

Abstract
Glycosaminoglycans (GAGs) play key roles in virtually all biologic responses through their interaction with proteins. A major challenge in understanding these roles is their massive structural complexity. Computational approaches are extremely useful in navigating this bottleneck and, in some cases, the only avenue to gain comprehensive insight. We discuss the state-of-the-art on computational approaches and present a flowchart to help answer most basic, and some advanced, questions on GAG–protein interactions. For example, 1) does my protein bind to GAGs?; 2) Where does the GAG bind?; 3) Does my protein preferentially recognize a particular GAG type?; 4) What is the most optimal GAG chain length?; 5) What is the structure of the most favored GAG sequence?; and 6) Is my GAG–protein system ‘specific’, ‘non-specific’, or a combination of both? Recent advances show the field is now poised to enable a non-computational researcher perform advanced experiments through the availability of various tools and online servers.
Keywords: Algorithm, Chemical biology, Drug discovery, Library screening, Molecular dynamics, Molecular docking, Non-specificity, Plasticity, Specificity
Graphical Abstract
Introduction
Glycosaminoglycans (GAGs) are structurally diverse biopolymers that are present on nearly all cell surfaces as a part of proteoglycans [1]. Nature appears to have engineered GAGs as the ‘go-to’ agents. It has sought out GAGs to induce numerous responses in the human body including the seemingly opposite, e.g., self-renewal as well as differentiation, growth as well as inhibition, or microbial invasion as well as defense. It has reached out to GAGs for a variety of purposes, of which activity modulation, signal transduction, protein sequestration, mechanosensing, and simply water retention are just a few. Not surprisingly, these ‘go-to’ agents are found outside the cells, on the cells and in the cells. They may occur as intact, long polymers and also as cleaved, much smaller oligomers. And GAGs display spatial and temporal dynamism that is unlikely to be rivalled by any other biomolecule.
The origin of the ‘go-to’ characteristics of GAGs is their massive structural possibilities, which enables interaction with numerous proteins [2,3]. In fact, GAGs are arguably the only biomolecules for which ‘specific’ as well as ‘non-specific’ (or ‘plastic’) interactions are known to be obligatory for their myriad functions [3]. For example, the highly specific antithrombin (AT)– heparin interaction is the foundation for the anticoagulant function of heparin [4]. In contrast, plasticity is obligatory for GAG modulation of chemokines [5]. Even more interestingly, the AT–heparin–thrombin (T) ternary system simultaneously requires specific as well as non-specific binding to engineer biological function [6,7]. Yet, this understanding possibly represents only a drop in the bucket.
There is a reason why nature might have chosen GAGs as the ‘go-to’ agents. If one considers distinct sequences possible for a hexamer of repeating units, then GAGs outnumber peptides and nucleic acids by nearly 102 and 106–fold, respectively. Thus, GAGs offer chemical space and information that is several orders greater than other biopolymers, although not all of these sequences may exist in nature.
Common GAGs, including heparan sulfate (HS), heparin (HP), chondroitin sulfate (CS), dermatan sulfate (DS) and hyaluronic acid (HA), are made up of repeating disaccharide units of glycosamine and uronic acid residues that are not uniformly sulfated and acetylated across the chain. The enzyme-driven biosynthesis of GAGs is a template-free process that is spatiotemporally regulated [8,9]. It is tempting to speculate that such a regulation evolved to facilitate the ‘go-to’ characteristics of GAGs.
The diversity of GAG structures coupled with their negative charge density engineers interactions with a hundreds of human proteins. Of these, only a few have been studied reasonably well including some serpins, coagulation proteases, growth factors, and chemokines [2,3,10]. Yet, each protein has been studied from the perspective of binding to HP-like sequences, which is a useful starting point but may be completely inappropriate in terms of biology, pharmacology and therapeutics. A case highlighting this is the interaction of GAGs with heparin cofactor II (HCII). Although generally believed to be non-specific, recent studies indicate highly selective binding of HS hexasaccharide to HCII [11].
A fundamental bottleneck in elucidating details of each GAG–protein system is the massive number of possible GAG sequences in vivo. We do not even have a small fraction of this number available on hand! The only means to access this library comprehensively is through in silico technologies. However, computational study of GAG–protein interactions has been thought of as ‘too dry’ (not involving water as solvent) and ‘too rigid’ (not invoking flexibility of bonds) [12,13]. Yet, recent advances incorporating both these aspects have shown that computational approaches offer major insight into the operation of GAG–protein systems. In fact, the field is now ready for non-computational researchers to perform advanced experiments through the availability of various tools and approaches.
A Brief History of Computational Advances
The first computational study of GAG-protein interactions published in 1989 presented two consensus sequences –XBBXBX– and –XBBBXXBX– (X = hydropathic residue; B = basic residue) that bind heparin [14]. Both molecular docking and molecular dynamics studies were used to deduce consensus sequences. Looking back, the computational tools used in the approach appear rudimentary. Docking involved manual placement of an oligosaccharide onto proteins, whereas MD trajectory was monitored for only 7.5 ps. Yet, the work is now recognized as having catapulted the field with the idea that there is some order in the apparently chaotic GAG recognition of proteins, which could be understood for the first time using computational approaches.
Several GAG–protein systems were studied following this work. Of particular note was the three-dimensional model building exercise of AT with HP pentasaccharide, despite the unavailability of the protein’s crystal structure [15]. Although this AT–pentasaccharide model did not match the crystal structure determined much later [16], it provided significant impetus to the idea of computationally predicting aspects of GAG–protein systems, e.g., the GAG binding site on proteins (Fig. 1A) [17–20], alternative modes of GAG binding (Fig. 1B) [18,21,22] the most optimal GAG sequence for a target protein (Fig. 1C) [11,23,24], and the structure of ternary protein1-GAG-protein2 system (Fig. 1D) [21,25,26]. The wealth of this biochemical/biological knowledge has been made possible by the development of robust computational tools over the past 3 decades. Force fields, such as GLYCAM, CHARMM and GROMOS, and parameters for charged moieties of GAGs have been developed [27,28]. Likewise, tools for automated docking [23,29,30], handling multiplicity of GAG conformations [11,24,31,32], inclusion of solvent molecules [33,34], calculating nature of interactions [2,33,35,36], etc. have been developed. In the process, technologies such as combinatorial virtual library screening (CVLS) [11,23,26,37], dynamic molecular docking (DMD) [33], and online web-servers for GAG docking (http://cluspro.bu.edu) [38,39] and modeling (http://www.glycosciences.de/modeling/), have been developed.
Figure 1.

A) Prediction of GAG binding site on a protein by ClusPro docking server (http://cluspro.bu.edu) [38,39]. A generic heparin sequence predicted to bind to FGF2 (cyan sticks) is compared with the crystal bound hexamer (magenta). B) Prediction of GAG binding modes through the DMD approach [31,33]. The study deduced most populated clusters of six DMD simulations with different GAG type and chain length binding to IL-10. Shown is schematic visualization of the two principally different GAG binding modes observed followed DMD. C) Prediction of the most favored GAG sequence that binds to HCII using dual-filter CVLS strategy that identifies ‘high-affinity & high-specificity’ sequences [11]. D) Prediction of two ternary GAG–protein complexes (AT–HS–T and HCII–HS–T) using CVLS approach [26]. Although the two serpins (AT and HCII) are strikingly similar, the position of T (thrombin) in the two is dramatically different and this matches with the 60° difference in mode of HS binding onto the two proteins. Figures to be reproduced after permission from respective publishers.
Finally, computational studies of GAG binding to proteins have been made more feasible through compilation of the available GAG–protein complex structures in the PDB by Samsonov and coworkers [29], who performed explicit computational analysis on the library. Since then, only 15 additional GAG–protein complex structures have become available in PDB, which highlights the difficulty of generating information through crystallography/NMR. But most importantly, the PDB structures do not represent the diversity of GAG sequences available in nature. This implies that computational approaches developed to date will be important in understanding GAG structural biology.
Key Questions Addressed by Computational Approaches
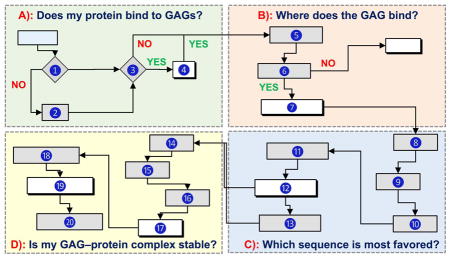
Several basic, and some advanced, questions can be addressed using computational approaches by following a rigorous line of sequential inquiry (Fig. 2). These include i) does my protein bind to GAGs? ii) what residues are involved? iii) which GAG sequence is the most preferred? iv) is my GAG–protein system ‘specific’ or ‘non-specific’? v) can I discover a GAG-based drug? and vi) can any mechanistic insights be computationally derived? This work describes how to answers these questions.
Figure 2.

A flowchart describing the use of computational approaches in addressing key questions on GAG–protein interactions (Panels A through D). Although shown in sequential format (A→B→C→D), it is not strictly necessary to rigorously follow this flowchart, especially if some information is already available for any of the steps. ➀ Sequence and atomic coordinates of a protein can be obtained from the protein data bank (www.rcsb.org). ➁ Homology model of a protein of unknown structure can be generated using programs such as Modeller (https://salilab.org/modeller/), Swiss-Model (https://swissmodel.expasy.org), etc. ➂ Consensus sequences include –XBBXBX-, -XBBBXXBX- (B = basic residue and X = hydropathic residue) [14], TXXBXXTBXXXTBB (T = turn),[41] CPC clif motif [43], clamp-like orientation of basic residues with beta sheet conformations [44]. ➃ If a protein satisfies step ➁, then it is likely to bind GAGs. ➄ a) Electrostatic potential (ESP) can be calculated using tools such as APBS from PyMol (https://www.pymol.org/), DeepView-Swiss-PdbViewer (http://spdbv.vital-it.ch/) and others. GRID search refers to protocol described by Goodford [50]. Site-mapping technique [30]. b) from step 5a we can identify basic site / subsite(s) ➅ Experimental evidence typically includes site directed mutagenesis, NMR, congenital mutation information, etc [52–54]. ➆ Putative GAG binding site(s) are identified based on results from ESP, GRID search, site-mapping techniques [17–20, 30]. ➇ This includes protonation, addition of hydrogens, modeling of missing residues and minimization of protein using a modeling software. GAG structures can be built using CHIMERA (https://www.cgl.ucsf.edu/chimera/) or GLYCAM (http://glycam.org/tools/molecular-dynamics/oligosaccharide-builder/build-glycan?id=8). ➈ Perform initial docking to site(s) of binding identified in step ➆ for various GAGs (HP, HS, CS, DS) of various lengths (dp2, dp4 and dp6) using either Autodock (http://autodock.scripps.edu), Autodock Vina (http://vina.scripps.edu), GOLD (https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/), DOCK (http://dock.compbio.ucsf.edu/), MOE (https://www.chemcomp.com/MOE-Structure_Based_Design.htm), or other programs(refer supplementary information) ➉ Here GAG length, radius of site of binding, number of iterations, number of docking runs, type of docking program, etc. are evaluated and the best protocol is implemented in production run. ⑪ Perform repeated molecular docking using the optimized program and parameters from step ➉ for a library of GAG sequences. A library of GAG sequences can be obtained from the Desai lab (built using SPL scripts) [23,24]. Based on need, this library could have 1,000 to more than 100,000 unique sequences. ⑫ Analysis includes ranking of docked poses by calculating either RMSD, energy, score, non-bonded interactions, etc. and identify the most favored GAG equence(s). ⑬ Although typically not considered part of a computational program, validation of results in solution experiments obtained in ⑫ is extremely important. ⑭ Utilize the most favored GAG–protein complex from ⑬ and prepare initial coordinates for MD, which includes selecting force field, ensuring charge neutralization, immersing in an explicit box of solvent molecules, and minimizing the system. ⑮ Equilibration implies allowing the system to reach physiological conditions such as constant temperature and pressure (NPT/NVE) conditions. ⑯ This includes performing MD run for ~1 ns to ~1 ms, based on need, and collecting trajectories of data. ⑰ – ⑲ Analysis of trajectories may involve RMSD convergence, direct and water mediated H-bond interactions and their occupancies, binding free energy calculations (MMPBSA/MMGBSA), FEP, LIE and single residue energy decomposition calculations. ⑳ This involves ascertaining that computational deduction of thermodynamic stability on the basis of steps ⑰ through ⑲ is supported by some results in solution.
Does My Protein Bind to GAGs?
This question is the most basic question and traditionally its answer has relied on performing solution experiments, such as affinity chromatography. These days a number of in silico tools have been developed to derive an answer in a matter of minutes. First, the primary sequence of a protein is analyzed for the presence of certain consensus sequence(s) (Fig. 2A). Starting from the two related Cardin and Weintraub consensus sequences (above) [14], several analog sequences have been presented as GAG-binding sequences, e.g., –XBBBXXBBBXXBBX– in von Willebrand factor and –TXXBXXTBXXXTBB– (T = turn) in αFGF, βFGF and TGFβ1 [40,41]. However, it is important to recognize that many GAG-binding proteins do not contain any of these contiguous, linear sequences. For example, chemokines have dissimilar GAG binding sites organized on multiple secondary structures [42]. So advanced approaches have emerged. One of these is the CPC clip motif [43], which relies on the presence of a polar residue, e.g., Gln or Asn, among two basic residues within a putative heparin-binding site for a dataset of 20 distinct proteins. Another projects that GAG-binding sites arise from non-contiguous basic patches of ~5 Å that are organized in a ‘clamp-like’ manner with an internal angle of ~100° [44]. And yet another posits that GAG binding sites have a spatial distance ~20 Å between basic residues [45]. Thus, the 3D structure of a protein, either downloaded from the PDB or built using homology modeling is analyzed for the above consensus sequence(s) and/or structural motif(s). The presence of these will enhance, but not guarantee, the probability of GAG binding (Fig. 2A).
Where Does the GAG Bind?
Although GAGs have been viewed as binding to any basic sub-site on proteins, the above discussion suggests otherwise. At the same time, the absence of a consensus sequence or structural motif does not automatically eliminate GAG recognition. This ambiguity lends significant importance to the task of computationally identifying a true putative GAG binding site. The question is addressed in sequential steps. First, protein surface electrostatic potential (ESP) is calculated using tools such as APBS, DeepView, etc. to locate electropositive sub-site(s) (Fig. 2B). This technique has been most widely used for identification of putative GAG binding site(s) since the beginning of computational studies and continues to be employed even today [18,25,29,31,46–49]. A tacit assumption in this analysis is that most GAG–protein interactions are predominantly electrostatic in nature. However, many GAG–protein systems utilize substantial non-Coulombic forces. In fact, a recent study with a subset of proteins shows that ESP at specific non-ionic residues adjacent to a constellation of basic residues is a better predictor of GAG-binding sites [7].
A slightly more involved approach to identify GAG-binding site is the preferential recognition of a probe atom, ion or molecule. This approach takes its roots from the GRID search algorithm [50], which uses polar or charged probes, e.g., predict position of H2O to identify areas of solvent displacement upon binding [34], H2O, N of -NH2, -OH, etc., to map interactions with a protein. This method has been adopted to predict energetically favorable locations of sulfate groups (−OSO3−) on target protein surface [17,21,22]. An even more involved approach employs combination of GRID, ESP, site mapping method, small saccharide docking and MD to more accurately identify GAG binding sites [23,30,31,39,51,]. In recent years, advances in computational technology have brought about online servers that help predict GAG binding sites on protein surfaces through docking [38,39].
In principle, the computational deduction of GAG binding site should be validated in solution experiments. Examples of such studies include site-directed mutagenesis of gremlin [52] and TSG-6 [53], and NMR studies of IL-8 [54]. On the flip side, literature presents cases where congenital mutations, site-directed mutagenesis or similarity with a known protein have helped pinpoint the site of GAG binding [11,15,26,54].
Which GAG sequence is the Most Favored?
This is ‘the’ key question to answer whether a target GAG–protein system is worth detailed biochemical/biophysical studies. In fact, computational tools present ‘the only’ approach to answering this question in an exhaustive manner. The question can take several forms, e.g., which GAG (HS, HP, CS, or DS) is the most preferred?; or what is the most optimal chain length (di-, tetra-, hexa-, etc.)?; or which sequence, from among the all possible, is the most favored?
The primary tool used in this approach is molecular docking and scoring, which is fundamentally a structure-based drug design tool, wherein a sequence’s orientation in a target binding site on protein surface is identified (Fig. 2C). A protein carrying more than one GAG binding sites may have more than one most favored GAG sequences. Advances in automated docking and scoring tools coupled with library generation scripts enable screening of 1000s of individual GAG sequences, a feat nearly impossible to achieve in solution. Several groups have used docking and scoring approaches to identify the optimal length of a GAG chain [22,24]. Likewise, the site, affinity and mode of binding have also been elucidated using this approach [17,20,49,55].
We have developed a combinatorial virtual library screening (CVLS) strategy that identifies ‘high affinity & high specificity’ sequence(s) by studying a library of 1000s of GAG sequences. The key idea here is to use dual filters – ‘in silico affinity’ (GOLD score) and ‘in silico specificity’ (consistency of binding) – to separate the needles (‘high affinity & high specificity’ sequences) from the chaff (the vast majority of non-specific sequences). The strategy can take care of prefered conformations of IdoA residues, chains of different lengths, explicit enumeration of natural as well as unnatural modifications of residues, and flexibility of rotatable bonds. The first demonstration of this strategy utilized a small library of only about 7,000 HS sequences binding to AT, which correctly identified possible high affinity variants of the well-known pentasaccharide [23,26]. The most recent application used a library of ~100,000 sequences to identify the most favored HS hexasaccharides binding to HCII [11]. Solution experiments were used to confirm the predictions. The work demonstrated for the first time that it is possible to design GAG sequences de novo, which suggests its potential in discoverying GAG-based drugs. Also recently, this method was applied to identify a specific tetrasaccharide sequence to stabilize CXCL13 in its dimeric form, which was supported by solution-based experiments [56].
Although GOLD has been the primary docking tool for CVLS studies, other programs should be applicable too, as shown by several groups [2,29,39,55,57], following optimization of parameters for the library to be screened. The dual filter CVLS strategy appears to carry an advantage of being able to elucidate GAG sequences that selectively bind the target binding site with high selectivity. This is one of major considerations for GAG–protein interactions because high in silico affinity (i.e., score) may also arise from non-selective or plastic recognition. For discovery of GAG-based drugs, this is a major liability. CVLS has the capability of weaning out the inconsistent, non-specific binders. This helps minimize the burden associated with synthesizing and screening a large library of GAGs.
In addition to explicit computational studies, several groups have combined biophysical/biochemical studies with computational approaches to understand the specificity of GAG–protein systems. For example, the role of divalent cation for binding heparin and heparan sulfate to endostatin was characterized by surface plasmon resonance (SPR) and molecular modeling [58]; a carbohydrate microarray technique was combined with modeling to understand GAG recognition by growth factor proteins (FGF2/FGFR1), malarial protein VAR2CSA and tumor necrosis factor-α (TNF-α) [13]; spectrofluorimetry and NMR were combined with molecular docking and dynamics to study recognition properties of IL-8 by various GAG [59]; and SPR and GAG polarity studies were combined to understand FGF-1 recognition [60]. Although these approaches are highly valuable, a serious limitations with these studies is that of the number of available GAG sequences. It is difficult to find a library of more than a few dozen diverse GAG sequences, which leaves lingering questions regarding the specificity/non-specificity of GAG–protein system. It appears that computational approaches of the CVLS type are more suitable for exhaustive analysis of the specificity of GAG–protein systems.
Is My GAG–Protein Complex Stable?
Since the time of the earliest of MD studies [14], efficient and reliable tools have been developed to enable much larger (number of atoms) and longer timescale (ns to ms) studies to afford insight into the dynamics of GAG–protein co-complexes in water. These tools include three comprehensive force fields including GLYCAM [61], CHARMM [62], and GROMOS [63], of which GLYCAM has an extensive library of glycans [64]. These developments enable elucidation of atomistic, molecular and ensemble properties of GAG–protein systems (see Table S1) [35,65–67]. MD studies are central to assessing the thermodynamic and kinetic stability of complexes, without which inferences drawn from docking and scoring approaches alone may sometimes lead to false positives or misinterpretations. Briefly, the co-complex structure obtained following molecular docking is used to generate the initial structure in the presence of explicit water using one of the three forcefields. The system is then minimized and equilibrated to physiological conditions for MD runs.
MD offers trajectories of structural coordinates as a function of time (Fig. 2D). Although the approach can simulate most conformational changes, large scale movements, such as domain movements in proteins arising from GAG binding (e.g., 10–100 Å), may not be simulated accurately. Once the trajectories are collected in sufficient resolution (e.g., one structure every ps or more), comparison of the macromolecular backbones from the initial ensemble affords insight into stability of the bound GAG. It is necessary to assess complex stability for 50 ns or more to understand the nature of forces (Coulombic, H-bond, etc.) [35,36,67], IdoA puckering [32,65], role of water molecules [34,36,68,69], free energy contribution of individual residues/groups [2,67,68] and conformational hinge movements [36]. It is heartening to note that tools have been developed, many available in public domain, for such analysis (see Supplementary Information).
H-Bonds and Ring Puckering
Through MD analysis it is possible to observe the formation, breakdown and re-formation of bonds, such as H-bonds, in MD trajectories. This helps compute the percent occupancy of a GAG donor or acceptor atom/group onto the protein binding site (Fig. 3A). An occupancy of 100% would imply persistence of a particular H-bond throughout the simulation time, which may correspond to specificity of interaction. In contrast, partial occupancy may imply plasticity/non-specificity (Fig. 3B) [5,35,68]. Such analysis has been used to deduce plasticity in GAG binding to FGF2 [35] and IL-8 [5] H-bond analysis studies have also presented transient formation of new interactions not invoked in X-ray structures [29,33,67]. Interestingly, the transient H-bond appeared to affect IdoA/IdoA2S puckering [67], which implies conformational mobility of GAG structure upon binding.
Figure 3.

The use of MD in understanding GAG–protein interactions. Analyzes of MD trajectories over a timescale of few picosec to hundreds of microsec affords a wealth of thermodynamic and kinetic information on GAG–protein co-complexes. Shown are results for an exemplary system, heparin octasaccharide (HS08) binding to CXCL5 in water (A-F). A) The observed intermolecular hydrogen bonds (H-bonds, broken black lines) between donors and acceptor atoms of GAG and side chains of amino acids at a given time frame. B) Percent occupancy of H-bonds between key amino acid residues with HS08 is shown (higher (red) to lower (blue)). C) A representative MD frame showing the bed of heterogeneous water molecules surrounding interacting regions. The distribution of water engineers GAG–water, water–water, protein–water, protein–GAG–water H-bonds. Water molecules are represented as red spheres and interactions are shown as faint black dashed lines. D) Significant number of interactions arise from water mediated H-bonds (i.e., not direct) between GAG and protein, as shown. E) Shown is the overall occupancy of water mediated H-bond interactions between GAG and protein. F) Single residue energy decomposition (SRED) of interacting amino acids in the co-complex as deduced by MM-PBSA/MM-GBSA method.
The Role of Water
MD simulations show radial distribution of water molecules around their polar atoms of GAGs in co-complexes [69], which implies that MD in a box of water presents a way of teasing out the role of water in GAG–protein interactions (Fig. 3C). In fact, a recent work shows that about half of GAG–protein interactions are mediated by water molecules, which may be bridging and non-bridging entities [68]. More importantly, studies on cathepsin K, CD44 and CXCL5 show that bridging as well as non-bridging interactions are important for GAG recognition (Fig. 3D) [34,68,69].
Free Energy of Binding
The MD trajectories also allow deduction of free energy of binding by averaging molecular mechanical contributions from bonded, non-bonded, and solvent interactions by using Poisson-Boltzmann (PBSA)/Generalized Born methods (GBSA). The first application of this technique to PECAM-1 and annexin A2 showed the presence of high and low affinity heparin-binding sites and explained affinity increases with chain length [36]. Further studies using DMD, a targeted MD simulation strategy, demonstrated good validation with the experimental observations [29,31,33]. Because the molecular mechanics calculations are pairwise additive functions, individual residue wise contributions can be easily computed. This has yielded insight into identifying major points of GAG recognition (Fig. 3F) [29,31,33]. Such information could be useful for rationally deducing sites for mutagenesis and functional analysis.
Changes in free energy on GAG binding to a protein could also be calculated by linear interaction energy (LIE) approximation method, which is the energy difference between the bound and free states of GAG. When used for hyaluronan binding to A and B forms of CD44, the LIE method gave good correlation agreement with the solution data [70]. Finally, the free energy difference approaches such as free energy of perturbation (FEP) [71] and thermodynamics integration (TI) [2] are particularly useful to understand the effect of single site mutations (Arg to Lys) or chemical modifications (SO3 to OH in GAG residue) in structure function relationship. These are typically far more challenging to do in solution.
Limitations, Challenges and Validation
Despite the advances and successes, it is important to appreciate the limitations of computational approaches. Parameters for sulfate groups in various micro-environments continue to be updated and this has direct implications for all docking and dynamics experiments [64]. Likewise, although IdoA exhibits multiple conformations [32,72], it is typically simulated in two conformations only (1C4 and 2SO) [11,23,55,72]. Challenges also exist in computationally handling the number of rotatable bonds in an oligosaccharide. This has limited most studies to chains shorter than hexasaccharides [24,31]. On the dynamics front, it has been challenging to study large systems in explicit solvent, e.g., full-length GAG, and approximations, such as coarse grain models [73], have had to be implemented. This implies that validation of computational results through solution-based experiments is important and essential. Although generally computational studies have attempted direct correspondence with solution results [5,31,35,67,68], many derivations remain predictions at the present time. This implies that the current state-of-art still leaves room for some computational predictions to turn out to be false positives or negatives. Another challenge is that despite advances in prediction algorithms, pose accuracy is not very good. Because a large number of systems rely primarily on electrostatic interactions, a multitude of poses are predicted by most computational approaches. In this connection, the CVLS algorithm appears to be especially useful in segregating specific systems, i.e., those with high level of consistency among poses, from non-specific system, i.e., those with poor pose consistency [11,23,24].
Future Inquiries
The liabilities should not deter non-computational researchers from undertaking computational studies, while these should embolden computational researchers’ the resolve to develop a more exact science. In fact, the field is at the doorstep of number of major advances in terms of biological insights. For example, computational approaches are likely to be the only means of understanding why nature has engineered such phenomenal diversity in GAGs? Beside the advantage of economy, it is highly likely that diversity affords much higher selectivity of protein recognition on a needs-basis than developing an evolutionary system geared to specific recognition of a particular set of proteins. It is also highly likely that non-selectivity, or plasticity, of protein recognition is a dominant component of GAG biology. Parsing these mechanisms at systems biology and biochemical/biophysical levels, difficult in solution-based experiments, is particularly suited at computational level. On the drug discovery front, a major area of inquiry is the computational design of GAG-based drugs. Theoretically, most GAG–protein systems should be ‘druggable’. However, other than AT–heparin system [15,65], none have been identified. Recent computational studies show that HCII–HS system appears to be a druggable system [11]. We predict that screening the library of natural and unnatural GAG sequences against a host of target proteins should afford many ‘high-specificity’ druggable systems. Finally, on the structural biology front, computational approaches will be used to understand larger ternary, quaternary and higher order systems in which GAG plays a key role. Such studies would hopefully help understand the role of two or more juxtaposed GAG chains on most proteoglycans in driving biology.
Supplementary Material
Highlights.
The state-of-the-art in computational approaches to understand GAG–proteins interactions is presented
The focus is to help non-computational researchers undertake computational studies
Computational tools can answer questions on the site and nature of GAG binding
It is possible to address whether a GAG–protein system is ‘specific’ and/or ‘non-specific’
Future tools will address structural biology of higher order GAG–protein complexes and design of GAG-based drugs
Acknowledgments
Funding
This work was funded by grants HL107152, HL090586 and HL128639 from the National Institutes of Health to URD.
Footnotes
- The authors have no conflicts of interest to declare
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balagurunathan K, Nakato H, Desai UR. Glycosaminoglycans. Chemistry and Biology. Methods Mol Biol. 2015;1229:1–625. doi: 10.1007/978-1-4939-1714-3. [DOI] [PubMed] [Google Scholar]
- 2.Babik S, Samsonov SA, Pisabarro MT. Computational drill down on FGF1-heparin interactions through methodological evaluation. Glycoconj J. 2017;34:427–440. doi: 10.1007/s10719-016-9745-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu D, Esko JD. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 2014;83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulloy B, Hogwood J, Gray E, Lever R, Page CP. Pharmacology of Heparin and Related Drugs. Pharmacol Rev. 2016;68:76–141. doi: 10.1124/pr.115.011247. [DOI] [PubMed] [Google Scholar]
- 5**.Joseph PR, Mosier PD, Desai UR, Rajarathnam K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem J. 2015;472:121–133. doi: 10.1042/BJ20150059. The paper presents a good combination of biophysical and computational experiments to understand the nature of interactions in monomer–dimer equilibrium. MD studies show that the GAG binding is plastic and pinpointed residues contributing to the dynamic nature of the process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosier PD, Krishnasamy C, Kellogg GE, Desai UR. On the specificity of heparin/heparan sulfate binding to proteins. Anion-binding sites on antithrombin and thrombin are fundamentally different. PLoS One. 2012;7:e48632. doi: 10.1371/journal.pone.0048632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Sarkar A, Desai UR. A simple method for discovering druggable, specific glycosaminoglycan-protein systems. Elucidation of key principles from heparin/heparan sulfate-binding proteins. PLoS One. 2015;10:e0141127. doi: 10.1371/journal.pone.0141127. The work shows that a unique constellation of non-ionic hydrogen-bonding residues among strongly electropositive residues in certain proteins results in highly specific interaction with heparin/heparan sulfate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghiselli G. Drug mediated regulation of glycosaminoglycan biosynthesis. Med Res Rev. 2017;37:1051–1094. doi: 10.1002/med.21429. [DOI] [PubMed] [Google Scholar]
- 9.Kreuger J, Kjellen L. Heparan sulfate biosynthesis: regulation and variability. J Histochem Cytochem. 2012;60:898–907. doi: 10.1369/0022155412464972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pomin VH, Mulloy B. Current structural biology of the heparin interactome. Curr Opin Struct Biol. 2015;34:17–25. doi: 10.1016/j.sbi.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 11**.Sankaranarayanan NV, Strebel TR, Boothello RS, Sheerin K, Raghuraman A, Sallas F, Mosier PD, Watermeyer ND, Oscarson S, Desai UR. A hexasaccharide containing rare 2-O-sulfate-glucuronic acid residues selectively activates heparin cofactor II. Angew Chem Int Ed. 2017;56:2312–2317. doi: 10.1002/anie.201609541. The paper reports the first screening of ~50,000 HS sequences against a protein target. A rare hexasaccharide was identified, synthesized and shown to be selective for heparin cofactor II in experimental studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 13.Rogers CJ, Clark PM, Tully SE, Abrol R, Garcia KC, Goddard WA, Hsieh-Wilson LC. Elucidating glycosaminoglycan–protein–protein interactions using carbohydrate microarray and computational approaches. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9747–9752. doi: 10.1073/pnas.1102962108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arterioscler Thromb Vasc Biol. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 15.Grootenhuis PDJ, Van Boeckel CAA. Constructing a molecular model of the interaction between antithrombin III and a potent heparin analog. J Am Chem Soc. 1991;113:2743–2747. [Google Scholar]
- 16.Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci. 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bitomsky W, Wade RC. Docking of glycosaminoglycans to heparin-binding proteins: Validation for aFGF, bFGF, and antithrombin and application to IL-8. J Am Chem Soc. 1999;121:3004–3013. [Google Scholar]
- 18.Stuckey JA, St Charles R, Edwards BF. A model of the platelet factor 4 complex with heparin. Proteins. 1992;14:277–287. doi: 10.1002/prot.340140213. [DOI] [PubMed] [Google Scholar]
- 19.Forster M, Mulloy B. Computational approaches to the identification of heparin-binding sites on the surfaces of proteins. Biochem Soc Trans. 2006;34:431–434. doi: 10.1042/BST0340431. [DOI] [PubMed] [Google Scholar]
- 20.Gandhi NS, Coombe DR, Mancera RL. Platelet endothelial cell adhesion molecule 1 (PECAM-1) and its interactions with glycosaminoglycans: 1. Molecular modeling studies. Biochemistry. 2008;47:4851–4862. doi: 10.1021/bi702455e. [DOI] [PubMed] [Google Scholar]
- 21.Lortat-Jacob H, Grosdidier A, Imberty A. Structural diversity of heparan sulfate binding domains in chemokines. Proc Natl Acad Sci. 2002;99:1229–1234. doi: 10.1073/pnas.032497699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sadir R, Baleux F, Grosdidier A, Imberty A, Lortat-Jacob H. Characterization of the stromal cell-derived factor-1α-heparin complex. J Biol Chem. 2001;276:8288–8296. doi: 10.1074/jbc.M008110200. [DOI] [PubMed] [Google Scholar]
- 23.Raghuraman A, Mosier PD, Desai UR. Finding a needle in a haystack: development of a combinatorial virtual screening approach for identifying high specificity heparin/heparan sulfate sequence(s) J Med Chem. 2006;49:3553–3562. doi: 10.1021/jm060092o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sankaranarayanan NV, Desai UR. Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology. 2014;24:1323–1333. doi: 10.1093/glycob/cwu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lam K, Rao VS, Qasba PK. Molecular modeling studies on binding of bFGF to heparin and its receptor FGFR1. J Biomol Struct Dyn. 1998;15:1009–1027. doi: 10.1080/07391102.1998.10508997. [DOI] [PubMed] [Google Scholar]
- 26.Raghuraman A, Mosier PD, Desai UR. Understanding dermatan sulfate-heparin cofactor II interaction through virtual library screening. ACS Med Chem Lett. 2010;1:281–285. doi: 10.1021/ml100048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ragazzi M, Ferro DR. Toward a realistic force field for the treatment of ionic sugars. J Mol Struct Theochem. 1997;395:107–122. [Google Scholar]
- 28.Huige CJM, Altona C. Force field parameters for sulfates and sulfamates based on ab initio calculations: Extensions of AMBER and CHARMm fields. J Comput Chem. 1995;16:56–79. [Google Scholar]
- 29.Samsonov SA, Pisabarro MT. Computational analysis of interactions in structurally available protein-glycosaminoglycan complexes. Glycobiology. 2016;26:850–861. doi: 10.1093/glycob/cww055. [DOI] [PubMed] [Google Scholar]
- 30.Agostino M, Gandhi NS, Mancera RL. Development and application of site mapping methods for the design of glycosaminoglycans. Glycobiology. 2014;24:840–851. doi: 10.1093/glycob/cwu045. [DOI] [PubMed] [Google Scholar]
- 31.Gehrcke JP, Pisabarro MT. Identification and characterization of a glycosaminoglycan binding site on interleukin-10 via molecular simulation methods. J Mol Graph Model. 2015;62:97–104. doi: 10.1016/j.jmgm.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Samsonov SA, Pisabarro MT. Importance of IdoA and IdoA(2S) ring conformations in computational studies of glycosaminoglycan-protein interactions. Carbohydr Res. 2013;381:133–137. doi: 10.1016/j.carres.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 33**.Samsonov SA, Gehrcke JP, Pisabarro MT. Flexibility and explicit solvent in molecular-dynamics-based docking of protein-glycosaminoglycan systems. J Chem Inf Model. 2014;54:582–592. doi: 10.1021/ci4006047. A novel dynamic molecular docking (DMD) method was implemented to understand the GAG – protein interactions. The method was validated with set of complexes and the observed results were compared to docking results. A high level of predictability was observed for the method with good correlation to experimental observations. [DOI] [PubMed] [Google Scholar]
- 34.Samsonov SA, Teyra J, Pisabarro MT. Docking glycosaminoglycans to proteins: analysis of solvent inclusion. J Comput Aided Mol Des. 2011;25:477–489. doi: 10.1007/s10822-011-9433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Sapay N, Cabannes E, Petitou M, Imberty A. Molecular modeling of the interaction between heparan sulfate and cellular growth factors: bringing pieces together. Glycobiology. 2011;21:1181–1193. doi: 10.1093/glycob/cwr052. Exhaustive sampling of GAG binding to growth factors was performed using different protocols. The experiments were successfully implemented for FGF2 and CXCL12α. Results showed good favorability for hexasaccharide binding to classical dimeric form of CXCL12α. [DOI] [PubMed] [Google Scholar]
- 36**.Gandhi NS, Mancera RL. Free energy calculations of glycosaminoglycan-protein interactions. Glycobiology. 2009;19:1103–1115. doi: 10.1093/glycob/cwp101. The MMPBSA/MMGBSA binding free energy calculations were implemented for the first time in protein-GAG complex systems. Observations showed increase in affinity with increase in GAG chain length. [DOI] [PubMed] [Google Scholar]
- 37.Sankaranarayanan NV, Sarkar A, Desai UR, Mosier PD. Designing “high-affinity, high-specificity” glycosaminoglycan sequences through computerized modeling. Methods Mol Biol. 2015;1229:289–314. doi: 10.1007/978-1-4939-1714-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mottarella SE, Beglov D, Beglova N, Nugent MA, Kozakov D, Vajda S. Docking server for the identification of heparin binding sites on proteins. J Chem Inf Model. 2014;54:2068–2078. doi: 10.1021/ci500115j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12:255–278. doi: 10.1038/nprot.2016.169. Online server, which is available to any user, helps identify putative heparin binding site(s) for a target protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sobel M, Soler DF, Kermode JC, Harris RB. Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J Biol Chem. 1992;267:8857–8862. [PubMed] [Google Scholar]
- 41.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 42.Johnson Z, Power CA, Weiss C, Rintelen F, Ji H, Ruckle T, Camps M, Wells TN, Schwarz MK, Proudfoot AE, et al. Chemokine inhibition – why, when, where, which and how? Biochem Soc Trans. 2004;32:366–377. doi: 10.1042/bst0320366. [DOI] [PubMed] [Google Scholar]
- 43.Torrent M, Nogues MV, Andreu D, Boix E. The “CPC clip motif”: a conserved structural signature for heparin-binding proteins. PLoS One. 2012;7:e42692. doi: 10.1371/journal.pone.0042692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng YY, Cheng CS, Lee TR, Chang WS, Lyu PC. A clamp-like orientation of basic residues set in a parallelogram is essential for heparin binding. FEBS Lett. 2016;590:3089–3097. doi: 10.1002/1873-3468.12361. [DOI] [PubMed] [Google Scholar]
- 45.Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J Biol Chem. 1993;268:19228–19231. [PubMed] [Google Scholar]
- 46.Kuhn LA, Griffin JH, Fisher CL, Greengard JS, Bouma BN, Espana F, Tainer JA. Elucidating the structural chemistry of glycosaminoglycan recognition by protein C inhibitor. Proc Natl Acad Sci. 1990;87:8506–8510. doi: 10.1073/pnas.87.21.8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alberdi E, Hyde CC, Becerra SP. Pigment epithelium-derived factor (PEDF) binds to glycosaminoglycans: analysis of the binding site. Biochemistry. 1998;37:10643–10652. doi: 10.1021/bi9802317. [DOI] [PubMed] [Google Scholar]
- 48.Rastegar-Lari G, Villoutreix BO, Ribba AS, Legendre P, Meyer D, Baruch D. Two clusters of charged residues located in the electropositive face of the von Willebrand factor A1 domain are essential for heparin binding. Biochemistry. 2002;41:6668–6678. doi: 10.1021/bi020044f. [DOI] [PubMed] [Google Scholar]
- 49.Ricard-Blum S, Feraud O, Lortat-Jacob H, Rencurosi A, Fukai N, Dkhissi F, Vittet D, Imberty A, Olsen BR, van der Rest M. Characterization of endostatin binding to heparin and heparan sulfate by surface plasmon resonance and molecular modeling: role of divalent cations. J Biol Chem. 2004;279:2927–2936. doi: 10.1074/jbc.M309868200. [DOI] [PubMed] [Google Scholar]
- 50.Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 51.Sage J, Mallevre F, Barbarin-Costes F, Samsonov SA, Gehrcke JP, Pisabarro MT, Perrier E, Schnebert S, Roget A, Livache T, et al. Binding of chondroitin 4-sulfate to cathepsin S regulates its enzymatic activity. Biochemistry. 2013;52:6487–6498. doi: 10.1021/bi400925g. [DOI] [PubMed] [Google Scholar]
- 52.Tatsinkam AJ, Mulloy B, Rider CC. Mapping the heparin-binding site of the BMP antagonist gremlin by site-directed mutagenesis based on predictive modelling. Biochem J. 2015;470:53–64. doi: 10.1042/BJ20150228. [DOI] [PubMed] [Google Scholar]
- 53.Mahoney DJ, Mulloy B, Forster MJ, Blundell CD, Fries E, Milner CM, Day AJ. Characterization of the interaction between tumor necrosis factor-stimulated gene-6 and heparin: Implications for the inhibition of plasmin in extracellular matrix microenvironments. J Biol Chem. 2005;280:27044–27055. doi: 10.1074/jbc.M502068200. [DOI] [PubMed] [Google Scholar]
- 54.Mobius K, Nordsieck K, Pichert A, Samsonov SA, Thomas L, Schiller J, Kalkhof S, Teresa Pisabarro M, Beck-Sickinger AG, Huster D. Investigation of lysine side chain interactions of interleukin-8 with heparin and other glycosaminoglycans studied by a methylation-NMR approach. Glycobiology. 2013;23:1260–1269. doi: 10.1093/glycob/cwt062. [DOI] [PubMed] [Google Scholar]
- 55.Mulloy B, Forster MJ. Application of drug discovery software to the identification of heparin-binding sites on protein surfaces: a computational survey of the 4-helix cytokines. Mol Simulation. 2008;34:481–489. [Google Scholar]
- 56.Monneau YR, Luo L, Sankaranarayanan NV, Nagarajan B, Vives RR, Baleux F, Desai UR, Arenzana-Seidedos F, Lortat-Jacob H. Solution structure of CXCL13 and heparan sulfate binding show that GAG binding site and cellular signalling rely on distinct domains. Open Biol. 2017;7:170133. doi: 10.1098/rsob.170133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh A, Kett WC, Severin IC, Agyekum I, Duan J, Amster IJ, Proudfoot AE, Coombe DR, Woods RJ. The Interaction of Heparin Tetrasaccharides with Chemokine CCL5 Is Modulated by Sulfation Pattern and pH. J Biol Chem. 2015;290:15421–15436. doi: 10.1074/jbc.M115.655845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ricard-Blum S, Feraud O, Lortat-Jacob H, Rencurosi A, Fukai N, Dkhissi F, Vittet D, Imberty A, Olsen BR, van der Rest M. Characterization of endostatin binding to heparin and heparan sulfate by surface plasmon resonance and molecular modeling: role of divalent cations. J Biol Chem. 2004;279:2927–2936. doi: 10.1074/jbc.M309868200. [DOI] [PubMed] [Google Scholar]
- 59.Pichert A, Samsonov SA, Theisgen S, Thomas L, Baumann L, Schiller J, Beck-Sickinger AG, Huster D, Pisabarro MT. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology. 2012;22:134–145. doi: 10.1093/glycob/cwr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Munoz-Garcia JC, Garcia-Jimenez MJ, Carrero P, Canales A, Jimenez-Barbero J, Martin-Lomas M, Imberty A, de Paz JL, Angulo J, Lortat-Jacob H, et al. Importance of the polarity of the glycosaminoglycan chain on the interaction with FGF-1. Glycobiology. 2014;24:1004–1009. doi: 10.1093/glycob/cwu071. [DOI] [PubMed] [Google Scholar]
- 61.Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mallajosyula SS, Guvench O, Hatcher E, Mackerell AD., Jr CHARMM additive all-atom force field for phosphate and sulfate linked to carbohydrates. J Chem Theory Comput. 2012;8:759–776. doi: 10.1021/ct200792v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pol-Fachin L, Rusu VH, Verli H, Lins RD. GROMOS 53A6GLYC, an improved GROMOS force field for hexopyranose-based carbohydrates. J Chem Theory Comput. 2012;8:4681–4690. doi: 10.1021/ct300479h. [DOI] [PubMed] [Google Scholar]
- 64*.Singh A, Tessier MB, Pederson K, Wang X, Venot AP, Boons GJ, Prestegard JH, Woods RJ. Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can J Chem. 2016;94:927–935. doi: 10.1139/cjc-2015-0606. New parameter set for different functional groups present in GAGs was developed. Simulations were performed with sulfated GAG disaccharides containing ΔUA residues. These new force field parameters were able to reproduce the solution NMR data for a majority of sequences studied. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verli H, Guimaraes JA. Insights into the induced fit mechanism in antithrombin-heparin interaction using molecular dynamics simulations. J Mol Graph Model. 2005;24:203–212. doi: 10.1016/j.jmgm.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Gandhi NS, Mancera RL. Molecular dynamics simulations of CXCL-8 and its interactions with a receptor peptide, heparin fragments, and sulfated linked cyclitols. J Chem Inf Model. 2011;51:335–358. doi: 10.1021/ci1003366. [DOI] [PubMed] [Google Scholar]
- 67.Nagarajan B, Sankaranarayanan NV, Patel BB, Desai UR. A molecular dynamics-based algorithm for evaluating the glycosaminoglycan mimicking potential of synthetic, homogenous, sulfated small molecules. PLoS One. 2017;12:e0171619. doi: 10.1371/journal.pone.0171619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sepuru KM, Nagarajan B, Desai UR, Rajarathnam K. Molecular basis of chemokine CXCL5-glycosaminoglycan interactions. J Biol Chem. 2016;291:20539–20550. doi: 10.1074/jbc.M116.745265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jana M, Bandyopadhyay S. Conformational flexibility of a protein-carbohydrate complex and the structure and ordering of surrounding water. Phys Chem Chem Phys. 2012;14:6628–6638. doi: 10.1039/c2cp24104h. The studies demonstrate the presence of heterogeneous water molecules present around GAGs in complexes with proteins. These water molecules demonstrate restricted motion when compared to bulk water. The paper presents the concept that these water molecules play important roles. [DOI] [PubMed] [Google Scholar]
- 70.Plazinski W, Knys-Dzieciuch A. Interactions between CD44 protein and hyaluronan: insights from the computational study. Mol Biosyst. 2012;8:543–547. doi: 10.1039/c2mb05399c. [DOI] [PubMed] [Google Scholar]
- 71.Sarkar A, Yu W, Desai UR, MacKerell AD, Mosier PD. Estimating glycosaminoglycan-protein interaction affinity: water dominates the specific antithrombin-heparin interaction. Glycobiology. 2016;26:1041–1047. doi: 10.1093/glycob/cww073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sattelle BM, Hansen SU, Gardiner J, Almond A. Free energy landscapes of iduronic acid and related monosaccharides. J Am Chem Soc. 2010;132:13132–13134. doi: 10.1021/ja1054143. [DOI] [PubMed] [Google Scholar]
- 73.Sattelle BM, Shakeri J, Cliff MJ, Almond A. Proteoglycans and their heterogeneous glycosaminoglycans at the atomic scale. Biomacromolecules. 2015;16:951–961. doi: 10.1021/bm5018386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.