

Abstract
It is now recognized that asthma incorporates a broad spectrum of syndromes with varying clinical manifestations. Future improvements in asthma treatment will require a clear characterization of these asthma phenotypes and the cellular mechanisms underlying these clinical manifestations. Herein, we will describe the current knowledge of asthma biology. This will include a review of the early pioneers in asthma and allergy, how this work led to our understanding of TH1 and TH2 cytokines, and the development of the “hygiene hypothesis.” We will discuss the utility and limitations of the TH1-TH2 model of asthma in animal and human studies, and how this knowledge addresses controversies surrounding the hygiene hypothesis and other competing models. We will discuss novel therapies that have been developed based on mechanistic understanding of asthma pathobiology, including successes and shortcomings of these therapies. We will review the early work that led to the recognition of “asthma phenotypes.” This will include the early discovery of various inflammatory subtypes in asthma and how these inflammatory subtypes correlate with response to therapy. Finally, we will describe recent discoveries in asthma biology that will include the role of the airway epithelium in asthma pathogenesis, novel cytokines important in asthma that may serve as novel therapeutic targets, and the identification of newly described innate immune cells and their role in asthma. Improved understanding of the complex biology underpinning the various asthma phenotypes is critical for our ability to optimize treatment for all patients that suffer from asthma and critical asthma syndromes.
Keywords: Asthma; TH1, TH2; Historical perspectives; Mechanisms; Asthma phenotypes; Innate lymphoid cells
Historical Perspective
Any understanding of the critical asthma syndrome (CAS) requires an accurate clinical diagnosis and an appreciation of its origins. The modern concept of asthma as an immunologic disorder has its foundations in clinical observations spanning two centuries (Fig. 1). This included early descriptions of “asthmatic sputum” associated with specific cell types, Charcot–Leyden crystals, and Curshmann spirals, inflammation of smaller airways, and paroxysms induced by environmental exposures [1]. In the early 1920s, specific mechanisms for allergic diseases including asthma, allergic rhinitis, and atopic dermatitis were identified to be mediated by serum substances known as reagins. This was first exemplified by the passive transfer of fish hypersensitivity from one individual to another [2]. Carl Prausnitz observed that his colleague Hans Küstner was exceptionally sensitive to cooked fish. To determine if this sensitivity was due to serum factors, he self-administered an intradermal injection of serum from his colleague and subsequently developed a new hypersensitivity to fish at the site of injection [3]. Subsequently, it was shown that this transfer of skin sensitization, the Prausnitz–Küstner reaction, was mediated by a newly identified antibody class, IgE, which mediated hypersensitivity reactions to a wide range of allergens in multiple tissues including the lung [4]. Today, approximately 60 % of asthma is linked to IgE-mediated reactions, and IgE remains one of the best predictors for the development of allergic asthma in humans. Further unpacking of these initial key discoveries has informed our modern understanding of asthma immunology and the importance of IgE in this process.
Fig. 1.
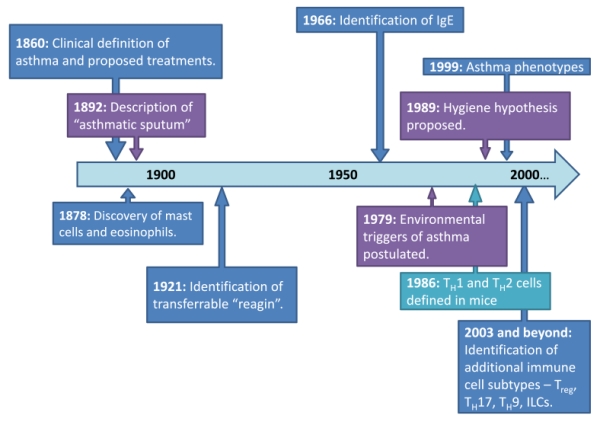
Timeline of asthma discoveries
Another set of groundbreaking discoveries was the identification of specialized lymphocyte populations that were capable of upregulating IgE production. Building on previous work on mouse helper T cells [5–9], Mosmann et al. clearly demonstrated that specific T cell lines had characteristic cytokine (lymphokine) and B cell-stimulating activities independent of the method of stimulation [10]. From 22 TH cell clones, they identified important phenotypic differences in response to antigen that classified each clone into one of two subgroups. One subgroup, dubbed TH1, produced interleukin-2 (IL-2) and interferon-gamma (IFNγ) but did not produce interleukin-4 (IL-4) or enhance IgE production. In contrast, the other subgroup, TH2, produced IL-4 and was able to enhance IgE antibody production, but did not produce IL-2 or IFNγ. This led to the TH1-TH2 paradigm that is well known to us today (Fig. 2).
Fig. 2.
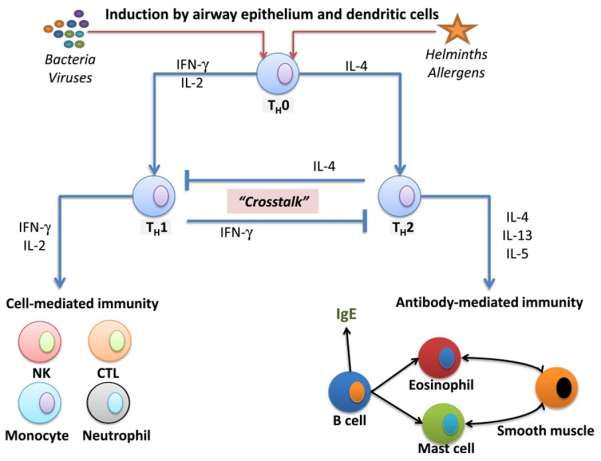
Classic Th1/Th2 paradigm: in response to pathogens and allergens, airway epithelium and dendritic cells promote differentiation of naïve TH0 cells into either TH1 or TH2 T-helper cells. Cytokines produced by TH1 cells (IL-2 and IFN-g) inhibit the differentiation of TH2 cells and activate cell-mediated immunity. This includes the recruitment and activation of natural killer (NK) cells, cytotoxic T lymphocytes (CTL), monocyte/macrophages, and neutrophils. Cytokines produced by TH2 cells include IL-4, IL-5, and IL-13, which will inhibit the differentiation of TH1 cells and activate antibody-producing B cells (IgE), eosinophils, mast cells, basophils, smooth muscle, and fibroblasts. This TH2-skewed response subsequently leads to histamine release, smooth muscle contraction, mucus cell secretion, and fibrosis characteristic of the asthmatic response. Although several features of this model are consistent with clinical data from mild asthmatics, recent evidence suggests several other regulatory factors are the primary targets for critical asthma syndromes (see Box 1)
Together, these findings have provided a framework that has substantially advanced our understanding of asthma today. This includes insights into the pathobiology of asthma and potential therapeutic targets that have proven useful in caring for the majority of patients with mild asthma. Several questions remain, however, including how asthma develops in the first place, the factors that determine asthma severity, the mechanisms responsible for recurrent exacerbations, or why some patients are unresponsive to current asthma medications including the cornerstone of therapy—the corticosteroids.
As we will outline below, recent discoveries belie a complex immune network that demonstrates that the TH1/TH2 paradigm is only part of the story. We will discuss the rationale for therapies that target TH2 cytokines and the limitations to this approach. Also, we will summarize newly described asthma phenotypes and recently discovered cytokines and cell types that potentially address the derangements seen in patients suffering from critical asthma syndromes.
The TH1-TH2 Model of Asthma
In the prevailing model of allergic or IgE-mediated asthma, various environmental exposures (i.e., house dust mite, pollens, animal dander, etc.) trigger airway epithelial cell and dendritic cell interactions that initiate the immune response. Dendritic cells process antigens derived from these environmental exposures and present them to T cells, which initiates a clonal expansion of TH2-type lymphocytes. The clonal expansion of TH2-type cells results in the elaboration of IL-4, IL-13, and IL-5 and class switching of B cells to produce IgE. In addition, TH2 cytokines upregulate mast cell and eosinophil proliferation, and there is subsequent recruitment and retention of these cells in the lung. Activated mast cells and eosinophils produce additional TH2 cytokines that perpetuate airway inflammation (Table 1).
Table 1.
Activated cell types and eosinophils that perpetuate airway inflammation
| Cell type | Products produced | Biologic effect |
|---|---|---|
| Airway epithelium | TSLP, IL-33, IL-25 | Respond to environmental insults through PRRs (see Box 2), activate dendritic cells |
| Dendritic cell | Processed antigen | Migrate to local lymph nodes and present antigen to expand TH2-type T cell population |
| TH2 lymphocyte | IL-4 | Promotes B cell isotype switching, production of eotaxin, induces airway goblet cell metaplasia, amplifies airway epithelial response to environmental insults |
| IL-13 | Shares receptor with IL-4 and has similar biologic effects, may play a more important role in asthma compared to IL-4 | |
| IL-5 | Promotes eosinophil growth, maturation, and activation | |
| B cell | Processed antigen, IgE | Activate T cells to reinforce IgE production, activate mast cells and eosinophils to release preformed products that characterize the early allergic response |
| Mast cell | Histamine, neutral proteases, lipid mediators | Increase vascular permeability and smooth muscle contraction resulting in airway hyperreactivity |
| IL-4, IL-13, IL-5 | Amplifies development and recruitment of mast cells to the lung | |
| Eosinophil | Major basic protein | Toxic to bacteria and helminthes, but damage host cell membranes as well, may increase recruitment of other inflammatory cells |
| IL-4, IL-13, IL-5 | Amplifies development and recruitment of eosinophils to the lung | |
| Airway smooth muscle | RANTES, eotaxin, IL-8, MCP, IL-5, ICAM/VCAM | Promotes activation and recruitment of inflammatory cells including mast cells and eosinophils, may promote matrix deposition and hypertrophy |
Similarly, there is mounting evidence that airway smooth muscle (ASM) cells from asthmatics respond abnormally to allergen. ASM cells primarily regulate airway diameter and bronchomotor tone, and can contribute significantly to airway hyperreactivity (AHR). They have been shown to secrete numerous cytokines capable of recruiting inflammatory cells, particularly mast cells, and it is thought that ASM myositis contributes significantly to the pathogenesis of asthma. Relevant to the TH1/TH2 paradigm, IL-5 appears to alter ASM contractility, and IL-4 receptor polymorphisms in asthmatics potentially alter ASM synthetic function and basement membrane deposition [11, 12]. Reasons for the differential response to otherwise innocuous antigens in asthmatics versus non-asthmatics remain unknown.
At the center of this altered reactivity to antigen, which leads to airway hyperreactivity, obstruction, and tissue damage, is the TH2-type T cell response. Early characterizations of inflammatory cell profiles in humans demonstrated increased numbers of T lymphocytes in the bronchial mucosa of asthmatic patients. Also, there were higher levels of CD4+ cells, airway eosinophils, and eosinophil cationic protein in asthmatics compared with non-asthmatic controls [13]. Subsequent studies in humans supported the concept that allergic individuals express skewed TH2-type inflammation. This included significantly elevated levels of IL-4, IL-13, and IL-5 in bronchoalveolar lavage and tissue biopsies from asthmatic individuals [14–16]. Whether this is directly responsible for the increased risk of acute asthma exacerbations secondary to environmental or infectious agents is uncertain. What is certain is that control of eosinophilic inflammation appears to correlate with better asthma control and a reduction in acute exacerbations [17].
Despite the circumstantial evidence that supports a role of TH2 cytokines and eosinophils in asthma pathogenesis, there was no direct evidence that CD4+ T cells were responsible for antigen-induced asthmatic responses in vivo or how these networks are involved in acute asthma exacerbations in humans. Initial work by Gavett et al. demonstrated that depletion of murine CD4+ T cells prevented AHR and pulmonary eosinophilia in A/J mice sensitized and challenged with sheep red blood cells. Although the model produced a significant degree of neutrophils, CD4+ cell depletion had a profound and specific effect on eosinophilic inflammation in the lung interstitium and bronchoalveolar lavage demonstrating a causal role for the CD4+ T cell [18]. In subsequent studies, deletion of IL-4 gene or neutralizing IL-4 in an ovalbumin (OVA) mouse model of asthma resulted in a reduced eosinophilic infiltration and lower amounts of IL-5 production [19]. Importantly, this work suggested that IL-4 was not responsible for eosinophilic infiltration into the airway, but instead enhanced IL-5 secretion that shifted the immune response to the TH2 phenotype. More recently, IL-13, which shares a receptor subunit with IL-4, has been implicated as the primary TH2 cytokine driving an asthma-like phenotype rather than IL-4 [20]. These findings provided the impetus to create targeted therapies to attenuate this robust TH2 response.
Results from clinical trials using targeted therapies against specific arms of the TH2 response have been mixed [21]. Fulfilling a research goal that first started in the 1920s, targeted therapy against IgE, omalizumab, was available for clinical use in 2002 for patients with poorly controlled disease. Omalizumab is a humanized antibody that prevents interactions of IgE with cells that carry the high-affinity Fcε Receptor I (i.e., mast cells). This prevents antigen-induced IgE bridging and subsequent cell activation that leads to early-phase and late-phase asthmatic responses. Omalizumab is associated with improved asthma symptoms and reduced exacerbations, confirming that targeted therapy against specific arms of the TH2 response is effective in treating some patients with critical asthma. In contrast, two different antibodies against IL-4, pascolizumab and altrakincept, were shown to be effective in blocking IL-4-mediated biologic effects in vitro, but did not improve outcomes in two different asthmatic patient populations. Additional therapeutic targets including targets against newly identified pathways (discussed below) are under active investigation (Fig. 3). The variable responses to targeted therapy reveal the complex immunobiology that underlies CAS. The early successes with targeted therapies indicate we are on the right track. However, continued efforts to fully characterize the multiple pathways that lead to the clinical manifestations of asthma, particularly patients suffering from critical asthma syndromes, is required for us to tailor therapies to each specific patient.
Fig. 3.
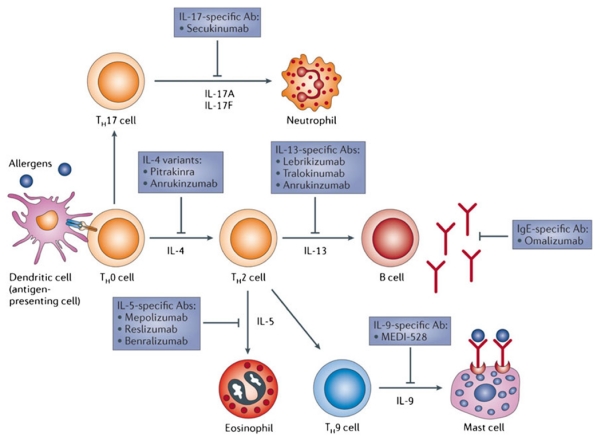
Adapted from Pelaia et al. [21]. Reproduced with permission. Targeted therapies against cytokines important in asthma
Although there is ample evidence that the TH2-type cell and resultant cytokine production is important in the allergic asthma response, it is not clear why atopic individuals have a skewed TH2 response to otherwise innocuous antigen exposure. Genetics undoubtedly play a role and atopic individuals have been recognized to come from allergic families since 1190 A.D. [22]. However, the observed pattern of inheritance suggests that multiple genes and possibly additional environmental exposures (i.e., aeroallergens, air pollutants, or viral infection) are required to manifest atopy [23].
Building on the work of Oscar Frick, Busse provided evidence that respiratory viral infections potentially alter the normal immune response towards a TH2 phenotype in susceptible individuals [22, 24]. In this hypothetical model, respiratory viruses interact with immune cells to enhance virus-specific IgE. This in turn sensitizes basophils and mast cells, which are able to amplify the TH2 response with resultant atopic symptoms. Whether or not infectious or noninfectious exacerbations promote the TH2 phenotype or perpetuate it is not known.
Based on an “epidemic” rise in atopic individuals after the industrial revolution, David Strachan presented epidemiological data that associated declining family size and improved hygiene with increased incidence of allergic rhinitis [25]. This alternative theory suggested that infection provided a paradoxically protective rather than injurious effect on immunologic responses to allergens. Repeated exposures to bacterial and viral infections reinforced the “natural” Th1 immune response. In the absence of repeated pathogen exposure, the Th1 immune response is then unable to suppress the TH2 response to innocuous allergens [26]. This theory became known as the “hygiene hypothesis.” Controversies remain between these two models. Most asthma exacerbations are provoked by viral infections [27], but it is unclear if this exuberant response to viral infection is caused by early viral infection, potentially prevented by early childhood exposures, or reflects an underlying predilection for dysregulated immune response to infection. Similarly, the relationship between asthma initiation and adult asthma symptoms remains unclear and is a field of active investigation.
By the mid-1990s, there was growing evidence that the TH1/TH2 paradigm reflects the extreme ends of the spectrum and incompletely explains the mechanisms underlying many patients with asthma [28]. Although the hygiene hypothesis held true for allergic rhinitis between two separate cohorts separated by 12 years, the data did not support significant predilections for asthma based on birth order or socioeconomic status [26] (Fig. 4). Similarly, not everyone with allergic rhinosinusitis develops asthma [29]. Worldwide studies definitively demonstrate that the burden of childhood asthma is similar between low-income and high-income countries [30], making it less likely that lack of pathogen exposure initiates a TH2-skewed immune response. Studies in mouse models of allergic asthma demonstrated that asthmatic airway hyperresponsiveness and eosinophilic inflammation were the result of two separate regulatory processes rather than a single TH2-mediated event [31, 32]. Improved clinical definitions of disease and the incorporation of more severe asthmatics in clinical studies have revealed important differences in the multiple inflammatory pathways that are responsible for the clinical heterogeneity of asthma [33], which suggest that asthma should be thought of as a syndrome rather than a single disease entity.
Fig. 4.
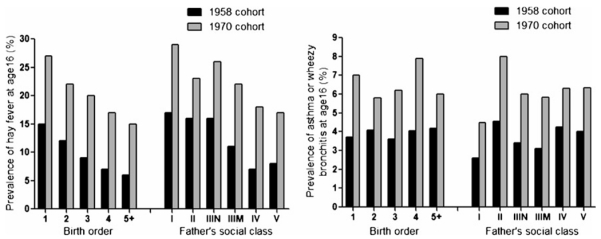
Adapted from Strachan [26]. Reproduced with permission. Relationship of birth order or socioeconomic status on the incidence of atopic disease
An important study by Wenzel and colleagues [34] defined two populations of severe asthma based on the pattern of inflammatory cells found in endobronchial biopsy samples. Based on their previous work that neutrophils were present in higher quantities in corticosteroid-resistant asthmatics [35], they hypothesized that subtypes of severe asthma could be separated into two immunopathologic categories based on the presence or absence of eosinophils in the airway. Patients referred to the investigators’ clinic who had an FEV1 <70 % on more than one occasion in the previous year and required ≥10 mg of prednisone during >75 % of the year underwent evaluation with bronchoscopy and endobronchial biopsy. Tissue samples were examined for tissue cell types and subbasement membrane thickness. In patients with severe asthma, 14 biopsy samples yielded solely neutrophils, while 20 patients had both eosinophils and neutrophils. The subbasement membrane was thicker in the eosinophil (+) severe asthmatics, but this group had significantly less episodes of respiratory failure, higher FEV1, and a higher ratio of FVC to slow vital capacity compared to the eosinophil (−) severe asthmatics.
Segregating patients based on clinical features using principal component or cluster analyses revealed three to five asthma phenotypes [36–38]. Haldar and colleagues described three major phenotypes divided by concordance of symptoms and eosinophilic inflammation. Patients with concordant disease were those patients that had respiratory symptoms that correlated with eosinophilic asthma. Patients with discordant disease fell into two categories: those with a high degree of symptoms and minimal eosinophilic inflammation and those with a high degree of eosinophilic inflammation with minimal symptoms (Fig. 5). It is postulated that those with concordant disease have an underlying immunopathology reflective of the classic TH2 model of inflammation. Asthmatics with discordant disease belie an underlying immunopathology that is distinct from the TH2 model, and therefore will be more resistant to TH2-targeted therapies and have more frequent critical asthma exacerbations. The nature of these biological differences is unclear and identifying clinical phenotype clusters is necessary to improve our ability to accurately provide targeted therapy.
Fig. 5.
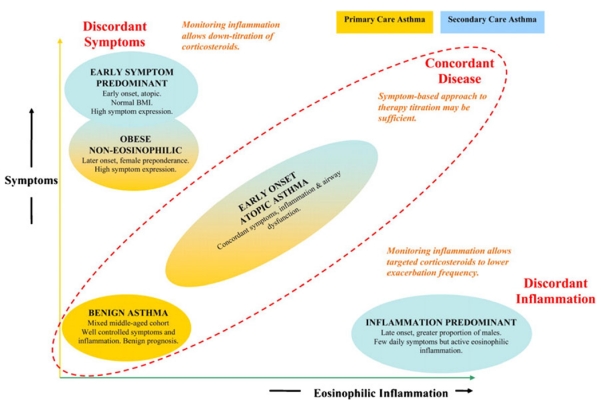
Adapted from Haldar et al. [36]. Reproduced with permission. Classification of phenotypes based on relationship between symptoms and eosinophilic inflammation. Those with discordant disease or higher symptom profiles should be referred to an asthma specialist
Despite the alternative pathways proposed for the development and pathogenesis of asthma, the allergic model of asthma as a TH2-dominant disease with elevated IgE production and eosinophilia should not be abandoned. The majority of asthma patients fit this classic paradigm well. This group of patients tends to respond favorably to corticosteroids and TH2-targeted therapies [39, 40]. Continued work in this area will likely lead to improved targets for drug therapy. For example, Woodruff and colleagues outlined “TH2-high” and “TH2-low” groups in patients with mild to moderate asthma. These patient groups were separated based on increased mRNA expression of IL-5 and IL-13 from bronchial biopsy specimens. The TH2-high subjects had higher airway hyperresponsiveness as measured by methacholine bronchial challenge, serum IgE levels, blood eosinophilia, intra-epithelial mast cells, and levels of eosinophils in bronchoalveolar lavage samples. Similarly, TH2-high patients had increased reticular basement membrane thickness and shifts in airway soluble mucin expression. In this 8-week study, TH2-high subjects had an average increase of 300 mL in FEV1 in response to inhaled corticosteroids and this was significantly greater than the increase in either the TH2-low or the placebo-control group [41]. This study was one of the first to show clear responses to therapy tailored to a specific molecular phenotype. However, whether such characterization is practical and cost-effective in the treatment of difficult-to-control asthma is unclear. In addition, patients with TH2-mediated asthma are felt to exhibit milder forms of asthma [39], are easier to treat, and are presumably are at lower risk for developing one of the critical asthma syndromes. Therefore, continued efforts to better understand the mechanisms underpinning the various asthma syndromes is desperately needed to improve therapeutic options for those patients at the highest risk of serious exacerbations.
Recent Discoveries
Where are we today in thinking about new drug targets for asthma and, perhaps, the critical asthma syndrome? Genome Wide Association Studies have revealed significant differences in gene expression and single nucleotide polymorphisms in asthmatics versus non-asthmatics that are potential targets for study [42] (Table 2). Several potential pathways have emerged based on current molecular techniques and improved understanding of the various cell types in the lung. It is now clear that early innate immune responses play a critical role in the development of asthma and atopy. The role of the airway epithelium, and how it informs the adaptive immune response, is increasingly appreciated as a critical component for asthma pathogenesis. Potential pathways differentially involved in the various asthmatic phenotypes include specific airway epithelium responses to allergens and infection, uptake and processing of antigen by dendritic cells, cross-talk between airway epithelium and dendritic cells, antigen presentation to immune activating cells, and regulation of cellular immune responses. The first interaction between the lung and the environment is through the airway epithelium, and pattern recognition receptors (PRRs) are one of the primary mechanisms by which the airway epithelium recognizes microorganisms and allergens (Box 1).
Table 2.
GWAS targets (adapted from Ramasamy)
| Gene locus | Asthma-associated SNP |
|---|---|
| IL-13 | rs1295686 |
| HLA-DQ | rs9273349 |
| IL-18R1 | rs3771166 |
| IL-1RL1 | rs1420101 |
| SMAD3 | rs744910 |
| IL-33 | rs3939286 |
| IL-33 | rs1342326 |
| RORA | rs11071559 |
Box 1.
PRRs function in airway epithelium to rapidly detect danger from the environment. This includes pathogen-associated products such as LPS or ssDNA, pathogen-associated molecular patterns (PAMPs), or products of cell damage such as ATP or lipid metabolites, damage-associated molecular patterns (DAMPs). The activation of PAMPs or DAMPs initiates airway epithelia to produce antimicrobial peptides and cytokines that interact with other immune cells in response to the microbe or injury. Examples include protease-activated receptors (PARs), Toll-like receptors (TLRs), NOD-like receptors (NLRs), and C-type lectins. PARs (i.e., PAR-2) induce airway epithelial cells to secrete cytokines in response to proteolytic allergens such as pollen and house dust mite. PAR-2 has been shown to be important for induction of TH2 immunity in response to proteolytic antigens. TLRs signal in response to bacterial or viral components to induce a number of proinflamatory cytokines that can exacerbate asthma symptoms. NLRs expressed by airway epithelium include NOD1 and NOD2. NLRs are associated with the production of TH2 cytokines, TH17-type immunity, and the inflammasome. C-type lectins are expressed on airway epithelial cells and recognize motifs found on inhaled fungus, house dust mite, pollens, and animal dander.
Activation of the airway epithelial cell is an essential component of dendritic cell activation and subsequent immune cell recruitment to the lung [43]. Although dendritic cells also express the same PRRs as epithelial cells, dendritic cells require epithelial recognition of the pathogen before they can be activated [44]. Recognition of protease-active allergens or microorganisms by PRRs in the airway epithelium results in the expression of multiple cytokines, nucleotides, and fatty acid metabolites that subsequently signal to immune cells responsible for recruiting TH1 or TH2 cytokines, neutrophils, and eosinophils [45] (Fig. 6).
Fig. 6.
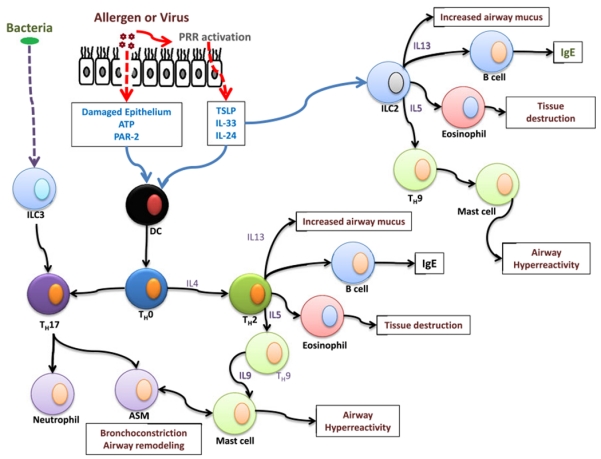
Overview of multiple pathways leading to asthma pathogenesis. Note that several features of asthma, traditionally attributed to the classic TH1/TH2 paradigm, potentially are mediated directly through innate lymphoid cells (ILCs)
One potentially important cytokine derived from airway epithelial cells in response to allergen is thymic stromal lymphopoietin (TSLP). TSLP was first identified as a promoter of pre-B cell growth in mouse thymic stromal tissue [46]. TSLP expression activates dendritic cell production of IL-8 and eotaxin-2, two cytokines known to recruit neutrophils and eosinophils, respectively. There is also evidence that TSLP derived from airway epithelial cells can stimulate CD4+ T cell production of TH2 cytokines [47]. A recent paradigm-shifting and novel discovery suggests that TSLP can directly activate TH2 cytokine production from a newly identified lymphoid cell type to produce independent of the adaptive immune system (see below). This suggests that TH2 cytokine production can occur in the airway without the need of dendritic cell activation or adaptive immune cells. Abnormal production of TSLP from the airway epithelium in response to allergens can promote pathologic recruitment of eosinophils or neutrophils. TSLP is essential for the development of allergic inflammation in several mouse models and human studies, suggesting that airway epithelial-derived factors can directly enhance the asthmatic phenotype [48–50].
Interleukin-33 (IL-33) is a member of the IL-1 cytokine family expressed by epithelial cells and can act directly on immune cells expressing the IL-33 (ST2) receptor. This receptor is preferentially expressed on Th2-type cells, suggesting a strong link to allergy [51]. IL-33 production from airway epithelial cells can be signaled by pathogen-associated or damage-associated molecular patterns (PAMPs or DAMPs) providing multiple mechanisms for allergens and viruses to activate IL-33 signals. For example, cell damage from protease-active allergens will activate DAMPs and virus infection will activate PAMPs which can both lead to upregulated IL-33 production from airway epithelial cells. Target cells of IL-33 include Th2-type cells, innate lymphoid cells (ILCs) (see below), mast cells, basophils, and dendritic cells. GWAS studies demonstrate that IL-33 and the IL-33 receptor are distinct targets for the development of asthma. In mouse models, the administration of IL-33 induces features of asthma independent of the adaptive immune system, which suggests derangements in innate immunity alone are sufficient to cause asthma [52].
Similar to IL-33, IL-25 is released from epithelial cells in response to allergen challenge in both murine models of asthma and in human asthma patients [53–55]. IL-25 signals through a heteroreceptor containing one subunit unique to IL-25 (IL-25R) and another subunit that is shared with IL-17B and IL-25 (IL-17RB). IL-25 is known to promote TH2 immune responses in the lung, and this effect appears to be through direct activation of ILCs, specifically group 2 ILCs. In addition, IL-25 has been shown to recruit fibroblasts and endothelial cells—suggesting a potential mechanism for airway remodeling in asthma [56]. IL-25 may also enhance production of TSLP and IL-33, acting to amplify the functions of these two cytokines. Targeted therapy against any of these three proteins may provide additional tools to prevent or treat patients suffering from CAS.
ILCs are newly identified members of the lymphoid lineage and have the capacity to produce many of the cytokines typically associated with the adaptive immune system, including TH1, TH2, and TH17 cytokines, without the need to respond in an antigen-specific manner (Box 2). Similar to previous characterization, innate lymphoid cells are classified into three major phenotypes according to the cytokines they produce [57]. Group 1 ILCs (ILC1, with NK cells reorganized into this group) function to mediate viral infection and produce high levels of IFNγ, perforin, or granzymes. Group 3 ILCs (ILC3) provide support for lymphoid tissue development and immunity to extracellular bacteria. The products of ILC3 include lymphotoxin, IL-17, a potent neutrophil chemoattractant, and IFNγ. Of particular importance to asthma are the group 2 ILCs (ILC2). These cells are differentially regulated by the RORα transcription factor, which has been associated with human asthma [58]. In addition, group 2 ILCs produce many of the cytokines implicated in asthma pathogenesis including IL-5, IL-9, and IL-13. And, they are directly regulated by TSLP, IL-25, and IL-33.
Box 2.
ILCs are newly identified group of immune cells that share several features with classical TH cell subtypes. They differentiate from ID2+ stem cells after stimulation by IL-7, IL-25, IL-33, and others into specific ILC subtypes. Similar to classical TH cell subtypes, ILCs are categories by the cytokines they produce. Group 1 ILCs include NK cells, which have been renamed, and produce IFNγ in response to viruses, inflammation, or tumor. Group 2 ILCs produce IL-5, IL-13, and IL-9 in response to helminths or tissue injury and are thought to be highly relevant to allergy and asthma. The nuocyte have been reclassified as a group 2 ILC. Group 3 ILCs provide host immunity by producing IL-17 and IFNγ in response to bacterial infection. Both IL-17 and IFNγ are known to recruit neutrophils to the site of injury and may have a particularly relevant role in severe asthma phenotypes that are characterized predominantly by neutrophilic inflammation.
The discovery of innate lymphoid cells provides a mechanism of how asthmatic symptoms can be seen in a mouse model lacking the adaptive immune system [52]. As modeled in Fig. 6, exposure of airway epithelial to protease-active allergens or viral infection results in the production of TSLP, IL-25, or IL-33. These cytokines can stimulate dendritic cells to move to the lymph tissues and stimulate TH2-like cells or directly act on ILC2 to immediately produce TH2 cytokines. This finding suggests deficits in immune signaling from the epithelium alone would be sufficient to drive an abnormal cytokine response from ILCs. This discovery may provide novel targets for therapy that do not require inhibiting the adaptive immune system.
Interleukin-17 (IL-17) and TH17 cells also play a role in allergic disease and asthma [59, 60]. This is an important subset of T cell effectors to consider in asthma and CAS since it represents one of several non-Th2 cellular responses important in disease pathogenesis. These TH17 cells are considered “adaptive lymphoid cells” that are derived from lymphoid precursors via the transcription factor retinoic acid receptor-related orphan receptor (RORγt) [61]. Th17 cells predominantly produce IL-17A, IL-17F, IL-6, IL-22, and TNF-α, all of which have been implicated in the development of airway diseases including asthma. Serum and airway mRNA and protein levels of IL-17 are elevated in asthmatics [62, 63]; IL-17 also correlates with airways hyperresponsiveness, asthma disease severity, and corticosteroid resistance. IL-17A induces the expression of two important mucin genes, Muc5AC and Muc5B, in human bronchial epithelial cells [64]. IL-17 also works in concert with other airway epithelial cytokines (e.g., IL-6, IL-8, ICAM-1) to promote airway inflammation relevant to asthma [65]. Thus, IL-17 mediates key pathogenic features of CAS. Lastly, IL-17 is thought to promote the influx of neutrophils into airways resulting in damage to airway resident cells and eventual adverse airway remodeling [60, 66, 67]. Related to this pathway is IL-23 which is a key molecular signal for TH17 cell propagation. IL-23 and IL-17 together may enhance TH2 cell-mediated airway eosinophilia in mouse models of allergic asthma [68]. Thus, targeting both IL-23 and IL-17 maturation, propagation, and signaling in CAS may have therapeutic benefits for this cohort of steroid-resistant asthmatics. However, the true effect of these T cell lineages in human asthma is more nuanced and complex since “innate lymphoid cells” such as NK cells are also involved in severe asthma [69]. Targeting the IL-17 signaling machinery (e.g., MAP kinase) and/or IL-17 receptors (IL17RA and IL17RC) is an area worthy of further research.
Immunotherapy has been held as the holy grail of asthma treatment since the very first discovery of IgE. In 1978, Ishizaka wrote that “a crucial role of IgE antibody in reaginic hypersensitivity and atopic diseases suggests strongly that prevention or suppression of IgE antibody formation is beneficial for atopic patients.” The seductive idea that we can identify a silver bullet to optimally treat critical asthma syndromes is the driving force for much of the research on the mechanisms underlying asthma. The limited success of anti-IgE therapy and moderate success of anti-IL-5 therapy should not dissuade us from pursuing immunotherapeutic options. In fact, the most recent findings suggest that clinical failures of TH2-type targeted therapies are likely due to phenotypic differences that we are only now beginning to understand.
Concluding Statement
Better illumination of the immunologic underpinnings of the various asthma phenotypes, the clinical features that characterize each phenotype, and potential biomarkers that are specific to each patient group will allow us to optimize therapy for each patient according to the specific treatment that they need. Harkening back to the curiosity that inspired Carl Prausnitz to discover the transferable factor that led to the first identification of IgE, we must remain curious about the differences in our patients that manifest as response or resistance to therapies. With this knowledge, we will be better equipped to care for all patients suffering from asthma and reduce the morbidity and mortality from critical asthma syndromes.
Contributor Information
Richart W. Harper, Division of Pulmonary, Critical Care, Sleep Medicine, Department of Internal Medicine, University of California, Davis, Davis, CA, USA; University of California, Davis, 4150 V Street, Suite 3400, Sacramento, CA 95817, USA
Amir A. Zeki, Division of Pulmonary, Critical Care, Sleep Medicine, Department of Internal Medicine, University of California, Davis, Davis, CA, USA
References
- 1.Holgate ST. A brief history of asthma and its mechanisms to modern concepts of disease pathogenesis. Allergy, Asthma Immunol Res. 2010;2:165–171. doi: 10.4168/aair.2010.2.3.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prausnitz C, Kustner H. Studien uber die Uberempfindlichkeit. Zentral-blatt f Bakt Originalien. 1921;160:169–1921. [Google Scholar]
- 3.Downie AW. Obituary—Carl Prausnitz (Giles) 11 October 1876-21 April 1963. J Pathol Bacteriol. 1966;92:241. doi: 10.1002/path.1700920130. [DOI] [PubMed] [Google Scholar]
- 4.Ishizaka K, Ishizaka T. Mechanisms of reaginic hypersensitivity and IgE antibody response. Immunol Rev. 1978;41:109–148. doi: 10.1111/j.1600-065x.1978.tb01462.x. [DOI] [PubMed] [Google Scholar]
- 5.Tada T, Takemori T, Okumura K, Nonaka M, Tokuhisa T. Two distinct types of helper T cells involved in the secondary antibody response: independent and synergistic effects of Ia− and Ia+ helper T cells. J Exp Med. 1978;147:446–458. doi: 10.1084/jem.147.2.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swierkosz JE, Marrack P, Kappler JW. Functional analysis of T cells expressing Ia antigens. I. Demonstration of helper T-cell heterogeneity. J Exp Med. 1979;150:1293–1309. doi: 10.1084/jem.150.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imperiale MJ, Faherty DA, Sproviero JF, Zauderer M. Functionally distinct helper T cells enriched under different culture conditions cooperate with different B cells. J Immunol. 1982;129:1843–1848. [PubMed] [Google Scholar]
- 8.Melchers I, Fey K, Eichmann K. Quantitative studies on T cell diversity. III. Limiting dilution analysis of precursor cells for T helper cells reactive to xenogeneic erythrocytes. J Exp Med. 1982;156:1587–1603. doi: 10.1084/jem.156.6.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Woods A, Becker-Dunn E, Bottomly K. Distinct functional phenotypes of cloned Ia-restricted helper T cells. J Exp Med. 1985;162:188–201. doi: 10.1084/jem.162.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 11.Lazaar AL, Panettieri RA Jr. Airway smooth muscle: a modulator of airway remodeling in asthma. J Allergy Clin Immunol. 2005;116:488–495. doi: 10.1016/j.jaci.2005.06.030. quiz 496. [DOI] [PubMed] [Google Scholar]
- 12.Panettieri RA Jr, Kotlikoff MI, Gerthoffer WT, Hershenson MB, Woodruff PG, Hall IP, Banks-Schlegel S. Airway smooth muscle in bronchial tone, inflammation, and remodeling: basic knowledge to clinical relevance. Am J Respir Crit Care Med. 2008;177:248–252. doi: 10.1164/rccm.200708-1217PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azzawi M, Bradley B, Jeffery PK, Frew AJ, Wardlaw AJ, Knowles G, Assoufi B, Collins JV, Durham S, Kay AB. Identification of activated T lymphocytes and eosinophils in bronchial biopsies in stable atopic asthma. Am Rev Respir Dis. 1990142:1407–1413. doi: 10.1164/ajrccm/142.6_Pt_1.1407. [DOI] [PubMed] [Google Scholar]
- 14.Walker C, Bode E, Boer L, Hansel TT, Blaser K, Virchow JC Jr. Allergic and nonallergic asthmatics have distinct patterns of T-cell activation and cytokine production in peripheral blood and bronchoalveolar lavage. Am Rev Respir Dis. 1992;146:109–115. doi: 10.1164/ajrccm/146.1.109. [DOI] [PubMed] [Google Scholar]
- 15.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 16.Del Prete GF, De Carli M, D’Elios MM, Maestrelli P, Ricci M, Fabbri L, Romagnani S. Allergen exposure induces the activation of allergen-specific Th2 cells in the airway mucosa of patients with allergic respiratory disorders. Eur J Immunol. 1993;23:1445–1449. doi: 10.1002/eji.1830230707. [DOI] [PubMed] [Google Scholar]
- 17.Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, Wardlaw AJ, Pavord ID. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 18.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 19.Coyle AJ, Le Gros G, Bertrand C, Tsuyuki S, Heusser CH, Kopf M, Anderson GP. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- 20.Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelaia G, Vatrella A, Maselli R. The potential of biologics for the treatment of asthma. Nat Rev Drug Disc. 2012;11:958–972. doi: 10.1038/nrd3792. [DOI] [PubMed] [Google Scholar]
- 22.Frick OL, German DF, Mills J. Development of allergy in children. I. Association with virus infections. J Allergy Clin Immunol. 1979;63:228–241. doi: 10.1016/0091-6749(79)90106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanarsdel PP Jr, Motulsky AG. Blood groups and secretion of blood group substances. Comparison of allergic with nonallergic persons in a Pacific Northwest college population. J Allergy. 1959;30:460–463. doi: 10.1016/0021-8707(59)90024-3. [DOI] [PubMed] [Google Scholar]
- 24.Busse WW. The relationship between viral infections and onset of allergic diseases and asthma. Clin Exp Allergy. 1989;19:1–9. doi: 10.1111/j.1365-2222.1989.tb02336.x. [DOI] [PubMed] [Google Scholar]
- 25.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strachan DP. Family size, infection and atopy: the first decade of the “hygiene hypothesis”. Thorax. 2000;55(Suppl 1):S2–S10. doi: 10.1136/thorax.55.suppl_1.s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boushey HA, Fahy JV. Basic mechanisms of asthma. Environ Health Perspect. 1995;103(Suppl 6):229–233. doi: 10.1289/ehp.95103s6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen JE, Maizels RM. Th1-Th2: reliable paradigm or dangerous dogma? Immunol Today. 1997;18:387–392. doi: 10.1016/s0167-5699(97)01102-x. [DOI] [PubMed] [Google Scholar]
- 29.Huurre TM, Aro HM, Jaakkola JJ. Incidence and prevalence of asthma and allergic rhinitis: a cohort study of Finnish adolescents. J Asthma: Off J Assoc Care Asthma. 2004;41:311–317. doi: 10.1081/jas-120026088. [DOI] [PubMed] [Google Scholar]
- 30.Bush A, Zar HJ. WHO universal definition of severe asthma. Curr Opin Allergy Clin Immunol. 2011;11:115–121. doi: 10.1097/ACI.0b013e32834487ae. [DOI] [PubMed] [Google Scholar]
- 31.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 32.Mehlhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, Martin TR, Oettgen HC. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci U S A. 1997;94:1344–1349. doi: 10.1073/pnas.94.4.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuijs MJ, Willart MA, Hammad H, Lambrecht BN. Cytokine targets in airway inflammation. Curr Opin Pharmacol. 2013;13:351–361. doi: 10.1016/j.coph.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, Chu HW. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–1008. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 35.Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. 1997;156:737–743. doi: 10.1164/ajrccm.156.3.9610046. [DOI] [PubMed] [Google Scholar]
- 36.Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, Wardlaw AJ, Green RH. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178:218–224. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D’Agostino R Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siroux V, Basagana X, Boudier A, Pin I, Garcia-Aymerich J, Vesin A, Slama R, Jarvis D, Anto JM, Kauffmann F, Sunyer J. Identifying adult asthma phenotypes using a clustering approach. Eur Respir J. 2011;38:310–317. doi: 10.1183/09031936.00120810. [DOI] [PubMed] [Google Scholar]
- 39.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet. 2008;372:1107–1119. doi: 10.1016/S0140-6736(08)61452-X. [DOI] [PubMed] [Google Scholar]
- 40.Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. [DOI] [PubMed] [Google Scholar]
- 41.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramasamy A, Kuokkanen M, Vedantam S, Gajdos ZK, Couto Alves A, Lyon HN, Ferreira MA, Strachan DP, Zhao JH, Abramson MJ, Brown MA, Coin L, Dharmage SC, Duffy DL, Haahtela T, Heath AC, Janson C, Kahonen M, Khaw KT, Laitinen J, Le Souef P, Lehtimaki T, Madden PA, Marks GB, Martin NG, Matheson MC, Palmer CD, Palotie A, Pouta A, Robertson CF, Viikari J, Widen E, Wjst M, Jarvis DL, Montgomery GW, Thompson PJ, Wareham N, Eriksson J, Jousilahti P, Laitinen T, Pekkanen J, Raitakari OT, O’Connor GT, Salomaa V, Jarvelin MR, Hirschhorn JN. Genome-wide association studies of asthma in population-based cohorts confirm known and suggested loci and identify an additional association near HLA. PloS One. 2012;7:e44008. doi: 10.1371/journal.pone.0044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Islam SA, Luster AD. T cell homing to epithelial barriers in allergic disease. Nat Med. 2012;18:705–715. doi: 10.1038/nm.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friend SL, Hosier S, Nelson A, Foxworthe D, Williams DE, Farr A. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp Hematol. 1994;22:321–328. [PubMed] [Google Scholar]
- 47.Liu YJ. TSLP in epithelial cell and dendritic cell cross talk. Adv Immunol. 2009;101:1–25. doi: 10.1016/S0065-2776(08)01001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, Comeau MR, Campbell DJ, Ziegler SF. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202:541–549. doi: 10.1084/jem.20041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005202:829–839. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, Corrigan C. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 51.Oboki K, Nakae S, Matsumoto K, Saito H. IL-33 and airway inflammation. Allergy, Asthma Immunol Res. 2011;3:81–88. doi: 10.4168/aair.2011.3.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, Hoshino T, Fujimoto J, Nakanishi K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Inter Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 53.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, Muchamuel T, Hurst SD, Zurawski G, Leach MW, Gorman DM, Rennick DM. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 54.Suzukawa M, Morita H, Nambu A, Arae K, Shimura E, Shibui A, Yamaguchi S, Suzukawa K, Nakanishi W, Oboki K, Kajiwara N, Ohno T, Ishii A, Korner H, Cua DJ, Suto H, Yoshimoto T, Iwakura Y, Yamasoba T, Ohta K, Sudo K, Saito H, Okumura K, Broide DH, Matsumoto K, Nakae S. Epithelial cell-derived IL-25, but not Th17 cell-derived IL-17 or IL-17F, is crucial for murine asthma. J Immunol. 2012;189:3641–3652. doi: 10.4049/jimmunol.1200461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corrigan CJ, Wang W, Meng Q, Fang C, Eid G, Caballero MR, Lv Z, An Y, Wang YH, Liu YJ, Kay AB, Lee TH, Ying S. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J Allergy Clin Immunol. 2011;128:116–124. doi: 10.1016/j.jaci.2011.03.043. [DOI] [PubMed] [Google Scholar]
- 56.Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, McKenzie AN, Lloyd CM. IL-25 drives re-modelling in allergic airways disease induced by house dust mite. Thorax. 2013;68:82–90. doi: 10.1136/thoraxjnl-2012-202003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells —how did we miss them? Nat Rev Immunol. 2013;13:75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 58.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson WO. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson KM, Manni ML, Biswas PS, Alcorn JF. Clinical consequences of targeting IL-17 and T17 in autoimmune and allergic disorders. Curr Allergy Asthma Rep. 2013;6:587–595. doi: 10.1007/s11882-013-0361-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halwani R, Al-Muhsen S, Hamid Q. T helper 17 cells in airway diseases: from laboratory bench to bedside. Chest. 2013;143:494–501. doi: 10.1378/chest.12-0598. [DOI] [PubMed] [Google Scholar]
- 61.Walker JA, McKenzie A. Development and function of group 2 innate lymphoid cells. Curr Opin Immunol. 2013;25:148–155. doi: 10.1016/j.coi.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, Ceuppens JL. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashimoto T, Akiyama K, Kobayashi N, Mori A. Comparison of IL-17 production by helper T cells among atopic and nonatopic asthmatics and control subjects. Int Arch Allergy Immunol. 2005;137(Suppl 1):51–54. doi: 10.1159/000085432. [DOI] [PubMed] [Google Scholar]
- 64.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 65.Kawaguchi M, Kokubu F, Kuga H, Matsukura S, Hoshino H, Ieki K, Imai T, Adachi M, Huang SK. Modulation of bronchial epithelial cells by IL-17. J Allergy Clin Immunol. 2001;108:804–809. doi: 10.1067/mai.2001.119027. [DOI] [PubMed] [Google Scholar]
- 66.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 67.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, Boulet LP, Hamid Q. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 68.Wakashin H, Hirose K, Maezawa Y, Kagami SI, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Lwakura Y, Puccetti P, Ivvamoto I, Nakajima H. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178:1023–1032. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]
- 69.Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, Wechsler ME, Israel E, Levy BD. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5:174ra126. doi: 10.1126/scitranslmed.3004812. [DOI] [PMC free article] [PubMed] [Google Scholar]