

Abstract
The Fibroblast Growth Factor Receptor (FGFR) kinases are promising therapeutic targets in multiple cancer types including lung and head and neck squamous cell carcinoma, cholangiocarcinoma and bladder cancer. Although several FGFR kinase inhibitors have entered clinical trials, single agent clinical efficacy has been modest and resistance invariably occurs. We therefore conducted a genome-wide functional screen to characterize mechanisms of resistance to FGFR inhibition in a FGFR1-dependent lung cancer cellular model. Our screen identified known resistance drivers, such as MET, and additional novel resistance mediators including members of the neurotrophin receptor pathway (NTRKs), the TAM family of tyrosine kinases (TYRO3, MERTK, AXL) and MAPK pathway, which were further validated in additional FGFR-dependent models. In an orthogonal approach, we generated a large panel of resistant clones by chronic exposure to FGFR inhibitors in FGFR1- and FGFR3-dependent cellular models, and characterized gene expression profiles employing the L1000 platform. Notably, resistant clones had enrichment for NTRK and MAPK signaling pathways. Novel mediators of resistance to FGFR inhibition were found to compensate for FGFR loss in part through reactivation of MAPK pathway. Intriguingly, co-inhibition of FGFR and specific receptor tyrosine kinases identified in our screen was not sufficient to suppress ERK activity or to prevent resistance to FGFR inhibition, suggesting a redundant re-activation of RAS-MAPK pathway. Dual blockade of FGFR and MEK, however, proved to be a more powerful approach in preventing resistance across diverse FGFR-dependencies, and may represent a therapeutic opportunity to achieve durable responses to FGFR inhibition in FGFR-dependent cancers.
Keywords: FGFR inhibitor, Targeted therapy, Resistance
Introduction
The fibroblast growth factor receptors (FGFR) are transmembrane receptor tyrosine kinases with growing relevance in cancer therapeutics. Focal amplification of FGFR1 has been identified in as many as 20% of squamous cell lung cancer (SqCLC) and approximately 10% of head and neck squamous cell carcinoma and 10% of breast cancer patients (1-5). Recurrent mutations, translocations or amplifications of FGFR2 have been identified in endometrial cancer, cholangiocarcinoma and gastric cancer, respectively (6-9). Furthermore, mutations and fusions of the FGFR3 gene have been described in approximately 15% of invasive bladder cancer while FGFR3 translocations have been identified in approximately 3% of glioblastoma multiforme (10-12). FGFR4 alterations have principally been identified in hepatocellular carcinoma, colorectal cancer, and rhabdomyosarcoma (13).
Preclinical studies have demonstrated the potential for FGFR kinases to serve as therapeutic targets across different cancer types and a number of selective FGFR inhibitors have entered clinical trials (3, 6, 14-16). Despite initial enthusiasm, clinical efficacy of these compounds as single agents has been modest, particularly in patients with FGFR1 amplification (15). Dramatic but short-lived responses in patients with urothelial cancer harboring FGFR3 mutations or fusions and cholangiocarcinoma with FGFR2 fusions have been observed more frequently, but resistance invariably occurs (15-17).
Intrinsic and acquired resistance to FGFR inhibitors has recently been described in several instances. Rapid acquisition of resistance to FGFR inhibition has been described in FGFR3-dependent bladder cancer models, by upregulation of ERBB2/3 (18, 19). Conversely, the FGF2-FGFR1 autocrine pathway has been shown to drive resistance to gefitinib in EGFR-dependent non-small cell lung cancer (NSCLC) (20, 21), suggesting an interplay between the EGFR and FGFR pathways. MET overexpression has further been shown to confer resistance to FGFR inhibitors in FGFR1-amplified lung cancer cell lines (22), while hepatocyte growth factor (HGF), the ligand for MET, can rescue from anti-FGFR therapy in gastric and bladder cancer lines dependent on FGFR2 or FGFR3, respectively (23). Furthermore, reactivation of PI3K signaling has been found to promote acquired resistance in FGFR2 and FGFR3 cellular models (24). Several in vitro studies have moreover described the emergence of gatekeeper mutations that shifts the ATP affinity to its binding site in the FGFR kinases (25). Gatekeeper mutations have also been recently described in tumors of patients with FGFR2-translocated cholangiocarcinoma treated with BGJ398, where despite initial response, tumors rapidly acquired FGFR2 mutations with marked inter- and intralesional heterogeneity (17). The sub-optimal clinical outcomes with FGFR inhibitors in multiple contexts underscores the need for upfront rational combination approaches which has the potential to overcome intrinsic resistance and suppress or delay the emergence of acquired resistance, prolonging the clinical benefit of FGFR inhibitors.
Given that clinical responses to FGFR inhibitors have been modest particularly in FGFR1-amplified cancers, broadening our understanding of how FGFR1-addicted tumor cells can overcome reliance on FGFR is critical to developing more effective therapeutic strategies. We therefore performed a genome-wide functional screen to identify genes whose overexpression is sufficient to confer resistance to FGFR inhibition, and performed a high-throughput gene expression profiling of a large panel of FGFR resistant clones to identify pathways enriched in resistance. We then tested whether co-inhibition of the resistance pathways identified could overcome or delay emergence of resistance to FGFR inhibitors.
Materials and Methods
Cell lines and chemical reagents
NCI-H2077, RT112, DMS114, NCI-H520 were cultured in RPMI media, supplemented with 10% FBS, and penicillin/streptomycin/L-glutamine (PSG). AN3 CA was cultured in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% FBS and PSG. All cell lines were cultured at 37 °C in a humidified chamber in the presence of 5% CO2. Cell lines were obtained from ATCC or Sigma-Aldrich, primarily in 2014 and 2016, and were not further authenticated. Cells were not passaged for more than 6 months and were routinely monitored in our laboratory for cellular morphology and microbial presence by microscopic observation. Cell lines used for ORF screen, gene expression and xenograft studies were tested and confirmed negative for Mycoplasma (MycoAlert PLUS, Lonza).
BGJ398(26), trametinib(27), LDC1267(28), LOXO-101 (ARRY-470)(29), imatinib(30), BKM120(31), AZD8931(32) and MGCD265 (glesatinib)(33) were purchased from Selleck. FIIN-3(34) and Torin2(35) were a generous gift from Dr. Nathanael Gray at Dana-Farber Cancer Institute (Boston, MA).
Pooled ORF screen
The ORF pooled barcoded library is derived from the Center for Cancer Systems Biology (CCSB)–Broad lentiviral expression library previously described (36) and expresses 17255 clones matching 12429 genes. NCI-H2077 cells were seeded at 3 million cells/well in 12-well plates and were transduced with the pooled lentiviral library in the presence of polybrene (8μg/mL) on Day -6 (Figure 1A). Two plates were seeded for replicate A and two for replicate B. An additional plate contained wells similarly transduced with eGFP-expressing lentivirus as a control, and several wells not transduced (non-infected controls). Plates were spun at 2000 rpm for 2 hours at 30 °C, and incubated overnight. A sufficient number of cells were infected to have a representation of 1000 cells per ORF (a 30% infection efficiency and 85% viability was assumed based on pre-screen optimization). On the following day, an in-line assay was set up to determine infection efficiency in a 6-well plate, and remaining cells were seeded in T175 flasks (Rep A, Rep B, eGFP). Transduced cells were selected for with puromycin (1 μg/mL). Infection efficiency was determined based on the in-line assay on Day -1 of the experimental protocol. Cells from Rep A and Rep B flasks were harvested on Day 0 and split into the following conditions: early time point sample (ETP) (20 million cells per sample), DMSO (20 million cells), BGJ398 100 nM (40 million cells), BGJ398 300 nM (40 million cells), FIIN-3 100 nM (40 million cells), FIIN-3 300 nM (40 million cells). The early time point sample was used to check library representation prior to treatment. Cells expressing eGFP were similarly harvested and split into treatment conditions, however were seeded at 4 million/flask into T75 flasks. The early time point sample was centrifuged, resuspended with 0.5 mL of PBS and stored at −20°C. Drug was first added on Day +1, and cells were passaged every three to four days. For flasks with fewer than 20 million cells remaining, all cells were reseeded when passaging. On Day +14, cells were harvested, counted, and stored at −20°C. DNA was extracted from all samples using the QIAamp DNA Blood Maxi Kit (Qiagen).
Figure 1. A Genome-wide Gain-of-Function Screen Identifies Candidate Mediators of Resistance to FGFR Inhibition.
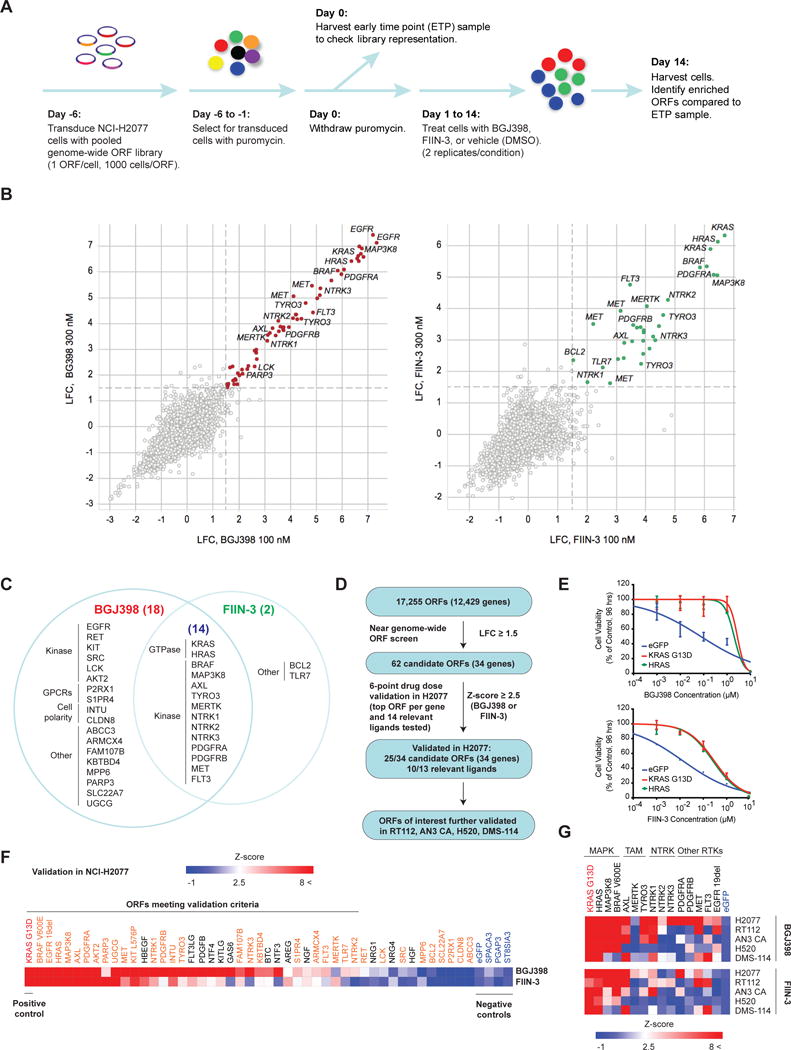
A) Schematic overview of the experimental approach. NCI-H2077 cells were transduced with a genome-wide Open Reading Frame (ORF) pooled barcoded library derived from the CCSB-Broad Lentiviral Expression Library. ORF-expressing cells were selected for, and then treated with BGJ398, FIIN-3 or DMSO as indicated. After 14 days of drug exposure, cells were harvested to assess for enriched ORFs compared to the early-time point (ETP) sample.
B) Scatter plot displaying log2-fold-change (LFC) compared to the ETP sample for BGJ398 (first panel) and FIIN-3 (second panel) at concentrations of 100 nM versus 300 nM for each ORF. Replicates were averaged to obtain the LFC for each condition. ORFs associated with LFC greater than or equal to 1.5 (dashed line) at both drug doses were considered mediators of resistance. Several of the top representative candidates are indicated.
C) Venn diagram outlining candidate resistance genes for BGJ398, FIIN-3 or for both drugs. The number of candidate genes for each group is displayed in parenthesis. Genes are organized into functional groups. (GPCR=G-protein-coupled receptor)
D) Summary of primary screening and validation studies.
E) Representative cell viability assay for HRAS wild type ORF validated for BGJ398 (top panel) and FIIN-3 (bottom panel) in NCI-H2077 cells. KRAS G13D and eGFP were used as positive and negative control, respectively.
F) Heat map displaying normalized IC50 (Z-score) for the validation of candidate drivers of resistance to BGJ398 and FIIN-3 in NCI-H2077 cells. Positive control KRAS G13D is displayed in red and negative controls in blue. Experimental ORFs are displayed in orange and ligands in black.
G) Heat map displaying normalized IC50 (Z-score) for a subset of candidate resistance genes in NCI-H2077, RT112, AN3 CA, NCI-H520 and DMS-114 cells for BGJ398 (top heat map) and FIIN-3 (bottom heat map).
PCR and sequencing were performed as previously described (37). Samples were sequenced on a HiSeq2000 (Illumina). For analysis, the read counts were normalized to reads per million and then log2 transformed. The log2 fold-change (LFC) of each ORF at Day +14 in drug was determined relative to the early time point for each biological replicate. Counts for every replicate were averaged given the high replicate reproducibility. Candidate mediators of resistance to BGJ398 and FIIN-3 were defined as ORFs that had a LFC greater than or equal to 1.5 following treatment compared to the ETP sample at both drug concentrations employed.
ORF screen validation
NCI-H2077 cells were seeded in 96-well plates at a density of 3000 cells/well and transduced with lentivirus corresponding to distinct candidate transcripts (Supplementary Methods). At 48 h post-infection, cells were subjected to 6-point dose response assays with BGJ398 and FIIN-3. eGFP and KRAS G13D transcripts were added to each batch of validation experiments as negative and positive controls, respectively. Following 96 h of drug exposure, cell viability was assessed using the CellTiter-Glo Luminescent Cell Viability assay (Promega). Validation studies of a subset of novel candidates of resistance were similarly performed in four additional cell lines, AN3 CA (FGFR2-mutated endometrial adenocarcinoma), RT112 (FGFR3-TACC3 translocated and FGFR3-amplificated bladder carcinoma), NCI-H520 (FGFR1-amplified squamous cell lung cancer) and DMS114 (FGFR1-amplified small cell lung cancer) (Supplementary Methods). All conditions were tested in triplicate, unless otherwise noted. Drug curves and IC50 values were generated using GraphPad Prism 6 (GraphPad Software).
Immunoblotting
Cells were lysed in RIPA buffer (Roche) containing protease inhibitors (Roche) and Phosphatase Inhibitor Cocktails I and II (CalBioChem). Protein concentrations were determined using a Bradford assay (Bio-Rad). Proteins were separated by SDS gel electrophoresis using NuPAGE 4-12% Bis-Tris gels (Life Technologies) in MOPS buffer. Resolved protein was transferred to nitrocellulose membranes, blocked in 10% milk and probed with primary antibodies recognizing p-FRS2 (3861), AKT (9272S), p-AKT (4060P), ERK (4695S), p-ERK (4370S), (all from Cell Signaling Technology), FRS2 (sc83-18, Santa Cruz), actin (A5441, Sigma-Aldrich) and vinculin (V9131, Sigma-Aldrich) in 5% milk or bovine serum albumin as recommended by the manufacturer. After incubation with the appropriate secondary antibody (Pierce anti-mouse IgG/IgM (31444, Thermo Scientific) and anti-rabbit IgG (31460, Thermo Scientific)), blots were imaged on film.
High-throughput generation and characterization of resistant FGFR-dependent cell lines
NCI-H2077 and RT112 cells were seeded in 24-well plates at a density of 7500 cells/well and allowed to adhere overnight. Cells were treated weekly with BGJ398 (1 μM). Once cells resumed a growth pattern that resembled the parental line, they were considered resistant to BGJ398, and were seeded into 96 well plates (4 replicates per resistant clone, seeded at 8000 cells per well) and allowed to adhere overnight. Cells were then treated with BGJ398 for 24 hours, media was then removed and cells were lysed using TCL Lysis Buffer (Qiagen, 100 μL/well, 30 minutes incubation at room temperature). Lysates were stored at −80 °C and subsequently sent to the Broad Institute (Cambridge, MA) for gene expression analysis using L1000 profiling platform. Parental NCI-H2077 and RT112 cell lines were similarly processed and served as controls.
The L1000 assay is a high-throughput, multiplexed mRNA expression profiling technique (38). It is based on the direct measurement of a reduced representation of the transcriptome (978 ‘landmark’ transcripts) and inference of the portion of the transcriptome not explicitly measured using an algorithm trained on several thousands of historical microarray-derived gene expression profiles. Robust z-score for each individual transcript was calculated based on quantile normalized data and subsequently utilized to query for enriched pathways.
To query for enrichment of biological pathways in resistant clones compared to controls, the Database for Annotation, Visualization and Integrated Discovery (DAVID) was utilized (39). The top 2500 upregulated genes in each cluster, as well as for all clones overall, was entered into DAVID for analysis. All p values of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway were adjusted by multiple testing correction (Benjamini-Hochberg FDR) (40) and signatures with adjusted p < 0.05 were considered statistically significant (Supplementary Methods).
Genomic analysis of resistant NCI-H2077 cells
NCI-H2077 cells were seeded in 10 cm plates and were treated BGJ398 starting at 10 nM. Every one to two weeks, once treated cells resumed growth rates similar to parental cells, the drug concentration was increased by 50-100 nM. After about four months, resistant cells were maintained in 5 μM of BGJ398. At that time, genomic DNA was isolated from the parental and resistant cells using the DNeasy Blood and Tissue Kit (Qiagen) and sent to the Center for Cancer Genome Discovery (CCGD) at the Dana-Farber Cancer Institute (Boston, MA) and the Partners Laboratory for Molecular Medicine (Cambridge, MA) for analysis using RNA baits targeting exons of 504 genes and select intronic regions of 15 genes to detect mutations, translocations and copy-number variations (Supplementary Methods).
Colony formation assays
Conventional 2D cell culture assays were employed to assay colon formation. One hundred thousand cells were seeded in 6-well plates, allowed to adhere overnight, and then incubated with media containing vehicle or drug as indicated for 4 weeks. Media (and drug) were replaced weekly. At 4 weeks, plates were fixed with 1% paraformaldehyde and then stained with 0.1% crystal violet as previously described (http://medicine.yale.edu/lab/kim/resources/protocols/cell/crystal_violet_stain.aspx) to assess colony formation. Results were quantified using an ImageJ Colony Area PlugIn (41).
Xenograft tumor studies
Xenograft studies were approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. NCI-H2077 xenograft models were established by subcutanoues (s.c.) injection of 0.5 × 106 cells in Matrigel (Corning) into both flanks of nude mice (NU/NU, #088 Charles River) when animals were 6-8 weeks of age. The xenograft studies were powered to include 5 mice (10 tumors per treatment group) providing 82% power to detect an underlying difference in progression free survival between 70% and 10% at eight weeks in Fisher’s exact test at a one-sided 0.05 level. When tumors reached approximately 200 mm3, as measured by caliper, mice were randomized to four groups of five female mice each, for each cell line: 1) vehicle, 2) BGJ398 3) Trametinib or 4) combination treatment. The animals were randomized to treatment using simple randomization by cage. Investigators were not blinded to group allocation. We employed an initial dose of BGJ398 15 mg/kg once daily (QD) by oral gavage and trametinib 1.5 mg/kg QD by oral gavage for the initial 3 weeks to assess toxicity as this combination has not been described previously. Given good tolerance, the dose of inhibitors was increased by approximately 33% at day 21 to BGJ398 20 mg/kg QD and trametinib 2.0 mg/kg QD. Caliper measurements were then performed weekly and continued for eight weeks.
Statistical analysis
Data are expressed as mean +/− standard deviation. Statistical significance was determined using Student’s t-test. Statistical analyses were performed in Prism 6 (GraphPad Software). Significance was set at P = 0.05.
Results
A Genome-wide Gain-of-Function Screen Identifies 34 Candidate Drivers of Resistance to FGFR Inhibition
To identify transcripts whose overexpression is sufficient to drive resistance to FGFR inhibition, we performed a genome-wide open-reading-frame (ORF) screen using a pooled barcoded lentiviral library (17,255 ORFs; representing 12,429 unique human genes and containing both wild-type and clinically relevant transcripts of common somatically altered oncogenes) in the FGFR1-amplified NSCLC cell line, NCI-H2077 (Figure 1A). NCI-H2077 was selected for this screen as it has marked baseline sensitivity to FGFR inhibitors with IC50 for BGJ398 and FIIN-3 of 17 and 3 nM, respectively, and is suitable to infection with the pooled lentiviral ORF library (Supplementary Figure S1A). We sought to identify which ORFs mediate resistance to two FGFR inhibitors: BGJ398, a first-generation reversible FGFR inhibitor currently in the clinic, and FIIN-3, a second-generation covalent inhibitor, to consider whether resistance mechanisms may vary for reversible versus covalent FGFR inhibitors (26, 34). BGJ398 is a potent inhibitor of FGFR1-3, but less so of FGFR4 (Supplementary Table 1). FIIN-3 is however a potent inhibitor of FGFR1-4. Additionally, FIIN-3 targets previously characterized FGFR gatekeeper mutations, as well as EGFR with lower potency (Supplementary Table 2) (34, 42, 43). The screen was performed using 100 and 300 nM of BGJ398 and FIIN-3; doses selected as they were sufficient to suppress growth in the NCI-H2077 model in near-scale optimization assays (Supplementary Figure S1B), while minimizing off-target effects. The screening library contained KRAS G13D, which has been shown to promote resistance to erlotinib, gefitinib and crizotinib in NSCLC (44, 45). Preliminary work from our lab prior to the ORF screen showed that KRAS G13D also induced resistance to FGFR inhibition, therefore we considered KRAS G13D as a positive control in the screen (Supplementary Figure S1C). The screen was also performed on a smaller scale in parallel with cells overexpressing eGFP (negative control), to ensure cells were responding as expected to drug. The screen was performed in duplicate.
Library representation following 14 days of drug treatment was compared with early time point representation (Figure 1A). Log2-fold change (LFC) of normalized read counts between these two time points was determined. Replicates were strongly correlated, thus LFC of replicates A and B were averaged together (Supplementary Figure S2A). Candidate mediators of resistance to BGJ398 and FIIN-3 were defined as ORFs that had a LFC greater than or equal to 1.5 following treatment compared to the early time point sample at both drug doses employed.
Sixty ORFs (corresponding to 32 genes) met this criterion for BGJ398-treated cells and 33 for FIIN-3 (corresponding to 16 genes), of which only two ORFs (TLR7, BCL6) were unique to FIIN-3 (Figure 1B, Supplementary Figure S2B, S3A, S3B). Candidate resistance genes reflected diverse protein classes (Figure 1C). Prominent among these were serine-threonine and tyrosine kinases, comprising 18 of the 34 genes. Genes from the MAPK pathway (HRAS, MAP3K8 and BRAF V600E) were the most effective in inducing resistance to both FGFR inhibitors, and EGFR was a potent inducer of resistance to BGJ398 (both wild-type and mutant forms, see Supplementary Figure S3A,B). Mutant EGFR only rescued from FIIN-3 at the lower dose employed in the screen, which is consistent with the pharmacological activity of FIIN-3 (Supplementary Table 1,2) (34). Similarly, RET kinase only rescued from BGJ398 and not FIIN-3, consistent with the off-target effects of FIIN-3 on RET kinase (34). Overall, we observed a high overlap between drivers of resistance for reversible and covalent FGFR inhibitors, with differences primarily being explained by their pharmacological activity.
Validation of Candidate Drivers of Resistance to FGFR Inhibition
We next validated the candidate drivers of resistance identified in the screen by expressing individual candidate ORFs in NCI-H2077 and evaluating their effect on cell viability upon treatment with BGJ398 and FIIN-3 in 6-point dose response assays. For genes represented by multiple ORFs (e.g. EGFR), we validated only the ORF with the highest LFC in the screen. We recognized that one limitation of our initial pooled screen may have been the inability to identify secreted factors which may induce resistance to FGFR inhibition, due to dilution effects. Therefore, we supplemented our validation screen with ORFs of key ligands of candidate receptor tyrosine kinases (RTKs) (e.g. PROS1, GAS6). The IC50 was determined for each ORF and the degree of shift in the IC50 for each ORF was compared to eGFP. ORFs associated with a standard deviation (SD) greater than 2.5 compared to controls for either drug were considered validated resistance drivers in NCI-H2077. 35 of 47 ORFs met this criterion and were considered to be validated (Figure 1D, 1E, 1F). Our screen confirmed known mediators of resistance to FGFR inhibitors such as wild-type AKT2 and MET, supporting the biological relevance of the screening results (19, 22, 46). Both wild-type and mutant EGFR (exon 19 deletion, L858R, etc.) rescued strongly from BGJ398 in the initial screen, however, only the top ORF was validated (EGFR exon 19 deletion, EGFR p.Glu746_Ala750del). Some of the mediators of resistance identified (AXL, MAP3K8, BRAF V600E, SRC, AKT2, HRAS, NTRK1 and NTRK2) have been previously identified as drivers of resistance to RAF/MEK inhibition in BRAF-mutant melanoma, PI3K inhibition in breast cancer, and ALK and EGFR inhibition in ALK-rearranged and EGFR mutated lung cancer, respectively (47-50). Novel resistance mechanisms to FGFR inhibition identified include members of the TAM kinase family (TYRO3, AXL and MERTK), neurotrophic tyrosine receptor kinases (NTRK1-3), PDGFRA and B, FLT3 and KIT L576P, as well as several MAPK family members (KRAS G13D, BRAF V600E, HRAS wild type and MAP3K8 wild type). As these are potential targets of existing pharmacological inhibitors, these kinases emerged as candidate mediators of resistance whose blockade in combination with FGFR inhibition might augment FGFR therapy either by blocking emergence of resistance or by combating resistance once it develops.
We further evaluated these novel and readily targetable mechanisms of resistance for their ability to overcome anti-FGFR therapy in additional FGFR-dependent cellular models, including FGFR1-amplified models of squamous cell lung carcinoma (NCI-H520) and small cell lung cancer (DMS114), FGFR2-mutated endometrial adenocarcinoma (AN3 CA) and FGFR3-TACC3 translocated bladder cancer (RT112). Cells were transduced with the ORFs of interest and were subjected to 10-point growth inhibition assays with BGJ398 and FIIN-3. eGFP-transduced cells were included as a negative control and KRAS G13D transduced cells as a positive control. Members of the MAPK families conferred resistance to FGFR inhibition broadly across different FGFR dependencies, with HRAS wild type overexpression conferring the greatest rescue (Figure 1G). Activating HRAS mutations have previously been shown to confer resistance to MET inhibition (51), and EGFR inhibitor in head and neck carcinoma (52), however to our knowledge HRAS overexpression has not previously been identified as a resistance mediator. BRAF V600E, which has also been shown to mediate resistance to first and newer generation EGFR inhibitors (53, 54) was similarly a strong mediator of resistance to FGFR inhibition, except for in DMS-114. MAP3K8 wild type, which has previously been shown to promote resistance in BRAF-mutant melanoma and ALK-dependent lung cancer (47, 49), was similarly a generalizable mediator of resistance to both BGJ398 and FIIN-3 across models, again with the exception of DMS-114. AXL, TYRO3 and NTRK1 also broadly conferred resistance across the various models tested, to both BGJ398 and FIIN-3. NTRK1, NTRK2, AXL and MERTK have also been reported to promote resistance to EGFR inhibition in EGFR-mutated NSCLC (50, 55). These findings raise the possibility of shared mediators of resistance to FGFR and EGFR inhibition in FGFR- and EGFR-dependent lung cancers.
We next explored biochemical effects of overexpression of the novel resistance genes including TAMs, NTRK1, HRAS, PDGFRA and MET upon FGFR inhibition with BGJ398 in NCI-H2077 (Figure 2), RT112 and AN 3CA (Supplementary Figure 4). In parental NCI-H2077 cells, BGJ398 blunts phosphorylation of AKT and only transiently suppresses phosphorylation of ERK with reactivation of ERK appearing within 12 hours (Figure 2A). Overexpression of AXL and TYRO3 led primarily to increased and maintained ERK activation despite FGFR blockade with BGJ398. PDGFRA overexpression caused sustained activation of AKT and had no significant effect on pERK (Figure 2B). Overexpression of NTRK1, MET and HRAS lead to strong activation of both ERK and AKT despite FGFR blockade, with HRAS having the strongest effect on both pAKT and pERK. A similar pattern of upregulation of ERK activity was observed in RT112 and AN 3CA cellular models following ectopic expression of candidate resistance mediators. Additionally, AKT activity upregulation was noted in RT112, in particular following HRAS and NTRK1 expression (Supplementary Figure 4).
Figure 2. A Subset of Validated Resistance ORFs Reactivate MEK/ERK and/or PI3K Signaling.
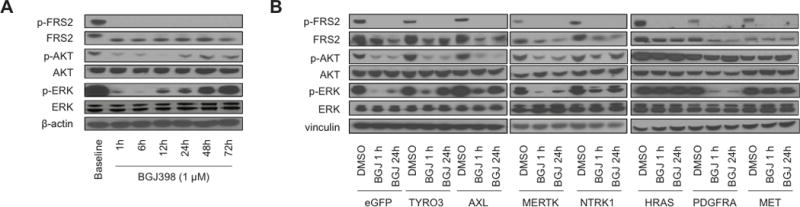
A) Protein lysates from NCI-H2077 cells after treatment with DMSO or BGJ398 1 μM probed at different time points for key members of the canonical FGFR signaling pathway.
B) ORF-expressing NCI-H2077 cells after treatment with DMSO or BGJ398 1 μM for 1 hr and 24 hr were probed for FRS2, ERK and AKT activity (BGJ, BGJ398).
RAS-MAPK Pathway is Enriched in Resistant FGFR Cellular Models
While our ORF screen identified a group of candidate kinases whose ectopic overexpression has the capacity to induce resistance to FGFR blockade in different FGFR-dependent models, these results do not necessarily indicate that a given kinase endogenously functions to mediate resistance. Therefore, we performed an orthogonal set of experiments to further identify candidate genes and pathways whose endogenous expression is altered with acquired resistance to FGFR blockade. We established a large panel of BGJ398-resistant clones of NCI-H2077 and RT112 by chronic exposure to high-dose BGJ398 (1 μM) in 24-well tissue culture plates (Figure 3A). mRNA from resistant clones and parental cells was analyzed by a high-throughput gene profiling method (L1000) (38) and the expression data was queried for enrichment in resistant clones compared to controls as detailed above (Supplementary Table 3) (39).
Figure 3. High-throughput generation and characterization of resistant FGFR-dependent cell lines.
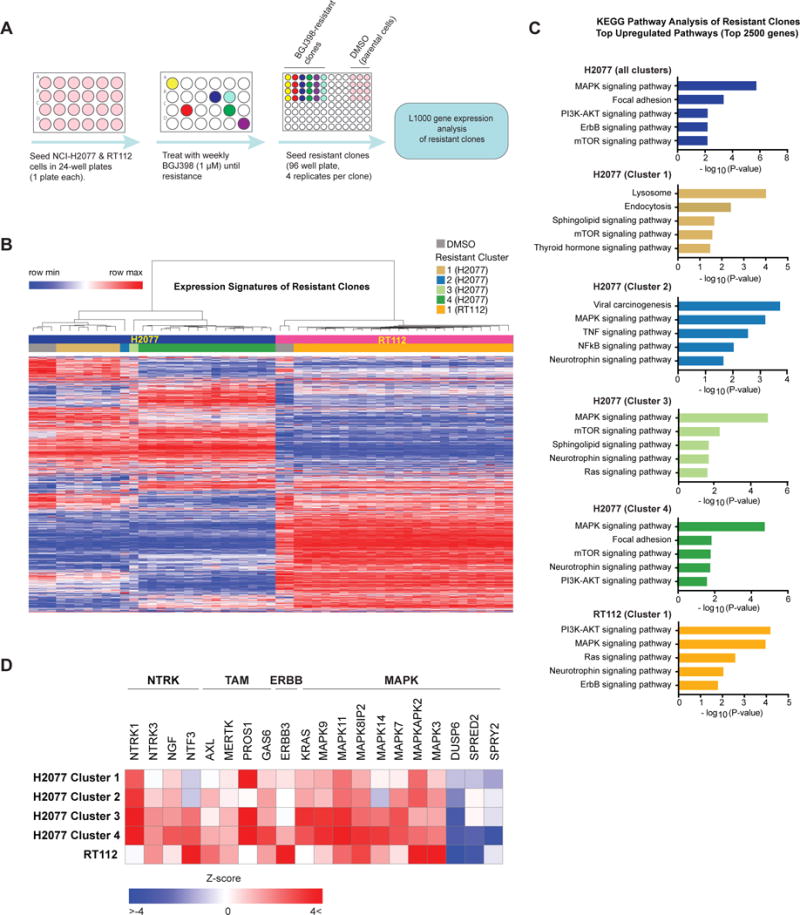
A) Schematic overview of the experimental approach. NCI-H2077 and RT112 were seeded in separate 24-well plates and subjected to weekly treatment with BGJ398 1 μM until resistance. At that time, resistant clones were seeded in quadruplicate in 96 wells plate, and mRNA was isolated for gene expression analysis by the L1000 platform.
B) Unsupervised hierarchical clustering of gene expression profiles of all BGJ398-resistant clones of NCI-H2077 and RT112 and respective DMSO controls. Clustering yielded two large groups of BGJ398-resistant NCI-H2077 clones, and two additional small clusters with distinct profiles. RT112 formed a single large cluster.
C) Pathway enrichment analysis of resistant clones compared to controls using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The top 2500 upregulated genes in each cluster were used for pathway enrichment analysis. Results were adjusted by multiple testing correction (Benjamini-Hochberg FDR) and signatures with adjusted p < 0.05 were considered statistically significant. Five significant results are shown per cluster.
D) Supervised analysis of gene expression of resistant clone clusters identified in Figure 3B, including several neurotrophin family receptors and their respective ligands, the TAM (TYRO3, MERTK, AXL) family members and their respective ligands, and positive and negative regulators of MAPK pathway.
Unsupervised hierarchical clustering of these signatures yielded two large clusters of FGFR-resistant NCI-H2077 clones, and two additional small clusters with distinct profiles (Figure 3B). Clustering by gene expression also largely matched clone cellular phenotype (i.e., clones with a similar morphology and similar time to resistance development clustered together) (Supplementary Figure S5A). Among the enriched pathways in resistant clones of NCI-H2077 compared to controls were four pathways associated with genes identified in our functional ORF resistance screen including PI3K and ERBB pathways, also consistent with prior reports of resistance to FGFR inhibitors (18, 19, 24) (Figure 3C). RAS-MAPK pathway was amongst the most enriched in NCI-H2077, while neurotrophin signaling pathway was also significantly upregulated in Clusters 2 and 3, providing further support for the NTRK family of kinases to serve as mediators of FGFR inhibitor resistance. Of note, resistant clones in cluster 1 displayed an expression pattern distinct from other clones with a more notable enrichment of the mTOR signaling pathway (Figure 3C). This is of particular interest as mTOR has been reported to be an essential gene in FGFR1-dependent cellular models and the combination of FGFR and mTOR inhibitors resulted in a synergistic effect in these models (56). Supervised analysis of gene expression changes in resistant clones further highlighted the NTRK and TAM families, as well as MAPK pathway family members, including both positive and negative regulators (Figure 3D, Supplementary Figure S5B). Of note, some of the genes that were strong mediators of resistance in the ORF screen were not overexpressed in the resistant clones (e.g. MAP3K8) suggesting that although these genes are able to mediate resistance when overexpressed they may be less likely to do so in more physiologic set-ups, such as chronic drug exposure experiments. Of note, unlike KRAS mutations, MAP3K8 mutations are a rare event in lung cancer, and MAP3K8 alterations have not yet been described in bladder cancer.
Unsupervised clustering of RT112 resistant to FGFR inhibition yielded one large cluster (Figure 3B). Among the pathways enriched in resistant clones compared to control we observed several of the same pathways identified in resistant NCI-H2077 (RAS-MAPK, ERBB, PI3K and NTRK), with RAS and MAPK signaling pathways being amongst the most enriched in resistant RT112 clones (Figure 3C, bottom panel). These gene expression studies paired with our functional genetic screen underscore the importance of RAS-MAPK reactivation in resistance to FGFR inhibition.
Resistant NCI-H2077 cells have a secondary NRAS mutation and arrest upon inhibition of the RAS-MAPK pathway
We next sought to complement our evaluation of resistance mechanisms to FGFR inhibitors by investigating whether secondary genomic events can induce acquired resistance to FGFR inhibitors. We addressed this question by generating a separate set of resistant NCI-H2077 cells with acquired resistance to FGFR inhibitors. Over several months, a separate set of NCI-H2077 cells were maintained in increasing concentrations of BGJ398, such that populations emerged that were resistant in ~5 μM of drug, hereafter referred to as H2077-R (Figure 4A). Genomic DNA was isolated from the parental and resistant cells and Illumina sequencing was performed. Notably, a NRAS Q61R mutation was identified in H2077-R at an approximate 30% allelic fraction with 148× coverage (Figure 4B, Supplementary Figure S6A). No high level amplification of known oncogenes were identified (Supplementary Figure S6B and S6C). NRAS Q61R is a known driver of cancer and a strong activator of the MAPK pathway (57). When treated with trametinib (27), a MEK inhibitor used to target MAPK pathway signaling, H2077-R cells were inhibited to a similar degree as NCI-H2077 cells (Supplementary Figure S6D). However, the combination of trametinib with a non-lethal concentration of BGJ398 resulted in significant growth suppression compared to BGJ398 treatment alone, suggesting a predominance of FGFR signaling through the MAPK pathway (Figure 4C).
Figure 4. NCI-H2077-R cells have a secondary NRAS mutation, rendering them more sensitive to inhibition of the MAPK pathway.
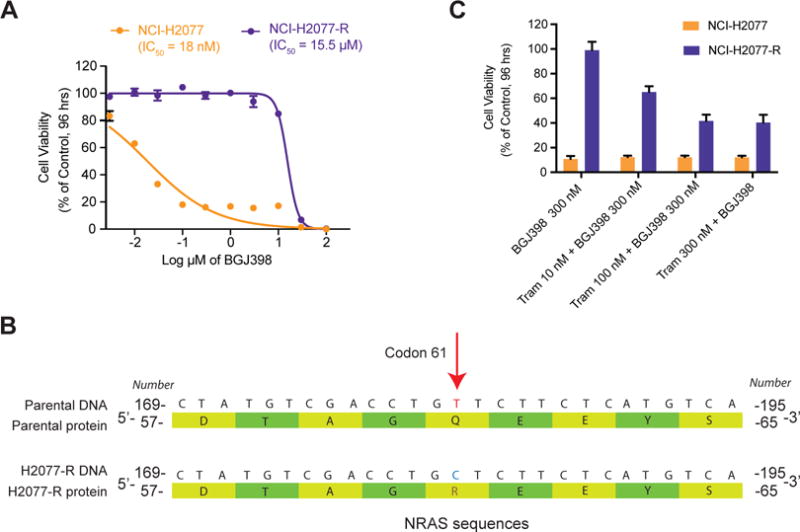
A) NCI-H2077 parental cells are sensitive to BGJ398. Treatment with increasing concentration of BGJ398 over time lead to the emergence of a resistant clone (H2077-R), which is insensitive to BGJ398.
B) Illumina sequencing analysis of 504 cancer and cancer-related genes revealed a canonical NRAS Q61R mutation in H2077-R cells (indicated by the red arrow over the sequencing reads). A schematic of the altered DNA and protein sequences is shown.
C) Cell viability assays of NCI-H2077 and H2077-R treated with BGJ398, trametinib (MEK inhibitor) or combination treatment, as indicated. Combination of non-lethal concentration of BGJ398 and increasing concentration of trametinib preferentially inhibited NCI-H2077-R cells, with modest additional effect on NCI-H2077 cells compared to BGJ398 treatment alone.
Despite escalating doses of trametinib, a subset of H2077-R cells survived even with combination treatment (Figure 4C), suggesting a cytostatic effect of trametinib, as previously described (58). We therefore performed flow cytometry to determine whether the cells treated with trametinib were predominantly in G1 phase, as would be expected if the drug had a cytostatic effect. As expected, more than 85% of NCI-H2077 and H2077-R cells were arrested in G1, with 2% or fewer cells in S phase, after treatment with 500 nM drug (Supplementary Figure S6E).
Upfront co-inhibition of FGFR and MEK enhances cellular response
The findings from our gain-of-function resistance screen and characterization of resistant clones generated by chronic exposure to FGFR inhibitors indicated a central role of RAS-MAPK signaling in mediating resistance to anti-FGFR therapy in diverse FGFR-dependent models. Therefore, we hypothesized that upfront blockade of MAPK and FGFR may suppress the emergence of resistance to FGFR inhibitors. To investigate this possibility, we performed colony formation assays in NCI-H2077 and RT112, treating with vehicle, FGFR inhibitor (BGJ398 or FIIN-3), MEK inhibitor (trametinib) or the combination of BGJ398 or FIIN-3 with trametinib for four weeks. We additionally investigated whether upfront co-inhibition of FGFR and novel mediators of resistance identified in our screen can similarly enhance the effect of anti-FGFR therapy. For these studies, we used the following inhibitors: LDC1267 (TAM family)(28), LOXO-101 (NTRK family)(29), imatinib (PDGFR and KIT)(30), MGCD265 (MET)(33), AZD8931 (pan-ERBB)(32) as well as Torin2 (mTOR)(35) and BKM120 (PI3K)(31) to explore the effects of PI3K/AKT/mTOR pathway inhibition (Supplementary Table 4).
Monotherapy with LDC1267, LOXO-101, imatinib, MGCD265, AZD8931, Torin2 and BKM120 had no appreciable impact on colony formation in NCI-H2077 and RT112 as compared to DMSO. BGJ398 and FIIN-3 had a significant impact on colony formation, with FIIN-3 further reducing colony outgrowth compared to BGJ398, which may be related to its dual FGFR/EGFR pharmacologic inhibition and underscoring the relevance of EGFR in the context of resistance to FGFR inhibitors. With the exception of imatinib, co-inhibition of FGFR and an additional kinase further decreased the outgrowth of cells to varying degrees. Consistent with prior reports in FGFR-mutant bladder carcinoma, FGFR2-mutant endometrial cancer and in FGFR1-dependent lung and head and neck squamous cell carcinoma, we also noted broad and potent suppression of colony formation with FGFR inhibitors combined with either ERBB or PI3K/AKT/mTOR pathway inhibitors (19, 24, 56, 59). However, the addition of trametinib to FGFR inhibitors yielded the most consistent and dramatic effect across the models tested, with only sparse colonies detected at 4 weeks (Figure 5A,B).
Figure 5. FGFR inhibition in combination with MEK inhibitor hinders the establishment of drug-resistant colonies in different FGFR-dependent models.
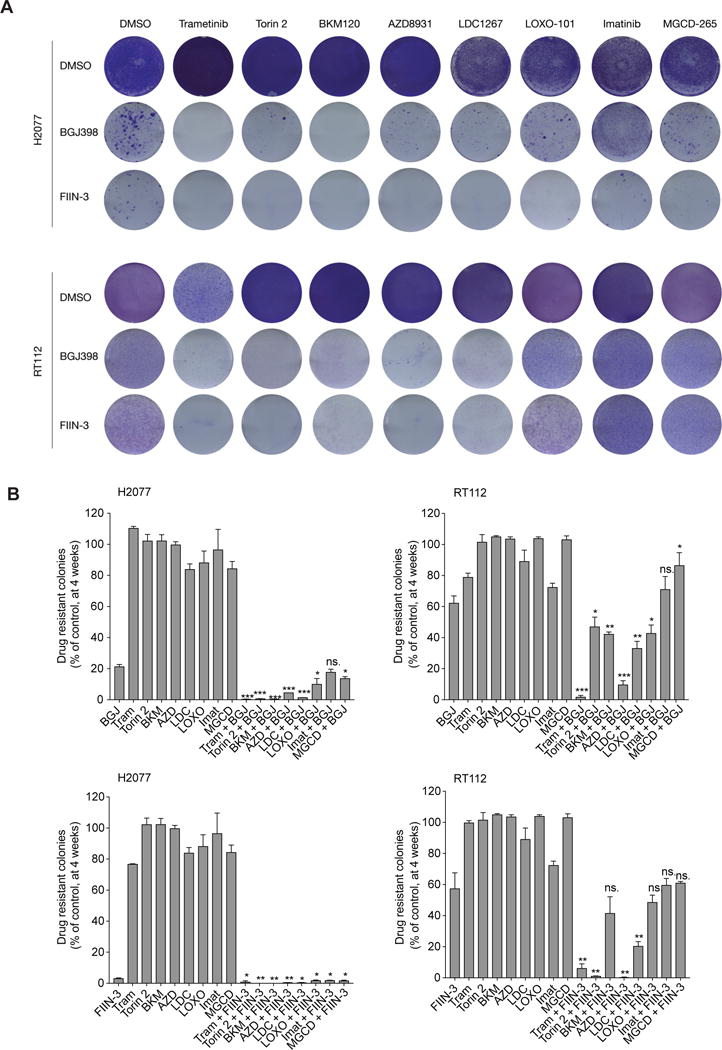
A) NCI-H2077 and RT112 cells were treated with DMSO, trametinib (MEK), Torin2 (mTOR), BKM120 (PI3K), AZD8931 (pan-ERBB), LDC1267 (TAM), LOXO-101 (NTRKs), imatinib (PDGFR), MGCD-265 (MET), BGJ398, FIIN-3 or either BGJ398 or FIIN-3 in combination with the above-mentioned kinase inhibitors. Colony formation was assayed by crystal violet staining at 4 weeks. DMSO was stained by 1 week. One representative well from a minimum of three biological replicates is shown per condition (Doses: BGJ398 at 300 nM for NCI-H2077 and 1 μM for RT112; trametinib at 100 nM; other tyrosine kinase inhibitors at 1 μM for both cell lines).
B) Quantification of colony formation in (A), shown as a percentage of the control for NCI-H2077 and RT112. Mean (3 biological replicates) +/− standard deviation (SD) shown (* p-value < 0.05, ** < 0.005, *** < 0.0005, two-sided t-test, comparing combination treatment to BGJ398 treatment). ns = not significant. (BGJ, BGJ398; Tram, Trametinib; BKM, BKM120; AZD, 8931; LDC, LDC1267; LOXO, LOXO-101; Imat, Imatinib; MGCD, MGCG-265).
We similarly evaluated the capability of BGJ398 plus trametinib to block the emergence of resistance in additional FGFR dependent models (AN3 CA, FGFR2-mutated endometrial cancer; NCI-H520, FGFR1-amplified squamous cell lung carcinoma; DMS114, FGFR1-amplified small cell lung cancer). Consistent with our prior observation, dual blockade of FGFR and MEK was highly effective in preventing the emergence of resistance across different dependencies and lineages (Figure 6A).
Figure 6. BGJ398 in combination with Trametinib hinders the establishment of drug-resistance.
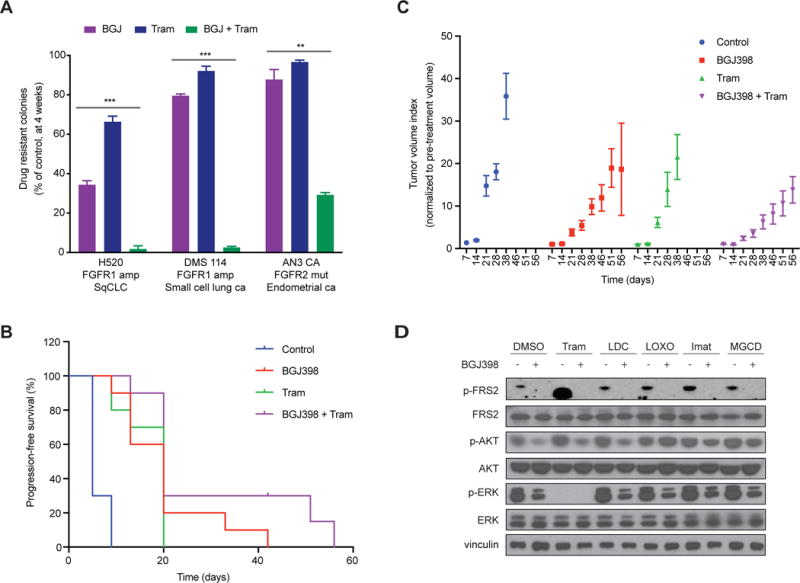
A) Quantification of colony formation for NCI-H520, DMS114 and AN3 CA cell lines treated for 4 weeks with BGJ398 (BGJ), trametinib (Tram) or combination (BGJ + Tram), shown as a percentage of the control. Mean (3 biological replicates) +/− standard deviation (SD) shown (* p-value < 0.05, ** < 0.005, *** < 0.0005, two-sided t-test).
B) Tumor volume index normalized to pre-treatment volume for NCI-H2077 xenografts treated with the indicated drugs for 8 weeks (n=5 mice in each treatment group, equivalent to 10 tumors in each group). Mean +/− SEM shown.
C) Progression-free survival for NCI-H2077 xenografts treated as in (B). Progression-free survival over time is shown as a percentage for each treatment group. Log-rank (Mantel-Cox) test analysis (p-value<0.0001).
D) Immunoblot analysis for FRS2, AKT and ERK activity for NCI-H2077 cells treated for 48 hours with DMSO, BGJ398, or additional kinase inhibitors as indicated, or combination treatment as indicated. (Tram, Trametinib; LDC, LDC1267; LOXO, LOXO-101; Imat, Imatinib; MGCD, MGCG-265).
To assess the efficacy and toxicity of the FGFR and MEK inhibitor combination in vivo, we performed xenograft studies using the NCI-H2077 cellular model (Figure 6B,C, and Supplementary Figure S7). Tumor-bearing mice were treated with a) vehicle, b) BGJ398, c) trametinib, or d) combination treatment. BGJ398 in combination with trametinib retarded tumor progression compared to FGFR inhibition or trametinib alone and significantly extended progression-free survival (log-rank test < 0.0001). Importantly, combination therapy was well tolerated, with no weight loss or behavioral changes observed (Supplementary Figure S7).
Next, we sought to determine the mechanisms by which co-inhibition of FGFR and MEK, or additional receptor tyrosine kinases, may prevent the emergence of resistance. To address this, we performed immunoblots to consider ERK and AKT activation in NCI-H2077 cells (Figure 6D). Dual blockade of FGFR and MEK strongly suppressed ERK phosphorylation, while co-blockade of TAMs, NTRKs, PDGFR and MET did not significantly affect phosphorylation of either ERK or AKT at 48 hours. These results support the colony formation results which showed superiority of dual blockade with FGFR and MEK in comparison to dual blockade with FGFR or an additional RTK, such as PDGFR, TAM, NTRK or MET. These findings corroborate the importance of effective RAS-MAPK signaling suppression in potentiating the effect of FGFR inhibition and preventing the emergence of resistance.
Together, our genetic and pharmacologic studies provide a rationale for upfront FGFR–MEK inhibitor polytherapy to enhance the initial therapeutic response and to suppress or delay the onset of acquired resistance in diverse FGFR-dependent models.
Discussion
The successful application of anti-FGFR therapy in the clinic has proven remarkably challenging with only modest efficacy of single-agent FGFR inhibitor therapy, particularly in FGFR1-amplified cancers (15, 16). Understanding the mechanisms leading to drug resistance is critical for the design of novel therapeutic strategies to improve efficacy. Here, we report the results of an unbiased genome-wide gain-of-function screen, employing barcoded ORFs in a pooled format, to systematically characterize mediators of resistance to FGFR inhibitors. Some of the potential drivers of resistance identified in our screen represent proteins or pathways previously implicated in resistance to FGFR inhibitors including MET, EGFR and AKT2 (18, 22, 24). These findings underscore the capacity of a functional genomic approach using appropriate cellular models to identify biologically relevant resistance mechanisms. Our screen also identified novel mechanisms of resistance involving members of the TAM, NTRK, PDGFR, FLT3, KIT and RAS-MAPK pathways. Overexpression of wild-type AXL, TYRO3, NTRK1 and MAPK genes consistently and strongly promoted resistance across cellular models with diverse FGFR dependencies and lineages (e.g. FGFR1-amplified lung cancer, FGFR3-TACC3 fusion bladder cancer, FGFR2-mutated endometrial cancer), with HRAS wild type having the strongest rescue effect.
MAPK activation was the most generalizable mechanism of resistance across models to both BGJ398 and FIIN-3, further supporting the rationale for upfront FGFR-MAPK blockade (Figure 1G). Our data further suggests that resistance mediated by receptor tyrosine kinases is, in contrast, more context dependent, with significant variability across different genomic backgrounds and lineages, underscoring the clinical challenge of rational combination with dual RTK inhibition. Interestingly, an exception to this context dependence was AXL (and to a lesser degree TYRO3 and NTRK1), which appeared to be a more broadly generalizable resistance mediator to FGFR inhibition. AXL has been linked to epithelial-mesenchymal transition, drug resistance and metastasis in several cancer types, including EGFR-dependent NSCLC, ALK-rearranged and lung cancer, as well as BRAF mutated melanoma (47, 49, 50, 60, 61). Interestingly, expression profiling of resistant clones of NCI-H2077 and RT112 showed high upregulation of the TAM ligands, GAS6 and PROS1. Likewise, NTRK1 overexpression strongly promoted resistance to FGFR inhibitors in different FGFR-dependent models, and the neurotrophin signaling pathway was also enriched in FGFR inhibitor resistant clones. Biochemical studies revealed that overexpression of these mediators of resistance result in persistent activation of ERK, despite FGFR inhibition, supporting a concept in which a subset of RTKs can promote resistance to FGFR inhibitors by sustaining MAPK activation when FGFR signaling is blocked. Furthermore, gene expression profiling and DNA sequencing analysis of resistant clones supported the key role of the RAS-MAPK family in mediating resistance to anti-FGFR therapy.
Our screen was performed in a pooled format which compared to arrayed screens greatly facilitates the screening process and makes the query of a near-genome scale pool of ORFs feasible. However, one limitation we noted was the inability to detect ligands as mediators of resistance in the pooled format, likely secondary to the dilution of ligands coming from only a small population of cells. We attempted to address this limitation in our validation studies by including relevant ligand ORFs for the RTKs identified in the initial pooled screen. Additionally, as our discovery genome-wide screen was performed in an FGFR1-amplified model, there may be additional mechanisms uniquely relevant to other FGFR-dependent cancers which were not detected in our study. For instance, ERBB2/3 was not identified as candidate mediator of resistance in our screen despite its contribution to resistance in FGFR3-dependent models, but not FGFR1 or FGFR2-dependent models, based on prior work from our group (62).
We observed a high overlap between drivers of resistance for reversible and covalent FGFR inhibitors, with differences primarily accounted by different spectrum of pharmacological activity. Activating EGFR mutants, for instance, were one of the strongest mediators of resistance to BGJ398, but only rescued FIIN-3 at low doses of this inhibitor. EGFR ligands (e.g. BTC, AREG) also mediated rescue from FGFR inhibition. Our data are consistent with prior literature demonstrating that the ERBB family can mediate rescue to FGFR inhibition (18, 23, 34, 62). The converse has also been shown to be true, with FGFR and FGFR ligands mediating rescue from EGFR inhibitors in the setting of EGFR-driven cancers (20, 63-65). These data support the further development of dual inhibitors of FGFR and EGFR, such as FIIN-3. Of further note, the number of candidate mediators of resistance to FIIN-3 was considerably lower than to BGJ398, and FIIN-3 monotherapy was more efficacious at suppressing colony formation than BGJ398. These findings can partly be explained by the additional targets of FIIN-3 (e.g. EGFR, RET), however may also reflect differences due to covalent vs reversible inhibition of FGFR in hampering resistance emergence. To our knowledge, this is the first study to consider an effect on resistance emergence between reversible and irreversible FGFR inhibitors. Further work is however necessary to investigate whether this is a phenomenon applicable to additional settings and the mechanism behind it.
As previously noted, BGJ398 is a potent inhibitor of FGFR1-3, but not FGFR4. FGFR4 is structurally distinct from FGFR1-3, explaining why the majority of advanced clinical stage FGFR1-3 inhibitors (BGJ398, AZD4547, JNJ493) are not potent FGFR4 inhibitors and, conversely, why clinical stage FGFR4 inhibitors (BLU-554, FGF401) are not potent FGFR1-3 inhibitors (66, 67). FIIN-3, however, has the unique ability to also inhibit FGFR4 with low nanomolar potency (in addition to FGFR1-3). We did not explore resistance mechanisms in FGFR4-dependent models in our work, however it is plausible that the resistance mechanisms identified to FIIN-3 are applicable to FGFR4-driven cancers. Future studies extending validation to FGFR4-dependent models would be valuable, in particular as to our knowledge resistance to FGFR4 inhibitors remains unexplored.
Gene expression profiling of resistant clones of NCI-H2077 and RT112 demonstrated enrichment of several pathways associated with genes identified in our functional ORF screen including RAS, MAPK, ERBB, PI3K and NTRK, with RAS-MAPK signaling pathway being one of the most enriched in both models, supporting the relevance of the RAS-MAPK family in mediating resistance to FGFR inhibition. Importantly, some of the differences in expression profiles of NCI-H2077 and RT112 may be accounted by differences in heterogeneity in the initial population. Our group has previously shown that RT112 rapidly becomes resistant to FGFR inhibition by upregulation of ERBB2/3 expression (19). However, the elevated ERBB2/3 expression may also represent selection of a pre-existing subclone. NCI-H2077 may represent a more homogenous population that requires additional mutation acquisition and thereby a longer time period to develop resistance. Based on our data, however, we can only hypothesize regarding the observed differences, as further characterization of the initial population at the single cell level would be required to address the question of upfront heterogeneity in the initial populations.
Concurrent upfront blockade of additional RTKs enhanced the efficacy of FGFR inhibitors modestly (Figure 5A,B). An exception to this was pan-ERBB inhibition, which yielded quite potent suppression of colony formation in combination with either BGJ398 or FIIN-3, in both RT112 and NCI-H2077, which is consistent with prior reports (19). We furthermore noted dramatic and consistent suppression of colony formation with FGFR inhibitors combined with PI3K/AKT/mTOR pathway inhibitors (19, 24, 56, 59). FGFR-MEK co-inhibition, however, led to the most significant decrease of colony formation across FGFR-dependent models, and led to sustained ERK inhibition, suggesting that re-activation of the MAPK pathway is critical to resistance establishment in FGFR-dependent cancers. Conversely, a recent study employing a short-hairpin RNA screen in KRAS-mutant lung cancer identified FGFR1 as a mediator of adaptive resistance to trametinib and showed that combinatorial blockade of MEK-FGFR was effective in preventing resistance (68). Similarly, Lee and colleagues have shown, across a diverse range of oncogenic dependencies, that MEK inhibition leads to STAT3 activation via FGFR signaling, leading to drug resistance (64). These data suggest that dual FGFR-MAPK inhibition may be of value across a broad range of cancer types.
Here we show that several RTKs can redundantly re-activate RAS-MAPK and combined upstream co-inhibition only partially prevents resistance to FGFR inhibitors. However, FGFR inhibition combined with downstream blockade of MEK provides a more robust and powerful approach in preventing resistance across diverse FGFR-dependencies, and may represent a therapeutic opportunity to achieve durable responses to FGFR inhibition.
Supplementary Material
Acknowledgments
We thank David Lahr from the Broad Institute (Cambridge, MA) for performing L1000 gene expression analysis.
Financial Support: This work was supported by NIH Grant R01CA196932 (A.J. Bass); by the American Society of Clinical Oncology Conquer Cancer Foundation - Young Investigator Award (B. Bockorny); by the Eliteforsk scholarship - Danish Council for Independent Research (M. Rusan); PhD stipend from Aarhus University, Denmark (M. Rusan); and by the Lundbeck Foundation Postdoctoral Research Grant (M. Rusan).
Conflict of interest disclosure statement: G.I.S is on advisory boards for Pfizer, Lilly, G1 Therapeutics, Vertex Pharmaceuticals and Roche and has research funding from Pfizer and Lilly. Dana-Farber Cancer Institute receives funding for the conduct of trials of FGFR inhibitors, including BGJ398, on which G.I.S. is an investigator. P.S.H. co-supervised this work while employed at Dana-Farber Cancer Institute but has since transitioned to a position at Novartis, which manufactures BGJ398.
References
- 1.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One. 2011;6(6):e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramos AH, Dutt A, Mermel C, Perner S, Cho J, Lafargue CJ, et al. Amplification of chromosomal segment 4q12 in non-small cell lung cancer. Cancer Biol Ther. 2009;8(21):2042–50. doi: 10.4161/cbt.8.21.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courjal F, Cuny M, Simony-Lafontaine J, Louason G, Speiser P, Zeillinger R, et al. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res. 1997;57(19):4360–7. [PubMed] [Google Scholar]
- 6.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105(25):8713–7. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68(7):2340–8. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- 8.Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26(50):7158–62. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Ding X, Wang S, Moser CD, Shaleh HM, Mohamed EA, et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 2016;380(1):163–73. doi: 10.1016/j.canlet.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–47. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang R, Wang L, Li Y, Hu H, Shen L, Shen X, et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer. Clin Cancer Res. 2014;20(15):4107–14. doi: 10.1158/1078-0432.CCR-14-0284. [DOI] [PubMed] [Google Scholar]
- 12.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231–5. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25(3):552–63. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013;73(16):5195–205. doi: 10.1158/0008-5472.CAN-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J Clin Oncol. 2017;35(2):157–65. doi: 10.1200/JCO.2016.67.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol. 2015;33(30):3401–8. doi: 10.1200/JCO.2014.60.7341. [DOI] [PubMed] [Google Scholar]
- 17.Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017;7(3):252–63. doi: 10.1158/2159-8290.CD-16-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrera-Abreu MT, Pearson A, Campbell J, Shnyder SD, Knowles MA, Ashworth A, et al. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013;3(9):1058–71. doi: 10.1158/2159-8290.CD-12-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Mikse O, Liao RG, Li Y, Tan L, Janne PA, et al. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene. 2015;34(17):2167–77. doi: 10.1038/onc.2014.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. 2013;11(7):759–67. doi: 10.1158/1541-7786.MCR-12-0652. [DOI] [PubMed] [Google Scholar]
- 21.Ware KE, Hinz TK, Kleczko E, Singleton KR, Marek LA, Helfrich BA, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. doi: 10.1038/oncsis.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SM, Kim H, Yun MR, Kang HN, Pyo KH, Park HJ, et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis. 2016;5(7):e241. doi: 10.1038/oncsis.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harbinski F, Craig VJ, Sanghavi S, Jeffery D, Liu L, Sheppard KA, et al. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012;2(10):948–59. doi: 10.1158/2159-8290.CD-12-0237. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Sustic T, Leite de Oliveira R, Lieftink C, Halonen P, van de Ven M, et al. A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur Urol. 2017 doi: 10.1016/j.eururo.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 25.Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, et al. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene. 2013;32(25):3059–70. doi: 10.1038/onc.2012.319. [DOI] [PubMed] [Google Scholar]
- 26.Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem. 2011;54(20):7066–83. doi: 10.1021/jm2006222. [DOI] [PubMed] [Google Scholar]
- 27.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 28.Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508–12. doi: 10.1038/nature12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19(11):1469–72. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1(7):493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 31.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–28. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 32.Hickinson DM, Klinowska T, Speake G, Vincent J, Trigwell C, Anderton J, et al. AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin Cancer Res. 2010;16(4):1159–69. doi: 10.1158/1078-0432.CCR-09-2353. [DOI] [PubMed] [Google Scholar]
- 33.Engstrom LD, Aranda R, Lee M, Tovar EA, Essenburg CJ, Madaj Z, et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin Cancer Res. 2017;23(21):6661–72. doi: 10.1158/1078-0432.CCR-17-1192. [DOI] [PubMed] [Google Scholar]
- 34.Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc Natl Acad Sci U S A. 2014;111(45):E4869–77. doi: 10.1073/pnas.1403438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, et al. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem. 2011;54(5):1473–80. doi: 10.1021/jm101520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8(8):659–61. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–91. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171(6):1437–52 e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 40.Benjamini Y, H Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. 1995;57(1):289–330. [Google Scholar]
- 41.Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9(3):e92444. doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res. 2012;18(3):748–57. doi: 10.1158/1078-0432.CCR-11-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ang D, Ballard M, Beadling C, Warrick A, Schilling A, O’Gara R, et al. Novel Mutations in Neuroendocrine Carcinoma of the Breast: Possible Therapeutic Targets. Diagn Mol Pathol. 2014 doi: 10.1097/PDM.0b013e3182a40fd1. [DOI] [PubMed] [Google Scholar]
- 44.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Datta J, Damodaran S, Parks H, Ocrainiciuc C, Miya J, Yu L, et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol Cancer Ther. 2017 doi: 10.1158/1535-7163.MCT-15-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27(3):397–408. doi: 10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer Discov. 2016;6(10):1134–47. doi: 10.1158/2159-8290.CD-16-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharifnia T, Rusu V, Piccioni F, Bagul M, Imielinski M, Cherniack AD, et al. Genetic modifiers of EGFR dependence in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2014;111(52):18661–6. doi: 10.1073/pnas.1412228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leiser D, Medova M, Mikami K, Nisa L, Stroka D, Blaukat A, et al. KRAS and HRAS mutations confer resistance to MET targeting in preclinical models of MET-expressing tumor cells. Mol Oncol. 2015;9(7):1434–46. doi: 10.1016/j.molonc.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hah JH, Zhao M, Pickering CR, Frederick MJ, Andrews GA, Jasser SA, et al. HRAS mutations and resistance to the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in head and neck squamous cell carcinoma cells. Head Neck. 2014;36(11):1547–54. doi: 10.1002/hed.23499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–33. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Chih-Hsin Yang J. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J Thorac Oncol. 2017;12(3):567–72. doi: 10.1016/j.jtho.2016.11.2231. [DOI] [PubMed] [Google Scholar]
- 55.Xie S, Li Y, Li X, Wang L, Yang N, Wang Y, et al. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget. 2015;6(11):9206–19. doi: 10.18632/oncotarget.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singleton KR, Hinz TK, Kleczko EK, Marek LA, Kwak J, Harp T, et al. Kinome RNAi Screens Reveal Synergistic Targeting of MTOR and FGFR1 Pathways for Treatment of Lung Cancer and HNSCC. Cancer Res. 2015;75(20):4398–406. doi: 10.1158/0008-5472.CAN-15-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janowski M. ras proteins and the ras-related signal transduction pathway. Radiat Environ Biophys. 1991;30(3):185–9. doi: 10.1007/BF01226615. [DOI] [PubMed] [Google Scholar]
- 58.Watanabe M, Sowa Y, Yogosawa M, Sakai T. Novel MEK inhibitor trametinib and other retinoblastoma gene (RB)-reactivating agents enhance efficacy of 5-fluorouracil on human colon cancer cells. Cancer Sci. 2013;104(6):687–93. doi: 10.1111/cas.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Packer LM, Geng X, Bonazzi VF, Ju RJ, Mahon CE, Cummings MC, et al. PI3K Inhibitors Synergize with FGFR Inhibitors to Enhance Antitumor Responses in FGFR2mutant Endometrial Cancers. Mol Cancer Ther. 2017;16(4):637–48. doi: 10.1158/1535-7163.MCT-16-0415. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–60. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Antony J, Huang RY. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017;77(14):3725–32. doi: 10.1158/0008-5472.CAN-17-0392. [DOI] [PubMed] [Google Scholar]
- 62.Wang J, Mikse O, Liao RG, Li Y, Tan L, Janne PA, et al. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene. 2014 doi: 10.1038/onc.2014.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget. 2014;5(15):5908–19. doi: 10.18632/oncotarget.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207–21. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 65.Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, et al. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS One. 2010;5(11):e14117. doi: 10.1371/journal.pone.0014117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015;5(4):424–37. doi: 10.1158/2159-8290.CD-14-1029. [DOI] [PubMed] [Google Scholar]
- 67.Graus Porta DWA, Fairhurst RA, Wartmann M, Stamm C, Reimann F, Buhles A, Kinyamu-Akunda J, Sterker D, Murakami M, Wang Y, Engelman J, Hofmann F, Sellers WR. NVP-FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of HCC. AACR: AACR. 2017 doi: 10.1158/1535-7163.MCT-18-1291. [DOI] [PubMed] [Google Scholar]
- 68.Manchado E, Weissmueller S, Morris JPT, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534(7609):647–51. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.