

Abstract
Background
The genetic code is known to be efficient in limiting the effect of mistranslation errors. A misread codon often codes for the same amino acid or one with similar biochemical properties, so the structure and function of the coded protein remain relatively unaltered. Previous studies have attempted to address this question quantitatively, by estimating the fraction of randomly generated codes that do better than the genetic code in respect of overall robustness. We extended these results by investigating the role of amino-acid frequencies in the optimality of the genetic code.
Results
We found that taking the amino-acid frequency into account decreases the fraction of random codes that beat the natural code. This effect is particularly pronounced when more refined measures of the amino-acid substitution cost are used than hydrophobicity. To show this, we devised a new cost function by evaluating in silico the change in folding free energy caused by all possible point mutations in a set of protein structures. With this function, which measures protein stability while being unrelated to the code's structure, we estimated that around two random codes in a billion (109) are fitter than the natural code. When alternative codes are restricted to those that interchange biosynthetically related amino acids, the genetic code appears even more optimal.
Conclusions
These results lead us to discuss the role of amino-acid frequencies and other parameters in the genetic code's evolution, in an attempt to propose a tentative picture of primitive life.
Background
One of the tantalizing questions raised by molecular biology is whether the basic structures of life as we know them arose through a Darwinian evolutionary process and, if so, what were the evolutionary pressures acting on them? One such structure that could have changed during evolution is the genetic code. The genetic code was initially believed to be universal throughout all living things [1], even though some variations in both nuclear and mitochondrial systems have recently been found (see [2] for a review). These variations are, however, limited and correspond essentially to the reassignment of one or a few codons to another amino acid. The genetic code may thus be considered as fairly universal.
The idea that the genetic code could have evolved to its present form has been repeatedly suggested [3]. For instance, it has been proposed that early codes were simpler, in that they coded for only a few amino acids, and that the number of amino acids coded in the genetic code increased as the code evolved [4,5,6,7]. Several hypotheses have been put forward to explain the evolution of the genetic code to its present form, and to find out what the genetic code is optimized for [6,8,9,10,11,12,13,14,15,16,17]. One possible scenario is that the genetic code evolved so as to minimize the consequence of errors during transcription and translation [9,10,11,12,13,18]. To test this hypothesis, some researchers have tried to estimate the percentage of optimal achievement of the natural code by quantifying the cost of single-base changes [19,20,21].
More recently, Haig and Hurst [22] and Freeland and Hurst [23] improved that approach by comparing the natural code with random codes. To this end, they defined a fitness function, Φ, that measures the efficiency of the code in limiting the consequences of transcription and translation errors. This function Φ supposedly evolved towards a minimum through evolution. To measure how close the natural code is to the actual minimum of Φ, they generated random genetic codes, and computed the fraction of those that are better - that is, have a smaller value of Φ - than the natural code. They found that only a very small fraction of the random codes are better than the natural code, and concluded that the natural code is therefore optimal in that it minimizes the effect of translation and transcription errors.
Haig and Hurst [22] tested several fitness functions, Φ, based on different physicochemical parameters, and found that single-base changes in the natural code had the smallest average effect when using, as a cost measure, the change in polarity or hydropathy between the corresponding amino acids. These parameters, although not unique, are clearly biologically relevant, as they are related to hydrophobicity, a property known to be important in protein conformation (see, for example, [24,25] for reviews). Changing, through a transcription or a translation error, a nonpolar amino acid into a polar one at some strategic position in the sequence of a protein can have dramatic consequences on its conformation. Using these parameters, and assuming that all point mutations occur with the same frequency, Haig and Hurst [22] found that the fraction of random codes that beat the natural code is of the order of 10-4.
It has been shown experimentally that individual translation errors occur more frequently at the first and third codon positions than at the second [10,26,27], and that there are transition/transversion biases [28,29,30,31]. Taking this into account, Freeland and Hurst [23] proposed a modified fitness function Φ, which models more accurately the probability of translation errors. They found that with this improved modeling, the fraction of random genetic codes that are better than the natural one decreases from 10-4 to 106. They retrieved from their calculations a well known property of the genetic code: single-base substitutions in the first and third codon position are strongly conservative with respect to changes in polarity [10,32]. Here, we highlight the importance of another parameter in the optimization of the genetic code, namely the frequency at which different amino acids occur in proteins. This frequency differs from protein to protein, and even from species to species, but there is a general pattern that prevails (Table 1). In Figure 1, we have plotted the number of codons coding for the same amino acid (synonyms) versus the amino-acid frequency. The correlation between these two quantities, first noted by King and Jukes [33], led us to suspect that the amino-acid frequency is an important parameter in the optimization of the genetic code, which should also be taken into account in the fitness function Φ. Our calculations indeed confirm that the genetic code is even more optimal with respect to translation errors if the amino-acid frequencies of Table 1 are properly incorporated in Φ.
Table 1.
The mean frequencies of the individual amino acids (p(a)) in the genomes of living organisms
| Amino acid | p(a) Archaea (%) | p(a) Bacteria (%) | p(a) Eukaryotes (%) | p(a) (%) |
| Ala | 7.85 (2.27) | 8.08 (2.61) | 6.48 (0.76) | 7.80 (2.38) |
| Arg | 5.92 (1.15) | 4.99 (1.61) | 5.24 (0.49) | 5.23 (1.43) |
| Asp | 5.47 (1.57) | 5.06 (0.42) | 5.31 (0.35) | 5.19 (0.81) |
| Asn | 3.40 (1.05) | 4.63 (1.97) | 4.76 (0.90) | 4.37 (1.73) |
| Cys | 0.89 (0.32) | 1.00 (0.31) | 1.86 (0.35) | 1.10 (0.44) |
| Glu | 7.79 (1.13) | 6.35 (1.21) | 6.64 (0.28) | 6.72 (1.24) |
| Gln | 1.90 (0.40) | 3.89 (0.95) | 4.28 (0.69) | 3.45 (1.19) |
| Gly | 7.49 (0.75) | 6.70 (1.46) | 5.88 (0.72) | 6.77 (1.32) |
| His | 1.70 (0.29) | 2.07 (0.39) | 2.41 (0.21) | 2.03 (0.41) |
| Ile | 7.59 (2.19) | 7.05 (2.26) | 5.48 (0.92) | 6.95 (2.16) |
| Leu | 9.65 (1.00) | 10.52 (0.66) | 9.35 (0.42) | 10.15 (0.86) |
| Lys | 6.04 (2.75) | 6.43 (2.78) | 6.30 (0.69) | 6.32 (2.53) |
| Met | 2.49 (0.47) | 2.19 (0.37) | 2.33 (0.21) | 2.28 (0.39) |
| Phe | 4.00 (0.74) | 4.57 (0.97) | 4.20 (0.59) | 4.39 (0.89) |
| Pro | 4.43 (0.92) | 3.99 (1.00) | 5.15 (0.75) | 4.26 (1.01) |
| Ser | 5.93 (1.11) | 6.18 (0.77) | 8.50 (0.47) | 6.46 (1.17) |
| Thr | 4.77 (0.89) | 5.15 (0.63) | 5.57 (0.32) | 5.12 (0.69) |
| Trp | 1.03 (0.20) | 1.10 (0.28) | 1.13 (0.12) | 1.09 (0.25) |
| Tyr | 3.68 (0.66) | 3.23 (0.64) | 3.03 (0.26) | 3.30 (0.63) |
| Val | 7.97 (0.85) | 6.87 (1.19) | 6.09 (0.42) | 7.01 (1.18) |
The frequencies p(a) were computed as averages over the frequencies observed in genomes of archaea (Aeropyrum pernix K1, Archaeoglobus fulgidus,Halobacterium sp. NRC-1, Methanococcus jannaschii, Methanobacterium thermoautotrophicum, Pyrococcus abyssi, Pyrococcus horikoshi and Thermoplasma acidophilum), bacteria (Aquifex aeolicus, Bacillus halodurans, Bacillus subtilis, Borrelia burgdorferi, Buchnera aphidicola, Campylobacter jejuni, Chlamydia trachomatis, Deinococcus radiodurans, Escherichia coli K-12, Haemophilus influenzae, Mycobacterium leprae, Mycobacterium tuberculosis, Mycoplasma genitalium, Mycoplasma pneumoniae, Pasteurella multocida, Pseudomonas aeruginosa, Rickettsia prowazekii, Thermotoga maritima, Treponema pallidum, Ureaplasma parvum, Vibrio cholerae and Xylella fastidiosa) and eukaryotes (Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens and Saccharomyces cerevisiae). The last column contains the average frequencies p(a) computed from all these genomes. The standard deviation of the distributions is given in parentheses.
Figure 1.
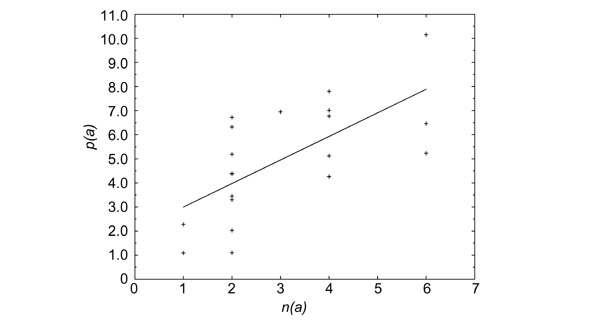
The relative frequency p(a) (in %) of amino acid a (right-hand column of Table 1), as a function of the number of synonyms n(a) that code for it. The linear regression line is indicated; the correlation coefficient is equal to 0.66.
In addition, we bring further improvements to Φ by using quantities other than polarity to measure the roles of the different amino acids in protein conformation and stability. It should be stressed that the biological relevance of the parameters used in Φ is crucial in the estimation of the relative robustness of the natural code. Indeed, one can always construct an artificial fitness function Φ such that the natural biological structure apparently lies at its minimum. Clearly, the hydrophobicity parameters used by Haig and Hurst [22] are biologically motivated, but we would like to do better by refining our cost measure. In particular, we have devised a mutation matrix describing the average cost of single amino-acid substitutions in protein stability, obtained by computer experiments. This mutation matrix combines many different physicochemical properties of the amino acids. For instance, it takes into account that mutating cysteine into any other amino acid may be very costly as it may break a disulfide bond. Such an effect would not be apparent if only a single property, say hydrophobicity, was taken into account. We show that, with a fitness function Φ depending on this mutation matrix and the amino-acid frequencies, only about two out of 109 randomly generated codes are better than the natural code. This suggests that the genetic code is even better optimized to limit translation errors than was previously thought.
Results
Fitness of the genetic code with respect to translation errors
Consider the natural genetic code. It is built out of 64 codons, each consisting of three consecutive DNA bases (A, G, C, T) or RNA bases (A, G, C, U). These 64 codons are divided into 21 sets of synonyms, which each code for one of the 20 natural amino acids or correspond to a stop signal; hence, to each codon, c, an amino acid (or stop signal) a is assigned through a function a(c). Consider now an error during transcription from DNA to RNA or during translation from RNA to protein, in which codon c is mistaken for codon c'. This error thus results in amino acid a(c) being replaced by amino acid a' = a(c'). The associated cost is estimated by a function g(a,a'), which measures the difference between the amino acids a and a' with respect to their physicochemical properties or their role in (de)stabilizing protein structures; when a or a' corresponds to a stop codon, we set g(a,a') = 0. Different cost functions g will be discussed in the next section. Following Freeland and Hurst [23], the fitness Φ of a code is measured by the average of the cost g over all codons c and all single-base errors c -> c'
![]()
where p(c' | c) is the probability of misreading codon c as codon c'. If one focuses on transcription errors only, as do Haig and Hurst in [22], then all p(c'| c) values must be taken as equal. But here we consider translation errors, as do Free-land and Hurst in [23], and hence p(c' | c) changes according to whether c and c' differ in the first, second or third base, and lead to a transition or a transversion. A transition is the substitution of a purine (A, G) into another purine, or a pyrimidine (C, U/T) into another pyrimidine, whereas a transversion interchanges purines and pyrimidines. On the basis of experimental data indicating that transitions are more common than transversions [28,29,30,31], and that errors on the third base are more frequent than errors on the first base, which are themselves more frequent than errors on the second base [10,26,27], Freeland and Hurst [23] have chosen the following values of p(c' |c), which we also use here:
p(c' | c) = 1/N if c and c' differ in the 3rd base only,
p(c' | c) = 1/N if c and c' differ in the 1st base only and cause a transition,
p(c' | c) = 0.5/N if c and c' differ in the 1st base only and cause a transversion,
p(c' | c) = 0.5/N if c and c' differ in the 2nd base only and cause a transition,
p(c' | c) = 0.1/N if c and c' differ in the 2nd base only and cause a transversion,
p(c' | c) = 0 if c and c' differ by more than 1 base.
where N is a normalization factor ensuring that Σc'p(c' | c) = 1. Obviously, these probabilities only roughly approximate the true transition/transversion and base position biases. However, the computed fitness of the genetic code has been shown to be relatively insensitive to their precise values [34].
Incorporating amino-acid frequencies in the fitness function
Let us now return to the correlations between the number of codons coding for an amino acid and the frequency of this amino acid (see Figure 1). King and Jukes [33], who first noted this correlation, suggested that most of the amino acids in genomes have arisen by random mutations that do not affect the properties and function of the proteins. As a consequence, the number of synonymous codons determines the frequency of amino acids.
An alternative interpretation, assuming a very different chain of causality, is that the amino-acid frequencies are fixed by their physicochemical properties. For instance, tryptophan would be a rare amino acid because its specific properties are seldom needed in proteins or because it is difficult to synthesize. The correlation between the amino-acid frequencies and number of synonymous codons (Figure 1) would then be interpreted as being due to an adjustment of the natural genetic code to the frequency of the amino acids. The conclusions reached using these two opposite interpretations are addressed in the Discussion.
Independent of the assumed chain of causality, it is natural to expect that a codon error replacing a frequent amino-acid type with another leads to more absolute errors and thus has more consequences, at least on average, than an error affecting a rare amino acid. The frequencies with which the different amino acids occur in proteins, which are similar in different organisms (Table 1), are only imperfectly taken into account in the fitness function ΦFH given by Equation (1), because of the imperfect correlation between amino-acid frequency and number of synonymous codons (Figure 1). To account properly for the amino-acid frequencies, we propose a modified fitness function Φfaa:
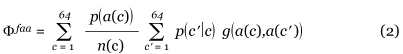
where p(a) is the relative frequency of amino acid a, and n(c) is the number of codons in the block to which c belongs. In other words, n(c) is the number of synonyms coding for the amino acid a(c) that c codes for. Note that Equation (2) supposes that there is no codon bias, that is, the different synonyms of a given amino acid appear with the same frequency.
To measure the effect of the amino-acid frequency on the value of the fitness function Φfaa, we define, for the sake of comparison, another fitness function Φequif where all the amino acids are supposed equally frequent, that is, p(a) = 1/20:
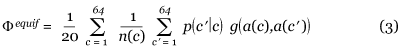
Cost of substituting an amino acid with another
The function g(a,a') in Equations (1) and (2) measures the cost - as far as protein stability and structure is concerned - of substituting amino acid a by a'. This cost depends on several physicochemical and energetic factors. Hydrophobic interactions are known to constitute a dominating energetic contribution to protein stability. Hence, a natural choice for g consists of taking the squared difference in hydrophobicity h of the amino acids a and a':
ghydro (a, a') = (h(a) - h(a'))2 (4)
There exist various hydrophobicity scales for amino acids. We have tested two of them. The first is the polarity scale defined by Woese et al. [35], which is the one used by Haig and Hurst [22] and Freeland and Hurst [23]. In the second scale, h(a) is the average solvent accessibility of amino acid a derived from a set of 141 well resolved and refined protein structures with low sequence identity (see Methods); solvent accessibilities are computed using SurVol [36]. We denote the associated cost functions as gpol and gaccess, respectively.
Although hydrophobic forces dominate in proteins, other types of interactions also contribute to protein stability (see [24,25] for reviews). We therefore also attempted to devise a better cost function g(a,a'), measuring more accurately the difference between amino acids a and a'. This new function is inspired by recent computations of the change in free energy of a protein when a single amino acid is mutated [37,38,39]. It is obtained by mutating in silico, in all proteins of the aforementioned set of 141 protein structures and at all positions, the wild-type amino acids into the 19 other possible ones, and evaluating the resulting changes in folding free energy with mean force potentials derived from the same structure dataset. The matrix elements M(a,a') are obtained as the average of all the computed folding free-energy changes, which correspond to a substitution a → a'. Details on the procedure and the value of the matrix elements M(a,a') are given in the Methods section. This matrix is taken as a cost function:
gmutate (a, a') = M(a, a') (5)
For the purpose of comparing gmutate with a reference matrix that exclusively reflects the structure of the genetic code, we define the cost function:
gcode (a,a') = 3 - Δ(a,a') (6)
where Δ (a,a') is zero when a and a' coincide, and otherwise equal to the minimum number of bases that must be changed to transform a codon coding for a into a codon coding for a'.
As a last cost function, used only for comparison with earlier work [34,40], we consider the point accepted mutations 74-100 (PAM74-100) substitution matrix [41], one of the most commonly used matrices in the context of protein-sequence alignment:
gPAM (a, a') = PAM74-100 (a,a') (7)
This matrix is derived from the pattern of amino-acid substitution frequencies observed within naturally occurring pairs of highly diverged homologous protein sequences. However, the use of this matrix for measuring the genetic code's fitness [34,40] has been criticized [42], as matrices derived from substitution patterns observed in homologous proteins reflect not only the similarity between amino acids with respect to their physicochemical and energetic properties, but also the facility with which one amino acid is mutated into another and thus their proximity in the genetic code. However, as the PAM74-100 matrix is derived from highly diverged protein sequences, this effect can be expected to be relatively limited. This is indeed the case, as the linear correlation coefficient between the nondiagonal entries of gPAM and gcode is only 0.43. Nevertheless, the correlation coefficient between gmutate and gcode is still much lower, namely 0.19, so that gmutate may in no way be suspected to include information on proximity in the genetic code. Finally, note that the correlation coefficient between gmutate and gPAM is equal to 0.60; these matrices thus share common features but contain also different information. This is not surprising considering that their derivations use very different starting points (protein three-dimensional structures in one case, sequence similarity in the other).
The genetic code versus random codes
To evaluate the robustness of the natural genetic code with respect to translation errors, we computed the fitness functions ΦFH, Φequif and Φfaa using Equations (1) to (3) for the natural genetic code, and compared it to the corresponding fitnesses of random codes. The random codes are obtained by maintaining the codon block structure of the natural genetic code, where each block corresponds to synonyms coding for the same amino acid (or stop signal). When generating a random code, the stop signal is kept assigned to the same block as in the natural genetic code, whereas the different amino acids are randomly interchanged among the 20 remaining blocks. Thus, each random code is simply specified by a different function a(c) in Equations (1) to (3). This is the procedure previously used by Haig and Hurst [22] and Freeland and Hurst [23].
Thus, in a first stage, we computed the fitness functions Φequif and Φfaa for the natural genetic code and for 109 randomly generated codes, using the three cost functions gpol, gaccess and gmutate. We then calculated the fraction f of random codes whose value of Φ is lower than that of the natural code. This fraction is supposedly a good estimate of the relative merit of the natural genetic code comparative to other codes. The results are given in Table 2. It appears that this fraction f is always smaller for Φfaa than for Φequif. This is especially true for the cost function gmutate where f is 300 times smaller. This result indicates that the natural code appears to be better optimized with respect to translation errors if the amino-acid frequencies are taken into account.
Table 2.
Fraction of random codes that are fitter than the genetic code
| f | ΦFH | Φequif | Φfaa |
| gpol | 9.8 × 10-7 | 1.5 × 10-6 | 6.5 × 10-7 |
| gaccess | 1.7 × 10-6 | 1.9 × 10-6 | 1.2 × 10-6 |
| gcode | 3.4 × 10-15* | 5.1 × 10-16* | 5.0 × 10-17* |
| gPAM | 3.8 × 10-6 | 2.2 × 10-6 | 2.0 × 10-9 |
| gmutate | 2.3 × 10-6 | 6.0 × 10-7 | 2.0 × 10-9 |
Fraction f of random codes that have a lower value of the fitness functionΦFH, Φequi or Φfaa) than the natural code, using each of the four cost functions gpol, gaccess, gcode, gPAM and gmutate. Values marked with an asterisk have been obtained by extrapolation as explained in text. The number of randomly generated codes is equal to 109 and the amino-acid frequencies used are the average ones listed in the right-hand column of Table 1.
To investigate this further, we have analyzed which of the cost functions gpol,gaccess or gmutate genetic code appears to be best optimized for. We compared the fraction f of better codes for each of the cost functions using the fitness function Φfaa. For the hydrophobicity functions gpol and gaccess, the result is roughly the same: f is about 0.5-1.0 in 106. The relative statistical error on this value is of the order of N-1/2, where N is the number of random codes better than the natural one that were found in our sample of 109 random codes; thus, N is about 650-1,200, and the error is insignificant. For the mutational cost function gmutate, f is several orders of magnitude lower, namely 2 in 109.
This result shows that the natural genetic code appears even more optimal if the cost function gmutate is used than if hydrophobicity-based cost functions are considered. As gmutate has been computed from protein stability changes effected by point mutations, we may conclude that the genetic code is optimized in such a way as to limit the effect of translation errors on the three-dimensional structure and stability of the coded proteins. Note that the improvement brought by the choice of gmutate results from the fact that it probably better accounts for the cost of a mutation than a mere difference of hydrophobicity; for example, glycine, proline and cysteine have close neighbors in hydrophobicity, whereas the cost of their mutation as accounted for by gmutate is high. This is due to their special role in determining protein structure: glycine and proline can adopt backbone torsion angles essentially inaccessible to other amino acids, and cysteine can form disulfide bonds.
To check the significance of this result, we have computed the fraction f of random codes that beat the natural one for random choices of the amino-acid frequencies, distinct from the natural frequencies p(a). We have generated 102 sets of random p(a) values, and, for each of them, estimated the fraction f (out of a sample of 106 random codes). The percentage of random amino-acid frequency sets that result in a lower fraction f than the natural frequencies is shown in Table 3. We find that a random assignment of the amino-acid frequencies does not decrease/in most (at least 97%) of the cases, and this tendency persists for all cost functions g. Thus, the probability that the decrease of f, observed in Table 2, when passing from Φequif to Φ faa, was due to chance is quite limited. We may therefore conclude that the genetic code is optimized so as to take into account the natural amino-acid frequencies.
Table 3.
Percentage of random amino-acid frequency assignments yielding lower fractions f than the natural one
| % | |
| gpol | 3 |
| gaccess | <1 |
| gPAM | <1 |
| gmutate | <1 |
Percentage of the sets of random amino-acid frequency assignments for which the fraction f of random codes that beat the natural code is lower than the corresponding fraction computed with the natural frequency p(a) values. This percentage is estimated for the four cost functions - gpol, gaccess, gmutate and gPAM - on the basis of 100 random frequencies and, for each of them, 106 random codes. For all cost functions except gpol, we were only able to give an upper bound (estimated to be equal to 1%), because our sample of random codes is too small and we did not find any random frequency set for which f is lower than that obtained with the natural frequencies.
We also investigated whether the result that the natural code is better optimized if amino-acid frequencies are taken into account does not depend crucially on the amino-acid frequencies used. For this purpose, we calculated the fraction f of random codes with lower Φfaa value than the genetic code for the four sets of amino-acid frequencies listed in Table 1, which are computed from genomes of eukaryotes, archaea, bacteria and from all these genomes together. The results turn out to be essentially insensitive to the chosen frequency set: the fractions f differ at most by a factor of two, which leaves all conclusions unchanged.
We have also included in Table 2 the results based on the fitness function ΦFH. It can be argued that this function partly takes, but imperfectly, the amino-acid frequencies into account. Indeed, for this fitness function each codon is assigned the same weight, which corresponds to each amino acid being assigned a frequency proportional to the number of synonyms n(a) coding for it. In the case of the natural genetic code, this frequency corresponds approximately to the amino-acid frequency as there is a correlation between n(a) and p(a), as shown in Figure 1. But for random codes, where the amino acids are randomly interchanged between the codon blocks, this correspondence breaks down. Thus, the way in which ΦFH takes amino-acid frequencies into account depends on the code considered. This explains why the fraction f of random codes better than the natural one is roughly of the same order using ΦFH and Φequif. Note that f is always larger for ΦFHthan for Φfaa, indicating again the importance of the amino acid frequencies in the optimality of the genetic code.
For sake of comparison, we have added in Table 2 the values of the fraction f of random codes with a lower Φfaa value than the natural one, using the cost functions gPAM and gcode, which include information about the structure of the genetic code. With gPAM, the fraction f is the same as with gmutate, whereas with gcode, we did not find any random code better than the natural one among the 109 random codes tested. To estimate f without having to generate a larger ensemble, we used the following procedure. We computed, from the values of Φfaa for the 109 random codes, the probability function π (Φ)faa to have a given value of Φfaa. We fitted log(π (Φ)faa) to a polynomial of fourth degree, and extrapolated this curve down to the value of Φfaa for the natural code. This provides an estimate of the fraction f of random codes that have a lower Φfaa value. Note that this estimate is essentially insensitive to the degree of the polynomial. We found with this procedure that/is of the order of 10-17 with gcode, and thus about 108 times smaller than with gPAM andgmutate.
It is not surprising that the fraction f of random codes that does better than the natural code is extremely small for gcode, as this matrix exclusively reflects the proximity of amino acids in the genetic code and renders the issue of the code's fitness tautologous. This has been suspected for the gPAM cost function too [42]. It can indeed be argued that gPAM contains information on the proximity of amino acids in the genetic code, superimposed on the desired measure of their similarity in preserving protein structure, because it is computed from amino-acid substitutions in families of evolutionarily related proteins, which are more frequent between amino acids that are closer in the genetic code. The fact that gPAM and gmutate yield similar f values (see Table 2) can be taken to indicate that this is not the case, and thus that both these cost functions can reliably be used to estimate the code's fitness against translation errors. This interpretation supports previous analyses that used the gPAM matrix [34,40]. It could, however, also be argued that gPAM describes protein structure less well than gmutate and includes somewhat more information about the genetic code (as monitored by correlation coefficients of 0.43 and 0.19 of gPAMand gmutate with respect to gcode). These two effects tend to compensate each other, and could be expected to yield similar f values for gPAM and gmutate. It therefore seems safer to use gmutate as cost function, because, owing to its very definition, it seems to capture important structural information and to be independent of the code's structure.
We also investigated how optimal the genetic code is with respect to amino-acid interchanges that do not affect codon degeneracy. We exhaustively generated all alternative codes that preserve the amino-acid degeneracy and computed the fraction f of these codes that do better than the natural code with respect to mistranslation. We found that f is of the order of 10-6 for the three fitness functions Φfaa, Φequif and ΦFH (Table 4). It is thus similar to the f value computed on the unrestricted set of alternative codes for Φequif and ΦFH, and much larger for Φfaa. This result simply reflects the correlation between the codon degeneracy and the amino-acid frequency (Figure 1). Indeed, this correlation implies that a much larger proportion of the better codes maintain the degeneracy, if the frequency of the amino acids is taken into account in the fitness function, as in Φfaa. In contrast, in Φequif and ΦFH the amino-acid frequency is not considered and f is of the same order with the restricted and unrestricted sets.
Table 4.
Fraction f for different sets of allowed amino-acid interchanges in the alternative codes
| f | ΦFH | Φequif | Φfaa |
| Unrestricted set | 2.3 × 10-6 | 6.0 × 10-7 | 2.0 × 10-9 (97%) |
| Biosynthesis-restricted set | 6.1 × 10-6 | 1.9 × 10-6 | 2.9 × 10-8 (98%) |
| Degeneracy-restricted set | 2.3 × 10-6 | 2.1 × 10-6 | 1.3 × 10-6 (97%) |
Fraction f of random codes that have a lower value of the fitness function (ΦFH, Φequif or Φfaa) than the natural code, using the cost function gmutate. For the unrestricted set, the f values were estimated from 109 randomly generated codes, where the only constraint is the preservation of the code's block structure (as in Table 2). For the biosynthesis-restricted set, only permutations of amino acids sharing the same metabolic pathway were considered, that is, interchanges of amino acids contained in one of the four sets {F, S, Y, C, W}, {L, P, H, Q, R}, {I, M, T, N, K}, {V, A, D, E, G} (single-letter amino-acid notation) [34]. As the number of alternative codes is reasonable (207,360,000), they have not been randomly chosen, but all have been tested. The degeneracy-restricted set contains results obtained by shuffling only amino acids with the same degeneracy in the natural code, corresponding to the sets {M, W}, {C, D, E, F, H, K, N, Q, Y}, {I}, {A, G, P, T, V}, {L, R, S}. Here also, all 522,547,200 possible codes have been systematically tested. The percentage of optimization of the natural code compared to the optimal alternative ones, as defined in the text, is given in parentheses for Φfaa. For the two restricted sets, for which all alternative codes were exhaustively generated, the Φfaa value of the optimal code was computed exactly. For the unrestricted set, the optimal Φfaa value was taken as the best of the unrestricted and two restricted sets.
It has been proposed that the genetic code has evolved from a simpler ancestral code, encoding only a few amino acids present at early times, and that new amino acids appearing as biosynthetic derivatives of the original ones were incorporated by subdivision and reassignment of their synonymous codons [4,5,6,7]. This so-called coevolution hypothesis is supported by the observation that biosynthetically related amino acids are close within the genetic code [6,43]. To investigate the optimality of the genetic code in the coevolution framework, we computed the fraction f of alternative codes that perform better than the natural code against translation errors and that differ from the natural code by shuffling amino acids belonging to the same biosynthetic pathway [34,44]. The allowed shufflings are given in the legend of Table 4. We found that f is equal to 2.9 × 10-8, whereas it is equal to 2 × 10-9 for the unrestricted set allowing all shufflings (Table 4). This means that the fraction f of better codes is somewhat larger in the biosynthesis-restricted set than in the complete set, and thus that the optimality rate is slightly lower. This result can also be viewed differently. When considering the total number of codes in the two sets (which are of the order of 108 and 1018), it means that there are only six codes that beat the natural code in the restricted set, whereas there are 109 such codes in the unrestricted set. This is particularly striking given that our definitions of cost functions and sets of biosynthetically related amino acids only constitute approximations [45]. We can thus conclude that the genetic code is quite robust against mistranslation in the space of all alternative codes, and is close to being fully optimal if historical biosynthetic constraints are taken into account.
A complementary measure of the optimality of the genetic code is its percentage of optimization, defined as 100%(Φcode - Φmean)/(Φbest - Φmean), where Φcode is the fitness of the genetic code, Φbest fitness of the best of all possible codes and Φmean the average fitness over all codes [20,21,46]. This measure indicates how close the fitness value of the genetic code is to the fitness value of the optimal code. Note however that this optimality measure has no absolute meaning and may not be compared among fitness functions Φ defined on the basis of different cost functions g; to illustrate this, consider the following example: if ghydro is defined by |h(a)-h(a')| instead of (h(a)-h(a'))2 (see Equation (4)), the fraction f of better codes remains unchanged but the percentage of optimality does change [34]. It is, however, meaningful to compare the percentage of optimization of the genetic code in the unrestricted and biosynthesis restricted sets with a same g function. For Φfaa, we find that this percentage is equal to 97% and 98% in the two sets, respectively. This indicates that the fitness value of the genetic code is not very far from that of the best possible code, whether focusing on the subset of alternative codes preserving biosynthetic proximities, or considering all possible codes.
Discussion
Our results confirm and specify those of Freeland and Hurst [22]: the genetic code seems structured so as to minimize the consequences of translation errors on the three-dimensional structure and stability of the coded proteins. We have shown that, using the cost function gmutate which best reflects the roles of various amino acids in protein structures, and taking amino-acid frequencies into account, about 2 out of 109 random codes do better than the natural code. But we have to keep in mind that there exist 20! ≈ 2 × 1018 possible codes preserving the codon block structure, which means that we can expect about 109 better codes overall [47]. Moreover, if the codon block structure is not preserved [46], the number of possible codes is larger by orders of magnitude, and therefore the number of codes better than the natural one will certainly be much larger.
However, if we preserved the block structure and in addition restricted the space of alternative codes by interchanging only amino acids belonging to the same biosynthetic pathway, we found that there are only six codes performing better than the natural code. The genetic code thus seems quite robust with respect to mistranslation compared to alternative codes, and almost fully optimal if the constraint is imposed that biosynthetically related amino acids are encoded in codons that are close within the genetic code. This does not prove, but is in agreement with, the coevolution hypothesis, which assumes that the genetic code has evolved from an simpler ancestral code of only a few amino acids, by subdivision and reassignment of synonymous codons [4,5,6,7], and that the present genetic code has kept imprints of this evolution.
So we can assert from our analysis that the genetic code has been optimized through evolution up to a certain point, even though it is probably not fully optimal, at least with respect to the parameters considered here [16], except perhaps if historical, biosynthesis-related, constraints are imposed. Our analysis does not, however, give us much information about the mechanism of this evolution as there is unfortunately no trace left of evolution of the code or amino-acid frequencies in early times. For instance, we do not know whether the relative frequency of occurrence of amino acids in proteins adapted so as to increase the optimality of the genetic code with respect to translation errors, or, on the contrary, whether the genetic code evolved to take into account pre-existing amino-acid frequencies. We can, however, argue that if the amino-acid frequencies adapted to the genetic code, as assumed by King and Jukes [33], a discrepancy in amino-acid composition between frequently and infrequently expressed genes might be detectable today (unless the period during which evolution took place was long enough for this discrepancy to vanish). If, alternatively, the genetic code adapted to the amino-acid frequencies, and thus if these frequencies acted as an evolutionary pressure, one can imagine two scenarios. Either the code optimized to take into account the prebiotic frequencies of the amino acids that became involved in it, or it optimized for the amino-acid frequencies of already formed proteins (or of a subset of them) that were important for life and maybe linked to the code's control. Perhaps can we assume, more realistically, that the genetic code and amino-acid frequencies evolved together during some evolutionary period, thereby approaching an optimal code/amino-acid relation.
More generally, the parameters that acted as evolutionary pressure on the genetic code probably included all the mechanisms that encode and maintain the genetic information, and were not just restricted to the frequency of amino acids and the preservation of protein structure. For example, the genetic code is obviously related to the translation apparatus, composed of the ribosomes and tRNAs, whose action we described schematically here by the probabilities p(c' | c) to misread codon c as c'. This apparatus was certainly less reliable at the beginning of evolution. All these mechanisms probably evolved together with the genetic code during the early stages of life.
Although the code still evolves today, as reflected by its departure from universality in some organisms, its evolution is very limited and concerns only the reassignment of a few codons [2]. As the same change sometimes recurs in different lineages, the code seems to have reached the bottom of a funnel in the evolutionary landscape that contains several roughly equivalent optimal codes. But apart from such restricted modifications, the code no longer evolves significantly, and has not undergone important modifications since an early stage in the development of life. This stability probably arose because even small modifications in the code would have entailed loss of functionality of genes that were already being expressed. Moreover, the advent of more sophisticated transcription/translation control mechanisms, which involve huge protein systems, could have decreased the evolutionary pressure on the genetic code. Even though our present information on the genetic code is insufficient to discriminate between evolutionary scenarios, our analysis enables us to put some constraints on the situation at the time when evolution of the code was pretty much frozen. In particular, it appears that the frequencies of the amino acids that were used in proteins synthesized at that time were similar to the present frequencies. We do not know what determines the present amino-acid frequencies, but presumably they result, at least in part, from the amino acids' physicochemical properties. For instance, the ratio of hydrophobic to hydrophilic amino acids is intrinsically related to the globular structure of proteins and certainly contributes to the pressure on amino-acid frequencies. Also, amino acids that are easily synthesized may be used more often. Thus, we can assert that some of the pressures that determine the present amino-acid frequencies were already present at the time the code took on its definitive form. In addition, the increased optimality of the genetic code with respect to gmutate implies that the three-dimensional structure of proteins probably played an equally important role in fixing the structure of the code. As the three-dimensional structure of a protein essentially determines its function, this suggests, more generally, that the protein function acted as a main evolutionary pressure on the code structure. Consequently, at the time when the genetic code took its present form, primitive life was presumably already synthesizing complex proteins. This provides a tentative picture of primitive life at that time: the translation apparatus was similar to the present one, and organisms were made of complex proteins whose amino-acid frequencies were comparable to the present ones.
Materials and methods
Derivation of the mutation matrix
The derivation is based on a dataset of 141 high-resolution protein structures determined by X-ray crystallography and listed in [48]. To avoid bias, these 141 proteins are chosen to present either less than 20% sequence identity or less than 25% sequence identity and no structural similarity.
The protein main chains are described by their heavy atoms, and each side chain is represented by a pseudo-atom Cμ. For a given amino-acid type, the Cμ has a well defined position relative to the main chain, corresponding to the geometric average of all heavy side-chain atoms of this type in the dataset [49]; for glycine, the Cμ pseudo-atom is positioned on the Cα. Side-chain degrees of freedom are thus neglected.
Each residue, at each position of each of the 141 proteins, is mutated in turn into the 19 non-wild-type amino acids. The mutations are made by keeping the main-chain structure unchanged, and substituting the Cμ of the given amino acid by that of the mutant amino acid. For each of these mutations, the change in folding free energy is evaluated using the database-derived potentials and the procedure detailed below. For each substitution of amino acid a into a', the average of all computed changes in folding free energy, at all protein positions, is computed and defined as minus the matrix element M(a,a'). We then symmetrize M by setting M(a,a') = [M(a,a')+M(a',a)]/2 and only consider the lower half of M(a < a'). This procedure does not define the diagonal elements of M. On the basis of the principle that the structural role of a given amino acid is fulfilled by no other amino acid better than by itself, we assign to all the diagonal element the same maximum value: M(a,a) = Max [M(a',a")] + 1. Then, to simplify M without modifying its structure, we center it around its mean value:
![]()
Finally, we multiply all matrix elements M(a,a') by 2 and replace them by the closest integer. The resulting half matrix is given in Figure 2.
Figure 2.

Mutation matrix M.
Database derived potentials
The potentials we use to evaluate the protein conformations are derived from observed frequencies of sequence and structure patterns in the aforementioned dataset of 141 proteins. We consider two types of potentials, called torsion [50,51] and Cμ -Cμ [49] potentials.
Torsion potentials describe only local interactions along the sequence. They take into account the propensities of single residues and residue pairs to be associated with a (ϕ, ψ, ω) backbone torsion angle domain. Seven (ϕ, ψ, ω) domains are considered, defined in [50]. We use two variants of the torsion potential, called torsionshort-range and torsionmiddle-range. Both are computed from propensities of a (ϕ, ψ, ω) domain ti, at position i along the sequence, or pairs of domains (ti, tj), at positions i and j, to be associated with an amino acid ak at position k. But we have k - 1 ≤ i,j ≤ k + 1 for the torsionshort-range potential and k - 8 ≤ i,j ≤ k + 8 for the torsionmiddle-range potential. The folding free energy Δ G(S,C) of a sequence S in the conformation C computed from these propensities is expressed as [52,53]:
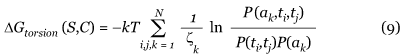
where P are normalized frequencies, N is the number of residues in the sequence S, k is the Boltzmann constant and T is a conformational temperature taken to be room temperature [54]. The normalization factor ζk ensures that the contribution to ΔG(S,C) of each residue in the window [k - 1, k + 1] for the torsionshort-range potential or [k - 8, k + 8] for the torsionmiddle-range potential is counted once. It is equal to the window width, except near the chain ends.
The Cμ-Cμ potentials are distance potentials dominated by nonlocal, hydrophobic interactions. They are based on propensities of pairs of amino acids (ai,aj) at position i and j along the sequence to be separated by a spatial distance dij, calculated between the pseudo-atoms Cμ. We consider two variants of Cμ-Cμ potentials. The first one, called Cμ-Cμlong_range potential, describes purely nonlocal interactions along the sequence, and only takes into account residues separated by at least 15 residues along the sequence, that is j ≥ i + 16. The second one, simply called Cμ-Cμ potential, though dominated by nonlocal interactions, possesses a local interaction component. The nonlocal component is obtained by considering together the frequencies of all residues separated by seven sequence positions and more, thus with j ≥ i + 8. The local component is obtained by computing separately the frequencies of residues separated by one to six positions along the sequence, for i + 1 < j < i + 8. Consecutive residues along the sequence are not considered. The folding free energies are expressed as:
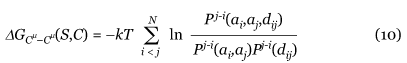
with j ≥ i + 16 and the normalized frequencies Pj-i independent of j - i for the Cμ-Cμlong_range potential, and i + 1 < j and the normalized frequencies Pj-i independent of j - i for j ≥ i + 8 for the Cμ-Cμ potential. The discretization of the spatial distances dij is performed by dividing the distances between 3 and 8 Å into 25 bins of 0.2 Å width and merging the distances greater than 8 Å. To increase the reliability of the statistics, these bins are smoothed by combining the counts in each bin with those of the 10 flanking bins at each side. The predominance of the central bin is preserved by weighting the counts from each flanking bin by a factor 1/n, where n is the position relative to the central bin; n is equal to 1 for the two closest bins and to 10 for the two most distant bins.
The so-defined folding free energies are reliable for common amino acids and structure motifs, but not for less common ones. To correct for the sparse data, we substitute the sequence-specific frequencies P(c,s), where s denotes a sequence pattern and c a structure motif, which appear in expressions (9) and (10) immediately above defining the torsion and Cμ-Cμ folding free energies, by a linear combination of these frequencies and the product of the separate frequencies of s and c, denoted P(s) and P(c) respectively [55]:
![]()
where ms is the number of occurrences of the sequence pattern s in the dataset, and σ a parameter. This expression ensures that the sequence-specific contribution dominates for rare sequence patterns and tends to zero for frequent ones. This behavior is modulated by the parameter σ, which we consider here equal to 50.
Evaluation of folding free energy changes
To estimate the stability changes caused by a single-site mutation, we compute the folding free energy changes as:
![]()
where Cm and Cw are the mutant and wild-type conformations and Sm and Sw the mutant and wild-type sequences, respectively. With this convention, ΔΔG is positive when the mutation is destabilizing, and negative when it is stabilizing. The conformations Cm and Cw of the mutant and wild-type protein are assumed to be nearly identical. More precisely, the backbone conformations are taken as identical and only the position of the Cμ pseudo-atom, which is amino-acid dependent, is different in the mutant and wild-type structures.
The folding free energies of the wild-type and mutant proteins are computed with linear combinations of the torsion and Cμ-Cμ potentials described in the previous section. Previous analyses [37,38,39] have shown that the combination that gives the best evaluation of the ΔΔG values depends on the solvent accessibility, A of the mutated residue; A is defined as the solvent-accessible surface in the protein structure, computed by SurVol [36], multiplied by 100 and divided by its solvent-accessible surface in an extended tripeptide Gly-X-Gly [56]. These analyses have revealed that the mutations can be divided in three subsets. When the mutated residue is at the surface, with a solvent accessibility A equal to or larger than 50%, the optimal folding free-energy change has been shown to be equal to:
![]()
When the mutated residue is half buried, half exposed to the solvent, with a solvent accessibility between 20 and 40%, the optimal folding free energy is:
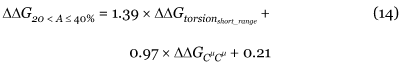
Finally, when the mutated residue is totally buried in the protein core, with a solvent accessibility less than or equal to 20%, the optimal folding free energy is:

When the mutated residue has a solvent accessibility comprised between 40 and 50%, we do not evaluate its folding free energy. We have indeed observed that in this case, the solvent accessibility of the mutated residue is not a good measure to guide the choice of the optimal potential.
Acknowledgments
Acknowledgements
We are grateful to J. Reisse and S. Freeland for useful discussions and comments. D.G., S.M. and M.R. are supported by the Belgian National Fund for Scientific Research.
References
- Woese CR. The Genetic Code: The Molecular Basis for Genetic Expression New York: Harper and Row; 1967.
- Knight RD, Freeland SJ, Landweber LF. Rewiring the keyboard: evolvability of the genetic code. Nat Rev Genet. 2001;2:49–58. doi: 10.1038/35047500. [DOI] [PubMed] [Google Scholar]
- Vogel G. Tracking the history of the genetic code. Science. 1998;281:329–331. doi: 10.1126/science.281.5375.329. [DOI] [PubMed] [Google Scholar]
- Orgel LE. A possible step in the origin of the genetic code. Israel J Chem. 1972;10:287–292. [Google Scholar]
- Dillon LS. The origins of the genetic code. Bot Rev. 1973;39:301–345. [Google Scholar]
- Wong JT. A co-evolution theory of the genetic code. Proc Natl Acad Sci USA. 1975;72:1909–1912. doi: 10.1073/pnas.72.5.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M, Winkler-Oswatitsch R. Transfer-RNA, an early gene? Naturwissenschaften. 1981;68:282–292. doi: 10.1007/BF01047470. [DOI] [PubMed] [Google Scholar]
- Speyer JF, Lengyel P, Basilio C, Wahba AJ, Gardner RS, Ochoa S. Synthetic polynucleotides and the amino acid code. Cold Spring Harbor Symp Quant Biol. 1963;28:559–567. [Google Scholar]
- Sonneborn TM. Degeneracy of the genetic code: extent, nature and the genetic implications. In Evolving Genes and Proteins Edited by Bryson V, Vogel HJ New York: Academic Press; 1965. pp. 377–397. [DOI] [PubMed]
- Woese CR. On the evolution of the genetic code. Proc Natl Acad Sci USA. 1965;54:1546–1552. doi: 10.1073/pnas.54.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelc SR. Correlation between coding-triplets and amino acids. Nature. 1965;207:597–599. doi: 10.1038/207597a0. [DOI] [PubMed] [Google Scholar]
- Epstein CJ. Role of the amino-acid "code" and of selection for conformation in the evolution of proteins. Nature. 1966;210:25–28. doi: 10.1038/210025a0. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Wittes RE. Genetic code: aspects of organization. Science. 1966;153:420–424. doi: 10.1126/science.153.3734.420. [DOI] [PubMed] [Google Scholar]
- Woese CR, Dugre DH, Saxinger WC, Dugre SA. The molecular basis for the genetic code. Proc Natl Acad Sci USA. 1966;55:966–974. doi: 10.1073/pnas.55.4.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshiro T, Kimura M. The role of robustness and changeability on the origin and evolution of genetic codes. Proc Natl Acad Sci USA. 1998;95:5088–5093. doi: 10.1073/pnas.95.9.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson OP, Haydon D. The genetic code: what is it good for? An analysis of the effects of selection pressure on genetic codes. J Mol Evol. 1999;49:539–550. doi: 10.1007/pl00006575. [DOI] [PubMed] [Google Scholar]
- Ronneberg TA, Landweber LF, Freeland SJ. Testing a biosynthetic theory of the genetic code: fact or artifact? Proc Natl Acad Sci USA. 2000;97:13690–13695. doi: 10.1073/pnas.250403097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Salemne FR, Miller MD, Jordan JR. Structural convergence during protein evolution. Proc Natl Acad Sci USA. 1977;74:2820–2824. doi: 10.1073/pnas.74.7.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JT-F. Role of minimization of chemical distances between amino acids in the evolution of the genetic code. Proc Natl Acad Sci USA. 1980;77:1083–1086. doi: 10.1073/pnas.77.2.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giulio M. The extension reached by the minimization of the polarity distances during the evolution of the genetic code. J Mol Evol. 1989;29:288–293. doi: 10.1007/BF02103616. [DOI] [PubMed] [Google Scholar]
- Haig D, Hurst LD. A Quantitative measure of error minimization in the genetic code. J Mol Evol. 1991;33:412–417. doi: 10.1007/BF02103132. [DOI] [PubMed] [Google Scholar]
- Freeland SJ, Hurst LD. The genetic code is one in a million. J Mol Evol. 1998;47:238–248. doi: 10.1007/pl00006381. [DOI] [PubMed] [Google Scholar]
- Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- Pace CN, Shirley BA, McNutt M, Gajiwala K. Forces contributing to the conformational stability of proteins. FASEB J. 1996;10:75–83. doi: 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- Friedman SM, Weinstein IB. Lack of fidelity in the translation of ribopolynucleotides. Proc Natl Acad Sci USA. 1964;52:988–996. doi: 10.1073/pnas.52.4.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 1989;53:273–298. doi: 10.1128/mr.53.3.273-298.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DW. Rates of transition and transversion in coding sequences since the human-rodent divergence. Genomics. 1994;20:386–396. doi: 10.1006/geno.1994.1192. [DOI] [PubMed] [Google Scholar]
- Kumar S. Patterns of nucleotide substitution in mitochondrial protein-coding genes of vertebrates. Genetics. 1996;143:537–548. doi: 10.1093/genetics/143.1.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama EN, Powell JR. Synonymous substitution rates in Drosophila : mitochondrial versus nuclear genes. J Mol Evol. 1997;45:378–391. doi: 10.1007/pl00006243. [DOI] [PubMed] [Google Scholar]
- Morton BR. Neighbouring base composition and transversion transition bias in a comparison of rice and maize chloroplast non coding regions. Proc Natl Acad Sci USA. 1995;92:9717–9721. doi: 10.1073/pnas.92.21.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alf-Steinberger C. The genetic code and error transmission. Proc Natl Acad Sci USA. 1969;64:584–591. doi: 10.1073/pnas.64.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JL, Jukes TH. Non-Darwinian evolution. Science. 1969;164:788–798. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- Freeland SJ, Knight RD, Landweber LF, Hurst LD. Early fixation of an optimal genetic code. Mol Biol Evol. 2000;17:511–518. doi: 10.1093/oxfordjournals.molbev.a026331. [DOI] [PubMed] [Google Scholar]
- Woese CR, Dugre DH, Dugre SA, Kondo M, Saxinger WC. On the fundamental nature and evolution of the genetic code. Cold Spring Harbor Symp Quant Biol. 1966;31:723–736. doi: 10.1101/sqb.1966.031.01.093. [DOI] [PubMed] [Google Scholar]
- Alard P. Calculs de surface d'énergie dans le domaine des macro-molécules. PhD thesis. 1991.
- Gilis D, Rooman M. Stability changes upon mutation of solvent-accessible residues in proteins evaluated by database-derived potentials. J Mol Biol. 1996;257:1112–1126. doi: 10.1006/jmbi.1996.0226. [DOI] [PubMed] [Google Scholar]
- Gilis D, Rooman M. Predicting protein stability changes upon mutation using database-derived potentials: solvent accessibility determines the importance of local versus non-local interactions along the sequence. J Mol Biol. 1997;272:276–290. doi: 10.1006/jmbi.1997.1237. [DOI] [PubMed] [Google Scholar]
- Gilis D, Rooman M. Prediction of stability changes upon single-site mutations using database-derived potentials. Theor Chem Acc. 1999;101:46–50. [Google Scholar]
- Ardell DH. On error-minimization in a sequential origin of the standard genetic code. J Mol Evol. 1998;47:1–13. doi: 10.1007/pl00006356. [DOI] [PubMed] [Google Scholar]
- Benner SA, Cohen MA, Gonnet GH. Amino acid substitution during functionally divergent evolution of protein sequences. Protein Eng. 1994;7:1323–1332. doi: 10.1093/protein/7.11.1323. [DOI] [PubMed] [Google Scholar]
- Di Giulio M. Genetic code origin and the strength of natural selection. J Theor Biol. 2000;205:659–661. doi: 10.1006/jtbi.2000.2115. [DOI] [PubMed] [Google Scholar]
- Taylor FJR, Coates D. The code within the codons. Biosystems. 1989;22:177–187. doi: 10.1016/0303-2647(89)90059-2. [DOI] [PubMed] [Google Scholar]
- Freeland S, Hurst LD. Load minimization of the genetic code: history does not explain the pattern. Proc Roy Soc Lond B. 1998;265:2111–2119. [Google Scholar]
- Amirnovin R. An analysis of the metabolic theory of the origin of the genetic code. J Mol Evol. 1997;44:473–476. doi: 10.1007/pl00006170. [DOI] [PubMed] [Google Scholar]
- Goldman N. Further results on error minimization in the genetic code. J Mol Evol. 1993;37:662–664. doi: 10.1007/BF00182752. [DOI] [PubMed] [Google Scholar]
- Di Giulio M. The origin of the genetic code. Trends Biochem Sci. 2000;25:44–44. doi: 10.1016/s0968-0004(99)01522-4. [DOI] [PubMed] [Google Scholar]
- Wintjens RT, Rooman MJ, Wodak SJ. Automatic classification and analysis of αα-turn motifs in proteins. J Mol Biol. 1996;255:235–253. doi: 10.1006/jmbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- Kocher J-PA, Rooman MJ, Wodak SJ. Factors influencing the ability of knowledge-based potentials to identify native sequence-structure matches. J Mol Biol. 1994;235:1598–1613. doi: 10.1006/jmbi.1994.1109. [DOI] [PubMed] [Google Scholar]
- Rooman MJ, Kocher J-PA, Wodak SJ. Prediction of protein backbone conformation based on 7 structure assignments: influence of local interactions. J Mol Biol. 1991;221:961–979. doi: 10.1016/0022-2836(91)80186-x. [DOI] [PubMed] [Google Scholar]
- Rooman MJ, Kocher J-PA, Wodak SJ. Extracting information on folding from the amino acid sequence: accurate predictions for protein regions with stable conformation in absence of tertiary interactions. Biochemistry. 1992;31:10226–10238. doi: 10.1021/bi00157a009. [DOI] [PubMed] [Google Scholar]
- Rooman MJ, Wodak SJ. Are database-derived potentials valid for scoring both forward and inverted protein folding? Protein Eng. 1995;8:849–858. doi: 10.1093/protein/8.9.849. [DOI] [PubMed] [Google Scholar]
- Rooman M, Gilis D. Different derivations of knowledge-based potentials and analysis of their robustness and context-dependent predictive power. Eur J Biochem. 1998;254:135–143. doi: 10.1046/j.1432-1327.1998.2540135.x. [DOI] [PubMed] [Google Scholar]
- Pohl FM. Empirical protein energy maps. Nat New Biol. 1971;234:277–279. doi: 10.1038/newbio234277a0. [DOI] [PubMed] [Google Scholar]
- Sippl M. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge based prediction of local structures in globular proteins. J Mol Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]