

Abstract
Whole-genome clustering of the two available genome sequences of Helicobacter pylori strains 26695 and J99 allows the detection of 110 and 52 strain-specific genes, respectively. This set of strain-specific genes was compared with the sets obtained with other computational approaches of direct genome comparison as well as experimental data from microarray analysis. A considerable number of novel function assignments is possible using database-driven sequence annotation, although the function of the majority of the identified genes remains unknown. Using whole-genome clustering, it is also possible to detect species-specific genes by comparing the two H.pylori strains against the genome sequence of Campylobacter jejuni. It is interesting that the majority of strain-specific genes appear to be species specific. Finally, we introduce a novel approach to gene position analysis by employing measures from directional statistics. We show that although the two strains exhibit differences with respect to strain-specific gene distributions, this is due to the extensive genome rearrangements. If these are taken into account, a common pattern for the genome dynamics of the two Helicobacter strains emerges, suggestive of certain spatial constraints that may act as control mechanisms of gene flux.
INTRODUCTION
About 20 years ago, a spirally shaped Gram-negative bacterial species, later to be known as Helicobacter pylori, was isolated from human gastric tissue (1). This organism, originally associated with peptic ulcer disease, is now also linked to a range of other diseases and disorders including gastric lymphoma of mucosa-associated lymphoid tissue (MALT) and adenocarcinoma of the stomach (2). It is estimated that at least half of the world population is chronically infected, although only a small percentage of the infected people develop symptoms. Human colonisation, pathogenicity and the evolution of infection all depend on strain diversity and host–bacterium interactions. For instance, there appears to be a strong correlation between disease and the presence of the cag pathogenicity island (PAI) (3,4). In addition, various virulence factors have a characteristic strain distribution (2), and some genes—like the cytotoxin encoding vacA gene (5)—clearly display a mosaic structure.
In view of the organism’s natural competence (6), such strain diversity may be partly accounted for by horizontal gene transfer. However, H.pylori resides in the stomach lumen for decades with very few or no co-inhabitant bacteria, except for transient bacteria, including other superinfecting H.pylori strains, which are not necessarily equipped or adapted to survive in the hostile environment. Due to this genetic isolation, interspecific gene transfer is probably quite infrequent. Not surprisingly, H.pylori has developed specialised intrastrain mechanisms to adapt swiftly to changes in the gastric environment, including increased mutation rates, slipped-strand synthesis, phase-variation, and plasmid- or transposon-mediated recombination. The overall result is a pool of genetic variants derived from a single ancestral strain within the same host (7). Such variations may allow migrational colonisation towards other compartments of the stomach, or may contribute to the spread and adaptive success in other hosts. From a medical viewpoint, it is important to understand the genetic drift in H.pylori populations and to investigate which genes are most likely involved in the development and final outcome of disease.
With the complete genome sequence of two H.pylori strains available (8,9), it has become feasible to identify strain-specific genes in silico. The computational approach we followed was based on whole-genome sequence clustering, resulting in the identification of 162 strain-specific genes. These genes were also compared against the full protein sequence database in order to detect homologies suggestive of possible function. Finally, these genes were subjected to positional analysis using directional statistics to assess the significance of the observed distribution patterns across the two strains. All our results are available on the World Wide Web (see data availability in Materials and Methods).
MATERIALS AND METHODS
Source of protein sequences
The protein sequences encoded in the entire genome for H.pylori strains J99 (8) and 26695 (9) were obtained from the corresponding web sites. These genome files contained 1495 and 1577 protein sequences for strains J99 and 26695, respectively. The Campylobacter jejuni (10) genome file, containing 1634 protein sequences, was downloaded from the Sanger Centre. Due to subsequent possible updates and in order to ensure reproducibility, we provide the original sequence files in FASTA format on our web site.
Identification of strain-specific genes by sequence clustering
We approach the problem of strain-specific gene identification as a clustering problem. Instead of performing a genome-to-genome comparison, we mix the two genomes into one dataset and detect clusters present in one strain only. To perform clustering, we have used the GeneRAGE algorithm, which allows automatic classification of protein sequence sets into families according to sequence similarity (11). The algorithm constructs a binary matrix holding all similarity relationships from an all-against-all protein sequence comparison performed with BLAST (v2.0) (12) and using the CAST algorithm (13) to filter sequences for low-complexity regions. The matrix is then processed for symmetry and transitivity relationships, using successive rounds of the Smith–Waterman dynamic programming algorithm (14). In this way, false relationships within the matrix as well as multi-domain protein families are detected (11). We have used a cut-off E-value threshold of 10–10 for all BLAST comparisons and the default Z-score values for symmetrification and multi-domain detection (10 and 3, respectively) (11).
From the resulting list of clusters, strain-specific genes were selected as those genes that are members of clusters which span one strain only, i.e. genes of single-member clusters or genes in multiple-member clusters with similarity to other genes within the same strain only. By definition, and due to the precisely specified protocol which includes multi-domain detection, proteins that share a domain with another protein across strains are not considered as strain specific. Similar procedures were followed for the comparison of H.pylori with C.jejuni, the only difference being the extension of strain-specific to a species-specific analysis. The total time that is necessary to fully cluster any genome pair of this size with GeneRAGE takes <5 h on a 4-CPU Sun Enterprise Server E450 with 2 GB of memory.
Large-scale annotation of strain-specific genes
We have analysed all 162 H.pylori strain-specific genes identified in this study with the GeneQuiz system (15). This system performs automatic functional annotation of large sets of genes, including whole genomes (16). The functional assignment is based on sequence similarity to protein sequence database entries and extraction of the most appropriate functional descriptions. The reliability of functional transfer with GeneQuiz is considered high when the query sequence is similar to a database entry with a BLAST E-value <10–10 or a FASTA score >130 (‘clear’ assignments) (15). We only considered ‘clear’ assignments in this analysis.
Positional analysis of strain-specific genes
For both H.pylori genomes, gene positions were mapped to the unit vector ![]() with polar angle θ = 2πx/L, where x is the mid-point position of each gene and L the length of the genome under consideration. Strain-specific genes were positioned clockwise along circular maps according to these polar coordinates. The two genome maps were aligned to each other with respect to the first gene of each genome (jhp0001 and HP0001), both positioned at ‘twelve o’clock’ (Fig. 1). Gene positional analysis was performed using circular statistics (17). For a given set of n gene positions, the trigonometric mean
with polar angle θ = 2πx/L, where x is the mid-point position of each gene and L the length of the genome under consideration. Strain-specific genes were positioned clockwise along circular maps according to these polar coordinates. The two genome maps were aligned to each other with respect to the first gene of each genome (jhp0001 and HP0001), both positioned at ‘twelve o’clock’ (Fig. 1). Gene positional analysis was performed using circular statistics (17). For a given set of n gene positions, the trigonometric mean ![]() can be defined as the arithmetic mean of their vectorial representation:
can be defined as the arithmetic mean of their vectorial representation:
Figure 1.

Distribution of strain-specific genes in the two H.pylori genome sequences. The outer circle represents the genome of strain 26695 (genes depicted by blue circles); the inner circle represents the genome of strain J99 (genes depicted by red circles). Gene identifiers are given. The inversion region between the two genomes is marked by triangles (green triangles indicate the eda-ftsZ markers and orange triangles indicate the pepF-lepA markers). The larger proportion of strain-specific genes within the inversion region is clearly visible (sets of strain-specific genes in these regions are listed in boxed areas).
![]() 1
1
The mean direction ![]() , then, is simply defined as the polar angle of the trigonometric mean, e.g.
, then, is simply defined as the polar angle of the trigonometric mean, e.g. ![]() , and may be regarded as equivalent to the well-known arithmetic mean value. However, for uniformly distributed data and symmetrical distributions (i.e.
, and may be regarded as equivalent to the well-known arithmetic mean value. However, for uniformly distributed data and symmetrical distributions (i.e. ![]() approaches
approaches ![]() ), the mean direction is undefined. Consequently, we perform a statistical test for uniformity to assess the significance of the mean direction
), the mean direction is undefined. Consequently, we perform a statistical test for uniformity to assess the significance of the mean direction ![]() with respect to the data distribution. This is done by introducing the mean resultant length
with respect to the data distribution. This is done by introducing the mean resultant length ![]() , defined as the norm of the trigonometric mean
, defined as the norm of the trigonometric mean ![]() . From equation 1, it follows that
. From equation 1, it follows that ![]() is bounded between 0 and 1; for genes confined to a small segment of the genome,
is bounded between 0 and 1; for genes confined to a small segment of the genome, ![]() becomes large, eventually approaching 1 for highly concentrated (e.g. closely linked or adjacent) genes, whereas
becomes large, eventually approaching 1 for highly concentrated (e.g. closely linked or adjacent) genes, whereas ![]() approaches 0 with increasing uniformity (or high symmetry) of gene distribution. The probability of uniformity,
approaches 0 with increasing uniformity (or high symmetry) of gene distribution. The probability of uniformity, ![]() , can be described using the following approximation:
, can be described using the following approximation:
![]() 2
2
with n the number of gene positions. For most applications, this approximation is sufficiently accurate, even for a small sample size (n > 30) (17).
The above description on how circular statistics can be used to define the mean direction of data and the assessment of its significance is somewhat limited, as it does not take into account multiple modes in the data distribution (i.e. in describing the data by its mean direction only, the data are implicitly considered to follow a unimodal distribution). In order to give a more precise description, we decided to decompose the data as a mixture of unimodal distributions (17). In this approach, each of the putative modes are approximated by a von Mises distribution, which is the most commonly used unimodal model for circular data and comparable with the Gaussian distribution for linear statitistics (17). Each of the von Mises distributions ![]() depends on two parameters, a mean direction µ and a dispersion factor σ, and has a weight ω in the mixture.
depends on two parameters, a mean direction µ and a dispersion factor σ, and has a weight ω in the mixture.
For a given number of modes k, we estimate the 3k parameters of a mixture of k von Mises (kVM) distributions by minimising the quadratic distance d2 between the cumulative distribution function (cdf) of the data and the mixture:
![]() 3
3
with θj sorted in increasing order and F being the cdf of the mixture: 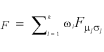 with
with ![]() . The minimisation is done using a modified downhill simplex method (18). The goodness-of-fit (gof) U2 is then calculated as:
. The minimisation is done using a modified downhill simplex method (18). The goodness-of-fit (gof) U2 is then calculated as:
![]() 4
4
where ![]() is the arithmetic mean of
is the arithmetic mean of ![]() (17). Then, using the kVM model with the estimated parameters for the data, we generate Nb parametric bootstrap samples Bi of same size n as the original gene set. For each sample, we perform the full parameter estimation and calculate the corresponding gof, U2Bi. The significance probability of the fit PVM is then simply estimated by comparing the gof obtained for the data with the gofs obtained for the bootstrap samples:
(17). Then, using the kVM model with the estimated parameters for the data, we generate Nb parametric bootstrap samples Bi of same size n as the original gene set. For each sample, we perform the full parameter estimation and calculate the corresponding gof, U2Bi. The significance probability of the fit PVM is then simply estimated by comparing the gof obtained for the data with the gofs obtained for the bootstrap samples:
![]() 5
5
where NU2 is the number of bootstrap samples for which the gof is larger then U2 (U2Bi > U2).
Finally, the gene distributions from the two genomes are compared in order to test whether their circular distributions are identical. We use Kuipers’s test, which is a modification of the Kolmogorov–Smirnov (K–S) test (17,18). Kuiper’s test is particularly well suited for data on circular genomes, because the arbitrarily assigned zero position does not affect the result of the test while the directional nature of gene positions is preserved. Kuiper’s V statistic is defined as follows:
![]()
6
where F1 and F2 are the cdfs of the two distributions being compared. Finally, we compute the probability PK to reject the null hypothesis that the two independent data sets are drawn from the same distribution using the following formula based on the asymptotic (n → + ∞) distribution of V (18):
![]() 7
7
Availability of data and results
Lists of H.pylori strain-specific genes, raw data, Perl scripts, and the results of our comparative analyses (BLAST runs, GeneRAGE clustering and functional annotations) are available on the World Wide Web (http://www.ebi.ac.uk/research/cgg/annotation/hpy/).
RESULTS AND DISCUSSION
Genome dynamics of H.pylori
The unrelated H.pylori strains 26695 and J99, both isolated from patients with gastritis, were sequenced by two independent groups (8,9, respectively). The availability of the genome sequences for the two strains has enabled many research groups to study the plasticity and genome dynamics of H.pylori. Both genomic sequences harbour the cag PAI but they display distinct vacA cytotoxin alleles, and the patient from which strain J99 was isolated had a duodenal ulcer. Ten segments in the J99 genome harbour DNA rearrangements, including a large inversion of 83 kb, and up to 7% of the genes are specific to each strain (8). The two strains display considerable difference in DNA sequence at the third codon position (3C), consistent with the view of extensive nucleotide variance as seen by genotyping methods (19,20). The 3C difference is mainly due to synonymous substitutions (21) and protein sequences in strains 26695 and J99 are relatively conserved, albeit that divergence at the protein level is still higher than that observed in other bacteria (22,23). Overall, the two strains are quite similar in genome organisation (8,24) and display virtually identical metabolic capacities (25,26). Also, they share 42 restriction–modification (R–M) systems with high homology (27), although different sets of these R–M genes are functionally active in each strain, with <30% of the type II R–M systems fully functional (28,29). In contrast, all strain-specific R–M genes are active (28,29), which is in agreement with the concept that H.pylori strain-specific genes may have been acquired through recent horizontal gene transfer and were selected for a specific function. The fact that many R–M genes are inactive, however, also suggests that, once acquired, they are easily lost through mutation. This and other mechanisms such as the mosaicity of the vacA locus (5) and the recombinational loss and gain of cag genes during chronic infection (7) point to a highly dynamic population structure in which descendants of the ancestral ‘founder’ strain (i.e. the original incoming strain) may evolve separately in different niches of the stomach where they encounter slightly different selection pressures during long-time colonisation (for instance pH fluctuations, or altered immune responses). In this respect, one should keep in mind that the H.pylori genome sequence merely represents the genetic signature of a strain at a given timepoint, thus only a fraction of all possible strain-specific genes within a population can be computationally determined. In addition, factors other than gene specificity, such as subtle gene dosage differences or drift in intergenic regions, may also contribute to strain-specific pathogenic properties. Nonetheless, the rigorous identification of strain-specific genes using sequence analysis algorithms can further our understanding of the biology and pathogenicity of H.pylori.
Identification of strain-specific genes in strains 26695 and J99
Using the procedures outlined in the Materials and Methods, a total of 161 clusters that contained one or more proteins of one strain only were finally retained. All but one of these clusters contained a single protein sequence (one cluster contained two paralogous 26695 sequences, HP0436 and HP0987). Thus, 110 strain-specific genes for strain 26695 and 52 genes for strain J99 were identified, bringing the total to 162 genes. This is slightly lower compared with the number of strain-specific genes reported by Alm et al. (8), who reported 117 genes for strain 26695 and 89 genes for strain J99 as strain specific, 206 genes in total. This discrepancy is most probably due to the different methodological approaches: in contrast to previous work (8), we have performed full clustering of the two genomes and identified the strain-specific genes within the merged dataset (see Materials and Methods). For the initial BLAST comparison, we have used the same cut-off E-value threshold of 10–10 to ensure that the results are comparable (8). Unfortunately, a list of 26695 or J99 strain-specific genes has not yet been published, but a comparative interactive database for both genomes is available (http://scriabin.astrazeneca-boston.com/hpylori/). By browsing this database gene by gene, we were able to confirm the strain specificity for all the genes identified by full clustering, except for four very short peptides (HP0359, HP0560, jhp0507 and jhp1021). These peptides are pairwise (HP0359 versus jhp1021 and HP0560 versus jhp0507) highly similar in sequence (>95% identity) and length (21–26 amino acids, respectively), having been incorrectly identified as strain specific, because of mutual scores (E-values) higher than the threshold value. Presumably, the BLAST operation on such short peptide sequences cannot be interpreted correctly and we consider these two pairs as false negatives. The only other exception consisted of a pair of small proteins that are very rich in histidine and glutamine residues (HP1432 and jhp1321). In our analysis, these two genes were retained as singletons (hence, they were given a strain-specific status) because their low-complexity regions (H- and Q-tracks)—spanning the entire length of the protein—were filtered out by CAST (see Materials and Methods).
It is possible that some of these strain-specific genes may not be protein coding. In fact, a significant number of them have lengths <100 residues, whereas the two strains exhibit a marked difference of length distributions for the identified genes (Fig. 2A). The two strains have similar distributions of strain-specific gene runs, with the three longest runs (two of eight genes and one of nine genes) present in 26695 (Fig. 2B).
Figure 2.

Summary of key features of strain-specific genes in H.pylori. (A) Percentile distribution of gene lengths: colour scale represents gene length intervals, shown in the legend. (B) Frequency distribution of strain-specific gene runs (i.e. ranging from single genes to sets of nine consecutive genes).
Full clustering versus direct genome comparison
We undertook a direct genome-to-genome BLAST analysis and compared the results with those obtained by the GeneRAGE algorithm (11), using the same E-value threshold. Query sequences that did not identify homologues with E-value scores lower than the threshold were considered as strain specific. This was done in a bidirectional manner, i.e. the J99 genome was used as a query against the 26695 genome, and vice versa. This resulted in 113 and 58 strain-specific genes for strains 26695 and J99, respectively, as compared with the 110 and 52 strain-specific genes identified by GeneRAGE (see Materials and Methods and Results above). For instance, gene jhp0050 of strain J99 does not appear to have a homologue in strain 26695 (at the E-value threshold of 10–10), whereas the gene HP0059 identifies jhp0050 (E-value 10–15). This asymmetry is detected by GeneRAGE (11), which then triggers a Smith–Waterman dynamic programming run and generates a highly significant Z-score of 14 (pairwise sequence identity is 23%). In our procedure, this pair is considered as a false negative case by BLAST, which is corrected by a more sensitive comparison method. Another example is the tnpA transposase gene HP0437 of the IS605 element in strain 26695 (Fig. 3), which has four identical homologues in this strain: HP0988, HP998, HP1096 and HP1535 (100% sequence identity). The direct sequence comparison using BLAST yields all the five genes as strain specific, whereas it also detects two more homologous genes HP0414 and HP1008 (with an E-value score of 3 × 10–13 and 27% sequence identity). The latter two genes HP0414 and HP1008 (which are in turn identical to each other and members of the same family of transposases to which HP0437 belongs to) identify gene jhp0827 from strain J99 (E-value score of 10–32 and 95% sequence identity), therefore, allowing the identification of the whole group of genes as non-strain specific. The inconsistent transitivity relationship by BLAST is detected by GeneRAGE (11) (Fig. 3) and subsequently supported by Smith–Waterman analysis (the Z-score between HP0414 and jhp0827 is 12 and considered as highly significant). In short, GeneRAGE appears to be more sensitive in detecting protein families that contain members with weak sequence similarities and, therefore, allows the reduction of the strain-specific genes from 171 (according to BLAST) to just 162 cases.
Figure 3.

Sequence alignment of representative members from three sets of related transposase genes: HP0437 represents set (A), HP0414 represents set (B) and jhp0827 represents set (C) (identifiers of the set members are shown in boxes). The three sets are detected by GeneRAGE as a single cluster, while BLAST fails to detect them as a single family due to the relatively low similarity between sets (A) and (C). For more details, please see text.
Comparison to experimental approaches
This is the first detailed report on the computational identification of strain-specific genes in H.pylori. Although Alm et al. (8) discuss a few strain-specific genes, a list of these genes was not provided. In addition, it is not clear which criteria were used to define a gene as strain specific. Experimental approaches for comparative genomics of H.pylori such as subtractive hybridisation (30) and gel-based proteome analysis (31) are very promising, but not particularly well suited for this particular problem.
Recently, Falkow and co-workers set out to characterise the genetic diversity among 15 H.pylori clinical isolates using a DNA microarray (32). This array contained 1660 PCR-derived probes based on the 26695 and J99 genomic sequences. Great care was taken to design the PCR primers for excluding ORF regions with high sequence similarity to avoid cross-hybridisation between members of the same paralogous gene family. A core of 1281 genes common to all 15 strains could be identified, and between 12 and 18% of each strain genome was found to correspond to strain-specific genes (the actual number may be higher as only 26695 and J99 sequences were used as reference). In detail (http://www.pnas.org/cgi/content/full/97/26/14668/DC1), only seven genes, all from strain 26695, were truly unique and gave a signal for 26695 DNA only, but not for any of the other DNAs. These are HP0335, HP0447–451 and HP0765 (except for the latter gene, we identified these genes as 26695 specific). Furthermore, 86 genes in strain J99 and 130 genes in strain 26695 were considered as being specific for either of these strains (32). The slightly lower numbers we detected (52 and 110, respectively) may be explained by the fact that the microarray study is entirely DNA based. Factors such as the degeneracy of the genetic code and the fact that hybridisation results do not necessarily reflect DNA similarity (e.g. effects of sequence bias or secondary structure formation on probe-to-template hybridisation) should be taken into account. Of the 162 strain-specific genes we identified, 19 were omitted from the microarray study because (i) PCR failed to yield a product, (ii) signals were too weak or (iii) there were too few readings across the 15 strains. Of the remaining 143 genes, 29 were not detected as strain specific with the microarray approach. Because all these genes are very short (55 residues on average), it may have been difficult to find a good probe for these genes. This eventually results in 114 genes that can be independently confirmed as strain specific by an experimental method (out of 143, or 80% of total). Reversely, 57 genes of strain J99 and 44 genes of strain 26695 were detected by the microarray as strain specific, whereas protein sequence clustering detects cross-strain similarities. A likely explanation is that probes where ‘pre-selected’ by excluding ORF regions with high cross-homology (32).
Function prediction
Based on the original annotation of the published genomes by Tomb et al. (9) and Alm et al. (8), the large majority of the 162 H.pylori strain-specific genes identified herein (146 genes, or 90%) code for proteins of yet unknown function. In an attempt to gain more information about their possible functional roles, we processed these genes with the automatic annotation system GeneQuiz (15). This analysis resulted in 30 genes whose function could be predicted with high reliability (‘clear’ assignments, see Materials and Methods). For 16 of these genes, a functional annotation had been given previously (8,9), all confirmed by GeneQuiz. The remaining 14 clear assigments, considered to be ‘putative new findings’, included vapD- and mcrB-related proteins, a protein (encoded by gene jhp0540) that, based on its similarity with capsule biosynthesis genes of Haemophilus influenzae (Hib orf3), Streptococcus pneumoniae (19bR) and C.jejuni (Cj1432c), may be involved in serotype specificity and capsule formation, and a helicase (HP0447) whose gene is located upstream of four strain-specific genes of unknown function (HP0448–51), possibly forming a genetic cassette.
An interesting case is the jhp0928 gene (2231 amino acids) that exhibits extensive similarity (26% amino acid sequence identity, 46% sequence similarity, BLAST E-value score of 10) to methylase gene homologs of the plant pathogens Agrobacterium tumefaciens (TiORF47; 1693 amino acids) and Agrobacterium rhizogenes (RiORF93; 1693 amino acids). The latter two genes are both located on plasmids that are able to introduce bacterial DNA into the chromosome of the host plant and both genes are associated with conjugation. The jhp0928 gene is located in the J99 plasticity zone and is accompanied by eight other strain-specific genes (jhp0924–27, jhp0930, jhp0932–34), none of which have a function assignment. According to the Pfam database (33), the protein encoded by gene jhp0928 contains at least four domains, including one methylase and three helicase domains. Remarkably, the cag PAIs of strains J99 and 26695 also contain six genes that are homologous to A.tumefaciens vir genes (involved in pilus formation and conjugation). It is possible that these cag genes and the jhp0928 gene are remnants of ancient gene transfers.
The hypothesis of frequent gene transfer in H.pylori is further supported by compositional analyses that attempt to identify horizontally transferred genes by taking into account unusual GC content and codon usage (34). A significant proportion (52 genes in total or 32%) of the 162 strain-specific genes have been identified as outliers in such analyses, compared with a much lower average over the entire genome (5%) (34) (data not shown).
The results of the GeneQuiz annotations can be interactively accessed via the World Wide Web (http://jura.ebi. ac.uk:8765/ext-genequiz/).
Identification of species-specific genes
We have also compared the two H.pylori genomes with the recently published genome of the closely related species C.jejuni (10). Using the procedures outlined above, we found 645 genes of strain 26695 and 558 genes of strain J99 to be species specific, i.e. without homologues in C.jejuni. Interestingly, the majority of strain-specific genes (108 of the 110 in strain 26695 and 49 of the 52 strain-specific genes in strain J99) do not have detectable homologues in C.jejuni and can thus be considered as species specific. This observation strengthens the hypothesis that the identified strain-specific genes are not exchanged across different species but appear to be confined within the Helicobacter group. In addition, half of these genes do not have any homologues in the protein database (see section on Function prediction). The remaining five strain-specific genes which do have a homologue in C.jejuni are HP0855 (algI), HP1045 (acoE), jhp0164, jhp0540 and jhp0840 (ackA), with sequence identity levels ranging from 26 to 69% between the two species. This species-specific feature detection is reminiscent of previous work (35). In that study, 594 H.pylori genes were detected as species specific with Escherichia coli and H.influenzae as ‘reference’ species. With the availability of a closest relative, we are able to detect 645 genes (for the same strain 26695), suggesting that the difference arises from stricter criteria of homology (E-value threshold 10–10 compared with 10–02 in that analysis).
Gene position analysis and directional statistics
Positional analysis showed that over half of the strain-specific genes (88 out of 162) were organised in an operonic fashion, with one-quarter (42 of 162) of the genes located on the chromosome as tandem pairs (Fig. 2B). Also, a large number of strain-specific genes are located at the plasticity zones (32 and 44% in 26695 and J99, respectively) (Fig. 1). The term ‘plasticity zone’ has been introduced by Alm et al. (8), who listed 46 and 48% of the 26695 and J99 strains in these regions, respectively. Interestingly, these plasticity zones partly coincide with an inversion between the two genomes (8), as demarcated by the eda-ftsZ and pepF-lepA gene markers (Fig. 1)—i.e. there are two separate plasticity zones in strain 26695.
To investigate whether the strain-specific genes along the two genomes are distributed in a particular fashion (in addition to the apparent grouping in the plasticity zones) and to compare the positional distributions between the two genomes, we undertook a thorough statistical analysis, taking into account the directional nature of gene positions on circular genomes in general and the positions of strain-specific genes in particular.
If there were no positional constraints on strain-specific genes, we would expect them to be uniformly distributed. When we test for uniformity as outlined in the Materials and Methods (equations 1 and 2) the null hypothesis of uniformity is clearly rejected for both genomes, with Pu = 10–91 for strain 26695 and Pu = 10–228 for strain J99, rendering the mean directions of both data sets highly significant (![]() = 424 kb for strain 26695 and
= 424 kb for strain 26695 and ![]() = 1042 kb for J99; see Materials and Methods). Both mean directions point towards the area of the genomes with the highest concentration of strain-specific genes (Fig. 1). Note that if we use the arithmetic mean
= 1042 kb for J99; see Materials and Methods). Both mean directions point towards the area of the genomes with the highest concentration of strain-specific genes (Fig. 1). Note that if we use the arithmetic mean ![]() for gene locations instead of the mean direction we obtain
for gene locations instead of the mean direction we obtain ![]() = 799 kb for strain 26695 and
= 799 kb for strain 26695 and ![]() = 951 kb for strain J99, values not corresponding to any meaningful mean position for the strain-specific gene sets. This non-uniformity is also obvious from the cumulative distribution functions (Fig. 4A).
= 951 kb for strain J99, values not corresponding to any meaningful mean position for the strain-specific gene sets. This non-uniformity is also obvious from the cumulative distribution functions (Fig. 4A).
Figure 4.

Cumulative distribution functions (cdfs) of strain-specific gene positions, measured in (A) kilobase coordinates (kb) and (B) angular distance from the origin of replication (ori)—the possible terminus of replication (ter) is set to 180°. Filled squares represent genes from strain J99 and open circles genes from 26695. (A) The line corresponds to the fitted multimodal models (2VM for J99 and 4VM for 26695, see Materials and Methods and Tables).
However, having highly significant mean directions in circular data does not necessarily guarantee that the data are distributed in a unimodal fashion. To test this, we fitted the data with a von Mises distribution (1VM model) (see Materials and Methods) and computed the corresponding significance probability using 100 bootstrap samples (i.e. Nb = 100). In both cases, the 1VM model is rejected with P1VM < 0.01 (Tables 1 and 2). To obtain acceptable descriptions of the 26695 and J99 strain-specific gene positions, multi-modal models consisting of a mixture of two or more von Mises distributions are needed (Tables 1 and 2).
Table 1. Decomposition of the H.pylori J99 strain-specific genes as a mixture of kVM distributions.
| Model |
gof |
P |
µ1 |
σ1 |
ω1 |
µ1 |
σ2 |
ω2 |
| 1VM | 0.334 | <0.01 | 1014 (±36) | 254 (±32) | 1.00 | – | – | – |
| 2VM | 0.016 | 0.70 | 1040 (±5.6) | 20.6 (±6.3) | 0.42 (±0.08) | 1054 (±361) | 511 (±83) | 0.58 (±0.08) |
For each model, the resulting gof (equation 4) and the significance probability of the fit PVM (equation 5) are given. In addition, the 3k parameters are listed: mean direction µ, a dispersion factor σ and the weight ω of each mode (see Materials and Methods for details). An estimate of the standard deviation of the parameters is also shown. The decomposition procedure is terminated when the probability exceeds 10% (PVM >0.1).
Table 2. Decomposition of the H.pylori 26695 strain-specific genes as a mixture of kVM distributions (details as in Table 1).
| Model |
gof |
P |
µ1 |
σ1 |
ω1 |
µ2 |
σ2 |
ω2 |
µ3 |
σ3 |
ω3 |
µ4 |
σ4 |
ω4 |
| 1VM | 0.22 | <0.01 | 420 (±265) | 598 (±64) | 1.00 | – | – | – | – | – | – | – | – | – |
| 2VM | 0.055 | 0.03 | 457 | 12 | 0.20 | 1353 | 578 | 0.80 | – | – | – | – | – | – |
| 3VM | 0.032 | 0.09 | 459 | 14 | 0.22 | 1050 | 10 | 0.09 | 1566 | 506 | 0.69 | – | – | – |
| 4VM | 0.020 | 0.25 | 325 | 10 | 0.11 | 464 | 14 | 0.24 | 1021 | 202 | 0.37 | 1623 | 166 | 0.28 |
Standard deviations cannot be estimated for certain parameters.
For strain J99, the 2VM model appears to be most suitable to describe the data (P2VM = 0.70), amounting to a very sharp mode centred on µ1 = 1040 kb with an approximate width of 40 kb. The width of a mode can be approximated by multiplying the dispersion parameter σ with a factor of two. For any given mode in this study (σ < 500 kb), this width corresponds to an interval [µ – σ, µ + σ] containing 75% of the data described by that mode. This first mode describes 42% of all data whereas the remaining 58% of the data correspond to another mode that is so wide (>1 Mb) that it actually should be interpreted as a uniform distribution (Table 1). This analysis identifies a very well defined ‘plasticity zone’ within the J99 genome corresponding to 23 strain-specific genes (from jhp0916 to jhp0973).
The situation for strain 26695 is slightly more complex. Increasing the number of modes in the fitting procedure only gradually improves the confidence level of the model (P2VM = 0.03, P3VM = 0.09, P4VM = 0.25). The 4VM decomposition identifies two well-localised modes (with widths of 20 and 28 kb) centred on µ1 = 352 and µ2 = 464 kb. The two other modes, however, are of intermediary width (∼330 and 400 kb), which makes it nearly impossible to assess the precise number of positional modes or to determine the ratio of localised over non-localised (i.e. uniformly distributed) 26695 strain-specific genes.
Additionally, the significance test using Kuiper’s V statistic (see Materials and Methods) clearly rejects the null hypothesis that both sets of strain-specific gene positions have the same distribution (PK < 10–5). Thus, both independent approaches, comparing the distributions of strain-specific genes, suggest that the two strains are considerably different in this respect.
Reconstruction of genome evolution
Many dynamic events such as recombination are responsible for extensive genome shuffling in bacterial evolution (36) even on relatively short timescales. Strain-specific genes provide an interesting snapshot of the evolutionary dynamics of a species and its genome structure. Previously, it has been realised that the two strains actually share a high proportion of common relative loci with one or more strain-specific genes (8) and that ‘(the) H.pylori (genome) may have limited flexibility for containing strain-specific genes’.
Although the use of positional statistics in our approach suggests that the two genomes are very different in terms of strain-specific gene distributions, these measures are strongly affected by genome shuffling, including large inversions such as the aforementioned inversion that splits the 26695 plasticity zone in two separate domains.
Recent work, both experimental (37) and computational (38,39), suggests that the origin of replication (ori) defines a major axis of symmetry in bacterial genome evolution, thus preserving distances to ori. In order to reduce the masking effect that genome rearrangements may have on gene distribution comparisons, we also performed a gene position analysis using the angular distances to the ori (40) rather than the actual position of the strain-specific genes (Fig. 4B). For the purpose of our analysis, we position ori at 1600 kb for strain 26695 and 1557 kb for strain J99 (40). Using this new scheme, the resulting cumulative distribution functions of the two genomes become strikingly similar, and the Kuiper’s test gives PK = 0.11. This value implies that the null hypothesis cannot be rejected and that both gene sets share a common distribution of distances with respect to the origin of replication.
Despite the independent evolutionary history of the two genomes and the resulting differences in genome organisation and content, it is interesting that the positioning of both sets of strain-specific genes, with reference to the origin of replication, follows a highly similar pattern. This suggests a certain degree of rigidity in the H.pylori genome, in which gene transfer takes place in preferred regions, due to either physical constraints or selective pressure. Given the high frequency of inter- and intrastrain gene transfers within H.pylori populations, such spatial constraints may represent mechanisms for the control of gene flux. Conclusive evidence for the existence of such constraints may be obtained from the availability of the genome sequences from additional H.pylori strains.
Computational detection of strain-specific genes
Our approach for the identification and analysis of strain-specific genes is simple, robust and generally applicable. Strain- and species-specific genes are easily detected with whole-genome clustering with high accuracy. Prediction of function is an important step and should be repeatedly performed with more up-to-date databases. Finally, the use of positional statistics assesses the distribution of strain-specific genes (or any other genomic feature) on circular genome sequences.
Several genome projects are now directed towards the sequencing of multiple strains (i.e. Chlamydia pneumoniae, Mycobacterium tuberculosis, E.coli, S.pneumoniae, etc.) and their number is expected to increase rapidly. Experimental methods, such as subtractive hybridisation (30) or whole-genome microarray analysis (32), in which very large numbers of strains can be examined without the need of sequencing, may provide valuable insights into the temporal distribution of strain-specific genes, within and between populations. In addition, computational methods for genome comparison and the detection of strain- and species-specific genes are necessary to address fundamental questions about genome organisation and evolution and to identify previously unknown virulence genes as potential targets for the development of narrow-spectrum antibiotics.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Anton Enright and other members of the Computational Genomics Group for comments. This work was supported by the European Molecular Biology Laboratory and the TMR Programme of the European Commission (DG-XII Science, Research and Development). C.O. thanks the UK Medical Research Council and IBM Research for additional support.
References
- 1.Marshall B.J. and Warren,J.R. (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet, i, 1311–1315. [DOI] [PubMed] [Google Scholar]
- 2.Covacci A., Telford,J.L., Del Giudice,G., Parsonnet,J. and Rappuoli,R. (1999) Helicobacter pylori virulence and genetic geography. Science, 284, 1328–1333. [DOI] [PubMed] [Google Scholar]
- 3.Blaser M.J., Perez-Perez,G.I., Kleanthous,H., Cover,T.L., Peek,R.M., Chyou,P.H., Stemmermann,G.N. and Nomura,A. (1995) Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res., 55, 2111–2115. [PubMed] [Google Scholar]
- 4.Censini S., Lange,C., Xiang,Z., Crabtree,J. E., Ghiara,P., Borodovsky,M., Rappuoli,R. and Covacci,A. (1996) cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl Acad. Sci. USA, 93, 14648–14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atherton J.C., Cao,P., Peek,R.M., Tummuru,M.K., Blaser,M.J. and Cover,T.L. (1995) Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem., 270, 17771–17777. [DOI] [PubMed] [Google Scholar]
- 6.Nedenskov-Sorensen P., Bukholm,G. and Bovre,K. (1990) Natural competence for genetic transformation in Campylobacter pylori. J. Infect. Dis., 161, 365–366. [DOI] [PubMed] [Google Scholar]
- 7.Kuipers E.J., Israel,D.A., Kusters,J.G., Gerrits,M.M., Weel,J., van Der Ende,A., van Der Hulst,R.W., Wirth,H.P., Hook-Nikanne,J., Thompson,S.A. and Blaser,M.J. (2000) Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J. Infect. Dis., 181, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alm R.A., Ling,L.S., Moir,D.T., King,B.L., Brown,E.D., Doig,P.C., Smith,D.R., Noonan,B., Guild,B.C., deJonge,B.L., Carmel,G., Tummino,P.J., Caruso,A., Uria-Nickelsen,M., Mills,D.M., Ives,C., Gibson,R., Merberg,D., Mills,S.D., Jiang,Q., Taylor,D.E., Vovis,G.F. and Trust,T.J. (1999) Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature, 397, 176–180. [DOI] [PubMed] [Google Scholar]
- 9.Tomb J.F., White,O., Kerlavage,A.R., Clayton,R.A., Sutton,G.G., Fleischmann,R.D., Ketchum,K.A., Klenk,H.P., Gill,S., Dougherty,B.A. et al. (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature, 388, 539–547. [DOI] [PubMed] [Google Scholar]
- 10.Parkhill J., Wren,B.W., Mungall,K., Ketley,J.M., Churcher,C., Basham,D., Chillingworth,T., Davies,R.M., Feltwell,T., Holroyd,S., Jagels,K., Karlyshev,A.V., Moule,S., Pallen,M.J., Penn,C.W., Quail,M.A., Rajandream,M.A., Rutherford,K.M., van Vliet,A.H., Whitehead,S. and Barrell,B.G. (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature, 403, 665–668. [DOI] [PubMed] [Google Scholar]
- 11.Enright A.J. and Ouzounis,C.A. (2000) GeneRAGE: a robust algorithm for sequence clustering and domain detection. Bioinformatics, 16, 451–457. [DOI] [PubMed] [Google Scholar]
- 12.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Promponas V.J., Enright,A.J., Tsoka,S., Kreil,D.P., Leroy,C., Hamodrakas,S., Sander,C. and Ouzounis,C.A. (2000) CAST: an iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics, 16, 915–922. [DOI] [PubMed] [Google Scholar]
- 14.Smith T.F. and Waterman,M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197. [DOI] [PubMed] [Google Scholar]
- 15.Andrade M.A., Brown,N.P., Leroy,C., Hoersch,S., de Daruvar,A., Reich,C., Franchini,A., Tamames,J., Valencia,A., Ouzounis,C. and Sander,C. (1999) Automated genome sequence analysis and annotation. Bioinformatics, 15, 391–412. [DOI] [PubMed] [Google Scholar]
- 16.Iliopoulos I., Tsoka,S., Andrade,M.A., Janssen,P., Audit,B., Tramontano,A., Valencia,A., Leroy,C., Sander,C. and Ouzounis,C.A. (2000) Genome sequences and great expectations. Genome Biol., 2, interactions0001.1–0001.3. [http://genomebiology.com/2000/2/1/interactions/0001/]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher N.I. (1993) Statistical Analysis of Circular Data. Cambridge University Press, Cambridge, UK.
- 18.Press W.H., Teukolsky,S.A., Vetterling,W.T. and Flannery,B.P. (1992) Numerical Recipes in C, 2nd Ed. Cambridge University Press, Cambridge, UK.
- 19.Akopyanz N., Bukanov,N.O., Westblom,T.U. and Berg,D.E. (1992) PCR-based RFLP analysis of DNA sequence diversity in the gastric pathogen Helicobacter pylori. Nucleic Acids Res., 20, 6221–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kansau I., Raymond,J., Bingen,E., Courcoux,P., Kalach,N., Bergeret,M., Braimi,N., Dupont,C. and Labigne,A. (1996) Genotyping of Helicobacter pylori isolates by sequencing of PCR products and comparison with the RAPD technique. Res. Microbiol., 147, 661–669. [DOI] [PubMed] [Google Scholar]
- 21.Achtman M., Azuma,T., Berg,D.E., Ito,Y., Morelli,G., Pan,Z.J., Suerbaum,S., Thompson,S.A., van der Ende,A. and van Doorn,L.J. (1999) Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol. Microbiol., 32, 459–470. [DOI] [PubMed] [Google Scholar]
- 22.Suerbaum S., Smith,J.M., Bapumia,K., Morelli,G., Smith,N.H., Kunstmann,E., Dyrek,I. and Achtman,M. (1998) Free recombination within Helicobacter pylori. Proc. Natl Acad. Sci. USA, 95, 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G., Humayun,M.Z. and Taylor,D.E. (1999) Mutation as an origin of genetic variability in Helicobacter pylori. Trends Microbiol ., 7, 488–493. [DOI] [PubMed] [Google Scholar]
- 24.Ge Z. and Taylor,D.E. (1999) Contributions of genome sequencing to understanding the biology of Helicobacter pylori. Annu. Rev. Microbiol., 53, 353–387. [DOI] [PubMed] [Google Scholar]
- 25.Marais A., Mendz,G.L., Hazell,S.L. and Megraud,F. (1999) Metabolism and genetics of Helicobacter pylori: the genome era. Microbiol. Mol. Biol. Rev., 63, 642–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doig P., de Jonge,B.L., Alm,R.A., Brown,E.D., Uria-Nickelsen,M., Noonan,B., Mills,S.D., Tummino,P., Carmel,G., Guild,B.C., Moir,D.T., Vovis,G.F. and Trust,T.J. (1999) Helicobacter pylori physiology predicted from genomic comparison of two strains. Microbiol. Mol. Biol. Rev., 63, 675–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobusato A., Uchiyama,I. and Kobayashi,I. (2000) Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene, 259, 89–98. [DOI] [PubMed] [Google Scholar]
- 28.Kong H., Lin,L.F., Porter,N., Stickel,S., Byrd,D., Posfai,J. and Roberts,R.J. (2000) Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res., 28, 3216–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin L.F., Posfai,J., Roberts,R.J. and Kong,H. (2001) Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc. Natl Acad. Sci. USA, 98, 2740–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akopyants N.S., Fradkov,A., Diatchenko,L., Hill,J.E., Siebert,P.D., Lukyanov,S.A., Sverdlov,E.D. and Berg,D.E. (1998) PCR-based subtractive hybridization and differences in gene content among strains of Helicobacter pylori. Proc. Natl Acad. Sci. USA, 95, 13108–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jungblut P.R., Bumann,D., Haas,G., Zimny-Arndt,U., Holland,P., Lamer,S., Siejak,F., Aebischer,A. and Meyer,T.F. (2000) Comparative proteome analysis of Helicobacter pylori. Mol. Microbiol., 36, 710–725. [DOI] [PubMed] [Google Scholar]
- 32.Salama N., Guillemin,K., McDaniel,T.K., Sherlock,G., Tompkins,L. and Falkow,S. (2000) A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc. Natl Acad. Sci. USA, 97, 14668–14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L. (2000) The Pfam protein families database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Vallve S., Romeu,A. and Palau,J. (2000) Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res., 10, 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huynen M., Dandekar,T. and Bork,P. (1998) Differential genome analysis applied to the species-specific features of Helicobacter pylori. FEBS Lett., 426, 1–5. [DOI] [PubMed] [Google Scholar]
- 36.Casjens S. (1998) The diverse and dynamic structure of bacterial genomes. Annu. Rev. Genet., 32, 339–77. [DOI] [PubMed] [Google Scholar]
- 37.Bergthorsson U. and Ochman,H. (1998) Distribution of chromosome length variation in natural isolates of Escherichia coli. Mol. Biol. Evol., 15, 6–16. [DOI] [PubMed] [Google Scholar]
- 38.Tillier E.R. and Collins,R.A. (2000) Genome rearrangement by replication-directed translocation. Nature Genet., 26, 195–197. [DOI] [PubMed] [Google Scholar]
- 39.Eisen J.A., Heidelberg,J.F., White,O. and Salzberg,S.L. (2000) Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol., 1, research0011.1–0011.9. [http://genomebiology.com/2000/1/6/research/0011/]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grigoriev A. (2000) Graphical genome comparison: rearrangements and replication origin of Helicobacter pylori. Trends Genet., 16, 376–378. [DOI] [PubMed] [Google Scholar]