tumor, whereas “b” is the diameter perpendicular to “a”. At the end of 4 weeks, mice were euthanized and tumors were removed for the histological analysis.
HNSCC tissues and immunohistochemistry
HNSCC tissues were obtained from Department of Otorhinolaryngology Head and Neck Surgery, Tianjin Huanhu Hospital (Tianjin, China) and from US Biomax Inc. (Rockville, MD) (tissue microarray slides #LP801 and T161). Tissues from healthy control (n = 28) and carcinoma (n = 194) were used for the IHC analysis.42 Staining intensity was graded as “−” (negative), “+” (weak), “++” (moderate), and “+++” (strong). Samples scored as “−” or “+” were categorized as low expression and those scored as “++” or “+++” as high expression. Slides were reviewed by 3 pathologists. Data from slides that different interpretations were made for the staining intensity by the pathologists were not included in the analysis. For calculation of the H score in IHC staining, we followed the method we reported previously.42,55
Statistical analysis
At least three independent experiments were performed in triplicate. Data were expressed as the means ± SD. Student's t-test and two-way ANOVA test were used for the statistical analysis. Differences were considered statistically significant if P value < 0.05.
Supplementary Material
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (Grant Nos: 81573087, 81572139, and 81772924) and the U.S. National Institutes of Health (Grant No:R01CA133053).
Footnotes
Conflict of Interest: The authors have no conflict of interest to report.
References
- 1.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 2.Cosenza M, Civallero M, Marcheselli L, Sacchi S, Pozzi S. Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis. 2017;22:827–840. doi: 10.1007/s10495-017-1364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer Cell. 2016;30:940–952. doi: 10.1016/j.ccell.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang BK, Langford TF, Sikes HD. Using Sensors and Generators of H2O2 to Elucidate the Toxicity Mechanism of Piperlongumine and Phenethyl Isothiocyanate. Antioxid Redox Signal. 2016;24:924–938. doi: 10.1089/ars.2015.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jutooru I, Guthrie AS, Chadalapaka G, Pathi S, Kim K, Burghardt R, et al. Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol Cell Biol. 2014;34:2382–2395. doi: 10.1128/MCB.01602-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan JK, Then SM, Mazlan M, Jamal R, Ngah WZ. Vitamin E, γ-tocotrienol, Protects Against Buthionine Sulfoximine-Induced Cell Death by Scavenging Free Radicals in SH-SY5Y Neuroblastoma Cells. Nutr Cancer. 2016;68:507–517. doi: 10.1080/01635581.2016.1153671. [DOI] [PubMed] [Google Scholar]
- 7.Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014;92:90–101. doi: 10.1016/j.bcp.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 8.Gersey ZC, Rodriguez GA, Barbarite E, Sanchez A, Walters WM, Ohaeto KC, et al. Curcumin decreases malignant characteristics of glioblastoma stem cells via induction of reactive oxygen species. BMC Cancer. 2017;17:99. doi: 10.1186/s12885-017-3058-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim EH, Deng CX, Sporn MB, Liby KT. CDDO-imidazolide induces DNA damage, G2/M arrest and apoptosis in BRCA1-mutated breast cancer cells. Cancer Prev Res (Phila) 2011;4:425–434. doi: 10.1158/1940-6207.CAPR-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satoh T, McKercher SR, Lipton SA. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free RadicBiol Med. 2013;65:645–657. doi: 10.1016/j.freeradbiomed.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasad S, Tyagi AK. Historical Spice as a Future Drug: Therapeutic Potential of Piperlongumine. Curr Pharm Des. 2016;22:4151–4159. doi: 10.2174/1381612822666160601103027. [DOI] [PubMed] [Google Scholar]
- 12.Pignanelli C, Ma D, Noel M, Ropat J, Mansour F, Curran C, et al. Selective Targeting of Cancer Cells by Oxidative Vulnerabilities with Novel Curcumin Analogs. Sci Rep. 2017;7:1105. doi: 10.1038/s41598-017-01230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Wang JW, Xiao X, Shan Y, Xue B, Jiang G, et al. Piperlongumine induces autophagy by targeting p38 signaling. Cell Death Dis. 2013;4:e824. doi: 10.1038/cddis.2013.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KS, Kim JA, Eom SY, Lee SH, Min KR, Kim Y. Inhibitory effect of piperlonguminine on melanin production in melanoma B16 cell line by downregulation of tyrosinase expression. Pigment Cell Res. 2006;19:90–98. doi: 10.1111/j.1600-0749.2005.00281.x. [DOI] [PubMed] [Google Scholar]
- 15.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Adams DJ, Dai M, Pellegrino G, Wagner BK, Stern AM, Shamji AF, et al. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc Natl Acad Sci U S A. 2012;109:15115–15120. doi: 10.1073/pnas.1212802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bezerra DP, Pessoa C, de Moraes MO, Saker-Neto N, Silveira ER, Costa-Lotufo LV. Overview of the therapeutic potential of piplartine (piperlongumine) Eur J Pharm Sci. 2013;48:453–463. doi: 10.1016/j.ejps.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Roh JL, Kim EH, Park JY, Kim JW, Kwon M, Lee BH. Piperlongumine selectively kills cancer cells and increases cisplatin antitumor activity in head and neck cancer. Oncotarget. 2014;5:9227–9238. doi: 10.18632/oncotarget.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bharadwaj U, Eckols TK, Kolosov M, Kasembeli MM, Adam A, Torres D, et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene. 2015;34:1341–1353. doi: 10.1038/onc.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel K, Chowdhury N, Doddapaneni R, Boakye CH, Godugu C, Singh M. Piperlongumine for Enhancing Oral Bioavailability and Cytotoxicity of Docetaxel in Triple-Negative Breast Cancer. J Pharm Sci. 2015;104:4417–4426. doi: 10.1002/jps.24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basak D, Punganuru SR, Srivenugopal KS. Piperlongumine exerts cytotoxic effects against cancer cells with mutant p53 proteins at least in part by restoring the biological functions of the tumor suppressor. Int J Oncol. 2016;48:1426–1436. doi: 10.3892/ijo.2016.3372. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Wu X, Zhou Y, Jiang H, Pan S, Sun B. Piperlongumine Suppresses Growth and Sensitizes Pancreatic Tumors to Gemcitabine in a Xenograft Mouse Model by Modulating the NF-kappa B Pathway. Cancer Prev Res (Phila) 2016;9:234–244. doi: 10.1158/1940-6207.CAPR-15-0306. [DOI] [PubMed] [Google Scholar]
- 23.Alpay M, Yurdakok-Dikmen B, Kismali G, Sel T. Antileukemic effects of piperlongumine and alpha lipoic acid combination on Jurkat, MEC1 and NB4 cells in vitro. J Cancer Res Ther. 2016;12:556–560. doi: 10.4103/0973-1482.151936. [DOI] [PubMed] [Google Scholar]
- 24.de Lima Moreira F, Habenschus MD, Barth T, Marques LM, Pilon AC, da Silva Bolzani V, et al. Metabolic profile and safety of piperlongumine. Sci Rep. 2016;6:33646. doi: 10.1038/srep33646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meegan MJ, Nathwani S, Twamley B, Zisterer DM, O'Boyle NM. Piperlongumine (piplartine) and analogues: Antiproliferative microtubule-destabilising agents. Eur J Med Chem. 2017;125:453–463. doi: 10.1016/j.ejmech.2016.09.048. [DOI] [PubMed] [Google Scholar]
- 26.Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014;588:2622–2627. doi: 10.1016/j.febslet.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 27.Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2010;29:1329–1338. doi: 10.1038/onc.2009.425. [DOI] [PubMed] [Google Scholar]
- 28.Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front Oncol. 2016;6:21. doi: 10.3389/fonc.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 30.Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8:8921–8946. doi: 10.18632/oncotarget.13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, et al. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013;4:e881. doi: 10.1038/cddis.2013.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shalom-Feuerstein R, Serror L, Aberdam E, Müller FJ, van Bokhoven H, Wiman KG, et al. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc Natl Acad Sci U S A. 2013;110:2152–2156. doi: 10.1073/pnas.1201753109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deben C, Lardon F, Wouters A, Op de Beeck K, Van den Bossche J, Jacobs J, et al. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016;375:313–322. doi: 10.1016/j.canlet.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 35.Tessoulin B, Descamps G, Moreau P, Maïga S, Lodé L, Godon C, et al. PRIMA-1Met induces myeloma cell death independent of p53by impairing the GSH/ROS balance. Blood. 2014;124:1626–1636. doi: 10.1182/blood-2014-01-548800. [DOI] [PubMed] [Google Scholar]
- 36.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell. 2014;54:960–974. doi: 10.1016/j.molcel.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou TC, Martin N. ComboSyn, Inc; Paramus, NJ: 2007. CompuSyn software for drug combinations and for general dose effect analysis, and user's guide. [ www.combosyn.com] [Google Scholar]
- 40.Tanaka N, Patel AA, Wang J, Frederick MJ, Kalu NN, Zhao M, et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAPAntiapoptotic Proteins. Clin Cancer Res. 2015;21:4831–4844. doi: 10.1158/1078-0432.CCR-15-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu HG, Zhai YX, Chen J, Lu Y, Wang JW, Quan CS, et al. LKB1 reduces ROS-mediated cell damage via activation of p38. Oncogene. 2015;34:3848–3859. doi: 10.1038/onc.2014.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng Q, Zhao RX, Chen J, Li Y, Li XD, Liu XL, et al. O-linked GlcNAcylation elevated by HPV E6 mediates viral oncogenesis. Proc Natl AcadSci U S A. 2016;113:9333–9338. doi: 10.1073/pnas.1606801113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzgerald AL, Osman AA, Xie TX, Patel A, Skinner H, Sandulache V, et al. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 2015;6:e1678. doi: 10.1038/cddis.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol. 2014;25:23–32. doi: 10.1016/j.semcancer.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Wang HB, Jin XL, Zheng JF, Wang F, Dai F, Zhou B. Developing piperlongumine-directed glutathione S-transferase inhibitors by an electrophilicity-based strategy. Eur J Med Chem. 2017;126:517–525. doi: 10.1016/j.ejmech.2016.11.034. [DOI] [PubMed] [Google Scholar]
- 46.Zou P, Xia Y, Ji J, Chen W, Zhang J, Chen X, et al. Piperlongumine as a direct TrxR1 inhibitor with suppressive activity against gastric cancer. Cancer Lett. 2016;375:114–126. doi: 10.1016/j.canlet.2016.02.058. [DOI] [PubMed] [Google Scholar]
- 47.Harshbarger W, Gondi S, Ficarro SB, Hunter J, Udayakumar D, Gurbani D, et al. Structural and Biochemical Analyses Reveal the Mechanism of Glutathione S-Transferase Pi 1 Inhibition by the Anti-cancer Compound Piperlongumine. J BiolChem. 2017;292:112–120. doi: 10.1074/jbc.M116.750299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bentz BG, Haines GK, 3rd, Radosevich JA. Glutathione S-transferase pi in squamous cell carcinoma of the head and neck. Laryngoscope. 2000;110:1642–1647. doi: 10.1097/00005537-200010000-00013. [DOI] [PubMed] [Google Scholar]
- 49.Singh M, Shah PP, Singh AP, Ruwali M, Mathur N, Pant MC, et al. Association of genetic polymorphisms in glutathione S-transferases and susceptibility to head and neck cancer. Mutat Res. 2008;638:184–194. doi: 10.1016/j.mrfmmm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Xi Y, Gao H, Callaghan MU, Fribley AM, Garshott DM, Xu ZX, et al. Induction of BCL2-Interacting Killer, BIK, is Mediated for Anti-Cancer Activity of Curcumin in Human Head and Neck Squamous Cell Carcinoma Cells. J Cancer. 2015;6:327–332. doi: 10.7150/jca.11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Junk DJ, Vrba L, Watts GS, Oshiro MM, Martinez JD, Futscher BW. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia. 2008;10:450–461. doi: 10.1593/neo.08120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu ZX, Liang J, Haridas V, Gaikwad A, Connolly FP, Mills GB, et al. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell Death Differ. 2007;14:1948–1957. doi: 10.1038/sj.cdd.4402207. [DOI] [PubMed] [Google Scholar]
- 54.Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene. 2005;24:3484–3491. doi: 10.1038/sj.onc.1208419. [DOI] [PubMed] [Google Scholar]
- 55.Zeng Q, Chen J, Li Y, Werle KD, Zhao RX, Quan CS, et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene. 2017;36:1245–1255. doi: 10.1038/onc.2016.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.